

Abstract
Background
Inositol‐requiring enzyme 1 (IRE1) is an endoplasmic reticulum (ER)‐resident transmembrane protein that senses ER stress and mediates an essential arm of the unfolded protein response (UPR). IRE1 reduces ER stress by upregulating the expression of multiple ER chaperones through activation of X‐box‐binding protein 1 (XBP1). Emerging lines of evidence have revealed that IRE1‐XBP1 axis serves as a multipurpose signal transducer during oncogenic transformation and cancer development. In this study, we explore how IRE1‐XBP1 signaling promotes chemoresistance in lung cancer.
Methods
The expression patterns of UPR components and MRP1 were examined by Western blot. qRT‐PCR was employed to determine RNA expression. The promoter activity was determined by luciferase reporter assay. Chemoresistant cancer cells were analyzed by viability, apoptosis. CUT & Tag (Cleavage under targets and tagmentation)‐qPCR analysis was used for analysis of DNA‐protein interaction.
Results
Here we show that activation of IRE1α‐XBP1 pathway leads to an increase in MDR‐related protein 1 (MRP1) expression, which facilitates drug extrusion and confers resistance to cytotoxic chemotherapy. At the molecular level, XBP1‐induced c‐Myc is necessary for SREBP1 expression, and SREBP1 binds to the MRP1 promoter to directly regulate its transcription.
Conclusions
We conclude that IRE1α‐XBP1 had important role in chemoresistance and appears to be a novel prognostic marker for lung cancer.
Keywords: chemoresistance, IRE1α, MRP1, SREBP1, XBP1
Our investigation revealed that activation of IRE1α‐XBP1 pathway leads to an increase in MDR‐related protein 1 (MRP1) expression, which facilitates drug extrusion and confers resistance to cytotoxic chemotherapy. At the molecular level, SREBP1 directly controls the transcription of MRP1 by binding to its promoter, and its expression is dependent on XBP1‐induced c‐Myc.
INTRODUCTION
Lung cancer continues to the most lethal cancer globally, despite recent advancements in targeted and immunotherapy treatment. Even though new therapeutic developments have significantly improved the prognosis for some non‐small cell lung cancer (NSCLC) patients, approximately 60% of NSCLC cases lack a targetable driver mutation, and only about 20% cases may benefit from PD‐1 or PD‐L1 therapy. 1 , 2 , 3 , 4 As a result, systemic chemotherapy is still primary treatment for NSCLC. However, cancer cells are naturally able to handle the additional stress, they may become resistant to chemotherapy (chemoresistance). Increased drug efflux, drug inactivation, and modifications to drug targets, can lead to drug elimination within tumors and adaptation to chemotherapeutics. 5 , 6
About one‐third of the protein in a cell is synthesized, folded, and transported by the endoplasmic reticulum (ER), which plays a significant role in maintaining the homeostasis of proteins within the cell. In addition, the ER plays a critical role in controlling several metabolic processes including carbohydrate metabolism, lipid biosynthesis, and calcium ion homeostasis. Numerous internal and external insults constantly threaten the integrity of the ER, and many of which can activate the unfolded protein response (UPR), a signaling pathway that is triggered to alleviate ER stress and restore metabolic and protein processing functions. 7 PERK, IRE1α, and ATF6 are three ER stress sensors that are activated during UPR. Among these UPR sensors, inositol‐regulated kinase 1 (IRE1, also known as ERN1) is most evolutionarily conserved and widely expressed in mammalian cells. Mammals have two IRE1 isoforms, designated IRE1α and IRE1β, with IRE1α found to be ubiquitously expressed, whereas IRE1β expression is restricted to intestinal cells. 8 IRE1α is a single transmembrane protein kinase that also displays nuclease activity. Ire1α‐dependent splicing activity is responsible for the accumulation of XBP transcription factor (XBP1s). 8 By activating XBP1, IRE1α increases the expression of numerous ER chaperones to reduce ER stress.
Due to the fact that cancer typically arises and progresses in a stressful microenvironment, cancer cells may utilize UPR activation as a survival strategy. 9 Previous studies have demonstrated that activation of the UPR is associated with several aspects of cancer progression in multiple types of cancer. 9 One crucial role of IRE1α‐XBP1s in cancer development is to trigger oncogenic c‐MYC signaling. 10 MYC is a basic helix‐loop‐helix leucine zipper (bHLH‐LZ) transcription factor capable of forming heterodimers with another bHLH‐LZ protein, MAX, binding to the E‐box element CACGTG, and regulating hundreds of expressed genes, either positively or negatively, depending on the cellular context. MYC affects basic cellular functions like growth, division, apoptosis, and energy metabolism. 11 , 12 It is regulated both transcriptionally and post‐transcriptionally. Overexpression of MYC in various cell types directly leads to malignant transformation and is a hallmark of many human cancers. 11
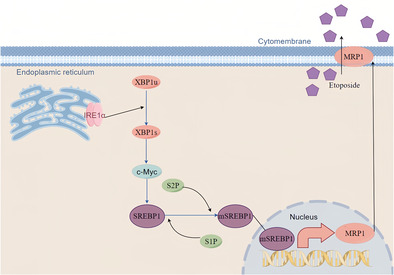
Sterol regulatory element‐binding proteins (SREBPs) are membrane‐bound transcription factors involved in lipid metabolism. 13 They possess a cytoplasmic N‐terminal domain belonging to the basic helix‐loop‐helix‐leucine zipper (bHLH‐LZ) family, which, upon binding to sterol response elements (SREs) in gene promoters, initiates gene transcription. The C‐terminal domain of SREBPs interacts with the SREBP‐cleavage activating protein (SCAP) in the ER membrane. In the presence of high cellular cholesterol levels, INSIG stabilizes and binds to SREBP‐SCAP, retaining this complex in the ER membrane. Conversely, under low cholesterol conditions, SCAP changes conformation, leading to INSIG release. Subsequently, the activated SREBP/SCAP complex translocate from the ER to the Golgi apparatus for proteolytic processing, liberating the active N‐terminal of SREBP. This enables SREBP to enter the nucleus, bind to SREs, and regulate gene expression. 13 Over the past three decades, SREBPs have been recognized for their multifunctional roles, integrating diverse cellular signals to modulate pathways crucial for lipid synthesis and various biological processes like ER stress, inflammation, autophagy, and apoptosis. Consequently, SREBPs play a significant role in the pathogenesis of diseases such as diabetes, fatty liver disease, chronic kidney disease (CKD), neurodegenerative disorders, and cancer. 14 , 15 , 16 Understanding how these signal pathways precisely regulate one another's functions, how they are interdependent will be critical. In this study, we have revealed the essential function of the IRE1α‐XBP1 axis in NSCLC chemoresistance. Mechanistically, UPR activation leads to an increase in ATP‐binding cassette (ABC) transporter MRP1 expression, which facilitates drug extrusion and confers resistance to cytotoxic chemotherapy. At the molecular level, ER stress‐induced c‐Myc is necessary for SREBP1 expression, and SREBP1 binds to the MRP1 promoter to directly regulate its transcription. Our findings identify a novel mechanism of NSCLC's chemoresistance, which is mediated by UPR activation.
