

Abstract

Cystobactamids have a unique oligoarylamide structure and exhibit broad-spectrum activity against Gram-negative and Gram-positive bacteria. In this study, the central α-amino acid of the cystobactamid scaffold was modified to address the relevance of stereochemistry, hydrogen bonding and polarity by 33 derivatives. As demonstrated by three matched molecular pairs, l-amino acids were preferred over d-amino acids. A rigidification to a six-membered system stabilized the bioactive conformation for the on-target Escherichia coli gyrase, but did not improve antimicrobial activity. Compound CN-CC-861, carrying a propargyl side chain, had more than 16-fold lower minimal inhibitory concentration (MIC) values against Enterococcus faecalis, Staphylococci and Acinetobacter strains, compared to known analogues. Moreover, CN-CC-861 retained activity against multidrug-resistant enterococci, displayed strong bactericidal activity, moderate-low frequencies of resistance and in vivo efficacy in a neutropenic thigh infection model with E. coli. Overall, the findings will guide the design of new promising structures with higher activities and broader spectrum.
Introduction
Infections, due to multidrug-resistant bacterial pathogens, are a major cause of morbidity and mortality.1 Among the “priority pathogens”, as designated by the WHO,2 many belong to the group of Gram-negative bacteria. Owing to their complex membrane structure, general rules for the development of permeable antibacterial drugs are just emerging.3−5 Consequently, no novel antibacterial scaffold with significant activity against Gram-negative bacteria was commercialized since the discovery of quinolones in the 1960s.6 Recent efforts to explore microbes as sources of novel antibacterials7 have led to the discovery of cystobactamids and the structurally related albicidins (Figure 1).8,9 Their unique hexapeptidic structures feature linearly connected para-amino benzoic acids (PABAs)10 as well as a central α-amino acid derived from asparagine. Although relatively large, this novel compound class shows remarkably high and resistance-breaking activity against Gram-positive and -negative bacteria. The compounds inhibit the bacterial gyrase and bacterial topoisomerase IV, with a binding mode that is unique and distinct from other classes.11
Figure 1.

Structures of natural cystobactamids 919–2 (1) and 861–2 (2), synthetic analogues CN 861 (3) and CN-DM-861 (4) and albicidin.
In recent years, the establishment of total syntheses for cystobactamids12,13 and albicidins14,15 opened the possibility to improve in vitro activity and spectrum. First, structure–activity relationship (SAR) investigations of the central amino acid revealed inter alia that the methoxy group in natural cystobactamid 861–2 can be omitted, and that the cyano group of albicidin can be replaced by a triazole among other residues,16−18 moieties that were kept in first congeners with in vivo activity16,19 such as CN-DM-861 (Figure 1). Due to the high relevance of the central α-amino acid, this work was devoted to the study of the SAR in a systematic manner through 33 new cystobactamid analogues. The design was guided by the biological assessment of the minimal inhibitory concentration (MIC) on a small panel of five bacterial strains. Highly active analogues were further evaluated on secondary panels comprising a higher variety of bacteria. Through this process, a new promising lead compound with a surprisingly simple functional group, enhanced broad-spectrum activity and resistance-breaking properties was found.
Results and Discussion
The introduction of the central α-amino acid variations were carried out at a late stage, as the longest linear sequence in the synthesis of cystobactamids currently involves about 15 steps. For this purpose, the central moiety was linked to complete, fully functionalized AB and CDE fragments (Figure 2).20 For the connection of the central α-amino acid to the CDE fragment, the former was converted to the respective acyl chloride.21 To avoid racemization, pyridine was employed in the subsequent amide coupling. Alternatively, the central amino acid was directly coupled to the CDE fragment with ethyl 2-ethoxyquinoline-1(2H)-carboxylate (EEDQ) or propanephosphonic acid anhydride (T3P).20 Upon the usage of EEDQ, a significant conversion of the amino acid to the ethyl ester was observed as side reaction due to the low nucleophilicity of the aniline. The assembly of the three fragments followed by global deprotection is exemplified for compound CN-CC-861 (Scheme 1). It is worth mentioning that the previously used phenylsilane as scavenger can be substituted by aniline during allyl deprotection, which leads to fewer side products.20 For alkyne-bearing analogues, the application of silanes was disadvantageous, since an unwanted reduction of the alkyne to the alkene was observed during deallylation. Both albicidin (5) as well as CN-DM-861 (4) have a hydrogen bond acceptor (HBA) in the side chain of the central amino acid (Figure 3). To explore its relevance, in a first step compounds 13–30 were investigated for their antibacterial activity (Table 1).
Figure 2.

Retrosynthesis of cystobactamids with modifications at the central amino acid. PG = protecting group.
Scheme 1. Fragment Connection to the Full-Length Cystobactamid and Global Deprotection Exemplified by the Synthesis of CN-CC-861.
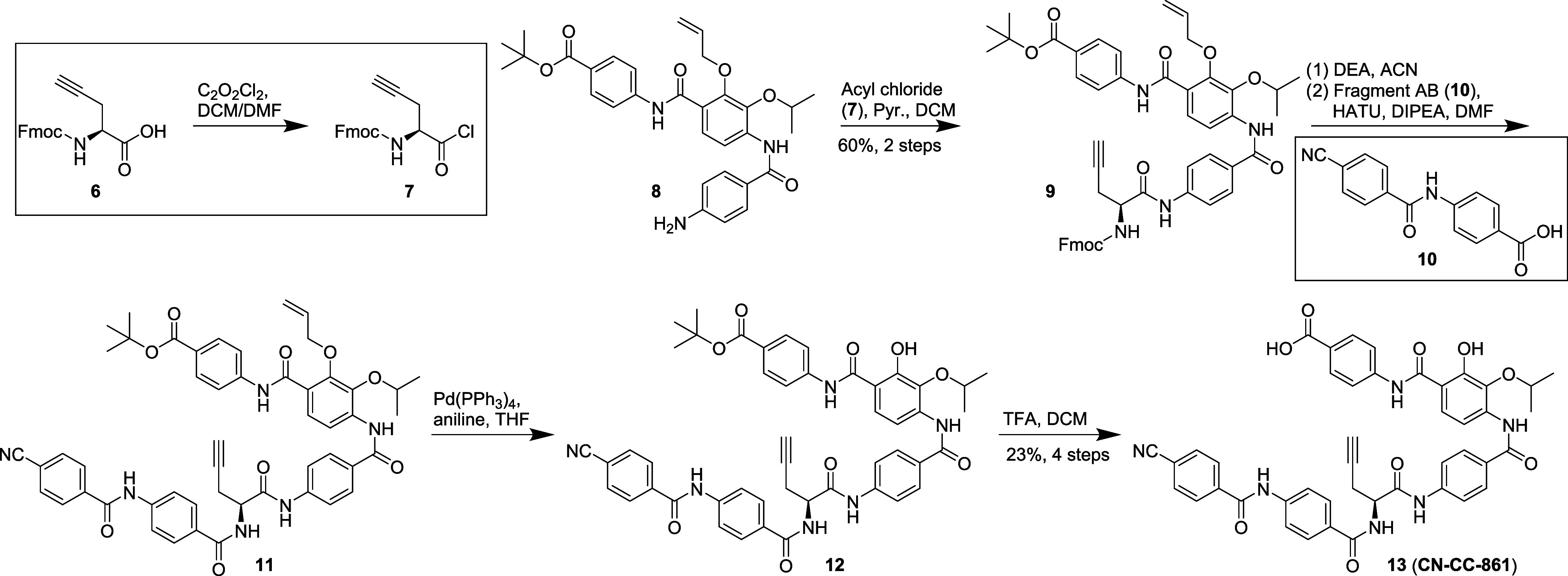
Figure 3.

Structures of the central α-amino acid moiety in CN-DM-861 (4) and albicidin (5) and their HBA (red) as well as the rigidified l-picolinic acid derivative 17.
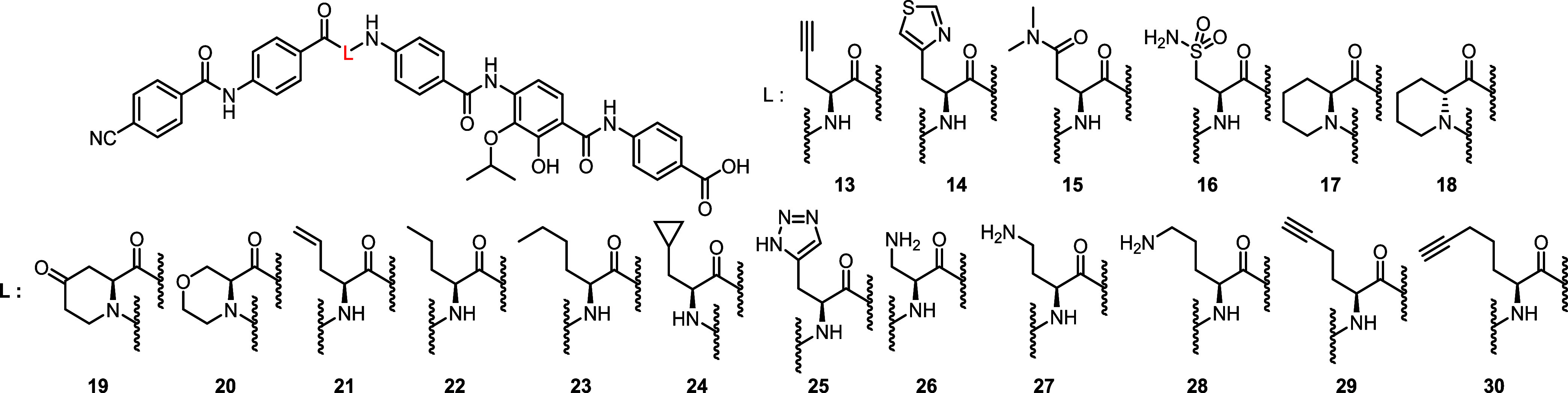
Table 1. MIC and IC50 Values of Cystobactamids CN-CC-861 (13)-30 with Modified Central Amino Acid Compared to CN-DM-861 and Ciprofloxacin (CIP).
| MIC [μg/mL] |
|||||||
|---|---|---|---|---|---|---|---|
| compound | E. coli WT (BW25113) | E. coli ΔacrB | E. coli LM705 (S83L, D87N, S80I, ΔacrR, ΔmarR) | Staphylococcus aureus (ATCC 29213) | P. aeruginosa WT (Pa14) | P. aeruginosa Pa14ΔmexAB | IC50 [μM] E. coli gyrase/TI IVa |
| 13 (CN-CC-861) | ≤0.03 | ≤0.03 | ≤0.03–0.2 | 0.02 | 0.5 | ≤0.03 | 0.23:0.47 |
| 14 | n.d. | ≤0.03 | 8 | 0.5 | >64 | 0.25 | 0.34 |
| 15 | 0.25 | ≤0.03 | 32 | 8 | >64 | 0.5 | 1.47 |
| 16 | 0.04 | 0.0125 | 0.25 | 1 | 16 | ≤0.03–1 | n.d. |
| 17 | 0.06 | ≤0.03 | >64 | 2 | 64 | 2 | 0.18:0.08 |
| 18 | 2 | ≤0.03 | >64 | 4 | 64 | 4 | 1.07:2.44 |
| 19 | ≤0.03 | ≤0.03 | 32 | 2 | 16 | 1 | 0.86 |
| 20 | 1 | ≤0.03 | >64 | 0.8 | >64 | 4–8 | n.d. |
| 21 | 0.04 | 0.0125 | 0.5 | ≤0.03 | 0.5 | 0.125 | 0.49 |
| 22 | 0.16 | 0.08 | 1 | ≤0.03 | >64 | 0.5 | 0.28 |
| 23 | ≤0.03 | ≤0.03 | 8 | 0.25 | >64 | 4 | 0.45 |
| 24 | 0.16 | 0.16 | 2 | 0.25 | 0.5 | 0.25 | n.d. |
| 25 | ≤0.03 | ≤0.03 | 0.5 | 0.5 | 1 | ≤0.03 | 0.33:0.37 |
| 26 | 0.125 | 0.006 | 0.05 | 0.5 | 0.25 | 0.25 | 0.86 |
| 27 | ≤0.03 | 0.125 | 3.2 | 3.2 | 4 | 1 | n.d. |
| 28 | ≤0.03 | 0.08 | >64 | >64 | >64 | 2 | 0.63 |
| 29 | 0.04 | 0.02 | 0.25 | ≤0.03 | 32 | 2 | 0.21 |
| 30 | ≤0.03 | ≤0.03 | 0.5 | 0.06 | 64 | 2 | 0.23 |
| CIP | 0.02 | 0.001 | >6.4 | 0.2 | 0.05 | ≤0.03 | 0.18:4.95 |
| CN-DM-861 (4) | 0.08 | 0.004 | 0.125 | 1 | 2 | 0.25 | 0.13:3.16 |
(a) n = 1. n.d.: not determined. IC50 values determined by the gyrase supercoiling inhibition assay. For selected compounds, IC50′s were also determined by a topoisomerase IV relaxation assay (values given after the ‘/’ sign).
The introduction of the thiazole as in 14 or the N,N-dimethyl asparagine as in 15 was well-tolerated but led to a loss of activity against the Pseudomonas aeruginosa wild type. Additionally, the exchange of the amide for a sulfonamide as in 16 was well tolerated. Notably, alkyne CN-CC-861 (13) depicted high activity against all strains with equal or better MIC values than the reference compound CN-DM-861 (4), including P. aeruginosa. The latter finding implies that the HBA/hydrogen bond donor (HBD) abilities of the original amide moiety were not essential for activity on the tested panel. We then rigidified the central amino acid. The six-membered l-picolinic acid derivative 17 was chosen to mimic a cyclic conformation in CN-DM-861 (4) resulting from an intramolecular hydrogen bond (Figure 3).
To determine the importance of the configuration, the respective d-enantiomer 18 was also synthesized and tested. The comparison of the activities indicated a slight preference for the l-configurated amino acid 17. More precisely, 17 shows a nearly 6-fold lower IC50 value on the Escherichia coli gyrase and more than a 30-fold lower IC50 on topoisomerase IV. The low IC50 value on gyrase of 0.18 μM, similar to CN-DM-861 (4), suggests that the rigidification in 17 stabilizes the bioactive conformation in the E. coli gyrase compared to its enantiomer 18, but also compared to the open chain analogs 22 and 23 with similar alkyl chain length, yet slightly higher IC50 values of 0.28 and 0.45 μM, respectively, that might reflect higher entropy costs upon binding. The hypothesis, regarding the stereochemical preference for 17vs18, was affirmed by docking the two compounds into the published E. coli gyrase binding site of the albicidin analogue Albi-1.22 Preceding docking studies with Albi-1 showed that the double deprotonated form adopts a comparable pose to its cryogenic electron microscopy (cryo-EM) structure (Figure S1). While the docked l-picolinic acid analogue 17 with the same ionization state was able to maintain the overall conformation of Albi-1, the best predicted pose of the respective d-picolinic acid 18 scored significantly worse (Table S6) and adopted a strongly deviating pose (Figure S2). Although both picolinic acid analogues were situated in the DNA binding region with their N-terminal fragment, the altered positioning of the central amino acid in 18 resulted in a completely different alignment of the B, C, D and E rings compared to Albi-1 (Figure 4).
Figure 4.
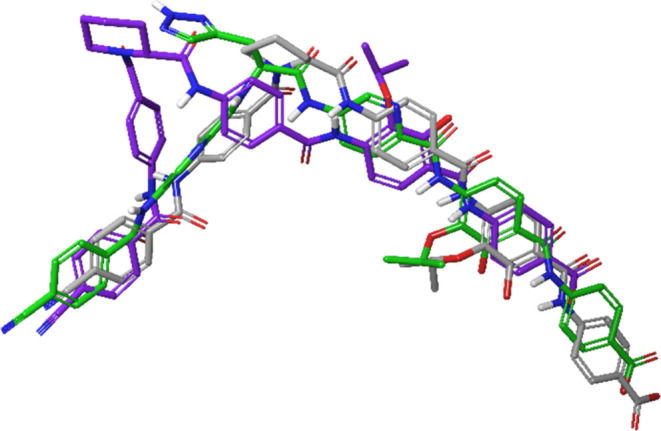
Overlay of the docking poses of Albi-1 (green ligand), compound 17 (gray ligand) and 18 (purple ligand) with Glide23 in the cryo-EM structure of E. coli gyrase holocomplex with 217 bp DNA (PDB: 7Z9K).22
An introduction of a 4-oxo functionality into the l-pipecolinic ring was well tolerated and led to equal or higher activity of compound 19 (Table 1). Morpholine derivative 20, on the other hand, performed worse than the piperidine 17 against the E. coli wild type. A rigidification and ring contraction to a 2-azabicyclo[2.1.1]hexane as in the constitutional isomer 31 substantially diminished the activity (Table 2). As the alkyne CN-CC-861 (13) demonstrated a positive impact of an aliphatic system on antibacterial activity, reduced analogues 21 and 22 were synthesized for comparison.
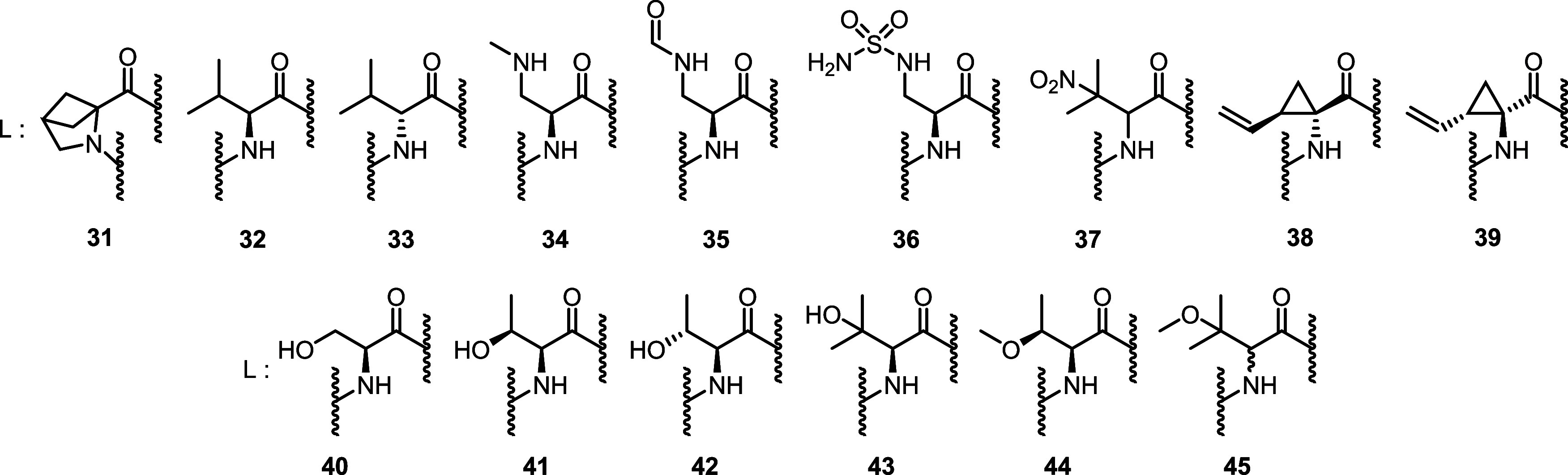
Table 2. MIC and IC50 Values of Cystobactamids 31-45 with Modified Central Amino Acid Compared to CN-CC-861 (13), CN-DM-861 (4) and Ciprofloxacin (CIP).
| MIC [μg/mL] |
|||||||
|---|---|---|---|---|---|---|---|
| compound | E. coli WT (BW25113) | E. coli ΔacrB | E. coli LM705 (S83L, D87N, S80I, ΔacrR, ΔmarR) | S. aureus (ATCC 29213) | P. aeruginosa WT (Pa14) | P. aeruginosa Pa14ΔmexAB | IC50 [μM] E. coli gyrase/TI IVa |
| 31 | 8 | 0.5 | >64 | 16 | >64 | 32 | n.d. |
| 32 | 1.6 | 0.2 | 0.5 | 0.05 | >6.4 | 6.4 | n.d. |
| 33 | 8 | 1 | >64 | >64 | >64 | >64 | n.d. |
| 34 | 0.06 | ≤0.03 | >64 | 0.25 | >64 | >64 | n.d. |
| 35 | 0.16 | 0.08 | ≤0.03 | 0.125 | 8 | 0.5 | n.d. |
| 36 | 0.125 | ≤0.03 | 0.25 | 2 | >64 | 0.5 | n.d. |
| 37 | 0.06 | ≤0.03 | 1 | ≤0.06 | 1 | 8 | n.d. |
| 38 | 0.06 | ≤0.03 | 0.125 | ≤0.03 | 8 | 0.125 | 0.18/0.27 |
| 39 | n.d. | n.d. | 2 | 0.5 | >64 | >64 | n.d. |
| 40 | n.d. | n.d | 0.06 | 0.125 | n.d. | n.d. | n.d. |
| 41 | 0.0125 | ≤0.003 | 0.06 | ≤0.03 | 8 | 0.5 | 0.09/0.09 |
| 42 | 0.04 | 0.006 | 0.5 | ≤0.03 | >64 | 0.5 | 0.14/0.67 |
| 43 | n.d. | n.d. | 0.125–0.25 | n.d. | n.d. | 0.04 | n.d. |
| 44 | 0.16 | 0.005 | 0.25 | ≤0.03 | 16 | 1 | n.d. |
| 45 | 0.06 | ≤0.03 | 2 | ≤0.03 | 64 | 0.25 | n.d. |
| CN-CC-861 (13) | ≤0.03 | ≤0.03 | ≤0.03–0.2 | 0.02 | 0.5 | ≤0.03 | 0.23/0.47 |
| CIP | 0.02 | 0.001 | >6.4 | 0.2–1 | 0.05 | ≤0.03 | 0.65/4.95 |
| CN-DM-861 (4) | 0.08 | 0.004 | 0.125–0.25 | 1 | 2 | 0.25 | 0.45/3.16 |
n.d.: not determined. IC50 values determined for selected compounds by the gyrase supercoiling inhibition assay/topoisomerase IV relaxation assay.
Alkene 21 turned out to be more potent than saturated analogue 22 against P. aeruginosa strains. However, cyclopropyl analogue 24, blending properties of unsaturated and saturated systems, showed activities comparable to 21 and 22. Branched derivative 32 based on l-valine proved to be less active (Table, Table S1) while d-valine analogue 33 was mostly inactive. Both triazole 25 and primary amine 26 possess an HBD in addition to at least one HBA, which probably contributes to their high activity comparable to alkyne CN-CC-861 (13). Similar to the extension of the alkyl chain from 22 to 23, neither the elongations of the alkyne from 13 to 29 and 30, nor of the amine side chain from 26 to 27 and 28 led to higher activities (Table 1). Therefore, we concluded that the optimal location of both substituents was at the β-position of the amino acid. Methylation as well as formylation and sulfamylation of the β-amine also resulted in considerably increased MIC values in 34, 35 and 36 (Table 2). A formal nitration of the β-hydrogen of valine resulted in 37, a racemic mixture with moderate activity in the primary panel, but high potency against additionally tested Acinetobacter baumannii strains (Table S2).
By integration of the α-carbon atom into a cyclopropane ring, 38 as rigidified analogue of the β-vinyl derivative 21 was obtained. While the 1S,2R derivative 38 retained broad-spectrum activity, the enantiomer 39, resembling the respective d-configuration, showed significantly reduced or total loss of activity on many strains (Table S2).
Finally, six additional derivatives 40–45 containing an oxygenated β-position were prepared with an increasing degree of 0–3 methyl substitutions at the β-carbon and oxygen. All derivatives exhibited robust broad-spectrum activity; an increasing level of methyl substitutions led to lower activities against the multidrug-resistant E. coli strain LM705, but improved activities against A. baumannii strains (Tables 2 and S3).
Based on the results of the small panel, a set of six promising cystobactamids underwent further MIC testing on an extended panel of clinically relevant Gram-negative and -positive pathogens (Table 3). Amino acid analogues CN-CC-861 (13), 25 and 26 depicted the broadest spectrum coverage. Particularly, alkyne CN-CC-861 (13) exhibited notable improvements compared to CN-DM-861 (4) in terms of enhanced activity against A. baumannii, Enterococcus faecalis and S. aureus. Amine 26 shared a very similar activity pattern to CN-DM-861 (4) with exceeding potency against Proteus mirabilis, but insufficient activity against P. aeruginosa ESBL2. Triazole 25 was lacking activity against Enterobacter aerogenes, but mostly retained potency against other strains with improvements against S. aureus. Derivatives CN-CC-861 (13), 21, 22 and 24 also impressed by their excellent activity against A. baumannii, including ciprofloxacin- and CN-DM-861 (4) resistant strains (Tables S1 and S4).
Table 3. MIC Values of Selected Cystobactamid Analogues on an Extended Panel of Pathogenic Bacteria Compared to CN-DM-861 and Ciprofloxacin (CIP).
| MIC (μg/mL) |
||||||||
|---|---|---|---|---|---|---|---|---|
| CN-DM-861 (4) | CIP | CN-CC-861 (13) | 21 | 22 | 24 | 25 | 26 | |
| gram-negative strains | ||||||||
| A. baumannii DSM 30008 | 0.5 | 0.2–0.32 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | 0.125 | 0.125 |
| Citrobacter freundii DSM 30039 | ≤0.03–0.125 | ≤0.03 | ≤0.03 | ≤0.03 | 0.06 | ≤0.03 | 0.06 | ≤0.03 |
| E. aerogenes DSM 30053 | 0.25–0.5 | 0.08–0.1 | 0.5 | 16 | >64 | >64 | >64 | 0.5 |
| Enterobacter cloacae DSM 30054 | 0.25–1 | 0.1–0.5 | 0.5 | 4 | 0.125 | 0.5 | 1 | 0.125 |
| E. coli DSM 1116 | ≤0.03 | 0.01 | ≤0.03 | ≤0.03 | 0.125 | ≤0.03 | ≤0.03 | ≤0.03 |
| E. coli WT-3 [gyrA(S83L,D87G)]a | 0.06–0.125 | 0.32–0.8 | ≤0.03 | 0.125 | 0.25 | 1 | 0.25 | 0.06 |
| Klebsiella pneumoniae DSM 30104 | 0.25–64 | 0.01–0.1 | >64 | >64 | >64 | >64 | 0.25 | >64 |
| P. aeruginosa ESBL1 DSM 24600 | 64 | 3.2–6.4 | 64 | >64 | >64 | >64 | 32 | >64 |
| P. aeruginosa ESBL2 DSM 46316 | 1 | 0.1–0.4 | 0.5 | 2 | >64 | >64 | 16 | 64 |
| P. mirabilis DSM 4479 | 32–64 | 0.02 | 64 | 64 | 0.25 | 0.125 | 32 | 1 |
| Proteus vulgaris DSM 2140 | 0.25–0.5 | ≤0.06 | 0.125 | 0.06 | 0.25 | 0.25 | 0.06 | 0.25 |
| Serratia marcescens DSM 30121 | 64 | 0.2 | >64 | >64 | >64 | >64 | 64 | 64 |
| gram-positive strains | ||||||||
| E. faecalis ATCC 29212 | 0.5 | 0.8 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | 0.5 |
| S. aureus ATCC 29213 | 0.25–1 | 0.4–0.8 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | 0.25 |
| Staphylococcus epidermidis DSM 28765 | ≤0.06 | 0.2–0.32 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | 0.06 |
| Streptococcus pneumoniae DSM 20566 | ≤0.03–0.125 | 0.8 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | ≤0.03 | 0.06 |
E. coli strain with the mentioned mutations in the gyrA subunit.
Microbiological Profile of CN-CC-861
Because of the superior efficacy of alkyne CN-CC-861 (13), the derivative was further evaluated against multidrug-resistant clinical strains (Tables 4 and S5). The results demonstrate that CN-CC-861 (13) surpassed the activity of CN-DM-861 (4) against most of the tested strains. The clinical isolate panels disclosed reduced median MICs of CN-DM-861 (4) and ciprofloxacin toward Gram-positive Enterococci and Staphylococci, whereas the antibacterial activity of CN-CC-861 (13) remained high against these strains. A high activity of CN-CC-861 (13) was also shown against several Acinetobacter strains that were less susceptible to CN-DM-861 (4). In contrast, moderate median MIC’s were observed for K. pneumoniae and P. aeruginosa. For P. aeruginosa, the pairwise testing of PA14 wild type versus the Pa14ΔmexAB strain demonstrated a significant increase of activity of many cystobactamid analogues in the efflux-deficient mutant. This implied contribution of efflux to the intrinsic resistance of P. aeruginosa. A hint for a cause behind the moderate activity against K. pneumoniae was obtained by a heptose lacking LPS mutant (K. pneumoniae KP10581; waaC::Tn30), which was susceptible (MIC = 0.25 μg/mL). This implies that the penetration of the outer membrane was a key issue hampering activity for K. pneumoniae.
Table 4. Antibiotic Activities of CN-DM-861 (4), CN-CC-861 (13) and Ciprofloxacin (CIP) against Susceptible and Multidrug-Resistant Bacteriaa.
| median
MIC (μg/mL) |
|||||
|---|---|---|---|---|---|
| genus | species | CN-DM-861 (4) | CN-CC-861 (13) | CIP | resistance phenotypes |
| Acinetobacter 8 strains | Acinetobacter johnsonii, Acinetobacter lwoffii, Acinetobacter ursingii and A. baumannii | 4 | 0.06–0.125 | 0.1–0.2 | 4× susceptible, 4× 3MRGN |
| Klebsiella 5 strains | Klebsiella oxytoca | 0.5 | 0.125 | 0.0125 | 2× susceptible, 2× 2MRGN, 1× 4MRGN |
| Klebsiella 5 strains | K. pneumoniae | 2 | 8 | >6.4 | 1× susceptible, 1× 2MRGN, 1× 3MRGN, 2× 4MRGN |
| Pseudomonas 11 strains | P. aeruginosa | >64 | 8 | 3.2 | 5× 3MRGN, 6× 4MRGN |
| Enterococcus 17 strains | not specified | 64 | 1 | 64 | 17× VRE |
| Staphylococcus 10 strains | S. aureus | 4–8 | 0.125 | >6.4 | 5× MSSA, 5× MRSA |
2/3/4 MRGN: Multidrug-resistant Gram-negative bacteria with resistance against 2, 3, or 4 of the 4 antibiotic groups acylureidopenicillins, third-generation cephalosporins, carbapenems or fluoroquinolones; example 3MRGN: Resistance against 3 of the 4 antibiotic groups. VRE: Vancomycin-resistant Enterococcus. MSSA: Methicillin-susceptible Staphylococcus aureus. MRSA: Methicillin-resistant Staphylococcus aureus.
