

SUMMARY
Signaling through classical death receptor Fas was mainly appreciated as a pro-death pathway until recent reports characterized pro-inflammatory outcomes of Fas-mediated activation in pathological contexts. How Fas signaling can switch to pro-inflammatory activation is poorly understood. Herein, we report that in macrophages and neutrophils, the Toll-like receptor (TLR) adapter CD14 determines the inflammatory output of Fas-mediated signaling. Our findings propose CD14 as a crucial chaperone of Fas receptor internalization in macrophages and neutrophils, resulting in Cd14−/− myeloid cells that are protected from FasL-induced apoptosis, activate nuclear factor κB (NF-κB), and release cytokines in response. As in TLR signaling, CD14 is also required for Fas to signal through the adaptor TRIF (TIR-domain-containing adapter-inducing interferon-β) and induce a pro-death complex. Our findings demonstrate that CD14 availability can determine the switch between Fas-mediated pro-death and pro-inflammatory outcomes by internalizing the receptor.
In brief
Activation of the Fas receptor can be pro-death or pro-inflammatory, but little is known about the switch between outcomes. Magri et al. demonstrate that TLR adaptor CD14 can mediate this switch between death and inflammation in myeloid cells by internalizing the Fas receptor to a pro-death complex and preventing inflammation.
Graphical Abstract
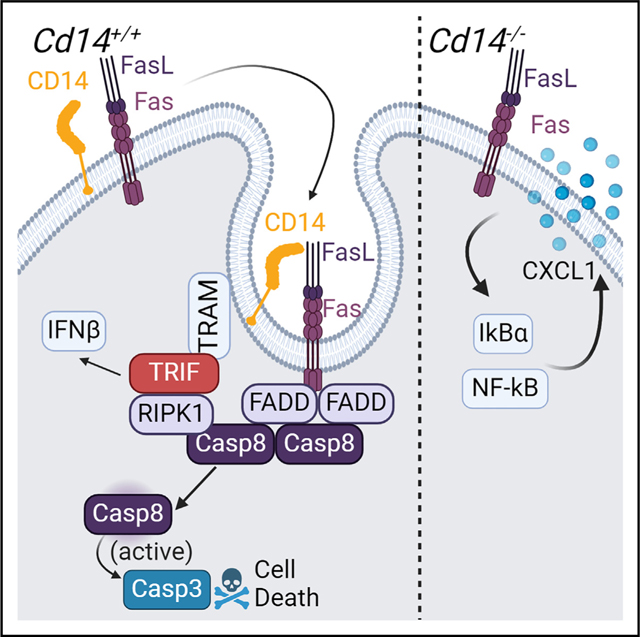
INTRODUCTION
The Fas receptor (Fas, FasR, CD95)-mediated signaling is traditionally regarded as an immunologically silent pro-death pathway. The apoptotic function of this receptor is crucial in the elimination of self-reactive or infected immune cells, maintaining immune-privileged tissues, and resolving inflammation.1–9 While Fas is viewed as a classic death receptor, recent studies broadened its function in various cell types and contexts. For example, activating Fas has been observed to induce interleukin (IL)-1β production in myeloid cells, chemotactic effects in the tumor environment, and inflammation in the context of viral infection and cardiovascular disease.10–17 Upon binding trimerized FasL, Fas clusters into lipid rafts on the cell surface and is internalized into endosomes, which is required for apoptotic signaling through Fas.18–21 This recruitment facilitates the oligomerization of FADD (Fas-associated protein with death domain), caspase-8 (CASP8), and maturation of CASP8,19 which further activates the executioners CASP3 and CASP7, leading to DNA fragmentation and apoptosis. Internalization of the Fas receptor is required for an immunologically “silent” cell death,22–24 which otherwise switches to pro-inflammatory activation of mitogen-activated protein kinase (MAPK) and nuclear factor κB (NF-κB) pathways if interrupted.19 Previous publications have identified pro-inflammatory complexes induced by Fas, but how this change of fate is regulated is unclear.10,13,25 How Fas internalization shifts the outcome of Fas-mediated activation toward cell death is poorly understood thus far and alludes to additional upstream regulators of the pathway.
In this regard, we recently reported that endosomal adaptor TRIF (TIR-domain-containing adapter-inducing interferon-β), characterized exclusively downstream of Toll-like receptors 3 and 4 (TLR3/4), is indispensable in activation of tumor necrosis factor receptor (TNFR).26 Initially shown to be required for the CASP8-dependent pro-death complex downstream of TLR4 in the context of Yersina pseudotuberculosis infection, TRIF was later reported to facilitate binding of RIPK1 (receptor interacting protein kinase 1) to FADD and CASP8, promoting the homodimerization and activation of CASP8.27–29 This complex has been described as the “TRIFosome” and is crucial to induce pyroptotic death in macrophages during Yersina infection.30 As TNFR-mediated cell death also requires activation of CASP8 through a similar complex, further investigations have pursued a role of TRIF in TNFR-mediated cell death to demonstrate that, indeed, TRIF is required for TNFR signaling in macrophages and neutrophils by facilitating the TRIFosome complex.26 Additionally, Fas-mediated death has been shown to require a death complex with similar components, including FADD, RIPK1, and CASP8.31–34 Given the similarity of TNFR- and Fas-mediated assembly of this pro-death complex, we hypothesized that TRIF may contribute to the Fas-mediated signaling.26,34
The lipopolysaccharide (LPS) coreceptor CD14 has been shown to facilitate TRIF-dependent assembly of the TRIFosome downstream of TNFR,9,10 thereby expanding the well-characterized role of CD14 in the internalization of TLRs into the endosomal compartment, allowing signaling through TRIF.35,36 Given these considerations, CD14 emerged as an attractive candidate for mediating Fas signaling, as both proteins aggregate in lipid rafts and promote signaling via trafficking to the endosome. As CD14 is well known for chaperoning several TLR receptors from the membrane to the endosomal compartment, we examined the role of CD14 in mediating Fas-dependent responses.35,37–39 Our findings present CD14 as a potential decision-maker between death and inflammation in Fas signaling, thus providing a more satisfying explanation for the hyperinflammatory phenotype of surface-bound Fas signaling and the context-dependent switch to inflammation.
RESULTS
TRIF is required for maximum Fas-mediated cell death in macrophages
We previously reported that TRIF interacts with RIPK1 to promote assembly of the pro-death complex downstream of TNFR.26 Given the similarity of the activation pathways downstream of TNFR1 and Fas and the requirement for RIPK1 in Fas-mediated activation and cell death complex assembly, we looked at the role of TRIF in macrophages activated with FasL.31–34 We mimicked ligand-induced lipid raft clustering and activation of the receptor by membrane-bound FasL by using multimeric Fas ligand (mFasL or FasL), a trimerized, collagen-bound form of FasL.10,40,41 Accordingly, we observed TRIF-dependent cell death in FasL-treated mouse neutrophils, peritoneal macrophages, and bone marrow-derived macrophages (BMDMs) (Figures 1A–1C). Neutrophils exhibited a subtle dependency on TRIF, likely due to lower expressions of TRIF in this cell type compared to macrophages (Figure S1A). In further support of the TRIF-dependent cell death, Trif−/− macrophages had attenuated cleavage and activation of CASP8 and CASP7 (Figure 1D). Given the role of TRIF as a known mediator of type I interferon (IFN-I) production and to dismiss the possibility of TRIF-mediated “tonic IFN” conferring the differences in viability, wild-type (WT; C57BL/6 [B6]) and Trif−/− macrophages were primed overnight with IFN-β at a low dose to mimic tonic IFN and then stimulated with FasL. Inducing this tonic IFN did not rescue the Trif−/− response to FasL (Figure 1E). These data establish a role of TRIF in activation of Fas-mediated cell death.
Figure 1. TRIF is required for maximum Fas-mediated cell death in macrophages.

(A–C) Cell death over time in FasL (100 ng/mL)-stimulated primary murine neutrophils, peritoneal macrophages, and bone marrow-derived macrophages (BMDMs) isolated from indicated genotypes as measured by propidium iodide incorporation over time. See also Figure S1.