MATERIALS AND METHODS
Cells, transfection, antibodies, plasmids, reagents
The A549 and H1299 cell lines, both of which are human lung adenocarcinoma, were cultured in DMEM (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS, Gibco BRL, Grand Island, NY, USA) at 37°C in a 5% CO2 humidified environment. To facilitate transfection, A549 and H1299 cells were seeded in six‐well plates (105 cells per well) and grown until they reached 70% confluence before transfecting plasmids using PolyJet DNA Transfection Reagent (SignaGen Laboratories, Gaithersburg, MD, USA) following the manufacturer's instructions. Anti‐ATF6 (K001590P), Anti‐MRP1 (K003560P), and Anti‐GPR78 (K000234M) antibodies were purchased from Solarbio (Beijing Solarbio Science & Technology, Beijing, China). Anti‐XBP1 (A1731) antibodies were obtained from ABclonal (ABclonal, Wuhan, China). Anti‐p‐IRE1α, Anti‐PARP (#9542), Anti‐SREBP1 (#95879), and Anti‐c‐Myc (#9402) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐β‐actin (#A1978) antibodies were obtained from Sigma (Victoria, BC, Canada). The plasmids of pCMV5‐Flag‐XBP1s (no. 63680), IRE1 alpha‐pcDNA3.EGFP (no. 13009), and pcDNA3.3‐c‐Myc were purchased from Addgene (Cambridge, MA, USA). 4‐Phenylbutyric acid (4‐PBA, P21005) was purchased from Sigma. Thapsigargin (TG, ab120286) from Abcam. Cisplatin solution (H20040813) was purchased from HANSOH PHARMA (Jiangsu, China). Doxorubicin Hydrochloride was obtained from Shenzhen Main Luck Pharmaceuticals Inc. (Shen zhen, China). Sequences of siRNA are listed in Table 1.
TABLE 1.
Sequences of siRNA.
MTT assay
Cell viability was determined using a 3‐(4,5‐dimethylthiazole‐2‐yl)‐2.5‐diphenyl tetrazolium bromide (MTT) (Beyotime Institute of Biotechnology, Shanghai, China) following the designated treatment, cells underwent a PBS wash and were subsequently incubated with fresh medium containing MTT solution (0.5 mg/mL) for 4 h at a temperature of 37°C. The resultant formazan crystals were dissolved using dimethyl sulfoxide (DMSO), and the absorbance was measured at a wavelength of 570 nm using a BioTek citation 5 plate reader (BioTek, Winooski, VT, USA).
Analysis of apoptosis
To assess cell apoptosis, the Annexin V‐FITC/PI assay was employed, utilizing the Annexin V‐FITC Apoptosis Detection Kit (KeyGEN Biotech, Jiangsu, China). Flow cytometric analyses were rigorously conducted using a BD FACSVerse Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Briefly, cells were harvested and stained with Annexin V‐FITC and PI at room temperature. The apoptotic index was derived from the percentage of apoptotic A549 and H1299 cells. To ensure robustness and reproducibility, this procedure was repeated across three independent experiments.
Western blot
After subjecting the cells to treatment, they were scraped and homogenized utilizing heated Laemmli sample buffer (S3401, Sigma, Victoria, BC, Canada). The resultant homogenate was then separated using SDS‐PAGE and subsequently transferred to a nitrocellulose (NC) membrane (GE Healthcare, Piscataway, NJ, USA). Upon application of different antibodies for detection, the signals were captured and scanned using the Chemiluminescence system AI680 (Protein Simple, San Jose, CA, USA).
Nuclear and cytoplasmic fractions were prepared using the cytoplasmic and nuclear protein extraction kit (Beyotime). Briefly, NSCLC cells were lysed with buffer A (50‐mM Tris–HCl [pH 8.0], 10‐mM NaCl, 5‐mM MgCl2, and 0.5% [v/v] NP‐40). After a 20‐min incubation period, cytosolic extracts were collected following centrifugation at 12000 g for 5 min. Nuclear pellets were washed with lysis buffer A and subsequently resuspended in lysis buffer B (20‐mM Hepes [pH 7.9], 0.5‐mM NaCl, 1‐mM EDTA, and 1‐mM dithiothreitol [DTT]). After an additional 20‐min incubation, nuclear extracts were obtained by centrifugation at 12000 g for 10 min.
Dual luciferase reporter assays
The reporter constructs were co‐transfected along with the Renilla luciferase vector into the cellular environment. Subsequently, the dual luciferase reporter assay system (Promega) was utilized to ascertain the expression levels of both firefly luciferase and Renilla luciferase. The summarized findings are exhibited as the mean value obtained from triplicate measurements, accompanied by the standard deviation (SD).
Quantitative real‐time RT–PCR analysis
Following cell treatment, Trizor (Invitrogen, Waltham, USA) extraction methodology was used to isolate cellular RNA. Subsequently, reverse transcription into complementary DNA (cDNA) utilizing the PrimeScript First Strand cDNA Synthesis Kit (TaKaRa Bio, DaLian, China) was performed. The TB Green Master Mix (procured from Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) was employed for fluorescence‐based detection on the Roche LightCycler 96 platform (Roche, Switzerland). Triplicate amplification runs were performed for each sample to ensure robustness of results. All RT‐PCR primer sequence are listed in Table 2.
TABLE 2.
Primers used for RT‐PCR amplifications.
| Gene | Genebank accession number | Primer (5′ >3′) |
|---|---|---|
| ABCC1 | NM_004996 | Forward: AGCCGGTGAAGGTTGTGTAC |
| Reverse: TGACGAAGCAGATGTGGAAG | ||
| β‐Actin | NM_001101.5 | Forward: CCAACCGCGAGAAGATGAC |
| Reverse: GAGGCGTACAGGGATAGCACA |
Hoechst 33342 efflux assays
Initiate the experiment by subjecting the cells to TG (30 nM) treatment for a duration of 12 h. Subsequently, a thorough washing of the cells was performed utilizing a 1xPBS solution proceeded by substituting the existing medium with fresh Hoechst medium (10 μg/mL). The cells were allowed to further incubate for 20 min at 37°C in the incubator. The calcium channel blocker verapamil (10 μM) was introduced the experimental setup following this incubation, and subsequently the incubation was continued for an additional 10 min. The fluorescence intensity assessments were conducted employing the BioTek citation 5 system (manufactured by BioTek, Winooski, VT, USA).