In order to assess the cidality of CN-CC-861 (13), time-kill curve experiments were performed with A. baumannii, K. pneumoniae and E. coli. CN-CC-861 (13) exerted a strong and rapid bactericidal activity that led to a reduction of colony-forming units (cfu) by three log10 units within 1–2 h in all three species at 4× MIC (Figure 5). A regrowth was observed to a stronger extent for K. pneumoniae (4× MIC or lower) than for E. coli (2× MIC or lower) or A. baumannii (1× MIC only). The generation of resistance to CN-CC-861 (13) was studied further in six different strains of A. baumannii, P. aeruginosa and E. coli (Table 5). At 4× MIC, resistant clones formed with frequencies between 4 × 10–8 and 2 × 10–10. Such frequencies of resistance were comparable but, on average, slightly higher than those found for ciprofloxacin. The MICs of resistant mutants were determined and turned out to decrease in the order E. coli (32–200 μg/mL) ≈ P. aeruginosa (8–256 μg/mL) > A. baumannii (2–25 μg/mL) (Table 5). These pronounced differences imply that there were no uniform mechanisms of resistance against CN-CC-861 (13), but strain dependent ones.
Figure 5.

Time-dependent killing of bacteria by CN-CC-861 (13). Time-kill curves for A. baumannii CIP-105742 (MIC = 0.03 μg/mL), E. coli ATCC-25922 (MIC = 0.016 μg/mL) and K. pneumoniae ATCC-13883 (MIC = 2 μg/mL). LOD = Limit of detection.
Table 5. Frequencies of Resistance (FoR) and MIC Shifts Associated with Resistant Mutants for Six Bacterial Strains against CN-CC-861 and Ciprofloxacin.
| FoR
(4× MIC) |
MIC
shift CysR mutants vs WTa |
|||
|---|---|---|---|---|
| CN-CC-861 (13) | CIP | CN-CC-861 (13) | CIP | |
| A. baumannii DSM 30008 | 2 × 10–10 | 7 × 10–10 | 6–25 | 1–2 |
| A. baumannii CIP105742 | 1 × 10–8 | 1 × 10–9 | <2 | 1–4 |
| E. coli MG1655/K12 | 4 × 10–8 | 2 × 10–10 | 32–128 | 0.5–4 |
| E. coli ATCC 25922 ΔtolC | 4 × 10–8 | 3 × 10–10 | 50–200 | 1–2 |
| P. aeruginosa Pa14ΔmexAB | 3 × 10–9 | 2 × 10–8 | >256 | 20–160 |
| P. aeruginosa PAO750 (ΔmexAB-oprM, ΔmexCD-oprJ, ΔmexEF-oprN, ΔmexJK, ΔmexXY) | 6 × 10–9 | 1 × 10–11 | 8–32 | 0.5–8 |
n = 9 for CN-CC-861 and n = 18 for CIP, with n being the number of clones tested in the MIC shift determination.
Physicochemical Profiling of Selected Cystobactamids
We first assessed experimental log D7.4 values: they were between 1.35 and 2.47, an expected range for antibacterials active against Gram-positive bacteria but rather high for molecules active against Gram-negatives (Table 6).24 Next, the thermodynamic solubility at pH 7.4 and pH 9.0 was measured. For the profiling, we selected analogues with promising antimicrobial activity. Additionally, we included cystobactamids with hydrophilic side chains to investigate their influence on physicochemical parameters, especially solubility and plasma protein binding. Several compounds with unpolar side chains, including CN-CC-861 (13), alkene 21 as well as alkanes 22 and 23, were neither detectable at pH 7.4 nor at pH 9.0. Surprisingly, the amines in 26 and 27 did not contribute to a higher solubility, presumably due to their zwitterionic character in the investigated pH range. On the other hand, solubility was clearly enhanced by polar side chains as the morpholine 20 as well as alcohol 41.
Table 6. Thermodynamic Solubility, Plasma Protein Binding and log D7.4 Values of Selected Cystobactamid Derivatives.
| aq solubility [μg/mL] |
plasma
protein binding [%] |
||||
|---|---|---|---|---|---|
| compound | pH 7.4 | pH 9.0 | Ma | Hb | log D7.4 |
| CN-CC-861 (13) | - | - | 100.0 ± 0.0 | 100.0 ± 0.0 | 2.26 |
| 26 | <1 | 14 | 99.89 ± 0.1 | 100.0 ± 0.0 | 1.41 |
| 24 | <1 | 270 | 100.0 ± 0.0 | 98.17 ± 2.7 | n.d. |
| 27 | <1 | 22 | n.d. | n.d. | 1.35 |
| 38 | <1 | 10 | 100.0 ± 0.0 | 100.0 ± 0.0 | 2.47 |
| 39 | <1 | 11 | 100.0 ± 0.0 | 100.0 ± 0.0 | n.d. |
| 20 | 37 | 980 | n.d. | n.d. | n.d. |
| 16 | <1 | 24 | 99.54 ± 0.7 | 99.95 ± 0.0 | 1.56 |
| 41 | 163 | 311 | 94.36 ± 2.0 | 99.39 ± 0.2 | n.d. |
| 42 | 1 | 46 | 86.43 ± 2.4 | 100.0 ± 0.0 | n.d. |
| CN-DM-861 (4) | <1 | 56 | 98.39 ± 2.3 | 99.71 ± 0.18 | 1.49 |
Mouse.
Human. n.d.: not determined.
Finally, the mouse and human plasma protein binding (PPB) was determined (Table 6). In line with the low solubility, the lipophilic amino acid analogues (e.g., CN-CC-861) increased the PPB to 100%, when compared to the PPB of 98.4% in mouse plasma for CN-DM-861 (4). Sulfonamide 16 retained high binding, while alcohol 41 as well as its diastereomer 42 showed a decreased bound fraction, although not for human plasma protein.
In Vivo Efficacy Study of CN-CC-861
Due to its high in vitro activity, CN-CC-861 (13) was profiled in an in vivo mouse model. The cytotoxicity assay indicated a low cytotoxicity against the HepG2 and CHO cell lines with IC50’s of 30 and ≥100 μM, respectively. The murine plasma stability was sufficient, with around 80% of the parent compound remaining after 4 h. Moreover, dosing 20 mg/kg four times at an interval of 6 h (q6h), summing up to a daily dose of 80 mg/kg, was well tolerated in healthy mice. To determine the in vivo efficacy, a neutropenic thigh infection model with E. coli ATCC 25922 was carried out with CN-CC-861 (13) (MIC = 0.03 μg/mL in dosing solution) and the lead structure CN-DM-861 (4) (MIC = 0.125 μg/mL in dosing solution) as a reference (Figure 6).
Figure 6.

In vivo efficacy study of cystobactamids and ciprofloxacin in a neutropenic mice thigh infection model (n = 5/group; n = 4 in pretreatment group) with E. coli ATCC 25922 (5 × 105 cfu/thigh). Dosing q6h i.v. starting 1 h after infection. LOD = Limit of detection.
The cystobactamids were administered to male CD-1 mice q6h starting with the first injection 1 h post infection. The animals were divided into three groups, and single doses of 5, 10, and 12.5/20 mg/kg were applied, respectively. Twenty-five hours after infection, bacterial burden in thigh was determined (Figure 6). However, as animals reached the human end point, vehicle-treated group was terminated 20 h and CN-CC-861 (13) groups 21 h post infection. While the two lower doses were not efficacious, a reduction of bacterial load by two log10 units compared to the vehicle control was observed for the highest dose of 80 mg/kg/day; here, the bacterial load reduction was close to stasis. However, compared to the previously published CN-DM-861 (4), where a reduction of the bacterial load was observed in all dosing groups,16 similar as observed in this study, albeit different end points, CN-CC-861 (13) did not exhibit similar efficacy. We attributed this lack of efficacy to the poor pharmacokinetic properties of CN-CC-861 (13) and in particular to the low solubility and the strong mouse protein binding (see above) that prevented the high in vitro potency of CN-CC-861 to fully translate into high in vivo efficacy.
Structure–Activity Relationships
For the determination of the structure–activity relationships, the broad-spectrum antibiotic coverage was the most important metric in this work. The functional inhibition data on E. coli gyrase were secondary parameters to investigate the inhibitor binding. While first-generation natural cystobactamids inhibited gyrase much stronger than topoisomerase (TI) IV, we note that the compounds described herein possess a more balanced gyrase vs TI IV profile, and that the absolute potency exceeded that of ciprofloxacin and other references (Tables 1 and 2). This might contribute to their superior antibiotic potency. However, target inhibition was tested only for the E. coli sequence, and because the values are species-dependent, we did not attempt to correlate them with the broad-spectrum antibiotic profile and therefore excluded them in the following SAR discussion.
Various residues in the central α-amino acid were tolerated, and an amide in the side chain was not mandatory for antibacterial activity (Figure 7). Positions 3–4 of the amino acid are ideal for functionalities such as a π-system or polar, neutral or cationic groups, while elongated side chains were less potent. Alkyne CN-CC-861 (13) showed superior in vitro activity, spectrum and resistance-breaking properties compared to other analogues. Basic amine side chains were tolerated, implying that the bacterial membrane allowed for the passage of zwitterionic compounds. The configuration of the α-carbon had a strong impact on the activity. As demonstrated by the pairs 17 and 18, 32 and 33, or 38 and 39, l-amino acids were preferred over the less active d-amino acids. This preference for l-amino acids was affirmed by docking studies of 17 and 18 in the published binding pocket of Albi-1.22 Herein, the rigidification to a six-membered system in 17 stabilized the bioactive conformation for E. coli gyrase. However, the target-based differences were not linked to an overall increased antimicrobial activity. Possible explanations include the bioactive conformation being different from the permeable conformation or involvement of additional targets in the mechanism of action.
Figure 7.

Structure–activity relationships at the central amino acid.
Conclusions
Through a series of structural modifications, the SAR at the central amino acid of cystobactamids could be elucidated. Several novel analogues with high broad-spectrum activity were discovered and characterized, including the alkyne derivative CN-CC-861 (13), that exhibited resistance-breaking properties. However, the modest performance of CN-CC-861 (13) in the in vivo thigh infection model indicates that future studies must focus on the physiochemical and pharmacokinetic optimization of cystobactamids. In summary, our findings indicate that the cystobactamid scaffold provides a remarkable basis for the development of broad-spectrum antibiotics.
Experimental Section
Chemistry
All nonaqueous reactions were carried out in dried glassware in dry solvents under inert conditions unless otherwise noted. Light sensitive reactions were carried out under light exclusion. Commercially available reagents were used without prior purification. Dry solvents (MeCN, DMF, Et2O) were taken from a MBraun solvent purification system. THF was freshly distilled over sodium (benzophenone as indicator). Petroleum ether was distilled (60 °C). Et3N was freshly distilled over KOH. Other commercially available (dry) solvents were purchased from Merck or Acros Organics.
For reactions under microwave irradiation a CEM Discover S-Class was used with a power maximum of 300 W.
Chromatographic separations by flash chromatography were carried out on a Grace Reveleris X2 (Büchi) with FlashPure EcoFlex cartridges (Büchi) or conducted with the flash purification system Sepacore (Büchi) or Biotage SP using prepacked cartridges (puriFlash by Interchim or chromobond by Macherey-Nagel). A Pure C-850 FlashPrep (Büchi) with FlashPure EcoFlex cartridges (Büchi) was utilized for reversed phase flash chromatography. For manual columns silica gel 60 0.04–0.063 mm; 230–400 mesh (Macherey-Nagel) was used.
Purifications by high performance liquid chromatography (HPLC) were performed by a Thermo Scientific Dionex UltiMate 3000 system with a Phenomenex Luna C18 column (250 mm × 21.2 mm, 5 μm) column under basic (10 mM NH4HCO3) or acidic (0.1% formic acid or acetic acid) conditions. Alternatively, semipreparative HPLC was performed by using a Waters Alliance 2695 HPLC-system with a 996 diode array detector (λ = 200–350 nm) and a Macherey-Nagel Nucleodur C18 ISIS column (5 μm, 250 mm, diameter = 8 mm). Mass detection was conducted with a Waters Quattro micro API mass spectrometer in negative ionization mode.
Thin-layer chromatography analytics were carried out on precoated silica gel 60 F254 plates (Merck) or on Macherey-Nagel aluminum plates coated with silica gel 60 F245. The sample was detected by ultraviolet (UV) light at 254 or 366 nm. Non-UV-absorbent samples were stained by a cerium-ammonium-molybdate, potassium permanganate, ninhydrin, vanillin and anisaldehyde solution.
NMR spectra were measured on different instruments, i.e., Bruker Advance-III HD 500 MHz and Bruker Advance-III HD 700 MHz spectrometer, Bruker Ascend 600 MHz with Avance Neo console, Ultrashield 500 MHz with Avance-III HD console, Ascend 400 MHz with Avance- III console, Ascend 400 MHz with Avance-III HD console or Ultrashield 400 MHz with Avance-I console. The chemical shifts for 1H, 13C and 19F spectra are reported in ppm at a temperature of 300 K. 19F spectra lack an internal reference. One dimensional 13C were measured with 1H decoupling. Multiplicities are specified with following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, hept./sept. = septet, oct = octet, m = multiplet, br = broad signal and combinations thereof.
HPLC-MS reaction controls were carried out by Agilent 1260 Infinity II LC connected to an Agilent 6130 (quadrupole MS) in ESI mode by a Phenomenex Gemini NX-C18 (50 mm × 2 mm, 3 μm) column. The gradient went from 0–100% acetonitrile to water with 0.1% formic acid in both solvents over three minutes at a flow rate of 1.5 mL/min. All isolated compounds were analyzed by HPLC to confirm a purity of ≥95%.
High-resolution mass spectra were measured at a Bruker maXis HD spectrometer in positive or negative ESI mode or at a Micromass LCT with lock-spray unit and injection via loop modus in a Waters (Alliance 2695) HPLC device. Alternatively, a Micromass Q-TOF was used in combination with a Waters Aquity UPLC device. The ionization occurred through electron spray ionization. Calculated and found masses are reported.
The specific optical rotation [α] was measured with a polarimeter type 341 from PerkinElmer at λ = 589.3 nm (sodium D line) in a 10 cm quartz cuvette. It is given in 10–1 cm2 g–1. The concentration c is given in 10 mg mL–1.
A Christ α 1–4 LCSbasic was used for the lyophilization of the products after purification by HPLC.
General Procedures
General Procedure 1: Amide Coupling with T3P
0.18 mmol of amine (1.00 equiv) and 0.27 mmol of the desired acid (1.50 equiv) were added to a dry flask and further dried under high vacuum. 0.61 mmol dry pyridine (3.40 equiv) and 0.4 mL dry EtOAc were added under nitrogen atmosphere. The reaction mixture was cooled down to 0 °C. 0.25 mL T3P solution (50 wt % in EtOAc, 0.42 mmol, 2.30 equiv) was added very slowly while keeping the temperature below 0 °C. The reaction was stirred at 0 °C overnight and controlled by liquid chromatography–mass spectrometry (LCMS). After completion, the reaction was quenched with 4 mL 1 M HCl and 12 mL brine and extracted with 3 × 6 mL EtOAc. The combined organic phases were washed with a sat. NaHCO3 solution and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography with petroleum ether and EtOAc or used without further purification.
General Procedure 2: Fmoc Deprotection
0.09 mmol (1.00 equiv) of the crude carbamate was dissolved in 0.8 mL ACN and 0.3 mL diethylamine (2.9 mmol, 31.5 equiv) at 0 °C and stirred for 1 h. The reaction was controlled by LCMS. The solvent was evaporated under reduced pressure and coevaporated with ACN 3 times. Optionally, the crude product was purified by RP flash or RP HPLC with ACN and water mixed with 0.1% HCOOH.
General Procedure 3: Amide Coupling with HATU
82.6 μmol of the desired carboxylic acid (1.20 equiv) and 78.9 μmol HATU (1.20 equiv) were added to a separate flask and dried under high vacuum. 0.5 mL dry DMF and 38 μL DIPEA (3.00 equiv) were added under nitrogen atmosphere and the reaction was stirred for 30 min. The solution was added to the amine or amine hydrochloride and stirred at 0 °C. The reaction was controlled by LCMS. After completion, the reaction was quenched with 8 mL of 0.1 M HCl and 4 mL brine. The inorganic layer was extracted with 3 × 6 mL of EtOAc. The organic phases were combined and washed with 2 × 5 mL brine. The solvent was removed under reduced pressure. The crude product was used without further purification.
General Procedure 4: Amide Coupling with HATU
65.5 μmol of the desired amine (hydrochloride) (1.00 equiv), 79.0 μmol HATU (1.20 equiv) and 79.0 μmol of the desired carboxylic acid (1.20 equiv) were added to a dry flask and further dried under high vacuum. 0.4 mL dry DMF and 35 μL DIPEA (3.1 equiv) were added under nitrogen atmosphere at 0 °C. The solution was stirred at 0 °C and controlled by LCMS. After completion, the reaction was quenched with 6 mL of 0.1 M HCl and 10 mL brine. The inorganic layer was extracted with 3 × 4 mL of EtOAc. The organic phases were combined and washed with 2 × 4 mL brine. The crude product was used without further purification.
General Procedure 5: Allyl Deprotection with Palladium and Phenylsilane
65.5 μmol of the desired allyl protected alcohol and 198 μmol phenylsilane (3.00 equiv) were added to a dry flask under nitrogen atmosphere. 1.2 mL dry THF and 6.5 μmol tetrakis(triphenylphosphine)palladium(0) (0.10 equiv) were added and the mixture was stirred for 3 h at rt. The reaction was controlled by LCMS. After completion, the solvent was removed under reduced pressure. Three ml 0.1 M HCl and 10 mL brine were added to the residue and extracted with 3 × 4 mL EtOAc. The combined organic phases purified by flash chromatography with petroleum ether and EtOAc mixed with 2% acetic acid.
General Procedure 6: Allyl Deprotection with Palladium and Aniline
65.5 μmol of the desired allyl protected alcohol and 198 μmol aniline (3.00 equiv) were added to a dry flask under nitrogen atmosphere. 1.2 mL dry THF and 6.5 μmol tetrakis(triphenylphosphine)palladium(0) (0.10 equiv) were added and the mixture was stirred for 3 h at rt. The reaction was controlled by LCMS. After completion, the solvent was removed under reduced pressure. Three ml 0.1 M HCl and 10 mL brine were added to the residue and extracted with 3 × 4 mL EtOAc. The combined organic phases were purified by flash chromatography with petroleum ether and EtOAc mixed with 2% acetic acid or with CH2Cl2 and methanol.
General Procedure 7: tert-Butyl Ester Deprotection with TFA
65.5 μmol of the desired tert-butyl protected acid (1.00 equiv) was added to a dry flask and further dried under high vacuum. 0.5 mL dry CH2Cl2 and, if necessary, 14 μL anisole (2 equiv) were added under nitrogen atmosphere and the solution was cooled down to 0 °C. 0.24 mL of trifluoroacetic acid (3.1 mmol, 54 equiv) was added under nitrogen atmosphere. The solution was stirred for 3 h at 0 °C and controlled by LCMS. After completion, the solvent was removed under reduced pressure. The residue was coevaporated with CH2Cl2 twice. The crude product was purified by RP-HPLC.
General Procedure 8: Amide Coupling CDE to Central AA with EEDQ
0.09 mmol of the desired aniline (1 equiv) and 0.14 mmol of the desired carboxylic acid (1.5 equiv) were added to a dry flask and were further dried under high vacuum. 0.15 mL dry CH2Cl2 was added under nitrogen atmosphere and the mixture was cooled down to 0 °C. 33.0 mg EEDQ (0.13 mmol, 1.5 equiv) dissolved in 0.15 mL dry CH2Cl2 was added to the stirring solution. The reaction was stirred at 0 °C for 30 min and slowly warmed up to rt, afterward. The reaction was controlled by LCMS. After completion, the reaction was quenched with 2 mL 1 M HCl and 6 mL brine and extracted with 3 × 3 mL CH2Cl2. The combined organic phases were washed with brine and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography.
General Procedure 9: Amide Coupling with Acid Chloride
0.11 mmol of the desired Fmoc-protected amino acid (1 equiv), 0.4 dry DCM and one drop of dry DMF were added to a dry flask under nitrogen atmosphere. The mixture was cooled to 0 °C and 17 μL oxalyl chloride (0.2 mmol, 1.4 equiv) was slowly added to the stirring mixture. The reaction was controlled by quenching a sample in methanol and running a TLC with petroleum ether and ethyl acetate. After completion, the solvent was evaporated under reduced pressure. The crude product was dried under high vacuum overnight and directly used without further purification.
0.09 mmol of aniline 8 (1 equiv) and 0.14 mmol of the preformed acid chloride (1.5 equiv) were added to a dry flask and further dried under high vacuum. 1.0 mL dry DCM was added under nitrogen atmosphere and the mixture was cooled down to 0 °C. Twenty-three μL pyridine (0.29 mmol, 3.1 equiv) was added under nitrogen atmosphere and the solution was kept at 0 °C for the whole reaction. The reaction was controlled over LCMS. After completion, the reaction was quenched with 2 mL 1 M HCl and 4 mL water. The aqueous phase was extracted with 3 × 4 mL of ethyl acetate. The organic phases were combined and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography with petroleum ether and ethyl acetate.
General Procedure 10: Fmoc Deprotection and Amide Coupling with HATU
68.0 μmol of the desired Fmoc protected central amino acid (1 equiv) was dissolved in 0.4 mL acetonitrile and 105 μL diethylamine (1 mmol, 15.1 equiv) at 0 °C and stirred for 1 h. The solvent was evaporated under reduced pressure. One ml acetonitrile was added to the residue and the solvent was removed under reduced pressure again. This was repeated twice. The crude residue was dried under high vacuum overnight [residue 1].
82.6 μmol of the desired fragment AB (1.2 equiv) and 78.9 μmol HATU (1.2 equiv) were added to a separate flask and dried under high vacuum. 0.5 mL dry DMF and 38 μL DIPEA (3 equiv) were added under nitrogen atmosphere and the reaction was stirred for 30 min. The solution was added to the residue [1] and stirred at 0 °C. The reaction was controlled over LCMS. After completion, the reaction was quenched with 8 mL of 0.1 M HCl and 4 mL brine. The inorganic layer was extracted with 3 × 6 mL of ethyl acetate. The organic phases were combined and washed with 2 × 5 mL brine. The solvent was removed under reduced pressure. The crude product was used without further purification.
General Procedure 11: Amide Coupling CDE to Central AA with IIDQ
0.09 mmol of the desired aniline (1 equiv) and 0.14 mmol of the desired carboxylic acid (1.5 equiv) were added to a dry flask and were further dried under high vacuum. 0.15 mL dry DCM was added under nitrogen atmosphere and the mixture was cooled down to 0 °C. 40 μL IIDQ (0.13 mmol, 1.5 equiv) dissolved in 0.15 mL dry DCM was added to the stirring solution. The reaction was kept at 0 °C for 30 min and slowly allowed to reach room temperature afterward. The reaction was stirred overnight and controlled over LCMS. The reaction was quenched with 2 mL 1 M HCl and 6 mL brine and extracted with 3 × 3 mL DCM. The combined organic phases were washed with brine and the solvent was removed under reduced pressure. The crude product was used in the next reaction.
tert-Butyl 4-[4-(4-Aminobenzamido)-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido]benzoate (8)
The compound was prepared according to the established literature procedure; see ref (20).
4-(4-Cyanobenzamido)benzoic Acid (10)
The compound was prepared according to the established literature procedure; see Dong, Y. et al.; Bioorg. Med. Chem. Lett.2014, 24, 3, 944–948.
tert-Butyl 4-(4-{4-[(2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)pent-4-ynamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (9)
After a suspension of the aniline 8 (1.53 g, 2.81 mmol, 1.00 equiv) in dry EtOAc (20.5 mL) was cooled down to 0 °C, (2S)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)pent-4-ynoic acid (1.32 g, 3.94 mmol, 1.40 equiv) and dry pyridine (770 μL, 9.56 mmol, 3.40 equiv) were added. T3P solution (50% in EtOAc, 3.35 mL, 5.62 mmol, 2.00 equiv) was added dropwise over 15 min and the resulting solution was stirred for 2.5 h at 0 °C. The reaction mixture was diluted with aq HCl (1 M, 30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with NaHCO3 solution (30 mL) and brine (30 mL), dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by flash chromatography (petroleum ether/EtOAc). Colorless solid, 2.11 g (87%).
1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.16 (s, 1H), 8.74 (s, 1H), 8.49 (d, 1H, J = 8.9 Hz), 8.07 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.8 Hz), 7.89 (d, 2H, J = 8.7 Hz), 7.77 (d, 2H, J = 7.5 Hz), 7.73 (d, 2H, J = 8.8 Hz), 7.69 (d, 2H, J = 8.8 Hz), 7.59 (d, 2H, J = 7.4 Hz), 7.40 (t, 2H, J = 7.5 Hz), 7.30 (t, 2H, J = 7.4 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 17.1 Hz), 5.64–5.57 (m, 1H), 5.49 (dq, 1H, J = 1.4 Hz, 17.1 Hz), 5.41 (dq, 1H, J = 1.1 Hz, 10.4 Hz), 4.75 (hept., 1H, J = 6.2 Hz), 4.69 (dt, 2H, J = 1.2 Hz, 5.9 Hz), 4.57–4.46 (m, 3H), 4.25 (t, 1H, J = 6.7 Hz), 2.94–2.68 (m, 2H), 2.17 (t, 1H, J = 2.6 Hz), 1.60 (s, 9H), 1.38 (d, 6H, J = 6.2 Hz).13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 168.5, 165.6, 164.3, 162.8, 156.7, 149.4, 143.6, 142.3, 141.5, 141.0, 139.1, 137.6, 132.3, 130.8, 130.4, 128.3, 128.0, 127.6, 127.5, 127.3, 125.1, 121.7, 120.3, 120.2, 120.0, 119.2, 115.8, 81.0, 79.0, 76.9, 75.1, 72.5, 67.7, 54.2, 47.2, 29.8, 28.4, 23.0.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}pent-4-ynamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (13)
Step 1: A solution of the Fmoc protected amine 9 (1.93 g, 2.24 mmol, 1.00 equiv) in ACN (9.7 mL) was cooled down to 0 °C and diethylamine (3.64 mL, 35.2 mmol, 15.75 equiv) was added. After stirring at 0 °C was continued for 1 h, all volatiles were removed under reduced pressure. The crude product was used without further purification. Step 2: The amino acid derivative (54.5 μmol) was coupled with the carboxylic acid 10 using general procedure 3. Step 3 and 4: The product was obtained by deprotection with general procedures 6 and 7. Yellowish solid, 10 mg (23% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.80 (br s, 1H), 12.30 (s, 1H), 10.71 (s, 1H), 10.58 (s, 1H), 9.40 (s, 1H), 8.77 (d, 1H, J = 7.5 Hz), 8.13 (d, 2H, J = 8.4 Hz), 8.05 (d, 2H, J = 8.4 Hz), 7.98–7.95 (m, 6H), 7.90 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 3H), 7.83 (d, 2H, J = 8.8 Hz), 7.70 (d, 1H, J = 8.8 Hz), 4.81 (dd, 1H, J = 7.6 Hz, 14.7 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 2.94 (t, 1H, J = 2.6 Hz), 2.79 (dddd, 2H, J = 2.6 Hz, 7.4 Hz, 11.1 Hz, 16.8 Hz), 1.27 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 169.7, 168.5, 166.9, 166.0, 164.5, 164.2, 154.2, 142.2, 142.0, 141.7, 138.7, 137.0, 136.4, 132.5, 130.2, 128.9, 128.6, 128.5, 128.4, 128.4, 126.2, 122.8, 120.7, 119.5, 119.0, 118.3, 114.0, 112.1, 80.6, 74.8, 73.2, 53.5, 22.3, 21.4. HRMS (ESI) calcd 793.2622 [M + H+], 793.2617 found. HPLC purity 98.1%.
tert-Butyl 4-(4-{4-[(2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-(1,3-thiazol-4-yl)propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (46)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-(1,3-thiazol-4-yl)propanoic acid using general procedure 9. Yellow orange solid, 62 mg (74%).1H NMR (700 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.53 (s, 1H), 8.88 (s, 1H), 8.73 (s, 1H), 8.49 (d, 1H, J = 8.9 Hz), 8.06 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.7 Hz), 7.87 (d, 2H, J = 8.7 Hz), 7.79–7.75 (m, 2H), 7.73 (d, 2H, J = 8.7 Hz), 7.69 (d, 2H, J = 7.9 Hz), 7.61–7.57 (m, 2H), 7.43–7.38 (m, 2H), 7.33–7.28 (m, 2H), 7.20 (s, 1H), 6.51 (d, 1H, J = 5.6 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 4.77–4.72 (m, 2H), 4.69 (d, 2H, J = 5.9 Hz), 4.49–4.39 (m, 2H), 4.23 (t, 1H, J = 6.6 Hz), 3.46–3.34 (m, 2H), 1.60 (s, 9H), 1.38 (d, 6H, J = 6.1 Hz).13C NMR (176 MHz, CDCl3, 300 K): δ (ppm) = 169.6, 165.6, 164.4, 162.8, 156.7, 153.6, 152.6, 149.4, 143.8, 142.3, 141.5, 141.5 141.5, 139.1, 137.7, 132.3, 130.8, 130.0, 128.2, 128.0, 127.6, 127.4, 127.3, 125.2, 121.7, 120.2, 120.2, 119.8, 119.2, 116.5, 115.8, 80.9, 76.9, 75.1, 67.5, 55.3, 47.3, 33.4, 28.4, 23.0. HRMS (ESI) calcd 922.3486 [M + H+], 922.3456 found. HPLC purity 95.9%.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-(1,3-thiazol-4-yl)propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (14)
The Fmoc protected amino acid 46 (67.5 μmol) was deprotected and coupled with carboxylic acid 10 using general procedure 10. The product was obtained by deprotection with general procedures 5 and 7. White solid, 9 mg (16% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.81 (br s, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.62 (br s, 1H), 10.56 (s, 1H), 9.40 (s, 1H), 9.06 (d, 1H, J = 1.9 Hz), 8.73 (d, 1H, J = 7.6 Hz), 8.12 (d, 2H, J = 8.5 Hz), 8.04 (d, 2H, J = 8.5 Hz), 7.98–7.94 (m, 4H), 7.90–7.87 (m, 4H), 7.87–7.84 (m, 3H), 7.82 (d, 2H, J = 8.8 Hz), 7.71 (d, 1H, J = 8.8 Hz), 7.49 (d, 1H, J = 1.9 Hz), 5.02 (dd, 1H, J = 7.8 Hz, 14.5 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 3.40–3.34 (m, 2H), 1.27 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 170.8, 168.5, 166.9, 165.9, 164.5, 164.2, 154.2, 153.8, 153.1, 142.4, 142.0, 141.6, 138.7, 137.0, 136.3, 132.5, 130.2, 129.1, 128.6, 128.4, 128.3, 126.3, 122.8, 120.7, 119.5, 119.0, 118.3, 115.9, 114.0, 112.4, 112.1, 74.8, 54.3, 32.9, 22.3. HRMS (ESI) calcd 852.2452 [M + H+], 852.2448 found. HPLC purity 95.7%.
tert-Butyl (2S)-3-(Dimethylcarbamoyl)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoate (49)
200 mg (3S)-4-(tert-butoxy)-3-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)-4-oxobutanoic acid (0.49 mmol, 1 equiv) and 185 mg HATU (0.49 mmol, 1.0 equiv) were added to a dry flask and further dried under high vacuum. The flask was cooled to 0 °C. 3.5 mL dry DMF and 0.09 mL DIPEA (66.8 mg, 0.52 mmol, 1.1 equiv) were added under nitrogen atmosphere. The reaction was stirred for 30 min at 0 °C. 0.27 mL 2 M dimethylamine in THF (0.54 mmol, 1.1 equiv) was added to the stirring solution. The mixture was stirred at 0 °C for the whole reaction. The reaction was controlled over TLC. After completion, 8 mL of 0.1 M HCl and 6 mL brine were added. The aqueous layer was extracted with 3 × 5 mL of ethyl acetate. The organic phases were combined and washed with 2 × 5 mL brine. The crude product was purified by flash chromatography (petroleum ether/ethyl acetate) and used directly in the next reaction. 239 mg (crude). HRMS (ESI) calcd 439.2233 [M + H+], 439.2223 found.
