

Abstract
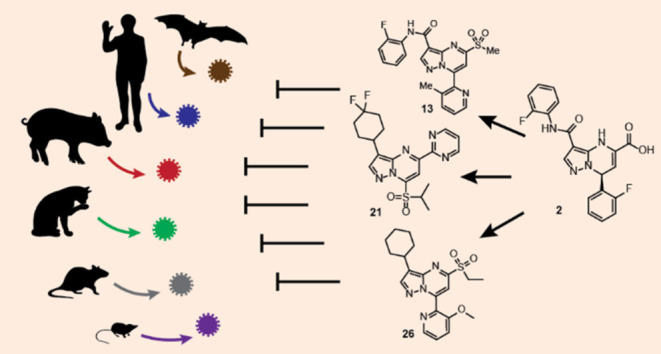
The COVID-19 pandemic highlights the ongoing risk of zoonotic transmission of coronaviruses to global health. To prepare for future pandemics, it is essential to develop effective antivirals targeting a broad range of coronaviruses. Targeting the essential and clinically validated coronavirus main protease (Mpro), we constructed a structurally diverse Mpro panel by clustering all known coronavirus sequences by Mpro active site sequence similarity. Through screening, we identified a potent covalent inhibitor that engaged the catalytic cysteine of SARS-CoV-2 Mpro and used structure-based medicinal chemistry to develop compounds in the pyrazolopyrimidine sulfone series that exhibit submicromolar activity against multiple Mpro homologues. Additionally, we solved the first X-ray cocrystal structure of Mpro from the human-infecting OC43 coronavirus, providing insights into potency differences among compound–target pairs. Overall, the chemical compounds described in this study serve as starting points for the development of antivirals with broad-spectrum activity, enhancing our preparedness for emerging human-infecting coronaviruses.
Introduction
The ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1 pandemic is the third global health crisis caused by a novel coronavirus in just the last twenty-two years, following the SARS-CoV-1 epidemic in 20022 and the Middle East respiratory syndrome coronavirus (MERS-CoV) epidemic in 2012.3 Recent examination of clinical evidence has also suggested that another human-infecting coronavirus, OC43, may have been the true etiologic agent for the “Russian Flu” pandemic in 1889–1891, which resulted in around a million deaths.4
Zoonotic emergence of novel coronaviruses represents a significant and ever-present threat to global health and the global economy. Cross-species transmission appears to be more prevalent in RNA viruses such as coronaviruses,5 and opportunities for spillover events may become more common with a rise in global poverty and as humans continue to encroach on new habitats and interact with nature in new ways.6,7 Examples of these situations include the following: incorporation of bat guano in fertilizer exposes guano miners, farmers, and livestock to reservoirs of α- and β-coronaviruses;8,9 trade of exotic pets and wildlife expose populations to potential spillover events at population-dense ports of entry,7,10 and the continued livestock intensification and the broad use of anti-infectives facilitate disease transmission as well as the emergence of treatment-resistant variants.11 Aside from potentially nucleating a pandemic event, the spread of new diseases through agricultural routes has the secondary effect of exacerbating food shortages and global poverty.12 Overall, there has been a steady increase in the number of emerging infectious diseases each decade going back to 1940, likely driven by socioeconomic, environmental, and ecological factors.13
The staggering costs to human life, well-being, and the global economy demand pre-emptive action to curtail the spread of future zoonotic coronaviruses before they become pandemic. One strategy is the creation of direct-acting broad-spectrum coronavirus antivirals14 which, when optimized against currently circulating coronaviruses, may be efficacious against related emerging coronaviruses and thus either stop the spread entirely or reduce the health burden sufficiently to allow time for the development of new vaccines.
While it is impossible to know the structure of, and thus design drugs against, a viral target for a virus that is not yet known, there are ways we can increase our chances of success against future coronavirus outbreaks. First is the selection of validated, well-conserved, and essential viral targets. The nonstructural protein 5 (nsp5), also known as the main protease or Mpro, is one of the best-characterized drug targets in coronaviruses.15−25 Mpro protease activity is necessary to process the large viral polyproteins pp1a and pp1ab into individual proteins required for replication.26,27 Mpro is attractive for several other reasons, one of which is the preferred residue at the P1 site is glutamine, a substrate specificity that is not shared by any known human proteases15 and thus decreases the chance of nonspecific inhibition of host proteases by Mpro inhibitors. Additionally, Mpro is a cysteine protease, which utilizes a catalytic histidine-cysteine dyad. The presence of a conserved reactive cysteine nucleophile in the active site enables the discovery of covalent inhibitors with pharmacological advantages such as increased potency, selectivity, and duration of inhibition.28 Inhibition of Mpro may also protect against direct protease-induced cell toxicity.29
While work on Mpro inhibitors, such as nirmatrelvir, the antiviral component of Paxlovid,19 has mainly focused on activity against human coronaviruses, activity against a broader set of coronaviruses has not been investigated. To that end, we developed a computational protocol to identify a set of Mpro homologues across all available Coronaviridae sequences that maximize structural diversity in the active site across a tractable number of proteins. We used this set to build a panel of RapidFire mass spectrometry (RFMS)-based biochemical enzymatic activity assays to drive medicinal chemistry optimization of our high-throughput screening hits against SARS-CoV-2 Mpro toward broad-spectrum activity across the viral family.
Results and Discussion
We constructed a pipeline to compile unique Mpro active site residue compositions across the 46,701 Coronaviridae family isolate sequences deposited in GenBank in preparation for clustering (Figure 1). These isolate sequences are available in the NCBI Nucleotide database30 where the majority have the positions of pp1ab annotated, but do not further identify the boundaries of the post-translationally processed proteins such as Mpro. As such, we compiled a data set of 35 diverse reference Mpro sequences from RefSeq and BV-BRC31 (Supporting Information, Table S1) and used them to search for and extract isolate Mpro sequences with the tBLASTn algorithm32 (Figure 1A(i–ii)). The resulting data set totaled 4533 viral isolate Mpro sequences as most deposited isolates did not span the full genome; quality control filtering and removal of identical sequences further reduced this number to 1192.
Figure 1.
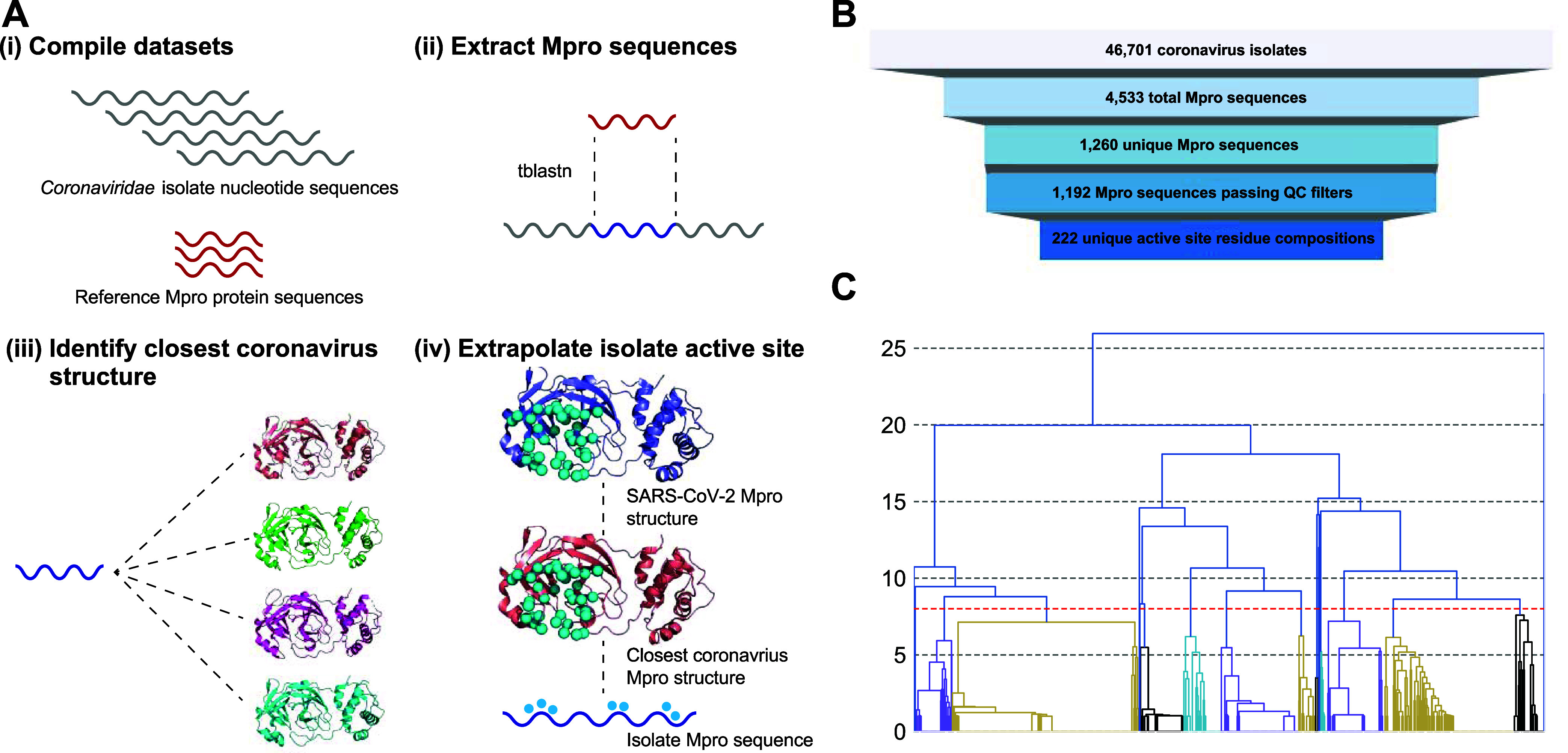
Mpro clustering procedure and results. (A) Active site curation flowchart; Mpro sequences are extracted from the NCBI Coronavirus Nucleotide database and active site residues are inferred through transitive structure alignment using SARS-CoV-2 as a reference. Dashed lines indicate sequence or structure alignments. Blue spheres are active site pocket residues. (B) Number of sequences following each step of the curation process. (C) Clustering results; trees rooted below the red-dashed line (distance cutoff of 8) represent the final clusters.
Next, we identified the active site pocket residues of these isolate Mpro sequences. To establish a reference set of these residues, we visually inspected multiple solved structures of SARS-CoV-2 Mpro in complex with various active site binders21,33,34 to enumerate the 38 amino acid positions comprising the active site pocket (Supporting Information, Figure S1). As the sequence identity between the isolate sequences and the SARS-CoV-2 sequence was too low to infer equivalent active site positions in isolates following a sequence alignment, we devised a strategy to make this assignment using transitive equivalence of positions when aligning isolates to their closest structure. First, all available crystal structures of coronavirus family Mpro proteins in the PDB (12 species total; Supporting Information, Table S2) were structurally aligned to the SARS-CoV-2 structure to identify with high-confidence equivalent active site positions in those solved structures. Then BLASTp was used to align the isolate Mpro sequences to these coronavirus structures to identify the closest structure to each isolate, followed by assigning active site residues based on the sequence alignment. The distribution of sequence identities between isolate Mpro sequences and their closest structure skews much higher than the identities of isolate sequences to SARS-CoV-2 Mpro (Supporting Information, Figure S2), thus allowing us to annotate active site residues in isolates with high confidence using these structures as intermediates. This process resulted in 222 unique active site residue sequences (Figure 1A(iii–iv),1B), each composed of 38 discontinuous residues that define the active site.
Next, a simple all-vs-all pairwise identity matrix was calculated across these 222 unique active site residue sequences and used as input to a hierarchical clustering method (Figure 1C). When constructing these clusters, one critical decision was the choice of distance cutoff to apply; a restrictive cutoff results in higher similarity between cluster members, while a more permissive cutoff has lower similarity but fewer clusters, making creating and testing an Mpro diversity panel more feasible. Here we empirically assigned a cutoff of eight residue differences, resulting in a final set of 19 clusters, which was considered an appropriate trade-off between intracluster diversity and experimental tractability. Notably, the median intercluster distance between any pair of cluster centroids was below 50%, while the mean intracluster distance between a centroid and other members of its cluster was at least 89% for all clusters (Supporting Information, Figure S3).
From each cluster we selected a representative member (Table 1). These representatives were typically the centroids of the cluster, defined as the sequences with the lowest average number of active site residue differences to all other members of the cluster. Some clusters had multiple identical centroids, in which case we prioritized sequences present in the RefSeq database or that were otherwise well-annotated. In other clusters we prioritized Mpro sequences from viruses that infect humans due to being more therapeutically relevant. These representatives were then considered for final Mpro diversity panel selection, protein production and enzymatic assay development. Here, because we were focused on covering diversity in the α- and β-coronaviruses, delta- and γ-coronavirus clusters 1, 7, and 12 were not considered. MERS-CoV Mpro was initially chosen as the representative for cluster 6; however, we observed compound-induced activation of this homologue in the enzymatic assay, which has previously been reported in the literature.35 Because this phenomenon results in inaccurate IC50 curves, MERS-CoV Mpro was replaced with HKU4 Mpro from the same cluster for panel inclusion. We also observed compound-induced activation for HKU9 Mpro (cluster 9) and thus this representative was excluded from the panel. Additionally, clusters 13–19 were omitted because they were represented by a single sequence; these were deprioritized to reduce the total volume of experimental work required. Finally, assays were developed for the other three human-infecting coronaviruses, HKU1 (cluster 3), SARS-CoV-1 (cluster 5), and NL63 (cluster 8), thus leading to multiple members in the panel for these clusters (Table 1). Full details, including annotation and cluster assignment, on these panel members, as well as all isolates, are available in the Supporting Information (Table S3 and Datasheet S1).
Table 1. Mpro Cluster and Panel Composition.
| cluster | size | representative | in panel | genus | host species | other panel members |
|---|---|---|---|---|---|---|
| 1 | 353 | IBV36,37 | no | γ | chicken | |
| 2 | 244 | PEDV38,39 | yes | α | pig | |
| 3 | 147 | HCoV-OC4340 | yes | β | human | HCoV-HKU141,42 |
| 4 | 107 | UU2343 | yes | α | cat | |
| 5 | 81 | SARS-CoV-21,44 | yes | β | human | SARS-CoV-12,45 |
| 6 | 71 | HKU446 | yes | β | bat | |
| 7 | 69 | HKU1547,48 | no | δ | pig | |
| 8 | 57 | HCoV-229E49,50 | yes | α | human | HCoV-NL6351,52 |
| 9 | 31 | HKU953 | no | β | bat | |
| 10 | 15 | Lucheng rat CoV54 | yes | α | rat | |
| 11 | 7 | Wencheng Shrew CoV55 | yes | α | Asian musk shrew | |
| 12 | 3 | SW148,56 | no | γ | Beluga whale | |
| 13 | 1 | Guangdong Chinese Water Skink CoV57 | no | N/A | skink | |
| 14 | 1 | Pacific salmon nidovirus58 | no | * | salmon | |
| 15 | 1 | Kanakana letovirus59 | no | * | lamprey | |
| 16 | 1 | Hipposideros bat CoV | no | N/A | bat | |
| 17 | 1 | HKU1947 | no | δ | night heron | |
| 18 | 1 | Shrew CoV60 | no | α | common shrew | |
| 19 | 1 | Bat CoV GCCDC161 | no | β | bat |
With the panel in hand, we successfully completed a high-throughput screen (HTS) of 1.9 million compounds against SARS-CoV-2 Mpro. We used an enzymatic cleavage assay with a Rhodamine-110 fluorescence intensity readout at a 10 μM single point, (50% inhibition cutoff) which resulted in ∼57,000 positive compounds (∼3% hit rate). In silico triaging based on physicochemical properties, structural flags for Pan-Assay INterference compoundS (PAINS), and promiscuous binders reduced this initial set down to 16,600 compounds which were then taken into an 8-point dose response assay. Approximately 8000 of these compounds were confirmed with an IC50 of <30 μM. The 8000 compound hit list was then manually triaged based on data quality, desirability of chemical matter and in silico projections of likely solubility, permeability, and covalent reactivity. This process led to the identification of ∼2000 compounds, which were classified as either covalent or noncovalent based on substructure flags to take forward into confirmation in a series of orthogonal assays. To remove fluorescence interference compounds, the hits were put through a SARS-CoV-2 Mpro RFMS cleavage inhibition assay. Most compounds (∼90%) were reconfirmed in this assay. Next, a surface plasmon resonance (SPR) binding assay was used to identify genuine Mpro binders in either a covalent or noncovalent binding mode. A relatively low percentage (0.66%) of compounds were SPR-positive; however, these compounds represented high-confidence hits.
