

Abstract

The methyl-d-erythritol phosphate (MEP) pathway has emerged as an interesting target in the fight against antimicrobial resistance. The pathway is essential in many human pathogens, including Plasmodium falciparum (Pf), but is absent in human cells. In the present study, we report on the discovery of a new chemical class targeting IspD, the third enzyme in the pathway. Exploration of the structure–activity relationship yielded inhibitors with potency in the low-nanomolar range. Moreover, we investigated the whole-cell activity, mode of inhibition, metabolic, and plasma stability of this compound class, and conducted in vivo pharmacokinetic profiling on selected compounds. Lastly, we disclosed a new mass spectrometry (MS)-based enzymatic assay for direct IspD activity determination, circumventing the need for auxiliary enzymes. In summary, we have identified a readily synthesizable compound class, demonstrating excellent activity and a promising profile, positioning it as a valuable tool compound for advancing research on IspD.
Introduction
Since the commercialization of penicillin in the 1940s, Sir Alexander Fleming warned the public about the dangers of antimicrobial resistance (AMR), resulting from overand misuse of anti-infectives. Now, decades later, his warnings are more relevant than ever with AMR reaching alarming levels.1,2 A recent example of newly developed resistance is the discovery of artemisinin-resistant strains of Plasmodium falciparum (Pf), one of the parasites that causes malaria, in Africa, Southeast Asia, the Pacific islands, and Latin America. This discovery is significant as artemisinin-based treatments have been the ‘gold standard’ for malaria treatments for many years, and resistance will have disastrous effects for malaria-prone regions.3 The 2-C-methylerythritol-d-erythritol-4-phosphate (MEP) pathway, needed for the biosynthesis of the isoprenoid precursors isopentenyl diphosphate (IDP) and dimethylallyl diphosphate (DMADP), is an essential biosynthetic pathway in most Gram-negative bacteria, Mycobacterium tuberculosis, and the Plasmodium parasites. Furthermore, the MEP pathway is absent in human cells, mitigating the risk of off-target side effects, hence making it a source of promising drug targets.4,5 Validation of the MEP pathway enzymes as drug target is based on fosmidomycin, an inhibitor of the second protein, IspC or DXR, of the MEP pathway. Several clinical trials have investigated fosmidomycin in combination therapy for malaria; however, none have achieved cure rates meeting the standards set by the World Health Organization. Nonetheless, a meta-analysis conducted by Fernandes et al. revealed a promising 85% cure rate in children, indicating significant potential for fosmidomycin in combination therapy and serving as incentive for further research in this field.6−8 Within the present study, we focused on targeting the third enzyme in the MEP pathway, known as, IspD, MEP cytidyltransferase, or ygbp. IspD catalyzes the formation of 4-diphosphocytidyl-2-C-methylerythritol (CDP-ME) from MEP and cytidine triphosphate (CTP) in the presence of Mg2+, releasing inorganic diphosphate (PPi) (Scheme 1).9 Previously reported inhibitors targeting PfIspD can be subdivided into three chemical classes, namely, benzoisothiazolones 1, identified by a combined approach of cheminformatics and high-throughput enzymatic screening, MMV008138 2 recognized through phenotypic screening of the library of GlaxoSmithKline and last, a biphenyl carboxylic acid fragment 3 recently discovered by our group in collaboration with BASF (Figure 1). Despite the potential of IspD as a drug target, the number of IspD inhibitors reported is rather low. Furthermore, the reported inhibitors are rather challenging to synthesize or lack whole-cell activity.5,10−14 A possible cause might be the lack of a crystal structure of PfIspD available in the Protein Data Bank (PDB). Here, we report the structure–activity relationship (SAR) study of a new urea-based compound class targeting PfIspD with low-nanomolar activity in vitro. Its synthesis is straightforward, in one step from the corresponding aniline and isocyanate. A high-throughput screening (HTS) approach on P. vivax IspD and subsequent confirmation of hits concomitantly on PfIspD and PfNF54 led to the discovery of the initial hit (4, Figure 2) endowed with an IC50 of 17 ± 2 μM against PfIspD but lacking whole-cell activity. Synthesis of a total of 34 derivatives shed a light on the SAR of this newly discovered class reaching IC50 values as low as 41 nM with a whole-cell activity in the low micromolar range. Throughout our study, we made efforts to maintain the easily synthesizable core of the urea class, ensuring the molecule’s accessibility, of particular improtance for antimalarials that are predominantly utilized in low- and middle-income countries.
Scheme 1. Reaction Catalyzed by IspD Starting from MEP and CTP Affording CDP-ME.
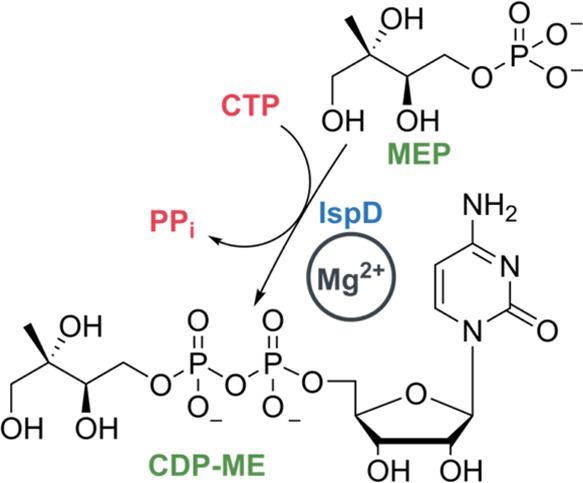
Mg2+ is the cofactor in the reaction.
Figure 1.
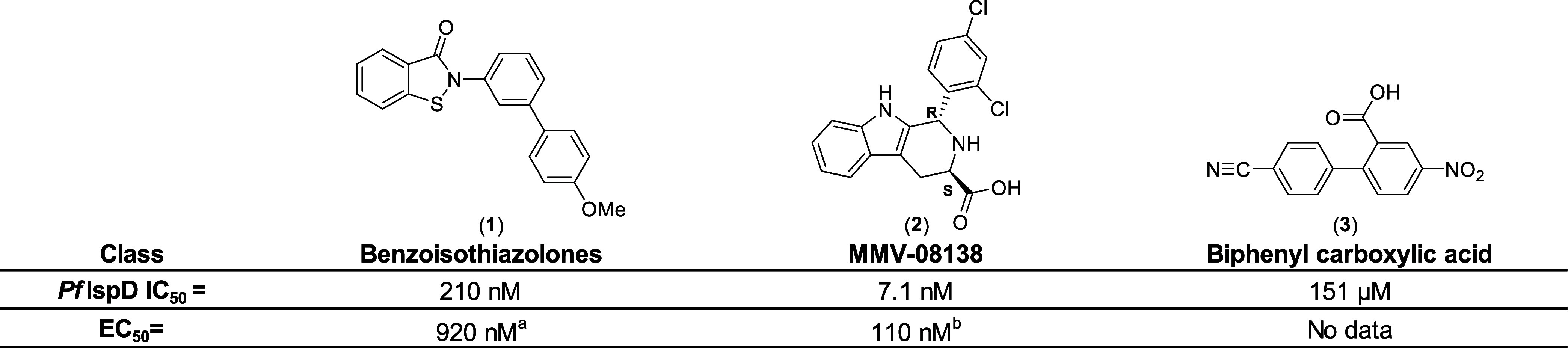
Currently known classes of inhibitors showing enzymatic activity against PfIspD. EC50 values were measured against different strains of Plasmodium falciparum. a: strain = 3D7; b: strain = W2; Benzoisothiazolones10 (1), MMV-0813811−14 (2), Biphenylcarboxylic acid5 (3).
Figure 2.
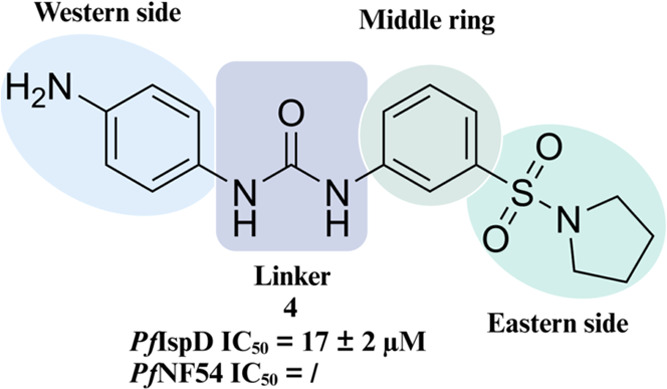
Initial hit compound 4 with an overview of the performed SAR study.
Results and Discussion
SAR Exploration
We commenced exploration of the initial hit by synthesizing derivatives with diverse moieties on the Western phenyl ring (Figure 2). Compounds 4–18 were synthesized as depicted in Scheme 2. Depending on commercial availability, we either generated isocyanates in situ by reacting the respective amine with triphosgene or purchased them. Nucleophilic addition between 3-(pyrrolidin-1-ylsulfonyl)aniline and the respective isocyanate afforded the desired compounds. At first, we directed modifications toward the primary amine and replaced it by moieties with different electronic effects (5–11) (Table 1). We observed that more electron-withdrawing substituents, such as a nitro (8) or nitrile group (10), had a pronounced effect on the potency, resulting in a 400-fold increase (e.g., 8, PfIspD IC50 = 41 ± 7 nM). While weaker electron-withdrawing substituents , such as the trifluoromethyl (11) and the chloride (5), had a lower effect on the potency (e.g., 11, PfIspD IC50 = 91 ± 19 nM). Lastly, weakly electron-donating groups, such as methyl (6), lead to an even smaller increase in potency (e.g., 6, PfIspD IC50 = 370 ± 80 nM), but were still significantly better than the initial hit compound 4. The smaller increase in potency of 9 (PfIspD IC50 = 330 ± 40 nM) could possibly be attributed to the size of the substituent as will be seen later. In addition, low-micromolar activity in the whole-cell assay was noted for these derivatives. Next, we explored various substitution patterns on the phenyl ring 12–14. Placement of trifluoromethyl in meta position (12, PfIspD IC50 = 415 ± 60 nM) did not improve upon its para-substituted counterpart (11, PfIspD IC50 = 91 ± 19 nM). Furthermore, having multiple substituents (13–14) did also not lead to improvements in PfIspD activity over the monosubstituted derivatives. To determine whether there was still room for growth on the Western side, we synthesized analogues 15–18 using the general synthetic route depicted in Scheme 2. Growth in this direction resulted in a significant loss in activity, which we interpret as a lack of space for further expansions. For the remainder of the SAR study, we selected 8 as scaffold for derivatization. Next, we focused on the urea linker itself (Table 2). Both positions of the urea bond were methylated successively as depicted in Scheme 3. To ensure selective methylation, 20 was synthesized by first transforming 3-(pyrrolidin-1-ylsulfonyl)aniline (19) into the corresponding isocyanate with triphosgene, followed by addition of deprotonated N-methyl-4-nitroaniline to the reaction mixture. On the other hand, 20 was synthesized via two steps. First, a reductive amination between 3-(pyrrolidin-1-ylsulfonyl)aniline and paraformaldehyde resulted in N-methyl-3-(pyrrolidin-1-ylsulfonyl)aniline to which 1-isocyanato-4-nitrobenzene was added, resulting in a nucleophilic addition affording 21. Unfortunately, methylation of either site of the urea bond led to complete loss of activity. A possible explanation for this observation could be the loss of hydrogen-bonding interactions or conformational changes imposed by the methylation. Next we explored the possibility of replacing the urea with a thiourea (23) by employing an isothiocyanate 22 in the synthesis instead of an isocyanate (Scheme 3, bottom). This modification resulted as well in a decrease in activity (23, PfIspD IC50 = 395 ± 60 nM) in comparison with its urea counterpart (8, PfIspD IC50 = 41 ± 7 nM). Subsequent modifications explored the Eastern side of the molecule (Figure 2). For the synthesis of these derivatives (24–28), we used the synthetic procedure as depicted in Scheme 4. Initially, we replaced the pyrrolidine with more flexible dimethyl (24) and diethyl (25) amine groups (Table 2). These derivatives did not manage to enhance activity (24, PfIspD IC50 = 180 ± 20 nM) over the pyrrolidinyl-containing parent compound (8, PfIspD IC50 = 41 ± 7 nM) (Table 2). Ring expansion toward piperidine (26) and morpholine (27) likewise failed to increase activity. On the other hand, when we substituted the pyrrolidinyl with a phenyl ring, the activity could be retained (28). In the next phase, we assessed the role of the sulfonyl linker in the compounds’ activity. To this end, we synthesized compounds 29 and 30. For the synthesis of compound 29, we began with amide-bond formation between 3-nitrobenzoic acid and pyrrolidine using propanephosphonic acid anhydride as the coupling reagent (Scheme S1). Subsequently, we reduced the nitro group to the corresponding amine, which was then reacted with 1-isocyanato-4-nitrobenzene, yielding the desired compound. For the synthesis of compound 30, we employed a similar reaction scheme starting from 3-nitrobenzyl bromide (Scheme S2). A nucleophilic substitution reaction with pyrrolidine produced, an intermediate, of which the nitro group was then reduced to the corresponding amine. This amine was reacted with 1-isocyanato-4-nitrobenzene to afford the final compound. As shown in Table 3, replacing the sulfonyl linker resulted in a significant decrease in activity toward PfIspD. Therefore, we reason that the sulfonyl linker is crucial for the activity of this compound class. Lastly, we explored a handful of compounds containing modifications on the middle ring (Table 4). Initially, these compounds were synthesized containing a morpholine (31–33) instead of a pyrrolidine, as anilines of these derivatives were commercially available. Interestingly, compound 31 (PfIspD IC50 = 395 ± 3.5 nM) showed enhanced activity over parent compound 27 (PfIspD IC50 = 600 ± 110 nM). Consequently, we decided to construct derivatives containing the pyrrolidine (34–40). A three-step synthesis led to compounds 34–40 (Scheme 5). As a first step, a nucleophilic substitution reaction took place between pyrrolidine and the respective 3-nitrobenzenesulfonyl chloride. Next, the nitro group was reduced to the amine, which was then reacted with 1-isocyanato-4-nitrobenzene, affording the desired urea compounds (Table 4). After examination of these compounds in our in vitro assays, we could not observe any increase in potency over 8, even not for the derivative containing the 4-fluoro moiety (34), which previously triggered a rise in potency. In summary, modifications directed to the Western side of the molecule are most beneficial for the activity of the compound class. Positioning electron-withdrawing substituents at the para position induced the most notable changes, enabling compounds 8 and 10 to reach IC50 values of 41 nM. Other substituents or further expansions at this position did not achieve such an increase in potency. In addition, an unsubstituted urea linker is detrimental for the activity of the compound class. Attempts to modify the middle ring turned out to be futile as any placement of a moiety led to a decrease in activity. On the Eastern side of the molecule, a pyrrolidinyl or phenyl ring led to the highest in vitro activity. Furthermore, we observed that the sulfonyl linker is essential for the in vitro activity of the compound class. Overall, compounds 8, 10, and 28 are seen as frontrunners of the urea class, exhibiting the best in vitro activity, while also showing modest activity in the whole-cell assay. Interestingly, compound 28 constitutes a potential starting point to further explore the urea class by placing substituents on the phenyl ring or by growing in this direction.