(D) Level of cleaved, active form of death caspases-8 and -7 over time in B6 and Trif−/− macrophages after FasL stimulation.
(E) Cell death over time in BMDMs isolated from indicated genotypes as measured by propidium iodide incorporation over time. BMDMs were stimulated with low dose (5 IU) IFN-β overnight to mimic tonic interferon signaling.
Data from cell death assays and western blots are representative of three or more independent experiments, and cell death data are presented as the means ± SD of triplicate wells (~5,000 cells per field of view [FOV]).
TRIF requires CD14 to access the Fas-mediated death complex
In the context of TLR4-mediated signaling, CD14 traffics the receptor to the endosome to promote activation of the TRIF-dependent complex. To test whether CD14 plays a similar role in the context of Fas-mediated signaling, we confirmed Fas-mediated colocalization of TRIF and RIPK1 (Figures 2A and S1B), which was dependent on CD14. While the role of RIPK1 in the Fas death complex has been established,31–34 we report RIPK1’s association with TRIF and dependency on CD14 downstream of Fas. Similar to the TNFR-mediated context, Fas stimulation promoted both oligomerized TRIF puncta, a hallmark of TRIF activation, and colocalization of those puncta with RIPK1 (Figures 2B and 2C).26 In contrast, no TRIF puncta were observed in Cd14−/− macrophages (Figures 2A–2C), which suggested that CD14 promoted the interaction of Fas with RIPK1 and TRIF. Trif−/− macrophages, initially included as a negative staining control, also showed no RIPK1 puncta, suggesting that TRIF is needed for RIPK1 activation (Figure 2A). The requirement for RIPK1 in Fas signaling varies with cell type, so we confirmed that RIPK1 and RIPK1 shuttle protein ZBP1 were required for maximum Fas-mediated cell death in primary murine macrophages. As expected, Fas-mediated cytotoxicity and CASP8 activation were significantly attenuated in Zbp1−/− and kinase-inactive RIP1 cells (Figures S1C and S1F).13,31,37–39 In further support of a TRIF-RIP1 complex, RIP1 cleavage was attenuated in Trif−/− macrophages, and other TRIF-dependent switches in cell death pathways were observed (Figure S1D). Likely due to decreased CASP8 activation, the cell death mode in Trif−/− macrophages became necroptotic as evidenced by the phosphorylation of MLKL (Figure S1D) in FasL-treated Trif−/− macrophages. Similarly, the CASP3/7 cleavage product of gasdermin D (GSDMD), p20, was absent in Trif−/− macrophages (Figure S1D).
Figure 2. TRIF requires CD14 to access the Fas-mediated death complex.

(A) Representative 40× images of TRIF or RIPK1 staining and colocalization in permeabilized macrophages of indicated genotypes stimulated with mFasL for 1 h.
(B and C) Quantification of total TRIF puncta (B) and TRIF puncta colocalized with RIP1K puncta (C). Each data point represents a FOV across 3 technical replicates, 5 FOVs per replicate, presented with mean ± SD.
(D) FADD-specific immunoprecipitation in B6 or Cd14−/− macrophages stimulated with mFasL for indicated timepoints and probed for death complex components.
(E–G) Cell death over time in primary murine neutrophils, peritoneal macrophages, and BMDMs isolated from indicated genotypes as measured by propidium iodide incorporation over time.
(H) Level of cleaved, active form of death caspases-8, -3, and -7 over time in B6 and Cd14−/− macrophages after FasL stimulation. See also Figure S1.
Data from cell death assays and western blotting are representative of three or more independent experiments, and cell death data are presented as the means ± SD of triplicate wells All blots from a specific image are run from a single set of lysates that were identically handled. Data from imaging experiments are representative of three or more independent experiments, and data points indicate FOVs, error bars indicate ± SD, n = 15 (~20 cells/FOV). ANOVA was used for comparison between groups: ns (p > 0.05), **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To further investigate the assembly of a pro-death complex downstream of Fas, we used antibodies to FADD to precipitate the death complex and used TRIF-adaptor protein TRAM as a proxy for TRIF because antibodies to TRIF are not available commercially. We observed RIPK1, the active form of CASP8, the Fas receptor, and TRAM in FADD-specific immunoprecipitate, which were all severely abrogated in CD14-deficient macrophages (Figure 2D). To directly test for RIPK1-Fas interactions, we identified the Fas receptor (and FADD) in RIPK1-specific immunoprecipitates (Figure S1E). While these immunoprecipitations suggested that CD14 promotes a Fas-mediated death complex in myeloid cells, a canonical CD14-independent mechanism of Fas-induced death was likely intact. The extent of CASP8 activation was assessed by detection of the p18 cleavage product of CASP8 and binding of TRIF via detection of TRAM. While quantities of TRAM in the FADD-specific lysate increased, levels of TRAM in the input control lysate decreased, suggesting that the majority of TRAM is in incorporated into the Fas death complex (Figure 2D). Mechanistically, CD14-dependent binding of TRAM to FADD positioned CD14 upstream of TRIF. Taken together, these results show that the incorporation of TRIF (TRAM) into the death complex was severely decreased in Cd14−/− macrophages, supporting a model in which CD14 internalizes the Fas receptor to an endosomal TRIF complex.
Given that for its activation, TRIF relies on CD14-dependent translocation of TLRs to the endosome,42 we hypothesized that a similar requirement for CD14 holds true in the context of Fas-mediated signaling. Such a role for CD14 was further supported by CD14-dependent activation of cell death downstream of TNFR.26 We chose to test this hypothesis in myeloid cells, as they are typically CD14-hi and can induce a pro-death or inflammatory response in certain contexts according to previous reports.10,12–17 Accordingly, treatment of peritoneal neutrophils and macrophages with FasL rapidly induced apoptosis in the WT but to a much lesser extent in Cd14−/− cells (Figures 2E–2G). In agreement with the delayed kinetics of cell death, both peritoneal macrophages and BMDMs from Cd14−/− mice exhibited delayed cleavage and activation of CASP8 and apoptotic executioners CASP3 and CASP7 (Figure 2H).
FasL intravenous injection induces inflammatory responses in Cd14−/− mice
To assess the role for CD14 Fas signaling in vivo, we administered a non-lethal dose of 0.3 μg mFasL via tail vein injection to WT and Cd14−/− mice. Administration of mFasL intravenously mimicked T cell-mediated killing and prevented liver-dependent lethality (analyzed via serum ALT) typically induced by intraperitoneal administration, thereby allowing for observation of effects on myeloid populations (Figures S3A and S3B). We harvested serum and spleens for analysis 2–4 h post-injection, as myeloid cells are in high concentrations in these compartments (Figure S3C). Interestingly, lactate dehydrogenase (LDH), a marker of tissue injury associated with cell death, was significantly increased in WT mice, while no increase was observed in serum from the Cd14−/− mice (Figure 3A). To provide evidence of cell death, spleens were fixed, sectioned, and stained with a TUNEL kit to identify dying cells by cleaved DNA (Figure 3B). In corroboration with the LDH assay, spleens from WT mice exhibited more TUNEL+ splenocytes than those from Cd14−/− mice (Figures 3B and 3C).
Figure 3. FasL intravenous injection induces inflammatory response in Cd14−/− mice.

(A) Serum levels of LDH 2–4 h post-mFasL tail vein injection of 0.3 μg/mL.
(B and C) TUNEL staining (B) and quantification of TUNEL+ cells per μm2 (C) in spleens 4 h post-tail vein injection of 0.3 μg/mL. Each point represents the four averaged FOVs of spleen section from an individual mouse, presented with mean ± SD (B6 n = 3, Cd14−/− n = 3, PBS data points represent data from B6 n = 3 and Cd14−/− n = 1).
(D) Serum and spleen levels of CXCL1, TNF, and IL-10 2–4 h post-mFasL injection of 0.3 μg/mL or PBS control. See also Figures S2 and S3.