To identify any possible false negative compounds arising from SPR artifacts, SPR-negative compounds were assayed using two additional orthogonal binding experiments. First, native mass spectrometry (nMS) was used to identify specific binders to Mpro (either as the monomer or active dimer), with active site binding being determined by competition with a known active site engager. Positive compounds were then taken into a 13C-labeled Mpro NMR binding experiment, using a known Mpro inhibitor as a positive control to define the fingerprint of chemical shift perturbations consistent with genuine active site engagement.
The hits from both the high-confidence SPR-positive and SPR-negative rescue workflows were then resynthesized. Their chemical structures were established, and their activity was confirmed in the SARS-CoV-2 Mpro RFMS cleavage inhibition assay. Reassaying the resynthesized hits resulted in further attrition to give only a select handful of compounds which were ultimately confirmed as active site binders through generation of X-ray cocrystal structures. Of these new hits, a scaffold bearing a dihydropyrazolo[1,5-a]pyrimidine core was prioritized based on its initial promising SAR and physiochemical property space (Compound 1, Figure 2, Table 1).
Figure 2.
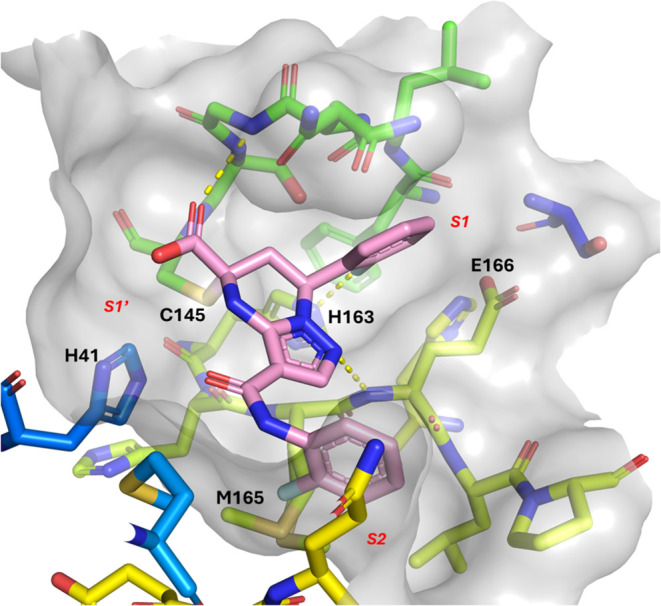
Cocrystal structure of 1 covalently bound to the catalytic C145 of SARS-CoV-2 Mpro (PDB ID 9C8O).
The cocomplex structure revealed that the fluorophenyl group of 1 occupies the S1 pocket and interacts with the side chain of H163 via fluorine hydrogen bonding. Hydrogen bonding between the carboxylic acid and the backbone nitrogen of C145 was observed, along with hydrogen bonding between the nitrogen of the pyrazole core and the backbone nitrogen of E166. Interestingly, the ligand was discovered to be covalently bound in the active site forming a C–S bond with the catalytic C145. The second fluorophenyl group, which is linked to the core via an amide bond, occupies the S2 region of the active site. The carbonyl of the amide linker participates in a water-mediated hydrogen bonding interaction with the backbone carbonyl of H41.
The enantiomers of 1 were separated to yield compound 2 which exhibited similar potency of 3.4 μM against SARS-CoV-2 Mpro (Table 2). As the enantiomer of compound 2 was inactive (>50 μM), compound 2 was assigned as (R)-7-(2-fluorophenyl)-3-((2-fluorophenyl)carbamoyl)-4,7-dihydropyrazolo[1,5-a]pyrimidine-5-carboxylic acid based on the cocrystal structure of compound 1 bound to SARS-CoV-2 Mpro (Figure 2). Exploration of the S1 pocket was carried out with the goal of optimizing the hydrogen bonding interaction between the aryl fluorine and H163–monitoring of lipophilic efficiency (LipE)62 was utilized to confirm that potency changes were not attributable to changes in lipophilicity alone. Heterocycles bearing hydrogen bond acceptors such as pyridine (3) showed comparable potency to the monofluoro phenyl group. However, a small improvement in potency was seen with hydrogen bond donor containing heterocycles such as pyridone (4), with a corresponding increase in lipophilic efficiency (LipE). No potency improvements were gained with dihalogenated phenyl groups such as compound 5. Replacements for the amide linker present in the S2 region were then explored; as the hydrogen bond donor was not engaging in any direct interactions with the protein, it was hypothesized that this linker could be removed entirely. Indeed, compounds 6–11 demonstrate that the amide linker at this position is not required in order to maintain biochemical potency, and LipE analysis indicates potency improvements are primarily driven by lipophilicity - as expected given the lipophilic nature of the S2 pocket.
Table 2. SARS-CoV-2 Mpro Biochemical Potency of Compounds with P2 Modifications.
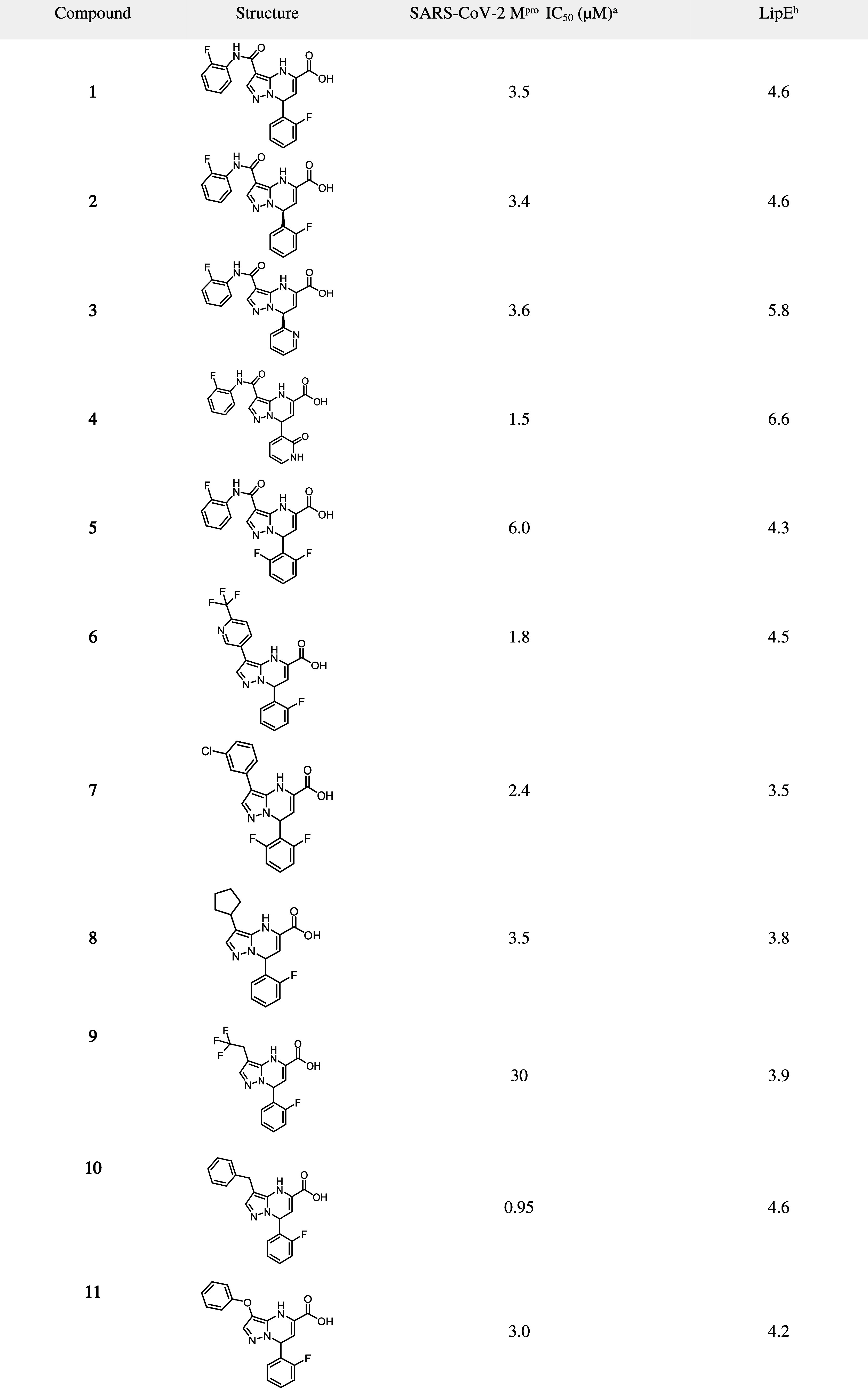
Shown are mean IC50 values derived from a minimum of three replicates;
cLogD was used to calculate LipE.
One of the main challenges that was faced while exploring SAR within this series was chemical instability, which resulted in many compounds being difficult to isolate or obtain in sufficient purity. This instability was thought to be caused by the high reactivity of the α-β unsaturation present in the core. However, the inherent reactivity of the core was also hypothesized to be responsible for the observed covalent mode of binding. Thus, although compounds synthesized with the aromatized (pyrazolopyrimidine) core showed improved chemical stability, this change caused a significant loss in SARS-CoV-2 activity (i.e., compound 12, Figure 3). As the carboxylic acid group of compounds 3 and 12 occupy a polar area of the active site adjacent to the catalytic residues (Figure 2), 3D-modeling hypothesized that inclusion of a sulfone group could facilitate hydrogen bonding interactions with residues located in the oxyanion hole and activate the core for covalent interaction with C145 via a SNAr mechanism. Indeed, replacing the carboxylic acid with a sulfone moiety rescued the potency of the aromatized (pyrazolopyrimidine) series, and even resulted in a dramatic improvement in potency (i.e., compound 13, Figure 3).
Figure 3.

Aromatization of the core leads to potency gains after replacement of the carboxylic acid moiety with a sulfone.
A cocrystal structure of a close analog (14) bound to SARS-CoV-2 Mpro confirmed this hypothetical binding mode, showing covalent binding to C145 (Figure 4). As also expected, the pyridine nitrogen in the S1 pocket interacts with the H163 side chain, as seen in previous series. The cyclohexyl group replaces the amide linked fluorophenyl group and occupies the lipophilic S2 pocket.
Figure 4.
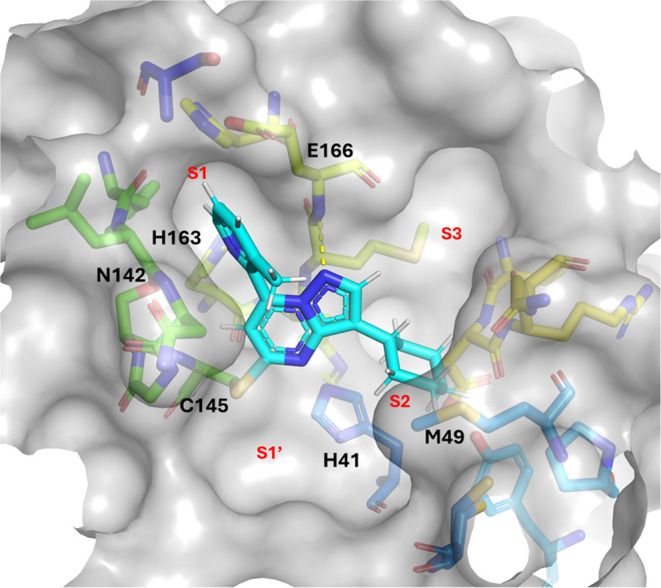
Co-structure of 14 covalently bound to the catalytic C145 of SARS-CoV-2 Mpro (PDB ID 9C8Q).
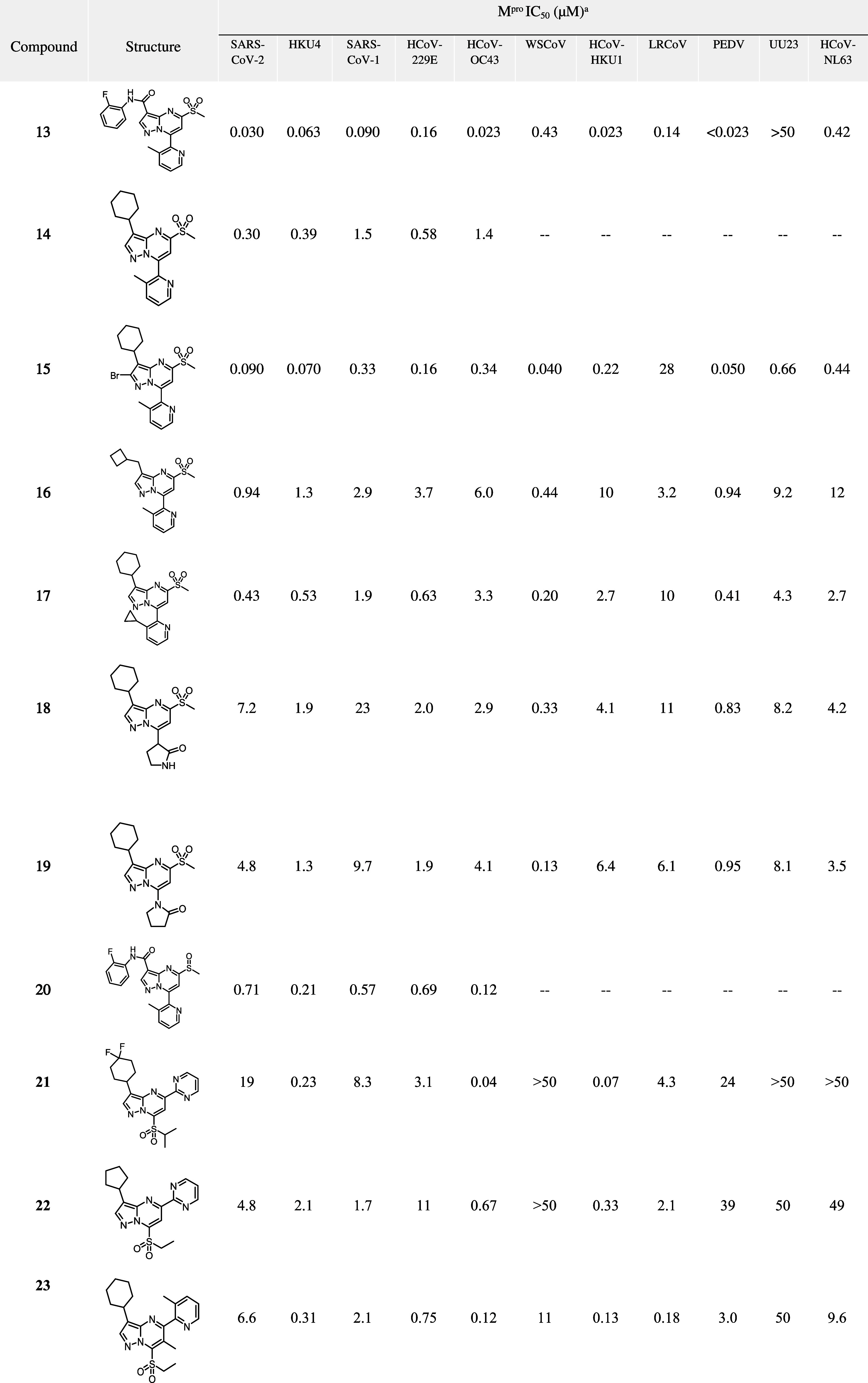
The more stable pyrazolopyrimidine core enabled further SAR exploration of the S1 region. Numerous compounds bearing a sulfone group were synthesized and tested for broad-spectrum activity in the full Mpro biochemical panel, with submicromolar potency being observed across multiple coronavirus species (Table 3). Substituted pyridines (13, 14, 16, 17, 20) were observed to be optimal in the S1 pocket and led to similar potency outcomes. However, replacement of pyridine with lactams 18 and 19 resulted in up to 100-fold loss in potency across the Mpro diversity panel. Bromination on the 2-position of the core (15) was also tolerated. Although various alternative potential covalent warheads (e.g., thioether, cyano, chloro) were not tolerated, sulfoxides (20) were active against SARS-CoV-2 Mpro- however work was focused on sulfones as they were consistently more potent than sulfoxides. Intriguingly, although structural isomers 21 and 22 lost activity against SARS-CoV-2 Mpro, good potency was noted against HCoV-OC43 Mpro. Structural isomer 23 was also potent against HCoV-OC43 Mpro but showed less activity against some of the other Mpro strains. To gain further insight into how compounds such as 21–23 bind to HCoV-OC43 Mpro, an X-ray cocrystal structure was obtained with 21 (Figure 5). As a result, we were able to determine that 21 is selective toward HCoV-OC43 Mpro over SARS-CoV-2 Mpro due to its large leaving group which results in a different binding orientation; steric hindrance prevents the 2-(methylsulfonyl)propane group from approaching close enough to catalytic C145 to form a covalent adduct in SARS-CoV-2 Mpro. However, it can form an activated complex at C142, which is uniquely present in HCoV-OC43 Mpro (the corresponding residue in SARS-CoV-2 Mpro, N142, is unreactive). Although compound 21 partially fills the lipophilic S2 region of HCoV-OC43 Mpro with the difluorocyclohexyl moiety, the pyridine moiety is not in the correct position to occupy the S1 pocket and as such is unable to make a hydrogen bonding interaction with H163 (as in SARS-CoV-2 Mpro). To the best of our knowledge, this work also represents the first disclosure of the structure of HCoV-OC43 Mpro.