Scheme 2. General Synthetic Procedure Followed for the Synthesis of 4–18.

Reagents and conditions: (a) triphosgene, Et3N, DCM, 0 °C to room temperature, 3 h, used without purification in the next reaction step; (b) 3-(pyrrolidin-1-ylsulfonyl)aniline, DMF, room temperature, overnight, 8–95% yield.
Table 1. In Vitro and Whole-Cell Activities for Compounds 4–18.

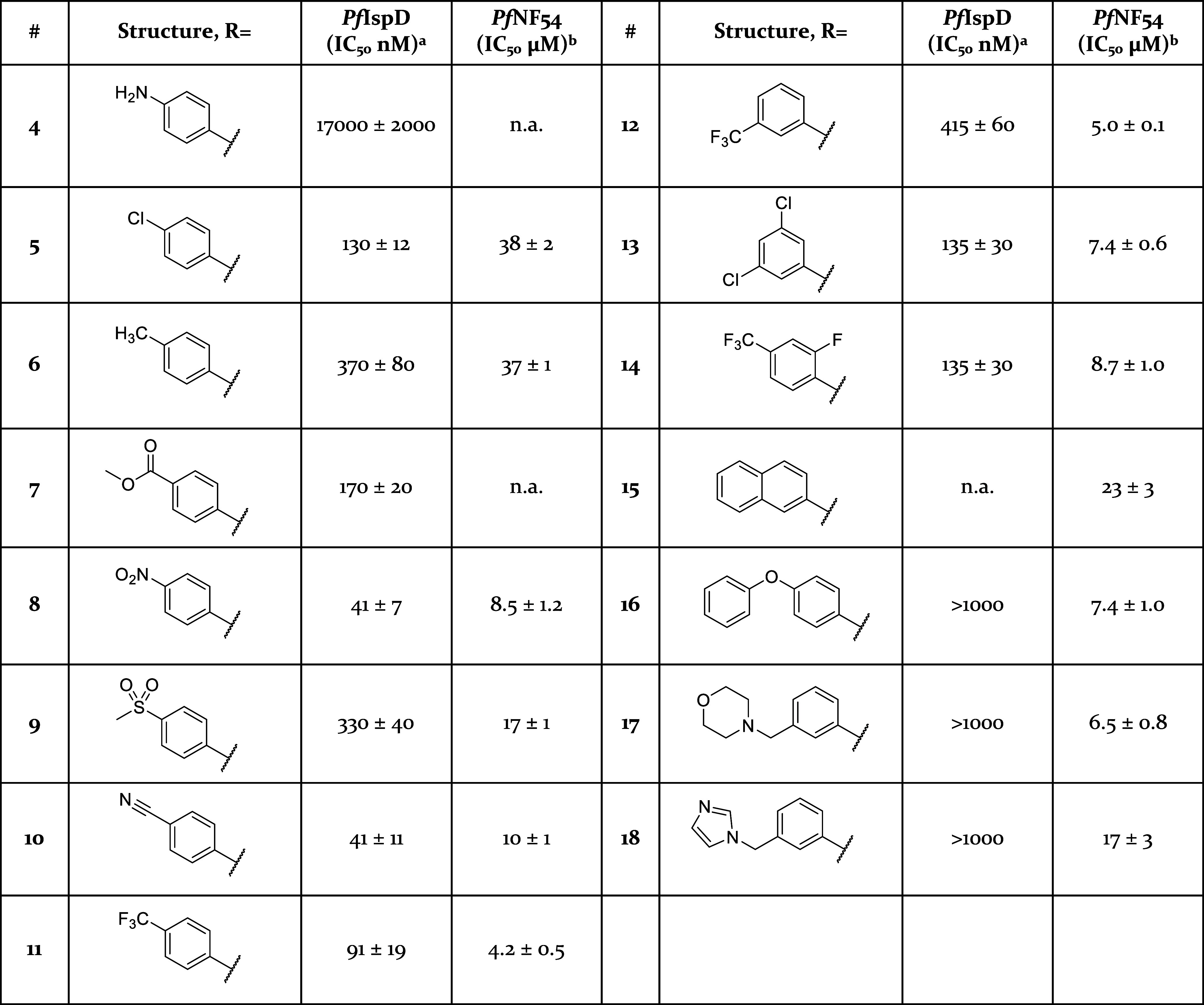
Assays were performed in replicate as independent experiments (n ≥ 2); values are shown as mean ± SD.
Assays were performed in replicate as independent experiments (n = 2); values are shown as mean ± SD n.a. indicates the absence of activity.
Table 2. In Vitro and Whole-Cell Activities for Compounds 20, 21, and 23–28.

Assays were performed in replicate as independent experiments (n ≥ 2); values are shown as mean ± SD.
Assays were performed in replicate as independent experiments (n = 2); values are shown as mean ± SD n.a. indicates the absence of activity.
Scheme 3. Synthetic Procedure Followed for the Synthesis of 20, 21, and 23.

Reagents and reactions conditions: (a) triphosgene, Et3N, DCM, 0 °C to room temperature, 3 h; (b) N-methyl-4-nitroaniline, NaH, DMF, room temperature, 1 h. 32% yield over two steps; (c) paraformaldehyde, NaBH4, MeOH, at room temperature for 2.5 h to 60 °C for 16 h, 57% yield; (d) 1-isocyanato-4-nitrobenzene, DMF, room temperature for 16 h, 12% yield. (e) 3-(pyrrolidin-1-ylsulfonyl)aniline, DMF, room temperature, 48 h, 35% yield.
Scheme 4. General Synthetic Procedure Followed for the Synthesis of 24–28.

Reagents and conditions: (a) 1-isocyanato-4-nitrobenzene, DMF, room temperature, overnight, 28–58% yield.
Table 3. In Vitro and Whole-Cell Activities for Compounds 8, 29, and 30.

Assays were performed in replicate as independent experiments (n ≥ 2); values are shown as mean ± SD.
Assays were performed in replicate as independent experiments (n = 2); values are shown as mean ± SD.
Table 4. In Vitro and Whole-Cell Activities for Compounds 31–40.

Assays were performed in replicate as independent experiments (n ≥ 2); values are shown as mean ± SD.
Assays were performed in replicate as independent experiments (n ≥ 2); values are shown as mean ± SD n.a. indicates the absence of activity.
Scheme 5. Synthesis of Compounds 34–40.

Reagents and reactions conditions: (a) respective 3-nitrobenzenesulfonyl chloride, pyrrolidine, triethylamine, acetonitrile, 0 °C, 5 min; (b) Fe powder, NH4Cl (166 mM in water), EtOH, 80 °C, 2.5 h; (c) 1-isocyanato-4-nitrobenzene, DMF, room temperature, 4 h, 5–37% yield over three steps.
Validating IspD as Target of the Urea Class
A discrepancy was observed in the activity trends of compounds from this class when comparing on target and whole-cell assays. For instance, compound 10 (PfIspD IC50 = 41 ± 7 nM) significantly outperforms compound 32 (PfIspD IC50 = 2400 ± 300 nM) in PfIspD activity. However, in the whole-cell assay, compound 32 (PfNF54 IC50 = 2.7 ± 0.2 μM) shows better performance than compound 10 (PfNF54 IC50 = 10 ± 1 μM). This inconsistency between PfIspD activity and whole-cell activity could be attributed in some part to off-target effects. To verify if IspD is contributing to the cellular activity, we measured the whole-cell activity of compound 10 against a wild-type (MR4) strain and two MMV008138-resistant strains (R2 and R3) of P. falciparum. As shown in Figure 3, both resistant strains appear more susceptible to compound 10 than the wild type. We hypothesize that this increased susceptibility is due to a less fit IspD enzyme in the resistant parasites. Mutations in IspD that confer resistance to MMV008138 may alter the enzyme, making resistant parasites more susceptible to other IspD inhibitors. This suggests that IspD is a target of this class of compounds. To confirm this observation, we repeated the assay, this time supplementing the parasites with 200 μM IDP, the end product of the MEP pathway. The addition of IDP equalized the sensitivity of the resistant strains and the wild type toward compound 10. This confirmed our hypothesis that IspD is indeed one of the in vivo targets of this compound class (Figure 4).
Figure 3.
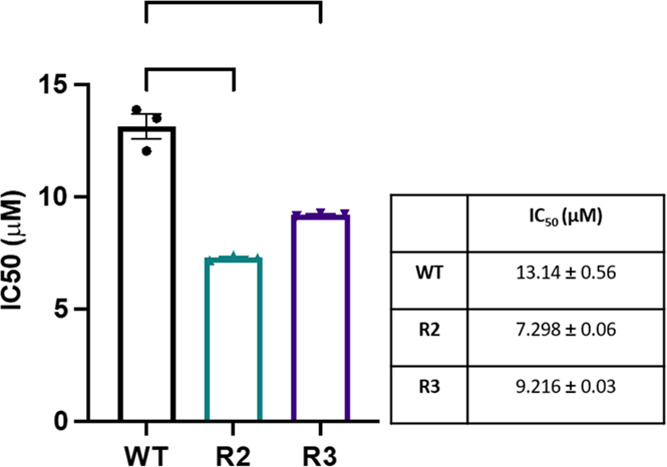
Comparison of the whole-cell activity of 10 toward one wild type strain (WT, MR4) and two MMV008138 resistant strains of P. falciparum. Statistical analysis—one-way anova, R2: P* < 0.0001, R3: P*—0.0003
Figure 4.

Difference in whole-cell activity of 10 between non supplemented and IDP supplemented conditions. WT = wild type; R2 and R3 are MMV008138 resistant strains.
LC-MS Based Activity Assay
To gain an idea on the mode of inhibition of our new compound class, we intended to do a characterization of the enzyme kinetics under a range of inhibitor concentrations. Our intention was to perform this experiment without the influence of auxiliary enzymes inherent to the photometric assay used for IC50 determinations.15 To achieve this goal, we sought to uncover a way to measure the progress of the enzymatic reaction without relying on any secondary reactions. In our exploration, we encountered the work of Li et al., who successfully profiled and quantified MEP metabolites in leaves using liquid chromatography-tandem mass spectrometry.16 With this information in hand, we sought to develop an IspD activity assay based on the LC-MS detection and quantification of both substrate and product. Initial experiments revealed a significantly more pronounced signal for CDP-ME compared to MEP, with the latter often indistinguishable from background noise. An explanation for this observation might be the difference in ease of ionization, with CDP-ME being more readily ionizable than MEP. Furthermore, we observed identical fragmentation for MEP and the MEP part of CDP-ME, resulting in an overestimation of the MEP concentration. Hence, we decided to continue the assay development relying on the quantification of the product, CDP-ME. Calibration curves measured for CDP-ME demonstrated a linear progression for a wide concentration range showing an R2 of 0.99 (Figure S1). Finally, an internal standard was chosen, initially several unreactive ATP derivatives, such as adenylyl-imidodiphosphate and adenosine-5′-[(α,β)-methyleno]triphosphate were tested, but those exhibited long elution times of 20 to 30 min. Ultimately, we chose 4-methyl-1-oxo-1-(p-tolylamino)pentane-2-sulfonic acid as our internal standard, as its elution time was in the range of that of CDP-ME and showed consistent results.17 To demonstrate the potential of our new assay, we determined the Michaelis constant (Km) of both substrates, and obtained similar results as previously published (Table 5).12,14,18 To our knowledge, this is the only reported IspD assay that is not dependent on auxiliary enzymes.
Table 5. Comparison of Michaelis Constants.
| KmCTP (μM) | KmMEP (μM) | |
|---|---|---|
| our resultsa | 58 ± 9 | 46 ± 3 |
| Wu et al.14 | not reported | 61 |
| Imlay et al.12 | 59 ± 4 | not reported |
| Ghavami et al.18 | 9 ± 3 | 12 ± 3 |
Assays were performed in replicate as independent experiments (n = 2); values are shown as mean ± SD.
Mode of Inhibition
Next, we measured the influence of 10 on the enzymatic kinetics of both substrates at different concentrations, ranging from 19.5 to 625 nM. The corresponding Lineweaver–Burk plots hint toward a noncompetitive inhibition of 10 toward CTP and uncompetitive toward MEP (Figures 5, S2, and S3). This finding indicates that compound 10 binds to PfIspD in a manner independent of CTP binding to the active site. In this way, it influences the catalytic activity of the enzyme without affecting CTP binding. This highlights an allosteric inhibition mechanism of the enzyme, which, has been observed before for Arabidopsis thaliana IspD by Witschel and co-workers but has never been observed previously for PfIspD.19 On the other hand, compound 10 selectively targets the PfIspD-MEP complex, influencing the catalytic activity of the enzyme as well as substrate binding. These findings unravel the selectivity of compound 10 against both substrates, revealing distinct modulatory effects dependent on the substrate specificity of PfIspD.
Figure 5.