Each point represents an individual mouse, presented with mean ± SD. ANOVA was used for comparison between groups: ns (p > 0.05), **p < 0.01, ***p < 0.001, and ****p < 0.0001.
While CD14 deficiency protected mice from Fas-mediated injury, it promoted the inflammatory response to FasL in the serum and spleen, with significant increases in CXCL1, IL-10, and TNF-α, which was not present in their WT counterparts, thereby suggesting a shift toward Fas-mediated inflammatory responses in the absence of CD14 (Figure 3D).
CD14 prevents Fas-mediated inflammation in myeloid cells
To further investigate the mechanism of Fas-mediated hyperinflammation in the absence of CD14, we set up in vitro activation of peritoneal neutrophils and macrophages with FasL. We observed no inflammatory response to the classically “silent” FasL stimulation in WT neutrophils, while Cd14−/− neutrophils exhibited significant elevation of Cxcl1 and Tnf mRNA by means of qPCR (Figure 4A). Cd14−/− neutrophils were also hyperinflammatory at the protein level, as they produced significant amounts of the chemokine CXCL1 compared to WT neutrophils (Figure 4B). In further support, Cd14−/− macrophages showed a similar inflammatory signature by qPCR, with significantly increased upregulation of Cxcl1, Tnf, and Il23 (Figure 4C). Primary Cd14−/− macrophages also produced significant amounts of CXCL1, while WT macrophages produced minimal CXCL1 (Figure 4D). To ensure that the hyperinflammatory state of Cd14−/− macrophages was not exclusively a result of increased viability, we stimulated macrophages deficient in CASP8 and RIPK3 with FasL. These double knockout (DKO) macrophages are resistant to most programmed cell death, yet they did not exhibit an inflammatory signature by qPCR (Figure S4A). Similarly, the addition of a CASP3/7 inhibitor did not increase the inflammatory response of WT macrophages to FasL (Figure S4B). To dismiss the possibility of a CD14-mediated effect due to LPS contamination, peritoneal macrophages from mice with TLR3 and TLR4 knocked out (TLR3/4 DKO) did not produce significant quantities of CXCL1, indicating that this function of CD14 can be decoupled from the associated TLRs (Figure 4D). As CXCL1 is a NF-κB-dependent cytokine, we examined the role of CD14 in the activation of NF-κB in peritoneal and BMDMs. Accordingly, we observed the rapid degradation of IκBα and the phosphorylation of p65 in Cd14−/−, but not WT, macrophages stimulated with FasL (Figure 4E), thus confirming a shift of Fas-mediated activation toward a pro-NF-κB response in CD14-deficient myeloid cells.
Figure 4. CD14 prevents Fas-mediated inflammation in myeloid cells.

(A) Relative CXCL1 and TNF mRNA levels normalized to ACTB in response to FasL over time in indicated neutrophils.
(B) Levels of CXCL1 produced by murine neutrophils in response to FasL over time. Data points indicate individual mice as source of isolated neutrophils.
(C) Relative CXCL1, TNF, and IL23 mRNA levels normalized to ACTB in response to FasL over time in indicated macrophages.
(D) Levels of CXCL1 produced by macrophages in response to FasL over time. Data points indicate individual mice as source of isolated macrophages.
(E) Levels of IκBα and total and phosphorylated p65 over time after FasL stimulation of macrophages. qPCR and ELISA data are presented as the means ± SD for duplicate wells from three or more independent experiments. See also Figure S4.
Data from western blotting are representative of three or more independent experiments. ANOVA was used for comparison between groups: ns (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
TRIF determines the inflammatory output of Fas through RIP1K activation
Similar to the Cd14−/− macrophages, Trif−/− macrophages-produced significantly more CXCL1 and activated the NF-κB pathway in response to FasL (Figures 5A and 5B). We hypothesized this was a result of TRIF controlling RIPK1 activity based on TRIF-dependent regulation of RIPK1-mediated cell death and inflammatory responses via RIP homotypic interaction motif (RHIM) domains.30,43 RIPK1 also been implicated in Fas signaling, but much less is known about its activity in this pathway.13,34 Our data support that TRIF recruits RIPK1 to a death complex, but in Trif−/−, RIPK1 is free to perform pro-inflammatory functions. In assessing the role of RIPK1 downstream of TRIF, we considered that phosphorylation of RIPK1 at S166 promotes RIPK1-dependent cell death, while phosphorylation at S321 induces inflammatory responses.44,45 Indeed, the phosphorylation patterns of RIPK1 differed between WT and knockout macrophages. Both Cd14−/− and Trif−/− macrophages exhibited abrogated phosphorylation at the pro-death S166 locus and a more sustained phosphorylation at the pro-inflammatory S321 site (Figure 5D). While RIPK1 clearly maintained some activity, cleavage of RIPK1 was decreased in Fas-stimulated Trif−/− macrophages, suggesting that RIPK1 was not incorporated in a death complex where it could be cleaved by CASP8 (Figure S1D). This supports the previously mentioned observation that reduced CASP8-RIPK1 interactions in Trif−/− macrophages induced phosphorylated MLKL, indicating a switch to necroptotic cell death (Figure S1D).
Figure 5. TRIF determines inflammatory output of Fas through RIP1K activation.

(A) Levels of CXCL1 produced by macrophages of indicated genotype in response to FasL over time. Data points indicate individual mice as source of isolated macrophages.
(B) IκBα and total and phosphorylated p65 over time in whole-cell lysates from B6, Cd14−/−, and Trif−/− macrophages after FasL stimulation.
(C, E, and F) Relative IFNB and ISG15 mRNA levels normalized to ACTB in response to FasL over time in indicated neutrophils and macrophages.
(D) Total and phosphorylated forms of RIPK1 over time in whole-cell lysates from B6, Cd14−/−, and Trif−/− macrophages after FasL stimulation.
(G) Spleen levels of IFN-β 4 h post-mFasL injection of 0.3 μg/mL; each point represents an individual mouse.
qPCR and ELISA data are presented as the means ± SD for duplicate wells from three or more independent experiments. Data from western blotting are representative of three or more independent experiments. ANOVA was used for comparison between groups: ns (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
This switch toward pro-inflammatory responses to FasL in the Cd14−/− and Trif−/− myeloid cells was reversed for IFN-β production and further supported by abrogation of IFN-stimulated gene (ISG) activation (Figures 5C–5F). We observed a similar effect in vivo, as the FasL-injected Cd14−/− mice had significantly lower amounts of IFN-β in the spleen compared to WT mice (Figure 5G). This abrogation of IFN responses in Cd14−/− and Trif−/− was expected, as IFN production is tightly controlled by TRIF-dependent activation of transcription factors like IRF3.46 As IFN responses are reduced, Cd14−/− and Trif−/− responses to FasL are more specifically pro-NF-κB rather than broadly inflammatory.
CD14 is required for the internalization of the Fas receptor in macrophages
The ability of CD14 to internalize TLRs into the endosomal compartment is central to its role in immune responses, which was recently found to also be the case for TNFR-mediated signaling.26,35 As Fas-mediated activation skews toward pro-inflammatory responses when internalization is inhibited, we hypothesized that the skewing of Fas responses toward inflammation in CD14-deficient cells was due to attenuated internalization of Fas in the absence of CD14.19 To test this, we primed WT and TLR3/4 DKO macrophages with LPS for 4 h before FasL stimulation to mimic an infectious context in which CD14 would bind a ligand, internalize, and be removed from the cell surface before Fas activation. As expected, LPS pre-treatment protected macrophages from Fas-mediated death regardless of the TLR availability (Figure S5A). However, it must be noted that LPS stimulation activated many pro-survival signaling pathways not specific to the apoptotic cascade. To better explore the role of CD14 internalization in Fas signaling, we considered that mouse CD14 contains a Glycosylphosphatidylinositol (GPI) anchor region, which facilitates its internalization and endosomal trafficking. Animals without a GPI anchor, such as chickens, have a membrane-bound form of CD14 and cannot internalize CD14.47 Accordingly, we compared the viability of Cd14−/− macrophages transduced with mouse or chicken CD14 to observe relative responses to FasL of cells transduced with “internalization-deficient” chicken CD14 (Figure S5B). Add back of both mouse and chicken CD14 maintained an NF-κB response to LPS by degrading IκBα, yet only BMDMs with chicken CD14 responded to FasL (Figures S5C and S5D). While adding back murine CD14 to Cd14−/− macrophages restored Fas-mediated death, add back of internalization-deficient chicken CD14 did not (Figure 6A). This led us to further investigate the receptor internalization function of CD14 in Fas signaling.