Table 3. Broad-spectrum Biochemical Activity of Pyrazolopyrimidine Sulfones in the Mpro Diversity Panel.
Shown are mean IC50 values derived from a minimum of three replicates.
Figure 5.
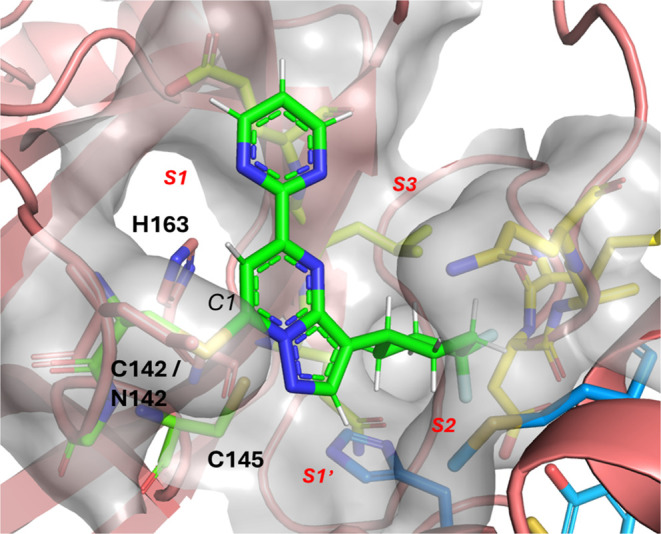
Cocrystal structure of 21 (green) bound to HCoV-OC43 Mpro (rainbow). Compound’s pyrimidine moiety covalently interacts with OC43 at residue C142, which is N142 in wt SARS-CoV-2 Mpro (salmon). (PDB ID 9C7W).
Although the pyrazolopyrimidine series displayed promising broad-spectrum activity, it was difficult to achieve desirable ADME properties while maintaining good potency (Table 4). This series generally displayed high liver microsomal clearance and was also highly reactive to glutathione (GSH). To improve GSH half-life, the sulfone methyl group was replaced with larger groups (ethyl, isopropyl) to sterically encumber the reactive site. Although this strategy was indeed successful, with longer GSH half-lives observed, there was a corresponding potency loss, particularly for tert-butyl sulfone analogs (compounds 24–27) and microsomal stability was not improved. Further modifications to the scaffold in the S2 and S1 areas were viewed as a plausible path forward for improving liver microsomal stability, but this generally came at a further loss in potency (compounds 28–31). As we were unable to achieve potent compounds with balanced properties in the pyrazolopyrimidine series, further development, including profiling in cellular assays, was not pursued. This novel series serves as proof-of-concept for the potential for the discovery of broad-spectrum Mpro inhibitors, as well as inspiration and novel structural insight into targeting other coronaviruses such as HCoV-OC43.
Table 4. SARS-CoV-2 Mpro Biochemical Potency and In Vitro ADME Data for Representative Compounds.
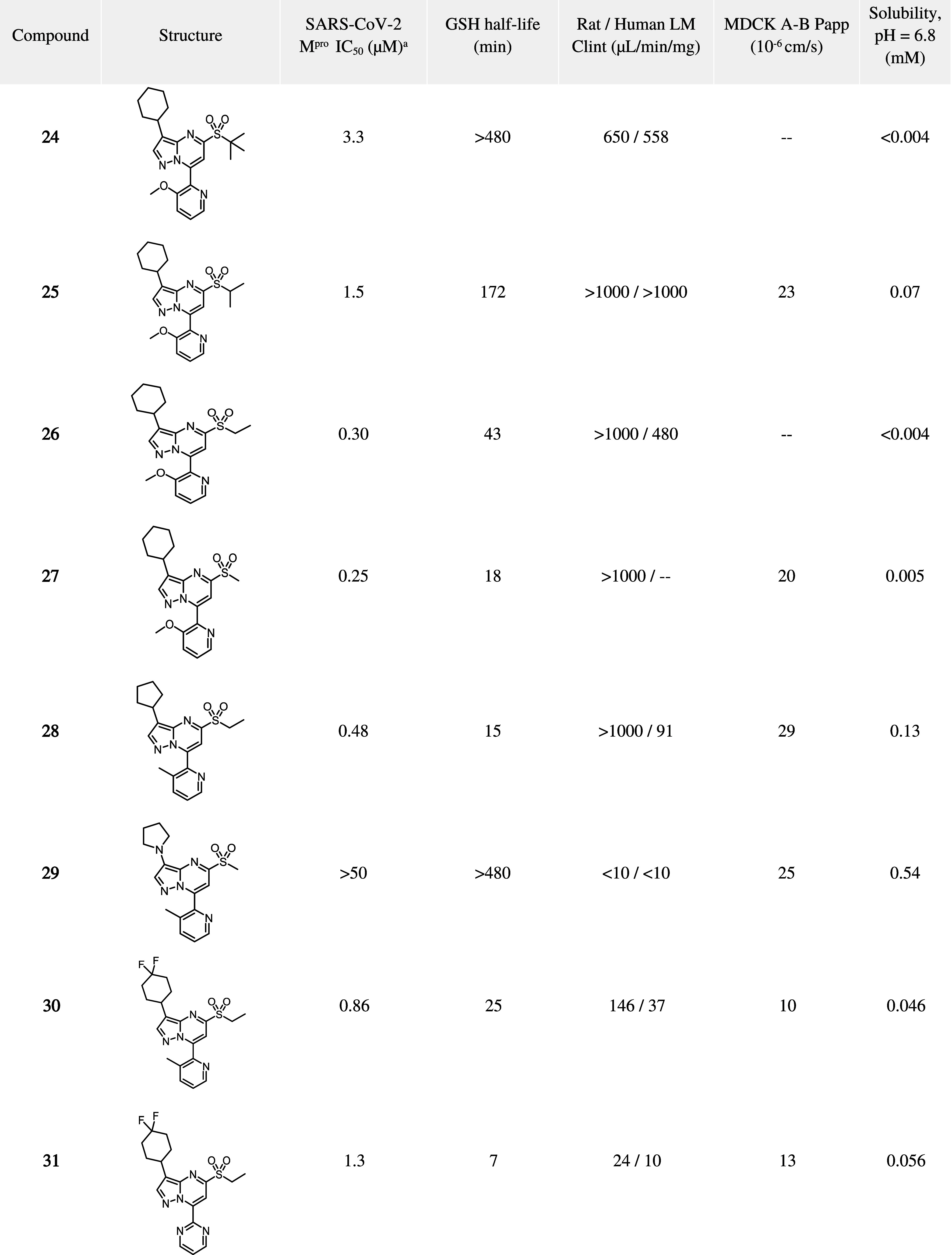
Shown are mean IC50 values derived from a minimum of three replicates.
Synthetic access to analogues bearing the dihydropyrazolo[1,5-a]pyrimidine core generally followed the synthetic strategy of compound 1 (Scheme 1).63,64 2-Fluoroaniline (32) was acylated to form 33 in 94% yield, then condensed with DMF-DMA, and cyclized with hydroxylamine to furnish the key aminopyrazole intermediate 35 (22% over two steps). Coupling 35 with (E)-4-(2-fluorophenyl)-2-oxobut-3-enoic acid readily constructed the core and final product in 33% yield. This modular strategy provided efficient access to a variety of analogues including compounds 3 to 11.
Scheme 1. Synthesis of Compound 1.
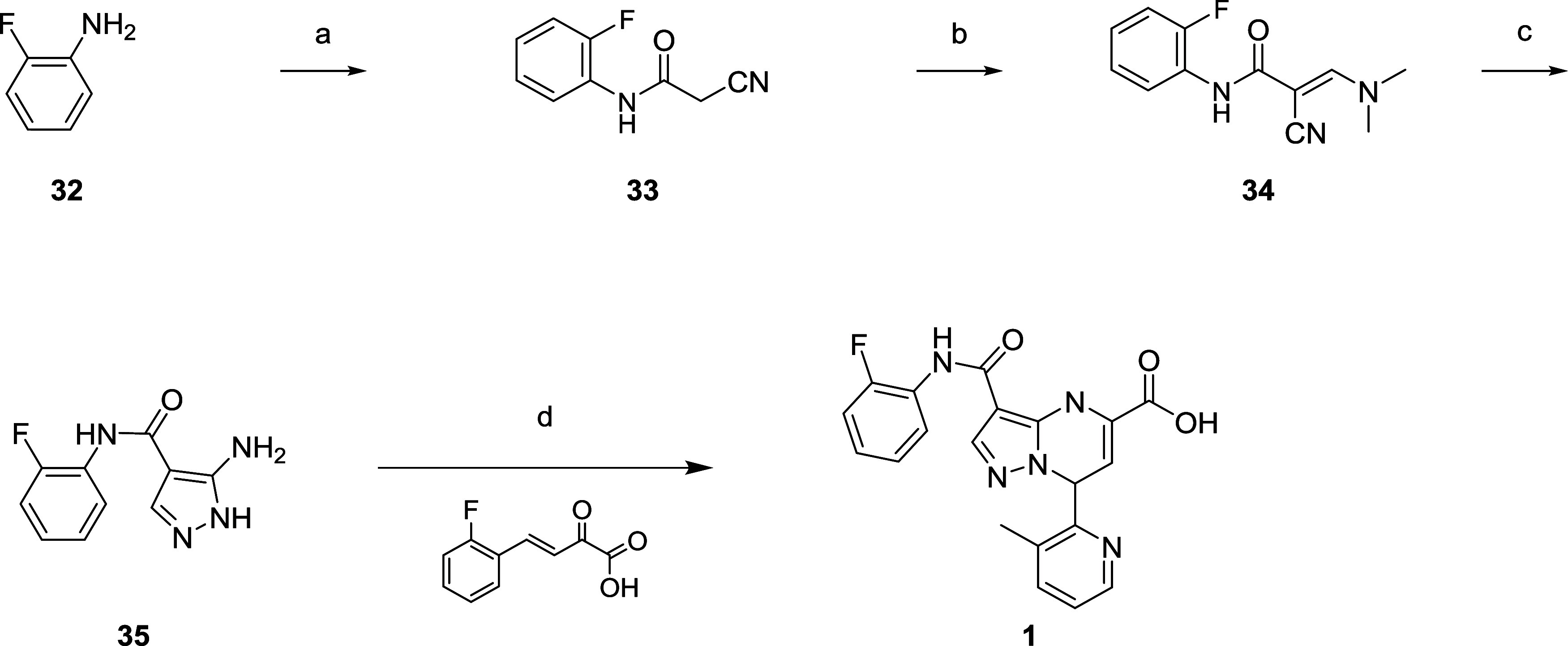
Reagents and conditions: (a) 2-cyanoacetic acid (1.0 equiv), EDCI·HCl (1.2 equiv), CH2Cl2, 25 °C, 94%; (b) DMF-DMA (2.0 equiv), toluene, 80 °C; (c) NH2NH2·H2O (2.0 equiv), EtOH, 25–80 °C, 22% over two steps; (d) (E)-4-(2-fluorophenyl)-2-oxobut-3-enoic acid (1.0 equiv), HOAc, 25–80 °C, 33%.
The route to access compound 13 originally involved further derivatization of intermediate 35 (Scheme 2). Condensation with diethyl malonate (88%) followed by treatment with POCl3 provided the dihalogenated core 37 in 71% yield. A regioselective Stille reaction with the corresponding pyridyl stannane afforded 38 with a yield of 8%. SNAr with NaSMe (5%) followed by oxidation furnished the sulfone, accessing 13 (34%).
Scheme 2. Synthesis of Compound 13.