(2S)-3-(Dimethylcarbamoyl)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoic Acid (50)
238 mg ester 49 (0.54 mmol, 1 equiv) was dissolved in 6 mL dry DCM under nitrogen atmosphere. 0.77 mL TFA (1147 mg, 10.1 mmol, 22.0 equiv) was added to the stirring mixture. The reaction was controlled over LCMS. After completion, the solvent was removed under reduced pressure. The excess of TFA was removed by coevaporation with DCM. 166 mg (89% over 2 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 7.89 (d, 2H, J = 7.5 Hz), 7.71 (d, 2H, J = 7.5 Hz), 7.42 (t, 2H, J = 7.5 Hz), 7.36 (d, 1H, J = 8.4 Hz), 7.33 (t, 2H, J = 7.4 Hz), 4.42–4.37 (m, 1H), 4.29 (dd, 2H, J = 3.0 Hz, 7.0 Hz), 4.22 (t, 1H, J = 7.0 Hz), 2.94 (s, 3H), 2.82 (s, 3H), 2.77 (dd, 1H, J = 7.3 Hz, 16.4 Hz), 2.72–2.69 (m, 1H). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 173.2, 169.2, 155.8, 143.8, 140.7, 127.6, 127.1, 125.3, 120.1, 65.7, 50.5, 46.6, 38.3, 36.6, 34.9, 34.6. HRMS (ESI) calcd 383.1607 [M + H+], 383.1601 found. HPLC purity 95.1%.
tert-Butyl 4-(4-{4-[(2S)-2-Amino-3-(dimethylcarbamoyl)propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (51)
Amine 8 (0.09 mmol) was coupled with carboxylic acid 50 using general procedure 9 and deprotected using general procedure 2. Faint yellow solid, 18 mg (29% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.16 (s, 1H), 8.73 (s, 1H), 8.48 (d, 1H, J = 8.9 Hz), 8.05 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.8 Hz), 7.87 (d, 2H, J = 8.8 Hz), 7.79 (d, 2H, J = 8.5 Hz), 7.72 (d, 2H, J = 8.8 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (ddd, 1H, J = 1.4 Hz, 2.8 Hz, 17.1 Hz), 5.40 (ddd, 1H, J = 1.0 Hz, 2.1 Hz, 10.4 Hz), 4.74 (hept., 1H, J = 6.2 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.97 (s), 3.05–2.87 (m, 8H), 1.59 (s, 9H), 1.37 (d, 6H, J = 6.2 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 172.4, 170.8, 165.5, 164.5, 162.8, 149.4, 142.3, 141.7, 139.1, 137.8, 132.3, 130.8, 129.6, 128.2, 127.6, 127.4, 121.6, 120.1, 119.4, 119.1, 115.8, 80.9, 76.9, 75.1, 52.5, 37.5, 37.3, 35.6, 28.4, 22.9. HRMS (ESI) calcd 688.3346 [M + H+], 688.3345 found. HPLC purity 98.4%.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-(dimethylcarbamoyl)propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (15)
The amine 51 (25.6 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 5 and 7. White solid, 7 mg (34% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.83 (br s, 1H), 12.29 (s, 1H), 10.70 (s, 1H), 10.60 (s, 1H), 10.46 (s, 1H), 9.39 (s, 1H), 8.62 (d, 1H, J = 7.1 Hz), 8.13 (d, 2H, J = 8.4 Hz), 8.04 (d, 2H, J = 8.4 Hz), 7.97 (d, 2H, J = 8.7 Hz), 7.95 (d, 2H, J = 8.8 Hz), 7.93 (d, 2H, J = 8.8 Hz), 7.89 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 3H), 7.83 (d, 2H, J = 8.7 Hz), 7.70 (d, 1H, J = 8.8 Hz), 5.00 (quart., 1H, J = 6.9 Hz), 4.54 (hept., 1H, J = 6.1 Hz), 3.01 (s, 3H), 2.95–2.88 (m, 2H), 2.85 (s, 3H), 1.26 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 170.8, 169.2, 168.5, 166.9, 165.7, 164.4, 164.2, 154.1, 142.7, 142.0, 141.6, 138.7, 137.1, 136.3, 132.5, 130.2, 129.2, 128.6, 128.4, 128.3, 128.2, 126.3, 122.8, 120.7, 119.5, 118.9, 118.3, 114.0, 112.4, 112.2, 74.9, 51.6, 36.6, 34.9, 34.5, 22.3. HRMS (ESI) calcd 840.2993 [M + H+], 840.2988 found. HPLC purity 98.8%.
(2R)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-oxo-3-(prop-2-en-1-yloxy)propane-1-sulfonic Acid (54)
200.0 mg carboxylic acid 53 (0.51 mmol, 1 equiv) was added to a dry vial and further dried at high vacuum. Two ml allyl alcohol (1.71 g, 29.4 mmol, 57.6 equiv) was added and the vial was cooled down to 0 °C. 0.23 mL chlorotrimethylsilane (1.81 mmol, 3.6 equiv) was added under argon atmosphere and the reaction was slowly allowed to warm up to room temperature. The reaction was stirred overnight and controlled by LCMS. After completion, the solvent was removed under reduced pressure. The residue was coevaporated with n-heptane. The crude product was dried under high vacuum. White solid, 221 mg (quant.). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 7.89 (d, 2H, J = 7.5 Hz), 7.69 (dd, 2H, J = 2.4 Hz, 7.4 Hz), 7.55 (d, 1H, J = 7.0 Hz), 7.42 (t, 2H, J = 7.4 Hz), 7.33 (tdd, 2H, J = 1.1 Hz, 2.5 Hz, 7.4 Hz), 5.89 (ddt, 1H, J = 5.3 Hz, 10.6 Hz, 17.3 Hz), 5.31 (dq, 1H, J = 1.7 Hz, 17.3 Hz), 5.17 (dq, 1H, J = 1.5 Hz, 10.6 Hz), 4.56–4.53 (m, 2H), 4.37–4.33 (m, 1H), 4.29–4.23 (m, 3H), 2.87 (ddd, 2H, J = 5.7 Hz, 13.8 Hz, 18.2 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 170.6, 155.6, 143.7, 140.7, 132.6, 127.7, 127.1, 125.2, 120.1, 117.5, 65.9, 64.9, 51.5, 50.7, 46.6. HPLC purity 96.7%.
Prop-2-en-1-yl (2R)-3-{Bis[(4-methoxyphenyl)methyl]sulfamoyl}-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoate (55)
210 mg sulfonic acid 54 (0.49 mmol, 1 equiv) was added to a dry vial and further dried under high vacuum. 1.6 mL dry DCM and 2 drops of dry DMF were added under argon atmosphere. The mixture was cooled to 0 °C and 65 μL oxalyl chloride (0.76 mmol, 1.56 equiv) was slowly added to the stirring mixture. The reaction was warmed up to room temperature and stirred for 2.5 h. The solvent was concentrated under reduced pressure and the residue was dried under high vacuum overnight. 150 mg bis(4-methoxybenzyl)amine (0.58 mmol, 1.2 equiv) and 1.6 mL dry DCM were added under argon atmosphere and the mixture was cooled down to 0 °C. 0.14 mL dry triethylamine (1.0 mmol, 2.1 equiv) was added and stirring was continued at 0 °C. After completion, 60 μL acetic acid (63 mg, 1.1 mmol, 2.2 equiv) and 1 mL acetone were added and the solution was concentrated under reduced pressure. The product was directly purified by chromatography. White solid, 158.4 mg (49%). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 7.76 (d, 2H, J = 7.5 Hz), 7.62 (d, 2H, J = 6.8 Hz), 7.39 (t, 2H, J = 7.5 Hz), 7.31 (tdd, 2H, J = 0.9 Hz, 3.2 Hz, 7.4 Hz), 7.21 (d, 4H, J = 8.6 Hz), 6.88 (d, 4H, J = 8.7 Hz), 5.98–5.89 (m, 2H), 5.35 (dd, 1H, J = 1.3 Hz, 17.2 Hz), 5.26 (ddd, 1H, J = 1.1 Hz, 2.3 Hz, 10.5 Hz), 4.75–4.69 (m, 3H), 4.42–4.34 (m, 2H), 4.24 (s, 4H), 3.80 (s, 6H), 3.41 (ddd, 2H, J = 5.0 Hz, 14.2 Hz, 18.5 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 169.0, 159.6, 155.9, 143.9, 143.9, 141.4, 131.5, 130.3, 127.9, 127.3, 125.4, 120.1, 119.5, 114.4, 67.8, 67.1, 55.5, 54.0, 50.8, 49.2, 47.2.
(2R)-3-{Bis[(4-methoxyphenyl)methyl]sulfamoyl}-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoic Acid (56)
200 mg ester 55 (0.3 mmol, 1 equiv) was added to a dry vial and further dried under high vacuum. Two mL dry THF, 110 μL phenylsilane (0.9 mmol, 3 equiv) and 6.9 mg tetrakis(triphenylphosphine)palladium(0) (6 μmol, 0.02 equiv) were added under argon atmosphere. The reaction was stirred for 3 h at room temperature and controlled over TLC. After completion, 0.53 mL saturated NaHCO3 solution (0.6 mmol, 2 equiv) was added and the mixture was stirred for 30 min. All volatiles were removed under reduced pressure. Twelve mL brine and 2 mL 1 M HCl was added. The aqueous layer was extracted with 3 × 6 mL ethyl acetate. The combined organic layers were concentrated under reduced pressure and the crude product was purified by flash chromatography (DCM/MeOH). The crude product was used without further purification.
tert-Butyl 4-(4-{4-[(2R)-2-Amino-3-{bis[(4-methoxyphenyl)methyl]sulfamoyl}propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (57·HCl)
Aniline 8 (0.20 mol) was coupled with carboxylic acid 56 using general procedure 1 and deprotected by general procedure 2. The crude product was precipitated from diethyl ether with 2 M HCl in diethyl ether. The solid was suspended in 20 mL saturated NaHCO3 solution and extracted with 4 × 6 mL DCM. The combined organic layers were concentrated under reduced pressure and dried under high vacuum. Yellowish solid, 117.5 mg (62%).1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.92 (s, 1H), 8.75 (s, 1H), 8.50 (d, 1H, J = 8.9 Hz), 8.07 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.7 Hz), 7.91 (d, 2H, J = 8.7 Hz), 7.77 (d, 2H, J = 8.7 Hz), 7.75–7.72 (m, 3H), 7.29 (d, 1H, J = 8.6 Hz), 7.22 (d, 4H, J = 8.7 Hz), 6.91–6.88 (m, 4H), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.50 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.41 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 4.75 (hept., 1H, J = 6.2 Hz), 4.69 (d, 2H, J = 5.9 Hz), 4.34 (d, 2H, J = 15.1 Hz), 4.24 (d, 2H, J = 15.2 Hz), 3.81 (s, 6H), 3.08–3.03 (m, 1H), 1.60 (s, 9H), 1.38 (d, 6H, J = 6.2 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 170.4, 165.6, 164.3, 162.8, 159.6, 149.4, 142.3, 141.0, 139.1, 137.7, 132.3, 130.8, 130.3, 128.4, 127.7, 127.4, 127.3, 121.7, 120.2, 119.5, 119.1, 115.8, 114.4, 114.1, 80.9, 75.1, 55.5, 49.3, 28.4, 23.0.
4-(4-{4-[(2R)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-sulfamoylpropanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (16)
The amine 57·HCl (117.5 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. Off-white solid, 34.4 mg (35% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.72 (br s, 1H), 12.29 (br s, 1H), 10.71 (s, 1H), 10.43 (s, 1H), 9.40 (s, 1H), 8.83 (d, 1H, J = 7.3 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.98–7.95 (m, 4H), 7.94 (d, 2H, J = 8.8 Hz), 7.90 (d, 2H, J = 8.8 Hz), 7.87–7.83 (m, 3H), 7.81 (d, 2H, J = 8.8 Hz), 7.68 (d, 1H, J = 8.6 Hz), 7.03 (br s, 2H), 5.05 (dd, 1H, J = 7.5 Hz, 12.6 Hz), 4.55 (quint., 1H, J = 5.9 Hz), 3.65 (ddd, 2H, J = 6.4 Hz, 14.3 Hz, 22.2 Hz), 1.26 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 168.7, 168.5, 166.9, 166.0, 164.5, 164.2, 142.1, 141.7, 138.7, 136.9, 136.4, 132.5, 130.2, 129.0, 128.7, 128.6, 128.5, 128.3, 126.2, 122.9, 120.6, 119.5, 119.4, 118.3, 114.1, 112.6, 74.7, 55.1, 50.9, 22.3. HRMS (ESI) calcd 848.2345 [M + H+], 848.2335 found. HPLC purity 97.7%.
tert-Butyl 4-(4-{4-[(2S)-Piperidine-2-amido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (59)
Amine 8 (0.09 mmol) was coupled with (2S)-1-{[(9H-Fluoren-9-yl)methoxy]carbonyl}piperidine-2-carboxylic acid using general procedure 9 and deprotected using general procedure 2. Yellowish solid, 24 mg (39% over 2 steps). 1H NMR (700 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.47 (s, 1H), 8.73 (s, 1H), 8.46 (d, 1H, J = 8.9 Hz), 8.04 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.7 Hz), 7.86 (d, 2H, J = 8.7 Hz), 7.76 (d, 2H, J = 8.7 Hz), 7.72 (d, 2H, J = 8.7 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.5 Hz, 16.3 Hz), 5.49 (d, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 4.74 (quart., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.52 (d, 1H, J = 7.8 Hz), 3.12 (d, 1H, J = 12.1 Hz), 2.81 (t, 1H, J = 11.1 Hz), 2.06 (dd, 1H, J = 2.9 Hz, 13.0 Hz), 1.85–1.81 (m, 1H), 1.67–1.63 (m, 2H), 1.59 (s, 9 H), 1.53–1.49 (m, 2H), 1.37 (d, 6H, J = 6.2 Hz). 13C NMR (176 MHz, CDCl3, 300 K): δ (ppm) = 172.0, 165.5, 164.4, 162.8, 149.4, 142.3, 141.7, 139.1, 137.7, 132.2, 130.8, 129.6, 128.2, 127.6, 127.4, 121.6, 120.1, 119.5, 119.2, 115.8, 80.9, 76.9, 75.1, 60.1, 45.4, 29.2, 28.4, 25.5, 23.5, 23.0. HRMS (ESI) calcd 657.3288 [M + H+], 657.3293 found. HPLC purity 99.3%.
4-(4-{4-[(2S)-1-[4-(4-Cyanobenzamido)benzoyl]piperidine-2-amido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (17)
The amine 59 (35.5 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 5 and 7. White solid, 9 mg (31% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.65 (s, 1H), 10.61 (s, 1H), 10.38 (s, 1H), 9.42 (s, 1H), 8.11 (d, 2H, J = 7.6 Hz), 8.04 (d, 2H, J = 8.2 Hz), 7.99–7.96 (m, 4H), 7.89–7.85 (m, 5H), 7.84–7.79 (m, 2H), 7.71 (d, 1H, J = 8.8 Hz), 7.50–7.43 (m, 1H), 5.24 (br s, 1H), 4.55 (hept., 1H, J = 6.1 Hz), 3.67–3.53 (m, 2H), 2.26–2.17 (m, 1H), 1.90–1.82 (m, 1H), 1.70 (dd, 1H, J = 3.2 Hz, 9.3 Hz), 1.52–1.42 (m, 2H), 1.27 (d, 6H, J = 6.1 Hz), 1.25–1.21 (m, 2H). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 170.6, 168.5, 166.9, 164.4, 164.2, 154.1, 142.4, 142.0, 139.9, 138.8, 137.0, 136.3, 132.5, 130.2, 128.6, 128.4, 127.8, 126.3, 122.8, 120.7, 120.0, 119.0, 118.3, 114.0, 112.4, 112.2, 74.9, 53.1, 45.7, 28.7, 27.3, 22.3, 20.2. HRMS (ESI) calcd 809.2935 [M + H+], 809.2930 found. HPLC purity 99.0%.
tert-Butyl 4-(4-{4-[(2R)-Piperidine-2-amido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (61)
Amine 8 (0.09 mmol) was coupled with (2R)-1-{[(9H-Fluoren-9-yl)methoxy]carbonyl}piperidine-2-carboxylic acid using general procedure 9 and deprotected using general procedure 2. White solid, 11 mg (18% over 2 steps). 1H NMR (700 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.36 (s, 1H), 8.74 (s, 1H), 8.48 (d, 1H, J = 8.9 Hz), 8.05 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.7 Hz), 7.88 (d, 2H, J = 8.7 Hz), 7.77 (d, 2H, J = 8.7 Hz), 7.73 (d, 2H, J = 8.8 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.0 Hz, 10.4 Hz), 4.75 (hept., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.47 (d, 1H, J = 7.4 Hz), 3.10 (d, 1H, J = 12.1 Hz), 2.83–2.77 (m, 1H), 2.05 (dd, 1H, J = 3.3 Hz, 13.1 Hz), 1.86–1.80 (m, 1H), 1.67–1.63 (m, 2H), 1.60 (s, 9 H), 1.53–1.49 (m, 2H), 1.38 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, CDCl3, 300 K): δ (ppm) = 172.3, 165.6, 164.4, 162.8, 149.4, 142.3, 141.7, 139.1, 137.7, 132.2, 130.8, 129.7, 128.3, 127.6, 127.4, 121.6, 120.2, 119.4, 119.2, 115.8, 80.9, 76.9, 75.1, 60.3, 45.5, 29.4, 28.4, 25.7, 23.6, 23.0. HRMS (ESI) calcd 657.3288 [M + H+], 657.3281 found. HPLC purity 99.9%.
4-(4-{4-[(2R)-1-[4-(4-Cyanobenzamido)benzoyl]piperidine-2-amido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (18)
The amine 61 (15.2 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 5 and 7. Beige solid, 7 mg (57% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.66 (s, 1H), 10.61 (s, 1H), 10.38 (br s, 1H), 9.42 (s, 1H), 8.11 (d, 2H, J = 7.7 Hz), 8.04 (d, 2H, J = 8.2 Hz), 7.98–7.96 (m, 4H), 7.89–7.84 (m, 5H), 7.84–7.80 (m, 2H), 7.71 (d, 1H, J = 8.8 Hz), 7.50–7.43 (m, 2H), 5.24 (br s, 1H), 4.55 (hept., 1H, J = 6.1 Hz), 2.25–2.20 (m, 1H), 1.90–1.81 (m, 1H), 1.70 (dd, 1H, J = 3.1 Hz, 9.4 Hz), 1.50–1.43 (m, 2H), 1.27 (d, 6H, J = 6.1 Hz), 1.25–1.21 (m, 3H). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 170.6, 168.5, 166.9, 164.4, 164.2, 154.1, 142.4, 142.0, 139.9, 138.8, 137.0, 136.3, 132.5, 130.2, 128.6, 128.4, 127.8, 126.3, 122.8, 120.7, 120.0, 119.0, 118.3, 114.0, 112.4, 112.2, 74.9, 53.1, 45.7, 28.7, 27.3, 22.3, 20.2. HRMS (ESI) calcd 809.2935 [M + H+], 809.2929 found. HPLC purity 98.4%.
tert-Butyl (2S)-2-[(4-{[4-({4-[(tert-Butoxy)carbonyl]phenyl}carbamoyl)-3-(prop-2-en-1-yloxy)-2-(propan-2-yloxy)phenyl]carbamoyl}phenyl)carbamoyl]-4-oxopiperidine-1-carboxylate (63)
50 mg Aniline 8 (0.09 mmol, 1 equiv) and 26.8 mg (S)-1-(tert-butoxycarbonyl)-4-oxopiperidine-2-carboxylic acid (0.11 mmol, 1.2 equiv) were added to a dry flask and further dried under high vacuum. 0.64 mL dry DCM and 38 μL triethylamine (27.6 mg, 0.27 mmol, 3 equiv) were added under nitrogen atmosphere and the solution was cooled down to 0 °C. 10.0 μL phosphoryl chloride (16.5 mg, 0.11 mmol, 1.2 equiv) was slowly added and the mixture was kept at 0 °C. The reaction was controlled over LCMS. After completion, the reaction was quenched with 4 mL water and 1 mL 1 M HCl. The aqueous phase was extracted with 3 × 4 mL of DCM. The organic phases were combined and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (petroleum ether/ethyl acetate) and directly used without further purification. 106 mg (crude).
tert-Butyl 4-(4-{4-[(2S)-4-Oxopiperidine-2-amido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (64)
70 mg crude carbamate 63 (0.09 mmol, 1 equiv) was dissolved in 0.4 mL tert-butyl acetate (3.0 mmol, 32.6 equiv) and 0.1 mL dry DCM under nitrogen atmosphere. Sixteen μL trifluoromethanesulfonic acid (27.1 mg, 0.18 mmol, 2.0 equiv) was slowly added to the stirring solution under nitrogen atmosphere. The reaction was controlled over LCMS. After completion, the reaction was quenched with Na2CO3 and 4 mL water. The aqueous phase was extracted with 3 × 2 mL ethyl acetate. The solvent was removed under reduced pressure and the residue was purified by RP HPLC. 9.5 mg (crude). HRMS (ESI) calcd 671.3081 [M + H+], 671.3075 found.
4-(4-{4-[(2S)-1-[4-(4-Cyanobenzamido)benzoyl]-4-oxopiperidine-2-amido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (19)
The amine 64 (14.0 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. White solid, 3 mg (25% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.29 (s, 1H), 10.68 (s, 1H), 10.60 (s, 1H), 10.58 (1H), 9.43 (s, 1H), 8.12 (d, 2H, J = 8.3 Hz), 8.05 (d, 2H, J = 8.4 Hz), 7.99–7.96 (m, 4H), 7.89 (d, 2H, J = 8.1 Hz), 7.87–7.84 (m, 3H), 7.81–7.77 (m, 2H), 7.70 (d, 1H, J = 8.8 Hz), 7.55 (d, 2H, J = 6.8 Hz), 5.15 (br s, 1H), 4.54 (hept., 1H, J = 6.1 Hz), 3.99–3.91 (m, 1H), 3.88–3.83 (m, 1H), 3.09 (dd, 1H, J = 6.9 Hz, 15.8 Hz), 2.75 (d, 1H, J = 12.5 Hz), 2.54 (dd, 2H, J = 4.5 Hz, 6.4 Hz), 1.27 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 206.3, 170.1, 170.0, 168.5, 166.9, 164.5, 164.2, 154.1,142.0, 140.4, 138.7, 137.0, 136.4, 132.5, 130.7, 130.2, 128.6, 128.4, 128.1, 126.3, 122.8, 120.7, 119.9, 119.0, 118.3, 114.0, 112.5, 112.3, 74.9, 55.0, 43.4, 41.1, 39.5, 22.3. HRMS (ESI) calcd 823.2728 [M + H+], 823.2722 found. HPLC purity 97.6%.
Methyl (2S)-2-[(2,2-Dimethoxyethyl)({[(9H-fluoren-9-yl)methoxy]carbonyl})amino]-3-hydroxypropanoate (67)
Methyl l-serinate hydrochloride (66·HCl) (500 mg, 3.21 mmol) was dissolved in MeOH (10 mL). Et3N (450 μL, 3.21 mmol, 1.00 equiv), 2,2-dimethoxyacetaldehyde (60% in H2O, 558 mg, 3.21 mmol, 1.00 equiv) and 10% Pd/C (45.0 mg) were added subsequently. The mixture was stirred under an H2 atmosphere for 17 h before filtration through a short plug of Celite. The filtrate was concentrated under reduced pressure. The crude product was dissolved in H2O (6 mL) and NaHCO3 (540 mg, 6.42 mmol, 2.00 equiv) and FmocCl (815 mg, 3.15 mmol, 1.00 equiv) were added. The mixture was diluted with EtOAc (7 mL) at 0 °C. After stirring for 1 h at 0 °C the mixture was warmed to rt and stirring was continued for 21 h. EtOAc was added and the phases were separated. The organic phase was washed with a 1 M HCl solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, PE/EtOAc = 2:1) to furnish tertiary amine (1.16 g, 2.69 mmol, 84%) as colorless oil. The analytical data are consistent with those reported in the literature (F. Sladojevich, A. Trabocchi, A. Guarna, J. Org. Chem.2007, 72, 4254–4257). [α]D24 = −29.1° (c 1.1Cl3). 1H NMR (400 MHz, CDCl3) = δ (3:2 mixture of rotamers) 7.78–7.76 (d, J = 7.4 Hz, 2H), 7.61–7.60 (d, J = 7.3 Hz, 1H), 7.56–7.53 (m, 1H), 7.43–7.29 (m, 4H), 4.77–4.69 (m, 2H), 4.63–4.45 (m, 2H), 4.24–4.22 (m, 1H), 3.96–3.94 (m, 1H), 3.86–3.80 (m, 1H), 3.70–3.60 (m, 4H), 3.48–3.44 (m, 2.5H), 3.22–3.11 (m, 4H), 2.99–2.94 (dd, J = 7.3, 15.1 Hz, 0.5H) ppm. HRMS (ESI+) calcd for C23H27NO7Na [M + Na]+: 452.1685; found: 452.1678.
4-(9H-Fluoren-9-yl)methyl 3-Methyl (3S)-3,4-Dihydro-2H-oxazine-3,4-dicarboxylate (68)
Alcohol 67 (459 mg, 1.07 mmol) was dissolved in PhMe (15 mL) and pTsOH·H2O (20.3 mg, 0.11 mmol, 0.10 equiv) was added. The reaction flask was equipped with a dropping funnel including MS (4 Å, 5.2 g) and a condenser. The mixture was stirred at 123 °C for 3 h, before it was filtered through a short plug of NaHCO3. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (PE/EtOAc = 4:1) to furnish the product (265 mg, 0.73 mmol, 68%) as colorless foam. The analytical data are consistent with those reported in the literature (F. Sladojevich, A. Trabocchi, A. Guarna, J. Org. Chem.2007, 72, 4254–4257). 1H NMR (400 MHz, CDCl3) = δ (3:2 mixture of rotamers) 7.79–7.75 (t, J = 7.6 Hz, 2H), 7.63–7.59 (m, 1H), 7.52–7.49 (dd, J = 2.7, 7.2 Hz, 1H), 7.44–7.39 (q, J = 6.9 Hz, 2H), 7.35–7.29 (m, 2H), 6.42–6.41 (dd, J = 1.1, 5.0 Hz, 0.4H), 6.42–6.41 (dd, J = 1.1, 5.0 Hz, 0.6H), 6.02–6.01 (d, J = 4.9 Hz, 0.4H), 5.98–5.97 (d, J = 5.0 Hz, 0.6H), 4.98 (s, 0.6H), 4.70–4.67 (dd, J = 1.2, 10.9 Hz, 0.6H), 4.61–4.40 (m, 3H), 3.34–4.30 (t, J = 7.2 Hz, 0.6H), 4.23–4.22 (t, J = 6.1 Hz, 0.4H), 4.01–3.98 (dd, J = 2.8, 11.1 Hz, 0.6H), 3.89–3.86 (dd, J = 2.8, 11.1 Hz, 0.4H), 3.79 (s, 1.9H), 3.71 (s, 1.1H) ppm. HRMS (ESI+) calcd for C21H19NO5Na [M + Na]+: 388.1161; found: 388.1161.
4-(9H-Fluoren-9-yl)methyl 3-Methyl (3S)-Morpholine-3,4-dicarboxylate (69)
Alkene 68 (160 mg, 0.44 mmol) was dissolved in MeOH (3.1 mL) and CH2Cl2 (1.6 mL). Pt/C (10%, 19.7 mg) was added and the mixture was stirred at rt under an H2 atmosphere for 15 h. The mixture was filtered through a short plug of Celite. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (dry load, PE/EtOAc = 4:1) to furnish the morpholine (149 mg, 0.41 mmol, 93%) as colorless foam.
The analytical data are consistent with those reported in the literature (F. Sladojevich, A. Trabocchi, A. Guarna, J. Org. Chem.2007, 72, 4254–4257). 1H NMR (400 MHz, CDCl3) = δ (mixture of rotamers) 7.78–7.75 (t, J = 7.0 Hz, 2H), 7.62–7.58 (m, 1H), 7.52–7.48 (t, J = 6.9 Hz, 1H), 7.43–7.38 (m, 2H), 7.35–7.28 (m, 2H), 4.66–4.65 (d, J = 2.8 Hz, 0.6H), 4.56–4.45 (m, 1.6H), 4.43–4.37 (m, 1.4H), 4.30–4.28 (m, 1.6H), 4.24–4.21 (t, J = 7.2 Hz, 0.5H), 3.91–3.84 (m, 1.4H), 3.78 (s, 1.5H), 3.73 (s, 1.5H), 3.69–3.65 (dd, J = 3.7, 11.7 Hz, 0.7H), 3.61–3.57 (dd, J = 3.9, 11.9 Hz, 0.5H), 3.51–3.41 (m, 1.8H), 3.32–3.25 (ddt, J = 3.7, 12.8 Hz, 0.5H) ppm. HRMS (ESI+) calcd for C21H21NO5Na [M + Na]+: 390.1317; found: 390.1317.