Figure 6. CD14 is required for the internalization of the Fas receptor in macrophages.

(A) Cell death of BMDMs transduced with indicated constructs measured over time by propidium iodide incorporation.
(B) Representative 40× images of CD14 or FasR staining and colocalization in permeabilized B6 macrophages stimulated with mFasL for 30 min.
(C) Quantification of CD14 and Fas puncta. Each data point represents a FOV across 3 technical replicates, ~10 FOV per replicate.
(D) Representative 40× images of extracellular FasR staining of non-permeabilized B6 and Cd14−/− macrophages after 1 h of mFasL stimulation.
(E) Quantification of extracellular FasR after 1 h of mFasL stimulation in B6 and Cd14−/− macrophages. Each data point represents a FOV across 3 experimental replicates, with 3 technical replicates per experiment.
(F) Cell death of PMA-differentiated U937-derived macrophages transduced with indicated constructs measured over time by propidium iodide incorporation.
(G) Relative Tnf mRNA levels normalized to ACTB in response to FasL in indicated U937-derived macrophages. See also Figure S6.
(H) Proposed model depicting role of CD14 in Fas-mediated death with and without pathogens present.
Data from cell death assays and qPCR are representative of three or more independent experiments, and cell death data are presented as the means ± SD of triplicate wells (~5,000 cells per FOV). Data from imaging experiments are representative of three or more independent experiments, and data points indicate FOVs, n = 15 (~20 cells/FOV). Image quantification is presented with mean ± SD. ANOVA was used for comparison between groups: ns (p > 0.05); **p < 0.01; ***p < 0.001; ****p < 0.0001.
To test this hypothesis, we costained CD14 and Fas on macrophages to detect CD14 and Fas colocalization into puncta upon FasL stimulation (Figures 6B and S2A). CD14 puncta per cell significantly increased upon FasL stimulation, and the number of CD14 puncta colocalized with Fas receptor puncta per cell also increased (Figures 6B and 6C).
To determine the functional outcome of internalization, we stained non-permeabilized peritoneal macrophages with a fluorescent anti-Fas antibody and quantified changes in surface Fas before and after stimulation with FasL. Using quantification software, we were able to identify and count “Fas+” macrophages and compare decreases in Fas+ macrophages after stimulation and internalization of the receptor (Figures 6D, 6E, and S2B). While we were able to capture a significant stimulation-dependent decrease in surface Fas staining in WT macrophages, we did not observe such a decrease in Cd14−/− macrophages (Figure 6E). These data suggested that the internalization of the Fas receptor was dependent on CD14.
Based on the CD14-dependent cell death in murine macrophages in response to FasL, we hypothesized that CD14 would have a similar role in human myeloid cells. Compared to more extensively studied human cell lines, like Jurkat lymphocytes, the human monocytic cell line U937 were relatively resistant to Fas-mediated cell death, which may be due to low CD14 levels.48,49 Indeed, even after PMA differentiation, U937-derived macrophages expressed very little CD14 (Figure S5E). In contrast, lentiviral transduction of human CD14 (“CD14 add back”) sensitized U937-derived macrophages to Fas-mediated cell death, while untransduced and empty vector controls remained protected (Figures 6F and S5E; Table S1). While untransduced and empty-vector-transduced U937-derived macrophages exhibited a minor early TNF response to FasL by qPCR, this response was abrogated in U937-derived macrophages that had received the CD14 add-back construct (Figure 6G). These data suggest that CD14 promotes Fas-mediated cell death and, to a lesser extent, limits an inflammatory response in human myeloid cells, consistent with our findings in murine myeloid cells.
DISCUSSION
The Fas receptor has been traditionally perceived as a potent inducer of apoptosis in a variety of cell types, yet recent publications have reported Fas-induced inflammation in pathological contexts, such as infection and tissue damage. This switch from pro-death to pro-inflammatory outcomes of Fas signaling is poorly understood, and our data propose an intriguing mechanism for this phenomenon in myeloid cells. As described herein, we have characterized a novel and unique function of CD14 in the regulation of silent cell death downstream of the Fas receptor. Previous studies have reported Fas-mediated inflammation in non-myeloid CD14-lo cell lines, using Fas agonist antibodies (which are poor inducers of internalization), or when apoptosis is inhibited.10,13,25 Our study describes a physiologically relevant context in which primary myeloid cells can respond to membrane-bound FasL with NF-κB activation and survival. When CD14 is on the cell surface of myeloid cells, it promotes the internalization of Fas, allowing access to a pro-death TRIF-dependent complex that can amplify apoptotic signaling. While some apoptotic signaling remains intact in Cd14−/− and Trif−/− myeloid cells, likely through classic death-inducing signaling complex (DISC) activation, utilizing CD14 to access a TRIF pro-death complex allows maximum apoptotic signaling. In the absence of CD14, the Fas receptor is retained at the membrane and has limited access to this TRIF-based death complex, which inhibits cell death and diverts signaling to a pro-inflammatory NF-κB pathway, leading to cytokine release. Additionally, we hypothesize that when TRIF is not present, RIPK1 is not appropriately shuttled into the DISC and, instead, associates with a pro-inflammatory complex.
Based on our findings, we propose that CD14 availability on the surface of myeloid cells dictates the switch between pro-death and pro-inflammatory Fas signaling (Figure 6H). The ability of CD14 to detect pathogens via LPS binding is well studied, and CD14 can detect the presence of damaged and dying cells via binding to ICAM3 and oxPAPC.50–52 Therefore, during infection or damage, CD14 ligands are plentiful, leading to the internalization and removal of CD14 from the cell surface. With CD14 removed from the cell surface, it cannot assist the Fas receptor into the endosome environment to access TRIF and the death complex. Instead, without CD14 on the surface, the Fas receptor would be at the cell surface. Therefore, when infiltrating cells such as natural killer (NK) or CD8+ T cells expose responding myeloid cells to FasL, low surface CD14 induces a pro-inflammatory and chemotactic response to FasL and prevents the elimination of the myeloid cells. As demonstrated by the above study, Fas-mediated CXCL1 release is highly regulated by CD14. CXCL1 is a potent chemokine that is essential for recruiting phagocytes and first responders, particularly neutrophils, that are necessary for the clearance of pathogens or damaged cells. When infection or damage resolves and there are no CD14 ligands present, CD14 returns to the cell surface, and myeloid cells can be eliminated via Fas-mediated apoptosis.
Knowing NK cells utilize FasL in an anti-tumor response, our data suggest a potentially novel role for CD14 in supporting the elimination of cancerous cells, particularly tumor-associated macrophages.53 While CD14 would promote apoptosis of these cells, it would also prevent the release of CXCL1 in the tumor environment. CXCL1 induces growth and angiogenesis within the tumor, and high levels of CXCL1 from tumor-associated macrophages have been associated with excessive tumor growth and poor prognosis.54 As CD14-dependent apoptosis limits the inflammatory response and prevents CXCL1 release, it could be a crucial player in anti-tumor defenses. Alternatively, the same characteristics make CD14 an interesting target in wound healing, a scenario where prevention of cell death and release of angiogenic chemokines would be extremely beneficial. Inhibiting CD14 in this context could prevent Fas-mediated cell death while inducing CXCL1 release to promote healing. CD14 inhibitors are currently in use therapeutically and offer a novel way of mediating the Fas cell death pathway.
Fas signaling is involved in numerous pathologies, but few treatments have been able to utilize this pathway for therapeutic benefit. Proposing CD14 as an upstream mediator of Fas provides rationale for further investigation into CD14 as a therapeutic target in these pathologies.