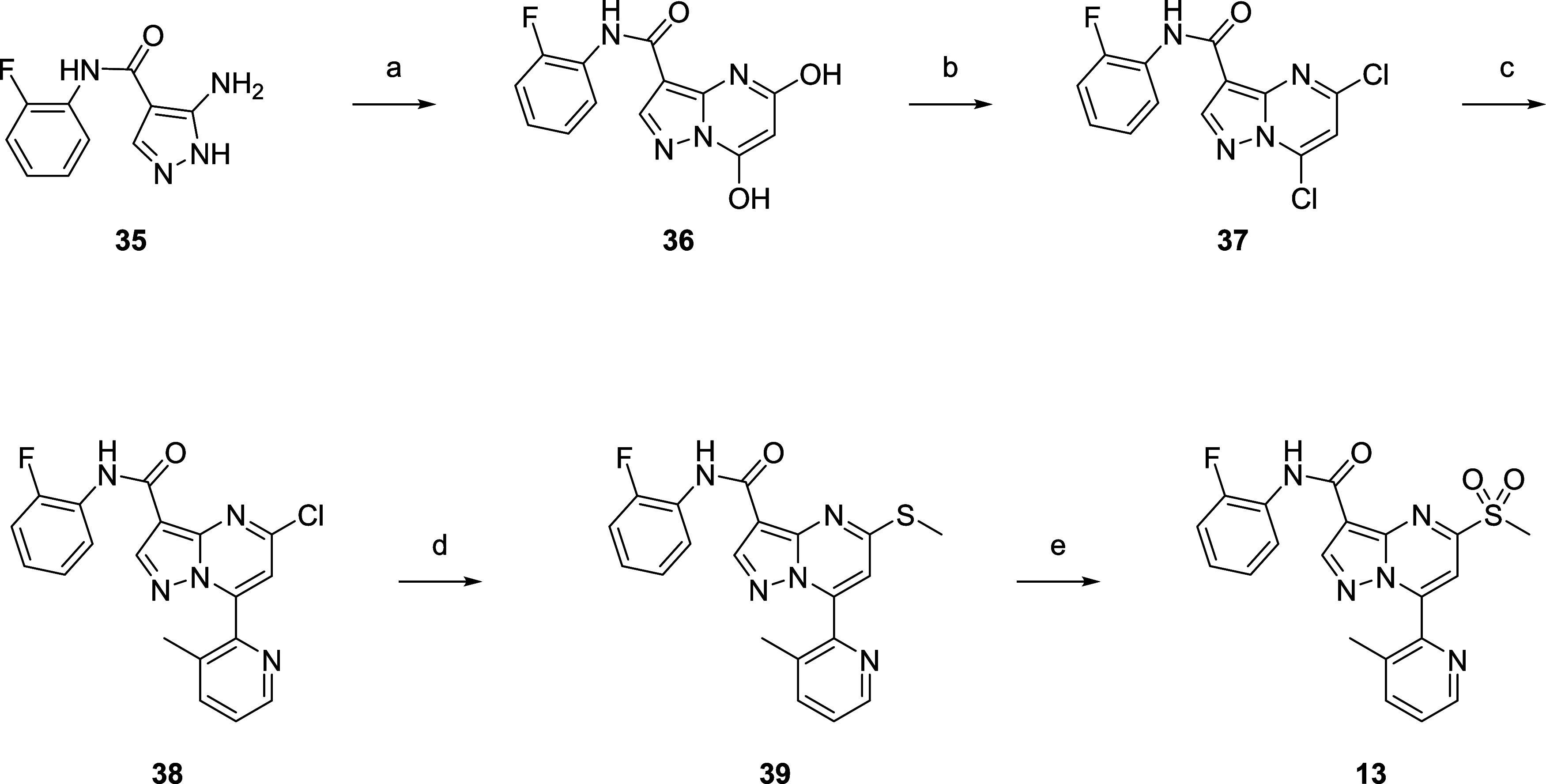
Reagents and conditions: (a) diethyl malonate (1.5 equiv), NaOEt (2.5 equiv), EtOH, 25–80 °C, 88%; (b) POCl3, 25–80 °C, 71%; (c) 3-methyl-2-(tributylstannyl)pyridine (0.8 equiv), Pd(PPh3)2Cl2 (0.1 equiv), CuI (0.1), dioxane, 130 °C, 8%; (d) NaSMe (1.1 equiv), THF/H2O = 1:1, 20 °C, 5%; (e) m-CPBA (3.5 equiv), CH2Cl2, 20 °C, 34%.
Compound 21 was synthesized from 1-(pyrimidin-2-yl)ethan-1-one (40) (Scheme 3). Condensation of 40 with CS2 followed by alkylation with i-PrI afforded 41 in 20% yield. Coupling of 41 with an aminopyrazole afforded the core 42 with the desired regioselectivity (30%), and oxidation of 42 readily afforded 21 in 36% yield.
Scheme 3. Synthesis of 21.

Reagents and conditions: (a) CS2 (1.0 equiv), i-PrI (2.2 equiv), NaH (2.5 equiv), THF, 25 °C, 20%; (b) 4-(4,4-difluorocyclohexyl)-1H-pyrazol-5-amine (1.0 equiv), piperidine (0.1 equiv), HOAc/H2O = 3/1, 25–110 °C, 30%; (c) m-CPBA (2.0 + 0.5 + 0.5 equiv), CH2Cl2, 0–25 °C, 36%.
A second generation synthesis of analogues of 13 was adapted from the synthesis of 21 and further elaborated to access compounds such as 24 (Scheme 4).65,66 Condensing 43 with CS2 followed by alkylation with methyl iodide afforded the dialkylated compound 44 in 75% yield. This intermediate reacted with the corresponding aminopyrazole selectively to form regioisomer 45 in 19% yield. Subsequent oxidation afforded methyl sulfone 27 (78%) which readily underwent a two-step sequence of an SNAr with a thiolate and oxidation (35% over two steps) to form 24.
Scheme 4. Synthesis of 24.
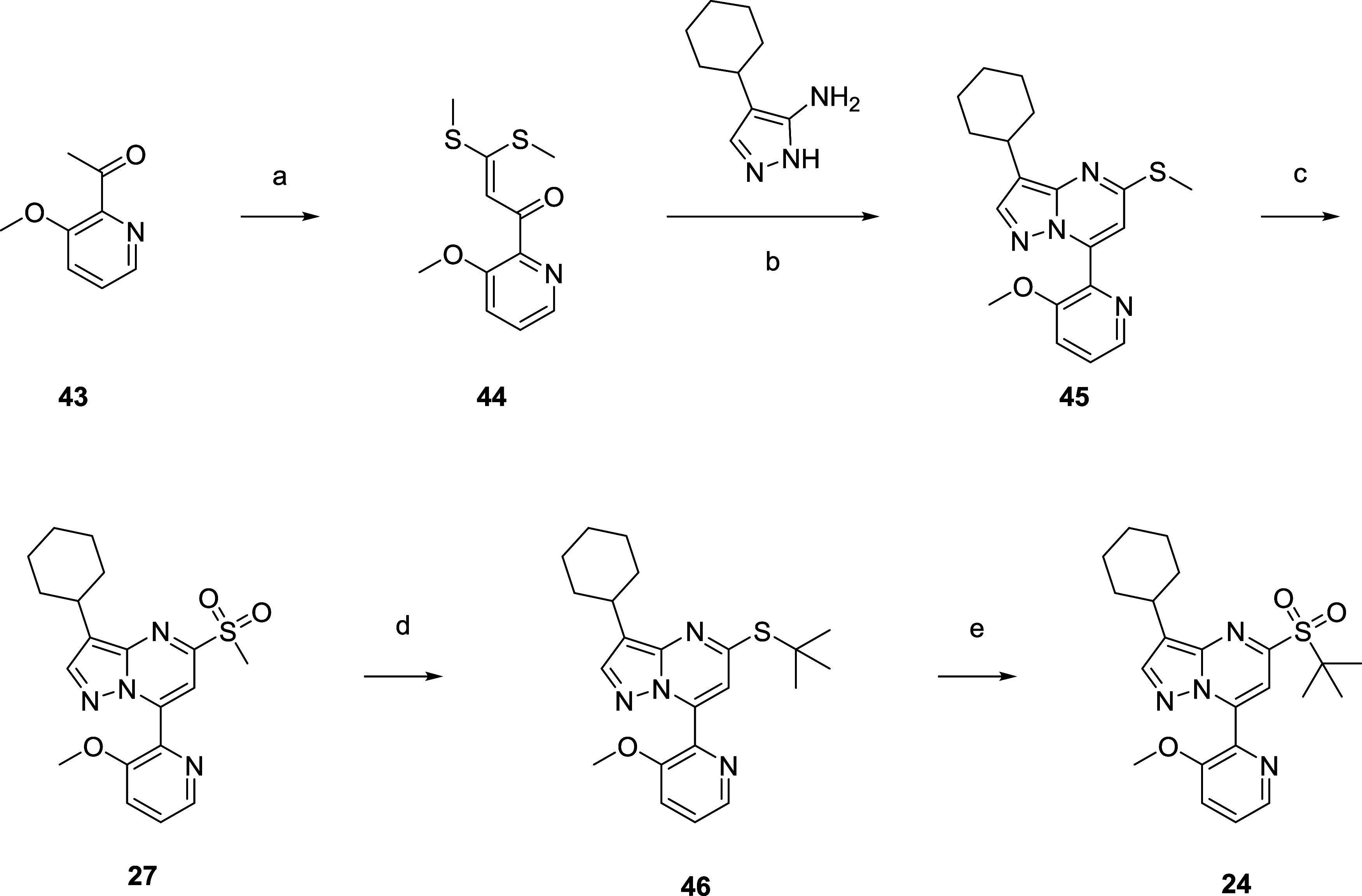
Reagents and conditions: (a) CS2 (1.0 equiv), MeI (2.2 equiv), NaH (2.5 equiv), THF, 25 °C, 75%; (b) 4-cyclohexyl-1H-pyrazol-5-amine (1.0 equiv), piperidine (0.1 equiv), HOAc/H2O = 3/1, 25–110 °C, 19%; (c) Oxone (3.0 eq +1.0 equiv), MeOH/H2O = 2/1, 25 °C, 78%; (d) t-BuSNa (3.0 equiv), t-BuOH, 25 °C; (e) Oxone (2.9 equiv), MeOH/H2O = 2/1, 25 °C, 35% over two steps.
Conclusions
A primary challenge in developing broad-spectrum antivirals is the amount of sequence diversity in the target binding site that can be present within and across viral species.67 Residues that contribute to compound engagement in one species may be different in another species and lead to reduced or abolished compound binding. It thus becomes critical to assess activity against multiple species to determine the degree of coverage for a set of compounds. This is clearly not feasible when the number of viral species under consideration is in the thousands. We therefore developed a strategy to cluster Mpro active site residues by similarity and select a centroid representative from each cluster, with the assumption being that if a compound is active against the representative, it is also likely to be active against most of its other cluster members. This heuristic dramatically reduces the total experimental workload and makes the drug discovery process much more tractable.
One important parameter in our approach was the choice of residue distance cutoff below which we separated species in our hierarchical clustering approach. We empirically evaluated how different cutoffs affected both the number of clusters as well as the degree of similarity within each cluster. A cutoff of eight differences was deemed optimal, corresponding to a maximum panel of 19 members, which was considered to be experimentally tractable in our antiviral program.
After identifying a hit from high-throughput screening, medicinal chemistry efforts produced compounds that showed submicromolar biochemical activity against Mpro from multiple coronaviruses. When considering both these compounds as well as availability of developed assays for these diverse coronaviruses, this work serves as an inspiration for further investigation, and provides a foundation for the discovery of future generations of broad-spectrum Mpro inhibitors.
Experimental Section
General Chemistry
All materials and reagents used were of the commercially available grade and used without further purification. Reaction progress was monitored by either TLC (Merck, 0.2 mm silica gel 60 F254 on glass plates) or LCMS systems (Shimadzu 20AD XR or Waters Acquity). Normal-phase column chromatography was carried out using prepacked silica gel cartridges on a Combiflash Rf separation system by Teledyne ISCO. All microwave reactions were conducted in a Biotage Initiator, irradiating at 0–400 W from a magnetron at 2.45 GH. 1H NMR, 19F, and 2D-spectra were determined on a Varian 400 or Bruker 300 or 400 and 500 MHz NMR spectrometers. Chemical shifts (d-values) are reported in ppm downfield from tetramethylsilane. The following abbreviations are used: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, bs = broad singlet. Mass spectra were acquired on LCMS systems using electrospray, chemical and electron impact ionization methods with a range of instruments of the following configurations: Agilent 1260 and 6125B, Shimadzu LC-20AD XR&MS 2020, Agilent 1200 and 6120B. [M+1]+ refers to the protonated molecular ion of the chemical species. Preparative HPLC was performed on Waters Prep HPLC systems or Gilson 281 Semipreparative HPLC systems using C18 reversed-phase columns Waters Xbridge Prep OBD C18, Phenomenex luna C18, eluting with gradient mixtures of water/acetonitrile containing a modifier 0.05% trifluoroacetic acid or formic acid. Enantiomers were purified by supercritical fluid chromatography (SFC) on a Waters Preparative SFC-100-MS system with ABSYS update, with a Waters 2998 Photodiode Array Detector and a Waters MS Single Quadrupole Detector using a DAICEL CHIRALPAK AD column. All compounds are >95% pure by HPLC (with the exception of compound 4, which was determined to be 93% purity).
2-Cyano-N-(2-fluorophenyl)acetamide (33)
Two batches were carried out in parallel. To a mixture of 2-fluoroaniline (32) (10.0 g, 89.99 mmol, 1.0 equiv) in CH2Cl2 (180 mL) was added 2-cyanoacetic acid (7.7 g, 89.99 mmol, 1.0 equiv) and EDCI·HCl (20.7 g, 107.99 mmol, 1.2 equiv) at 25 °C. Then the mixture was stirred at 25 °C for 1 h under Ar. TLC (Petroleum ether/Ethyl acetate = 3/1, Rf = 0.30) indicated the starting material was consumed completely and one new spot was formed. Two parallel reactions were worked up together and concentrated. The residue was diluted with CH2Cl2 (50 mL) and aq. HCl solution (0.1 M, 100 mL). The mixture was extracted with CH2Cl2 (50 mL × 2). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give the title compound (30 g, 94%) as a white solid was used for the next step directly. 1H NMR (400 MHz, DMSO-d6) δ = 10.14 (br s, 1H), 7.98–7.78 (m, 1H), 7.32–7.23 (m, 1H), 7.23–7.13 (m, 2H), 3.99 (s, 2H).
(E)-2-Cyano-3-(dimethylamino)-N-(2-fluorophenyl)acrylamide (34)
Two batches were carried out in parallel. A mixture of 2-cyano-N-(2-fluorophenyl)acetamide (33) (10.0 g, 56.13 mmol, 1.0 equiv) and DMF-DMA (13.4 g, 112.25 mmol, 2.0 equiv) in toluene (180 mL) was stirred at 80 °C for 3 h under N2. TLC (Petroleum ether/Ethyl acetate = 1/1, Rf = 0.30) indicated the starting material was consumed completely and one new spot was formed. Two parallel reactions were worked up together. The reaction mixture was concentrated to give the title compound (26 g, crude) as a yellow solid which would be used directly in the next step. 1H NMR (400 MHz, DMSO-d6) δ = 8.69–8.63 (m, 1H), 7.84 (s, 1H), 7.76–7.64 (m, 1H), 7.28–7.18 (m, 1H), 7.18–7.09 (m, 2H), 3.29 (s, 3H), 3.22 (s, 3H).
5-Amino-N-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (35)
Three parallel reactions were carried out. To the mixture of (E)-2-cyano-3-(dimethylamino)-N-(2-fluorophenyl)acrylamide (34) (11.3 g, 48.45 mmol, 1.0 equiv) in EtOH (100 mL) was added NH2NH2·H2O (4.8 g, 96.89 mmol, 2.0 equiv) at 25 °C. The mixture was stirred at 80 °C for 3 h under N2. TLC (Ethyl acetate, Rf = 0.20) indicated the starting material was consumed completely and one new spot was formed. Three parallel reactions were worked up together. The reaction mixture was concentrated to give the crude product. The crude product was purified by RP-MPLC (neutral) to give the title compound (7.0 g, 22%) as a white solid. LCMS (ESI): m/z = 221.0 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 11.86 (br s, 1H), 9.31 (s, 1H), 7.99 (s, 1H), 7.69–7.54 (m, 1H), 7.28–7.14 (m, 3H), 5.82 (br s, 2H).