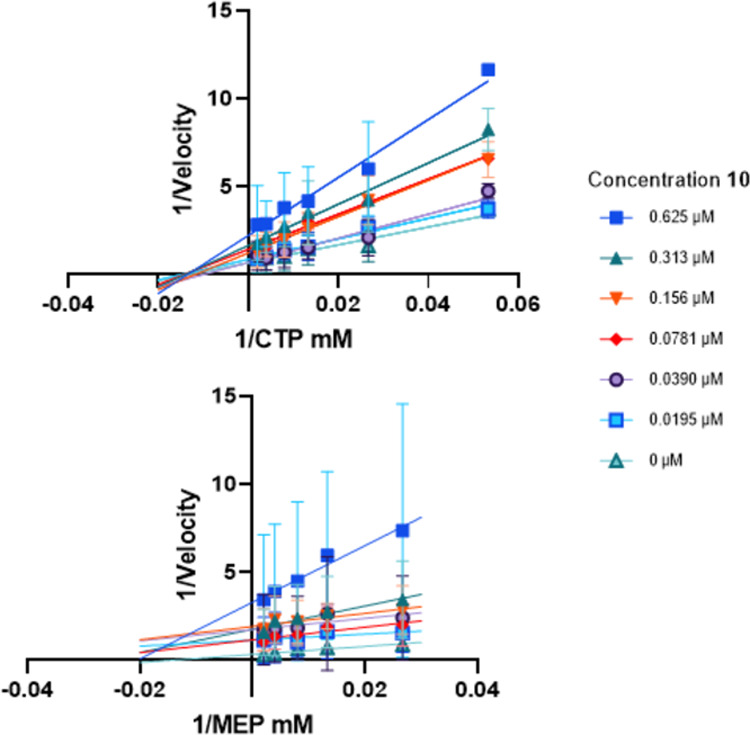
Inhibition of PfIspD by 10 is characterized as noncompetitive with CTP, while uncompetitive with MEP. Lineweaver–Burk plots of both substrates at varying concentrations of 10. Above: CTP was varied; below: MEP concentration was varied.
Metabolic and Plasma Stability
To gain an initial understanding of selected in vitro ADMET properties of the urea class, the metabolic and plasma stability of selected compounds (5, 8, 10, and 28), was determined in liver S9 fractions and plasma of both mouse and human (Table 6). Clearance in mouse S9 liver fraction was high to moderate, with compounds 5, 6, and 28 showing lower clearance than 8. In human S9, clearance showed a similar trend with generally lower turnover. No metabolism during 120 min was observed in human S9 fractions for 28. Regarding plasma stability, all selected compounds showed complete stability in both species (Table 6). Lastly, we also assessed the cytotoxicity toward HepG2 cells. No significant cytotoxicity was observed for 28 up to 100 μM, while the other compounds showed an CC50 range of 29–62 μM.
Table 6. In Vitro Metabolic and Plasma Stability of Compounds 5, 8, 10, and 28.
| model system | 5 | 8 | 10 | 28 | |
|---|---|---|---|---|---|
| mouse liver S9 | T1/2 [min]a | 23 ± 3 | 11 ± 3 | 20 ± 5 | 23 ± 3 |
| Clint [μL/min/mg]a | 31 ± 4 | 66 ± 20 | 36 ± 10 | 31 ± 4 | |
| human liver S9 | T1/2 [min]a | 91 ± 19 | 53 ± 11 | 69 ± 12 | >120 |
| Clint [μL/min/mg]a | 8 ± 2 | 14 ± 3 | 10 ± 2 | <5 | |
| mouse plasma | T1/2 [min]a | >150 | >150 | >150 | >150 |
| % at 2.5 ha | >100 | >100 | >100 | >100 | |
| human plasma | T1/2 [min]a | >150 | >150 | >150 | >150 |
| % at 2.5 ha | >100 | >100 | >100 | >100 | |
| PPB mouse [%] | 98.9 ± 0.1 | 98.2 ± 0.3 | 96.9 ± 0.1 | 99.7 ± 0.1 | |
| PPB human [%] | 99.9 ± 0.1 | 98.1 ± 0.1 | 96.3 ± 0.3 | 99.7 ± 0.1 | |
| HepG2 cytotoxicity | CC50 [μM]a | 29 ± 7 | 62 ± 15 | 40 ± 5 | >100 |
Assays were performed in duplicate as independent experiments (n = 2); values are shown as mean ± SD.
Pharmacokinetic (PK) Profiling
As several compounds exhibited promising initial ADME properties, we embarked on in vivo PK studies with compounds 10 and 28 in mice. We administered both compounds in a cassette format via the intravenous (IV) route at 1 mg/kg. Whereas compound 10 exhibited a short half-life of only 0.5 h, 28 had a half-life of around 1.6 h suggesting additional clearance mechanisms in vivo for compound 10 compared to 28 as the latter had a lower observed plasma clearance compared to 10. Furthermore, both compounds had a similar volume of distribution of around 2.5–2.9 L/kg, suggesting that compounds might also distribute into tissue (Table 7). Moreover, 28 was still detectable until 5 h and had higher exposure levels (Figure 6). However, when considering the different plasma protein binding properties of compounds 10 and 28, compound 10 exhibited an fAUC of around 7.71 ng/mL·h compared to 28 with an fAUC of around 1.86 ng/mL·h. When looking at terminal organ concentrations, 28 was found at around 203 ng/g tissue in liver, whereas 10 was not detected. This demonstrated that 28 had already favorable PK properties for further development as it distributed well to tissues. Nevertheless, it also showed that further optimization of the plasma protein binding level might be necessary.
Table 7. In Vivo PK Data for Compounds 10 and 28.
| in vivo PKa | 10 | 28 |
|---|---|---|
| T1/2 [h] | 0.05 ± 0.1 | 1.55 ± 0.5 |
| C0 [ng/mL] | 912.6 ± 400.2 | 433.1 ± 256.6 |
| AUC0–t [ng/mL·h] | 246.4 ± 63.3 | 717.4 ± 135.9 |
| MRT [h] | 0.6 ± 0.2 | 2.24 ± 0.7 |
| Vz_obs [L/kg] | 2.5 ± 0.8 | 2.9 ± 1.2 |
| Cl_obs [mL/min/kg] | 53.0 ± 8.5 | 20.9 ± 2.0 |
| liver ng/g | ND | 202.6 ± 45.8 |
Assays were performed in replicate as independent experiments (n = 2 mice); values are shown as mean ± SD. AUC0–t = area under the concentration–time curve from time zero to time t; MRT = mean residence time; Vz_obs = observed volume of distribution; Cl_obs = observed clearance (based on observed last time point with measurable concentration); ND = not detected.
Figure 6.

PK plasma profile over time of 10 and 28 at 1 mg/kg IV.
With our SAR, we accomplished a 400-fold increase in inhibitory activity of IspD, while also achieving activity in a whole-cell assay (Figure 7). Modifications directed to the Western side of the molecule were most impactful toward the increase in potency, and achieving whole-cell activity. Furthermore, trying to grow at this side of the molecule indicated that there is no space in the binding pocket to further expand in this direction. Adjustments directed at the urea linker, taught us that an unsubstituted urea bond is key for the activity. From there on, modifications directed to the middle ring and Eastern side of the scaffold did not result in further enhancement of the potency. Furthermore, by exploring different linkers between the middle ring and the Eastern ring, we noticed that the sulfonyl linker is essential for the potency of the compound class. Unfortunately, we observed a discrepancy between the PfIspD activity and cellular activities of the compounds. Despite this, we successfully confirmed that IspD is a target for the compound class. Development of the new LC-MS based activity assay allowed us to gain an idea of the mode of inhibition of this new compound class without the use of auxiliary enzymes. Ultimately, this led to the confirmation of noncompetitive inhibition of 10 toward CTP and uncompetitive toward MEP. Therefore, the whole SAR could potentially teach us something about the structure of the allosteric pocket of PfIspD. Structural information of the allosteric pocket could facilitate future research toward PfIspD inhibitors. Especially as the active site of IspD appears to be challenging to target due to its polar character and solvent-exposure. Lastly, the metabolic clearance and plasma stability experiments demonstrated moderate to good values for some of the representative compounds of the urea class, which were confirmed by in vivo PK studies, revealing compound 28 with the best PK properties. Overall, due to its potent inhibitory activity, ease to synthesize, interesting mode of inhibition, and good ADME profile, the urea class has a great potential for further development in the anti-infective field.
Figure 7.

Initial hit compound and the best performing urea class derivatives. n.a. = no activity.
Experimental Section
General
Purity of all compounds used in biochemical assays was ≥95%. Be aware, in contact with water, triphosgene is converted to the extremely toxic phosgene gas. Starting materials and solvents were purchased from commercial suppliers, and used without further purification. All chemical yields refer to purified compounds and were not optimized. Column chromatography was performed using the automated flash chromatography system CombiFlashRf (Teledyne Isco) equipped with RediSepRf silica columns. Preparative reversed-phase high-performance liquid chromatography (RP-HPLC) was performed either using an UltiMate 3000 Semi-Preparative System (Thermo Fisher Scientific) equipped with nucleodurC18 Gravity (250 mm × 16 mm, 5 μm) column or using a Pure C-850 Flash/Prep (Buchi) equipped with Nucleodur C18 HTec (250 mm × 40 mm, particle size 5 μm). Low-resolution mass spectrometry and purity control of final compounds was carried out using an Ultimate 3000-MSQ LCMS system (Thermo Fisher Scientific) consisting of a pump, an autosampler, MWD detector, and an ESI quadrupole mass spectrometer. 1H and 13C NMR spectra were recorded as indicated on a Bruker Avance Neo 500 MHz (1H, 500 MHz; 13C, 126 MHz) with prodigy cryoprobe system. Chemical shifts were recorded as δ values in ppm units and referenced against the residual solvent peak (DMSO-d6, δ = 2.50, 39.52 and acetone-d6: δ = 2.05, 29.84, CD3OD: δ = 3.27, 47.6). Splitting patterns describe apparent multiplicities and are designated as s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), m (multiplet). Coupling constants (J) are given in Hertz (Hz). High-resolution mass spectra were recorded on a ThermoFisher Scientific (TF, Dreieich, Germany) Q Exactive Focus system equipped with heated electrospray ionization (HESI)-II source. For the LC-MS based IspD assay, a TF UltiMate 3000 binary RSLC UHPLC (Thermo Fisher, Dreieich, Germany) equipped with a degasser, a binary pump, an autosampler, and a thermostatted column compartment and a MWD, coupled to a TF TSQ Quantum Access Max mass spectrometer with heated electrospray ionization source (HESI-II) was used. The separation was performed with a SeQuant ZIC-HILIC 5 μM polymeric HPLC column (100 mm × 2.1 mm) with a precolumn at flow rate of 0.225 μL/min with a mobile phase composed of 50 mM ammonium acetate pH 8.5 (elute A), ACN (eluent B) under the following conditions: 0–30 s 80% B, 30–105 s 70% B, 105–135 s 70–40% B, 135–300 s hold, and 300–420 s 80% B with 225 μL/min flow rate and a total run time of 7 min. The divert valve was set to 0.49 min. The injection volume was 5 μL. The temperature of the autosampler was set to 6 °C. The following MS settings were used: electrospray ionization (ESI); negative mode for CDP-ME and MEP; collision gas pressure: 1.5 Torr; spray voltage: 10 V. The mass spectrometer was operated in the SRM mode with the following masses: 520.116 (fragment: 322.135–322.145) m/z for CDP-ME (tube lens offset 93 V and collision energy 23 eV); 215.006 (fragment: 79.395–79.405, 97.395–97.405) m/z for MEP (tube lens offset 94 V and collision energy 23 and 47 eV, respectively); 284.07 (fragment: 106.19, 177.03–150.15) m/z for 4-methyl-1-oxo-1-(p-tolylamino)pentane-2-sulfonic acid (tube lens offset 28 and 21 respectively V and collision energy 28 and 21 eV, respectively) with a scan width of 0.010 m/z and a scan time of 0.1 s, respectively. Observed retention times were as follows: CDP-ME, MEP, and 4-methyl-1-oxo-1-(p-tolylamino)pentane-2-sulfonic acid 4.90, 4.72, and 1.04 min, respectively (Figure S4). MS-peak areas were determined using TF Xcalibur Software then CDP-ME and MEP peak areas were normalized by the internal standard peak area. All PK plasma samples were analyzed via HPLC-MS/MS using an Agilent 1290 Infinity II HPLC system coupled to an AB Sciex QTrap 6500plus mass spectrometer. HPLC conditions were as follows: column: Agilent Zorbax Eclipse Plus C18, 50 mm × 2.1 mm, 1.8 μm; temperature: 30 °C; injection volume: 5 μL; flow rate: 700 μL/min; solvent A: water + 0.1% formic acid; solvent B: acetonitrile + 0.1% formic acid; gradient for 10 and 26: 99% A at 0 min, 99 – 0% A from 0.1 to 4.0 min, 0% A until 4.5 min. Mass spectrometric conditions were as follows: Scan type: Q1 and Q3 masses for glipizide, 10 and 28 can be found in Table S4; peak areas of each sample and of the corresponding internal standard were analyzed using Multi-Quant 3.0 software (AB Sciex).
Chemistry
General Procedure 1 (GP-1) for the Synthesis of Analogues 5–12
To a flask containing 3-(pyrrolidin-1-ylsulfonyl)aniline (1 equiv), and DMF (150 equiv, unless otherwise stated), the respective isocyanate (1 equiv) was added at 0 °C. The resulting mixture was stirred at room temperature overnight, after which, water was added, and the resulting mixture was extracted with EtOAc (3×, 20 mL). The combined organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, concentrated in vacuo, and purified.
General Procedure 2 (GP-2) for the Synthesis of Analogues 13–18
To a flask that contains triphosgene (0.5 equiv) in DCM (150 equiv, unless otherwise stated) at 0 °C under argon atmosphere, a solution of DCM (150 equiv, unless otherwise stated) containing the respective amine (1.2 equiv) and trimethylamine (1.2 equiv) was added, and the resulting mixture was stirred at room temperature for 3 h. Next, a flask was charged with 3-(pyrrolidin-1-ylsulfonyl)aniline (1 equiv), NaH 60% (1.2 equiv) and DMF (150 equiv), the resulting mixture was stirred for 1 h under argon atmosphere, after which, the solution was added dropwise to the flask containing the triphosgene reaction mixture, and the resulting solution was stirred at room temperature overnight. Water (20 mL) was added, and the mixture was extracted with EtOAc (3×, 20 mL), washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, concentrated in vacuo, and purified.