Cloning and DNA construction
The SREBP1 promoter is inserted into the pGL3‐BASIC (Promega, Madison, WI, USA) fluorescent plasmid from the Hind III and Kpn I sites. Site‐specific mutagenesis utilizing the overlapping PCR extension technique was employed to introduce point mutations into the SREBP1 promoter, with the longest SREBP1 promoter serving as the template. The primers are listed in Table 3.
TABLE 3.
Primers used for PCR amplifications.
| Gene | Genebank accession number | Primer (5′ >3′) |
|---|---|---|
| ABCC1 | NM_004996 |
Forward: AATACTACTTTCTGATGTCGGGGGTTGTGA Reverse: TCACAACCCCCGACATCAGAAAGTAGTATT |
| ABCC1 | NM_004996 |
Forward: AATACTACTTCTTACCTACTGGGGTTGTGA Reverse: TCACAACCCCCGACATCAGAAAGTAGTATT |
| ABCC1 | NM_004996 |
Forward: CGGGGTACCAACAATCCTATCTACCTTCCTCCT Reverse: CCCAAGCTTCCACACACCCTGCGACCACTTTTCAA |
| SREBF1 | NM_004176 |
Forward: TTTGTTCTCTGTGTTCTCCAGCAGTT Reverse: AAAGTTCCTCGGAAACTGGGTTCC |
Cleavage under targets and tagmentation (CUT&Tag)
We employed Cleavage Under Targets and Tagmentation (CUT&Tag), an immunotethering technique, to verify the in vivo binding of SREBP1 to the promoter of MRP1. The experiment was conducted following the manufacturer's instructions for the Novo CUT&Tag High‐Sensitivity Kit (N259‐YH01, Novoprotein, China). After harvesting and washing of cells in Wash Buffer. The cells, bound to ConA magnetic beads, were subsequently resuspended in 50 μL of precooled Primary Antibody Buffer containing 1 μg of specific primary antibody (SREBP1). IgG served as the control antibody. Following the incubation with the primary antibody, the samples were washed to remove unbound primary antibodies and then resuspended in Wash Buffer containing the secondary antibody. These samples were incubated at 37°C for 1 h. Subsequently, the cells were resuspended in Tagmentation Buffer and incubated again at 37°C for 1 h. Beads were added to each tube to extract the DNA. The purified DNA obtained from this process was utilized as a template for standard PCR and quantitative real‐time PCR (qRT‐PCR). The standard PCR products were analyzed on a 1.2% agarose gel to visualize qRT‐PCR results. The primer for CUT&Tag are listed in Table 4.
TABLE 4.
Primers used for CUT&tag qRT‐PCR amplifications.
| Gene | Genebank accession number | Primer (5′ >3′) |
|---|---|---|
| ABCC1 | NM_004996 | Forward: GGCTTAATCACTTACCATCT |
| Reverse: TTCCTAAGGCTGTGTTCTT |
Statistical analysis
Statistical assessments were executed utilizing analysis of variance (ANOVA) via GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA, USA). The results are expressed as the mean ± standard deviation (S.D.) derived from three distinct and replicated experimental iterations. Statistical significance was deemed achieved when the p‐value resulting from a two‐tailed test fell below 0.05.
RESULTS
Activation of IRE1α‐XBP1 axis of UPR induces MRP1 expression and chemoresistance in NSCLC cells
We first examined the relationship between ER stress and chemotherapy resistance in NSCLC. We pretreated NSCLC cell lines A549 and H1299 with ER stress inducer Thapsigargin (TG) for 2 h, then treated the cells with chemotherapeutic drug etoposide, and found that the TG‐treated cells showed enhanced etoposide resistance at different time points and different concentrations (Figure 1a,b). Apoptosis experiments also confirmed that TG could induce NSCLC resistance to etoposide‐induced apoptosis (Figure 1c). ER stress maintains cell homeostasis through three classical pathways. By bioinformatics analysis, we found that one of the pathways, IRE1α encoded by ERN1 and chemotherapeutic drug efflux protein ABCC1 gene were positively correlated (Figure 1d). ABCC1 encodes the MDR‐related protein 1 (MRP1) protein, which is highly expressed in lung cancer (Figure 1e) and negatively correlated with the prognosis in NSCLC (Figure 1f). Western blot results showed that TG not only induced ER stress but also dose‐dependently induced MRP1 protein expression, this was paralleled with IRE1α‐XBP1 downstream target Bip/GPR78 expression (Figure 1g). Moreover, overexpression of IRE1α cDNA in A549 and H1299 cells significantly increased MRP1 expression (Figure 1h). IRE1α's function is to induce the splicing and nuclear translocation of transcription factor XBP1. Thus, the control and TG‐treated cells were fractionated to cytoplasm and nuclear fractions, and the spliced XBP1s levels was examined by Western blot. As expected, TG‐treated cells had significant nuclear XBP1s protein (Figure 1i). Furthermore, overexpression of active XBP1s cDNA also significantly increased MRP1 expression (Figure 1j). Using the ER stress inhibitor 4‐PBA to pretreat cells, we found that 4‐PBA reversed the TG‐induced MRP1 in parallel with Bip/GPR78 expression (Figure 1k). Significantly, TG‐induced chemoresistance, as indicated by increased cell survival, was reversed by depletion of either IRE1α, XBP1, or MRP1 (Figure 1l–n). These results demonstrate that the IRE1α‐XBP1 pathway leads to the induction of MRP1 expression, which in turn increases chemoresistance.
FIGURE 1.