(3S)-4-{[(9H-Fluoren-9-yl)methoxy]carbonyl}morpholine-3-carboxylic Acid (70)
Ester 69 (147 mg, 0.40 mmol) was dissolved in 1,4-dioxane (1 mL) and a 5 M HCl solution (1 mL) was added. The mixture was stirred at 110 °C for 16 h. A 5% Na2CO3 solution was added at rt and the aqueous phase was washed with Et2O, acidified with conc. HCl and extracted with CH2Cl2 (3×). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The acid (127 mg, 0.36 mmol, 90%) was obtained as colorless amorphous solid, which was used in the next step without further purification. The peaks in the 1H NMR spectrum are shifted compared to the literature (F. Sladojevich, A. Trabocchi, A. Guarna, J. Org. Chem.2007, 72, 4254–4257), what might be caused by 1,4-dioxane impurities. [α]D25 = −35.2° (c 1.02Cl2). 1H NMR (400 MHz, CDCl3) = δ (mixture of rotamers) 7.78–7.76 (d, J = 7.6 Hz, 1H), 7.75–7.71 (t, J = 6.8 Hz, 1H), 7.60–7.57 (m, 1H), 7.53–7.48 (m, 1H), 7.42–7.36 (m, 2H), 7.34–7.28 (m, 2H), 4.71–4.70 (d, J = 3.0 Hz, 0.5H), 4.59–4.50 (m, 1.5H), 4.47–4.40 (m, 1H), 4.32–4.21 (m, 2H), 3.93–3.90 (m, 1H), 3.81–3.73 (m, 1.5H), 3.68–3.63 (m, 1H), 3.60–3.56 (dd, J = 4.0, 11.9 Hz, 0.5H), 3.52–3.40 (m, 1.5H), 3.31–3.23 (ddt, J = 3.4, 13.4 Hz, 0.5H) ppm. HRMS (ESI+) calcd for C20H18NO5 [M – H]−: 352.1185; found: 352.1185.
(9H-Fluoren-9-yl)methyl (3S)-3-[(4-{[4-({4-[(tert-Butoxy)carbonyl]phenyl}carbamoyl)-3-(prop-2-en-1-yloxy)-2-(propan-2-yloxy)phenyl]carbamoyl}phenyl)carbamoyl]morpholine-4-carboxylate (71)
Step 1: Carboxylic acid 70 (90.0 mg, 0.25 mmol) was stirred in SOCl2 (1 mL) at 80 °C for 30 min. The solvent was removed under reduced pressure to furnish the acyl chloride (101 mg, quant.) as yellow oil, which was used in the next step without further purification. Step 2: Amine 8 (139.0 mg, 0.25 mmol) and 2,6-lutidine (136 μL, 1.17 mmol, 4.6 equiv) were dissolved in CH2Cl2 (2.5 mL). The acid chloride (94.7 mg, 0.25 mmol, 1.00 equiv) in CH2Cl2 (2.5 mL) was added at 0 °C and the mixture was stirred at rt for 18 h. Afterward the mixture was washed with a 1 M HCl solution, a 5% Na2CO3 solution and brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (PE/EtOAc = 1:1) to furnish the product (142 mg, 0.16 mmol, 63%) as yellow amorphous solid. [α]D25 = −72.0° (c 0.32Cl2). 1H NMR (400 MHz, DMSO-d6) = (mixture of rotamers) 10.53 (s, 1H), 10.45 (s, 0.5H), 10.39 (s, 0.5H), 9.58 (s, 0.5H), 9.54 (s, 0.5H), 8.05–8.03 (d, J = 8.4 Hz, 1H), 7.99–7.98 (d, J = 8.4 Hz, 1H), 7.92–7.88 (m, 3H), 7.84–7.80 (m, 5H), 7.76–7.74 (d, J = 8.5 Hz, 2H), 7.71–7.68 (t, J = 7.7 Hz, 1H), 7.57–7.55 (m, 1H), 7.45–7.27 (m, 4H), 7.09–7.03 (m, 1H), 6.06–5.98 (m, 1H), 5.39–5.36 (d, J = 17.2 Hz, 1H), 5.21–5.19 (d, J = 10.5 Hz, 1H), 4.61 (s, 2H), 4.56–4.54 (m, 1H), 4.49 (bs, 1H), 4.40–4.20 (m, 4H), 3.92–3.86 (m, 1H), 3.79–3.71 (m, 2H), 3.62–3.58 (m, 1H), 3.50–3.38 (m, 2H), 1.55 (s, 9H), 1.27–1.25 (m, 6H) ppm. 13C NMR (101 MHz, DMSO-d6) = (mixture of rotamers) 169.0, 164.6, 164.6, 164.3, 156.0, 155.5, 149.5, 143.7, 143.0, 140.7, 135.6, 133.7, 130.1, 128.5, 127.7, 127.6, 127.2, 127.0, 126.0, 125.1 (t, J = 12.4 Hz), 123.6, 120.1, 118.8, 118.7, 117.8, 80.3, 76.3, 74.3, 67.8–67.1, 65.9, 65.5, 59.7, 55.2, 54.6, 46.6, 46.5, 27.9, 22.3 ppm. HRMS (ESI) calcd for C51H52N4O10 Na[M + Na]+: 903.3581; found: 903.3572.
tert-Butyl 4-(4-{4-[(3S)-4-[4-(4-Cyanobenzamido)benzoyl]morpholine-3-amido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (72)
Step 1: Carbamate 71 (132 mg, 0.15 mmol) was dissolved in MeCN/piperidine (4:1, 1.8 mL). The solution was stirred at rt for 90 min and then concentrated under reduced pressure. The residue was coevaporated with MeCN (3×) to furnish the crude product, which was used in the next step without further purification. Step 2: DIPEA (88 μL, 0.50 mmol, 5.00 equiv) was added dropwise to a stirred solution of HATU (95.7 mg, 0.25 mmol, 2.50 equiv) and carboxylic acid 10 (67.0 mg, 0.25 mmol, 2.50 equiv) in DMF (2.5 mL). The solution was stirred for 5 min and was then transferred to a stirred solution of the amine (66.3 mg, 0.10 mmol) in DMF (1.4 mL). The reaction mixture was stirred at rt for 16 h. The mixture was diluted with EtOAc and washed with a 1 M HCl solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, MeOH in CH2Cl2 = 0.5, 1, 3%) to furnish the product (63.8 mg, 0.07 mmol, 70%) as yellow amorphous solid. [α]D27 = −8.0° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = δ 10.67 (s, 1H), 10.53 (s, 1H), 10.47 (s, 1H), 9.55 (s, 1H), 8.12–8.11 (m, 2H), 8.05–8.04 (d, J = 8.2 Hz, 2H), 8.01–8.00 (m, 2H), 7.90–7.88 (m, 4H), 7.84–7.69 (m, 6H), 7.50 (bs, 1H), 7.42–7.40 (d, J = 8.5 Hz, 1H), 6.06–5.98 (m, 1H), 5.40–5.35 (dq, J = 1.7, 17.2 Hz, 1H), 5.22–5.19 (dq, J = 1.6, 10.5 Hz, 1H), 5.03 (bs, 0.5H), 4.62–4.61 (d, J = 5.4 Hz, 2H), 4.53–4.46 (sept, J = 6.1 Hz, 1H), 4.43–4.40 (m, 1H), 4.35–4.18 (m, 0.5H), 3.96–3.80 (m, 3H), 3.54–3.42 (m, 2H), 1.55 (s, 9H), 1.27–1.26 (d, J = 6.0 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = δ 170.7, 168.7, 164.6, 164.5, 164.4, 164.3, 149.5, 143.0, 142.6, 142.1, 140.2, 138.7, 135.6, 133.7, 132.5, 130.1, 128.6, 128.5, 128.1, 127.2, 126.0, 123.6, 119.9, 119.0, 118.8, 118.3, 117.8, 114.0, 80.3, 76.2, 74.3, 67.9, 65.6, 53.3, 45.5, 27.9, 22.3 ppm. HRMS (ESI) calcd for C51H50N6O10Na [M + Na]+: 929.3486; found: 929.3494.
tert-Butyl 4-(4-{4-[(3S)-4-[4-(4-Cyanobenzamido)benzoyl]morpholine-3-amido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (73)
Allyl ether 72 (60.9 mg, 0.07 mmol) was dissolved in THF (3 mL). Aniline (20 μL, 0.22 mmol, 3.30 equiv) and Pd(PPh3)4 (7.8 mg, 7 μmol, 0.10 equiv) were added subsequently and the resulting mixture was stirred at rt for 90 min. The mixture was concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, MeOH in CH2Cl2 = 2, 3%) to furnish the product (53.0 mg, 0.06 mmol, 91%) as yellow amorphous solid. [α]D27 = −10.6° (c 0.2, THF). 1H NMR (600 MHz, DMSO-d6) = 12.28 (s, 1H), 10.67–10.20 (m, 3H), 9.43 (s, 1H), 8.11 (bs, 2H), 8.05–8.04 (d, J = 7.9 Hz, 2H), 7.98 (bs, 2H), 7.94–7.92 (d, J = 8.8 Hz, 2H), 7.90–7.73 (m, 7H), 7.70–7.68 (d, J = 8.8 Hz, 1H), 7.50–7.38 (m, 2H), 5.03 (bs, 0.5H), 4.58–4.52 (sept, J = 6.1 Hz, 1H), 4.43–4.18 (m, 1.5H), 3.98–3.80 (m, 3H), 3.54–3.50 (m, 2H), 1.55 (s, 9H), 1.27–1.26 (d, J = 5.7 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 170.7, 168.7, 168.4, 164.5, 164.4, 164.2, 154.2, 142.2, 142.0, 140.2, 138.7, 137.0, 136.4, 132.5, 131.5, 131.4, 130.5, 129.9, 128.6, 128.4, 128.1, 126.8, 122.8, 120.7, 119.9, 118.9, 118.3, 114.0, 112.5, 112.2, 80.5, 74.8, 67.9, 66.0, 53.3, 45.5, 27.8, 22.3 ppm. HRMS (ESI) calcd for C48H46N6O10Na [M + Na]+: 889.3173; found: 889.3176.
4-(4-{4-[(3S)-4-[4-(4-Cyanobenzamido)benzoyl]morpholine-3-amido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (20)
tert-Butyl ester 73 (48.0 mg, 0.06 mmol) was dissolved in precooled TFA (3 mL) at 0 °C with stirring. The solution was warmed up to rt over 30 min. Et2O was added at 0 °C. The precipitate was filtered off, washed with an excess of Et2O and dried in vacuo to furnish the product (35.4 mg, 0.04 mmol, 79%) as colorless amorphous solid. [α]D27 = −7.9° (c 0.1, MeOH). 1H NMR (600 MHz, DMSO-d6) = 12.82 (bs, 1H), 12.28 (s, 1H), 10.67 (s, 1H), 10.60 (s, 1H), 10.49–10.20 (m, 1H), 9.44 (s, 1H), 8.11 (bs, 2H), 8.05–8.04 (d, J = 7.9 Hz, 2H), 7.98–7.96 (m, 5H), 7.89–7.73 (m, 8H), 7.71–7.69 (d, J = 8.8 Hz, 1H), 7.50–7.38 (m, 2H), 5.04 (bs, 0.7H), 4.58–4.52 (sept, J = 6.1 Hz, 1H), 4.41–4.17 (m, 2H), 3.96–3.80 (m, 4.3H), 1.28–1.27 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 170.7, 168.7, 168.5, 166.9, 164.4, 164.2, 154.1, 142.2, 142.0, 140.2, 138.7, 137.0, 136.4, 132.5, 130.2, 128.6, 128.4, 128.1, 126.3, 122.8, 120.7, 119.9, 118.9, 118.3, 114.0, 112.5, 112.3, 74.9, 67.9, 66.0, 53.3, 45.5, 22.3 ppm. HRMS (ESI) calcd for C44H37N6O10 [M-H]−: 809.2571; found: 809.2578.
tert-Butyl 4-(4-{4-[(2S)-2-Aminopent-4-enamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (74)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)pent-4-enoic acid using general procedure 9 and deprotected using general procedure 2. Yellow to orange solid, 32 mg (51% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.85 (s, 1H), 8.75 (s, 1H), 8.49 (d, 1H, J = 8.9 Hz), 8.05 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.8 Hz), 7.89 (d, 2H, J = 8.8 Hz), 7.79 (d, 2H, J = 8.8 Hz), 7.73 (d, 2H, J = 8.8 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.84–5.74 (m, 1H), 5.49 (dq, 1H, J = 1.4 Hz, 17.1 Hz), 5.40 (ddd, 1H, J = 1.0 Hz, 2.2 Hz, 10.4 Hz), 5.22–5.18 (m, 2H), 4.75 (hept., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.59 (dd, 1H, J = 3.5 Hz, 8.3 Hz), 2.75–2.67 (m, 1H), 2.45–2.38 (m, 1H), 1.59 (s, 9H), 1.38 (dd, 6H, J = 1.7 Hz, 6.1 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 173.1, 165.5, 164.4, 162.8, 149.4, 142.3, 141.5, 139.1, 137.8, 134.0, 132.3, 130.8, 129.7, 128.3, 127.6, 127.4, 121.6, 120.2, 119.6, 119.6, 119.3, 115.8, 80.9, 76.9, 75.1, 54.5, 39.3, 28.4, 23.0. HRMS (ESI) calcd 643.3132 [M + H+], 643.3126 found. HPLC purity 97.3%.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}pent-4-enamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (21)
The amine 74 (49.8 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. Beige solid, 13 mg (35% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.70 (s, 1H), 10.60 (s, 1H), 10.50 (s, 1H), 9.40 (s, 1H), 8.62 (d, 1H, J = 7.5 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.98–7.95 (m, 6H), 7.89 (d, 2H, J = 8.8 Hz), 7.87–7.85 (m, 3H), 7.82 (d, 2H, J = 8.7 Hz), 7.71 (d, 1H, J = 8.8 Hz), 5.89 (ddt, 1H, J = 6.9 Hz, 10.2 Hz, 17.1 Hz), 5.20 (dd, 1H, J = 1.8 Hz, 17.2 Hz), 5.09 (dd, 1H, J = 1.9 Hz, 10.2 Hz), 4.70 (quart., 1H, J = 7.5 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 2.62 (t, 2H, J = 7.1 Hz), 1.27 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 171.0, 168.5, 166.9, 166.0, 164.4, 164.2, 154.1, 142.3, 142.0, 141.6, 138.7, 137.0, 136.3, 134.3, 132.5, 130.2, 129.1, 128.6, 128.4, 128.4, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 117.8, 114.0, 112.4, 112.1, 74.8, 54.1, 35.7, 22.3. HRMS (ESI) calcd 795.2779 [M + H+], 795.2774 found. HPLC purity 99.0%.
tert-Butyl 4-(4-{4-[(2S)-2-Aminopentanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (76)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)pentanoic acid using general procedure 8 and deprotected using general procedure 2. The product was a yellowish solid. Twenty-seven mg (42% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.29 (br s, 1H), 10.15 (s, 1H), 8.72 (s, 1H), 8.37 (d, 1H, J = 8.1 Hz), 7.97–7.92 (m, 3H), 7.82–7.73 (m, 4H), 7.69 (d, 2H, J = 8.6 Hz), 6.10 (dq, 1H, J = 5.8 Hz, 10.9 Hz), 5.46 (d, 1H, J = 17.1 Hz), 5.38 (d, 1H, J = 10.4 Hz), 4.74–4.61 (m, 3H), 4.20–4.02 (m, 1H), 2.02–1.90 (m, 1H), 1.86–1.74 (m, 1H), 1.58 (s, 9H), 1.52–1.40 (m, 2H), 1.33 (d, 6H, J = 5.6 Hz), 0.93–0.86 (m, 3H). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 170.6, 165.3, 164.5, 162.7, 149.3, 142.0, 141.4, 139.1, 137.4, 132.1, 130.6, 129.6, 128.0, 127.4, 127.2, 121.6, 120.0, 119.7, 119.1, 115.7, 80.8, 76.7, 74.9, 54.8, 35.1, 28.2, 22.7, 18.5, 13.7. HRMS (ESI) calcd 645.3288 [M + H+], 645.3284 found.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}pentanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (22)
The amine 76 (40.3 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. Beige solid, 14 mg (47% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.60 (s, 1H), 10.47 (s, 1H), 9.40 (s, 1H), 8.57 (d, 1H, J = 7.4 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.4 Hz), 7.98–7.95 (m, 6H), 7.89 (d, 2H, J = 8.8 Hz), 7.87–7.85 (m, 3H), 7.82 (d, 2H, J = 8.8 Hz), 7.71 (d, 1H, J = 8.9 Hz), 4.61 (dd, 1H, J = 7.4 Hz, 14.7 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 1.87–1.78 (m, 2H), 1.55–1.48 (m, 1H), 1.43–1.36 (m, 1H), 1.27 (dd, 6H, J = 0.9 Hz, 6.1 Hz), 0.95 (t, 3H, J = 7.4 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 171.9, 168.5, 166.9, 166.1, 164.4, 164.2, 154.1, 142.5, 142.0, 141.5, 138.7, 137.1, 136.3, 132.5, 130.2, 129.2, 128.6, 128.4, 128.4, 128.3, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 74.9, 54.4, 33.5, 22.3, 19.2, 13.7. HRMS (ESI) calcd 797.2935 [M + H+], 797.2929 found. HPLC purity 95.8%.
tert-Butyl 4-(4-{4-[(2S)-2-Aminohexanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (78)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)hexanoic acid using general procedure 9 and deprotected using general procedure 2. Slightly yellow solid, 19 mg (30% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.98 (s, 1H), 8.74 (s, 1H), 8.46 (d, 1H, J = 8.9 Hz), 8.03 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.7 Hz), 7.86 (d, 2H, J = 8.4 Hz), 7.78 (d, 2H, J = 8.4 Hz), 7.72 (d, 2H, J = 8.8 Hz), 6.13 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dd, 1H, J = 1.4 Hz, 17.2 Hz), 5.40 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 4.74 (hept., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.8 Hz), 3.75–3.63 (m, 1H), 2.03–1.94 (m, 1H), 1.73–1.63 (m, 1H), 1.59 (s, 9H), 1.45–1.39 (m, 2H), 1.39–1.33 (m, 8H), 0.91 (t, 3H, J = 7.0 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 173.0, 165.6, 165.5, 162.8, 149.5, 142.3, 141.6, 139.1, 137.7, 132.3, 130.8, 129.7, 128.3, 127.5, 127.5, 121.7, 120.2, 119.5, 119.2, 115.8, 80.9, 76.9, 75.1, 55.6, 34.1, 28.4, 27.9, 22.9, 22.6, 14.0. HRMS (ESI) calcd 659.3445 [M + H+], 659.3439 found.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}hexanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (23)
The amine 78 (33.2 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. Beige solid, 11 mg (50% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.60 (s, 1H), 10.47 (s, 1H), 9.40 (s, 1H), 8.57 (d, 1H, J = 7.4 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.98–7.95 (m, 6H), 7.89 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 3H), 7.82 (d, 2H, J = 8.8 Hz), 7.71 (d, 1H, J = 8.8 Hz), 4.59 (dd, 1H, J = 7.3 Hz, 14.7 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 1.84 (dd, 2H, J = 7.3 Hz, 14.5 Hz), 1.50–1.44 (m, 1H), 1.38–1.33 (m, 3H), 1.27 (d, 6H, J = 6.1 Hz), 0.90 (t, 3H, J = 7.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 171.9, 168.5, 166.9, 166.1, 164.4, 164.2, 154.1, 142.5, 142.0, 141.5, 138.7, 137.1, 136.3, 132.5, 130.2, 129.2, 128.6, 128.4, 128.4, 128.3, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 74.9, 54.7, 31.2, 28.1, 22.3, 21.9, 13.9. HRMS (ESI) calcd 811.3092 [M + H+], 811.3086 found. HPLC purity 97.3%.
tert-Butyl 4-(4-{4-[(2S)-3-Cyclopropyl-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (80)
Amine 8 (400 mg, 0.73 mmol), (2S)-3-cyclopropyl-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoic acid (438 mg, 1.25 mmol, 1.70 equiv) and EEDQ (290 mg, 1.17 mmol, 1.60 equiv) were dissolved in precooled CHCl3 (3.7 mL) at 0 °C. The mixture was stirred for 19 h while warming to rt. The solution was concentrated on silica and purified by column chromatography (20% Et2O in CH2Cl2, then 1% MeOH in CH2Cl2) to furnish the carbamate (547 g, 0.62 mmol, 85%) was yellow amorphous solid, which contained minor impurities. The compound was used in the next step without further purification. HRMS (ESI) calcd for C52H54N4O9Na [M + Na]+: 901.3788; found: 901.3812.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-cyclopropylpropanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (81)
Step 1: Carbamate 80 (500 mg, 0.57 mmol) was dissolved in MeCN (5.3 mL). Et2NH (1.3 mL) was added. The solution was stirred at rt for 30 min. The mixture was concentrated under reduced pressure. The residue was diluted with MeCN and concentrated three times. The crude product was dried in vacuo to furnish the amine, which was used in the next step without further purification. Step 2: DIPEA (435 μL, 2.49 mmol, 5.00 equiv) was added dropwise to a stirred solution of HATU (473 mg, 1.24 mmol, 2.50 equiv) and carboxylic acid 10 (331 mg, 1.24 mmol, 2.50 equiv) in DMF (12 mL). The solution was stirred for 30 min and was then transferred to a stirred solution of the amine (327 mg, 0.50 mmol) in DMF (7 mL). The reaction mixture was stirred at rt for 19 h. The mixture was diluted with EtOAc and washed with a 0.1 M HCl solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, MeOH in CH2Cl2 = 1, 5%) to furnish the product (405.2 mg, 0.45 mmol, 90%) as yellow amorphous solid. [α]D22 = +13.6° (c 0.3, MeOH). 1H NMR (500 MHz, DMSO-d6) = δ 10.69 (s, 1H), 10.53 (s, 1H), 10.46 (s, 1H), 9.51 (s, 1H), 8.62–8.61 (d, J = 7.1 Hz, 1H), 8.14–8.12 (d, J = 8.6 Hz, 2H), 8.06–8.04 (d, J = 8.6 Hz, 2H), 7.99–7.97 (m, 4H), 7.90–7.88 (d, J = 7.8 Hz, 4H), 7.84–7.80 (m, 5H), 7.42–7.40 (d, J = 8.5 Hz, 1H), 6.05–5.99 (m, 1H), 5.39–5.36 (dq, J = 1.7, 17.2 Hz, 1H), 5.21–5.19 (dq, J = 1.7, 10.5 Hz, 1H), 4.71–4.67 (q, J = 7.5 Hz, 1H), 4.61–4.60 (d, J = 5.4 Hz, 2H), 4.53–4.47 (sept, J = 6.1 Hz, 1H), 1.96–1.92 (m, 1H), 1.60–1.57 (m, 1H), 1.55 (s, 9H), 1.27–1.26 (d, J = 6.1 Hz, 6H), 0.91–0.88 (m, 1H), 0.49–0.38 (m, 2H), 0.25–0.14 (m, 2H) ppm. 13C NMR (500 MHz, DMSO-d6) = δ 171.7, 166.0, 164.6, 164.5, 164.4, 164.3, 149.5, 143.0, 142.5, 142.4, 141.5, 138.7, 135.7, 133.6, 132.5, 130.1, 129.3, 128.6, 128.5, 128.4, 128.3, 127.1, 126.0, 123.6, 119.5, 118.9, 118.8, 118.7, 118.3, 117.8, 114.0, 80.3, 76.2, 74.3, 55.3, 36.3, 27.9, 22.3, 8.2, 4.7, 4.0 ppm. HRMS (ESI) calcd for C52H52N6O9Na [M + Na]+: 927.3693; found: 927.3712.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-cyclopropylpropanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (82)
Allyl ether 81 (374 mg, 0.41 mmol) was dissolved in THF (18 mL). Aniline (124 μL, 1.36 mmol, 3.30 equiv) and Pd(PPh3)4 (47.7 mg, 0.04 mmol, 0.10 equiv) were added subsequently and the resulting mixture was stirred at rt for 2 h. The mixture was concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, MeOH in CH2Cl2 = 2, 5%) to furnish the product (286 mg, 0.33 mmol, 80%) as yellow amorphous solid. [α]D22 = −10.2° (c 0.3, MeOH). 1H NMR (500 MHz, DMSO-d6) = δ 12.29 (s, 1H), 10.69 (s, 1H), 10.62 (bs, 1H), 10.48 (s, 1H), 9.39 (s, 1H), 8.62–8.61 (d, J = 7.4 Hz, 1H), 8.14–8.12 (d, J = 8.6 Hz, 2H), 8.05–8.04 (d, J = 8.6 Hz, 2H), 7.98–7.95 (m, 4H), 7.93–7.92 (d, J = 8.9 Hz, 2H), 7.90–7.88 (d, J = 8.9 Hz, 2H), 7.86–7.84 (m, 3H), 7.82–7.81 (d, J = 8.9 Hz, 2H), 7.71–7.70 (d, J = 8.9 Hz, 1H), 4.71–4.68 (q, J = 7.5 Hz, 1H), 4.57–4.53 (sept, J = 6.1 Hz, 1H), 1.96–1.91 (m, 1H), 1.60–1.58 (m, 1H), 1.55 (s, 9H), 1.27–1.26 (dd, J = 1.7, 6.1 Hz, 6H), 0.91–0.87 (m, 1H), 0.49–0.38 (m, 2H), 0.25–0.13 (m, 2H) ppm. 13C NMR (151 MHz, DMSO-d6) = δ 171.7, 168.5, 166.0, 164.5, 164.4, 164.2, 154.1, 142.5, 142.0, 141.5, 138.7, 137.0, 136.3, 132.5, 129.9, 129.3, 128.6, 128.4, 128.4, 128.3, 126.8, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.1, 80.5, 74.8, 55.3, 36.3, 27.8, 22.3, 8.1, 4.6, 4.0 ppm. HRMS (ESI) calcd for C49H48N6O9Na [M + Na]+: 887.3380; found: 887.3381.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-cyclopropylpropanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (24)
tert-Butyl ester 82 (250 mg, 0.29 mmol) was dissolved in precooled TFA (10 mL) at 0 °C with stirring. The solution was warmed up to rt over 30 min. Et2O was added at 0 °C. The precipitate was filtered off, washed with an excess of Et2O and dried in vacuo to furnish the acid (205 mg, 0.25 mmol, 88%) as colorless amorphous solid. [α]D22 = +3.8° (c 0.1, MeOH). 1H NMR (600 MHz, DMSO-d6) = δ 12.82 (bs, 1H), 12.29 (s, 1H), 10.69 (s, 1H), 10.60 (s, 1H), 10.48 (s, 1H), 9.40 (s, 1H), 8.62–8.61 (d, J = 7.4 Hz, 1H), 8.14–8.12 (d, J = 8.6 Hz, 2H), 8.06–8.04 (d, J = 8.6 Hz, 2H), 7.98–7.95 (m, 6H), 7.90–7.88 (d, J = 9.1 Hz, 2H), 7.86–7.84 (m, 3H), 7.82–7.81 (d, J = 8.8 Hz, 2H), 7.72–7.70 (d, J = 8.9 Hz, 1H), 4.71–4.68 (q, J = 6.9 Hz, 1H), 4.58–4.52 (sept, J = 6.1 Hz, 1H), 1.96–1.91 (m, 1H), 1.60–1.55 (m, 1H), 1.27–1.26 (dd, J = 1.5, 6.1 Hz, 6H), 0.90–0.87 (m, 1H), 0.49–0.37 (m, 2H), 0.25–0.13 (m, 2H) ppm. 13C NMR (151 MHz, DMSO-d6) = 171.7, 168.5, 166.9, 166.0, 164.4, 164.2, 154.1, 142.5, 142.0, 141.5, 138.7, 137.0, 136.3, 132.5, 130.2, 129.2, 128.6, 128.4, 128.4, 128.3, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 74.9, 55.3, 36.3, 22.3, 8.1, 4.6, 4.0 ppm. HRMS (ESI) calcd for C45H39N6O9 [M – H]−: 807.2779; found: 807.2767. HPLC purity 98,0%.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-(2H-1,2,3-triazol-4-yl)propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (25)
7.4 mg alkyne 13 (9.3 μmol, 1 equiv), 0.15 mg copper(II) sulfate (0.9 μmol, 0.1 equiv), 1.1 mg sodium ascorbate (5.6 μmol, 0.6 equiv) and 2.0 mg TBTA (3.7 μmol, 0.4 equiv) were added to a dry flask and further dried under high vacuum. 0.2 mL DMSO and 0.1 mg THF were added under nitrogen atmosphere. 6.1 mg sodium azide was dissolved in 1.0 mL water and 0.1 mL of the solution was added to the mixture (9.3 μmol, 1 equiv) under nitrogen atmosphere. The reaction was stirred at room temperature and controlled over LCMS. After completion, the crude product was purified by RP HPLC. White solid, 4 mg (49%). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 15.00 (br s, 0.3 H), 14.64 (s, 0.6 H), 12.82 (br s, 1H), 12.29 (s, 1H), 10.70 (s, 1H), 10.60 (s, 1H), 10.55 (s, 1H), 9.41 (s, 1H), 8.77 (d, 1H, J = 7.5 Hz), 8.13 (d, 2H, J = 8.4 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.98–7.95 (m, 4H), 7.92 (d, 2H, J = 8.8 Hz), 7.88 (d, 2H, J = 8.7 Hz), 7.87–7.85 (m, 3H), 7.81 (d, 2H, J = 8.6 Hz), 7.71 (d, 1H, J = 8.8 Hz), 7.65 (s, 0.6 H), 4.93 (dd, 1H, J = 7.7 Hz, 14.3 Hz), 4.55 (hept., 1H, J = 6.0 Hz), 3.30–3.21 (m, 2H), 1.27 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 170.6, 168.5, 166.9, 166.0, 164.5, 164.2, 154.1, 143.3, 142.3,142.0, 141.6, 138.7, 137.0, 136.3, 132.9, 132.5, 130.2, 129.0, 128.6, 128.5, 128.4, 128.4, 126.3, 122.8, 120.7, 119.5, 119.0, 118.3, 114.0, 112.4, 112.2, 74.9, 54.3, 27.5, 22.3. HRMS (ESI) calcd 836.2792 [M + H+], 836.2788 found. HPLC purity 95.0%.
tert-Butyl 4-(4-{4-[(2S)-2-Amino-3-{[(prop-2-en-1-yloxy)carbonyl]amino}propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (83)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-{[(prop-2-en-1-yloxy)carbonyl]amino}propanoic acid using general procedure 9 and deprotected using general procedure 2. Yellow solid, 14 mg (21% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.16 (s, 1H), 9.99 (br s, 1H), 8.74 (s, 1H), 8.46 (d, 1H, J = 8.9 Hz), 8.04 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.7 Hz), 7.88 (d, 2H, J = 8.6 Hz), 7.77 (d, 2H, J = 8.6 Hz), 7.72 (d, 2H, J = 8.8 Hz), 6.13 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.89 (ddd, 1H, J = 5.7 Hz, 11.0 Hz, 16.1 Hz), 5.49 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 5.29 (dd, 1H, J = 1.5 Hz, 17.2 Hz), 5.21 (dd, 1H, J = 1.2 Hz, 10.4 Hz), 4.74 (quart., 1H, J = 6.2 Hz), 4.69 (d, 2H, J = 5.9 Hz), 4.57 (d, 2H, J = 4.7 Hz), 3.78–3.72 (m, 1H), 3.71–3.65 (m, 2H), 1.59 (s, 9H), 1.37 (d, 6H, J = 6.2 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 171.4, 165.6, 164.4, 162.8, 157.8, 149.4, 142.3, 141.2, 139.1, 137.7, 132.6, 132.3, 130.8, 130.0, 128.3, 127.6, 127.5, 121.7, 120.2, 119.5, 119.2, 118.2, 115.8, 80.9, 76.9, 75.1, 66.2, 56.4, 44.7, 28.4, 23.0. HRMS (ESI) calcd 716.3296 [M + H+], 716.3289 found. HPLC purity 99.9%.