7-(2-Fluorophenyl)-3-((2-fluorophenyl)carbamoyl)-4,7-dihydropyrazolo[1,5-a]pyrimidine-5-carboxylic acid (1)
To a solution of 5-amino-N-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (35) (200 mg, 0.91 mmol, 1.0 equiv) in AcOH (3 mL) was added (E)-4-(2-fluorophenyl)-2-oxobut-3-enoic acid (211 mg, 0.91 mmol, 1.0 equiv) at 25 °C. The solution was stirred at 80 °C for 16 h under N2. LCMS showed the starting material was consumed completely and the desired product was detected. The reaction mixture was quenched by H2O (5 mL) and then the solid was precipitated and collected by filtration. The crude material was purified by prep-HPLC (column: Phenomenex Luna C18 100 × 30 mm × 3 μm; mobile phase: [A: H2O (0.04% HCl); B: ACN]; B%: 40.00%- 70.00%, 8.00 min) to afford the title compound (70 mg, 33%) as a white solid. LCMS (ESI): m/z = 397.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 9.72 (s, 1H), 8.42 (d, J = 1.5 Hz, 1H), 8.08 (s, 1H), 7.63 (dt, J = 2.1, 7.7 Hz, 1H), 7.41–7.35 (m, 1H), 7.32–7.28 (m, 1H), 7.27–7.23 (m, 2H), 7.22 (d, J = 1.8 Hz, 1H), 7.19 (s, 1H), 7.18–7.16 (m, 1H), 7.15 (br d, J = 1.5 Hz, 1H), 6.47 (d, J = 4.1 Hz, 1H), 5.72 (br s, 1H).
N-(2-Fluorophenyl)-5,7-dihydroxypyrazolo[1,5-a]pyrimidine-3-carboxamide (36)
To the mixture of 5-amino-N-(2-fluorophenyl)-1H-pyrazole-4-carboxamide (35) (20.0 g, 90.82 mmol, 1.0 equiv) in EtOH (200 mL) was added diethyl malonate (21.8 g, 136.82 mmol, 1.5 equiv) and NaOEt (77.3 g, 227.06 mmol, 2.5 equiv) at 25 °C. The mixture was stirred at 80 °C under N2 for 16 h. LCMS showed the starting material was consumed completely and one main peak with desired mass was detected. The mixture was diluted with water (400 mL), adjusted to pH = 4 with 2 M HCl at 0 °C. The reaction mixture was filtered and the filter cake was washed with H2O (100 mL × 3). The filter cake was dried under vacuum to give the title compound (23 g, 88%) as a white solid. LCMS (ESI): m/z = 288.9 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 11.86–10.78 (m, 1H), 9.96 (br s, 1H), 8.45 (br s, 1H), 7.67 (br s, 1H), 7.44–7.03 (m, 4H), 5.11 (br s, 1H).
5,7-Dichloro-N-(2-fluorophenyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (37)
Three batches were carried out in parallel. N-(2-fluorophenyl)-5,7-dihydroxypyrazolo[1,5-a]pyrimidine-3-carboxamide (36) (1.0 g, 3.47 mmol, 1.0 equiv) and POCl3 (4 mL) were combined at 25 °C under N2. The mixture was stirred at 80 °C for 16 h under N2. LCMS showed the starting material was consumed completely and one main peak with desired mass was detected. Three batches were worked up together. The reaction mixture was quenched with aq. NaHCO3 (50 mL) and filtered. The filter cake was washed by H2O (10 mL × 3) and dried under vacuum to give the title compound (2.4 g, 71%) as a white solid. LCMS (ESI): m/z = 324.9 [M + H]+.
5-Chloro-N-(2-fluorophenyl)-7-(3-methylpyridin-2-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (38)
To a solution of 5,7-dichloro-N-(2-fluorophenyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (37) (240 mg, 0.06 mmol, 1.0 equiv), 3-methyl-2-(tributylstannyl)pyridine (225 mg, 0.59 mmol, 0.8 equiv) and CuI (14 mg, 0.01 mmol, 0.1 equiv) in dioxane (3 mL) was added Pd(PPh3)2Cl2 (52 mg, 0.01 mmol, 0.1 equiv). Then the mixture was stirred at 130 °C for 2 h under N2. TLC (Petroleum ether/Ethyl acetate = 1/1, Rf-P1 = 0.30) indicated the starting material was consumed completely and three new spots were shown on TLC. The reaction mixture was quenched by H2O (20 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layers were washed with brine (20 mL × 3), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by flash silica gel chromatography (12 g SepaFlash Silica Flash Column, Eluent of 0–30% Ethyl acetate/Petroleum ether gradient @ 80 mL/min). The crude product was further purified by prep-HPLC (column: 3_Phenomenex Luna C18 75 × 30 mm × 3 μm; mobile phase: [A: H2O (0.2% FA), B: ACN], B%: 40.00–70.00%, 8.00 min) to give the title compound (22 mg, 8%) as a white solid. LCMS (ESI): m/z = 382.1 [M + H]+;1H NMR (400 MHz, DMSO-d6) δ = 9.91 (d, J = 2.5 Hz, 1H), 8.75 (s, 1H), 8.62 (d, J = 4.1 Hz, 1H), 8.38 (dt, J = 1.4, 8.0 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.68 (s, 1H), 7.61 (dd, J = 4.7, 7.8 Hz, 1H), 7.37 (ddd, J = 1.2, 8.2, 11.2 Hz, 1H), 7.29–7.22 (m, 1H), 7.21–7.14 (m, 1H), 2.16 (s, 3H).
N-(2-Fluorophenyl)-7-(3-methylpyridin-2-yl)-5-(methylthio)pyrazolo[1,5-a]pyrimidine-3-carboxamide (39)
To a stirred solution of 5-chloro-N-(2-fluorophenyl)-7-(3-methylpyridin-2-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (38) (200 mg, 0.52 mmol, 1.0 equiv) in THF (2 mL) and H2O (2 mL) was added NaSMe (202 mg, 20%, 0.14 mmol, 1.1 equiv). The reaction was stirred at 20 °C for 12 h. LCMS showed the reagent was consumed completely and desired mass was detected. The reaction mixture was filtered, and the filter cake was washed with methyl tert-butyl ether (10 mL × 3). The filter cake was dried under vacuum. The crude product was purified by prep-HPLC (column: Phenomenex Luna C18 100*30 mm*3um; mobile phase: [A: H2O (0.2% FA); B: ACN]; B%: 30.00–60.00%, 8.00 min) to afford the title compound (10 mg, 28%) as a yellow solid. LCMS (ESI): m/z = 394.1 [M + H]+;1H NMR (400 MHz, DMSO-d6) δ = 9.96 (br d, J = 2.0 Hz, 1H), 8.59 (br d, J = 4.4 Hz, 1H), 8.56 (s, 1H), 8.49 (t, J = 7.8 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 7.57 (dd, J = 4.8, 7.7 Hz, 1H), 7.43 (s, 1H), 7.38 (dd, J = 8.9, 11.3 Hz, 1H), 7.29–7.21 (m, 1H), 7.20–7.11 (m, 1H), 2.80 (s, 3H), 2.14 (s, 3H).
N-(2-Fluorophenyl)-7-(3-methylpyridin-2-yl)-5-(methylsulfonyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (13)
To a stirred solution of N-(2-fluorophenyl)-7-(3-methylpyridin-2-yl)-5-(methylthio)pyrazolo[1,5-a]pyrimidine-3-carboxamide (39) (80 mg, 0.20 mmol, 1.0 equiv) in CH2Cl2 (2 mL) was added m-CPBA (144 mg, 85%, 0.71 mmol, 3.5 equiv). The reaction was stirred at 20 °C for 1 h. LCMS showed the starting material was consumed completely and ∼59% of desired mass was detected. The reaction mixture was quenched by Na2SO3 aq. (20 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layers were washed with brine (20 mL × 3), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The crude product was purified by prep-HPLC (column: Phenomenex luna C18 100 × 40 mm × 5 μm; mobile phase: [A: H2O (0.2% FA); B: ACN]; B%: 25.00–65.00%, 8.00 min) to afford the title compound (29 mg, 33%) as a yellow solid. LCMS (ESI): m/z = 426.0 [M + H]+;1H NMR (400 MHz, DMSO-d6) δ = 9.91 (d, J = 2.4 Hz, 1H), 8.97 (s, 1H), 8.65 (d, J = 3.9 Hz, 1H), 8.44–8.36 (m, 1H), 7.99–7.89 (m, 2H), 7.64 (dd, J = 4.7, 7.8 Hz, 1H), 7.40 (dt, J = 1.1, 9.7 Hz, 1H), 7.31–7.24 (m, 1H), 7.23–7.15 (m, 1H), 3.63 (s, 3H), 2.17 (s, 3H).
(Z)-3-(Isopropylthio)-3-mercapto-1-(pyrimidin-2-yl)prop-2-en-1-one (41)
To the mixture of NaH (2.5 g, 61.41 mmol, 2.5 equiv) in THF (30 mL) was added 1-(pyrimidin-2-yl)ethan-1-one (40) (3.0 g, 24.56 mmol, 1.0 equiv) at 25 °C under N2. The reaction mixture was stirred at 25 °C for 0.5 h. CS2 (1.9 g, 24.56 mmol, 1.0 equiv) was added to the reaction mixture over 0.5 h followed by addition of i-PrI (9.2 g, 54.04 mmol, 2.2 equiv) and then the mixture was stirred for 15 h at 25 °C. TLC (Petroleum ether/Ethyl acetate = 1/1, Rf-p1 = 0.30) indicated the starting material was consumed completely and three new spots were shown on TLC. The reaction mixture was quenched by aq. NH4Cl (40 mL). The reaction mixture was extracted with CH2Cl2 (40 mL × 3). The combined organic layers were washed with brine (30 mL × 3), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (120 g SepaFlash Silica Flash Column, Eluent of 30–80% Ethyl acetate/Petroleum ether gradient @ 60 mL/min) to give the title compound (1.4 g, 20%) as a brown solid. LCMS (ESI): m/z = 241.0 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 8.98 (s, 1H), 8.97 (s, 1H), 7.64 (t, J = 4.9 Hz, 1H), 7.51 (s, 1H), 4.00–3.94 (m, 1H), 1.40 (s, 3H), 1.38 (s, 3H).
3-(4,4-Difluorocyclohexyl)-7-(isopropylthio)-5-(pyrimidin-2-yl)pyrazolo[1,5-a]pyrimidine (42)
A solution of (Z)-3-(isopropylthio)-3-mercapto-1-(pyrimidin-2-yl)prop-2-en-1-one (41) (250 mg, 1.04 mmol, 1.0 equiv) in HOAc/H2O (2.8 mL, 3/1) was added 4-(4,4-difluorocyclohexyl)-1H-pyrazol-5-amine (209 mg, 1.04 mmol, 1.0 equiv) and piperidine (9 mg, 0.10 mmol, 0.1 equiv) under N2 at 25 °C. Then the mixture was stirred at 110 °C for 16 h. TLC (Petroleum ether/Ethyl acetate = 2:1, Rf-P1 = 0.50) indicated the starting material was consumed completely and several new spots were detected. The reaction mixture was quenched by aq. NaHCO3 (4 mL) and extracted with CH2Cl2 (4 mL × 3). The combined organic layers were washed with brine (3 mL × 3), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (10 g SepaFlash Silica Flash Column, Eluent of 25–60% Ethyl acetate/Petroleum ether gradient @20 mL/min) to give the title compound (120 mg, 30%) as a yellow solid. LCMS (ESI): m/z = 390.2 [M + H]+;1H NMR (400 MHz, DMSO-d6) δ = 9.06 (d, J = 4.9 Hz, 2H), 8.26 (s, 1H), 7.92 (s, 1H), 7.65 (t, J = 4.9 Hz, 1H), 4.05–3.97 (m, 1H), 3.24–3.14 (m, 1H), 214–2.09 (m, 4H), 1.98 (s, 2H), 1.91–1.82 (m, 2H), 1.51 (s, 3H), 1.50 (s, 3H).
3-(4,4-Difluorocyclohexyl)-7-(isopropylsulfonyl)-5-(pyrimidin-2-yl)pyrazolo[1,5-a]pyrimidine (21)
To the mixture of 3-(4,4-difluorocyclohexyl)-7-(isopropylthio)-5-(pyrimidin-2-yl)pyrazolo[1,5-a]pyrimidine (42) (100 mg, 0.26 mmol, 1.0 equiv) in CH2Cl2 (4 mL) was added m-CPBA (104 mg, 0.51 mmol, 2.0 equiv) at 0 °C. The reaction was stirred at 25 °C for 0.5 h under N2. Then the m-CPBA (26 mg, 0.13 mmol, 0.5 equiv) was added to the mixture at 0 °C and stirred at 25 °C for 0.5 h. Then the m-CPBA (26 mg, 0.13 mmol, 0.5 equiv) was added to the mixture at 0 °C stirred at 25 °C for 0.5 h. LCMS showed the starting material was consumed completely and 29% with desired mass was detected. The reaction solution was quenched by aq. Na2SO3 (5 mL) and extracted with CH2Cl2 (5 mL × 3). The combined organic layers were washed with brine (2 mL × 3), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by prep-HPLC (column: Waters Xbridge BEH C 18 100 × 30 mm × 10 μm; mobile phase: [A: H2O (10 mM NH4HCO3); B: ACN]; B%: 35.00–65.00%, 8.00 min) to give the title compound (38.5 mg, 36%) as yellow solid. LCMS (ESI): m/z = 358.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 9.11 (d, J = 4.8 Hz, 2H), 8.52 (s, 1H), 8.50 (s, 1H), 7.70 (t, J = 4.8 Hz, 1H), 4.43–4.33 (m, 1H), 3.30–3.23 (m, 1H), 2.18–2.11 (m, 5H), 2.07 (br d, J = 14.2 Hz, 1H), 1.97–1.87 (m, 2H), 1.31 (d, J = 6.8 Hz, 6H).
1-(3-Methoxypyridin-2-yl)-3,3-bis(methylthio)prop-2-en-1-one (44)
To a solution of NaH (860 mg, 21.50 mmol, 2.5 equiv) in THF (20 mL) was added slowly 1-(3-methoxypyridin-2-yl)ethan-1-one (43) (1.3 g, 8.60 mmol, 1.0 equiv) at 25 °C under N2, and the reaction mixture was stirred at 25 °C for 0.5 h. CS2 (655 mg, 8.60 mmol, 1.0 equiv) was added to the reaction mixture over 0.5 h followed by addition of MeI (2.7 g, 18.92 mmol, 2.2 equiv). The mixture was stirred for 12 h at 25 °C. TLC (Ethyl acetate, Rf-P1 = 0.35) indicated the starting material was consumed completely and two new spots were shown on TLC. The reaction mixture was quenched by aq. NH4Cl (30 mL) and extracted with CH2Cl2 (40 mL × 3). The combined organic layers were washed with brine (40 mL × 3), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (25 g SepaFlash Silica Flash Column, Eluent of 0–70% Ethyl acetate/Petroleum ether gradient @ 80 mL/min) to the title compound (1.7 g, 75%) as a yellow solid. LCMS (ESI): m/z = 256.1 [M + H]+; 1H NMR (400 MHz, CDCl3) δ = 8.22 (dd, J = 1.6, 4.0 Hz, 1H), 7.38–7.29 (m, 2H), 7.01 (s, 1H), 3.90 (s, 3H), 2.52 (s, 6H).