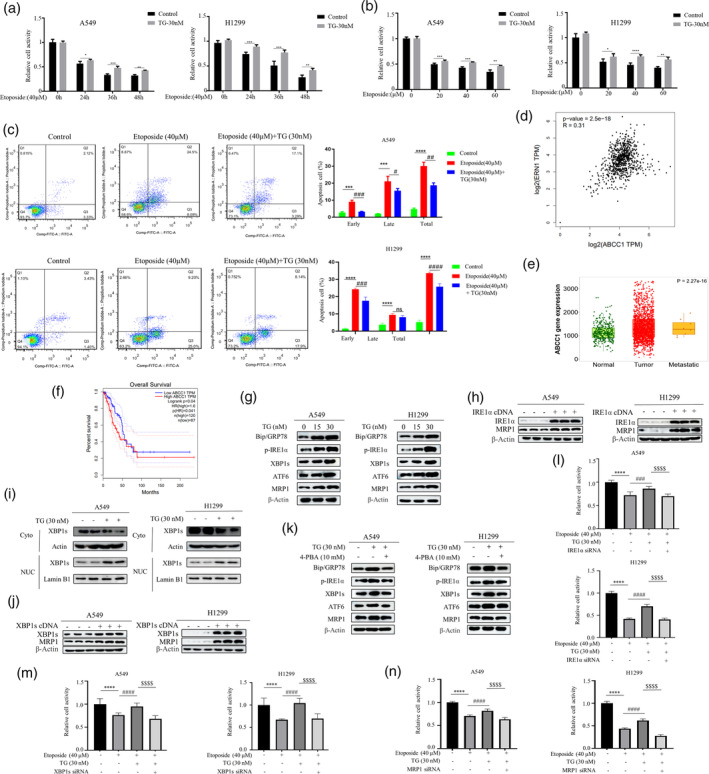
Endoplasmic Reticulum Stress Orchestrates Chemotherapy Resistance Through the IRE1α‐XBP1‐MRP1 Axis. (a) The A549 and H1299 cells were treated with etoposide (40 μM) for different time point (left panel) and various concentrations (right panel) in the presence or absence of TG (30 nM). MTT assay was used to detect cell viability (*p < 0.05, **p < 0.01, ***p < 0.001 for difference from control cells). (b) The A549 and H1299 cells were pretreated with TG (30 nM) for 2 h and then were treated with etoposide (20, 40, and 60 μM) for 48 h. Cell viability was assessed using the MTT assay. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 for difference from control cells). (c) H1299 and A549 cells were treated with etoposide (40 μM) in the presence or absence of TG (30 nM). Flow cytometry analysis revealed that TG could induce resistance to etoposide in NSCLC. The bars represent the mean ± S.D. of triplicates (***p < 0.001, ****p < 0.0001 for difference from control cells, # p < 0.05, ## p < 0.01, ### p < 0.001, #### p < 0.0001 for difference from etoposide‐treated cells by ANOVA with Dunnett's correction for multiple comparisons, ns means no statistical difference). (d) The correlation between ABCC1 with UPR related gene ERN1 (IRE1) expression from GEPIA data (GEPIA‐LUAD). (e) The results of the TCGA database statistical analysis reveal that the MRP1 protein encoded by ABCC1 is highly expressed in cancer cells. (f) The results of the GEPIA database reveal a correlation between the expression of MRP1 and the prognosis of lung cancer cell. (g) Western blot revealed that treating H1299 and A549 cells with TG (15, 30 nM) for 48 h resulted in a dose‐dependent enhancement of MRP1 protein expression. (h) The cells were transfected with IRE1α and XBP1s cDNA for 48 h. Western blot analysis demonstrated an increase in the expression level of MRP1 upon overexpression of IRE1α and XBP1s. (i) Cells treated with TG (30 nM) for 12 h or untreated subjected to subcellular fractionation. Equivalent celly lysates from cytoplasmic and nuclear fraction were analyzed by Western blot for subcellular localization of XBP1s. (j) After transfecting cells with XBP1s cDNA, Western blot analysis revealed the expression level of XBP1s and MRP1. (k) Western blot analyzed the expression levels of Bip/GRP78, phosphorylated IRE1α, XBP1s, ATF6, and MRP1 after treatment A549 and H1299 cells with 4‐PBA (10 mM) in the presence or absence of TG (30 nM). (l–n) The A549 and H1299 cells were first transfected with siRNA of IRE1α, XBP1s or MRP1. At 48 h post‐transfection, cells were treated with etoposide for 24 h in the presence and absence of TG followed by MTT assay (****p < 0.001 for difference from control cells, #### p < 0.0001 for the difference from etoposide‐treated cells, $$$$ p < 0.001 for difference from etoposide and TG‐treated cells by ANOVA with Dunnett's correction for multiple comparisons).
Induction of MRP1 mediates ER stress‐induced chemoresistance
MRP1 belongs to the ABC transporter protein family. It has been established that the expression of ABC transporter protein family contributes to drug efflux, leading to cellular resistance to cytotoxic chemotherapy. Therefore, we first conducted Hoechst efflux experiments in TG‐treated cells to detect the efflux of the fluorescent dye Hoechst 33342. As seen in Figure 2a, the fluorescent intensity in TG‐treated cells decreased, indicating that TG was able to efflux the Hoechst dye. The decreased apoptosis and increased cell survival of TG‐treated cells upon exposure to the established MRP/ABCC substrates Doxorubicin and Cisplatin is also in line with MRP1 mediating cellular efflux of a variety of xenobiotics (Figure 2b,c). Crucially, depletion of MRP1 by siRNA rescued increased cell apoptosis in TG‐treated cells, manifested by decreased cell survival (Figure 2d) and increased levels of cleaved‐PARP (Figure 2e). Overall, the data indicate that TG promotes chemoresistance by upregulating MRP1.
FIGURE 2.
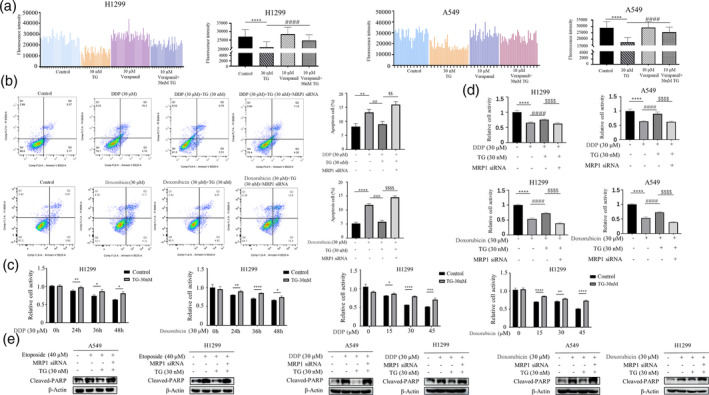
Reversal of MRP1 Enhances Cellular Sensitivity to Drugs. (a) After treating cells with TG (30 nM) for 12 h, cells were either left untreated or treated with the ABC transporter inhibitor Verapamil (10 μM). Representative histogram profiles of untreated cells (control), cells treated with 30 nM TG, 10 μM Verapamil, and the combined treatment of TG and Verapamil. The quantitative results are presented in the right panel, with data represented as the mean ± S.D. of triplicate measurements. Statistical analysis was performed using ANOVA with Dunnett's correction for multiple comparisons, indicating ****p < 0.0001 for the difference from control cells and #### p < 0.0001 for the difference from TG‐treated cells. (b) H1299 and A549 cells were first transfected with MRP1 siRNA, At 48 h post‐transfection, cells were treated with Diaminodichloroplatinum (DDP, 30 μM) or Doxorubicin (30 μM) in the presence or absence of TG (30 nM). Flow cytometry analysis revealed MRP1 siRNA rescued increased cell apoptosis in TG‐treated cells depletion of MRP1 by siRNA rescued increased cell apoptosis in TG‐treated cells (**p < 0.01 for difference from control cells, ## p < 0.01 for the difference from DDP or Doxorubicin‐treated cells, $$ p < 0.01 for difference from DDP or Doxorubicin and TG‐treated cells by ANOVA with Dunnett's correction for multiple comparisons). (c) The A549 and H1299 cells were treated with DDP or Doxorubicin for different time point (Left panel) and various concentrations (Right panel) in the presence or absence of TG (30 nM). MTT assay was used to detect cell viability (*p < 0.05, **p < 0.01, ***p < 0.001 for difference from control cells). (d) After transfecting with MRP1 siRNA for 48 h, cells were treated with DDP (30 μM) or Doxorubicin (30 μM) for 36 h in the presence or absence of TG (30 nM). MTT assay was used to detect cell viability. (****p < 0.001 for difference from control cells, #### p < 0.0001 for the difference from DDP or Doxorubicin‐treated cells, $$$$ p < 0.001 for difference from DDP or Doxorubicin and TG‐treated cells by ANOVA with Dunnett's correction for multiple comparisons). (e) The A549 and H1299 cells were first transfected with MRP1 siRNA. At 48 h post‐transfection, cells were treated with etoposide, DDP, and Doxorubicin in the presence and absence of TG followed by Western blot analysis of Cleaved‐PARP.