General Procedure 3 (GP-3) for the Synthesis of Analogues 22–29
To a flask containing 1-isocyanato-4-nitrobenzene and DMF (150 equiv, unless otherwise stated), the respective aniline (1 equiv) was added at room temperature. The resulting mixture was stirred at room temperature overnight, after which, water was added and the resulting mixture was extracted with EtOAc (3×, 20 mL). The combined organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, concentrated in vacuo, and purified.
General Procedure 4 (GP-4) for the Synthesis of Analogues 30–34
To a flask containing acetonitrile (100 equiv), trimethylamine (2 equiv), and pyrrolidine (1 equiv) at 0 °C, the respective 3-nitrobenzenesulfonyl chloride (1 equiv) was added. Next, the resulting solution was stirred at 0 °C for 5 min, after which, the solvent was evaporated. To the residue, EtOH (140 equiv), an aqueous solution of NH4Cl at a concentration of 166 mM (0.50 equiv), and Fe powder (5 equiv) were added, the resulting reaction mixture was stirred at 80 °C for 2.5 h. Next, the organic solvent was evaporated in vacuo, water (20 mL) was added, and the solution was extracted with EtOAc (3×, 20 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Subsequent, the residue was solubilized with DMF (45 equiv), and 1-isocyanato-4-nitrobenzene (1.5 equiv) was added. The resulting reaction mixture was stirred at room temperature for 1 h, after which, DMF was removed on reduced pressure and the residue was purified.
1-(4-Aminophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (4)
A mixture of 1-(4-nitrophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (8) (0.15 g, 0.4 mmol), Fe (0.11 mg, 1.9 mmol), and ammonium chloride (0.01 g, 0.2 mmol) was dissolved in an ethanol/water (2:1) mixture. The mixture was heated to 100◦C for 2 h. Excess ethanol was evaporated in vacuo, and the remaining residue was washed with water (3×, 20 mL), and then filtered. The obtained solid was then purified using preparative HPLC affording 4 as a white powder (0.1 g, 72% yield).
1H NMR (500 MHz, DMSO-d6) δ 10.60 (s, 1H), 10.51 (s, 1H), 8.22 (d, J = 9.1, 2H), 8.08 (s, 1H), 7.82 (d, J = 9.1, 2H), 7.75 (d, J = 7.9, 1H), 7.61 (t, J = 7.8, 1H), 7.56 (d, J = 7.8, 1H), 3.17 (t, J = 6.6, 4H), 1.66 (t, J = 6.6, 4H). 13C NMR (126 MHz, DMSO-d6) δ 179.7, 145.8, 142.6, 140, 136.2, 129.7, 127.5, 124.5, 123.3, 122.2, 121.8, 47.9, 24.8. High-resolution mass spectrometry (HR-MS) (ESI) calculated for C17H21N4O3S [M + H]+: 361.1256, found: 361.1327
1-(4-Chlorophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (5)
According to GP-1, using 1-chloro-4-isocyanatobenzene (0.08 g, 0.49 mmol), affording after purification by flash chromatography (CH2Cl2/MeOH, 10/0 → 9.5/0.5), and washing with MeOH, 5 was afforded as a white powder (22 mg, 11% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.1 (s, 2H), 8.1 (t, J = 2.0, 1H), 7.7–7.6 (m, 1H), 7.6–7.5 (m, 3H), 7.4 (d, J = 7.7, 1H), 7.4–7.3 (m, 2H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.9, 141, 138.9, 137, 130.3, 129.1, 126.1, 122.7, 120.8, 120.5, 116.9, 48.3, 25.2. HR-MS (ESI) calculated for C17H19ClN3O3S [M + H]+, 380.0757, found: 380.0822. HPLC purity: 98%.
1-(3-(Pyrrolidin-1-ylsulfonyl)phenyl)-3-(p-tolyl)urea (6)
According to GP-1, using p-tolyl isocyanate (0.06 mL, 0.48 mmol), afforded after purification by flash chromatography (cyclohexane/EtoAc = 1:1) 6 as white solid (0.15 g, 95% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.0 (s, 1H), 8.7 (s, 1H), 8.1 (t, J = 2.0, 1H), 7.6–7.5 (m, 1H), 7.5 (t, J = 7.9, 1H), 7.4–7.3 (m, 3H), 7.1 (d, J = 8.3, 2H), 3.2–3.1 (m, 4H), 2.3 (s, 3H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 153, 137.2, 137, 131.5, 130.2, 129.7, 122.5, 120.6, 119.1, 116.7, 48.3, 25.2, 20.8. HR-MS (ESI) calculated for C18H22N3O3S [M + H]+, 360.1304, found: 360.1361. HPLC purity: 99%.
Methyl 4-(3-(3-(Pyrrolidin-1-ylsulfonyl)phenyl)ureido)benzoate (7)
According to GP-1, using methyl 4-isocyanatobenzoate (0.088 g, 0.5 mmol), and CH2Cl2 (10 mL), to afford after filtration of the precipitate, 7 as a white solid (0.15 g, 75% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.2 (br s, 2H), 8.1 (t, J = 1.8 Hz, 1H), 7.9 (d, J = 8.7 Hz, 2H), 7.7–7.6 (m, 3H), 7.6 (t, J = 7.9 Hz, 1H), 7.4 (d, J = 7.6 Hz, 1H), 3.8 (s, 3H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 166.4, 152.7, 144.5, 140.7, 137.1, 130.9, 130.4, 123.2, 122.8, 121.1, 118.1, 117.1, 52.3, 48.3, 25.2. HR-MS (ESI) calculated for C19H22N3O5S [M + H]+: 404.1202, found: 404.1275. HPLC purity: 98%
1-(4-Nitrophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (8)
According to GP-1, using 1-isocyanato-4-nitrobenzene (0.1 g, 0.61 mmol), to afford after purification by flash chromatography (CH2Cl2/MeOH, 10/0 → 9.5/0.5), 8 as a yellow powder (0.02 g, 8% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.6 (br s, 1H), 9.4 (br s, 1H), 8.2 (br d, J = 8.9 Hz, 2H), 8.1 (br s, 1H), 7.7 (br d, J = 8.4 Hz, 2H), 7.7 (br d, J = 7.6 Hz, 1H), 7.6 (br t, J = 7.9 Hz, 1H), 7.4 (br d, J = 7.6 Hz, 1H), 3.2–3.1 (m, 4H), 1.7 (br s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.1, 146.1, 141.2, 140, 136.6, 129.9, 125.1, 122.5, 120.9, 117.9, 117.8, 116.8, 47.9, 24.7. HR-MS (ESI) calculated for C17H19N4O5S [M + H]+: 391.0998, found: 391.1066. HPLC purity: 100%.
1-(4-(Methylsulfonyl)phenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (9)
According to GP-1, using 1-isocyanato-4-(methylsulfonyl)benzene (0.99 g, 0.5 mmol) and, DCM (10 mL), to afford after filtration of the precipitate 9 as a white powder (0.15 g, 70% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.29 (br s, 2H), 8.1–8.0 (m, 1H), 7.8 (d, J = 8.9 Hz, 2H), 7.7 (d, J = 8.9 Hz, 2H), 7.7–7.6 (m, 1H), 7.5 (t, J = 7.9 Hz, 1H), 7.4 (d, J = 7.8 Hz, 1H), 3.2–3.1 (m, 7H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.7, 144.7, 140.6, 137.1, 133.9, 130.4, 128.8, 122.9, 121.2, 118.5, 117.2, 48.3, 44.4, 25.2. HR-MS (ESI) calculated for C18H22N3O5S2 [M + H]+: 424.0923, found: 424.0997. HPLC purity: 99%.
1-(4-Cyanophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (10)
According to GP-1, using 4-isocyanatobenzonitrile (0.1 g, 0.69 mmol), to afford after purification by flash chromatography (CH2Cl2/MeOH, 10/0 → 9.5/0.5) and recrystallization with CH2Cl2, and diethyl ether, 10 as a yellow powder (0.075 g, 29% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.3 (br s, 2H), 8.1 (t, J = 1.8 Hz, 1H), 7.8–7.7 (m, 2H), 7.7–7.6 (m, 2H), 7.7–7.6 (m, 1H), 7.6 (t, J = 7.9 Hz, 1H), 7.4 (d, J = 7.8 Hz, 1H), 3.2–3.1 (m, 4H), 1.7–1.64 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.6, 144.4, 140.6, 137.1, 133.8, 130.4, 123, 121.3, 119.7, 118.8, 117.2, 104.1, 49.1, 48.3, 25.2. HR-MS (ESI) calculated for C18H19N4O3S [M + H]+: 371.1010, found: 371.1157. HPLC purity: 98%.
1-(3-(Pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea (11)
According to GP-1, using 1-isocyanato-4-(trifluoromethyl)benzene (0.09 g, 0.5 mmol), and DCM (10 mL), to afford after filtration of the precipitate, 11 as an off-white powder (0.09 g, 0.213 mmol, 43% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.2 (s, 1H), 8.1 (t, J = 1.8 Hz, 1H), 7.7–7.6 (m, 1H), 7.6–7.5 (m, 1H), 7.5 (t, J = 7.9 Hz, 1H), 7.4 (d, J = 7.8 Hz, 1H), 3.2–3.1 (m, 1H), 1.7–1.6 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 152.8, 143.6, 140.7, 137.1, 130.4, 126.7–126.5 (m), 122.9, 121.1, 118.7, 117.1, 48.3, 25.2. 19F NMR (470 MHz DMSO-d6) δ −60.1. HR-MS (ESI) calculated for C18H19F3N3O3S [M + H]+: 414.1021, found: 414.1087. HPLC purity: 99%.
1-(3-(Pyrrolidin-1-ylsulfonyl)phenyl)-3-(3-(trifluoromethyl)phenyl)urea (12)
According to GP-1, using 1-isocyanato-3-(trifluoromethyl)benzene (0.07 g, 0.35 mmol), to afford after purification by flash chromatography (EtOAc/petroleum ether, 3/7 → 5/5), recrystallization using MeOH, and washing with CH2Cl2 (3×, 5 mL), 12 as a white crystalline powder (0.01 g, 8% yield). 1H NMR (500 MHz, CD3OD) δ 8.1 (t, J = 1.9 Hz, 1H), 7.9 (s, 1H), 7.7 (ddd, J = 8.0, 2.2, 1.2 Hz, 1H), 7.6 (dd, J = 8.2, 1.6 Hz, 1H), 7.6–7.5 (m, 1H), 7.5–7.4 (m, 2H), 7.3–7.2 (m, 1H), 3.3–3.2 (m, 4H), 1.8–1.7 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 153.3, 140.2, 140, 137.2, 129.4 (d, J = 4.4), 125.3, 123.1, 122.7, 122, 121.1, 118.8, 117.4, 115.2, 47.8, 24.8. 19F NMR (470 MHz DMSO-d6) δ −61.3. HR-MS (ESI) calculated for C18H19F3N3O3S [M + H]+: 414.1021, found: 414.1088. HPLC purity: 98%.
1-(3,5-Dichlorophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (13)
According to GP-2, using 1,3-dichloro-5-isocyanatobenzene (0.09 g, 0.48 mmol) to afford 13 after evaporation of the solvent as white solid (0.14 g, 77%). 1H NMR (500 MHz, DMSO-d6) δ 9.4–9.1 (m, 2H), 8.1 (t, J = 1.8 Hz, 1H), 7.7–7.6 (m, 1H), 7.6–7.5 (m, 3H), 7.4 (d, J = 7.8 Hz, 1H), 7.2 (t, J = 1.8 Hz, 1H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.7, 142.5, 140.6, 137.1, 134.6, 130.4, 123, 121.7, 121.2, 117.2, 117.1, 48.3, 25.2. HR-MS (ESI) calculated for C17H18Cl2N3O3S [M + H]+: 414.0368, found: 414.0430. HPLC purity: 99%
1-(2-Fluoro-4-(trifluoromethyl)phenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (14)
According to GP-2, 2-fluoro-4-(trifluoromethyl)aniline (0.14 g, 0.79 mmol), to afford after purification by flash chromatography (cyclohexane/EtoAc = 1:1), 14 as white powder (0.04 g, 15% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.6 (br s, 1H), 9.0 (br s, 1H), 8.4 (t, J = 8.3 Hz, 1H), 8.1 (s, 1H), 7.8–7.7 (m, 1H), 7.6–7.5 (m, 3H), 7.5–7.4 (m, 1H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 151.9, 151.7, 145.0, 139.7, 136.5, 129.9, 122.1, 121.9–121.6, 120.7, 120.1, 116.2, 47.7, 24.5. 19F NMR (470 MHz DMSO-d6) δ −60.2, −127.7. HR-MS (ESI) calculated for C18H18F4N3O3S [M + H]+: 432.0927, found: 432.0995. HPLC purity: 99%.
1-(Naphthalen-2-yl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (15)
According to GP-2, naphthalen-2-amine (0.1 g, 0.70 mmol), to afford after washing with MeOH (5×, 5 mL), 15 as an off-white solid (0.02 g, 8% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.2 (s, 1 H), 9.0 (s, 1 H), 8.2 (s, 1 H), 8.1 (s, 1 H), 7.9 (br d, J = 8.85, 2 H), 7.8 (br d, J = 9.77, 1 H), 7.6 (d, J = 8.06, 1 H), 7.6 (t, J = 7.93, 1 H), 7.5 (dd, J = 8.77, 2.06, 1 H), 7.5 (t, J = 7.48, 1 H), 7.4–7.3 (m, 2 H), 3.2–3.1 (m, 4 H), 1.7–1.6 (m, 4 H). 13C NMR (126 MHz, DMSO-d6) δ 152.7–152.8, 152.7, 140.7, 138.9–142.1, 137.2, 136.7, 133.8, 130, 129.4, 128.6, 127.6, 127.2, 126.6, 124.3, 122.3, 120.5, 120, 116.5, 114, 48, 24. HR-MS (ESI) calculated for C21H22N3O3S [M + H]+: 396,1304, found: 396.1364. HPLC purity: 99%.