3-Cyclohexyl-7-(3-methoxypyridin-2-yl)-5-(methylthio)pyrazolo[1,5-a]pyrimidine (45)
A solution of 1-(3-methoxypyridin-2-yl)-3,3-bis(methylthio)prop-2-en-1-one (44) (100 mg, 0.39 mmol, 1.0 equiv) in HOAc (0.9 mL) and H2O (0.3 mL) was added dropwise into a solution of 4-cyclohexyl-1H-pyrazol-5-amine (65 mg, 0.39 mmol, 1.0 equiv) and piperidine (3 mg, 0.04 mmol, 0.1 equiv) at 25 °C under N2. The mixture was stirred at 110 °C for 12 h. LCMS showed the starting material was consumed completely and one main peak with desired mass was detected. The mixture was diluted with water (10 mL), adjusted to pH = 7 with saturated aqueous NaHCO3 and extracted with ethyl acetate (15 mL × 3). The combined organic layers were washed with brine (15 mL × 3), dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by prep-HPLC (column: Phenomenex Luna C18 75*30 mm*3um; mobile phase: [A: H2O (0.2% FA); B: ACN]; B%: 50.00–90.00%, 8.00 min) to afford the title compound (26 mg, 19%) as a white solid. LCMS (ESI): m/z = 355.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 8.29 (dd, J = 1.1, 4.6 Hz, 1H), 7.87 (s, 1H), 7.72–7.67 (m, 1H), 7.60 (dd, J = 4.6, 8.5 Hz, 1H), 6.91 (s, 1H), 3.75 (s, 3H), 2.86 (tt, J = 3.5, 11.7 Hz, 1H), 2.62 (s, 3H), 2.04–1.95 (m, 2H), 1.84–1.76 (m, 2H), 1.72 (br d, J = 12.0 Hz, 1H), 1.59 (dq, J = 3.0, 12.3 Hz, 2H), 1.47–1.35 (m, 2H), 1.33–1.22 (m, 1H).
3-Cyclohexyl-7-(3-methoxypyridin-2-yl)-5-(methylsulfonyl)pyrazolo[1,5-a]pyrimidine (27)
To a mixture of 3-cyclohexyl-7-(3-methoxypyridin-2-yl)-5-(methylthio)pyrazolo[1,5-a]pyrimidine (45) (200 mg, 0.56 mmol, 1.0 equiv) in MeOH/H2O (3 mL, 2/1) was added Oxone (285 mg, 1.69 mmol, 3.0 equiv) at 25 °C. The reaction mixture was stirred at 25 °C for 12 h under N2. Then additional Oxone (95 mg, 0.56 mmol, 1.0 equiv) was added to the mixture at 25 °C. The reaction was stirred at 25 °C for 4 h under N2. TLC (Petroleum ether/Ethyl acetate = 3/1, Rf = 0.50) indicated that starting material still remained and one new spot was formed. The mixture was diluted with H2O (2 mL) and extracted with ethyl acetate (3 mL × 3). The combined organic layers were washed with brine (2 mL), dried over Na2SO4, filtered, and concentrated. The crude was purified by MPLC (Petroleum ether/EtOAc = 20–50%) to give the title compound (170 mg, 78%) as a yellow solid. LCMS (ESI): m/z = 387.2 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ = 8.36 (dd, J = 1.2, 4.6 Hz, 1H), 8.33 (s, 1H), 7.80–7.76 (m, 1H), 7.68 (dd, J = 4.6, 8.6 Hz, 1H), 7.55 (s, 1H), 3.78 (s, 3H), 3.48 (s, 3H), 3.01 (s, 1H), 2.05–1.98 (m, 2H), 1.85–1.78 (m, 2H), 1.74 (br d, J = 12.7 Hz, 1H), 1.61 (dq, J = 2.8, 12.4 Hz, 2H), 1.51–1.39 (m, 2H), 1.36–1.26 (m, 1H).
5-(tert-Butylthio)-3-cyclohexyl-7-(3-methoxypyridin-2-yl)pyrazolo[1,5-a]pyrimidine (46)
To a mixture of 3-cyclohexyl-7-(3-methoxypyridin-2-yl)-5-(methylsulfonyl)pyrazolo[1,5-a]pyrimidine (27) (100 mg, 0.26 mmol, 1.0 equiv) in t-BuOH (1.5 mL) was added t-BuSNa (87 mg, 0.78 mmol, 3.0 equiv) at 25 °C. The reaction mixture was stirred at 25 °C for 16 h under N2. LCMS showed the starting material was consumed completely and one main peak with desired mass was detected. The mixture was diluted with H2O (3 mL) and extracted with ethyl acetate (3 mL × 3). The combined organic mixture was washed with brine (2 mL), dried over Na2SO4, filtered and concentrated to give the title compound (120 mg, crude) as brown oil which would be used for the next step directly. LCMS (ESI): m/z = 397.2 [M + H]+.
5-(tert-Butylsulfonyl)-3-cyclohexyl-7-(3-methoxypyridin-2-yl)pyrazolo[1,5-a]pyrimidine (24)
To the mixture of 5-(tert-butylthio)-3-cyclohexyl-7-(3-methoxypyridin-2-yl)pyrazolo[1,5-a]pyrimidine (46) (120 mg, 0.26 mmol, 1.0 equiv) in MeOH/H2O (3 mL) was added Oxone (127 mg, 0.76 mmol, 2.9 equiv) at 25 °C. The reaction mixture was stirred at 25 °C for 16 h under N2. LCMS showed the starting material was consumed completely and 70% with desired mass was detected. The mixture was diluted with H2O (4 mL) and extracted with ethyl acetate (3 mL × 3). The combined organic mixture was washed with brine (2 mL), dried over Na2SO4, filtered, and concentrated to give crude product. The crude product was purified by prep-HPLC (column: Phenomenex luna C 18 100 × 40 mm × 5 μm; mobile phase: [A: H2O (0.04% HCl); B: ACN]; B%: 45.00–75.00%, 8.00 min) to give the title compound (39 mg, 35%) as a yellow solid. LCMS (ESI): m/z = 429.2 [M + H]+;1H NMR (400 MHz, DMSO-d6) δ = 8.36 (dd, J = 1.1, 4.6 Hz, 1H), 8.34 (s, 1H), 7.78 (dd, J = 1.0, 8.6 Hz, 1H), 7.68 (dd, J = 4.6, 8.6 Hz, 1H), 7.50 (s, 1H), 3.79 (s, 3H), 2.99 (tt, J = 3.4, 11.7 Hz, 1H), 2.06–1.98 (m, 2H), 1.84–1.77 (m, 2H), 1.77–1.70 (m, 1H), 1.63 (dq, J = 2.9, 12.4 Hz, 2H), 1.44 (br d, J = 3.3 Hz, 2H), 1.41 (s, 9H), 1.32–1.21 (m, 1H).
Curation of Mpro Sequences in Coronavirus Isolates
Two sequence data sets were assembled for identifying Mpro boundaries in unannotated coronavirus genomes (Figure 1A(i)). The first included RefSeq Mpro protein sequences, identified through having “3C” or “nsp5” in their FASTA headers and totaling between 250 and 350 amino acids in length (Supporting Information, Table S2). These were derived from the NCBI Protein Database and from the Bacterial and Viral Bioinformatics Resource Center (BV-BRC)31 platform. The second data set comprised isolate sequences in the NCBI Nucleotide database annotated as being in the Coronaviridae family (Taxon ID 11118), excluding SARS-CoV-2 sequence isolates other than their RefSeq genomes (Supporting Information, Table S1). Protein sequences were used as queries in a tBLASTn32 search against the translated isolate nucleotide sequences to identify the Mpro regions and amino acid sequences in the viral genomes (Figure 1A(ii)). This data set was filtered to remove identical sequences, sequences less than 250 amino acids in length, and sequences with stop codons or 10 or more ambiguous “X” amino acids. All data were obtained in September 2022.
Identification of Active Site Residues in Isolate Mpro Sequences
SARS-CoV-2 Mpro active site residues were identified through enumerating residues within 5 Å of ligands present in selected solved structures (PDB accessions 5r84,34 7mb4,21 7vu633) followed by manual refinement (Supporting Information, Figure S1). Coronavirus Mpro solved structures in the PDB were compiled and structurally aligned to the SARS-CoV-2 Mpro structure using the PyMOL “align” algorithm, annotating equivalent active site residues in these structures through the resulting 3D-superposition. Next, the genome isolate Mpro sequences identified in the previous step were aligned to the structure sequences using BLASTp,32 and for each isolate, the structure with the lowest e-value was selected as its reference structure. The alignment between each isolate Mpro sequence and its reference structure was used to infer the equivalent active site residues for that sequence, resulting in an array of noncontiguous sequences residue sequences (Figure 1A(iii–iv)).
Clustering Mpro Active Site Residues
An all-by-all distance matrix was constructed by counting the number of identical amino acids across each pair of active sites. This distance matrix was used as input to a standard agglomerative, bottom-up, average linkage hierarchical procedure to cluster the active site residues. Final clusters were generated by identifying subtrees rooted below the empirically chosen horizontal line of eight amino acid differences, and active site leaves of each subtree were grouped as members of a cluster.
Protein Expression and Purification
Genes encoding Mpro homologues were codon optimized and synthesized into a PET-based expression plasmid encoding an N-terminal His-SUMO tag. SUMO was chosen for two reasons: first, activity of Mpro requires an authentic N-terminus and SUMO cleavage leaves no “scar”, unlike other proteases; second, the protein, when fused to SUMO, is inactive, leading to less cytotoxicity in the expression host. We note that we originally tried to use TEV protease, however the cleavage sequence was too similar to the native Mpro cleavage sequence, and these tags were often self-cleaved. The synthesis was outsourced to Twist Biosciences.
Chemically competent Hi-Control BL21(DE3) Escherichia coli cells were transformed with expression plasmids using standard techniques, and a single colony was used to start an overnight culture in Lysogeny Broth (LB) media68 (with 50 μg/mL kanamycin). This culture was used to inoculate a 1 L culture in Terrific Broth (TB)69 supplemented with 50 mM sodium phosphate pH 7.0 and 50 μg/mL kanamycin. These cells were grown in Fernbach flasks at 37 °C while shaking at 200 rpm until the OD600 reached between 1–1.5, at which point the temperature was reduced to 19 °C and the cells were induced with 0.5 mM Isopropyl ß-d-1-thiogalactopyranoside (IPTG). Cells were allowed to grow overnight before harvesting by centrifugation (6000g for 30 min) and either stored at −70 °C or immediately processed (storage did not affect the yield or quality of the protein; data not shown).
Cell pellets were resuspended in lysis buffer (50 mM Tris pH 8.0, 400 mM NaCl, 1 mM TCEP) and lysed by 3–5 passes through a cell homogenizer. The lysate was clarified by centrifugation (45,000g for 30 min) and then was flowed through a 5 mL Ni-Sepharose Excel column (pre-equilibrated with 5 column volumes (CVs) of lysis buffer) on an AKTA FPLC. The material was flowed using a sample pump with a flow rate of 5 mL/min.
Following column loading, the column was washed with lysis buffer until the A280 stabilized. The material was then slowly eluted with a linear gradient of elution buffer (50 mM Tris pH 8.0, 400 mM NaCl, 1 mM TCEP, 500 mM imidazole) over 40 column volumes.
Fractions containing Mpro protein were determined by SDS-PAGE analysis and were pooled. ESI-LCMS analysis was used to confirm the correct molecular weight in all cases (occasionally, a mass corresponding to the loss of the N-terminal methionine was observed; this resolved after cleavage of the N-terminal tag).
100 μg of ULP1 was added to the pooled fractions from the IMAC purification to cleave the His-SUMO tag. Cleavage proceeded at room temperature overnight while dialyzing into 3 L of lysis buffer using a 3500 MWCO dialysis cassette. In most cases, substantial precipitate was observed at this step. However, the protein which remained soluble did not further precipitate throughout the rest of the purification or in downstream applications, and by all observations was very stable.
The protein was then flowed over 5 mL of Ni-NTA resin pre-equilibrated with lysis buffer to remove additional contaminants, the His-SUMO tag, and the ULP1. The flow-through was collected and concentrated to approximately 5 mL prior to purification via SEC. A Superdex 75 16/60 column was pre-equilibrated with fresh SEC Buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP) and the protein was flowed through the column at 1 mL/min while collecting 1.0 mL fractions. Fractions were analyzed by SDS-PAGE and those containing pure Mpro were pooled, concentrated, aliquoted, and stored at −70 °C. Typical yields observed were between 40 and 80 mg of final protein per L of bacterial culture.
X-ray Crystallography
Crystals of SARS-CoV-2 Mpro (1-306) were grown by hanging-drop vapor diffusion at 18 °C. One μL of protein solution (12.4 mg/mL in 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP) and 1 μL reservoir solution (0.1 M MIB pH 7.0, 20% PEG 1500) were mixed above a reservoir of 300 μL. Crystals grew overnight. They were supplemented with 25% glycerol and flash frozen in liquid nitrogen. Crystals of HCoV-OC43 Mpro (1-298) was grown by hanging-drop vapor diffusion at 18 °C. One μL of protein solution (10 mg/mL in 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP) and 1 μL reservoir solution (0.1 M HEPES pH 7.5, 10% polyethylene glycol 8000, 8% ethylene glycol) were mixed above a reservoir of 300 μL. Crystals grew overnight. They were supplemented with 25% glycerol and flash frozen in liquid nitrogen. X-ray diffraction data was collected at Advanced Light Source (beamline 2.0.1.). Data integration and scaling were performed using XDS from AutoProc. The structure was solved by molecular replacement with Phaser using SARS-CoV-2 Mpro as a search model. Subsequence structure refinement was performed using PHENIX suite, and manual model building with Coot. Structural figures were prepared with PyMOL. Data collection and refinement statistics are summarized with Table 5.
Table 5. Crystallographic Statistics (Values in Parentheses are for the Highest Resolution Shell).
| HCoV-OC43 Mpro + 21 | SARS-CoV-2 Mpro + 14 | SARS-CoV-2 Mpro + 1 | |
|---|---|---|---|
| PDB code | 9C7W | 9C8Q | 9C8O |
| data collection | |||
| resolution range | 95.21–2.08 (2.19–2.08) | 48.37–2.03 (2.14–2.03) | 48.53–2.00 (2.00–2.10) |
| space group | P 21 21 21 | C1 21 1 | P1 21 1 |
| mol. in the ASU | 4 | 1 | 2 |
| unit cell [a, b, c (Å)] | 67.25 129.80 140.07 90 90 90 | 44.57 53.53 115.03 | 44.78 53.87 114.05 |
| 90.00 101.12 90.00 | 90.00 101.31 90.00 | ||
| total reflections | 403,833 | 97,369 | 116,464 |
| unique reflections | 73,411 | 33,713 | 34,629 |
| multiplicity (high shell) | 5.5 (3.8) | 2.9 (2.4) | 3.3 4 (3.4) |
| completeness (%) | 98.7 (92.3) | 97.4 (93.3) | 94.9 (96.9) |
| mean I/σ (I) | 7.0 (1.6) | 9.2 (4.4) | 8.4 (2.3) |
| Wilson B-factor | 39.16 | 23.98 | 31.86 |
| R-merge (high shell) | 0.111 (0.702) | 0.045 (0.128) | 0.060 (0.0374) |
| R-meas (high shell) | 0.135 (0.902) | 0.062 (0.177) | 0.082 (0.495) |
| R-pim (high shell) | 0.076 (0.560) | 0.036 (0.121) | 0.044 (0.267) |
| CC1/2 (high shell) | 0.983 (0.503) | 0.992 (0.541) | 0.998 (0.872) |
| refinement | |||
| reflections used in refinement | 72,700 | 62,254 | 34,617 |
| reflections used for R-free | 3582 | 3286 | 1698 |
| R-work | 0.2142 | 0.2067 | 0.2611 |
| R-free | 0.2459 | 0.2563 | 0.2911 |
| CC(work) | 0.939 | 0.964 | 0.941 |
| CC(free) | 0.927 | 0.937 | 0.920 |
| number of non-hydrogen atoms | 9794 | 2372 | 4567 |
| protein atoms | 9070 | 2350 | 4567 |
| solvent | 662 | 509 | 371 |
| RMS (bonds) | 0.008 | 0.008 | 0.037 |
| RMS (angles) | 1.00 | 1.00 | 0.73 |
| Ramachandran favored (%) | 1165 (98%) | 303 (98%) | 592 (98%) |
| Ramachandran allowed (%) | 28 (2%) | 5 (2%) | 13 (2%) |
| Ramachandran outliers (%) | 1 (0%) | 0 (0%) | 1 (0%) |
| Rotamer outliers (%) | 9 (1%) | 3 (1%) | 2 (0%) |
| average B-factor | 39.09 | 23.17 | 33.71 |
Coronavirus Main Protease Enzymatic Reactions and Compound Testing
Primary screening for SARS-CoV-2 main protease inhibitors was performed using a Rhodamine-110 fluorescent short peptide substrate cleavage assay. Briefly, compounds and controls were dry spotted into 1536-well plates, followed by 50 nM of SARS-CoV-2 Mpro in a volume of 2.5 μL, and incubated for 20 min at room temperature. Following incubation, 2.5 μL of 25 μM Rhodamine-110 peptide substrate was dispensed, and reactions were allowed to incubate at room temperature for 90 min. Fluorescence intensity was then quantified using a PHERAstar plate reader at an excitation wavelength of 485 nm and an emission wavelength of 520 nm.