SREBP1 mediates ER stress‐induced MRP1 expression
Our next goal was to investigate the molecular mechanism by which ER stress promotes MRP1 expression. We first used quantitative PCR to measure the mRNA level of MRP1 in TG‐treated A549 cells and found that TG dose‐dependently promoted MRP1 mRNA expression (Figure 3a). We also found that TG enhanced the activity of the MRP1 promoter (Figure 3b). This suggests that TG induces MRP1 expression at the transcriptional level. Therefore, we hypothesized that XBP1 might bind to the MRP1 promoter to directly promote its transcription. However, this hypothesis was not supported as we did not find any XBP1 binding sites in the MRP1 promoter sequence by bioinformatics analysis. Our previous work has shown that ER stress can induce metabolic reprogramming in lung cancer cells. 17 We found two putative sterol regulatory element‐binding protein 1 (SREBP1) binding sites on the MRP1 promoter (Figure 3c). SREBP1 plays a key role in tumor lipid metabolism. Therefore, we next want to explore whether SREBP1 mediates ER stress‐induced MRP1 expression. Overexpression of the full‐length SREBP1 cDNA not only increased the cleaved and active SREBP1, but also greatly increased the expression of the MRP1 protein (Figure 3d). Similarly, knocking down SREBP1 greatly reduced MRP1 expression (Figure 3e). We simultaneously found that TG and overexpression of IRE‐1α could induce active cleavage of SREBP1 and MRP1 expression. Importantly, TG and overexpression of IRE‐1α‐induced MRP1 expression was reversed by SREBP1 siRNA (Figure 3f,g). To further explore this, we also probed for the subcellular location of SREBP1. After IRE‐1α overexpression, SREBP1 expression in the nucleus was found to be significantly higher, as shown in Figure 3h. These results indicate that SREBP1 mediates ER stress‐induced MRP1 induction. To prove that SREBP1 directly regulates MRP1 transcription, we cloned the MRP1 promoter containing two predicted SREBP1 binding sites (1412) into a luciferase reporter gene (Figure 3c). As expected, overexpression of SREBP1 strongly increased the activity of the MRP1 promoter (Figure 3i), while knock‐down of SREBP1 markedly reduced the activity of the MRP1 promoter (Figure 3j). Markedly, TG and overexpression of IRE‐1α‐increased the activity of the MRP1 promoter was reversed by SREBP1 siRNA transfection (Figure 3k). Additionally, we used site‐directed mutagenesis to mutate the binding site of SREBP1, which is distant from the transcription start site, to non‐binding site (1412‐Mut) (Figure 3l upper panel). As shown in Figure 3m, overexpression of SREBP1 also significantly enhanced the activity of the MRP1 promoter, and crucially, mutation of the SREBP1 binding site abolished the responsiveness of the MRP1 promoter activity to SREBP1 overexpression. At the same time, we subcloned the reporter construct to contain only one SREBP1 binding site (408) (Figure 3l lower panel). Surprisingly, overexpression of SREBP1 did not increase the fluorescence intensity of the reporter gene, which contained only one SREBP1 binding site (Figure 3n). In order to confirm that SREBP1 binds the promoter of MRP1 directly, we conducted CUT&Tag (Cleavage under targets and tagmentation)‐qPCR analysis, a recently developed genome‐wide immunotethering assay for protein‐DNA interaction. When SREBP1 was expressed ectopically in cells, the region covering the SREBP1 binding site was specifically immunoprecipitated with ChIP‐grade SREBP1 antibody (Figure 3o). In addition, CUT&Tag‐qPCR assay did not reveal that XBP1 binds this region of MRP1 promoter (Figure 3o). Therefore, we concluded that SREBP1 binds to one of the sites to directly regulate transcription of MRP1. More importantly, SREBP1 siRNA was able to reverse TG‐induced resistance to etoposide (Figure 3p), indicating SREBP1 is relevant mechanism of TG‐induced chemoresistance.
FIGURE 3.