Limitations of this study
This study has not fully investigated the downstream activity of RIPK1 in Cd14−/− or Trif−/− conditions. The role of CD14 or TRIF in primary human myeloid cells has not been assessed.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information, resources and reagents should be directed to the Corresponding Author, Alexander Poltorak (alexander.poltorak@tufts.edu). This study did not generate new unique reagents.
Materials availability
All unique reagents in this study are available from the lead contact and will be provided upon request. This paper does not report original code.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not include original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
C57BL/6 (B6), Tlr4−/−, Tlr3−/−, Ticam1−/−(Trif−/−), Cd14−/−, and Lpr mice were obtained from the Jackson Laboratory. Rip3−/− Casp8−/− mice were generated by Dr. Douglas Green (St. Jude Children’s Research Hospital) and were a gift from Dr. Katherine Fitzgerald (University of Massachusetts Medical School). Tlr3−/−Tlr4−/− mice were generated by in-house breeding. Ripk1K45A/K45A (RIP1Kinase inactive) mice were a gift from Dr. Alexei Degterev (Tufts University School of Medicine) and Zbp1−/− mice were a gift from Dr. Siddarth Balachandran (Fox Chance Cancer Center). 10–12-week-old female mice were used for in vivo experiments. All mice were housed in a pathogen-free facility at the Tufts University School of Medicine and experiments were performed in accordance with regulations and approval of the Tufts University Institutional Animal Care and Use Committee.
Primary cell cultures
To generate the BMDMs used in this study, bone marrow was isolated from the long bones of male and female mice, propagated in RPMI containing 20% fetal bovine serum (FBS), 2% penicillin-streptomycin, and 30% L cell supernatant on non-tissue culture–treated petri dishes for 7 days at 37°C. Once differentiated, BMDMs were plated for experiments at a density of 1 × 106 cm2 in RPMI containing 20% FBS and 1% penicillin-streptomycin at 37°C. To isolate neutrophils, mice were primed with 1 mL of thioglycolate for 8 h. To isolate peritoneal macrophages, mice were primed with 1 mL of thioglycolate for 4 days. Peritoneal cells enriched for neutrophils or macrophages were isolated in cold PBS and plated for experiments at a density of 1 × 106 cm2 in RPMI containing 20% FBS and 1% penicillin-streptomycin at 37°C.
Human cell line
Human monocyte cell line U937 was maintained in RPMI containing 20% FBS, 1% L-glutamine, and 1% penicillin-streptomycin at 37°C. U937 monocytes were differentiated into macrophages in 100 ng/mL PMA for 24 h and washed to remove non-adherent cells. 100 ng/mL of mFasL was used in U937-derived macrophage in vitro experiments.
METHOD DETAILS
Reagents
Multimeric FasL was purchased from Adiopgen and used at 100 ng/mL for BMDMs, peritoneal cells, and U937-derived macrophages. 200 ng/mL of mFasL was used on transduced BMDMs. Recombinant mouse IFNb (12404–1) was purchased from PBL Assay Science and used at 5 IU. zVAD.fmk was purchased from Millipore and used at 50uM. Lipopolysaccharide (LPS) (Escherichia coli 0111:B4) was purchased from Millipore Sigma and used at 10 ng/ml.
Tail vein administration of mFasL
10–12-week-old female mice were intravenously injected with 0.3 μg of recombinant MultimericFasL (Adiopgen) in sterile PBS (200 μL) or an equal volume of sterile PBS for control mice. Temperature was monitored hourly by rectal thermometer. At indicated time points after administration of FasL or when mouse body temperature reached <30°C, mice were euthanized by CO2 asphyxiation, and the spleen and blood were harvested for quantification of cytokine levels using enzyme-linked immunosorbent assay (ELISA) and serum LDH levels using the CyQuant LDH Cytotoxicity Assay Kit (C20300). For histology and TUNEL staining, spleens were isolated from mFasL- or PBS-injected mice after 4 h, fixed in 10% formalin for 48 h, and transferred to 75% ethanol. Sectioning of spleens was performed by iHisto. TUNEL staining was completed using the In Situ Cell Death Detection Kit (Roche Diagnostics) according to manufacturer’s instructions. TUNEL+ cells per square micrometer were calculated on the BioTek Lionheart Automated microscope. All experiments were performed in accordance with regulations and approval of the Tufts University Institutional Animal Care and Use Committee.
ELISA
mTNF (DY410), IL-10 (DY417), and CXCL1 (DY453) DuoSet ELISA Kits were used according to the manufacturer’s instructions to quantify serum and tissue cytokine levels. For IFN-β ELISAs, 384-well ELISA plates were coated overnight at 4°C with monoclonal rat anti-mouse IFN-β antibody (Santa Cruz Biotechnology, sc-57201; 1:500 dilution in 0.1 M carbonate buffer) and blocked with 10% FBS in PBS for 2 h at 37°C. Samples were incubated on plates overnight at 4°C before washing with 0.005% Tween 20 in PBS and adding polyclonal rabbit anti-mouse IFN-β antibody (R&D Systems, 32400–1; 1:2000 dilution in 10% FBS in PBS) overnight at 4°C. After washing, goat anti-rabbit–horseradish peroxidase antibody (Cell Signaling Technologies, 7074; 1:2000 dilution in 10% FBS in PBS) was added for 2 to 3 h at room temperature. TMB (3,3′,5,5′,-tetramethylbenzidine) substrate was added, and re-action was stopped with 2 N of H2SO4.
Kinetic microscopy
The Cytation3 and Lionheart automated microscopes were used for kinetic macrophage and neutrophil imaging assays, and built-in environmental control was maintained at 37°C and 5% CO2 for the duration of the assay. Cells were seeded at a density of 1 × 106 cm2 in RPMI on 1.17-mm-thick glass bottom imaging plates. To generate kinetic cell death curves, cells were imaged at 30-min intervals at 4× magnification to capture about 5000 cells per field of view. Propidium iodide incorporation was detected at 617 nm, and propidium iodide–positive nuclei were counted. Wells treated with 0.1% Triton X-100 were used as controls for 100% cell death.
FADD and RIPK1 immunoprecipitation
Cells were plated on 10-cm dishes, stimulated as indicated, and harvested in IP lysis buffer [0.5% Triton X-100, 50 mM Tris base (pH 7.4), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, and 1× protease inhibitor cocktail]. Lysed cells were rotated for 60 min at 4°C with intermittent vortexing and centrifuged at 5000g for 5 min, and the supernatant was incubated with α- FADD or RIPK1 antibody–conjugated Protein G–agarose beads (Cell Signaling Technology, 37478). Samples were washed three times in IP lysis buffer, and protein complexes were eluted with 1× Laemmli buffer containing 5% β-mercaptoethanol at 90°C for 15 min.
Immunoblotting
After indicated treatments, cells were lysed in 1× Laemmli buffer containing 5% β-mercaptoethanol, boiled for 10 min, and incubated on ice for 10 min e. Protein lysates were resolved on a 10% or 15% Bis-Tris SDS gel, and transferred to a nitrocellulose membrane using the Pierce Power transfer system. Membranes were blocked with 5% BSA in PBS-T for 1 h. Primary antibodies were diluted to 1:1000 in 1% BSA in PBS-T, and membranes were incubated with indicated primary antibodies (key resources table) overnight at 4C. Infrared secondary antibodies (680 or 700 nm) were diluted 1:30,000 in 1% BSA in PBS-T, and membranes were incubated with indicated secondary antibodies (key resources table) for 45 min at room temperature. Membranes were imaged using an Odyssey CLx Imaging System, and image analysis was performed using Image Studio software.