4-(4-{4-[(2S)-3-Amino-2-{[4-(4-cyanobenzamido)phenyl]formamido}propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (26)
The amine 83 (19.8 μmol) was coupled with carboxylic acid 10 using general procedure 4. After deprotection of the tert-butyl ester using general procedure 7, the crude product was purified by RP HPLC. The product was obtained by deprotection with general procedure 5 followed by RP HPLC purification. White solid, 2 mg (15% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 10.73 (s, 1H), 9.31 (s, 1H), 8.83 (d, 1H, J = 6.9 Hz), 8.13 (d, 2H, J = 8.4 Hz), 8.05 (d, 2H, J = 8.4 Hz), 7.99 (d, 2H, J = 8.7 Hz), 7.97–7.94 (m, 4H), 7.92 (d, 2H, J = 8.8 Hz), 7.84–7.81 (m, 4H), 7.77 (d, 1H, J = 7.5 Hz), 7.59–7.52 (m, 1H), 4.90 (d, 1H, J = 5.2 Hz), 4.68–4.59 (m, 1H), 3.21 (dd, 1H, J = 8.2 Hz, 13.0 Hz), 1.25 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 168.4, 168.2, 167.0, 166.5, 164.5, 164.0, 141.9, 138.6, 136.7, 132.5, 130.3, 129.1, 128.9, 128.6, 128.6, 128.2, 123.0, 120.2, 119.5, 119.4, 118.3, 114.1, 52.7, 40.0, 22.4. HRMS (ESI) calcd 784.2731 [M + H+], 784.2727 found. HPLC purity 95.1%.
tert-Butyl 4-(4-{4-[(2S)-2-Amino-4-{[(tert-butoxy)carbonyl]amino}butanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (85)
Amine 8 (0.09 mmol) was coupled with (2S)-4-{[(tert-Butoxy)carbonyl]amino}-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)butanoic acid using general procedure 8 and deprotected using general procedure 2. Yellow solid, 38 mg (52% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.16 (s, 1H), 10.11 (br s, 1H), 8.74 (s, 1H), 8.48 (d, 1H, J = 8.9 Hz), 8.05 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.8 Hz), 7.89 (d, 2H, J = 8.8 Hz), 7.80 (d, 2H, J = 8.6 Hz), 7.72 (d, 2H, J = 8.8 Hz), 6.13 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.0 Hz, 10.4 Hz), 4.89 (t, 1H, J = 6.3 Hz), 4.74 (hept., 1H, J = 6.2 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.60–3.48 (m, 1H), 3.37–3.29 (d, 2H, J = 6.0 Hz), 1.83–1.74 (m, 1H), 1.59 (s, 9H), 1.42 (s, 9H), 1.38 (d, 6H, J = 6.1 Hz).13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 173.7, 165.5, 164.4, 162.8, 157.0, 149.4, 142.3, 141.7, 139.1, 137.8, 132.3, 130.8, 129.6, 128.2, 127.6, 127.4, 121.6, 120.1, 119.4, 119.1, 115.8, 80.9, 80.0, 76.9, 75.1, 53.2, 37.1, 36.1, 28.5, 28.4, 22.9. HRMS (ESI) calcd 746.3765 [M + H+], 746.3759 found.
4-(4-{4-[(2S)-4-Amino-2-{[4-(4-cyanobenzamido)phenyl]formamido}butanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (27)
The amine 85 (49.6 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 5 and 7. Slightly beige solid, 19 mg (51% over 3 steps). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 10.73 (s, 1H), 10.52 (br s, 1H), 9.22 (s, 1H), 8.81 (d, 1H, J = 6.7 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.04 (d, 2H, J = 8.5 Hz), 7.98 (d, 2H, J = 8.7 Hz), 7.96–7.89 (m, 6H), 7.84–7.80 (m, 4H), 7.72 (d, 1H, J = 8.8 Hz), 7.49 (d, 1H, J = 8.5 Hz), 4.76–4.67 (m, 2H), 3.00–2.93 (m, 2H), 2.25–2.19 (m, 1H), 2.17–2.09 (m, 1H), 1.24 (d, 6H, J = 6.2 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 170.4, 168.0, 167.2, 166.3, 164.5, 163.8, 158.0, 143.0, 142.0, 141.7, 138.6, 136.9, 136.0, 132.5, 130.3, 129.0, 128.9, 128.6, 128.5, 128.1, 125.6, 123.2, 119.8, 119.5, 119.1, 118.3, 114.1, 113.7, 73.3, 52.2, 36.4, 29.4, 22.4. HRMS (ESI) calcd 798.2888 [M + H+], 798.2884 found. HPLC purity 97.0%.
tert-Butyl 4-(4-{4-[(2S)-2-Amino-5-{[(prop-2-en-1-yloxy)carbonyl]amino}pentanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (87)
Aniline 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)-5-{[(prop-2-en-1-yloxy)carbonyl]amino}pentanoic acid and deprotected using general procedure 11. Yellow solid, 14 mg (19% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.14 (br s, 1H), 8.72 (s, 1H), 8.39 (d, 1H, J = 8.7 Hz), 7.96 (m, 3H), 7.84–7.74 (m, 4H), 7.70 (d, 2H, J = 8.7 Hz), 6.16–6.07 (m, 1H), 5.85 (ddd, 1H, J = 5.5 Hz, 10.7 Hz, 22.5 Hz), 5.47 (dd, 1H, J = 1.1 Hz, 17.1 Hz), 5.39 (d, 1H, J = 10.4 Hz), 5.25 (d, 1H, J = 17.2 Hz), 5.15 (d, 1H, J = 10.3 Hz), 4.74–4.64 (m, 3H), 4.53 (d, 2H, J = 5.3 Hz), 4.25–4.15 (m, 1H), 3.36–3.24 (m, 1H), 3.22–3.09 (m, 1H), 2.12–1.99 (m, 1H), 1.98–1.86 (m, 1H), 1.82–1.65 (m, 2H), 1.59 (s, 9H), 1.35 (d, 6H, J = 6.1 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 165.5, 164.5, 162.8, 157.4, 149.5, 142.2, 141.5, 139.3, 137.5, 132.7, 132.3, 130.8, 130.0, 128.2, 127.5, 127.4, 121.8, 120.1, 119.8, 119.2, 117.9, 115.8, 81.0, 76.9, 75.1, 66.0, 54.1, 53.6, 39.9, 28.4, 26.1, 22.9. HRMS (ESI) calcd 744.3609 [M + H+], 744.3603 found. HPLC purity 98.8%.
4-(4-{4-[(2S)-5-Amino-2-{[4-(4-cyanobenzamido)phenyl]formamido}pentanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (28)
The amine 87 (17.5 μmol) was coupled with carboxylic acid 10 using general procedure 4. After deprotection of the tert-butyl ester using general procedure 7, the crude product was purified by RP HPLC. The product was obtained by deprotection with general procedure 5 followed by RP HPLC purification. White solid, 6.4 mg (48% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 10.72 (s, 1H), 10.53 (s, 1H), 9.08 (br s, 1H), 8.67 (d, 1H, J = 7.6 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.4 Hz), 7.98 (d, 2H, J = 8.8 Hz), 7.92–7.88 (m, 6H), 7.82 (d, 2H, J = 8.7 Hz), 7.79 (d, 2H, J = 8.6 Hz), 7.62–7.58 (m, 1H), 7.36–7.30 (m, 1H), 4.84 (br s, 1H), 4.66 (dd, 1H, J = 7.9 Hz, 14.2 Hz), 2.86 (t, 2H, J = 7.7 Hz), 1.95–1.85 (m, 2H), 1.77–1.64 (m, 2H), 1.22 (dd, 6H, J = 2.5 Hz, 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 171.2, 167.8, 167.2, 166.1, 164.5, 163.6, 144.1, 142.0, 141.7, 138.7, 137.1, 135.4, 132.5, 130.4, 129.2, 129.1, 128.6, 128.5, 128.0, 128.0, 123.4, 119.5, 119.2, 118.9, 118.3, 114.1, 72.1, 53.8, 38.7, 28.5, 24.0, 22.5. HRMS (ESI) calcd 812.3044 [M + H+], 812.3040 found. HPLC purity 97.7%.
tert-Butyl 4-(4-{4-[(2S)-2-Aminohex-5-ynamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (89)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)hex-5-ynoic acid and deprotected using general procedure 8 and deprotected using general procedure 2. Yellow solid, 35 mg (56% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.88 (br s, 1H), 8.74 (s, 1H), 8.46 (d, 1H, J = 8.9 Hz), 8.03 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.7 Hz), 7.87 (d, 2H, J = 8.8 Hz), 7.77 (d, 2H, J = 8.7 Hz), 7.72 (d, 2H, J = 8.8 Hz), 6.13 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 4.74 (hept., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.71 (dd, J = 4.4 Hz, 8.4 Hz), 2.44–2.39 (m, 2H), 2.26 (ddd, 1H, J = 4.5 Hz, 7.3 Hz, 11.8 Hz), 2.03 (t, 1H, J = 2.6 Hz), 1.81 (td, 1H, J = 6.5 Hz, 14.8 Hz), 1.59 (s, 9H), 1.37 (dd, 6H, J = 1.2 Hz, 6.1 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 172.8, 165.5, 164.4, 162.8, 149.4, 142.3, 141.5, 139.1, 137.7, 132.3, 130.8, 129.7, 128.2, 127.5, 127.4, 121.6, 120.1, 119.4, 119.1, 115.7, 83.1, 80.9, 76.9, 75.0, 70.1, 55.1, 33.0, 28.4, 22.9, 15.7. HRMS (ESI) calcd 655.3132 [M + H+], 655.3126 found.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}hex-5-ynamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (29)
The amine 89 (51.9 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. Beige solid, 21 mg (55% over 3 steps). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 12.81 (br s, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.60 (s, 1H), 10.47 (s, 1H), 9.40 (s, 1H), 8.63 (d, 1H, J = 7.4 Hz), 8.13 (d, 2H, J = 8.6 Hz), 8.04 (d, 2H, J = 8.4 Hz), 7.99–7.95 (m, 6H), 7.90 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 3H), 7.82 (d, 2H, J = 8.8 Hz), 7.71 (d, 1H, J = 8.9 Hz), 4.67 (dd, 1H, J = 7.4 Hz, 14.5 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 2.85 (t, 1H, J = 2.6 Hz), 2.43–2.30 (m, 2H), 2.07 (dd, 2H, J = 7.6 Hz, 14.7 Hz), 1.27 (dd, 6H, J = 6.1 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 171.1, 168.5, 166.9, 166.3, 164.4, 164.2, 154.1, 142.4, 142.0, 141.6, 138.7, 137.0, 136.3, 132.5, 130.2, 129.1, 128.6, 128.5, 128.4, 128.3, 126.3, 122.8, 120.7, 119.5, 119.0, 118.3, 114.0, 112.4, 112.1, 83.4, 74.9,71.8, 53.9, 30.3, 22.3, 15.2. HRMS (ESI) calcd 807.2779 [M + H+], 807.2773 found. HPLC purity 98.8%.
tert-Butyl 4-(4-{4-[(2S)-2-aminohept-6-ynamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (91)
Amine 8 (0.09 mmol) was coupled with (2S)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)hept-6-ynoic acid using general procedure 8 and deprotected using general procedure 2. Yellow solid, 41 mg (63% over 2 steps). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 9.88 (br s, 1H), 8.74 (s, 1H), 8.46 (d, 1H, J = 8.9 Hz), 8.04 (d, 1H, J = 8.9 Hz), 7.97 (d, 2H, J = 8.7 Hz), 7.87 (d, 2H, J = 8.7 Hz), 7.77 (d, 2H, J = 8.6 Hz), 7.72 (d, 2H, J = 8.8 Hz), 6.13 (ddt, 1H, 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dd, 1H, J = 1.3 Hz, 17.1 Hz), 5.40 (dd, 1H, J = 1.1 Hz, 10.4 Hz), 4.74 (hept., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 3.66–3.59 (m, 1H), 2.29–2.24 (m, 2H), 2.13–2.04 (m, 1H), 1.98 (t, 1H, J = 2.6 Hz), 1.82–1.73 (m, 1H), 1.73–1.65 (m, 2H), 1.59 (s, 9H), 1.37 (dd, 6H, J = 0.9 Hz, 6.1 Hz). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 173.0, 165.5, 164.5, 162.8, 149.4, 142.3, 141.5, 139.1, 137.7, 132.3, 130.8, 129.7, 128.3, 127.5, 127.4, 121.6, 120.1, 119.4, 119.2, 115.8, 83.7, 80.9, 76.9, 75.0, 69.3, 55.2, 33.9, 28.4, 24.8, 22.9, 18.4. HRMS (ESI) calcd 669.3288 [M + H+], 669.3283 found.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}hept-6-ynamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (30)
The amine 91 (55.3 μmol) was coupled with carboxylic acid 10 using general procedure 4. The product was obtained by deprotection with general procedures 6 and 7. Beige solid, 14 mg (29% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.70 (s, 1H), 10.61 (s, 1H), 10.49 (s, 1H), 9.40 (s, 1H), 8.62 (d, 1H, J = 7.3 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.99–7.95 (m, 6H), 7.89 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 3H), 7.82 (d, 2H, J = 8.8 Hz), 7.71 (d, 1H, J = 8.8 Hz), 4.61 (dd, 1H, J = 7.3 Hz, 14.6 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 2.81 (t, 1H, J = 2.6 Hz), 2.26–2.23 (m, 2H), 1.97–1.89 (m, 2H), 1.71–1.65 (m, 1H), 1.58–1.52 (m, 1H), 1.27 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 171.6, 168.5, 166.9, 166.1, 164.5, 164.2, 154.1, 142.4, 142.0, 141.6, 138.7, 137.1, 136.3, 132.5, 130.2, 129.1, 128.6, 128.5, 128.4, 128.4, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 84.2, 74.9, 71.6, 54.3, 30.7, 25.0, 22.3, 17.6. HRMS (ESI) calcd 821.2935 [M + H+], 821.2929 found. HPLC purity 98.7%.
tert-Butyl 1-[(4-{[4-({4-[(tert-Butoxy)carbonyl]phenyl}carbamoyl)-3-(prop-2-en-1-yloxy)-2-(propan-2-yloxy)phenyl]carbamoyl}phenyl)carbamoyl]-2-azabicyclo[2.1.1]hexane-2-carboxylate (93)
The aniline 8 (0.18 mol) was coupled with 2-(tert-butoxycarbonyl)-2-azabicyclo[2.1.1]hexane-1-carboxylic acid using general procedure 1. Yellow to orange solid, 88 mg (64%). 1H NMR (500 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 8.75 (br s, 1H), 8.50 (d, 1H, J = 8.9 Hz), 8.07 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.8 Hz), 7.90 (d, 2H, J = 8.8 Hz), 7.76–7.72 (m, 4H), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (dq, 1H, J = 1.4 Hz, 17.1 Hz), 5.41 (dq, 1H, J = 1.0 Hz, 10.4 Hz), 4.75 (quint. 1H, J = 6.2 Hz), 4.69 (dt, 2H, J = 1.2 Hz, 5.9 Hz), 3.58 (br s, 2H), 2.83 (t, 1H, J = 3.2 Hz), 2.31–2.28 (m, 2H), 1.73 (dd, 2H, J = 1.9 Hz, 4.7 Hz), 1.60 (s, 9H), 1.39 (d, 6H, J = 6.2 Hz), 1.36 (s, 9H). 13C NMR (126 MHz, CDCl3, 300 K): δ (ppm) = 167.1, 165.6, 164.4, 162.3, 158.5, 149.4, 142.3, 141.8, 139.1, 137.8, 132.2, 130.8, 129.6, 128.4, 127.6, 127.4, 121.6, 120.2, 119.2, 119.2, 115.8, 81.7, 80.9, 75.1, 73.5, 54.2, 42.7, 34.8, 28.4, 28.3, 23.0. HPLC purity 95.0%.
4-[4-(4-{2-[4-(4-Cyanobenzamido)benzoyl]-2-azabicyclo[2.1.1]hexane-1-amido}benzamido)-2-hydroxy-3-(propan-2-yloxy)benzamido]benzoic Acid (31)
Step 1:83 mg carbamate 93 (0.11 mmol, 1 equiv) was added to a dry flask and further dried under high vacuum. 0.9 mL dry DCM was added under argon atmosphere and the solution was cooled down to 0 °C. 0.5 mL trifluoroacetic acid (6.5 mmol, 59.3 equiv) was added under argon atmosphere. The solution was stirred for 3 h at 0 °C and controlled over LCMS. After completion, 1 mL DCM was added and the solvent was concentrated under reduced pressure. The residue was coevaporated with DCM three times. The crude product was dried under high vacuum.
Step 2:35 mg carboxylic acid 10 (0.13 mmol, 1.2 equiv) and 46 mg HATU (0.12 mmol, 1.1 equiv) were added to a dry vial and dried under high vacuum. 0.8 mL dry DMF and 76 μL dry DIPEA (0.44 mmol, 4 equiv) were added under argon atmosphere and the reaction was stirred for 30 min at 0 °C. The solution was added to the crude amine trifluoroacetate (0.11 mmol, 1 equiv) in a dry vial under argon atmosphere and stirred at 0 °C. The reaction was controlled over LCMS. After full consumption of the amine, 40 μL aniline was added under argon atmosphere and the reaction was stirred for another 30 min at 0 °C. Six mg tetrakis(triphenylphosphine)-palladium(0) (5.2 μmol, 0.05 equiv) and 1.5 mL dry THF were added under argon atmosphere. The reaction was controlled over LCMS. After full deprotection, the solvent was concentrated under reduced pressure. The DMF was coevaporated with n-heptane. The crude product was purified by RP-HPLC. Off-white solid, 43.5 mg (49% over 3 steps). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 12.81 (br s, 1H), 12.29 (br s, 1H), 10.70 (br s, 1H), 9.86 (s, 1H), 9.37 (br s, 1H), 8.11 (d, 2H, J = 8.5 Hz), 8.04 (d, 2H, J = 8.4 Hz), 7.98–7.92 (m, 4H), 7.89 (d, 2H, J = 8.6 Hz), 7.87–7.82 (m, 5H), 7.78 (d, 2H, J = 8.3 Hz), 7.68 (d, 1H, J = 8.8 Hz), 4.55 (hept., 1H, J = 6.1 Hz), 3.64 (br s, 2H), 2.82–2.80 (m, 1H), 2.22–2.17 (m, 2H), 1.78 (d, 2H, J = 3.4 Hz), 1.26 (d, 6H, J = 6.2 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 171.9, 168.5, 167.1, 167.0, 164.6, 164.3, 154.4, 142.9, 142.2, 141.5, 138.8, 137.1, 136.5, 132.6, 130.3, 130.0, 129.3, 128.7, 128.2, 128.0, 126.2, 122.9, 120.7, 119.6, 119.3, 118.4, 114.1, 112.6, 112.2, 74.8, 72.2, 55.1, 41.4, 34.5, 22.4. HRMS (ESI) calcd 807.2773 [M + H+], 807.2776 found. HPLC purity 97.0%.
tert-Butyl 4-(4-{4-[(2S)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (95)
Amine 8 (200 mg, 0.37 mmol) and Fmoc-l-valine (174 mg, 0.51 mmol, 1.40 equiv) were dissolved in EtOAc (800 μL) and pyridine (180 μL, 2.20 mmol, 6.00 equiv) was added. T3P (50% in EtOAc, 900 μL, 1.47 mmol, 4.00 equiv) was added dropwise at 0 °C. The mixture was stirred for 4 h while warming to rt. The mixture was diluted with EtOAc and the organic phase was washed with 1 M HCl solution, sat. NaHCO3 solution, brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (dry load, 40% EtOAc in hexane) to furnish the product (324 mg, quant.) as yellowish amorphous solid, which was directly used in the next step.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (96)
Step 1: Carbamate 95 (324 mg, 0.37 mmol, 1.00 equiv) was dissolved in MeCN (4 mL) and piperidine (1 mL) was added. The mixture was stirred at rt for 3 h, before it was concentrated under reduced pressure. The residue was coevaporated with MeCN (3×) to furnish the amine, which was used in the next step without further purification. Step 2: Acid 10 (136 mg, 0.51 mmol, 1.40 equiv) and HATU (195 mg, 0.51 mmol, 1.40 equiv) were dissolved in DMF (9 mL) and DIPEA (200 μL, 1.10 mmol, 3.00 equiv) was added. The mixture was stirred for 5 min before it was transferred to a stirred solution of the amine (236 mg, 0.37 mmol, 1.00 equiv) in DMF (5 mL). The resulting mixture was stirred at rt for 23 h. EtOAc was added and the organic phase was washed with 0.1 M HCl solution, brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (2% MeOH in CH2Cl2) to furnish the product (168 mg, 0.19 mmol, 51% over three steps) as yellowish amorphous solid. [α]D22 = +5.5° (c 0.1, MeOH). 1H NMR (600 MHz, DMSO-d6) = δ 10.69 (s, 1H), 10.53 (s, 1H), 10.52 (s, 1H), 9.52 (s, 1H), 8.49–8.47 (d, J = 8.1 Hz, 1H), 8.14–8.12 (d, J = 8.6 Hz, 2H), 8.05–8.04 (d, J = 8.6 Hz, 2H), 7.99–7.96 (m, 4H), 7.90–7.88 (m, 4H), 7.84–7.81 (m, 5H), 7.41–7.40 (d, J = 8.5 Hz, 1H), 6.05–5.98 (m, 1H), 5.39–5.35 (dq, J = 1.6, 17.2 Hz, 1H), 5.21–5.19 (dq, J = 1.6, 10.5 Hz, 1H), 4.61–4.60 (d, J = 5.5 Hz, 1H), 4.53–4.47 (sept, J = 6.1 Hz, 1H), 4.45–4.42 (t, J = 8.3 Hz, 1H), 2.28–2.20 (m, 1H), 1.55 (s, 9H), 1.26–1.25 (d, J = 6.1 Hz, 6H), 1.04–0.99 (dd, J = 6.7 Hz, 6H) ppm. 13C NMR (151 MHz, DMSO-d6) = δ 171.2, 166.2, 164.6, 164.5, 164.4, 164.3, 149.5, 143.0, 142.5, 142.1, 141.5, 138.7, 135.7, 133.6, 132.5, 130.1, 129.3, 128.6, 128.5, 128.4, 127.1, 126.0, 123.6, 119.5, 118.9, 118.8, 118.7, 118.3, 117.8, 114.0, 80.3, 76.2, 74.8, 60.3, 30.0, 27.9, 22.3, 19.3, 19.2 ppm. HRMS (ESI+) calcd for C51H52N6O9Na [M + Na]+: 915.3693; found: 915.3715.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (97)
Allyl ether 96 (152 mg, 0.17 mmol, 1.00 equiv) was dissolved in THF (9 mL). Aniline (50.0 μL, 0.56 mmol, 3.30 equiv) and Pd(PPh3)4 (20.0 mg, 0.02 mmol, 10 mol %) were added and the mixture was stirred at rt for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by column chromatography (dry load, 3% MeOH in CH2Cl2) to obtain a brown amorphous solid (169 mg), which was stirred in CH2Cl2. The mixture was filtered and the precipitate was washed with CH2Cl2 to furnish the product (78.4 mg, 0.09 mmol, 54%) as beige amorphous solid. [α]D22 = +7.8° (c 0.2, MeOH). 1H NMR (600 MHz, DMSO-d6) = δ 12.29 (s, 1H), 10.69 (s, 1H), 10.61 (s, 1H), 10.54 (s, 1H), 9.39 (s, 1H), 8.49–8.48 (d, J = 8.1 Hz, 1H), 8.14–8.12 (d, J = 8.5 Hz, 2H), 8.05–8.04 (d, J = 8.6 Hz, 2H), 7.97–7.96 (m, 4H), 7.93–7.92 (d, J = 8.7 Hz, 2H), 7.89–7.88 (d, J = 8.8 Hz, 2H), 7.86–7.82 (m, 5H), 7.71–7.70 (d, J = 8.7 Hz, 1H), 4.58–4.52 (sept, J = 6.1 Hz, 1H), 4.45–4.42 (t, J = 8.3 Hz, 1H), 2.26–2.21 (m, 1H), 1.55 (s, 9H), 1.27–1.26 (dd, J = 1.2, 6.1 Hz, 6H), 1.04–0.99 (dd, J = 6.7 Hz, 6H) ppm. 13C NMR (151 MHz, DMSO-d6) = δ 171.2, 168.5, 166.2, 164.5, 164.4, 164.2, 154.1, 142.2, 142.0, 141.5, 138.7, 137.0, 136.3, 132.5, 129.9, 129.3, 128.6, 128.5, 128.4, 126.8, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 80.5, 74.8, 60.3, 30.0, 27.8, 22.3, 19.3, 19.2 ppm. HRMS (ESI+) calcd for C48H47N6O9 [M – H]−: 851.3405; found: 851.3419.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (32)
Ester 97 (34.7 mg, 0.04 mmol, 1.00 equiv) was dissolved in precooled TFA (2 mL) at 0 °C. The mixture was stirred for 30 min while warming to rt. Et2O was added at 0 °C. The precipitate was filtered off and washed with an excess of Et2O to furnish the product (21.4 mg, 0.03 mmol, 66%) as beige amorphous solid. [α]D22 = +5.7° (c 0.1 MeOH). 1H NMR (500 MHz, DMSO-d6) = δ 12.82 (s, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.60 (s, 1H), 10.54 (s, 1H), 9.40 (s, 1H), 8.49 (d, J = 8.0 Hz, 1H), 8.14–8.12 (d, J = 8.5 Hz, 2H), 8.06–8.04 (d, J = 8.4 Hz, 2H), 7.97–7.95 (m, 6H), 7.89–7.82 (m, 7H), 7.72–7.70 (d, J = 8.9 Hz, 1H), 4.59–4.51 (sept, J = 6.1 Hz, 1H), 4.46–4.42 (t, J = 8.4 Hz, 1H), 2.28–2.19 (sept, J = 6.7 Hz, 1H), 1.27–1.26 (d, J = 6.1 Hz, 6H), 1.04–0.99 (dd, J = 6.7, 18.5 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = δ 171.2, 168.5, 166.9, 166.2, 164.4, 164.2, 154.1, 142.2, 142.0, 141.5, 138.7, 138.6, 137.0, 136.3, 132.5, 130.2, 129.3, 128.6, 128.5, 128.4, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 74.8, 60.3, 30.0, 22.3, 19.3, 19.2 ppm. HRMS (ESI+) calcd for C44H39N6O9 [M – H]−: 795.2779; found: 795.2773.
tert-Butyl 4-(4-{4-[(2R)-2-{[(tert-Butoxy)carbonyl]amino}-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (98)
Aniline 8 (200 mg, 0.37 mmol) and Boc-d-Val (112 mg, 0.51 mmol, 1.40 equiv) were dissolved in EtOAc (1 mL) and pyridine (87 μL, 1.10 mmol, 3.00 equiv) was added. T3P (50% in EtOAc, 440 μL, 0.73 mmol, 2.00 equiv) was added at 0 °C and the mixture was stirred at rt for 4 h. EtOAc was added and the organic phase was washed with a 1 M HCl solution, a sat. NaHCO3 solution, brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (1% MeOH in CH2Cl2) to furnish the product (232 mg, 0.31 mmol, 85%) as yellowish amorphous solid. [α]D22 = +1.6° (c 0.2, MeOH). 1H NMR (600 MHz, DMSO-d6) = 10.52 (s, 1H), 10.30 (s, 1H), 9.51 (s, 1H), 7.98–7.96 (d, J = 8.6 Hz, 2H), 7.90–7.88 (d, J = 8.6 Hz, 2H), 7.84–7.82 (m, 3H), 7.78–7.77 (d, J = 8.6 Hz, 2H), 7.41–7.40 (d, J = 8.5 Hz, 1H), 6.76–6.74 (d, J = 8.7 Hz, 1H), 6.05–5.99 (m, 1H), 5.39–5.38 (d, J = 17.2 Hz, 1H), 5.21–5.19 (d, J = 10.3 Hz, 1H), 4.61–4.60 (d, J = 5.4 Hz, 1H), 4.53–4.47 (sept, J = 6.2 Hz, 1H), 3.96–3.94 (t, J = 7.8 Hz, 1H), 2.02–1.99 (m, 1H), 1.55 (s, 9H), 1.40 (s, 9H), 1.27–1.26 (d, J = 6.1 Hz, 6H), 0.92–0.91 (d, J = 6.5 Hz, 6H) ppm. 13C NMR (151 MHz, DMSO-d6) = 171.38, 164.6, 164.5, 164.3, 155.7, 149.5, 143.0, 142.5, 142.1, 135.7, 133.6, 130.1, 128.5, 128.3, 127.1, 126.0, 123.6, 118.9, 118.8, 118.7, 117.8, 80.3, 78.1, 76.2, 74.3, 60.8, 30.2, 28.1, 27.9, 22.3, 19.2, 18.5 ppm. HRMS (ESI) calcd for C41H52N4O9Na [M + Na]+: 767.3632; found: 767.3619.
tert-Butyl 4-(4-{4-[(2R)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (99)
Step 1: Carbamate 98 (220 mg, 0.30 mmol) was dissolved in HCl (4 M in 1,4-dioxane, 7.30 mL, 29.5 mmol, 100 equiv) at 0 °C. The mixture was warmed up to rt and stirred for 15 min. The solution was transferred to a stirred suspension of EtOAc (190 mL) and a sat. NaHCO3 solution (190 mL). The aqueous phase was extracted with EtOAc and the combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was used in the next step without further purification. Step 2: Compound 99 (129 mg, 0.14 mmol, 49%) was synthesized from carboxylic acid 10 (94.4 mg, 0.35 mmol) and the amine (190 mg, 0.30 mmol) in accordance to the procedure of compound 96. [α]D21 = −4.9° (c 0.2, MeOH).
tert-Butyl 4-(4-{4-[(2R)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (100)
Compound 100 (88.3 mg, 0.10 mmol, 75%) was synthesized from allyl ether 99 (124 mg, 0.14 mmol) in accordance to the procedure of compound 97. [α]D21 = −3.1° (c 0.1, MeOH).