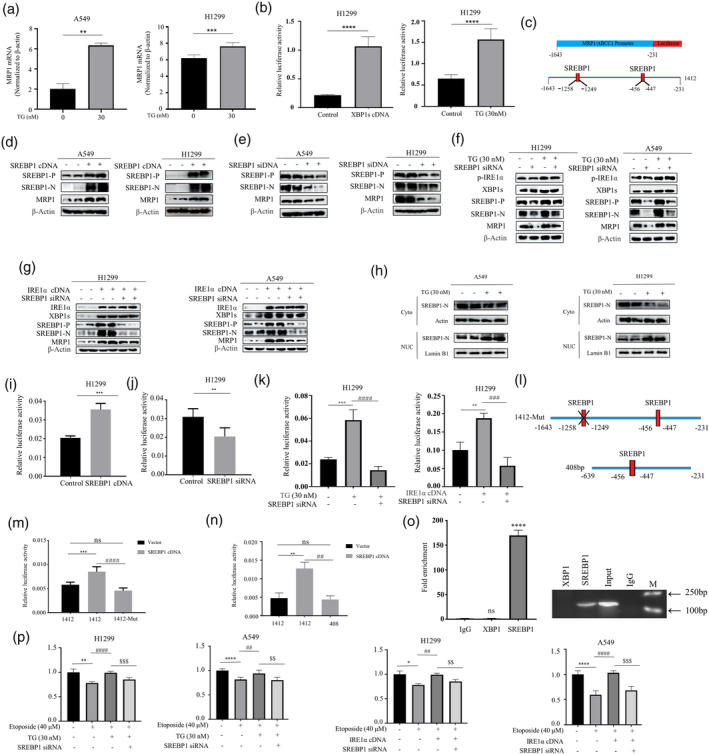
SREBP1 Mediated Regulation of MRP1 Expression Induced by Endoplasmic Reticulum Stress. (a) After treating cells with TG (30 nM), the mRNA levels of MRP1 were assessed by real‐time qPCR. (b) TG (30 nM) and exogenous XBP1 cDNA significantly increased the promoter activity of MRP1. (c) There are two putative binding sites for SREBP1 on the MRP1 promoter. (d) Following 48‐h transfection of SREBP1 cDNA, Western blot results revealed the expression of full‐length SREBP1, cleaved (active) SREBP1, and MRP1. (e) H1299 and A549 cells were transfected for 48‐h with SREBP1 siRNA or control siRNA. Western blot results revealed the expression of full‐length SREBP1, cleaved SREBP1 (active), and MRP1. (f) After transfection cells with SREBP1 siRNA, cells were further treated with TG (30 nM) for an additional 12 h. Western blot analysis revealed the expression of p‐IRE1α, XBP1s, SREBP‐P, SREBP‐N, and MRP1. (g) 48 h before Western blot experiments, cells were co‐transfected with IRE1α cDNA and SREBP1 siRNA. Western blot analysis revealed the expression of IRE1α, XBP1s, SREBP‐P, SREBP‐N, and MRP1. (h) Western blot analysis of cytosolic and nuclear extracts of SREBP1from TG (30 nM) or untreated cells. (i) H1299 cells were co‐transfected with MRP1 promoter reporter construct 1412 and SREBP1 cDNA plasmid. 48 h after transfection, luciferase activity was determined and normalized using the dual luciferase reporter system (***p < 0.001 for difference from control cells). (j) H1299 cells were co‐transfected with MRP1 promoter reporter construct 1412 and SREBP1 siRNA. Transfection of SREBP1 siRNA significantly reduced the promoter activity of MRP1. (k) The cells were co‐transfected with MRP1 promoter and SREBP1 siRNA in the presence and absence of TG (30 nM) (Left panel) or co‐transfection with IRE1α cDNA (Right panel). Luciferase activity was determined and normalized using the dual luciferase reporter system (**p < 0.01, ***p < 0.001 for difference from control cells, ### p < 0.001 for the difference from TG or transfected with IRE1α cDNA cells by ANOVA with Dunnett's correction for multiple comparisons). (l) Schematic representation of two designated constructs of MRP1 promoter (1412‐Mut and 408). (m) Cells were co‐transfected either MRP1 promoter reporter construct 1412 or mutated MRP1 promoter reporter (1412‐Mut) with SREBP1 cDNA. After 48‐h transfection, luciferase activity was determined and normalized using the dual luciferase reporter system. (***p < 0.001 for difference of wild‐type MRP1 promoter, #### p < 0.0001 for difference of mutated MRP1 promoter). (n) The cells were either co‐transfected MRP1 promoter reporter construct 1412 or the short length MRP1 promoter construct 408 with SREBP1 cDNA. Luciferase activity was determined and normalized using the dual luciferase reporter system. (o) qRT‐PCR were taken to analyze the CUT&Tag results. qRT‐PCR products were run on a gel (Right panel). qRT‐PCR results were quantified and are indicated (Left panel) (***p < 0.001 for difference from IgG‐added cells). (p) After transfecting with SREBP1 siRNA alone or co‐transfected with IRE1α cDNA for 48 h, cells were treated with etoposide (40 μM) in the presence or absence of TG (30 nM). MTT assay was used to detect cell viability. (****p < 0.001 for difference from control cells, #### p < 0.0001 for the difference from etoposide‐treated cells, $$$$ p < 0.001 for difference from SREBP1 siRNA and TG‐treated cells by ANOVA with Dunnett's correction for multiple comparisons).
Induction of c‐Myc by IRE1‐XBP1 is required for SREBP1‐induced chemoresistance
The results so far suggest that ER stress‐induced SREBP1 contributes to chemotherapy resistance by increasing the expression of MRP1. It remained unclear how IRE1‐XBP1 upregulates SREBP1. Unexpectedly, no XBP1 binding sites were found on the SREBP1 promoter. According to the literature review, we found that XBP1 can activate c‐Myc in prostate cancer cells and natural killer cells, 10 , 18 while c‐Myc can activate SREBP1 in hepatocellular carcinoma cells. 19 Therefore, we aim to determine whether c‐Myc mediates induction of SREBP1 by XBP1 in lung cancer cells. We found that overexpression of c‐Myc indeed induced cleavage of SREBP1 and promoter activity of SREBP1 (Figure 4a,b). Importantly, TG dose‐dependently upregulated c‐Myc expression in lung cancer cells (Figure 4c). Likewise, overexpression of IRE1α and XBP1 cDNA also promoted c‐Myc expression (Figure 4d,e). Importantly, the SREBP1 induced by IRE1α and XBP1 cDNA could be reversed by c‐Myc siRNA (Figure 4f,g). Crucially, depletion of c‐Myc by siRNA rescued the etoposide‐induced cell death in TG‐treated cells (Figure 4h), indicating that induction of c‐Myc by ER stress is functionally relevant for SREBP1‐induced chemoresistance.
FIGURE 4.