1-(4-Phenoxyphenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (16)
According to GP-2, using 4-phenoxyaniline (0.17 g, 0.94 mmol), to afford after purification with flash chromatography (cyclohexane/EtOAc = 7:3) 16 as a white solid (0.18 g, 44%). 1H NMR (500 MHz, DMSO-d6) δ 9.1 (s, 1H), 8.8 (s, 1H), 8.1 (t, J = 2.0, 1H), 7.6 (ddd, J = 8.2, 2.3, 1.0, 1H), 7.5 (t, J = 7.9, 1H), 7.5–7.4 (m, 2H), 7.4–7.3 (m, 3H), 7.1 (t, J = 7.5, 1H), 7.0 (m, 4H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 158.1, 153.1, 151.4, 141.2, 137, 135.8, 130.4, 130.3, 123.3, 122.6, 120.9, 120.7, 120.2, 118.1, 116.8, 48.3, 25.2. HR-MS (ESI) calculated for C23H24N3O4S [M + H]+: 438.1409, found: 438.1474. HPLC purity: 99%.
1-(3-(Morpholinomethyl)phenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (17)
According to GP-2, 3-(morpholinomethyl)aniline (0.1 g, 0.52 mmol), to afford after washing with MeOH (5×, 5 mL), 17 as a yellow crystalline powder (0.09 g, 31% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.1 (s, 1 H), 8.8 (s, 1 H), 8.1 (t, J = 1.91, 1 H), 7.6 (dd, J = 8.16, 1.14, 1 H), 7.5 (t, J = 7.93, 1 H), 7.4 (s, 1 H), 7.4 – 7.3 (m, 2 H), 7.3–7.2 (m, 1 H), 7.0–6.9 (m, 1 H), 3.6 (t, J = 4.50, 4 H), 3.4 (s, 2 H), 3.2–3.1 (m, 4 H), 2.4 (br s, 4 H), 1.7–1.6 (m, 4 H). 13C NMR (126 MHz, DMSO-d6) δ 152.4, 140.6, 139.3, 138.6, 136.5, 129.8, 128.6, 122.8, 122.1, 120.2, 118.8, 117.2, 116.3, 66.2, 62.5, 53.2, 47.8, 24.7. HR-MS (ESI) calculated for C22H29N4O4S [M + H]+: 445,1831, found: 445,1899. HPLC purity: 98%.
1-(3-((1H-Imidazol-1-yl)methyl)phenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (18)
According to GP-2, using 3-((1H-imidazol-1-yl)methyl)aniline (0.1 g, 0.58 mmol), to afford after washing with MeOH (5×, 5 mL), 18 as a white solid (0.01 g, 4% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.2 (br s, 1H), 8.9 (br s, 1H), 8.1 (s, 1H), 7.8 (s, 1H), 7.6–7.5 (m, 1H), 7.5 (t, J = 7.9 Hz, 1H), 7.4–7.3 (m, 2H), 7.4–7.3 (m, 1H), 7.3 (t, J = 7.9 Hz, 1H), 7.2–7.1 (m, 1H), 6.9 (s, 1H), 6.9–6.8 (m, 1H), 5.2 (s, 2H), 3.1 (br t, J = 6.6 Hz, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.4, 140.5, 139.7, 138.4, 136.4, 129.7, 129, 122, 120.9, 120.1, 117.6, 117.1, 116.1, 49.4, 47.7, 24.6. HR-MS (ESI) calculated for C21H24N5O3S [M + H]+: 426,1522, found: 426.1582. HPLC purity: 98%.
1-Methyl-1-(4-nitrophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (20)
To a flask containing triphosgene (0.08 g, 0.27 mmol), and DCM (2 mL) at 0 °C under argon atmosphere, a solution containing 3-((1H-imidazol-1-yl)methyl)aniline (0.13 g, 0.89 mmol), trimethylamine (247 mL, 1.78 mmol), and DCM (2 mL), was added. The resulting solution was stirred at room temperature for 2 h. To a different flask, N-methyl-4-nitroaniline (0.13 g, 0.89 mmol), sodium hydride (0.03 mg, 1.19 mmol), and DMF (2.5 mL) were added, the resulting solution was stirred at room temperature for 2 h, after which, it was added dropwise to the solution containing triphosgene. The resulting mixture was stirred at room temperature for 1 h, next water was added and the mixture was extracted with EtOAc (5×, 20 mL). The combined organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, concentrated in vacuo, and purified by column chromatography, (CH2Cl2/MeOH, 10/0 → 9.5/0.5), and recrystallization (CH2Cl2/diethyl ether), to afford 19 as a yellow solid (0.12 g, 32% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.2 (s, 1H), 8.3–8.2 (m, 1H), 8.0 (t, J = 1.9 Hz, 1H), 7.8 (dd, J = 8.1, 1.4 Hz, 1H), 7.7–7.6 (m, 1H), 7.5 (t, J = 8.0 Hz, 1H), 7.4 (d, J = 7.8 Hz, 1H), 3.4 (s, 1H), 3.2–3.1 (m, 1H), 1.7–1.6 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 154.7, 150.6, 143.9, 141.1, 136.7, 130, 125.4, 124.9, 124.3, 121.5, 118.8, 48.3, 37.4, 25.2. HR-MS (ESI) calculated for: C18H21N4O5S [M + H]+: 405.1154, found: 405.1214. HPLC purity: 99%.
1-Methyl-3-(4-nitrophenyl)-1-(3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (21)
A flask was charged with 3-(pyrrolidin-1-ylsulfonyl)aniline (0.1 g, 0.44 mmol), paraformaldehyde (0.09 g), and MeOH (5 mL). The resulting solution was stirred at room temperature for 2.5 h, after which, NaBH4 (0.03 g, 0.88 mmol) was added. The resulting mixture was stirred at 60 °C for 16 h, next, water (20 mL) was added, and the mixture was extracted with EtOAc (5×, 20 mL). The combined organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, concentrated in vacuo, and purified by flash chromatography (EtOAc/petroleum benzyne 3/7 → 6/4) affording N-methyl-3-(pyrrolidin-1-ylsulfonyl)aniline (ii) (0.06 g, 0.25 mmol, 57% yield) which was added to a flask containing 1-isocyanato-4-nitrobenzene (0.07 g, 0.28 mmol), and DMF (5 mL). The solution was stirred at room temperature overnight, next, water (20 mL) was added, and the resulting mixture was extracted with EtOAc (5×, 20 mL). The combined organic layers were washed with saturated aqueous NaCl solution, dried over MgSO4, filtered, concentrated in vacuo, purified by flash chromatography (CH2Cl2/MeOH, 10/0 → 9.5/0.5), and recrystallized (MeOH, CH2Cl2 and diethyl ether) to afford 20 as an off-white solid (0.02 g, 12% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.2 (br s, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.8–7.6 (m, 6H), 3.4 (s, 3H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H) 13C NMR (126 MHz, DMSO-d6) δ 154.5, 147.4, 144.9, 141.6, 137.2, 130.7 (d, J = 10.9), 125.1, 124.9, 119.1, 48.3, 38.1, 25.1. HR-MS (ESI) calculated for: C18H21N4O5S [M + H]+: 405.1154, found: 405.1215. HPLC purity: 100%.
1-(4-Nitrophenyl)-3-(3-(pyrrolidin-1-ylsulfonyl)phenyl)thiourea (23)
To 3-(pyrrolidin-1-ylsulfonyl)aniline (0.03 g, 0.11 mmol) dissolved in DCM (5 mL) was added 1-isothiocyanato-4-nitrobenzene (0.02 g, 0.11 mmol) at 0 °C. The reaction was then stirred at room temperature for 2 days. The reaction was quenched by the addition of saturated aqueous solution of NaHCO3 (20 mL), and extracted with DCM. The organic solvent was dried over MgSO4, filtered, and then removed in vacuo and the reaction was purified using preparative HPLC affording 21 (0.02 g, 35% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.0 (s, 1H), 7.9 (t, J = 1.8 Hz, 1H), 7.8 (d, J = 8.5 Hz, 2H), 7.8 (dd, J = 8.2, 1.3 Hz, 1H), 7.6 (d, J = 8.4 Hz, 2H), 7.5 (t, J = 8.0 Hz, 1H), 7.4–7.3 (m, 1H), 3.2–3.1 (m, 4H), 1.7–1.62 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 153.7, 145.4, 140.5, 136, 129.5, 127.3, 126.5, 123.9, 121, 118.4, 80.1, 75.1, 47.9, 24.7. HR-MS (ESI) calculated for C17H19N4O4S2 [M + H]+: 407.0770, found: 407.0839. HPLC purity: 95%.
N,N-Dimethyl-3-(3-(4-nitrophenyl)ureido)benzenesulfonamide (24)
According to GP-3 using, 3-amino-N,N-dimethylbenzenesulfonamide (0.1 g, 0.5 mmol), and DCM (10 mL), to afford after filtration of the precipitate, 22 as a white solid (0.09 g, 49% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.6–9.3 (m, 2H), 8.2–8.1 (m, 2H), 8.1 (t, J = 1.8 Hz, 1H), 7.8–7.7 (m, 2H), 7.6 (dd, J = 7.9, 1.5 Hz, 1H), 7.6 (t, J = 7.9 Hz, 1H), 7.4 (d, J = 7.8 Hz, 1H), 2.7–2.6 (m, 6H). 13C NMR (126 MHz, DMSO-d6) δ 152.5, 146.6, 141.7, 140.5, 135.7, 130.4, 125.6, 123.2, 121.7, 118.3, 117.5, 38.1. HR-MS (ESI) calculated for C15H17N4O5S [M + H]+: 365.0841, found: 365.0915. HPLC purity: 99%.
N,N-Diethyl-3-(3-(4-nitrophenyl)ureido)benzenesulfonamide (25)
According to GP-3 using, 3-amino-N,N-diethylbenzenesulfonamide (0.114 g, 0.5 mmol), and DCM (10 mL), to afford after filtration of the precipitate, 23 as a yellow solid (0.11 g, 58% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.4 (br s, 2H), 8.2–8.1 (m, J = 9.2 Hz, 2H), 8.1–8.0 (m, 1H), 7.8–7.7 (m, 2H), 7.6 (dd, J = 8.2, 1.0 Hz, 1H), 7.5 (t, J = 7.9 Hz, 1H), 7.4 (d, J = 7.8 Hz, 1H), 3.2 (q, J = 7.2 Hz, 4H), 1.1 (t, J = 7.2 Hz, 6H) 13C NMR (126 MHz, DMSO-d6) δ 152.5, 146.6, 141.7, 140.8, 140.5, 130.4, 125.6, 122.7, 120.8, 118.3, 116.7, 42.3, 14.6. HR-MS (ESI) calculated for C17H21N4O5S [M + H]+: 393.1154, found: 393.1221. HPLC purity: 99%.
1-(4-Nitrophenyl)-3-(3-(piperidin-1-ylsulfonyl)phenyl)urea (26)
According to GP-3 using, 3-(piperidin-1-ylsulfonyl)aniline (0.120 g, 0.5 mmol), and DCM (10 mL), to afford after filtration of the precipitate, 24 as an off-white solid (0.112 g, 55% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.6–9.3 (m, 2H), 8.2–8.1 (m, 2H), 8.1–8.0 (m, 1H), 7.8–7.7 (m, 2H), 7.7 (dd, J = 7.9, 1.6 Hz, 1H), 7.6 (t, J = 7.9 Hz, 1H), 7.4–7.3 (m, 1H), 2.9–2.8 (m, 4H), 1.6–1.5 (m, 4H), 1.4–1.3 (m, 2H) 13C NMR (126 MHz, DMSO-d6) δ 152.5, 146.5, 141.7, 140.5, 136.6, 130.4, 125.6, 123.1, 121.6, 118.3, 117.3, 47.1, 25.2, 23.3. HR-MS (ESI) calculated for C18H21N4O5S [M + H]+: 405.1154, found: 405.1213. HPLC purity: 99%.
1-(3-(Morpholinosulfonyl)phenyl)-3-(4-nitrophenyl)urea (27)
According to GP-3 using, (3-orpholinosulfonyl)aniline (0.12 g, 0.5 mmol), and CH2Cl2 (5 mL), to afford after filtration of the precipitate, 25 as a white powder (0.06 g, 31% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.6–9.3 (m, 2H), 8.2–8.1 (m, 2H), 8.1–8.0 (m, 1H), 7.8–7.7 (m, 2H), 7.7–7.6 (m, 1H), 7.6–7.5 (m, 1H), 7.4 (d, J = 7.6 Hz, 1H), 3.7–3.6 (m, 4H), 2.9–2.8 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.6, 141.7, 140.6, 135.4, 130.5, 125.6, 123.5, 121.8, 118.3, 117.5, 65.8, 46.4. HR-MS (ESI) calculated for C17H19N4O6S [M + H]+: 407.0947, found: 407.1006. HPLC purity: 99%.
1-(4-Nitrophenyl)-3-(3-(phenylsulfonyl)phenyl)urea (28)
According to GP-3 3-(pyrrolidin-1-ylsulfonyl)aniline (0.16 g, 0.69 mmol) to afford after purification by flash chromatography (CH2Cl2/MeOH, 10/0 → 9.5/0.5), and washing the residue with MeOH, and CH2Cl2, 26 as a yellow powder (0.08 g, 28% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.7–9.6 (m, 1H), 9.5–9.4 (m, 1H), 8.3–8.2 (m, 1H), 8.2–8.1 (m, 2H), 8.0–7.9 (m, 2H), 7.8–7.7 (m, 3H), 7.7–7.5 (m, 5H). 13C NMR (126 MHz, DMSO-d6) δ 152.5, 146.5, 142.1, 141.8, 141.5, 140.8, 134.3, 130.9, 130.3, 127.8, 125.6, 123.8, 121.5, 118.5, 118.3, 117. HR-MS (ESI) calculated for: C19H16N3O5S [M + H]+: 398.0732, found: 398.0800. HPLC purity: 98%.