4-(4-{4-[(2R)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (33)
33 (52.1 mg, 0.07 mmol, 65%) was synthesized from ester 100 (85.7 mg, 0.10 mmol) in accordance to the procedure of compound 32. [α]D21 = −6.1° (c 0.1 MeOH).
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-(methylamino)propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (34)
Amine 8 (0.15 mmol) was coupled with (2S)-3-{[(tert-butoxy)carbonyl](methyl)amino}-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoic acid using general procedure 1, deprotected and coupled with carboxylic acid 10 using general procedure 10 and deprotected by general procedures 6 and 7. Off-white solid, 28 mg (24% over 5 steps). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 10.72 (s, 1H), 9.26 (s, 1H), 8.81 (d, 1H, J = 6.2 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.04 (d, 2H, J = 8.4 Hz), 7.99 (d, 2H, J = 8.8 Hz), 7.96–7.89 (m, 6H), 7.84–7.80 (m, 4H), 7.75 (d, 1H, J = 8.8 Hz), 7.54 (d, 1H, J = 8.8 Hz), 4.96–4.89 (m, 1H), 4.68 (hept. 1H, J = 6.1 Hz), 3.25 (ddd, J = 6.7 Hz, 12.5 Hz, 20.8 Hz), 2.55 (s, 3H), 1.25 (d, 6H, J = 6.1 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 169.1, 168.1, 167.3, 166.3, 164.5, 163.9, 142.7, 142.0, 141.8, 138.6, 136.7, 136.2, 132.5, 130.2, 129.0, 128.6, 128.5, 128.2, 126.1, 123.1, 120.0, 119.5, 119.2, 118.3, 114.1, 113.4, 73.7, 52.3, 50.3, 34.3, 22.4. HRMS (ESI) calcd 798.2882 [M + H+], 798.2883 found. HPLC purity 98.0%.
(2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-formamidopropanoic Acid (104)
The compound was prepared according to the established literature procedure; see M. Serafini, A. Griglio, S. Viarengo, S. Aprile, T. Pirali, Org. Biomol. Chem.2017, 15, 6604–6612. 100 mg Amine 103 (0.31 mmol, 1 equiv) was dissolved in 0.3 mL formic acid. The flask was cooled down to 0 °C and 0.23 mL acetic anhydride (2.44 mmol, 8 equiv) was added. The flask was sealed and the reaction was stirred overnight. After completion, the 1 mL water was added. The residue was lyophilized to remove the solvent. White solid, 97.8 mg (90%). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 12.85 (br s, 1H), 8.10 (t, 1H, J = 5.6 Hz), 8.03 (d, 1H, J = 1.1 Hz), 7.90 (d, 2H, J = 7.6 Hz), 7.72 (d, 2H, J = 7.4 Hz), 7.61 (d, 1H, J = 8.3 Hz), 7.42 (t, 2H, J = 7.4 Hz), 7.33 (t, 2H, J = 7.5 Hz), 4.32–4.28 (m, 2H), 4.23 (t, 1H, J = 6.9 Hz), 4.10 (td, 1H, J = 5.1 Hz, 8.2 Hz), 3.55 (dt, 1H, J = 5.4 Hz, 13.3 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 171.9, 161.6, 156.0, 143.8, 140.7, 127.7, 127.1, 125.3, 120.1, 65.7, 53.7, 46.6, 38.3. HPLC purity 98.2%.
tert-Butyl 4-(4-{4-[(2S)-2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-formamidopropanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (35)
The aniline 8 (0.18 mol) was coupled with carboxylic acid 104 using general procedure 1, coupled with 10 using general procedure 10 and deprotected by general procedures 6 and 7. Off-white solid, 16 mg (11% over 4 steps). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 12.77 (br s, 1H), 12.31 (br s, 1H), 10.72 (s, 1H), 10.48 (s, 1H), 9.36 (s, 1H), 8.64 (d, 1H, J = 7.2 Hz), 8.30 (dd, 1H, J = 5.6 Hz, 6.6 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.10 (d, 1H, J = 1.3 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.97–7.93 (m, 6H), 7.91 (d, 2H, J = 8.9 Hz), 7.85 (d, 2H, J = 8.8 Hz), 7.83–7.79 (m, 3H), 7.63 (d, 1H, J = 7.9 Hz), 4.70 (q, 1H, J = 6.5 Hz), 4.63–4.55 (m, 1H), 3.64 (t, 2H, J = 6.1 Hz), 1.26 (d, 6H, J = 6.1 Hz). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 169.3, 168.4, 166.9, 165.9, 164.5, 164.1, 162.1, 142.3, 142.2, 141.7, 138.7, 136.7, 136.5, 132.5, 130.2, 129.0, 128.6, 128.6, 128.4, 128.2, 122.9, 120.5, 119.6, 119.1, 118.3, 114.1, 74.4, 54.9, 38.8, 22.4. HRMS (ESI) calcd 812.2675 [M + H+], 812.2675 found. HPLC purity 99.4%.
Prop-2-en-1-yl (2S)-3-[({[(tert-Butoxy)carbonyl]amino}sulfonyl)amino]-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoate (106)
The compound was prepared according to the established literature procedure; see R.H. Crampton, M. Fox, S. Woodward, Tetrahedron: Asymmetry2013, 24, 599–605. In a dry flask, 50.0 μL tert-butanol (0.52 mmol, 1.05 equiv) was dissolved in 0.7 mL dry toluene under argon atmosphere and cooled down to 0 °C. 45.0 μL chlorosulfonyl isocyanate (0.52 mmol, 1.04 equiv) was added dropwise. The mixture was stirred for 45 min at 0 °C. 0.12 mL dry pyridine (1.49 mmol, 3 equiv) was added dropwise under argon atmosphere and a precipitate formed. The supernatant was added to 200 mg amine hydrochloride 105·HCl (0.5 mmol, 1 equiv) under argon atmosphere at 0 °C. The remaining precipitate was transferred after addition of 1.6 mL dry acetonitrile. The reaction was warmed up to room temperature and stirred overnight. After completion, 0.1 mL methanol was added and the solvent was concentrated under reduced pressure. The crude product was purified by flash chromatography. White solid, 148 mg (55%). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 10.98 (br s, 1H), 7.90 (d, 2H, J = 7.5 Hz), 7.70 (d, 2H, J = 7.9 Hz), 7.66 (t, 1H, J = 6.3 Hz), 7.42 (t, 2H, J = 7.4 Hz), 7.34 (t, 2H, J = 7.2 Hz), 5.88 (ddd, 1H, J = 5.2 Hz, 10.5 Hz, 22.1 Hz), 5.31 (d, 1H, J = 17.3 Hz), 5.19 (d, 1H, J = 10.5 Hz), 4.62–4.54 (m, 2H), 4.36–4.22 (m, 4H), 3.31–3.26 (m, 2H), 1.42 (s, 9H). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 169.8, 155.9, 150.6, 143.7, 140.7, 132.3, 127.7, 127.1, 125.2, 120.2, 117.7, 81.5, 65.8, 65.1, 53.9, 46.6, 31.3, 27.7. HPLC purity 96.1%.
(2S)-3-[({[(tert-Butoxy)carbonyl]amino}sulfonyl)amino]-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoic Acid (107)
144.0 mg ester 106 (0.26 mmol, 1 equiv) was added to a dry vial and further dried under high vacuum. 1.7 mL dry THF and 0.1 mL phenylsilane (0.81 mmol, 3.1 equiv) and 8 mg tetrakis(triphenylphosphine)palladium(0) (7 μmol, 0.026 equiv) were added under argon atmosphere. The reaction was stirred for 3 h at room temperature and controlled over LCMS. After completion, the crude product was directly purified by flash chromatography (DCM/MeOH). Brown solid, 113 mg (84%). 1H NMR (500 MHz, DMSO-d6, 300 K): δ (ppm) = 12.85 (br s, 1H), 10.99 (br s, 1H), 7.90 (d, 2H, J = 7.5 Hz), 7.72 (d, 2H, J = 7.4 Hz), 7.65–7.60 (m, 1H), 7.42 (t, 2H, J = 7.5 Hz), 7.34 (t, 2H, J = 7.4 Hz), 4.34–4.12 (m, 4H), 3.29–3.21 (m, 2H), 1.42 (s, 9H). 13C NMR (126 MHz, DMSO-d6, 300 K): δ (ppm) = 171.5, 155.9, 150.7, 143.8, 140.7, 128.8, 127.7, 127.1, 125.2, 120.1, 81.4, 65.8, 53.7, 46.6, 43.9, 27.7. HPLC purity 97.6%.
tert-Butyl 4-(4-{4-[(2S)-3-[({[(tert-Butoxy)carbonyl]amino}sulfonyl)amino]-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (108)
The aniline 8 (0.16 mol) was coupled with carboxylic acid 107 using general procedure 1. Yellow solid, 97 mg (57%). 1H NMR (700 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 8.99 (br s, 1H), 8.73 (s, 1H), 8.48 (d, 1H, J = 8.9 Hz), 8.06 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.6 Hz), 7.87 (d, 2H, J = 8.6 Hz), 7.76 (d, 2H, J = 7.6 Hz), 7.73 (d, 2H, J = 8.7 Hz), 7.68 (d, 2H, J = 8.5 Hz), 7.61–7.58 (m, 3H), 7.39 (dd, 2H, J = 6.4 Hz, 13.3 Hz), 7.32–7.27 (m, 2H), 6.19 (br s, 1H), 6.14 (ddt, 1H, J = 5.9 Hz, 10.5 Hz, 16.4 Hz), 6.05 (br s, 1H), 5.49 (dd, 1H, J = 1.2 Hz, 17.1 Hz), 5.40 (d, 1H, J = 10.4 Hz), 4.75 (quint., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 4.56–4.51 (m, 1H), 4.50–4.44 (m, 2H), 4.23 (t, 1H, J = 7.0 Hz), 3.68–3.54 (m, 2H), 1.60 (s, 9H), 1.51 (s, 9H), 1.36 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, CDCl3, 300 K): δ (ppm) = 168.5, 165.6, 164.3, 162.8, 157.5, 150.4, 149.4, 143.6, 143.5, 142.3, 141.5, 140.9, 139.1, 137.6, 132.3, 130.8, 130.5, 128.3, 128.1, 127.6, 127.5, 127.3, 125.2, 121.8, 120.3, 120.2, 120.1, 119.2, 115.8, 84.8, 81.0, 75.1, 68.1, 54.7, 47.1, 45.3, 28.4, 28.1, 23.0. HPLC purity 95.3%.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-(sulfamoylamino)propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (36)
The Fmoc protected amino acid 108 (88.0 μmol) was deprotected and coupled with carboxylic acid 10 using general procedure 10. The product was obtained by deprotection with general procedures 5 and 7. Off-white solid, 38.5 mg (51% over 3 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.29 (br s, 1H), 10.71 (s, 1H), 10.62 (br s, 1H), 10.39 (s, 1H), 9.41 (s, 1H), 8.48 (d, 1H, J = 7.2 Hz), 8.13 (d, 2H, J = 8.4 Hz), 8.05 (d, 2H, J = 8.3 Hz), 7.98–7.95 (m, 6H), 7.91 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 3H), 7.82 (d, 2H, J = 8.7 Hz), 7.70 (d, 1H, J = 8.8 Hz), 6.86 (t, 1H, J = 6.5 Hz), 6.72 (s, 2H), 4.77 (q, 1H, J = 6.5 Hz), 4.55 (hept., 1H, J = 6.0 Hz), 3.46–3.40 (m, 2H), 1.26 (d, 6H, J = 6.1 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 169.3, 168.5, 166.9, 165.9, 164.5, 164.2, 154.2, 142.3, 142.0, 141.7, 138.7, 137.0, 136.4, 132.5, 130.2, 129.0, 128.6, 128.5, 128.4, 128.3, 126.3, 122.8, 120.7, 119.5, 119.1, 118.3, 114.1, 112.5, 112.2, 74.8, 54.6, 43.9, 22.3. HRMS (ESI) calcd 863.2454 [M + H+], 863.2456 found. HPLC purity 99.5%.
2-Amino-3-methyl-3-nitrobutanoic Acid (112)
To a solution of KOH (14.1 g, 251 mmol, 2.20 equiv) in H2O (280 mL), 2-nitropropane (20.0 mL, 223 mmol, 1.95 equiv)3 (25% in H2O, 0.93 mol, 150 mL, 8.54 equiv) and a solution of glyoxylic acid monohydrate (10.5 g, 114 mmol, 1.00 equiv) in H2O (40.0 mL) were added. The mixture was stirred for 2 h at ambient temperature. Then, the reaction was terminated by adjusting the pH to 0 with conc. HCl whereby a blue coloration of the solution appeared. The aqueous phase was washed with CH2Cl2 (3×) and the solvent was removed under reduced pressure. The residue was diluted in EtOH, filtered and the filtrate was concentrated to about half under reduced pressure, mixed in equal parts with Et2O, filtered again and aniline (10–30 mL) was added to the filtrate until a turbidity of the solution appeared. The turbid solution was stored at 2–8 °C for 16 h. It was then filtered, the solid was washed with EtOH and dried under high vacuum for 16 h. 2-Amino-3-methyl-3-nitrobutanoic acid (10.0 g, 61.8 mmol, 54%) was obtained as a colorless solid. 1H NMR (D2O, 400 MHz): δ 4.35 (s, 1H), 1.81 (s, 3H), 1.77 (s, 3H) ppm. 13C NMR (D2O, 100 MHz): δ 170.2, 88.3, 60.8, 24.6, 23.5 ppm.
2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-methyl-3-nitrobutanoic Acid (113)
Amine 112 (1.03 g, 6.43 mmol) was dissolved in 1,4-dioxane (30 mL) and a 10% Na2CO3 solution was added at 0 °C, followed by a solution of FmocCl (1.83 g, 7.07 mmol, 1.10 equiv) in 1,4-dioxane (30 mL) via a dropping funnel. The mixture was stirred for 18 h while warming to rt. Afterward, the mixture was diluted with H2O and Et2O. The aqueous phase was washed with Et2O (2×), acidified with a 6 M HCl solution and extracted with EtOAc (2×). The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was coevaporated with PhMe (4×), MeOH (3×) and CH2Cl2 (4×) to furnish the product (1.40 g, 3.65 mmol, 57%) as colorless solid. 1H NMR (400 MHz, DMSO-d6) = δ 13.40 (s, 1H), 8.12–8.09 (d, J = 9.9 Hz, 1H), 7.91–7.89 (d, J = 7.6 Hz, 2H), 7.73 (d, J = 6.5 Hz, 2H), 7.42 (t, J = 7.4 Hz, 2H), 7.32 (dt, J = 1.0 Hz, 7.4 Hz, 2H), 4.98 (d, J = 9.9 Hz, 1H), 4.40 (dd, J = 7.2, 10.4 Hz, 1H), 4.35–4.31 (m, 1H), 4.27–4.23 (m, 1H), 1.58 (s, 3H), 1.50 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) = δ 169.8, 156.6, 143.6, 143.6, 140.7, 140.7, 127.8, 127.8, 127.1, 127.1, 125.3, 125.3, 120.2, 120.2, 88.4, 66.1, 59.2, 46.6, 24.7, 21.1 ppm. HRMS (ESI) calcd for C20H19N2O6 [M – H]−: 383.1243, found: 383.1244.
tert-Butyl 4-(4-{4-[2-({[(9H-Fluoren-9-yl)methoxy]carbonyl}amino)-3-methyl-3-nitrobutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (114)
Amine 8 (100 mg, 0.18 mmol) and the carboxylic acid 113 (98.6 mg, 0.26 mmol, 1.40 equiv) were dissolved in EtOAc (400 μL) and pyridine (45 μL, 0.55 mmol, 3.00 equiv) was added. T3P (50% in EtOAc, 200 μL, 0.33 mmol, 1.80 equiv) was added dropwise at 0 °C. The mixture was stirred at 0 °C for 2 h. H2O was added and the aqueous phase was extracted with EtOAc (3×). The combined organic phases were washed with a sat. NaHCO3 solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (dry load, PE/EtOAc = 2:1) to furnish the product (127 mg, 0.14 mmol, 76%) as yellowish solid. 1H NMR (500 MHz, DMSO-d6) = 10.73 (s, 1H), 10.53 (s, 1H), 9.56 (s, 1H), 8.25 (d, J = 8.6 Hz, 1H), 7.99 (d, J = 8.7 Hz, 2H), 7.90–7.88 (m, 4H), 7.84–7.75 (m, 7H), 7.43–7.39 (m, 3H), 7.34–7.29 (m, 2H), 6.06–5.98 (m, 1H), 5.39–5.35 (dq, J = 1.6, 17.2 Hz, 1H), 5.21–5.17 (m, 2H), 4.60 (d, J = 5.5 Hz, 2H), 4.50 (sept, J = 6.1 Hz, 1H), 4.42–4.36 (m, 1H), 4.29–4.23 (m, 2H), 1.67 (s, 3H), 1.64 (s, 3H), 1.55 (s, 9H), 1.26 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 166.8, 164.6, 164.5, 164.2, 156.2, 149.5, 143.7, 143.7, 143.0, 142.7, 141.4, 140.7, 135.6, 133.7, 130.1, 129.1, 128.5, 127.7, 127.7, 127.2, 127.1, 127.1, 126.0, 125.4, 125.4, 123.6, 120.1, 120.1, 119.4, 119.1, 118.8, 117.8, 89.0, 80.3, 76.3, 74.3, 66.2, 60.5, 46.6, 27.9, 23.2, 22.3 ppm. HRMS (ESI) calcd for C51H53N5O11Na [M + Na]+: 934.3639; found: 934.3634.
tert-Butyl 4-{4-[4-(2-Amino-3-methyl-3-nitrobutanamido)benzamido]-2-hydroxy-3-(propan-2-yloxy)benzamido}benzoate (115)
The carbamate 114 (400 mg, 0.44 mmol) was dissolved in piperidine (1.3 mL) and MeCN (5.3 mL) and the mixture was stirred at rt for 2 h, before it was concentrated under reduced pressure and coevaporated with MeCN (3×) to furnish the product (quant.) as yellow solid, which was used in the next step without further purification.
tert-Butyl 4-{4-[4-(2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methyl-3-nitrobutanamido)benzamido]-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido}benzoate (116)
DIPEA (230 μL, 1.32 mmol, 3.00 equiv) was added dropwise to a stirred solution of HATU (200 mg, 0.53 mmol, 1.20 equiv) and the carboxylic acid 10 (140 mg, 0.53 mmol, 1.20 equiv) in DMF (11 mL). The solution was stirred for 5 min and then transferred to a stirred solution of the amine 115 (303 mg, 0.44 mmol) in DMF (6 mL) at 0 °C. The reaction mixture was stirred at 20 h while warming to rt. The mixture was diluted with EtOAc and washed with a 1 M HCl solution, brine, dried over MgSO4 and concentrated under reduced pressure. The crude residue was purified by column chromatography (2% MeOH in CH2Cl2) to furnish the product (315 mg, 0.34 mmol, 77%) as orange amorphous solid. 1H NMR (500 MHz, DMSO-d6) δ = 10.71 (s, 1H), 10.70 (s, 1H), 10.53 (s, 1H), 9.56 (s, 1H), 8.77 (d, J = 9.2 Hz, 1H), 8.13 (d, J = 8.5 Hz, 2H), 8.05 (d, J = 8.5 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H), 7.91–7.88 (m, 4H), 7.84–7.79 (m, 5H), 7.41 (d, J = 8.4 Hz, 1H), 6.05–5.98 (m, 1H), 5.70 (d, J = 9.3 Hz, 1H), 5.37 (dq, J = 1.6, 17.2 Hz, 1H), 5.20 (dq, J = 1.5, 10.5 Hz, 1H), 4.61 (d, J = 5.4 Hz, 2H), 4.49 (sept, J = 6.1 Hz, 1H), 1.76 (s, 3H), 1.69 (s, 3H), 1.55 (s, 9H), 1.26 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) δ = 168.6, 166.8, 166.7, 164.6, 164.5, 164.2, 149.5, 143.0, 142.6, 141.9, 141.5, 138.6, 135.6, 133.7, 132.5, 130.1, 129.1, 128.9, 128.7, 128.6, 128.4, 127.6, 127.2, 126.0, 123.6, 119.9, 119.5, 119.4, 118.8, 117.8, 114.1, 89.0, 80.3, 76.2, 74.3, 58.5, 27.9, 23.2, 22.9, 22.3 ppm. HRMS (ESI) calcd for C51H51N7O11Na [M + Na]+: 960.3544; found: 960.3557.
tert-Butyl 4-{4-[4-(2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methyl-3-nitrobutanamido)benzamido]-2-hydroxy-3-(propan-2-yloxy)benzamido}benzoate (117)
The allyl ether 116 (56.8 mg, 0.06 mmol) was dissolved in THF (2.7 mL). Aniline (18 μL, 0.20 mmol, 3.30 equiv) and Pd(PPh3)4 (7.0 mg, 6 μmol, 0.10 equiv) were added subsequently and the resulting mixture was stirred at rt for 90 min. The mixture was concentrated under reduced pressure. The crude residue was purified by column chromatography (dry load, 2% MeOH in CH2Cl2) to furnish the product (57.3 mg, 0.05 mmol, 84%) as yellow solid, which was of 80% purity. The compound was used in the next step without further purification. HRMS (ESI) calcd for C48H46N7O11 [M – H]−: 896.3255; found: 896.3212.
4-{4-[4-(2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methyl-3-nitrobutanamido) benzamido]-2-hydroxy-3-(propan-2-yloxy)benzamido}benzoic Acid (37)
The tert-butyl ester (19.8 mg, 0.02 mmol) was dissolved in precooled TFA (1.1 mL) at 0 °C with stirring. The solution was warmed up to rt over 30 min. Et2O was added at 0 °C. The precipitate was filtered off, washed with excess of Et2O and dried in vacuo to furnish the product (11.4 mg, 0.01 mmol, 61%) as colorless solid. 1H NMR (500 MHz, DMSO-d6) δ = 12.82 (s, 1H), 12.28 (s, 1H), 10.71 (bs, 2H), 10.60 (s, 1H), 9.44 (s, 1H), 8.77 (d, J = 9.3 Hz, 1H), 8.13 (d, J = 8.6 Hz, 2H), 8.05 (d, J = 8.6 Hz, 2H), 7.98–7.94 (m, 6H), 7.91 (d, J = 8.9 Hz, 2H), 7.86–7.84 (m, 3H), 7.81 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.9 Hz, 1H), 5.70 (d, J = 9.3 Hz, 1H), 4.53 (sept, J = 6.1 Hz, 1H), 1.76 (s, 3H), 1.70 (s, 3H), 1.26 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) δ = 168.5, 166.9, 166.8, 166.7, 164.5, 164.2, 154.1, 142.0, 141.9, 141.6, 138.6, 137.0, 136.4, 132.5, 130.2, 129.1, 128.9, 128.7, 128.6, 128.3, 126.3, 122.8, 120.7, 119.6, 119.4, 118.3, 114.1, 112.5, 112.3, 89.0, 74.9, 58.5, 23.2, 22.9, 22.3 ppm. HRMS (ESI) calcd for C44H38N7O11 [M – H]−: 840.2629; found: 840.2632.
tert-Butyl 4-(4-{4-[(1S,2R)-1-{[(tert-Butoxy)carbonyl]amino}-2-ethenylcyclopropane amido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (118)
After a suspension of the aniline 8 (150 mg, 0.275 mmol, 1.00 equiv) in dry EtOAc (2.00 mL) was cooled down to 0 °C, (1S,2R)-1-{[(tert-butoxy)carbonyl]amino}-2-ethenylcyclopropane-1-carboxylic acid (62 mg, 0.275 mmol, 1.00 equiv) and dry pyridine (75 μL, 0.935 mmol, 3.40 equiv) were added. T3P solution (50% in EtOAc, 327 μL, 0.550 mmol, 2.00 equiv) was added dropwise over 15 min and the resulting solution was stirred for 2.5 h at 0 °C. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with NaHCO3 solution (10 mL) and brine (10 mL), dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by flash chromatography (petroleum ether/EtOAc). Colorless solid, 156 mg (75%). 1H NMR (500 MHz, CDCl3): δ (ppm) = 1.36–1.39 (m, 6H), 1.51 (s, 10H), 1.60 (s, 9H), 1.91 (s, 1H), 2.17 (q, J = 8.6 Hz, 1H), 4.69 (d, J = 5.8 Hz, 2H), 4.75 (h, J = 6.1 Hz, 1H), 5.12 (dd, J = 10.3 Hz, 1.8 Hz, 1H), 5.30 (dd, J = 17.1 Hz, 2.0 Hz, 1H), 5.35–5.44 (m, 2H), 5.49 (dd, J = 17.1 Hz, 1.4 Hz, 1H), 5.64 (s, 1H), 6.09–6.18 (m, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.73 (d, J = 8.7 Hz, 2H), 7.86 (d, J = 8.7 Hz, 2H), 7.96–7.99 (m, 2H), 8.06 (d, J = 8.9 Hz, 1H), 8.48 (d, J = 9.0 Hz, 1H), 8.73 (s, 1H), 9.45 (s, 1H), 10.18 (s, 1H). 13C NMR (126 MHz, CDCl3): δ (ppm) = 20.0, 22.9–23.0 (m), 28.4, 28.4, 34.0, 43.1, 75.1, 80.9, 81.9, 115.8, 118.0, 119.2, 119.5, 120.2, 121.6, 127.4, 127.6, 128.3, 129.6, 130.8, 132.3, 133.5, 137.8, 139.1, 142.3, 149.4, 157.3, 162.8, 164.4, 165.6, 168.2.
(1S,2R)-1-{[4-({4-[(4-Carboxyphenyl)carbamoyl]-3-(prop-2-en-1-yloxy)-2-(propan-2-yloxy)phenyl}carbamoyl)phenyl]carbamoyl}-2-ethenylcyclopropan-1-aminium Chloride (119·HCl)
To the Boc-protected amine 118 (140 mg, 0.185 mmol, 1.00 equiv) was added HCl in dioxane (4.00 M, 1.85 mL, 7.42 mmol, 40.0 equiv) at 0 °C and stirred for 8 h. Afterward, all volatiles were removed, the residue transferred on a fritted funnel, washed with Et2O (3 × 1.5 mL) and dried in vacuo. Colorless solid, 90 mg (76%).
4-(4-{4-[(1S,2R)-1-[4-(4-Cyanobenzamido)benzamido]-2-ethenylcyclopropaneamido] benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoic Acid (120)
To a solution of the carboxylic acid 10 (47 mg, 0.177 mmol, 1.25 equiv) in dry DMF (518 μL) were added HATU (54 mg, 0.142 mmol, 1.00 equiv) and DIPEA (99 μL, 0.567 mmol, 4.00 equiv) at 0 °C. After 30 min, the amine hydrochloride 119·HCl (90 mg, 0.142 mmol, 1.00 equiv) dissolved in dry DMF (518 μL) was added and stirring was continued at 0 °C for 30 min before the mixture was stirred overnight at rt. HCl solution (0.5 M, 10 mL) was added to the reaction mixture and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with HCl solution (0.5 M, 10 mL), water (10 mL) and brine (10 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (CH2Cl2/MeOH). Colorless solid, 47 mg (39%). 1H NMR (700 MHz, DMSO–d6): δ (ppm) = 1.25 (dd, J = 6.1–4.3 Hz, 6H), 1.31–1.35 (m, 1H), 1.91–1.94 (m, 1H), 2.44 (q, J = 8.8 Hz, 1H), 4.49 (hept, J = 6.2 Hz, 1H), 4.61 (d, J = 5.5 Hz, 2H), 5.08–5.11 (m, 1H), 5.20 (dq, J = 10.4–1.5 Hz, 1H), 5.31 (d, J = 17.1 Hz, 1H), 5.37 (dq, J = 17.2–1.7 Hz, 1H), 5.68 (dt, J = 17.2–9.7 Hz, 1H), 6.02 (ddt, J = 16.1/10.7/5.5 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.5 Hz, 2H), 7.82 (dd, J = 14.1–8.5 Hz, 3H), 7.90 (d, J = 8.6 Hz, 2H), 7.93 (d, J = 8.6 Hz, 2H), 7.96 (d, J = 8.7 Hz, 2H), 7.99 (d, J = 8.6 Hz, 2H), 8.03–8.06 (m, 2H), 8.13 (d, J = 8.4 Hz, 2H), 9.03 (s, 1H), 9.51 (s, 1H), 10.00 (s, 1H), 10.52 (s, 1H), 10.71 (s, 1H), 12.45 (s, 1H). 13C NMR (176 MHz, DMSO–d6): δ (ppm) = 21.1, 21.4, 22.4, 42.4, 74.3, 76.3, 114.1, 117.1, 117.8, 118.3, 118.9, 119.0, 119.5, 120.7, 123.7, 125.5, 127.2, 128.3, 128.5, 128.7, 128.8, 129.1, 130.4, 132.6, 133.7, 134.7, 135.7, 138.7, 141.8, 142.2, 142.6, 143.1, 149.6, 164.3, 164.5, 164.6, 166.9–167.1 (m), 168.4, 172.1.