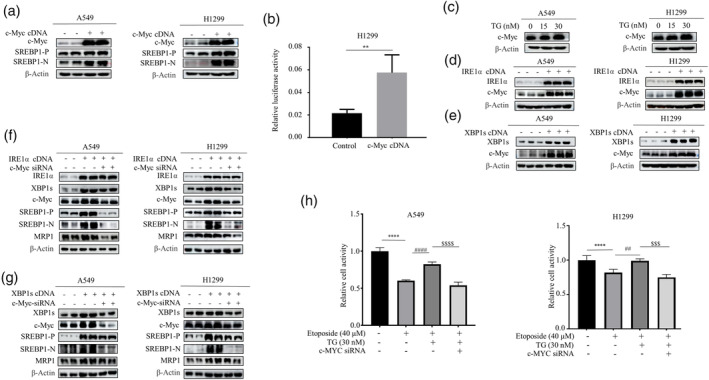
C‐Myc Orchestration of ER Stress‐Induced SREBP1 Activation in Lung Cancer Chemoresistance. (a) Western blot results revealed that transfection of c‐Myc cDNA increased the expression levels of SREBP1. (b) A549 cells were co‐transfected with the MRP1 promoter reporter construct and the c‐Myc cDNA plasmid. luciferase activity was determined and normalized using the dual luciferase reporter system (**p < 0.01, indicating differences relative to the untreated control group, assessed through ANOVA for multiple comparisons). (c) Following a 12‐h treatment with TG (15, 30 nM), Western blotting revealed the expression levels of c‐Myc. (d and e) 48 h after transfection cells with either IRE1α or XBP1s cDNA, Western blot was performed to show an elevation in the expression of IRE1α, XBP1s and c‐Myc. (f and g) Cell lysates were collected for Western blotting 48 h after co‐transfecting IRE1α‐ cDNA (f) or XBP1s‐ cDNA (g), and c‐Myc siRNA. Western blot results revealed the expression of IRE1α, XBP1s, c‐Myc, SREBP1 and MRP1. (h) The A549 and H1299 cells were first transfected with c‐Myc siRNA. After 48 h transfection, cells were treated with etoposide in the presence and absence of TG followed by MTT assay (****p < 0.001 for difference from control cells, #### p < 0.001 for the difference from etoposide‐treated cells, $$$$ p < 0.001 for difference from c‐Myc siRNA and TG‐treated cells by ANOVA with Dunnett's correction for multiple comparisons).
DISCUSSION
Lung cancer continues to be the leading cause of cancer‐related deaths worldwide, with NSCLC accounting for over 80% of cases. Significant resistance to chemotherapy contributes to the poor prognosis of advanced lung cancer. Therefore, tremendous efforts have been undertaken to discover novel genes and signaling networks involved in chemotherapy resistance. In recent years, there has been a growing recognition of the contribution of UPR signaling to tumor development. It is now widely believed that ER stress and UPR activation plays a crucial role in the progression of various cancers and also in their resistance to chemotherapy. In this report, we used well‐known ER stressor Thapsigargin (TG) to induce acute ER Stress. Our research has revealed that the IRE1α‐XBP1 pathway plays a crucial part in chemoresistance in NSCLC. Mechanistically, SREBP1 is induced under acute ER stress conditions to promote MRP1 expression, thus facilitating drug efflux and provides resistance against cytotoxic chemotherapy.
The main product of IRE1α's RNase activity is the transcription factor XBP1s, which has been linked to carcinogenesis previously. XBP1s is overexpressed in various cancers including lung cancer. Overexpression of XBP1s can directly promote tumorigenesis in cancer cells, this is seen in chronic lymphocytic leukemia, 20 and targeting the activation of IRE1α and subsequent XBP1 splicing in preclinical models of multiple myeloma has shown promising results. 21 Furthermore, it has been demonstrated that XBP1s contributes to the in vivo promotion of TNBC tumor recurrence. 22 Despite considerable understanding of its role in other malignancies, the potential function of the IRE1α‐XBP1s signaling in lung cancer remains unclear. In this study, it is demonstrated for the first time that the IRE1α‐XBP1 pathway induces MRP1 expression, which confers resistance to cytotoxic chemotherapy. Overexpression of IRE1α cDNA or active XBP1 cDNA in NSCLC cells significantly increased MRP1 expression, and depletion of either IRE1α or XBP1 reversed TG‐induced chemoresistance. Furthermore, we have recently discovered that the IRE1α‐XBP1 axis plays a fundamental role in reprogramming lung cancer cell metabolism, 17 suggesting that IRE1α‐XBP1 signaling may have particular relevance in ER‐associated tumorigenesis in NSCLC.
The capacity of the human ABC transporter multidrug resistance protein 1 (MRP1), encoded by the ABCC1 gene, to confer multidrug resistance in lung cancer cells led to its initial discovery. MRP1 mediates the active efflux of different antitumor drugs against the concentration gradient, which lowers drug accumulation and causes drug resistance in tumor cells. 6 , 23 Recent reports have also suggested that MRP1 can recognize and transport novel “targeted” anticancer drugs, such as small‐molecule tyrosine kinase inhibitors (TKIs). 23 Crucially, most studies consistently validate MRP1's role in mediating drug resistance commonly seen in NSCLC, prostate cancer, and some breast cancers. 24 Furthermore, high‐level expression of ABCC1 is a strong independent predictor of a poor prognosis in primary neuroblastomas based on both retrospective and prospective studies. Consistently, it has been demonstrated that ABCC is highly expressed in lung cancer and has an inverse correlation with the prognosis in NSCLC (Figure 1e,f). Most importantly, our study also showed that TG‐induced chemoresistance to the known MRP/ABCC substrates could be reversed by depletion of MRP1 by siRNA (Figure 2b,d,e), indicating that the pivotal role of MRP1 in ER stress‐induced chemoresistance in NSCLC. Interestingly, previous studies reported that MRP1 expression is associated with the expression of the MYCN oncogene. 25 , 26 Thus, our results expand upon these publish work that c‐Myc first activates SREBP1 expression, and SREBP1 binds to the MRP1 promoter to directly regulate its transcription. In this context, inhibition of SREBP activity may be a useful addition to spectrum of lung cancer treatments available after chemotherapy.
Previous studies show that c‐Myc is directly activated by IRE1α through the XBP1s signal. 10 Consistent with this, our results demonstrate for the first time that XBP1s‐induced c‐MYC is required for SREBP1 expression. The bHLH‐zip transcription factors SREBP (1 and 2) have distinct functions in controlling lipid homeostasis. Genes related to fatty acid metabolism are mainly regulated by SREBP1, whereas lipoprotein uptake and de novo cholesterol synthesis genes are primarily regulated by SREBP2. 27 Although it is becoming evident that increased SREBP activity is a crucial component of cancer metabolism programs, 28 the molecular mechanisms by which SREBPs promote tumor progression are still unknown. In this study, using site‐directed mutagenesis and the recently developed CUT&Tag‐qPCR analysis, we were able to show that SREBP1 binds to MRP1 promoter to directly control MRP1 transcription, indicating that SREBP1 mediates ER stress‐induced chemoresistance in NSCLC cells. It is also noteworthy that MYC coordinates with SREBP1 to regulate fat synthesis, which is necessary for tumor initiation and development in vitro as well as in vivo. 29 Nevertheless, whether c‐Myc works with SREBP1 to regulate chemotherapy resistance was unclear prior to this study. Here, we report that the XBP1s/c‐Myc/SREBP1 axis promotes MRP1 expression and chemotherapy resistance.