KEY RESOURCES TABLE.
| REAGENTor RESOURCE | SOURCE | identifier |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| NF-κB p65 | Cell Signaling Technology | Cat#8242; RRID: AB_10859369 |
| Phospho-NF-κB p65 | Cell Signaling Technology | Cat#3033; RRID:AB_331284 |
| IκBα | Cell Signaling Technology | Cat#4814; RRID:AB_390781 |
| RIPK1 | Cell Signaling Technology | Cat#3493; RRID:AB_2305314 |
| Cleaved Caspase 8 | Cell Signaling Technology | Cat#8592; RRID:AB_10891784 |
| Cleaved Caspase 3 | Cell Signaling Technology | Cat#9665; RRID:AB_2069872 |
| GAPDH | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| MLKL | Abcam | Cat#ab243142 |
| Phospho-MLKL | Abcam | Cat#ab196436; RRID:AB_2687465 |
| FADD | MilliporeSigma | Cat#05486; RRID:AB_2100627 |
| Fas Receptor | Abcam | Cat#ab82419; RRID:AB_1658628 |
| TRAM | Santa Cruz Biotechnology | Cat#sc-376076; RRID:AB_10989356 |
| RIPK1 phospho-site S166 | Cell Signaling Technology | Cat#31122; RRID:AB_2799000 |
| RIPK1 phosho-site S321 | Cell Signaling Technology | Cat#83613; RRID:AB_2800023 |
| Gasdermin D | Abcam | Cat#ab209845; RRID:AB_2783550 |
| FLAG | Cell Signaling Technology | Cat#14793; RRID:AB_2572291 |
| CD14 (mouse) | Cell Signaling Technology | Cat#93882; RRID:AB_2800216 |
| CD14 (human) | Cell Signaling Technology | Cat#56082; RRID:AB_2799504 |
| Anti-rabbit IgG DyLight 800 conjugate | Cell Signaling Technology | Cat#5151; RRID:AB_10697505 |
| Anti-mouse IgG DyLight 800 conjugate | Cell Signaling Technology | Cat#5257; RRID:AB_10693543 |
| IFN-β | Santa Cruz Biotechnology | Cat#57201; RRID:AB_2122911 |
| IFN-β | R&D Systems | Cat#32400-1; RRID:AB_2121718 |
| TRIF | Abcam | Cat#ab13810; RRID:AB_2201578 |
| RIPK1 | 3D Biosciences | Cat#610459; RRID:AB_397832 |
| CD14 | Abcam | Cat#ab221678; RRID:AB_2935854 |
| Fas | 3D Biosciences | Cat#554258; RRID:AB_395330 |
| Alexa Fluor 555 goat anti-mouse IgG | Invitrogen | Cat#150114 |
| Alexa Fluor 405 goat anti-rabbit IgG | Invitrogen | Cat#175652 |
| Alexa Fluor 488 goat anti-rabbit IgG | Invitrogen | Cat#11008 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Multimeric FasL | Adipogen | Cat#AG-40B-0130-3010 |
| Lipopolysaccharide (LPS, Escherichia coli 0111:B4) | Sigma | Cat#L4391 |
| zVAD.fmk | Millipore | Cat#627610 |
| Recombinant mouse IFNb | PBL Assay Science | Cat#12404-1 |
| Caspase 3/7 inhibitor | Cayman Chemical Company | Cat#14464 |
| MMuLV reverse transcriptase | New England Biolabs | Cat#M0253 |
| RNase Inhibitor, Human Placenta | New England Biolabs | Cat#M0307 |
| Random Primers 9 | New England Biolabs | Cat#S1254 |
| Deoxynucleoside Triphosphate (dNTP mix) | New England Biolabs | Cat#N0447 |
| SYBER Green PCR Master Mix | Applied Biosystems | Cat#4309155 |
| TRIzol | Invitrogen | Cat#15596026 |
|
| ||
| Critical commercial assays | ||
|
| ||
| CyQuant LDH Cytotoxicity Assay Kit | Cat#C20300 | |
| Pointe Scientific ALT (SGPT) Liquid Reagents | Fisher Scientific | Cat#23-666-089 |
| In Situ Cell Death Detection Kit (TUNEL) | Roche Diagnostics | Cat#11684795910 |
| TNF DuoSet ELISA Kit | R&D | Cat#DY410 |
| CXCL1 DuoSet ELISA Kit | R&D | Cat#DY453 |
| IL10 DuoSet ELISA Kit | R&D | Cat#DY417 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| U937 | ATCC | Cat#CRL-1593.2 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 (B6) mice | Jackson Laboratories | 000664 |
| Tlr4−/− mice | Jackson Laboratories | 007227 |
| Tlr3−/− mice | Jackson Laboratories | 005217 |
| Ticam1−/− (Trif−/−) mice | Jackson Laboratories | 005037 |
| Cd14−/− mice | Jackson Laboratories | 003726 |
| Lpr mice | Jackson Laboratories | 000482 |
| Tlr3−/− Tlr4−/− mice | This paper | N/A |
| RIP3−/− Casp8−/− mice | Dr. D. Green | N/A |
| Ripk1K45A/K45A (RIP1Kinase inactive) mice | Dr. A Degterev | N/A |
| Zbp1−/− mice | Dr. S. Balachandran | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| TNF (F) 5’-CTGTAGCCCAC GTCGTAGC-3’ | Integrated DNA Technologies | N/A |
| TNF (R) 5’-TTGAGATCCATGCCGTTG-3’ | Integrated DNA Technologies | N/A |
| IFN β (F) 5’-CAGCTCCAAGAAAGGACGAAC-3’ | Integrated DNA Technologies | N/A |
| IFN β (R) 5’-GGCAGTGTAACTCTTCTGCAT-3’ | Integrated DNA Technologies | N/A |
| CXCL1 (F) 5’-TGAGCTGCGCTGTCAGTG-3’ | Integrated DNA Technologies | N/A |
| CXCL1 (R) 5’-AGAAGCCAGCGTTCACCAGA-3’ | Integrated DNA Technologies | N/A |
| Ticam1 (F) 5’-AGACCCTACAGCCAGGTCT-3’ | Integrated DNA Technologies | N/A |
| Ticam1 (R) 5’-TGGAGACATGTTCCGAACCG-3’ | Integrated DNA Technologies | N/A |
| ISG15 (F) 5’-TTCTGGGCAATCTGCTTCTT-3’ | Integrated DNA Technologies | N/A |
| ISG15 (R) 5’-GAGCTAGAGCCTGCAGCAAT-3’ | Integrated DNA Technologies | N/A |
| Beta Actin (F) 5’CACTGTCGAGTCGCGTCCA-3’ | Integrated DNA Technologies | N/A |
| Beta Actin (R) 5’-GACCCATTCCCAACCATCACA-3’ | Integrated DNA Technologies | N/A |
| IL23 (F) 5’-CACCTCCCTACTAGGACTCAG-3’ | Integrated DNA Technologies | N/A |
| IL23 (R) 5’-TGGGCATCTGTTGGGTCT-3’ | Integrated DNA Technologies | N/A |
| Human GAPDH (F) 5’ GCCACATCGCTCAGACAC-3’ | Integrated DNA Technologies | N/A |
| Human GAPDH (R) 5’-GCCCAATACGACCAAATCC-3’ | Integrated DNA Technologies | N/A |
| Human TNF (F) 5’-CCCCAGGGACCTCTCTCTAATC-3’ | Integrated DNA Technologies | N/A |
| Human TNF (R) 5’-GGTTTGCTACAACATGGGCTACA-3’ | Integrated DNA Technologies | N/A |
| See Table S1 for gBlock Fragment Sequences | Integrated DNA Technologies | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| psPAX plasmid | Addgene | Plasmid 12260 |
| VSV-G pseudotyping vector pMD2.G | Addgene | Plasmid 12259 |
| pLEX lentiviral vector | Addgene | Plasmid 25896 |
|
| ||
| Software and algorithms | ||
|
| ||
| BioTek Gen5 Microplate Reader and Imager Software | Agilent | N/A |
| GraphPad Prism 9.0 | Graphpad | N/A |
| Image Studio | LI-COR | N/A |
|
| ||
| Other | ||
|
| ||
| Protein G Agarose Beads | Cell Signaling Technology | Cat#37478 |
RNA isolation and analysis
A total of 5 × 105 BMDMs, peritoneal macrophages, neutrophils, or U937-derived macrophages were plated on 24-well tissue culture–treated plates. Cells were lysed with TRIzol (Invitrogen), and RNA extraction was carried out according to the manufacturer’s instructions. Reverse transcription was performed using MMuLV reverse transcriptase, ribonuclease inhibitor, random primers 9, and deoxynucleoside triphosphate (dNTP) mix (New England Biolabs) to synthesize cDNA. cDNA was analyzed for relative mRNA levels using SYBR Green (Applied Biosystems) and indicated primers (key resources table) primers. β-actin (murine cells) or GAPDH (human cells) were used to normalize mRNA levels.