4-(4-{4-[(1S,2R)-1-[4-(4-Cyanobenzamido)benzamido]-2-ethenylcyclopropaneamido] benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (38)
To a solution of the allyl ether 120 (41 mg, 0.048 mmol, 1.00 equiv) in dry THF (365 μL) were added aniline (18 μL, 0.194 mmol, 4.00 equiv) and Pd(PPh3)4 (2 mg, 0.04 equiv) and the resulting mixture was stirred at rt for 3 h. After completion, HCl solution (0.5 M, 10 mL) was added and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with HCl solution (0.5 M, 10 mL) and brine (10 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by RP-HPLC (H2O with 10 mM NH4HCO3/Acetonitrile) and freeze-dried. Colorless solid, 21 mg (54%). 1H NMR (700 MHz, DMSO–d6): δ (ppm) = 1.24–1.26 (m, 6H), 1.31–1.35 (m, 1H), 1.92 (t, 1H), 2.44 (q, J = 8.7 Hz, 1H), 4.54 (hept, J = 6.1 Hz, 1H), 5.10 (d, J = 12.1 Hz, 1H), 5.31 (d, J = 17.1 Hz, 1H), 5.67 (dt, J = 17.1/9.7 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.82–7.87 (m, 3H), 7.90 (d, J = 8.6 Hz, 2H), 7.93 (d, J = 8.8 Hz, 2H), 7.95–7.97 (m, 2H), 7.99 (d, J = 8.6 Hz, 2H), 8.03–8.06 (m, 2H), 8.11–8.15 (m, 2H), 9.03 (s, 1H), 9.39 (s, 1H), 10.01 (s, 1H), 10.67 (s, 1H), 10.71 (s, 1H), 12.28 (s, 1H), 12.80 (s, 1H). 13C NMR (176 MHz, DMSO–d6): δ (ppm) = 21.4, 22.3, 32.2, 42.4, 74.8, 111.9, 112.5, 114.1, 117.2, 118.3, 119.5, 119.6, 120.7, 122.9, 126.3, 128.2, 128.5, 128.7, 129.1, 130.2, 132.6, 134.7, 136.4, 137.0, 138.7, 141.8, 142.1, 142.3, 154.2, 164.2, 164.5, 166.9, 167.0, 168.5, 168.5. HPLC purity 99.0%.
tert-Butyl 4-(4-{4-[(1R,2S)-1-{[(tert-Butoxy)carbonyl]amino}-2-ethenylcyclopropane amido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-loxy)benzamido)benzoate (121)
After a suspension of the aniline 8 (150 mg, 0.275 mmol, 1.00 equiv) in dry EtOAc (2.00 mL) was cooled down to 0 °C, (1R,2S)-1-{[(tert-butoxy)carbonyl]amino}-2-ethenylcyclopropane-1-carboxylic acid (62 mg, 0.275 mmol, 1.00 equiv) and dry pyridine (75 μL, 0.935 mmol, 3.40 equiv) were added. T3P solution (50% in EtOAc, 327 μL, 0.550 mmol, 2.00 equiv) was added dropwise over 15 min and the resulting solution was stirred for 2.5 h at 0 °C. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with NaHCO3 solution (10 mL) and brine (10 mL), dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by flash chromatography (petroleum ether/EtOAc). Colorless solid, 71 mg (34%).
(1R,2S)-1-{[4-({4-[(4-Carboxyphenyl)carbamoyl]-3-(prop-2-en-1-yloxy)-2-(propan-2-yloxy)phenyl}carbamoyl)phenyl]carbamoyl}-2-ethenylcyclopropan-1-aminium Chloride (122·HCl)
To the Boc-protected amine (68 mg, 0.090 mmol, 1.00 equiv) was added HCl in dioxane (4.00 M, 901 μL, 3.60 mmol, 40.0 equiv) at 0 °C and stirred for 8 h. Afterward, all volatiles were removed, the residue transferred on a fritted funnel, washed with Et2O (3 × 1.5 mL) and dried in vacuo. Colorless solid, 50 mg.
4-(4-{4-[(1R,2S)-1-[4-(4-Cyanobenzamido)benzamido]-2-ethenylcyclopropaneamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoic Acid (123)
To a solution of the carboxylic acid 10 (58 mg, 0.216 mmol, 1.25 equiv) in dry DMF (300 μL) were added HATU (66 mg, 0.173 mmol, 1.00 equiv) and DIPEA (121 μL, 0.693 mmol, 4.00 equiv) at 0 °C. After 30 min, the amine hydrochloride 122·HCl (110 mg, 0.173 mmol, 1.00 equiv) dissolved in dry DMF (650 μL) was added and stirring was continued at 0 °C for 30 min before the mixture was stirred overnight at rt. HCl solution (0.5 M, 10 mL) was added to the reaction mixture and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with HCl solution (0.5 M, 10 mL), water (10 mL) and brine (10 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography (CH2Cl2/MeOH). Colorless solid, 61 mg (41%). 1H NMR (500 MHz, DMSO–d6): δ (ppm) = 1.25 (dd, J = 6.1–3.1 Hz, 6H), 1.31–1.35 (m, 1H), 1.93 (dd, J = 7.7–5.1 Hz, 1H), 2.45 (q, J = 8.7 Hz, 1H), 4.48 (p, J = 6.2 Hz, 1H), 4.59–4.63 (m, 2H), 5.06–5.12 (m, 1H), 5.20 (dq, J = 10.5–1.4 Hz, 1H), 5.28–5.40 (m, 2H), 5.68 (ddd, J = 17.1/10.5/9.1 Hz, 1H), 5.97–6.07 (m, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.76–7.84 (m, 5H), 7.88–8.00 (m, 8H), 8.04–8.06 (m, 2H), 8.11–8.15 (m, 2H), 9.03 (s, 1H), 9.52 (s, 1H), 10.00 (s, 1H), 10.53 (s, 1H), 10.71 (s, 1H), 12.72 (s, 1H). 13C NMR (126 MHz, DMSO–d6): δ (ppm) = 21.4, 22.2–22.4 (m), 32.2, 42.4, 74.3, 76.3, 114.1, 117.1, 117.8, 118.3, 118.9, 119.0, 119.4–119.6 (m), 120.7, 123.6, 125.5, 127.2, 128.3, 128.5, 128.7, 128.9, 129.1, 130.4, 132.6, 133.7, 134.7, 135.6, 138.6, 141.8, 142.1, 142.6, 143.0, 151.1, 164.3, 164.5, 164.6, 166.9, 167.0, 168.4. LC-MS: m/z = 847.5 (calcd 847.31 for C48H43N6O9+ [M + H+]).
4-(4-{4-[(1R,2S)-1-[4-(4-Cyanobenzamido)benzamido]-2-ethenylcyclopropaneamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (39)
To a solution of the allyl ether 123 (48 mg, 0.057 mmol, 1.00 equiv) in dry THF (428 μL) were added aniline (21 μL, 0.227 mmol, 4.00 equiv) and Pd(PPh3)4 (3 mg, 0.04 equiv) and the resulting mixture was stirred at rt for 3 h. After completion, HCl solution (0.5 M, 10 mL) was added and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with HCl solution (0.5 M, 10 mL) and brine (10 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by RP-HPLC (H2O with 10 mM NH4HCO3/Acetonitrile) and freeze-dried. Colorless solid, 24 mg (51%). 1H NMR (700 MHz, DMSO–d6): δ (ppm) = 1.25–1.27 (m, 6H), 1.34 (dt, J = 9.4/4.2 Hz, 1H), 1.91–1.94 (m, 1H), 2.44 (q, J = 8.7 Hz, 1H), 4.54 (hept, J = 6.1 Hz, 1H), 5.08–5.11 (m, 1H), 5.31 (dd, J = 17.1–1.8 Hz, 1H), 5.64–5.71 (m, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.77–7.80 (m, 2H), 7.83–7.87 (m, 3H), 7.89–7.92 (m, 2H), 7.92–7.95 (m, 2H), 7.96–7.98 (m, 2H), 7.98–8.01 (m, 2H), 8.04–8.06 (m, 2H), 8.12–8.15 (m, 2H), 9.04 (s, 1H), 9.39 (s, 1H), 10.02 (s, 1H), 10.64 (s, 1H), 10.71 (s, 1H), 12.29 (s, 1H), 12.82 (s, 1H). 13C NMR (176 MHz, DMSO–d6): δ (ppm) = 21.4, 22.3–22.3 (m), 32.2, 42.4, 74.8, 112.1, 112.5, 114.1, 117.1, 118.3, 119.5, 119.6, 120.7, 122.9, 126.3, 128.2, 128.5, 128.6, 129.1, 130.2, 132.5, 134.7, 136.3, 137.0, 138.6, 141.8, 142.0, 142.3, 154.2, 164.2, 164.5, 166.9, 167.0, 168.4, 168.5. HPLC purity 99.0%.
tert-Butyl 4-(4-{4-[(2S)-3-(tert-Butoxy)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (124)
Under Ar atmosphere, aniline 8 (100 mg, 1.0 equiv) and (2S)-3-(tert-butoxy)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)propanoic acid (77 mg, 1.1 equiv) were dissolved in dry EtOAc (1.6 mL). Then dry pyridine (77 μL, 5.0 equiv) and T3P (50% sol. in EtOAc, 0.22 mL, 2.0 equiv) were added and the reaction was stirred at rt while being screened by LCMS. After 2 h, T3P (50% sol. in EtOAc, 0.11 mL, 1.0 equiv) was added again and after 3 h the mixture was partitioned between EtOAc (30 mL) and aq HCl solution (0.1 M, 30 mL). The aqueous phase was extracted with EtOAc (2 × 15 mL). The combined organic phases were dried over Na2SO4 and dried in vacuo. The residue was purified by flash chromatography (solid loading, 80 × theor. prod mass, Cyclohexane/EtOAc, 70:30). Beige sticky foam, 148 mg (89%). 1H NMR (500 MHz, CDCl3): δ = 10.19 (s, 1H), 9.01 (s, 1H), 8.75 (s, 1H), 8.50 (d, J = 8.9, 1H), 8.07 (d, J = 8.9, 1H), 7.98 (d, J = 8.9, 2H), 7.91 (d, J = 8.7, 2H), 7.78 (d, J = 7.9, 2H), 7.74 (d, J = 8.9, 2H), 7.69 (d, J = 8.9, 2H), 7.62 (d, J = 7.3, 2H), 7.41 (t, J = 7.5, 2H), 7.37–7.27 (m, 2H), 6.14 (ddt, J = 17.1, 10.4, 5.8, 1H), 5.85–5.77 (m, 1H), 5.50 (dq, J = 17.1, 1.5, 1H), 5.41 (dq, J = 10.4, 1.1, 1H), 4.80–4.72 (m, 1H), 4.70 (dt, J = 6.0, 1.4, 2H), 4.47 (d, J = 7.2, 2H), 4.40 (s, 1H), 4.26 (t, J = 6.9, 1H), 3.99–3.88 (m, 1H), 3.48 (t, J = 8.8, 1H), 1.60 (s, 9H), 1.39 (d, J = 6.3, 6H), 1.28 (d, J = 17.7, 9H). 13C NMR (126 MHz, CDCl3): δ = 169.0, 165.6, 164.3, 162.7, 149.4, 143.8, 143.8, 142.3, 141.5, 141.2, 139.1, 137.7, 132.3, 130.8, 130.1, 128.4, 128.0, 127.6, 127.4, 127.2, 125.2, 121.7, 120.2, 120.2, 119.6, 119.1, 115.8, 80.9, 75.3, 75.1, 67.4, 61.8, 55.0, 47.3, 28.4, 27.7, 23.0. HRMS (ESI) calcd [M + H]+: 911.4226; found: 911.4226.
tert-Butyl 4-(4-{4-[(2S)-3-(tert-butoxy)-2-{[4-(4-cyanobenzamido)phenyl]formamido}propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (125)
Carbamate 124 (57 mg, 1.0 equiv) was dissolved in ACN (1.0 mL) and HNEt2 (0.10 mL, 15 equiv) was added. The reaction was stirred at rt while being screened by LCMS. After 1 h, the solvent was removed under reduced pressure and by coevaporation with ACN (2×). The crude product was used without further purification. Under Ar atmosphere, the crude carboxylic acid 10 (18.3 mg, 1.1 equiv) and HATU (26 mg, 1.1 equiv) were added to the amine and the mixture was dissolved in dry DMF (0.65 mL). DIPEA (32 μL, 3.0 equiv) was added and the reaction was stirred at rt for 2 h 10 min. The reaction solution was directly used in the next step. Dry THF (0.65 mL), aniline (17 μL, 3.0 equiv) and Pd(PPh3)4 (spatula tip) were added to the reaction solution. The reaction was then stirred at rt while being screened by LCMS. After 2 h, the mixture was partitioned between EtOAc (20 mL)/aq HCl solution (0.1 M, 20 mL) and the aqueous phase was extracted with EtOAc (2 × 15 mL). The combined organic layers were dried over Na2SO4 and all volatiles were removed under reduced pressure. The residue was purified by flash chromatography (solid loading, 100 × theor. prod mass, cyclohexane/acetone, 75:25 → 70:30 → 60:40). Colorless solid, crude 51 mg.
4-(4-{4-[(2S)-3-(tert-Butoxy)-2-{[4-(4-cyanobenzamido)phenyl]formamido}propanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (40)
Under Ar atmosphere, to a solution of tert-Butyl ester 125 (51 mg, 1.0 equiv) in dry CH2Cl2 (0.85 mL) were added anisole (12.4 μL, 2.0 equiv) and TFA (0.22 mL, 50 equiv). After 2 h, the flask was flushed with Ar to concentrate the solution by evaporating the CH2Cl2. After 3.25 h, TFA (0.22 mL, 50 equiv) was added again and after 4.5 h the reaction mixture was stirred at 40 °C for 15 min. All volatiles were removed under reduced pressure, by coevaporation with ACN and by lyophilization overnight. The material was purified by RP-HPLC (H2O with 10 mM NH4HCO3/Acetonitrile) and freeze-dried. Colorless solid, 22 mg (45% over 4 steps). 1H NMR (500 MHz, DMSO): δ = 12.84 (s, 1H), 12.30 (s, 1H), 10.72 (s, 1H), 10.66 (br s, 1H), 10.46 (s, 1H), 9.41 (s, 1H), 8.43 (d, J = 7.3, 1H), 8.13 (d, J = 8.5, 2H), 8.05 (d, J = 8.5, 2H), 7.99–7.94 (m, 6H), 7.90 (d, J = 9.0, 2H), 7.88–7.81 (m, 5H), 7.69 (d, J = 8.9, 1H), 5.14 (t, J = 5.7, 1H), 4.69 (q, J = 6.0, 1H), 4.54 (hept, J = 6.3, 1H), 3.89–3.79 (m, 2H), 1.26 (d, J = 6.1, 6H). 13C NMR (126 MHz, DMSO): δ = 169.9, 168.5, 166.9, 166.0, 164.5, 164.2, 154.2, 142.4, 142.1, 141.6, 138.7, 137.0, 136.4, 132.6, 130.2, 129.2, 128.7, 128.4, 128.4, 128.3, 126.2, 122.9, 120.7, 119.5, 118.8, 118.3, 114.1, 112.5, 112.1, 74.8, 61.5, 57.1, 22.3. HRMS (ESI) calcd [M + H]+: 785.2566; found: 785.2564. HPLC purity 98.2%.
tert-Butyl 4-(4-{4-[(2S,3S)-3-(tert-Butoxy)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl} amino)butanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (126)
The amine 8 (0.18 mol) was coupled with (2S,3S)-3-(tert-butoxy)-2-({[(9H-fluoren-9-yl) methoxy]carbonyl}amino)butanoic acid using general procedure 1. Yellowish solid, 124.9 mg (74%). 1H NMR (700 MHz, CDCl3, 300 K): δ (ppm) = 10.17 (s, 1H), 8.74 (s, 1H), 8.50 (d, 1H, J = 8.9 Hz), 8.27 (br s, 1H), 8.07 (d, 1H, J = 8.9 Hz), 7.98 (d, 2H, J = 8.7 Hz), 7.90 (d, 2H, J = 8.7 Hz), 7.77 (dd, 2H, J = 2.7 Hz, 7.5 Hz), 7.73 (d, 2H, J = 8.8 Hz), 7.70 (d, 2H, J = 8.7 Hz), 7.61–7.58 (m, 2H), 7.40 (td, 2H, J = 2.7 Hz, 7.4 Hz), 7.30 (t, 2H, J = 7.3 Hz), 6.14 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 5.49 (ddd, 1H, J = 1.4 Hz, 2.7 Hz, 17.1 Hz), 5.41 (ddd, 1H, J = 1.0 Hz, 2.1 Hz, 10.4 Hz), 4.76 (hept., 1H, J = 6.1 Hz), 4.69 (d, 2H, J = 5.9 Hz), 4.52 (dd, 1H, J = 7.0 Hz, 10.6 Hz), 4.46–4.42 (m, 1H), 4.24 (t, 1H, J = 6.8 Hz), 3.97–3.91 (m, 1H), 1.60 (s, 9H), 1.39 (dd, 6H, J = 1.6 Hz, 6.2 Hz), 1.19 (s, 9H). 13C NMR (176 MHz, CDCl3, 300 K): δ (ppm) = 165.6, 164.3, 162.8, 149.4, 143.9, 143.8, 142.3, 141.5, 141.1, 139.1, 137.7, 132.3, 130.8, 130.1, 128.4, 127.9, 127.6, 127.4, 127.3, 125.1, 121.7, 120.2, 119.6, 119.2, 115.8, 80.9, 75.4, 75.1, 47.3, 28.6, 28.4, 23.0. HPLC purity 96.8%.
4-(4-{4-[(2S,3S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-hydroxybutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (41)
Step 1: The amino acid derivative 126 (129.3 μmol) was deprotected using general procedure 2. The crude product was used without further purification. Step 2: The crude amine (129.3 μmol) was coupled with the carboxylic acid 10 using general procedure 4. Steps 3 and 4: The product was obtained by deprotection with general procedures 6 and 7. White solid, 39 mg (38% over 4 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.82 (br s, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.61 (br s, 1H), 10.40 (br s, 1H), 9.38 (s, 1H), 8.45 (d, 1H, J = 8.2 Hz), 8.13 (d, 2H, J = 8.5 Hz), 8.05 (d, 2H, J = 8.5 Hz), 7.98–7.94 (m, 6H), 7.89 (d, 2H, J = 8.8 Hz), 7.87–7.84 (m, 5H), 7.71 (d, 1H, J = 8.8 Hz), 5.16 (d, 1H, J = 5.2 Hz), 4.57–4.50 (m, 2H), 4.14–4.09 (m, 1H), 1.27 (dd, 6H, J = 0.9 Hz, 6.1 Hz), 1.23 (d, 3H, J = 6.2 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 170.5, 168.5, 166.9, 165.9, 164.4, 164.2, 154.1, 142.5, 142.0, 141.6, 138.7, 137.1, 136.3, 132.5, 130.2, 129.2, 128.6, 128.4, 128.3, 128.2, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.1, 74.9, 66.5, 60.7, 22.3, 21.0. HPLC purity 99.8%.
tert-Butyl 4-(4-{4-[(2S,3R)-3-(tert-Butoxy)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl} amino)butanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (128)
The aniline 8 (0.37 mol) was coupled with (2S,3R)-3-(tert-Butoxy)-2-({[(9H-fluoren-9-yl)methoxy]carbonyl}amino)butanoic acid using general procedure 1. Yellowish solid, 312.5 mg (92%). 1H NMR (700 MHz, CDCl3, 300 K): δ (ppm) = 10.18 (s, 1H), 9.48 (br s, 1H), 8.76 (br s, 1H), 8.52 (d, 1H, J = 8.9 Hz), 8.09 (d, 1H, J = 8.9 Hz), 8.00 (d, 2H, J = 8.7 Hz), 7.92 (d, 2H, J = 8.7 Hz), 7.79 (d, 2H, J = 7.6 Hz), 7.75 (d, 2H, J = 8.7 Hz), 7.68 (d, 2H, J = 8.7 Hz), 7.64 (d, 2H, J = 7.3 Hz), 7.43 (t, 2H, J = 7.2 Hz), 7.34 (t, 2H, J = 7.8 Hz), 6.15 (ddt, 1H, J = 5.9 Hz, 10.4 Hz, 16.3 Hz), 6.04 (d, 1H, J = 5.0 Hz), 5.51 (dq, 1H, J = 1.4 Hz, 17.1 Hz), 5.42 (dq, 1H, J = 1.1 Hz, 10.4 Hz), 4.77 (hept., 1H, J = 6.2 Hz), 4.71 (dt, 2H, J = 1.2 Hz, 5.9 Hz), 4.46 (d, 2H, J = 7.0 Hz), 4.38 (t, 1H, J = 4.3 Hz), 4.32–4.29 (m, 1H), 4.27 (t, 1H, J = 7.1 Hz), 1.61 (s, 9H), 1.42–1.39 (m, 15H), 1.12 (d, 6H, J = 6.4 Hz). 13C NMR (176 MHz, CDCl3, 300 K): δ (ppm) = 168.2, 165.6, 164.3, 162.8, 156.2, 149.4, 144.0, 143.8, 142.3, 141.5, 141.3, 139.1, 137.7, 132.3, 130.8, 130.0, 128.4, 127.9, 127.7, 127.4, 127.2, 125.3, 121.7, 120.2, 120.2, 119.6, 119.2, 115.8, 80.9, 76.9, 75.1, 67.1, 59.3, 47.3, 28.6, 28.4, 23.0. HPLC purity 95.3%.
4-(4-{4-[(2S,3R)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-hydroxybutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (42)
Step 1: The amino acid derivative 128 (99.6 μmol) was deprotected using general procedure 2. The crude product was used without further purification. Step 2: The crude amine (167.8 μmol) was coupled with the carboxylic 10 acid using general procedure 4. Step 3 and 4: The product was obtained by deprotection with general procedures 6 and 7. White to off-white solid, 49 mg (37% over 4 steps). 1H NMR (700 MHz, DMSO-d6, 300 K): δ (ppm) = 12.78 (br s, 1H), 12.31 (br s, 1H), 10.72 (s, 1H), 10.40 (s, 1H), 9.36 (br s, 1H), 8.16–8.13 (m, 3H), 8.05 (d, 2H, J = 8.4 Hz), 7.99–7.94 (m, 6H), 7.92 (d, 2H, J = 8.8 Hz), 7.85 (d, 2H, J = 8.7 Hz), 7.83–7.80 (m, 3H), 7.66 (d, 1H, J = 8.0 Hz), 5.08 (d, 1H, J = 6.5 Hz), 4.61–4.56 (m, 2H), 4.23–4.18 (m, 1H) 1.26 (dd, 6H, J = 1.8 Hz, 6.1 Hz), 1.20 (d, 3H, J = 6.3 Hz). 13C NMR (176 MHz, DMSO-d6, 300 K): δ (ppm) = 169.9, 168.4, 166.9, 166.2, 164.1, 142.3, 141.7, 138.7, 136.8, 136.4, 132.5, 130.2, 129.2, 128.6, 128.5, 128.3, 126.1, 122.9, 120.5, 119.6, 118.9, 118.3, 114.1, 112.7, 74.5, 66.8, 60.6, 22.3, 20.4. HPLC purity 97.2%.
Methyl (2R)-2-{[(tert-Butoxy)carbonyl]amino}-3-hydroxypropanoate (131)
d-Serine (500 mg, 4.76 mmol) was suspended in MeOH (10 mL) and SOCl2 (2.1 mL, 28.5 mmol, 6.00 equiv) was added dropwise at 0 °C. The solution was stirred at ambient temperature for 19 h. The solvent was removed under reduced pressure and coevaporated with Et2O (3×). The residue was dissolved in CH2Cl2 (20 mL) and cooled to 0 °C. Then Et3N (1.80 mL, 12.8 mmol, 2.70 equiv) and Boc2O (1.10 g, 5.23 mmol, 1.10 equiv) were added carefully and the reaction mixture was allowed to warm to rt. The mixture was stirred for 22 h before the solvent was removed under reduced pressure. The residue was purified by column chromatography (MeOH in CH2Cl2 = 2, 5, 10%) to furnish the product (945 mg, 4.31 mmol, 91%) as a yellow oil. The analytical data are consistent with those reported in the literature (F. W. Foss, A. H. Snyder, M. D. Davis, M. Rouse, M. D. Okusa, K. R. Lynch, T. L. Macdonald, Bioorg. Med. Chem.2007, 15, 663–677). 1H NMR (400 MHz, CDCl3) = δ 5.43 (s, 1H), 4.39 (m, 1H), 3.99–3.88 (m, 2H), 3.79 (s, 3H), 2.24 (s, 1H), 1.46 (s, 9H) ppm.
tert-Butyl N-(1,3-Dihydroxy-3-methylbutan-2-yl)carbamate (132)
Ester 131 (940 mg, 4.30 mmol) was suspended in Et2O (23 mL). MeMgBr (3 M in Et2O, 8.60 mL, 25.7 mmol, 6.00 equiv) was added at −78 °C. The emulsion was allowed to warm to rt and stirred at rt for 3 h. The reaction mixture was cooled to 0 °C and a sat. NH4Cl solution was added. The aqueous phase was extracted with EtOAc (3×). The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure to furnish the alcohol (866 mg, 3.95 mmol, 92%) as yellow oil. The analytical data are consistent with those reported in the literature (J. E. Dettwiler, W. D. Lubell, J. Org. Chem.2003, 68, 177–179). 1H NMR (400 MHz, CD3OD) = δ 3.82–3.79 (dd, J = 4.1, 11.2 Hz, 1H), 3.62–3.57 (m, 1H), 3.51–3.48 (m, 1H), 1.45 (s, 9H), 1.23 (s, 3H), 1.15 (s, 3H) ppm.
2-{[(tert-Butoxy)carbonyl]amino}-3-hydroxy-3-methylbutanoic Acid (133)
Diol 132 (860 mg, 3.92 mmol) was dissolved in MeCN (15 mL). Phosphate buffer (pH 7, 14 mL) and TEMPO (61.3 mg, 0.39 mmol, 0.10 equiv) were added. The solution was warmed to 35 °C and NaClO2 (2 M in H2O, 4.00 mL, 7.84 mmol, 2.00 equiv) and NaOCl (0.04 M in H2O, 2.00 mL, 0.08 mmol, 2 mol %) were added simultaneously over 2 h. The mixture was stirred at 35 °C for 24 h. Citric acid (10%) was added until pH 2. The aqueous phase was extracted with EtOAc (3×) and the combined organic phases were concentrated under reduced pressure. The residue was dissolved in a sat. NaHCO3 solution (80 mL). The aqueous phase was washed with EtOAc (2×) and afterward treated with a 1 M H3PO4 solution (100 mL) until pH 2 was reached. The acidic phase was extracted with EtOAc (3×). The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure to furnish the carboxylic acid (719 mg, 3.08 mmol, 79%) as a colorless amorphous solid. The analytical data are consistent with those reported in the literature (J. E. Dettwiler, W. D. Lubell, J. Org. Chem.2003, 68, 177–179). 1H NMR (400 MHz, CD3OD) = δ 4.08 (m, 1H), 1.45 (s, 9H), 1.29 (s, 3H), 1.25 (s, 3H) ppm.