1-(4-Nitrophenyl)-3-(3-(pyrrolidine-1-carbonyl)phenyl)urea (29)
To a flask containing 3-nitrobenzoic acid (0.25 g, 1.5 mmol), pyrrolidine (0.21 g, 3.0 mmol), trimethylamine (0.33 g, 3.3 mmol) and DCM (2 mL), a solution of propanephosphonic acid anhydride in EtOAc (50%, 1.4 g, 2.2 mmol) was added. The resulting solution was stirred at room temperature for 2 h, after which, water was added, and the solution was extracted with DCM (3×, 15 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. To the residue, EtOH (5 mL), an aqueous solution of NH4Cl at a concentration of 166 mM in water (3.83 mL, 0.64 mmol) and Fe powder (0.18 g, 3.2 mmol) were added, the resulting reaction mixture was stirred at 80 °C for 2.5 h. Next, the organic solvent was evaporated in vacuo, water (20 mL) was added, and the solution was extracted with DCM (3×, 15 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Subsequent, the residue was solubilized with DCM (15 mL), and 1-isocyanato-4-nitrobenzene (0.15 g, 0.89 mmol) was added. The resulting reaction mixture was stirred at room temperature for 1 h, after which, DMF was removed under reduced pressure, and the residue was purified using preparative RP-HPLC to yield 29 as a yellow solid (0.04 g, 9% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.5 (br s, 1H), 9.1 (br s, 1H), 8.2 (d, J = 9.0 Hz, 2H), 7.7–7.6 (m, 3H), 7.5–7.4 (m, 1H), 7.4 (t, J = 7.8 Hz, 1H), 7.1 (d, J = 7.5 Hz, 1H), 3.5 (t, J = 6.9 Hz, 2H), 3.4 (t, J = 6.4 Hz, 2H), 1.9–1.8 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 168.0, 152.0, 146.3, 141.1, 138.9, 137.8, 128.8, 125.1, 121.0, 119.8, 117.9, 117.2, 49.0, 45.9, 26.0, 23.9. HR-MS (ESI) calculated for: C18H19N4O4 [M + H]+: 355.1328, found: 355.14202. HPLC purity: 100%
1-(4-Nitrophenyl)-3-(3-(pyrrolidin-1-ylmethyl)phenyl)urea (30)
To a flask containing 3-nitrobenzyl bromide (0.2 g, 0.92 mmol), trimethylamine (0.09 g, 0.92 mmol) and DCM (2.5 mL), pyrrolidine (0.07 g, 0.92 mmol) was added. Next the solution was stirred at room temperature for 2 h, after which, water was added and the resulting solution was extracted with DCM (3×, 15 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. To the residue, EtOH (5 mL), an aqueous solution of NH4Cl at a concentration of 166 mM in water (4.85 mL, 0.64 mmol), and Fe powder (0.22 g, 4.02 mmol) were added, the resulting reaction mixture was stirred at 80 °C for 2.5 h. Next, the organic solvent was evaporated in vacuo, water (20 mL) was added, and the solution was extracted with DCM (3×, 15 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Subsequent, the residue was solubilized with DCM (15 mL), and 1-isocyanato-4-nitrobenzene (0.11 g, 0.68 mmol) was added. The resulting reaction mixture was stirred at room temperature for 1 h, after which, DMF was removed on reduced pressure, and the residue was purified using preparative RP-HPLC to yield 30 as a yellow solid (0.03 g, 9% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.2 (br s, 1H), 9.7 (br s, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.7 (d, J = 9.2 Hz, 2H), 7.6 (s, 1H), 7.4 (br d, J = 8.1 Hz, 1H), 7.3 (t, J = 7.8 Hz, 1H), 7.0 (d, J = 7.5 Hz, 1H), 3.7 (s, 2H), 2.6 (br s, 4H), 1.8 (br s, 4H). 13C NMR (126 MHz, DMSO-d6) δ 164.3, 151.9, 146.5, 140.5, 139.1, 128.4, 124.8, 122.5, 118.6, 117.3, 117.1, 58.7, 53.0, 22.6. HR-MS (ESI) calculated for: C18H21N4O3 [M + H]+: 341.1535, found: 341.16151. HPLC purity: 98%
1-(4-Fluoro-3-(morpholinosulfonyl)phenyl)-3-(4-nitrophenyl)urea (31)
According to GP-3 using, 4-fluoro-3-(morpholinosulfonyl)aniline (0.08 g, 0.46 mmol), to afford after purification by flash chromatography (CH2Cl2/MeOH, 10/0 → 9.5/0.5), and washing the residue with MeOH, 27 as a yellow powder (0.05 g, 23% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.5 (br s, 1H), 9.3 (br s, 1H), 8.2–8.1 (m, 2H), 8.1 (dd, J = 6.0, 2.7 Hz, 1H), 7.8–7.7 (m, 3H), 7.5 (t, J = 9.5 Hz, 1H), 3.7–3.6 (m, 4H), 3.1–3.0 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 154.9, 152.9, 152.5, 146.5, 141.7, 136.4, 126, 125.6, 123.7, 120.6, 118.7, 118.5, 118.3, 66, 46. 19F NMR(470 MHz DMSO-d6) δ −116.4. HR-MS (ESI) calculated for: C17H18FN4O6S [M + H]+: 425.0853, found: 425.0913. HPLC purity: 96%.
1-(4-Chloro-3-(morpholinosulfonyl)phenyl)-3-(4-nitrophenyl)urea (32)
According to GP-3 using, 4-chloro-3-(morpholinosulfonyl)aniline (0.1 g, 0.36 mmol), to afford after filtration, and washing (MeOH, CH2Cl2 and diethyl ether), 28 as yellow solid (0.01 g, 7% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.7–9.4 (m, 2H), 8.2 (d, J = 2.4 Hz, 1H), 8.3–8.2 (m, 2H), 7.8–7.7 (m, 3H), 7.7– 7.6 (m, 1H), 3.7–3.6 (m, 4H), 3.2–3.1 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 151.96, 145.98, 141.33, 138.69, 134.93, 132.74, 125.12, 123.85, 123.31, 120.89, 117.90, 65.72, 45.74. HR-MS (ESI) calculated for: C17H18ClN4O6S [M + H]+: 441.0557, found: 441.0623. HPLC purity: 99%.
1-(4-Methyl-3-(morpholinosulfonyl)phenyl)-3-(4-nitrophenyl)urea (33)
According to GP-3 using, 4-methyl-3-(morpholinosulfonyl)aniline (0.1 g, 39 mmol), affording after purification by flash chromatography (CH2Cl2/MeOH, 10/90 → 95/05), and recrystallization (MeOH, CH2Cl2, and diethyl ether), 29 as a white solid (0.02 g, 9% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.5 (br s, 1H), 9.28 (br s, 1H), 8.3–8.2 (m, 2H), 8.1 (d, J = 2.4 Hz, 1H), 7.8–7.7 (m, 2H), 7.6 (dd, J = 8.2, 2.3 Hz, 1H), 7.4 (d, J = 8.4 Hz, 1H), 3.7–3.6 (m, 4H), 3.4 (s, 9H), 3.1–3.0 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 152.5, 146.7, 141.6, 137.9, 135.2, 134, 131.3, 125.7, 123.5, 119.8, 118.2, 66, 45.8, 20.2. HR-MS (ESI) calculated for: C18H21N4O6S [M + H]+: 421.1104, found: 421.1174. HPLC purity: 99%.
1-(4-Fluoro-3-(pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-nitrophenyl)urea (34)
According to GP-4 using, 1-((2-fluoro-5-nitrophenyl)sulfonyl)pyrrolidine (0.1 g, 0.42 mmol), to afford after purification by preparative RP-HPLC 30 as an orange solid (0.01 g, 6% yield). 1H NMR (500 MHz, DMSO-d6): δ 10.2–9.8 (m, 1H), 8.8–8.6 (m, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.8 (d, J = 2.3 Hz, 1H), 7.7 (d, J = 9.2 Hz, 2H), 7.7 (d, J = 2.3 Hz, 1H), 7.0 (d, J = 9.0 Hz, 1H), 3.5–3.4 (m, 4H), 1.9–1.8 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 153.1, 150.7, 146.6, 140.7, 129.3, 127.1, 124.9, 123.4, 117.3, 115.2, 49.8, 24.9. 19F NMR (470 MHz DMSO-d6) δ −73.5. HR-MS (ESI) calculated for: C17H17FN4O5S [M + H]+: 409.0904, found: 409.0967. HPLC purity: 96%.
1-(2-Fluoro-5-(pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-nitrophenyl)urea (35)
According to GP-4 using, 4-fluoro-3-nitrobenzenesulfonyl chloride (0.1 g, 0.42 mmol), to afford after purification by preparative RP-HPLC 31 as a gray solid (0.01 g, 5% yield). 1H NMR (500 MHz, DMSO-d6): δ 9.6 (br s, 1H), 9.2 (br s, 1H), 8.2 (d, J = 2.4 Hz, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.7 (d, J = 9.2 Hz, 2H), 7.7 (dd, J = 9.2, 2.3 Hz, 1H), 7.3 (d, J = 9.2 Hz, 1H), 3.4–3.3 (m, 4H), 2.0–1.9 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 152.1, 146.2, 144.9, 141.0, 130.3, 128.4, 125.0, 120.7, 120.0, 117.5, 51.8, 25.1. 19F NMR (470 MHz DMSO-d6) δ −115.9. HR-MS (ESI) calculated for: C17H17FN4O5S [M + H]+: 409.0904, found: 409.0967. HPLC purity: 98.
1-(2-Chloro-5-(pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-nitrophenyl)urea (36)
According to GP-4 using, 4-chloro-3-nitrobenzenesulfonyl chloride (0.15 g, 0.59 mmol), to afford after purification by preparative RP-HPLC 32 as a yellow solid (0.02 g, 7% yield). 1H NMR (500 MHz, DMSO-d6): δ 10.3–10.1 (m, 1H), 8.9–8.7 (m, 1H), 8.7 (d, J = 2.0 Hz, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.8 (d, J = 8.4 Hz, 1H), 7.7 (d, J = 9.2 Hz, 2H), 7.5 (dd, J = 8.4, 2.0 Hz, 1H), 3.2–3.1 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 152.1, 146.0, 142.0, 136.7, 135.9, 130.8, 126.8, 125.7, 122.5, 119.8, 118.0, 118.3, 48.4, 25.2. HR-MS (ESI) calculated for: C17H17ClN4O5S [M + H]+: 425.0608, found: 425.0686. HPLC purity: 97%.
1-(2-Methyl-5-(pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-nitrophenyl)urea (37)
According to GP-4 using, 4-methyl-3-nitrobenzenesulfonyl chloride (0.15 g, 0.59 mmol), to afford after purification by preparative RP-HPLC 33 as a yellow solid (0.07 g, 28% yield). 1H NMR (500 MHz, DMSO-d6): δ 10.0–9.7 (m, 1H), 8.4 (d, J = 1.7 Hz, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.7 (d, J = 9.2 Hz, 2H), 7.5 (d, J = 7.9 Hz, 1H), 7.4 (dd, J = 8.7, 2.0 Hz, 1H), 3.2–3.1 (m, 4H), 2.4 (s, 3H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 151.9, 145.9, 141.0, 137.4, 133.7, 132.5, 130.9, 125.1, 125.0, 121.4, 118.8, 117.8, 117.4, 47.6, 24.5, 17.8. HR-MS (ESI) calculated for: C18H20N4O5S [M + H]+: 405.1154, found: 405.1219. HPLC purity: 98%.
1-(2,5-Dimethyl-3-(pyrrolidin-1-ylsulfonyl)phenyl)-3-(4-nitrophenyl)urea (38)
According to GP-4 using, 2,5-dimethyl-3-nitrobenzenesulfonyl chloride (0.15 g, 0.60 mmol), to afford after purification by preparative RP-HPLC 34 as a yellow solid (0.09 g, 37% yield). 1H NMR (500 MHz, DMSO-d6): δ 9.9–9.7 (m, 1H), 8.5–8.2 (m, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.8–7.7 (m, 1H), 7.7 (d, J = 9.2 Hz, 2H), 7.5–7.4 (m, 1H), 3.2 (m, 4H), 2.4 (s, 3H), 2.4 (s, 3H), 1.9–1.8 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 152.4, 146.5, 141.2, 138.7, 137.6, 135.6, 128.3, 126.2, 125.4, 125.0, 117.6, 47.4, 25.2, 20.9, 14.1. HR-MS (ESI) calculated for: C19H22N4O5S [M + H]+: 419.1311, found: 419.1373. HPLC purity: 100%.
1-(4-Nitrophenyl)-3-(4-(pyrrolidin-1-yl)-3-(pyrrolidin-1-ylsulfonyl)phenyl)urea (39)
To a flask containing acetonitrile (1.5 mL), trimethylamine (0.12 g, 1.17 mmol), and pyrrolidine (0.04 g, 0.59 mmol) at room temperature, 2-chloro-5-nitrobenzenesulfonyl chloride (0.15 g, 0.59 mmol) was added. Next the resulting solution was stirred at room temperature for 5 min, after which, the solvent was evaporated. To the residue, EtOH (5.4 mL, 0.1 mmol), an aqueous solution of NH4Cl at a concentration of 166 mM in water (2.0 mL, 0.34 mmol), and Fe powder (0.19 g, 3.44 mmol) were added, the resulting reaction mixture was stirred at 80 °C for 2.5 h. Next, the organic solvent was evaporated in vacuo, water (20 mL) was added, and the solution was extracted with EtOAc (3×, 20 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Subsequent, the residue was solubilized with DMF (2 mL), and 1-isocyanato-4-nitrobenzene (0.14 g, 0.86 mmol) was added. The resulting reaction mixture was stirred at room temperature for 1 h, after which, DMF was removed on reduced pressure, and the residue was purified using preparative RP-HPLC to yield 35 as a yellow solid (0.01 g, 13% yield). 1H NMR (500 MHz, DMSO-d6): δ 9.6–9.3 (m, 1H), 9.3–9.0 (m, 1H), 8.2 (d, J = 9.2 Hz, 2H), 8.0 (d, J = 2.6 Hz, 1H), 7.7 (d, J = 9.2 Hz, 1H), 7.6 (dd, J = 8.9, 2.6 Hz, 1H), 7.4 (d, J = 8.9 Hz, 1H), 3.3–3.2 (m, 4H), 3.2–3.1 (m, 4H), 1.9–1.8 (m, 4H), 1.8–1.7 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 152.0, 146.3, 144.3, 141.0, 134.1, 133.6, 125.1, 123.8, 123.4, 120.8, 117.6, 53.6, 47.7, 25.4, 24.2. HR-MS (ESI) calculated for: C21H25N5O5S [M + H]+: 460.1576, found: 460.1653. HPLC purity: 99%.