The RFMS assay was run in clear 384-deep well small volume polypropylene plates (Greiner Bio-One, Monroe, NC; cat# 784201). All solutions were prepared with ultrapure Milli-Q purified water (EMD Millipore, Burlington, MA). Reaction buffer consisted of HEPES, pH 7.3 (VWR International, Radnor, PA; cat# J848), sodium chloride (Quality Biological, Gaithersburg, MD; cat# 351-036-101), EDTA (Quality Biological; cat# 351-027-101), and Pluronic F-127 (EMD Millipore; cat# 540025). Buffer was filtered through a 0.2 μm pore size bottle top filter (ThermoFisher, Waltham, MA; cat# 567-0020).
Evaluation of compound mediated inhibition of coronavirus Mpro enzyme activity was determined in an RFMS assay using an Agilent RapidFire 365 autosampler (Agilent, Santa Clara, CA) coupled to an Agilent 6400 Triple Quad LC/MS mass spectrometer. Mpro enzyme was diluted in assay buffer (50 mM HEPES, pH 7.3, 150 mM NaCl, 1 mM EDTA, 0.01% Pluronic F-127); final assay concentrations are listed in the Supporting Information (Table S5) for specific Mpro proteins. Five μL of 2X Mpro enzyme dilution was added into the assay ready plates dry-spotted with compounds (using 8 or 10 point, 3-fold serial dilutions with a top concentration of 50 μM dissolved in DMSO) via an Echo 555 liquid handler (Labcyte, San Jose, CA), and incubated at room temperature for 15 min prior to addition of substrate. To initiate the reaction, 2X substrate (final assay concentrations listed in Table S5) was added to the compound plate and incubated at room temperature (incubation times are listed in the Supporting Information, Table S5). Conditions were kept similar for each Mpro enzyme reaction with enzyme, substrate concentrations and reaction times adjusted to produce 10–20% substrate turnover in the absence of compound. After incubation, the reaction was quenched with 35 μL of stop solution (5% acetic acid, 0.2 nM 13C,15N-AVLQ, diluted in H2O) and then submitted to the RFMS.
Samples on the RapidFire were loaded onto a C18 SPE cartridge (Agilent) in H2O with 0.1% formic acid at a flow rate of 1.5 mL/min. Samples were then eluted with 75:20:5 acetonitrile: H2O: isopropyl alcohol with 0.1% formic acid at a flow rate of 1.0 mL/min.
Multiple reaction monitoring (MRM) transitions corresponding to the substrate and the related products, along with 13C,15N-AVLQ (for signal normalization), were monitored using MassHunter Workstation LC/MS Data Acquisition (Agilent), and peaks integrated using Agilent RapidFire Integrator. Corresponding peptides (Vivitide, Gardner, MA) monitored for each Mpro enzyme are listed in the Supporting Information (Table S6).
Determination of the Km value for the peptide substrate for each Mpro was conducted as described above for compound testing using RFMS using a dilution series of peptide substrate concentration across the indicated incubation time for each enzyme. The experimental values were graphed as velocity as a function of peptide substrate concentration. These values were fitted to a Michaelis–Menten plot using Prism 3.0 (Graphpad, San Diego, CA; Supporting Information, Figure S4).
Acknowledgments
We acknowledge the synthetic contributions of our colleagues at WuXi AppTech. We thank Viktor Hornak, Patrick Rudewicz, Manjunatha Ujjini, and Gu Feng (Novartis) for helpful discussions, as well as Vineet Menachery and Xuping Xie (University of Texas at Galveston, Medical Branch). We also thank Vincent Leonard (Novartis) for inspiring the original clustering concept. We are grateful to the Global Health Alliance Management and Partnering, Legal, and Finance team (Thomas Krucker, Elianna Amin, Daniel Raymond, Marcus Hall, Tracey Heinrich, and Jean Claude Poilevey) for their operational and administrative support.
Glossary
Abbreviations
- BLAST
basic local alignment search tool
- BV-BRC
bacterial and viral bioinformatics resource center
- CCR2
CC chemokine receptor 2
- CCL2
CC chemokine ligand 2
- CCR5
CC chemokine receptor 5
- COVID-19
coronavirus disease 2019
- CoV
coronavirus
- CV
column volume
- DMF-DMA
N,N-dimethylformamide dimethyl acetal
- EDCI
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide
- FA
formic acid
- FPLC
fast protein liquid chromatography
- GSH
glutathione
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HCoV
human coronavirus
- IBV
infectious bronchitis virus
- IMAC
immobilized metal affinity chromatography
- IPTG
isopropyl ß-d-1-thiogalactopyranoside
- LB
lysogeny broth
- LM
liver microsomal
- LRCoV
Lucheng rat coronavirus
- MDCK
Madin-Darby canine kidney
- MERS-CoV
middle eastern respiratory syndrome coronavirus
- MIB
malonic acid:imidazole:boric acid
- MPLC
medium pressure liquid chromatography
- Mpro
main protease
- MRM
multiple reaction monitoring
- MWCO
molecular weight cutoff
- NCBI
National Center for Biotechnology Information
- nMS
native mass spectrometry
- PAINS
pan-assay interference compounds
- PEDV
porcine epidemic diarrhea virus
- RFMS
RapidFire mass spectrometry
- RP
reverse phase
- SARS-CoV
severe acute respiratory syndrome coronavirus
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SNAr
nucleophilic aromatic substitution
- SEC
size exclusion chromatography
- SPE
solid phase extraction
- SPR
surface plasmon resonance
- SUMO
small ubiquitin-like modifier
- TB
terrific broth
- TCEP
tris(2-carboxyethyl)phosphine
- TEV
tobacco etch virus
- WSCoV
Wencheng shrew coronavirus
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01404.
Machine-readable datasheet file with full cluster composition including isolate accessions, cluster assignment, and annotation (XLSX)
Molecular formula strings (CSV)
Additional details regarding coronaviridae isolate curation and clustering, lab strain panel composition, and biochemical assay parameters, as well as experimental procedures and 1H NMR for all mentioned compounds (PDF)
Author Contributions
# D.D., S.M., and V.M.M. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number U19AI171413. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Wu F.; Zhao S.; Yu B.; Chen Y. M.; Wang W.; Song Z. G.; Hu Y.; Tao Z. W.; Tian J. H.; Pei Y. Y.; Yuan M. L.; Zhang Y. L.; Dai F. H.; Liu Y.; Wang Q. M.; Zheng J. J.; Xu L.; Holmes E. C.; Zhang Y. Z. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579 (7798), 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T. G.; Erdman D.; Goldsmith C. S.; Zaki S. R.; Peret T.; Emery S.; Tong S.; Urbani C.; Comer J. A.; Lim W.; Rollin P. E.; Dowell S. F.; Ling A.-E.; Humphrey C. D.; Shieh W.-J.; Guarner J.; Paddock C. D.; Rota P.; Fields B.; DeRisi J.; Yang J.-Y.; Cox N.; Hughes J. M.; LeDuc J. W.; Bellini W. J.; Anderson L. J. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348 (20), 1953–1966. 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Zaki A. M.; van Boheemen S.; Bestebroer T. M.; Osterhaus A. D. M. E.; Fouchier R. A. M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367 (19), 1814–1820. 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Brüssow H.; Brüssow L. Clinical Evidence That the Pandemic from 1889 to 1891 Commonly Called the Russian Flu Might Have Been an Earlier Coronavirus Pandemic. Microbiol. Biotechnol. 2021, 14, 1860–1870. 10.1111/1751-7915.13889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. L.; Duchêne S.; Holmes E. C. Comparative Analysis Estimates the Relative Frequencies of Co-Divergence and Cross-Species Transmission within Viral Families. PLoS Pathog. 2017, 13 (2), e1006215 10.1371/journal.ppat.1006215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daszak P.; Cunningham A. A.; Hyatt A. D. Anthropogenic Environmental Change and the Emergence of Infectious Diseases in Wildlife. Acta Trop. 2001, 78 (2), 103–116. 10.1016/S0001-706X(00)00179-0. [DOI] [PubMed] [Google Scholar]
- Chomel B. B.; Belotto A.; Meslin F. X. Wildlife, Exotic Pets, and Emerging Zoonoses. Emerging Infect. Dis. 2007, 13 (1), 6–11. 10.3201/eid1301.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee S.; Sintunawa C.; Kaewpom T.; Khongnomnan K.; Olival K. J.; Epstein J. H.; Rodpan A.; Sangsri P.; Intarut N.; Chindamporn A.; Suksawa K.; Hemachudha T. Group C Betacoronavirus in Bat Guano Fertilizer, Thailand. Emerging Infect. Dis. 2013, 19 (8), 1349–1351. 10.3201/eid1908.130119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyjinda Y.; Rodpan A.; Chartpituck P.; Suthum K.; Yaemsakul S.; Cheun-Arom T.; Bunprakob S.; Olival K. J.; Stokes M. M.; Hemachudha T.; Wacharapluesadee S. First Complete Genome Sequence of Human Coronavirus HKU1 from a Nonill Bat Guano Miner in Thailand. Microbiol. Resour. Announce. 2019, 8 (6), 7–8. 10.1128/MRA.01457-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. M.; Anthony S. J.; Switzer W. M.; Epstein J. H.; Seimon T.; Jia H.; Sanchez M. D.; Huynh T. T.; Galland G. G.; Shapiro S. E.; Sleeman J. M.; McAloose D.; Stuchin M.; Amato G.; Kolokotronis S. O.; Lipkin W. I.; Karesh W. B.; Daszak P.; Marano N. Zoonotic Viruses Associated with Illegally Imported Wildlife Products. PLoS One 2012, 7, e29505 10.1371/journal.pone.0029505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. A.; Grace D.; Kock R.; Alonso S.; Rushton J.; Said M. Y.; McKeever D.; Mutua F.; Young J.; McDermott J.; Pfeiffer D. U. Zoonosis Emergence Linked to Agricultural Intensification and Environmental Change. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (21), 8399–8404. 10.1073/pnas.1208059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomley F. M.; Shirley M. W. Livestock Infectious Diseases and Zoonoses. Philos. Trans. R. Soc., B 2009, 364 (1530), 2637–2642. 10.1098/rstb.2009.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. E.; Patel N. G.; Levy M. A.; Storeygard A.; Balk D.; Gittleman J. L.; Daszak P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451 (7181), 990–993. 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Zhang L.; Chen S.; Ouyang H.; Ren L. Possible Targets of Pan-Coronavirus Antiviral Strategies for Emerging or Re-Emerging Coronaviruses. Microorganisms 2021, 9 (7), 1479 10.3390/microorganisms9071479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved a-Ketoamide Inhibitors. Science 2020, 368 (6489), 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582 (7811), 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Trauner D.; Fischer C.; Veprek N.; Peitsinis Z.; Rühmann P.; Yang C.; Spradlin J.; Dovala D.; Nomura D.; Zhang Y. De Novo Design of SARS-CoV-2 Main Protease Inhibitors. Synlett 2022, 33, 458. 10.1055/a-1582-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biering S. B.; Van Dis E.; Wehri E.; Yamashiro L. H.; Nguyenla X.; Dugast-Darzacq C.; Graham T. G. W.; Stroumza J. R.; Golovkine G. R.; Roberts A. W.; Fines D. M.; Spradlin J. N.; Ward C. C.; Bajaj T.; Dovala D.; Schulze-Gamen U.; Bajaj R.; Fox D. M.; Ott M.; Murthy N.; Nomura D. K.; Schaletzky J.; Stanley S. A. Screening a Library of FDA-Approved and Bioactive Compounds for Antiviral Activity against SARS-CoV-2. ACS Infect. Dis. 2021, 7 (8), 2337–2351. 10.1021/acsinfecdis.1c00017. [DOI] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374 (6575), 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Flynn J. M.; Samant N.; Schneider-Nachum G.; Barkan D. T.; Yilmaz N. K.; Schiffer C. A.; Moquin S. A.; Dovala D.; Bolon D. N. A. Comprehensive Fitness Landscape of SARS-CoV-2 Mpro Reveals Insights into Viral Resistance Mechanisms. eLife 2022, 11, e77433 10.7554/eLife.77433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaqra A. M.; Zvornicanin S. N.; Huang Q. Y. J.; Lockbaum G. J.; Knapp M.; Tandeske L.; Bakan D. T.; Flynn J.; Bolon D. N. A.; Moquin S.; Dovala D.; Kurt Yilmaz N.; Schiffer C. A. Defining the Substrate Envelope of SARS-CoV-2 Main Protease to Predict and Avoid Drug Resistance. Nat. Commun. 2022, 13 (1), 3556 10.1038/s41467-022-31210-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. M.; Huang Q. Y. J.; Zvornicanin S. N.; Schneider-Nachum G.; Shaqra A. M.; Yilmaz N. K.; Moquin S. A.; Dovala D.; Schiffer C. A.; Bolon D. N. A. Systematic Analyses of the Resistance Potential of Drugs Targeting SARS-CoV-2 Main Protease. ACS Infect. Dis. 2023, 9 (7), 1372–1386. 10.1021/acsinfecdis.3c00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J.; Solowiej J. E.; Steppan C. M.; Taggart B. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63 (21), 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon P.; Zammit C. M.; Shao Q.; Dovala D.; Boike L.; Henning N. J.; Knapp M.; Spradlin J. N.; Ward C. C.; Wolleb H.; Fuller D.; Blake G.; Murphy J. P.; Wang F.; Lu Y.; Moquin S. A.; Tandeske L.; Hesse M. J.; McKenna J. M.; Tallarico J. A.; Schirle M.; Toste F. D.; Nomura D. K. Discovery of Potent Pyrazoline-Based Covalent SARS-CoV-2 Main Protease Inhibitors**. ChemBioChem 2023, 24 (11), e202300116 10.1002/cbic.202300116. [DOI] [PubMed] [Google Scholar]