High magnification imaging
To observe TRIF, RIPK1, CD14, and FAS localizations, we seeded macrophages at a density of 1 × 106 cm2 in RPMI on 1.17- mm-thick glass-bottom imaging plates. Cells were stimulated with mFasL for indicated times, fixed in 4% paraformaldehyde for 15 min, blocked in 1× PBS (5% FBS and 0.2% Triton X-100), and incubated overnight with anti-TRIF (Abcam, ab13810), anti-RIPK1 (BD Bio-sciences, 610459), anti-CD14 (Abcam, ab221678), or anti-Fas (BD Pharmigen, 554258) antibody, followed by a 2-h incubation in the presence of Alexa Fluor 555–conjugated goat anti-mouse (ab150114) and Alexa Fluor 405–conjugated goat anti-rabbit (ab175652) antibodies. For non-permeabilized samples, blocking buffer without Triton X-100 was used. The Lionheart automated microscope was used to image cells at ×60 or ×40 magnification to capture about 20 cells per field of view in quintuplicate. Gen5 3.10 imaging software was used to quantify positive staining for each protein, and a mask was generated to calculate puncta/cell.
Lentivirus constructs and transduction
For CD14 reconstitution experiments, mouse, chicken, and human CD14 gBlock sequences were ordered from Integrated DNA Technologies and ligated into a pLEX lentiviral vector. Lentiviral particles were generated by transfection with packaging vector psPAX2 (plasmid 12260; Addgene) and the VSV-G pseudotyping vector pMD2.G (plasmid 12259; Addgene) into the 239T cell line. Generated lentiviral particles were used to transduce Cd14−/− BMDMs on day 4 of differentiation, followed by puromycin selection (4 μg/mL, 48 h) starting on day 6. Generated lentiviral particles were used to transduce U937 monocytes, followed by puromycin selection (1 μg/ml, 24 h). After puromycin selection, U937 monocytes were differentiated into macrophages with PMA over 24 h, washed, and replated for assays.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 9 software. qPCR experiments represent the mean of duplicate or triplicate wells and error bars represent SD of three or more independent experiments. Data from imaging experiments are representative of three or more independent experiments. Immunoblots are representative of three or more independent experiments. For in vivo data, points represent individual mice. For ELISA data on peritoneal cells, points represent individual mice peritoneal cells were harvested from. Significance was determined using a one-way or two-way analysis of variance (ANOVA) as appropriate: ns (not significant), p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Supplementary Material
Highlights.
Fas-mediated death of myeloid cells requires TLR-associated protein CD14 TRIF
Without CD14, Fas activation induces NF-κB-mediated cytokine production in vitro and in vivo
CD14 promotes internalization of Fas from the cell surface and formation of a death complex
ACKNOWLEDGMENTS
We thank Dr. Douglas Green, Dr. Siddarth Balachandran, and Dr. Alexei Degterev for sharing the various mouse strains used in this study. Illustrations were created with BioRender.com. This work was supported by NIH grants AI167245 and AI056234 to A.P. and NIH grant AI176055–01A1 to S.B.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114685.
REFERENCES
- 1.Green DR, and Ferguson TA (2001). The role of fas ligand in immune privilege. Nat. Rev. Mol. Cell Biol. 2, 917–924. 10.1038/35103104. [DOI] [PubMed] [Google Scholar]
- 2.Akane K, Kojima S, Mak TW, Shiku H, and Suzuki H (2016). CD8+CD122+CD49dlow regulatory T cells maintain T-cell homeostasis by killing activated T cells via Fas/FasL-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 113, 2460–2465. 10.1073/pnas.1525098113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, and Henson PM (2011). Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 184, 547–560. 10.1164/rccm.201011-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kearns MT, Barthel L, Bednarek JM, Yunt ZX, Henson PM, and Janssen WJ (2014). Fas ligand-expressing lymphocytes enhance alveolar macrophage apoptosis in the resolution of acute pulmonary inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 307, L62–L70. 10.1152/ajplung.00273.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krzyzowska M, Baska P, Grochowska A, Orlowski P, Nowak Z, and Winnicka A (2014). Fas/FasL pathway participates in resolution of mucosal inflammatory response early during HSV-2 infection. Immunobiology 219, 64–77. 10.1016/j.imbio.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stone-braker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, et al. (2007). Elimination of Antigen-Presenting Cells and Autoreactive T Cells by Fas Contributes to Prevention of Autoimmunity. Immunity 26, 629–641. 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, and Luo ZQ (2007). Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. USA 104, 5121–5126. 10.1073/pnas.0611030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caulfield AJ, Walker ME, Gielda LM, and Lathem WW (2014). The Pla protease of Yersinia pestis degrades Fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15, 424–434. 10.1016/j.chom.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, and Neish AS (2008). Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 3, 233–244. 10.1016/j.chom.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, Lavelle EC, Leverkus M, and Martin SJ (2013). Fas/CD95-Induced Chemokines Can Serve as “Find-Me” Signals for Apoptotic Cells. Mol. Cell 49, 1034–1048. 10.1016/j.molcel.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z, Fitzgerald M, Meisinger T, Batra R, Suh M, Greene H, Penrice AJ, Sun L, Baxter BT, and Xiong W (2019). CD95-ligand contributes to abdominal aortic aneurysm progression by modulating inflammation. Cardiovasc. Res. 115, 807–818. 10.1093/cvr/cvy264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraga-Silva TFC, Cipriano UG, Fumagalli MJ, Correa GF, Fuzo CA, Dos-Santos D, Mestriner FLAC, Becari C, Teixeira-Carvalho A, Coelho-Dos-Reis J, et al. (2023). Airway epithelial cells and macrophages trigger IL-6-CD95/CD95L axis and mediate initial immunopathology of COVID-19. iScience 26, 108366. 10.1016/j.isci.2023.108366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidovich P, Higgins CA, Najda Z, Longley DB, and Martin SJ (2023). cFLIPL acts as a suppressor of TRAIL- and Fas-initiated inflammation by inhibiting assembly of caspase-8/FADD/RIPK1 NF-κB-activating complexes. Cell Rep. 42, 113476. 10.1016/j.celrep.2023.113476. [DOI] [PubMed] [Google Scholar]
- 14.Seino K, Kayagaki N, Okumura K, and Yagita H (1997). Antitumor effect of locally produced CD95 ligand. Nat. Med. 3, 165–170. 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- 15.Maeda K, Nakayama J, Taki S, and Sanjo H (2022). TAK1 Limits Death Receptor Fas-Induced Proinflammatory Cell Death in Macrophages. J. Immunol. 209, 1173–1179. 10.4049/jimmunol.2200322. [DOI] [PubMed] [Google Scholar]
- 16.Park DR, Thomsen AR, Frevert CW, Pham U, Skerrett SJ, Kiener PA, and Liles WC (2003). Fas (CD95) Induces Proinflammatory Cytokine Responses by Human Monocytes and Monocyte-Derived Macrophages. J. Immunol. 170, 6209–6216. 10.4049/jimmunol.170.12.6209. [DOI] [PubMed] [Google Scholar]