1-(4-Nitrophenyl)-3-(2-(pyrrolidin-1-yl)-5-(pyrrolidin-1-ylsulfonyl)phenyl)urea (40)
To a flask containing acetonitrile (1.5 mL), trimethylamine (0.12 g, 1.17 mmol), and pyrrolidine (0.04 g, 0.59 mmol) at room temperature, 4-chloro-3-nitrobenzenesulfonyl chloride (0.15 g, 0.59 mmol) was added. Next the resulting solution was stirred at room temperature for 5 min, after which, the solvent was evaporated. To the residue, EtOH (5.4 mL, 0.1 mmol), an aqueous solution of NH4Cl at a concentration of 166 mM in water (2.0 mL, 0.34 mmol), and Fe powder (0.19 g, 3.44 mmol) were added, the resulting reaction mixture was stirred at 80 °C for 2.5 h. Next, the organic solvent was evaporated in vacuo, water (20 mL) was added, and the solution was extracted with EtOAc (3×). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Subsequently, the residue was solubilized with DMF (2 mL) and 1-isocyanato-4-nitrobenzene (0.14 g, 0.86 mmol) was added. The resulting reaction mixture was stirred at room temperature for 1 h, after which, DMF was removed on reduced pressure and the residue was purified using preparative RP-HPLC to yield 36 as a yellow solid (0.02 g, 9% yield) 1H NMR (500 MHz, DMSO-d6): δ 9.8 (br s, 1H), 8.2 (br s, 1H), 8.2 (d, J = 9.2 Hz, 2H), 7.8 (d, J = 2.1 Hz, 1H), 7.7 (d, J = 9.3 Hz, 2H), 7.4 (dd, J = 8.7, 2.3 Hz, 1H), 7.0 (d, J = 8.9 Hz, 1H), 3.3 (m, 4H), 3.1–3.0 (m, 4H), 2.0–1.9 (m, 4H), 1.7–1.6 (m, 4H). 13C NMR (126 MHz, DMSO-d6): δ 152.9, 147.4, 146.7, 141.1, 126.0, 125.7, 125.3, 125.2, 124.5, 117.6, 115.8, 50.2, 47.9, 25.0, 24.7. HR-MS (ESI) calculated for: C21H25N5O5S [M + H]+: 460.1576, found: 460.1652. HPLC purity: 97%.
Photometric In Vitro Assay
Dilution series (1:2) of inhibitors in DMSO covered the concentration range of approximately 200–0.01 μM. After finishing the dilution series, the final volume of compound solution in DMSO per well was 3 μL. For the IspD assay, 30 μL aliquots of a solution containing 100 mM Tris-HCl, pH 7.6, 0.02% NaN3, 1 mM MEP and 1 mM CTP were added to microplate wells preloaded with 3 μL of DMSO containing test compounds. The reaction was started by addition of 27 μL aliquots of buffer: 100 mM Tris-HCl, pH 7.6, containing 10 mM MgCl2, 60 mM KCl, 10 mM dithiothreitol, 0.02% NaN3, 1 mM NADH, 2 mM phosphoenolpyruvate, 2 mM ATP, 1 U mL–1 pyruvate kinase, 1 U mL–1 lactate dehydrogenase, 1.5 U mL–1Escherichia coli IspE, 0.01 μM PfIspD. The reaction was monitored photometrically (340 nm) at room temperature for 30–60 min on a plate reader (Spectramax M2, Molecular Devices, Biberach an der Riss, Germany). Initial rates were estimated using Softmax Pro 6.1 software (Molecular Devices, Biberach an der Riss, Germany). IC50 values were determined with a nonlinear regression method using the program Dynafit.21
Whole-Cell Assay
PfNF54 wild type parasites cultured in RPMI 1640 medium supplemented with 25 mM HEPES, 24 mM sodium bicarbonate (pH 7.3), 0.36 mM hypoxanthine, 100 μg/mL neomycin and 0.5% Albumax II were used to test for compound activity on parasite multiplication using a [3H]-hypoxanthine incorporation assay.22 Compounds were dissolved in DMSO (10 mM), diluted in hypoxanthine-free culture medium and titrated in duplicate over a 64-fold range (6 step 2-fold dilutions) in 96 well plates. 100 μL Asexual parasite culture (prepared in hypoxanthine-free medium) were added to each well and mixed with the compound to obtain a final hematocrit of 1.25% and a final parasitemia of 0.3%. After incubation for 48 h, 0.25 μCi of [3H]-hypoxanthine was added per well and plates were incubated for an additional 24 h. Parasites were then harvested onto glass-fiber filters using a Microbeta FilterMate cell harvester (Perkin-Elmer, Waltham), and radioactivity was counted using a MicroBeta2 liquid scintillation counter (Perkin-Elmer, Waltham). The results were recorded and expressed as a percentage of the untreated controls. Fifty percent inhibitory concentrations (EC50) were estimated by linear interpolation.23
P. falciparum Culture
P. falciparum strain 3D7 (wild-type, WT) was sourced from MR4 as part of the BEI Resources Repository, NIAID, NIH (www.mr4.org). P. falciparum strains resistant to 1R,3S-MMV008138 (R2 and R3), containing mutations in PfIspD, were generated in strain 3D7. Parasites were cultured in a 2% suspension of human erythrocytes in RPMI 1640 (Sigma) medium supplemented with 27 mM sodium bicarbonate, 11 mM glucose, 5 mM HEPES, 1 mM sodium pyruvate, 0.37 mM hypoxanthine, 0.01 mM thymidine, 10 μg/mL gentamicin, and 0.5% Albumax (Gibco). Cultures were maintained at 37 °C, 5% O2/5% CO2/90% N2 atmosphere.
Drug Sensitivity Assays
Asynchronous cultures of P. falciparum, wild type (3D7) and 1R,3S-MMV008138-resistant strains (R2 and R3) were diluted to 0.5% parasitemia and 2% hematocrit. The parasites were then exposed to varying concentrations (0–240 μM) of PfIspD-targeting compound 10 for 72 h in a 96-well plate with 100 μL culture volumes per well. After 72 h, parasite growth was quantified using PicoGreen assay (Invitrogen) to measure the DNA content as previously described.10 Fluorescence measurement was taken using a CLARIOstar Plus microplate reader (BMG Labtech) at 485 nm excitation/528 nm emission. To determine the effectiveness of 10, half maximal inhibitory concentrations (IC50) values were calculated using nonlinear regression analysis in GraphPad Prism software. The reported IC50 value represent the mean ± standard error of the mean (SEM) from at least three independent replicates.
IDP Rescue Assay
For studying the reversal of growth inhibition, Pf3D7 (WT), and 1R,3S-MMV008138 resistant parasites (R2 and R3) were grown in the presence of varying concentrations of 10, and the presence or absence of 200 μM isopentenyl pyrophosphate (IDP; Echelon Biosciences) for 72 h. The parasite growth was quantified by measuring DNA content using PicoGreen assay (Invitrogen). IC50 values were calculated by nonlinear regression analysis using GraphPad Prism software and reflect the mean ± standard error of the mean (SEM) from at least three independent experiments. Statistical comparison of the values was done via one-way ANOVA.
LC-MS Based In Vitro Assay
Dilution series (1:2) of inhibitors in DMSO covered the concentration range of approximately 200–0.01 μM. After finishing the dilution series, the final volume of compound solution in DMSO per well was 2.0 μL. During the assay, the following buffer was used: 100 mM Tris-HCl pH 7.6, 1 mM DTT. To start the assay, aliquots of buffer (49 μL) containing: 306.1 μM CTP, 2.0 mM MgCl2 and 0.1 μM PfIspD, were added to a 96-well plate (Nunc V). Next 2 μL of the inhibitor dilutions (in DMSO) are added and the plate is allowed to incubate at 37 °C for 10 min. Then another 49 μL of buffer containing 306.1 μM MEP was added to start the reaction. The plates were incubated at 37 °C for 40 min, after which, the protein was denaturated by heating up the plate to 95 °C for 5 min. The plate was then centrifuged at 4000 rpm at 4 °C for 5 min to precipitate all solids present in the solution. To another 96-well plate, 190 μL of ice cold 3:1:1 ACN, isopropanol, water mixture was added. Thereafter, 10 μL of each of the supernatants from the assay plate were added. The plate was centrifuged again at 4000 rpm at 4 °C for 5 min, after which, 50 μL of the supernatant was transferred to a plate capable to measured in the MS and covered with a silicon cover. LC-MS conditions and data analysis methods we used were described above.
Determination of Enzyme Kinetics
A volume of 80 μL Buffer A containing 100 mM Tris-HCl pH 7.6, 1 mM DTT, 1 mM MgCl2, 50 nM PfIspD were added to well A1 96-well plate while 50 μL were added to the rest of the wells. A volume of 20 μL of 10 mM CTP was added to the first well, then a serial dilution was conducted by moving 50 μL. To start the reaction, we then added on buffer A 50 μL of Buffer B containing 100 mM Tris-HCl pH 7.6, 1 mM DTT, and 1 mM MEP. The assay plate was incubated at 37 °C for 40 min, after which, the protein was denaturated by heating up the plate to 95 °C for 5 min. The plate was then centrifuged at 4000 rpm at 4 °C for 5 min to precipitate the protein. To another 96-well plate, 190 μL of ice cold 3:1:1 ACN, isopropanol, water mixture was added containing 100 nM 4-methyl-1-oxo-1-(p-tolylamino)pentane-2-sulfonic acid, adenylyl-imidodiphosphate and adenosine-5′-[(α,β)-methyleno]triphosphate as internal standard.15 Thereafter, 10 μL of each of the supernatants from the assay plate were added to the plate containing the mixture with our internal standard. The plate was centrifuged again at 4000 rpm at 4 °C for 5 min, after which, 50 μL of the supernatant was transferred to an LC-MS plate and closed with a silicon cover. LC-MS conditions and data analysis methods we used were described above. The peak area for each conditions were used to calculate the Michaelis–Menten kinetic parameters using Graphpad Prism v 9. Measurements were performed in duplicates, repeated at least two times from two to three independent experiments.
Determination of Mode of Inhibition of 10
Dilution series (1:2) of inhibitors in DMSO covered the concentration range of approximately 200–0.01 μM. After finishing the dilution series, the final volume of compound solution in DMSO per well was 2.0 μL. During the assay, the following buffer was used: 100 mM Tris-HCl pH 7.6, 1 mM DTT. To study the inhibition mode against CTP, aliquots of buffer (49 μL) containing: 0, 37.5, 75, 125, 250, 500 μM CTP, 2.0 mM MgCl2 and 0.1 μM PfIspD, were added to a 96-well plate (Nunc V). Next 2 μL of the inhibitor dilutions (in DMSO) are added and the assay plate was incubated at 37 °C for 10 min. Then another 49 μL of buffer containing 500 μM MEP was added to start the reaction. To study the Mode of inhibition toward MEP, similar steps were followed as in case of CTP with using 0, 37.5, 75, 125, 250, 500 μM MEP and 500 μM CTP The assay plate was incubated at 37 °C for 40 min, after which, the protein was denaturated by heating up the plate to 95 °C for 5 min. The assay plate was then centrifuged at 4000 rpm at 4 °C for 5 min to precipitate the protein. To another 96-well plate, 190 μL of ice cold 3:1:1 ACN, isopropanol, water mixture was added containing 100 nM 4-methyl-1-oxo-1-(p-tolylamino)pentane-2-sulfonic acid as internal standard.15 Thereafter, 10 μL of each of the supernatants from the assay plate were added to the plate containing the mixture with our internal standard. The plate was centrifuged again at 4000 rpm at 4 °C for 5 min, after which, 50 μL of the supernatant was transferred to an LC-MS plate and closed with a silicon cover. LC-MS conditions and data analysis methods we used were described above.
Metabolic Stability in Liver S9 Fractions
For the evaluation of combined phase I and phase II metabolic stability, the compound (1 μM) was incubated with 1 mg/mL pooled mouse liver S9 fraction (Xenotech, Kansas City) or human liver S9 fraction (Corning, New York, USA), 2 mM NADPH, 1 mM UDPGA, 10 mM MgCl2, 5 mM GSH and 0.1 mM PAPS at 37 °C for 240 min. The metabolic stability of testosterone, verapamil and ketoconazole were determined in parallel to confirm the enzymatic activity of mouse S9 fractions, for human S9 testosterone, diclofenac and propranolol were used. The incubation was stopped after defined time points by precipitation of aliquots of S9 enzymes with 2 volumes of cold acetonitrile containing internal standard (150 nM diphenhydramine). Samples were stored on ice until the end of the incubation and precipitated protein was removed by centrifugation (4 °C, 15 min, 4000g). Concentration of the remaining test compound at the different time points was analyzed by HPLC-MS/MS (TSQ Quantum Access MAX, Thermo Fisher, Dreieich, Germany) and used to determine half-life (t1/2).
Stability in Mouse and Human Plasma
To determine stability in mouse plasma, the compound (1 μM) was incubated with pooled CD-1 mouse or human plasma (Neo Biotech, Nanterre, France). Samples were taken at defined time points by mixing aliquots with 4 volumes of acetonitrile containing internal standard (125 nM diphenhydramine). Samples were stored on ice until the end of the incubation and precipitated protein was removed by centrifugation (4 °C, 15 min, 4000g, 2 centrifugation steps). Concentration of the remaining test compound at the different time points was analyzed by HPLC-MS/MS (TSQ Quantum Access MAX, Thermo Fisher, Dreieich, Germany). The plasma stability of procain, propantheline and diltiazem were determined in parallel to confirm the enzymatic activity.
Plasma Protein Binding Plasma protein binding was determined using the Rapid Equilibrium Dialysis (RED) system (Thermo Fisher Scientific, Waltham MA, USA). Compounds were diluted to 10 μM in 50% murine (CD-1) or human plasma (Neo Biotech, Nanterre, France) in PBS pH 7.4 and added to the respective chamber according to the manufacturer’s protocol, followed by addition of PBS pH 7.4 to the opposite chamber. Samples were taken immediately after addition to the plate as well as after 2, 4 and 5 h by mixing 10 μL with 80 μL ice-cold acetonitrile containing 12.5 nM diphenhydramine as internal standard, followed by addition of 10 μL plasma to samples taken from PBS and vice versa. Samples were stored on ice until the end of the incubation and precipitated protein was removed by centrifugation (15 min, 4 °C, 4,000 g, 2 centrifugation steps). Concentration of the remaining test compound at the different time points was analyzed by HPLC-MS/MS (Vanquish Flex coupled to a TSQ Altis Plus, Thermo Fisher, Dreieich, Germany). The amount of compound bound to protein was calculated using the equation PPB [%] = 100 – 100 x (amount in buffer chamber/amount in plasma chamber).