In summary, the research presented here highlights the key role of ER stress in coordinating multiple signaling pathways involved in the regulation of drug resistance programs. This process involves the activation of c‐Myc and SREBP to control the expression of MRP1 in response to acute ER stress. ER stress can induce resistance programs to a variety of chemotherapeutic drugs, which may have profound implications for treatment. In particular, the IRE1α‐XBP1 pathway may indicate potential susceptibility in lung cancer and be suitable for targeted therapy.
AUTHOR CONTRIBUTIONS
XY, GF, ZZ, and CZ performed the in vitro experiments. WZ, CX, HY, WH, and FY coordinated in all experiments and done supervision. WZ designed this study and the experiments, wrote the manuscript. All authors read and approved the final manuscript.
FUNDING INFORMATION
This work was supported by National Natural Science Foundation of China (81872371), Program for Excellent Sci‐tech Innovation Teams of Universities in Anhui Province (2023AH010073), and Natural Science Foundation of the Higher Education Institutions of Anhui Province (KJ2018A0248). College Students Innovation and Entrepreneurship Training Program (S202310368049 and S202310368007).
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest.
ACKNOWLEDGMENTS
Yuzhou Xu, Feng Gui, Zhe Zhang, and Zhongyang Chen are team members of Precision Medicine Interesting Group.
Xu Y, Gui F, Zhang Z, Chen Z, Zhang T, Hu Y, et al. IRE1α‐XBP1s axis regulates SREBP1‐dependent MRP1 expression to promote chemoresistance in non‐small cell lung cancer cells. Thorac Cancer. 2024;15(29):2116–2127. 10.1111/1759-7714.15442
Yuzhou Xu, Feng Gui and Zhe Zhang contributed equally to this work.
Contributor Information
Yuchen Fu, Email: 20190041@wnmc.edu.cn.
Xinde Chen, Email: 2894946115@qq.com.
Zhihao Wu, Email: zwu2ster@wnmc.edu.cn.
DATA AVAILABILITY STATEMENT
The data used or analyzed in this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Shea M, Costa DB, Rangachari D. Management of advanced non‐small cell lung cancers with known mutations or rearrangements: latest evidence and treatment approaches. Ther Adv Respir Dis. 2016;10(2):113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sundar R, Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol. 2015;7(2):85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang C, Leighl NB, Wu YL, Zhong WZ. Emerging therapies for non‐small cell lung cancer. J Hematol Oncol. 2019;12(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olaussen KA, Postel‐Vinay S. Predictors of chemotherapy efficacy in non‐small‐cell lung cancer: a challenging landscape. Ann Oncol. 2016;27(11):2004–2016. [DOI] [PubMed] [Google Scholar]
- 6. Cole SP. Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol. 2014;54:95–117. [DOI] [PubMed] [Google Scholar]
- 7. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang S, Xing Y, Liu Y. Emerging roles for the ER stress sensor IRE1alpha in metabolic regulation and disease. J Biol Chem. 2019;294(49):18726–18741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein‐folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–597. [DOI] [PubMed] [Google Scholar]
- 10. Sheng X, Nenseth HZ, Qu S, Kuzu OF, Frahnow T, Simon L, et al. IRE1alpha‐XBP1s pathway promotes prostate cancer by activating c‐MYC signaling. Nat Commun. 2019;10(1):323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lourenco C, Resetca D, Redel C, Lin P, MacDonald AS, Ciaccio R, et al. MYC protein interactors in gene transcription and cancer. Nat Rev Cancer. 2021;21(9):579–591. [DOI] [PubMed] [Google Scholar]
- 12. van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301–309. [DOI] [PubMed] [Google Scholar]
- 13. Efeyan A, Comb WC, Sabatini DM. Nutrient‐sensing mechanisms and pathways. Nature. 2015;517(7534):302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dorotea D, Koya D, Ha H. Recent insights into SREBP as a direct mediator of kidney fibrosis via lipid‐independent pathways. Front Pharmacol. 2020;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geng F, Zhong Y, Su H, Lefai E, Magaki S, Cloughesy TF, et al. SREBP‐1 upregulates lipophagy to maintain cholesterol homeostasis in brain tumor cells. Cell Rep. 2023;42(7):112790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao QS, Lin XY, Wang G. Targeting SREBP‐1‐mediated lipogenesis as potential strategies for cancer. Front Oncol. 2022;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao X, Yu C, Yin F, Xu W, Pan Y, Yang B, et al. IRE1alpha‐XBP1 regulates PDK1‐dependent induction of epithelial‐mesenchymal transition in non‐small cell lung cancer cells. Exp Cell Res. 2022;421(1):113376. [DOI] [PubMed] [Google Scholar]
- 18. Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c‐Myc. Nat Immunol. 2019;20(7):865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J, Ding C, Chen Y, Hu W, Yu C, Peng C, et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c‐Myc/SREBP1 pathway. Cancer Lett. 2021;502:154–165. [DOI] [PubMed] [Google Scholar]
- 20. Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla‐Ibarz JA, et al. Inhibition of ER stress‐associated IRE‐1/XBP‐1 pathway reduces leukemic cell survival. J Clin Invest. 2014;124(6):2585–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12(9):703–719. [DOI] [PubMed] [Google Scholar]
- 22. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple‐negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508(7494):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brozik A, Hegedus C, Erdei Z, Hegedus T, Ozvegy‐Laczka C, Szakacs G, et al. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol. 2011;7(5):623–642. [DOI] [PubMed] [Google Scholar]
- 24. Deeley RG, Westlake C, Cole SP. Transmembrane transport of endo‐ and xenobiotics by mammalian ATP‐binding cassette multidrug resistance proteins. Physiol Rev. 2006;86(3):849–899. [DOI] [PubMed] [Google Scholar]
- 25. Haber M, Smith J, Bordow SB, Flemming C, Cohn SL, London WB, et al. Association of high‐level MRP1 expression with poor clinical outcome in a large prospective study of primary neuroblastoma. J Clin Oncol. 2006;24(10):1546–1553. [DOI] [PubMed] [Google Scholar]
- 26. Pajic M, Murray J, Marshall GM, Cole SP, Norris MD, Haber M. ABCC1 G2012T single nucleotide polymorphism is associated with patient outcome in primary neuroblastoma and altered stability of the ABCC1 gene transcript. Pharmacogenet Genomics. 2011;21(5):270–279. [DOI] [PubMed] [Google Scholar]
- 27. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100(21):12027–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dysregulation of sterol response element‐binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004;64(6):2212–2221. [DOI] [PubMed] [Google Scholar]
- 29. Gouw AM, Margulis K, Liu NS, Raman SJ, Mancuso A, Toal GG, et al. The MYC oncogene cooperates with sterol‐regulated element‐binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 2019;30(3):556–572 e555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used or analyzed in this study are available from the corresponding author upon reasonable request.