- 17.Bossaller L, Chiang P-I, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VAK, Mocarski ES, Subramanian D, Green DR, Silverman N, et al. (2012). Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1b and IL-18 Maturation via Caspase-8 in an RIP3-Independent Manner. J. Immunol. 189, 5508–5512. 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S, Carmona A, Skowronek A, Yu F, Collins MO, Naik S, Murzeau CM, Tseng PL, and Erdmann KS (2019). Apoptotic signalling targets the post-endocytic sorting machinery of the death receptor Fas/CD95. Nat. Commun. 10, 3105. 10.1038/s41467-019-11025-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schüze S, Peter ME, and Chan AC (2006). The role of receptor internalization in CD95 signaling. EMBO J. 25, 1009–1023. 10.1038/sj.emboj.7601016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabandhu K, Huault S, Garmy N, Fantini J, Stebe E, Mailfert S, Marguet D, and Hueber AO (2008). The extracellular glycosphingolipid-binding motif of Fas defines its internalization route, mode and outcome of signals upon activation by ligand. Cell Death Differ. 15, 1824–1837. 10.1038/cdd.2008.115. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabandhu K, Hérincs Z, Huault S, Dost B, Peng L, Conchonaud F, Marguet D, He HT, and Hueber AO (2007). Palmitoylation is required for efficient Fas cell death signaling. EMBO J. 26, 209–220. 10.1038/sj.emboj.7601456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ram DR, Ilyukha V, Volkova T, Buzdin A, Tai A, Smirnova I, and Poltorak A (2016). Balance between short and long isoforms of cFLIP regulates Fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci. USA 113, 1606–1611. 10.1073/pnas.1517562113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, and Salvesen GS (2003). A unified model for apical caspase activation. Mol. Cell 11, 529–541. 10.1016/S1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 24.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, and Peter ME (1998). Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17, 1675–1687. 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tummers B, Mari L, Guy CS, Heckmann BL, Rodriguez DA, Rühl S, Moretti J, Crawford JC, Fitzgerald P, Kanneganti TD, et al. (2020). Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 52, 994–1006.e8. 10.1016/j.immuni.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muendlein HI, Connolly WM, Cameron J, Jetton D, Magri Z, Smirnova I, Vannier E, Li X, Martinot AJ, Batorsky R, and Poltorak A (2022). Neutrophils and macrophages drive TNF-induced lethality via TRIF/CD14-mediated responses. Sci. Immunol. 7, eadd0665. 10.1126/sciimmunol.add0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, and Poltorak A (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis duringYersinia infection. Proc. Natl. Acad. Sci. USA 115, E10888–E10897. 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FKM, Kelliher MA, et al. (2014). Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. USA 111, 7391–7396. 10.1073/pnas.1403477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, and Poltorak A (2020). CFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 367, 1379–1384. 10.1126/science.aay3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muendlein HI, Connolly WM, Magri Z, Smirnova I, Ilyukha V, Gautam A, Degterev A, and Poltorak A (2021). ZBP1 promotes LPS-induced cell death and IL-1β release via RHIM-mediated interactions with RIPK1. Nat. Commun. 12, 86. 10.1038/s41467-020-20357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, and Tschopp J (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. http://immunol.nature.com. [DOI] [PubMed] [Google Scholar]
- 32.Geserick P, Hupe M, Moulin M, Wong WWL, Feoktistova M, Kellert B, Gollnick H, Silke J, and Leverkus M (2009). Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187, 1037–1054. 10.1083/jcb.200904158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanger BZ, Leder P, Lee TH, Kim E, and Seed B (1995). RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81, 513–523. 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 34.Morgan MJ, Kim Y-S, and Liu Z.g. (2009). Membrane-Bound Fas Ligand Requires RIP1 for Efficient Activation of Caspase-8 within the Death-Inducing Signaling Complex. J. Immunol. 183, 3278–3284. 10.4049/jimmunol.0803428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, and Kagan JC (2011). CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell 147, 868–880. 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciesielska A, Matyjek M, and Kwiatkowska K (2021). TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 78, 1233–1261. 10.1007/s00018-020-03656-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Blüml S, Grebien F, Bruckner M, Pasierbek P, Aumayr K, Planyavsky M, et al. (2010). CD14 is a coreceptor of toll-like receptors 7 and 9. J. Exp. Med. 207, 2689–2701. 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raby AC, Holst B, Le Bouder E, Diaz C, Ferran E, Conraux L, Guillemot JC, Coles B, Kift-Morgan A, Colmont CS, et al. (2013). Targeting the TLR co-receptor CD14 with TLR2-derived peptides modulates immune responses to pathogens. Sci. Transl. Med. 5, 185ra64–12. 10.1126/scitranslmed.3005544. [DOI] [PubMed] [Google Scholar]
- 39.Lee HK, Dunzendorfer S, Soldau K, and Tobias PS (2006). Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity 24, 153–163. 10.1016/j.immuni.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 40.Bando M, Miyake Y, Shiina M, Wachi M, Nagai K, and Kataoka T (2002). Actin cytoskeleton is required for early apoptosis signaling induced by anti-Fas antibody but not Fas ligand in murine B lymphoma A20 cells. Biochem. Biophys. Res. Commun. 290, 268–274. 10.1006/bbrc.2001.6199. [DOI] [PubMed] [Google Scholar]
- 41.Chaigne-Delalande B, Mahfouf W, Daburon S, Moreau JF, and Legembre P (2009). CD95 engagement mediates actin-independent and -dependent apoptotic signals. Cell Death Differ. 16, 1654–1664. 10.1038/cdd.2009.111. [DOI] [PubMed] [Google Scholar]
- 42.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. (2005). CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6, 565–570. 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 43.Kaiser WJ, and Offermann MK (2005). Apoptosis Induced by the Toll-Like Receptor Adaptor TRIF Is Dependent on Its Receptor Interacting Protein Homotypic Interaction Motif. J. Immunol. 174, 4942–4952. 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 44.Laurien L, Nagata M, Schünke H, Delanghe T, Wiederstein J, Kumari S, Schwarzer R, Corona T, Kruger M, Bertrand M, et al. (2020). Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 11, 1747. 10.1038/s41467-020-15466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al. (2014). RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202. 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 47.Wu Z, Rothwell L, Hu T, and Kaiser P (2009). Chicken CD14, unlike mammalian CD14, is trans-membrane rather than GPI-anchored. Dev. Comp. Immunol. 33, 97–104. 10.1016/j.dci.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 48.Zamani F, Zare Shahneh F, Aghebati-Maleki L, and Baradaran B (2013). Induction of CD14 expression and differentiation to monocytes or mature macrophages in promyelocytic cell lines: New approach. Adv. Pharmaceut. Bull. 3, 329–332. 10.5681/apb.2013.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oyaizu N, Kayagaki N, Yagita H, Pahwa S, and Ikawa Y (1997). Requirement of cell-cell contact in the induction of Jurkat T cell apoptosis: The membrane-anchored but not soluble form of fasL can trigger anti-CD3-induced apoptosis in Jurkat T cells. Biochem. Biophys. Res. Commun. 238, 670–675. 10.1006/bbrc.1997.7357. [DOI] [PubMed] [Google Scholar]
- 50.Moffatt OD, Devitt A, Bell ED, Simmons DL, and Gregory CD (1999). Macrophage Recognition of ICAM-3 on Apoptotic Leukocytes. J. Immunol. 162, 6800–6810. 10.4049/jimmunol.162.11.6800. [DOI] [PubMed] [Google Scholar]
- 51.Zanoni I, Tan Y, Di Gioia M, Springstead JR, and Kagan JC (2017). By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity 47, 697–709.e3. 10.1016/j.immuni.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim OH, Kang GH, Hur J, Lee J, Jung Y, Hong IS, Lee H, Seo SY, Lee DH, Lee CS, et al. (2022). Externalized phosphatidylinositides on apoptotic cells are eat-me signals recognized by CD14. Cell Death Differ. 29, 1423–1432. 10.1038/s41418-022-00931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf NK, Kissiov DU, and Raulet DH (2023). Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 23, 90–105. 10.1038/s41577-022-00732-1. [DOI] [PubMed] [Google Scholar]
- 54.Wang N, Liu W, Zheng Y, Wang S, Yang B, Li M, Song J, Zhang F, Zhang X, Wang Q, and Wang Z (2018). CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-κB/SOX4 signaling. Cell Death Dis. 9, 880. 10.1038/s41419-018-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not include original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.