- Flynn J. M.; Zvornicanin S. N.; Tsepal T.; Shaqra A. M.; Kurt Yilmaz N.; Jia W.; Moquin S.; Dovala D.; Schiffer C. A.; Bolon D. N. A. Contributions of Hyperactive Mutations in Mpro from SARS-CoV-2 to Drug Resistance. ACS Infect. Dis. 2024, 10, 1174. 10.1021/acsinfecdis.3c00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S. R.; Hughes S. A.; Bonilla P. J.; Turner J. D.; Leibowitz J. L.; Denison M. R. Coronavirus Polyprotein Processing. Arch. Virol., Suppl. 1994, 9, 349–358. 10.1007/978-3-7091-9326-6_35. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Shieh C. K.; Gorbalenya A. E.; Koonin E. V.; La Monica N.; Tuler J.; Bagdzhadzhyan A.; Lai M. M. C. The Complete Sequence (22 Kilobases) of Murine Coronavirus Gene 1 Encoding the Putative Proteases and RNA Polymerase. Virology 1991, 180 (2), 567–582. 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Resnick S. J.; Iketani S.; Hong S. J.; Zask A.; Liu H.; Kim S.; Melore S.; Lin F.-Y.; Nair M. S.; Huang Y.; Lee S.; Tay N. E. S.; Rovis T.; Yang H. W.; Xing L.; Stockwell B. R.; Ho D. D.; Chavez A. Inhibitors of Coronavirus 3CL Proteases Protect Cells from Protease-Mediated Cytotoxicity. J. Virol. 2021, 95 (14), e0237420 10.1128/JVI.02374-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers E. W.; Bolton E. E.; Brister J. R.; Canese K.; Chan J.; Comeau D. C.; Connor R.; Funk K.; Kelly C.; Kim S.; Madej T.; Marchler-Bauer A.; Lanczycki C.; Lathrop S.; Lu Z.; Thibaud-Nissen F.; Murphy T.; Phan L.; Skripchenko Y.; Tse T.; Wang J.; Williams R.; Trawick B. W.; Pruitt K. D.; Sherry S. T. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50 (D1), D20–D26. 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson R. D.; Assaf R.; Brettin T.; Conrad N.; Cucinell C.; Davis J. J.; Dempsey D. M.; Dickerman A.; Dietrich E. M.; Kenyon R. W.; Kuscuoglu M.; Lefkowitz E. J.; Lu J.; Machi D.; Macken C.; Mao C.; Niewiadomska A.; Nguyen M.; Olsen G. J.; Overbeek J. C.; Parrello B.; Parrello V.; Porter J. S.; Pusch G. D.; Shukla M.; Singh I.; Stewart L.; Tan G.; Thomas C.; VanOeffelen M.; Vonstein V.; Wallace Z. S.; Warren A. S.; Wattam A. R.; Xia F.; Yoo H.; Zhang Y.; Zmasek C. M.; Scheuermann R. H.; Stevens R. L. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A Resource Combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51 (1 D), D678–D689. 10.1093/nar/gkac1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C.; Coulouris G.; Avagyan V.; Ma N.; Papadopoulos J.; Bealer K.; Madden T. L. BLAST+: Architecture and Applications. BMC Bioinf. 2009, 10, 421 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoh Y.; Uehara S.; Nakahara K.; Nobori H.; Yamatsu Y.; Yamamoto S.; Maruyama Y.; Taoda Y.; Kasamatsu K.; Suto T.; Kouki K.; Nakahashi A.; Kawashima S.; Sanaki T.; Toba S.; Uemura K.; Mizutare T.; Ando S.; Sasaki M.; Orba Y.; Sawa H.; Sato A.; Sato T.; Kato T.; Tachibana Y. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499. 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douangamath A.; Fearon D.; Gehrtz P.; Krojer T.; Lukacik P.; Owen C. D.; Resnick E.; Strain-Damerell C.; Aimon A.; Ábrányi-Balogh P.; Brandão-Neto J.; Carbery A.; Davison G.; Dias A.; Downes T. D.; Dunnett L.; Fairhead M.; Firth J. D.; Jones S. P.; Keeley A.; Keserü G. M.; Klein H. F.; Martin M. P.; Noble M. E. M.; O’Brien P.; Powell A.; Reddi R. N.; Skyner R.; Snee M.; Waring M. J.; Wild C.; London N.; von Delft F.; Walsh M. A. Crystallographic and Electrophilic Fragment Screening of the SARS-CoV-2 Main Protease. Nat. Commun. 2020, 11 (1), 5047 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar S.; Johnston M. L.; John S. E. S.; Osswald H. L.; Nyalapatla P. R.; Paul L. N.; Ghosh A. K.; Denison M. R.; Mesecar A. D. Ligand-Induced Dimerization of Middle East Respiratory Syndrome (MERS) Coronavirus Nsp5 Protease (3CLpro): Implications for Nsp5 Regulation and the Development of Antivirals. J. Biol. Chem. 2015, 290 (32), 19403–19422. 10.1074/jbc.M115.651463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach J. R. A Filtrable Virus, the Cause of Infectious Laryngotracheitis of Chickens. J. Exp. Med. 1931, 54 (6), 809–816. 10.1084/jem.54.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.; Yu H.; Yang H.; Xue F.; Wu Z.; Shen W.; Li J.; Zhou Z.; Ding Y.; Zhao Q.; Zhang X. C.; Liao M.; Bartlam M.; Rao Z. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82 (5), 2515–2527. 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasey D.; Cartwright S. F. Virus-like Particles Associated with Porcine Epidemic Diarrhoea. Res. Vet. Sci. 1978, 25 (2), 255–256. 10.1016/S0034-5288(18)32994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John S. E.; Anson B. J.; Mesecar A. D. X-Ray Structure and Inhibition of 3C-like Protease from Porcine Epidemic Diarrhea Virus. Sci. Rep. 2016, 6 (1), 25961 10.1038/srep25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K.; Dees J. H.; Becker W. B.; Kapikian A. Z.; Chanock R. M. Recovery in Tracheal Organ Cultures of Novel Viruses from Patients with Respiratory Disease. Proc. Natl. Acad. Sci. U.S.A. 1967, 57 (4), 933–940. 10.1073/pnas.57.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y.; Lau S. K. P.; Chu C.; Chan K.; Tsoi H.; Huang Y.; Wong B. H. L.; Poon R. W. S.; Cai J. J.; Luk W.; Poon L. L. M.; Wong S. S. Y.; Guan Y.; Peiris J. S. M.; Yuen K. Characterization and Complete Genome Sequence of a Novel Coronavirus, Coronavirus HKU1, from Patients with Pneumonia. J. Virol. 2005, 79 (2), 884–895. 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Li S.; Xue F.; Zou Y.; Chen C.; Bartlam M.; Rao Z. Structure of the Main Protease from a Global Infectious Human Coronavirus, HCoV-HKU1. J. Virol. 2008, 82 (17), 8647–8655. 10.1128/JVI.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. W.; Egberink H. F.; Halpin R.; Spiro D. J.; Rottie P. J. M. Spike Protein Fusion Peptide and Feline Coronavirus Virulence. Emerging Infect. Dis. 2012, 18 (7), 1089–1095. 10.3201/eid1807.120143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L.; Ye F.; Feng Y.; Yu F.; Wang Q.; Wu Y.; Zhao C.; Sun H.; Huang B.; Niu P.; Song H.; Shi Y.; Li X.; Tan W.; Qi J.; Gao G. F. Both Boceprevir and GC376 Efficaciously Inhibit SARS-CoV-2 by Targeting Its Main Protease. Nat. Commun. 2020, 11 (1), 4417 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Yang M.; Ding Y.; Liu Y.; Lou Z.; Zhou Z.; Sun L.; Mo L.; Ye S.; Pang H.; Gao G. F.; Anand K.; Bartlam M.; Hilgenfeld R.; Rao Z. The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and Its Complex with an Inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003, 100 (23), 13190–13195. 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y.; Lau S. K. P.; Li K. S. M.; Poon R. W. S.; Wong B. H. L.; Tsoi H. wah.; Yip B. C. K.; Huang Y.; Chan K. hung.; Yuen K. yung. Molecular Diversity of Coronaviruses in Bats. Virology 2006, 351 (1), 180–187. 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y.; Lau S. K. P.; Lam C. S. F.; Lau C. C. Y.; Tsang A. K. L.; Lau J. H. N.; Bai R.; Teng J. L. L.; Tsang C. C. C.; Wang M.; Zheng B.-J.; Chan K.-H.; Yuen K.-Y. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J. Virol. 2012, 86 (7), 3995–4008. 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvornicanin S. N.; Shaqra A. M.; Huang Q. J.; Ornelas E.; Moghe M.; Knapp M.; Moquin S.; Dovala D.; Schiffer C. A.; Kurt Yilmaz N. Crystal Structures of Inhibitor-Bound Main Protease from Delta- and Gamma-Coronaviruses. Viruses 2023, 15 (3), 781 10.3390/v15030781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamre D.; Procknow J. J. A New Virus Isolated from the Human Respiratory Tract. Proc. Soc. Exp Biol. Med. 1966, 121 (1), 190–193. 10.3181/00379727-121-30734. [DOI] [PubMed] [Google Scholar]
- Anand K.; Anand K.; Ziebuhr J.; Wadhwani P. Coronavirus main proteinase (3CL pro) Structure: Basis for Design of Anti-SARS Drugs. Science 2014, 1763 (2003), 1763–1768. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- van der Hoek L.; Pyrc K.; Jebbink M. F.; Vermeulen-Oost W.; Berkhout R. J. M.; Wolthers K. C.; Wertheim-van Dillen P. M. E.; Kaandorp J.; Spaargaren J.; Berkhout B. Identification of a New Human Coronavirus. Nat. Med. 2004, 10 (4), 368–373. 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Chen C.; Tan W.; Yang K.; Yang H. Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Sci. Rep. 2016, 6 (March), 22677 10.1038/srep22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y.; Wang M.; Lau S. K. P.; Xu H.; Poon R. W. S.; Guo R.; Wong B. H. L.; Gao K.; Tsoi H.; Huang Y.; Li K. S. M.; Lam C. S. F.; Chan K.; Zheng B.; Yuen K. Comparative Analysis of Twelve Genomes of Three Novel Group 2c and Group 2d Coronaviruses Reveals Unique Group and Subgroup Features. J. Virol. 2007, 81 (4), 1574–1585. 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X. D.; Wang W.; Hao Z. Y.; Wang Z. X.; Guo W. P.; Guan X. Q.; Wang M. R.; Wang H. W.; Zhou R. H.; Li M. H.; Tang G. P.; Wu J.; Holmes E. C.; Zhang Y. Z. Extensive Diversity of Coronaviruses in Bats from China. Virology 2017, 507 (February), 1–10. 10.1016/j.virol.2017.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Lin X.-D.; Liao Y.; Guan X.-Q.; Guo W.-P.; Xing J.-G.; Holmes E. C.; Zhang Y.-Z. Discovery of a Highly Divergent Coronavirus in the Asian House Shrew from China Illuminates the Origin of the Alphacoronaviruses. J. Virol. 2017, 91 (17), e00764-17 10.1128/JVI.00764-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihindukulasuriya K. A.; Wu G.; St Leger J.; Nordhausen R. W.; Wang D. Identification of a Novel Coronavirus from a Beluga Whale by Using a Panviral Microarray. J. Virol. 2008, 82 (10), 5084–5088. 10.1128/jvi.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M.; Lin X.-D.; Chen X.; Tian J.-H.; Chen L.-J.; Li K.; Wang W.; Eden J.-S.; Shen J.-J.; Liu L.; Holmes E. C.; Zhang Y.-Z. The Evolutionary History of Vertebrate RNA Viruses. Nature 2018, 556 (7700), 197–202. 10.1038/s41586-018-0012-7. [DOI] [PubMed] [Google Scholar]
- Mordecai G. J.; Miller K. M.; Di Cicco E.; Schulze A. D.; Kaukinen K. H.; Ming T. J.; Li S.; Tabata A.; Teffer A.; Patterson D. A.; Ferguson H. W.; Suttle C. A. Endangered Wild Salmon Infected by Newly Discovered Viruses. eLife 2019, 8, e47615 10.7554/eLife.47615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. K.; Mifsud J. C. O.; Costa V. A.; Grimwood R. M.; Kitson J.; Baker C.; Brosnahan C. L.; Pande A.; Holmes E. C.; Gemmell N. J.; Geoghegan J. L. Slippery When Wet: Cross-Species Transmission of Divergent Coronaviruses in Bony and Jawless Fish and the Evolutionary History of the Coronaviridae. Virus Evol. 2021, 7 (2), veab050 10.1093/ve/veab050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Lu L.; Du J.; Yang L.; Ren X.; Liu B.; Jiang J.; Yang J.; Dong J.; Sun L.; Zhu Y.; Li Y.; Zheng D.; Zhang C.; Su H.; Zheng Y.; Zhou H.; Zhu G.; Li H.; Chmura A.; Yang F.; Daszak P.; Wang J.; Liu Q.; Jin Q. Comparative Analysis of Rodent and Small Mammal Viromes to Better Understand the Wildlife Origin of Emerging Infectious Diseases 06 Biological Sciences 0604 Genetics 11 Medical and Health Sciences 1108 Medical Microbiology. Microbiome 2018, 6 (1), 178 10.1186/s40168-018-0554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F.; Duong V.; Lim X. F.; Hul V.; Chawla T.; Keatts L.; Goldstein T.; Hassanin A.; Tu V. T.; Buchy P.; Sessions O. M.; Wang L. F.; Dussart P.; Anderson D. E. Presence of Recombinant Bat Coronavirus GCCDC1 in Cambodian Bats. Viruses 2022, 14 (2), 176 10.3390/v14020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. W.; Gallego R. A.; Edwards M. P. Lipophilic Efficiency as an Important Metric in Drug Design. J. Med. Chem. 2018, 61 (15), 6401–6420. 10.1021/acs.jmedchem.8b00077. [DOI] [PubMed] [Google Scholar]
- Sakhno Y. I.; Murlykina M. V.; Zbruyev O. I.; Kozyryev A. V.; Shishkina S. V.; Sysoiev D.; Musatov V. I.; Desenko S. M.; Chebanov V. A. Ultrasonic-Assisted Unusual Four-Component Synthesis of 7-Azolylamino-4,5,6,7-Tetrahydroazolo[1,5-a]Pyrimidines. Beilstein J. Org. Chem. 2020, 16, 281–289. 10.3762/bjoc.16.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebanov V. A.; Sakhno Y. I.; Desenko S. M.; Chernenko V. N.; Musatov V. I.; Shishkina S. V.; Shishkin O. V.; Kappe C. O. Cyclocondensation Reactions of 5-Aminopyrazoles, Pyruvic Acids and Aldehydes. Multicomponent Approaches to Pyrazolopyridines and Related Products. Tetrahedron 2007, 63 (5), 1229–1242. 10.1016/j.tet.2006.11.048. [DOI] [Google Scholar]
- Patel A. S.; Kapuriya N. P.; Naliapara Y. T. A Concise [3 + 3] Heteroaromatization Synthetic Strategy Afford Dicarboxamide Functionalized Novel Pyrazolo[1,5-a]Pyrimidines. J. Heterocycl. Chem. 2017, 54 (5), 2635–2643. 10.1002/jhet.2860. [DOI] [Google Scholar]
- Thomas A.; Chakraborty M.; Ila H.; Junjappa H. Cyclocondensation of Oxoketene Dithioacetals with 3-Aminopyrazoles: A Facile Highly Regioselective General Route to Substituted and Fused Pyrazolo]Pyrimidines. Tetrahedron 1990, 46 (2), 577–586. 10.1016/S0040-4020(01)85438-7. [DOI] [Google Scholar]
- Geraghty R. J.; Aliota M. T.; Bonnac L. F. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 13 (4), 667 10.3390/v13040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertani G. Studies on Lysogenesis. I. The Mode of Phage Liberation by Lysogenic Escherichia Coli. J. Bacteriol. 1951, 62, 293–300. 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartof K. D. Improved Media for Growing Plasmid and Cosmid Clones. Bethesda Res. Lab. Focus. 1987, 9, 12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.