Pharmacokinetic (PK) Studies
For pharmacokinetic experiments, outbred male CD-1 mice (Charles River, Germany), 4 weeks old, were used. The animal studies were conducted in accordance with the recommendations of the European Community (Directive 2010/63/EU, first January 2013). All animal procedures were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). Animals were excluded from further analysis if sacrifice was necessary according to the humane end points established by the ethical board. All experiments were approved by the ethical board of the Niedersächsisches Landesamt für Verbraucherschutz and Lebensmittelsicherheit, Oldenburg, Germany. Compounds 10 and 28 were administered at 1 mg/kg intravenously in a cassette format (n = 2). At the time points 0.25, 0.5, 1, and 3 post administration, up to 25 μL of blood were collected from the lateral tail vein. At 5 h post administration, mice were euthanized to collect blood from the heart as well as to remove spleen and liver aseptically. Whole blood was collected into Eppendorf tubes coated with 0.5 M EDTA and immediately spun down at 15,870g for 10 min at 4 °C. Then, plasma was transferred into a new Eppendorf tube spleen and liver were homogenized using a Polytron tissue homogenizer. Spleen, liver, and plasma samples were stored at −80 °C until analysis. First, a calibration curve was prepared by spiking different concentrations of 10 and 28 into mouse plasma, homogenized spleen or homogenized liver from CD-1 mice. Glipizide was used as an internal standard. In addition, quality control samples (QCs) were prepared for 10 and 26 in the same matrices. For 10 and 28 the same extraction procedure was used: 7.5 μL of a plasma sample (calibration samples, QCs or PK samples) was extracted with 22.5 μL of acetonitrile containing 12.5 ng/mL of glipizide as an internal standard for 5 min at 2000 rpm on an Eppendorf MixMate vortex mixer. Then samples were spun down at 13.000 rpm for 10 min. Supernatants were transferred to standard HPLC-glass vials. For liver and spleen, 20 μL of a sample (calibration samples, QCs or PK samples) were extracted with 10 μL water containing 10% formic acid, and 22.5 μL acetonitrile with 12.5 ng/mL of glipizide as internal standard. Samples were extracted for 5 min at 800 rpm on an Eppendorf MixMate vortex mixer and spun down for 5 min at 4000 rpm. Peaks of PK samples were quantified using the calibration curve. The accuracy of the calibration curve was determined using QCs independently prepared on different days (Table S4). PK parameters were determined using a noncompartmental analysis with PKSolver.24
Cytotoxicity Study
An MTT-based assay was employed to evaluate the viability of HepG2 after challenge with selected inhibitors and performed as described previously.25
Acknowledgments
D.W. Thank you Simone Amann, Jeannine Jung, and Jannine Seelbach for the great work. K.R. receives funding from the German Center for Infection Research (DZIF, TTU 09.719). K.R. thanks Andrea Ahlers, Kimberley Vivien Sander, Janine Schreiber and Jennifer Wolf for excellent technical assistance. Figure 2 and Scheme 1 and the graphical abstract were created with BioRender.com.
Glossary
Abbreviations Used
- AMR
antimicrobial resistance
- AUC0-t
area under the concentration–time curve from time zero to time t
- CDP-ME
4-diphosphocytidyl-2-C-methylerythritol
- Cl_obs
clearance (based on observed last time point with measurable concentration)
- CTP
cytidine triphosphate
- CuAAC
copper-catalyzed azide-alkyne cycloaddition
- DDA
data-dependent acquisition
- DIA
data-independent acquisition
- DCM
dichloromethane
- DMADP
dimethylallyl diphosphate
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- Eq
equivalents
- ESI
electron spray ionization
- FA
formic acid
- EC50
fifty percent inhibitory concentrations
- HESI
heated electrospray ionization
- HTS
high throughput screening
- HPLC
high pressure liquid chromatography
- IDP
isopentenyl diphosphate
- IV
intravenous
- Km
Michaelis constant
- LCMS
liquid chromatography-mass spectrometry
- MEP
2-C-methylerythritol-D-erythritol-4-phosphate
- MRT
mean residence time
- MWD
multiple wave detector
- n.a.
no activity
- ND
not detected
- NMR
nuclear magnetic resonance
- Pf
Plasmodium falciparum
- PPi
inorganic diphosphate
- PK
pharmacokinetic
- SAR
structure–activity relationship
- SD
standard deviation
- Vz_obs
volume of distribution associated with the terminal phase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00212.
Activities of selected compounds against auxiliary enzymes; calibration curve for CDP-ME; kinetic parameters concerning MEP and CTP; LC-MS chromatograms of MEP, CDP-ME, and the internal standard; Q1 and Q3 masses for glipizide, 10 and 28; supplementary reaction schemes; activities of representative compounds against different bacterial pathogens; 1H, 13C NMR spectra, and HPLC traces of all final compounds (PDF)
Molecular formula strings, activity data against PfIspD and whole-cell activity against PfNF54 (CSV)
Author Present Address
‡‡ Department of Pharmacy and Biotechnology, Alma Mater Studiorum—University of Bologna, 40126 Bologna, Italy
Author Contributions
Second authors with equal contribution. D.W., E.D., M.M.H., M.W., and A.K.H.H. coordinated the project; synthesis and characterization of the compounds was performed by D.W., E.D., M.M.H., and M.W.; HTS and biological evaluation of derivatives against PfIspD was performed by B.I. and M.F.; evaluation of the potency against PfNF54 was performed by P.B. and M.R.; development of the LC-MS based IspD assay and kinetic characterization was performed by A.A. and L.B.; ADMET and PK profiling experiments were executed by A.K. and K.R.; D.W. wrote the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 860816. MepAnti.
The authors declare no competing financial interest.
Supplementary Material
References
- Dadgostar P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. 10.2147/IDR.S234610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassini A.; Högberg L. D.; Plachouras D.; Quattrocchi A.; Hoxha A.; Simonsen G. S.; Colomb-Cotinat M.; Kretzsch-mar M. E.; Devleesschauwer B.; Cecchini M.; Ouakrim D. A.; Oliveira T. C.; Struelens M. J.; Suetens C.; Monnet D. L.; et al. At-tributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect. Dis. 2019, 19 (1), 56–66. 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov M. Resistance to front-line malaria drugs con-firmed in Africa. Nature 2021, 597, 604 10.1038/d41586-021-02592-6. [DOI] [Google Scholar]
- Masini T.; Kroezen B. S.; Hirsch A. K. H. Druggability of the enzymes of the non-mevalonate-pathway. Drug Discovery Today 2013, 18 (23), 1256–1262. 10.1016/j.drudis.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Diamanti E.; Hamed M. M.; Lacour A.; Bravo P.; Illari-onov B.; Fischer M.; Rottmann M.; Witschel M.; Hirsch A. K. H. Targeting the IspD Enzyme in the MEP Pathway: Identifica-tion of a Novel Fragment Class. ChemMedChem 2022, 17, e202100679 10.1002/cmdc.202100679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombo-Ngoma G.; Remppis J.; Sievers M.; Manego R. Z.; Endamne L.; Kabwende L.; Veletzky L.; Nguyen T. T.; Groger M.; Lötsch F.; Mischlinger J.; Flohr L.; Kim J.; Cattaneo C.; Hutchinson D.; Duparc S.; Moehrle J.; Velavan T. P.; Lell B.; Ramharter M.; Adegnika A. A.; Mord-müller B. B.; Kremsner P. G. Efficacy and Safety of Fosmidomycin–Piperaquine as Nonartemisinin-Based Combination Therapy for Uncomplicated Falciparum Malaria: A Single-Arm, Age De-escalation Proof-of-Concept Study in Gabon. Clin. Infect. Dis. 2018, 66 (12), 1823–1830. 10.1093/cid/cix1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knak T.; Abdullaziz M. A.; Höfmann S.; Alves Avelar L. A.; Klein S.; Martin M.; Fischer M.; Tanaka N.; Kurz T. Over 40 Years of Fosmidomycin Drug Research: A Comprehensive Review and Future Opportunities. Pharmaceuticals 2022, 15, 1553 10.3390/ph15121553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes J. F.; Lell B.; Agnandji S. T.; Obiang R. M.; Bassat Q.; Kremsner P. G.; Mordmüller B.; Grobusch M. P. Fosmidomycin as an antimalarial drug: a meta-analysis of clinical trials. Future Microbiol. 2015, 10 (8), 1375–1390. 10.2217/FMB.15.60. [DOI] [PubMed] [Google Scholar]
- Frank A.; Groll M. The Methylerythritol Phosphate Pathway to Isoprenoids. Chem. Rev. 2017, 117 (8), 5675–5703. 10.1021/acs.chemrev.6b00537. [DOI] [PubMed] [Google Scholar]
- Price K. E.; Armstrong C. M.; Imlay L. S.; Hodge D. M.; Pidathala C.; Roberts N. J.; Park J.; Mikati M.; Sharma R.; Lawrenson A. S.; Tolia N. H.; Berry N. G.; O’Neill P. M.; John A. R. O. Molecular Mechanism of Action of Antimalarial Benzo-isothiazolones: Species-Selective Inhibitors of the Plasmodium spp. MEP Pathway enzyme, IspD. Sci. Rep. 2016, 6, 36777 10.1038/srep36777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew J.; Ding S.; Kunz K. A.; Stacy E. E.; Butler J. H.; Haney R. S.; Merino E. F.; Butschek G. J.; Rizopoulos Z.; Totrov M.; Cassera M. B.; Carlier P. R. Malaria Box-Inspired Discovery of N-Aminoalkyl-β-carboline-3-carboxamides, a Novel Orally Active Class of Antimalarials. ACS Med. Chem. Lett. 2022, 13 (3), 365–370. 10.1021/acsmedchemlett.1c00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay L. S.; Armstrong C. M.; Masters M. C.; Li T.; Price K. E.; Edwards R. L.; Mann K. M.; Li L. X.; Stallings C. L.; Berry N. G.; O’Neill P. M.; Odom A. R. Plasmodium IspD (2-C-Methyl-d-erythritol 4-Phosphate Cytidyltransferase), an Essential and Druggable Antimalarial Target. ACS Infect. Dis. 2015, 1 (4), 157–167. 10.1021/id500047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.; Ghavami M.; Butler J. H.; Merino E. F.; Slebodnick C.; Cassera M. B.; Carlier P. R. Probing the B- & C-rings of the antimalarial tetrahydro-β-carboline MMV008138 for steric and conformational constraints. Bioorg. Med. Chem. Lett. 2020, 30 (22), 127520 10.1016/j.bmcl.2020.127520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Herrera Z.; Ebert D.; Baska K.; Cho S. H.; Derisi J. L.; Yeh E. A. Chemical Rescue Screen Identifies a Plasmodium falciparum Apicoplast Inhibitor Targeting MEP Isoprenoid Precursor Biosynthesis. Antimicrob. Agents Chemother. 2015, 59 (1), 356–364. 10.1128/AAC.03342-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honold A.; Lettl C.; Schindele F.; Illarionov B.; Haas R.; Witschel M.; Bacher A.; Fischer M. Inhibitors of the Bifunctional 2-C-Methyl-D-erythritol 4-Phosphate Cytidylyl Transferase/2-C-Methyl-D-erythritol-2,4-cyclopyrophosphate Synthase (IspDF) of Helicobacter pylori. Helv. Chim. Acta 2019, 102, e1800228 10.1002/hlca.201800228. [DOI] [Google Scholar]
- Li Z.; Sharkey T. D. Metabolic profiling of the methylerythritol phosphate pathway reveals the source of post-illumination isoprene burst from leaves. Plant, Cell Environ. 2013, 36, 429–437. 10.1111/j.1365-3040.2012.02584.x. [DOI] [PubMed] [Google Scholar]
- Konstantinović J.; Kany A. M.; Alhayek A.; Abdelsamie A.; Sikandar A.; Voos K.; Yao Y.; Andreas A.; Shafiei R.; Loretz B.; Schönauer E.; Bals R.; Brandstetter H.; Hartmann R. W.; Ducho C.; Lehr C.; Beisswenger C.; Müller R.; Rox K.; Haupenthal J.; Hirsch A. K. H. Inhibitors of the Elastase LasB for the Treatment of Pseudomonas aeruginosa Lung Infections. ACS Cent. Sci. 2023, 9 (12), 2205–2215. 10.1021/acscentsci.3c01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami M.; Merino F. E.; Yao Z.; Elahi R.; Simpson M. E.; Fernández-Murga M. L.; Butler J. H.; Casasanta M. A.; Krai P. M.; Totrov M. M.; Slade D. J.; Carlier P. R.; Cassera M. B. Biological Studies and Target Engagement of the 2-C-Methyl-d-Erythritol 4-Phosphate Cytidylyltransferase (IspD)-Targeting Antimalarial Agent (1R,3S)-MMV008138 and Analogs. ACS Infect. Dis. 2018, 4 (4), 549–559. 10.1021/acsinfecdis.7b00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witschel M. C.; Höffken H. W.; Seet M.; Parra L.; Mietzner T.; Thater F.; Niggeweg R.; Röhl F.; Illarionov B.; Rohdich F.; Kaiser J.; Fischer M.; Bacher A.; Diederich F. Inhibitors of the Herbicidal Target IspD: Allosteric Site Binding. Angew. Chem., Int. Ed. 2011, 50, 7931–7935. 10.1002/anie.201102281. [DOI] [PubMed] [Google Scholar]
- Kuzmič P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996, 237, 260–273. 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- Snyder C.; Chollet J.; Santo-Tomas J.; Scheurer C.; Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp. Parasitol. 2007, 115 (3), 296–300. 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Huber W.; Koella J. C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55 (4), 257–261. 10.1016/0001-706X(93)90083-N. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Huo M.; Zhou J.; Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Haupenthal J.; Baehr C.; Zeuzem S.; Piiper A. RNAse A-like enzymes in serum inhibit the anti-neoplastic activity of siRNA targeting polo-like kinase 1. Int. J. Cancer 2007, 121, 206–210. 10.1002/ijc.22665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.