tert-Butyl 4-(4-{4-[(2S)-2-{[(tert-Butoxy)carbonyl]amino}-3-hydroxy-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (134)
Amine 8 (200 mg, 0.37 mmol) and carboxylic acid 133 (120 mg, 0.51 mmol, 1.40 equiv) were dissolved in EtOAc (800 μL) and pyridine (89 μL, 1.10 mmol, 3.00 equiv) was added. T3P (50% in EtOAc, 400 μL, 0.66 mmol, 1.80 equiv) was added at 0 °C and the mixture was stirred for 22 h while warming to rt. H2O was added and the aqueous phase was extracted with EtOAc (3×). The combined organic phases were washed with a sat. NaHCO3 solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (MeOH in CH2Cl2 = 0, 1%) to furnish the product (189 mg, 0.25 mmol, 68%) as yellow amorphous solid. [α]D22 = −2.7° (c 0.2, MeOH). 1H NMR (500 MHz, DMSO-d6) = 10.53 (s, 1H), 10.10 (s, 1H), 9.51 (s, 1H), 7.98–7.96 (d, J = 8.8 Hz, 2H), 7.90–7.88 (d, J = 8.8 Hz, 2H), 7.84–7.81 (m, 3H), 7.79–7.77 (d, J = 8.7 Hz, 2H), 7.41–7.40 (d, J = 8.4 Hz, 1H), 6.66–6.64 (d, J = 9.0 Hz, 1H), 6.06–5.98 (m, 1H), 5.39–5.35 (dq, J = 1.7, 17.1 Hz, 1H), 5.22–5.19 (dq, J = 1.7, 10.5 Hz, 1H), 4.87 (s, 1H), 4.61–4.60 (d, J = 5.5 Hz, 1H), 4.53–4.46 (sept, J = 6.1 Hz, 1H), 4.12–4.08 (m, 1H), 1.55 (s, 9H), 1.40 (s, 9H), 1.26–1.25 (d, J = 6.1 Hz, 6H), 1.21 (s, 3H), 1.16 (s, 3H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.8, 164.6, 164.6, 164.3, 155.4, 149.5, 143.0, 142.5, 142.1, 135.7, 133.7, 130.1, 128.4, 128.3, 127.1, 126.0, 123.6, 118.9, 118.9, 118.8, 117.8, 80.4, 78.4, 76.3, 74.3, 70.8, 63.2, 28.2, 27.9, 27.4, 26.4, 22.3 ppm. HRMS (ESI) calcd for C41H52N4O10Na [M + Na]+: 783.3581; found: 783.3585.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-hydroxy-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (135)
Step 1: Carbamate 134 (184 mg, 0.24 mmol) was dissolved in HCl (4 M in 1,4-dioxane, 6.00 mL, 24.2 mmol, 100 equiv) at 0 °C. The mixture was warmed up to rt and stirred for 15 min. The solution was transferred to a stirred suspension of EtOAc (160 mL) and a sat. NaHCO3 solution (160 mL). The aqueous phase was extracted with EtOAc and the combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was used in the next step without further purification. Step 2: DIPEA (62 μL, 0.36 mmol, 3.00 equiv) was added dropwise to a stirred solution of HATU (54.1 mg, 0.14 mmol, 1.20 equiv) and carboxylic acid 10 (37.9 mg, 0.14 mmol, 1.20 equiv) in DMF (3.0 mL). The solution was stirred for 5 min and was then transferred to a stirred solution of the amine (78.3 mg, 0.12 mmol) in DMF (1.7 mL). The reaction mixture was stirred at rt for 21 h. The mixture was diluted with EtOAc and washed with a 1 M HCl solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (MeOH in CH2Cl2 = 1, 2, 3%) to furnish the product (50.3 mg, 0.06 mmol, 47%) as colorless amorphous solid. [α]D23 = +6.0° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 10.71 (s, 1H), 10.53 (s, 1H), 10.28 (s, 1H), 9.52 (s, 1H), 8.14–8.12 (d, J = 8.8 Hz, 2H), 8.08–8.06 (d, J = 8.9 Hz, 1H), 8.06–8.04 (d, J = 8.6 Hz, 2H), 7.99–7.97 (d, J = 8.8 Hz, 2H), 7.96–7.95 (d, J = 8.9 Hz, 2H), 7.92–7.88 (m, 4H), 7.83–7.80 (m, 5H), 7.41–7.40 (d, J = 8.5 Hz, 1H), 6.05–5.97 (m, 1H), 5.39–5.35 (dq, J = 1.7, 17.2 Hz, 1H), 5.21–5.18 (dq, J = 1.7, 10.5 Hz, 1H), 5.07 (s, 1H), 4.67–4.66 (d, J = 8.7 Hz, 1H), 4.61–4.60 (d, J = 5.5 Hz, 2H), 4.53–4.46 (sept, J = 6.1 Hz, 1H), 1.54 (s, 9H), 1.31 (s, 3H), 1.27 (s, 3H), 1.26–1.25 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.4, 166.1, 164.6, 164.6, 164.5, 164.3, 149.6, 143.0, 142.6, 142.1, 141.7, 138.7, 135.7, 133.7, 132.6, 130.1, 129.3, 128.7, 128.5, 128.4, 127.1, 126.1, 123.7, 119.6, 119.0, 118.9, 118.3, 117.8, 114.1, 80.4, 76.3, 74.3, 70.9, 62.3, 27.9, 27.4, 27.2, 22.4 ppm. HRMS (ESI) calcd for C51H52N6O10Na [M + Na]+: 931.3643; found: 931.3638.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-hydroxy-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (136)
Allyl ether 135 (47.4 mg, 0.05 mmol) was dissolved in THF (2.6 mL). Aniline (16 μL, 0.17 mmol, 3.30 equiv) and Pd(PPh3)4 (6.0 mg, 5 μmol, 0.10 equiv) were added subsequently and the resulting mixture was stirred at rt for 2 h. The mixture was concentrated under reduced pressure. The crude product was purified by column chromatography (MeOH in CH2Cl2 = 1, 2, 3%) to furnish the product (37.7 mg, 0.04 mmol, 83%) as yellow amorphous solid. [α]D23 = +4.9° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 12.28 (s, 1H), 10.71 (s, 1H), 10.61 (s, 1H), 10.30 (s, 1H), 9.40 (s, 1H), 8.14–8.13 (d, J = 8.6 Hz, 2H), 8.08–8.04 (m, 3H), 7.97–7.90 (m, 8H), 7.86–7.81 (m, 5H), 7.71–7.69 (d, J = 8.9 Hz, 1H), 5.05 (s, 1H), 4.69–4.67 (d, J = 8.7 Hz, 1H), 4.58–4.51 (sept, J = 6.1 Hz, 1H), 1.55 (s, 9H), 1.31 (s, 3H), 1.27–1.26 (s, 9H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.4, 168.5, 166.1, 164.5, 164.5, 164.2, 154.1, 142.2, 142.0, 141.7, 138.7, 137.0, 136.3, 132.5, 129.9, 129.3, 128.6, 128.4, 128.4, 126.8, 122.8, 120.7, 119.6, 118.9, 118.3, 114.1, 112.4, 80.5, 74.9, 70.9, 62.3, 27.8, 27.3, 27.1, 22.3 ppm. HRMS (ESI) calcd for C48H47N6O10Na [M – H]−: 867.3354; found: 867.3352.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-hydroxy-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (43)
Ester 136 (35.8 mg, 0.04 mmol) was dissolved in precooled TFA (2 mL) at 0 °C with stirring. The solution was warmed up to rt over 30 min. Et2O was added at 0 °C. The precipitate was filtered off, washed with an excess of Et2O and dried in vacuo to furnish the product (13.0 mg, 0.02 mmol, 39%) as beige amorphous solid. [α]D23 = +9.5° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 12.82 (s, 1H), 12.29 (s, 1H), 10.71 (s, 1H), 10.60 (s, 1H), 10.30 (s, 1H), 9.40 (s, 1H), 8.14–8.13 (d, J = 8.5 Hz, 2H), 8.08–8.04 (m, 3H), 7.98–7.95 (m, 5H), 7.92–7.90 (d, J = 8.9 Hz, 2H), 7.86–7.81 (m, 5H), 7.72–7.70 (d, J = 9.0 Hz, 1H), 5.05 (s, 1H), 4.69–4.67 (d, J = 8.7 Hz, 1H), 4.58–4.51 (sept, J = 6.2 Hz, 1H), 1.31 (s, 3H), 1.27–1.26 (s, 9H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.4, 168.5, 166.9, 166.1, 164.5, 164.2, 154.1, 142.2, 142.0, 141.7, 138.7, 137.0, 136.3, 132.5, 130.2, 129.3, 128.6, 128.4, 128.4, 126.3, 122.8, 120.7, 119.6, 118.9, 118.3, 114.1, 112.4, 112.2, 74.9, 70.9, 62.3, 27.3, 27.1, 22.3 ppm. HRMS (ESI) calcd for C44H39N6O10 [M – H]−: 811.2728; found: 811.2729.
(2S,3S)-2-{[(tert-Butoxy)carbonyl]amino}-3-hydroxybutanoic Acid (138)
A mixture of NaHCO3 (543 mg, 6.46 mmol, 1.50 equiv) and Boc2O (1.43 g, 6.55 mmol, 1.60 equiv) in MeOH (8.5 mL) was added to a solution of l-allothreonine (137) (500 mg, 4.20 mmol) in H2O (8.5 mL). The mixture was stirred at rt for 18 h and afterward acidified with a 0.5 M HCl solution. The aqueous phase was extracted with EtOAc (3×). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure to furnish the carbamate (963 mg), which was used in the next step without further purification.
Methyl (2S,3S)-2-{[(tert-Butoxy)carbonyl]amino}-3-methoxybutanoate (139)
Amino acid derivative 138 (920 mg, 4.20 mmol) was dissolved in MeCN (4.6 mL) and Ag2O (4.86 g, 21.0 mmol, 5.00 equiv) was added. MeI (4.20 mL, 67.2 mmol, 16.00 equiv) was added at 0 °C and the resulting mixture was stirred at rt for 48 h. The mixture was filtered through Celite and the plug was washed with EtOAc. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (dry load, PE/EtOAc = 8:1) to furnish product 295 (486 mg, 1.97 mmol, 47% over two steps) as colorless oil. The analytical data are consistent with those reported in the literature (G. B. Martinez, A. Griffin, P. Charifson, K. Reddy, K. M. Kahlig, B. Marron, WO2020227101 (A1), 2020). 1H NMR (400 MHz, CDCl3) = δ 5.28–5.27 (d, J = 7.2 Hz, 1H), 4.44–4.41 (dd, J = 3.6, 4.9 Hz, 1H), 3.76 (s, 3H), 3.64–3.62 (m, 1H), 3.36 (s, 3H), 1.44 (s, 9H), 1.21–1.19 (d, J = 6.4 Hz, 1H) ppm.
(2S,3S)-2-{[(tert-Butoxy)carbonyl]amino}-3-methoxybutanoic Acid (140)
Ester 139 (479 mg, 1.94 mmol) was dissolved in THF (3.6 mL) and H2O (1.8 mL). LiOH·H2O (488 mg, 11.6 mmol, 6.00 equiv) was added and the mixture was stirred at rt for 3 h. THF was removed under reduced pressure and the residue was acidified with a 2 M HCl solution. The aqueous phase was extracted with EtOAc (3×). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure to furnish the acid (490 mg, quant.) as yellow oil, which was used in the next step without further purification. The analytical data are consistent with those reported in the literature (G. B. Martinez, A. Griffin, P. Charifson, K. Reddy, K. M. Kahlig, B. Marron, WO2020227101 (A1), 2020). 1H NMR (400 MHz, CDCl3) = δ 8.34 (bs, 1H), 5.31–5.29 (d, J = 7.5 Hz, 1H), 4.44–4.43 (m, 1H), 3.69 (m, 1H), 3.38 (s, 3H), 1.45 (s, 9H), 1.26–1.24 (d, J = 6.3 Hz, 3H) ppm.
tert-Butyl 4-(4-{4-[(2S,3S)-2-{[(tert-Butoxy)carbonyl]amino}-3-methoxybutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (141)
Amine 8 (100 mg, 0.18 mmol), carboxylic acid 140 (72.7 mg, 0.31 mmol, 1.70 equiv) and EEDQ (72.5 mg, 0.29 mmol, 1.60 equiv) were dissolved in precooled CHCl3 (1 mL) at 0 °C. The mixture was stirred at 18 h while warming to rt. The mixture was concentrated under reduced pressure and the residue was purified by column chromatography (1. dry load, 20% Et2O in CH2Cl2, 2. dry load, 1% MeOH in CH2Cl2) to furnish the product (74.3 mg, 0.10 mmol, 53%) as yellow amorphous solid, which contained small impurities. The compound was used in the next step without further purification. 1H NMR (500 MHz, DMSO-d6) = δ 10.53 (s, 1H), 10.33–10.25 (m, 1H), 9.51–9.49 (m, 1H), 7.98–7.94 (m, 2H), 7.90–7.88 (m, 2H), 7.83–7.79 (m, 5H), 7.42–7.40 (d, J = 8.4 Hz, 1H), 7.08–7.07 (d, J = 8.6 Hz, 1H), 6.07–5.98 (m, 1H), 5.39–5.36 (dd, J = 1.6, 17.2 Hz, 1H), 5.21–5.19 (dd, J = 1.5, 10.5 Hz, 1H), 4.61–4.60 (d, J = 5.4 Hz, 1H), 4.54–4.46 (sept, J = 6.1 Hz, 1H), 4.23–4.20 (t, J = 8.1 Hz, 1H), 3.59–3.57 (m, 1H), 3.23 (s, 3H), 1.55 (s, 9H), 1.39 (s, 9H), 1.27–1.26 (d, J = 6.1 Hz, 6H), 1.13–1.12 (d, J = 5.9 Hz, 3H) ppm. HRMS (ESI) calcd for C41H52N4O10Na [M + Na]+: 783.3581; found: 783.3583.
tert-Butyl 4-(4-{4-[(2S,3S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methoxybutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (142)
Step 1: Carbamate 141 (70.9 mg, 0.09 mmol) was dissolved in HCl (4 M in 1,4-dioxane, 2.3 mL, 9.32 mmol, 100 equiv) at 0 °C. The mixture was warmed up to rt and stirred for 15 min. The solution was transferred to a stirred suspension of EtOAc (60 mL) and a sat. NaHCO3 solution (60 mL). The aqueous phase was extracted with EtOAc and the combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was used in the next step without further purification. Step 2: DIPEA (49 μL, 0.28 mmol, 3.00 equiv) was added dropwise to a stirred solution of HATU (42.5 mg, 0.11 mmol, 1.20 equiv) and carboxylic acid 10 (29.8 mg, 0.11 mmol, 1.20 equiv) in DMF (2.3 mL). The solution was stirred for 5 min and was then transferred to a stirred solution of the amine (61.6 mg, 0.09 mmol) in DMF (1.3 mL). The reaction mixture was stirred at rt for 16 h. The mixture was diluted with EtOAc and washed with a 1 M HCl solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, 2% MeOH in CH2Cl2) to furnish the product (31.4 mg, 0.04 mmol, 37%) as colorless amorphous solid. [α]D23 = +3.3° (c 0.1, MeOH). 1H NMR (600 MHz, DMSO-d6) = 10.69 (s, 1H), 10.52 (s, 1H), 10.50 (s, 1H), 9.51 (s, 1H), 8.53–8.52 (d, J = 8.3 Hz, 1H), 8.13–8.12 (d, J = 8.4 Hz, 2H), 8.05–8.04 (d, J = 8.4 Hz, 2H), 7.99–7.95 (m, 4H), 7.90–7.88 (m, 4H), 7.84–7.82 (m, 4H), 7.41–7.40 (d, J = 8.4 Hz, 1H), 6.05–5.98 (m, 1H), 5.39–5.36 (dd, J = 1.6, 17.2 Hz, 1H), 5.21–5.19 (dd, J = 1.3, 10.5 Hz, 1H), 4.61–4.60 (d, J = 5.3 Hz, 2H), 4.74–4.72 (t, J = 8.3 Hz, 1H), 4.53–4.48 (sept, J = 6.1 Hz, 1H), 3.85–3.81 (m, 1H), 3.29 (s, 3H), 1.55 (s, 9H), 1.27–1.25 (d, J = 6.1 Hz, 6H), 1.23–1.22 (d, J = 6.0 Hz, 3H) ppm. 13C NMR (151 MHz, DMSO-d6) = 170.1, 166.0, 164.6, 164.5, 164.4, 164.3, 149.5, 143.0, 142.5, 142.3, 141.6, 138.7, 135.6, 133.6, 132.5, 130.1, 129.1, 128.6, 128.5, 128.4, 128.4, 127.1, 126.0, 123.6, 119.5, 118.9, 118.8, 118.8, 118.3, 117.8, 114.0, 80.3, 76.3, 76.2, 74.3, 58.5, 56.5, 27.9, 22.3, 16.2 ppm. HRMS (ESI) calcd for C51H52N6O10Na [M + Na]+: 931.3643; found: 931.3643.
tert-Butyl 4-(4-{4-[(2S,3S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methoxybutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (143)
Allyl ether 142 (29.0 mg, 0.03 mmol) was dissolved in THF (1.6 mL). Aniline (10 μL, 0.11 mmol, 3.30 equiv) and Pd(PPh3)4 (3.7 mg, 0.003 mmol, 0.10 equiv) were added subsequently and the resulting mixture was stirred at rt for 90 min. The mixture was concentrated under reduced pressure. The crude product was purified by column chromatography (dry load, MeOH in CH2Cl2 = 2, 3%) to furnish product 149 (19.7 mg, 0.02 mmol, 71%) as beige amorphous solid. [α]D25 = +2.3° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 12.29 (s, 1H), 10.69 (s, 1H), 10.61 (bs, 1H), 10.52 (s, 1H), 9.39 (s, 1H), 8.54–8.52 (d, J = 8.3 Hz, 1H), 8.14–8.12 (d, J = 8.6 Hz, 2H), 8.05–8.04 (d, J = 8.6 Hz, 2H), 7.96–7.92 (m, 6H), 7.89–7.84 (m, 7H), 7.72–7.70 (d, J = 8.9 Hz, 1H), 4.74–4.71 (t, J = 8.3 Hz, 1H), 4.58–4.51 (sept, J = 6.1 Hz, 1H), 3.85–3.80 (m, 1H), 3.29 (s, 3H), 1.55 (s, 9H), 1.27–1.26 (d, J = 6.1 Hz, 6H), 1.24–1.22 (d, J = 6.0 Hz, 3H) ppm. 13C NMR (126 MHz, DMSO-d6) = 170.1, 168.5, 166.0, 164.5, 164.4, 164.2, 142.4, 142.0, 141.6, 138.7, 137.0, 136.3, 132.5, 129.9, 129.1, 128.6, 128.5, 128.4, 126.8, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.1, 80.5, 76.3, 74.8, 58.5, 56.5, 27.8, 22.3, 16.2 ppm. In the 13C NMR spectrum is one signal around 154 ppm missing due to the minimal analytical amount. HRMS (ESI) calcd for C48H48N6O10Na [M + Na]+: 891.3330; found: 891.3321.
4-(4-{4-[(2S,3S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methoxybutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (44)
tert-Butyl ester 143 (17.6 mg, 0.02 mmol) was dissolved in precooled TFA (1.2 mL) at 0 °C with stirring. The solution was warmed up to rt over 30 min. Et2O was added at 0 °C. The precipitate was filtered off, washed with an excess of Et2O and dried in vacuo to furnish the acid (10.1 mg, 0.01 mmol, 61%) as gray amorphous solid. [α]D25 = +4.2° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 12.82 (bs, 1H), 12.30 (s, 1H), 10.69 (s, 1H), 10.60 (s, 1H), 10.52 (s, 1H), 9.40 (s, 1H), 8.54–8.52 (d, J = 8.4 Hz, 1H), 8.14–8.12 (d, J = 8.5 Hz, 2H), 8.05–8.04 (d, J = 8.5 Hz, 2H), 7.98–7.95 (m, 6H), 7.89–7.84 (m, 7H), 7.72–7.70 (d, J = 8.9 Hz, 1H), 4.74–4.71 (t, J = 8.3 Hz, 1H), 4.58–4.51 (sept, J = 6.1 Hz, 1H), 3.85–3.80 (m, 1H), 3.29 (s, 3H), 1.27–1.26 (d, J = 6.1 Hz, 6H), 1.24–1.22 (d, J = 6.1 Hz, 3H) ppm. 13C NMR (126 MHz, DMSO-d6) = 170.1, 168.5, 166.9, 166.0, 164.4, 164.2, 154.1, 142.4, 142.0, 141.6, 138.7, 137.1, 136.3, 132.5, 130.2, 129.1, 128.6, 128.5, 128.4, 128.4, 126.3, 122.8, 120.7, 119.5, 118.8, 118.3, 114.0, 112.4, 112.2, 76.3, 74.9, 58.5, 56.5, 22.3, 16.2 ppm. HRMS (ESI) calcd for C44H39N6O10 [M – H]−: 811.2728; found: 811.2733.
(2S)-2-{[(tert-Butoxy)carbonyl]amino}-3-methoxy-3-methylbutanoic Acid (144)
Tertiary alcohol 133 (300 mg, 1.29 mmol) in THF (2.0 mL) was added to NaH (60% in mineral oil, 154 mg, 3.86 mmol, 3.00 equiv) in THF (2.4 mL) at 0 °C. The mixture was stirred at rt for 1 h. MeI (96 μL, 1.54 mmol, 1.20 equiv) was added and the mixture was stirred at rt for 19 h. H2O was added and the aqueous phase was extracted with Et2O (3×). The aqueous phase was acidified with 6 M HCl until pH 2 and extracted with EtOAc (4×). The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (MeOH in CH2Cl2 = 0, 2%) to furnish product (162 mg, 0.65 mmol, 51%) as yellow gum. [α]D21 = +5.8° (c 1.3, MeOH). 1H NMR (500 MHz, CD3OD) = δ 4.20 (s, 1H), 3.23 (s, 3H), 1.45 (s, 9H), 1.28 (s, 3H), 1.26 (s, 3H) ppm. 13C NMR (126 MHz, CD3OD) = δ 174.1, 157.9, 80.8, 77.1, 61.7, 50.0, 28.7, 22.7 (d, J = 8.0 Hz) ppm. HRMS (ESI+) calcd for C11H20NO5 [M – H]−: 246.1341; found: 246.1351.
tert-Butyl 4-(4-{4-[(2S)-2-{[(tert-Butoxy)carbonyl]amino}-3-methoxy-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (145)
Amine 8 (100 mg, 0.18 mmol) and carboxylic acid 144 (63.5 mg, 0.26 mmol, 1.40 equiv) were dissolved in EtOAc (400 μL) and pyridine (44 μL, 0.55 mmol, 3.00 equiv) was added. T3P (50% in EtOAc, 200 μL, 0.33 mmol, 1.80 equiv) was added at 0 °C and the mixture was stirred at 0 °C for 3 h. H2O was added and the aqueous phase was extracted with EtOAc (3×). The combined organic phases were washed with a sat. NaHCO3 solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography (dry load, PE/EtOAc = 2:1) to furnish the product (70.2 mg, 0.09 mmol, 49%) as brown amorphous solid. [α]D22 = −15.2° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 10.53 (s, 1H), 10.16 (s, 1H), 9.51 (s, 1H), 7.98–7.97 (d, J = 8.7 Hz, 2H), 7.90–7.88 (d, J = 8.7 Hz, 2H), 7.84–7.81 (m, 3H), 7.79–7.78 (d, J = 8.7 Hz, 2H), 7.41–7.40 (d, J = 8.4 Hz, 1H), 6.76–6.74 (d, J = 8.7 Hz, 1H), 6.05–5.99 (m, 1H), 5.39–5.38 (dd, J = 1.6, 17.2 Hz, 1H), 5.21–5.19 (dd, J = 1.4, 10.5 Hz, 1H), 4.61–4.60 (d, J = 5.4 Hz, 1H), 4.53–4.47 (sept, J = 6.2 Hz, 1H), 4.31–4.30 (d, J = 9.0 Hz, 1H), 3.17 (s, 3H), 1.55 (s, 9H), 1.40 (s, 9H), 1.26–1.25 (d, J = 6.1 Hz, 6H), 1.19 (s, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.4, 164.6, 164.5, 164.3, 155.4, 149.5, 143.0, 142.5, 142.0, 135.6, 133.6, 130.1, 128.5, 128.4, 127.1, 126.0, 123.6, 118.9, 118.8, 118.8, 117.8, 80.5, 78.5, 76.2, 76.0, 74.3, 59.7, 49.2, 28.1, 27.9, 22.3, 22.1 ppm. HRMS (ESI) calcd for C42H54N4O10Na [M + Na]+: 797.3738; found: 797.3726.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methoxy-3-methylbutanamido]benzamido}-2-(prop-2-en-1-yloxy)-3-(propan-2-yloxy)benzamido)benzoate (146)
Step 1: Carbamate 145 (65.0 mg, 0.08 mmol) was dissolved in HCl (4 M in 1,4-dioxane, 2.10 mL, 9.32 mmol, 100 equiv) at 0 °C. The mixture was warmed up to rt and stirred for 15 min. The solution was transferred to a stirred suspension of EtOAc (60 mL) and a sat. NaHCO3 solution (60 mL). The aqueous phase was extracted with EtOAc and the combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was used in the next step without further purification. Step 2: DIPEA (44 μL, 0.25 mmol, 3.00 equiv) was added dropwise to a stirred solution of HATU (38.3 mg, 0.10 mmol, 1.20 equiv) and carboxylic acid 10 (26.8 mg, 0.10 mmol, 1.20 equiv) in DMF (2.1 mL). The solution was stirred for 5 min and was then transferred to a stirred solution of the amine (56.6 mg, 0.08 mmol) in DMF (1.2 mL). The reaction mixture was stirred at rt for 21 h. The mixture was diluted with EtOAc and washed with a 1 M HCl solution, brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (MeOH in CH2Cl2 = 1, 2, 3%) to furnish the product (40.3 mg, 0.04 mmol, 52%) as colorless amorphous solid. [α]D22 = +5.4° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 10.71 (s, 1H), 10.53 (s, 1H), 10.33 (s, 1H), 9.52 (bs, 1H), 8.14–8.12 (d, J = 8.6 Hz, 2H), 8.07–8.04 (m, 3H), 7.99–7.97 (d, J = 8.8 Hz, 2H), 7.96–7.94 (d, J = 8.9 Hz, 2H), 7.91–7.88 (m, 3H), 7.84–7.80 (m, 5H), 7.41–7.40 (d, J = 8.5 Hz, 1H), 6.06–5.98 (m, 1H), 5.39–5.35 (dq, J = 1.7, 17.2 Hz, 1H), 5.22–5.19 (dq, J = 1.7, 10.5 Hz, 1H), 4.89–4.87 (d, J = 8.7 Hz, 1H), 4.61–4.60 (d, J = 5.5 Hz, 1H), 4.53–4.46 (sept, J = 6.1 Hz, 1H), 3.23 (s, 3H), 1.55 (s, 9H), 1.31 (s, 6H), 1.26–1.25 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.1, 166.2, 164.6, 164.5, 164.4, 164.3, 149.5, 143.0, 142.5, 142.0, 141.7, 138.7, 135.6, 133.7, 132.5, 130.4, 130.1, 129.2, 128.6, 128.6, 128.5, 127.1, 126.0, 123.6, 119.5, 119.0, 118.9, 118.8, 118.3, 117.8, 114.0, 80.5, 76.2, 76.1, 74.3, 60.4, 49.3, 27.9, 22.3, 21.7 ppm. HRMS (ESI) calcd for C52H54N6O10Na [M + Na]+: 945.3799; found: 945.3806.
tert-Butyl 4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methoxy-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoate (147)
Allyl ether 146 (38.5 mg, 0.04 mmol) was dissolved in THF (2 mL). Aniline (13 μL, 0.14 mmol, 3.30 equiv) and Pd(PPh3)4 (4.8 mg, 4 μmol, 0.10 equiv) were added subsequently and the resulting mixture was stirred at rt for 2 h. The mixture was concentrated under reduced pressure. The crude product was purified by column chromatography (MeOH in CH2Cl2 = 1, 2, 3%) to furnish the product (27.0 mg, 0.03 mmol, 73%) as colorless amorphous solid. [α]D22 = −3.8° (c 0.1, MeOH).
1H NMR (600 MHz, DMSO-d6) = 12.29 (s, 1H), 10.70 (s, 1H), 10.60 (bs, 1H), 10.35 (s, 1H), 9.39 (bs, 1H), 8.14–8.13 (d, J = 8.6 Hz, 2H), 8.07–8.04 (m, 3H), 7.96–7.89 (m, 8H), 7.86–7.81 (m, 5H), 4.89–4.88 (d, J = 8.9 Hz, 1H), 4.55 (bs, 1H), 3.23 (s, 3H), 1.55 (s, 9H), 1.31 (s, 6H), 1.27–1.26 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (151 MHz, DMSO-d6) = 169.1, 168.4, 166.2, 164.6, 164.4, 164.1, 142.1, 141.7, 138.7, 136.3, 132.5, 131.5, 131.4, 129.9, 129.2, 128.8, 128.7, 128.6, 128.5, 128.4, 122.9, 120.6, 119.5, 118.9, 118.3, 114.0, 80.5, 76.1, 74.9, 60.4, 49.3, 27.8, 22.3, 21.7 ppm. HRMS (ESI) calcd for C49H50N6O10Na [M + Na]+: 905.3486; found: 905.3470.
4-(4-{4-[(2S)-2-{[4-(4-Cyanobenzamido)phenyl]formamido}-3-methoxy-3-methylbutanamido]benzamido}-2-hydroxy-3-(propan-2-yloxy)benzamido)benzoic Acid (45)
Ester 147 (25.0 mg, 0.03 mmol) was dissolved in precooled TFA (1.5 mL) at 0 °C with stirring. The solution was warmed up to rt over 30 min. Et2O was added at 0 °C. The precipitate was filtered off, washed with an excess of Et2O and dried in vacuo to furnish the product (5.3 mg, 0.01 mmol, 23%) as beige amorphous solid. [α]D22 = +0.8° (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6) = 12.82 (s, 1H), 12.29 (s, 1H), 10.71 (s, 1H), 10.60 (s, 1H), 10.35 (s, 1H), 9.40 (s, 1H), 8.14–8.12 (d, J = 8.5 Hz, 2H), 8.07–8.04 (m, 3H), 7.98–7.94 (m, 6H), 7.91–7.89 (d, J = 8.8 Hz, 2H), 7.86–7.81 (m, 5H), 7.71–7.70 (d, J = 8.9 Hz, 1H), 4.89–4.87 (d, J = 8.9 Hz, 1H), 4.58–4.51 (sept, J = 6.1 Hz, 1H), 3.23 (s, 3H), 1.31 (s, 6H), 1.27–1.26 (d, J = 6.1 Hz, 6H) ppm. 13C NMR (126 MHz, DMSO-d6) = 169.1, 168.5, 166.9, 166.2, 164.4, 164.2, 154.1, 142.1, 142.0, 141.7, 138.7, 137.0, 136.3, 132.5, 130.2, 129.2, 128.6, 128.6, 128.5, 126.3, 122.8, 120.7, 119.5, 118.9, 118.3, 114.0, 112.4, 112.2, 76.1, 74.9, 60.4, 49.3, 22.3, 21.7 ppm. HRMS (ESI) calcd for C45H41N6O10 [M – H]−: 825.2884; found: 825.2889.
Determination of Thermodynamic (Equilibrium) Solubility
The selected compound is suspended in phosphate buffer (50 mM, pH 7.4) at a target concentration of 1 mg/mL. After overnight stirring at room temperature, protected from light, suspensions are filtered through a 0.45 μm PTFE membrane. An aliquot of the resulting supernatant is quantified using UPLC UV method against a reference solution obtained by preparation of a DMSO stock solution. The solubility value is expressed as μL.
Determination of Log D7.4 Values
Log D values at pH 7.4 were determined by a standardized HPLC method as essentially derived from Genieser et al.25 The calculation of the log D value for measured compounds is performed by comparison of the retention times with standard compounds of known distribution coefficients between 1-octanol and water at pH 7.4.
Determination of Minimal Inhibitory Concentrations (MIC)
Method 1
All microorganisms were handled according to standard procedures. The microorganisms were obtained from the German Collection of Microorganisms and Cell Cultures (Deutsche Sammlung für Mikroorganismen and Zellkulturen, DSMZ), the American Type Culture Collection (ATCC) or were part of the internal strain collection. The cystobactamids were prepared as DMSO stock solutions with a concentration of 5 mg/mL. The Minimum inhibitory concentrations (MIC) were determined by standard procedures.8 Single colonies of the bacterial strains were suspended in Müller-Hinton broth and grown overnight at appropriate temperature. On the following day, the bacterial count was adjusted by dilution to achieve a final inoculum of approximately 5 × 105 – 1 × 106 CFU/mL in cation-adjusted Müller-Hinton broth. Serial dilutions of the tested cystobactamids and reference antibiotics (0.03–64 μg/mL) were prepared in sterile 96-well plates and the bacterial suspension was added. The microorganisms were grown overnight at appropriate temperature under shaking conditions. The growth inhibition was assessed visually. The determined MIC value represented the lowest concentration of antibiotic at which there was no visible growth. This determination method was applied for the testing of the compounds in Table 3.
Method 2
All microorganisms were handled according to standard procedures. The microorganisms were obtained from the German Collection of Microorganisms and Cell Cultures (Deutsche Sammlung für Mikroorganismen and Zellkulturen, DSMZ), the American Type Culture Collection (ATCC), Evotec or were part of the internal strain collection. The cystobactamids were prepared as DMSO stock solutions with a concentration of 5 mg/mL. The Minimum inhibitory concentrations (MIC) were determined by standard procedures.26 The bacterial cultures were streaked out on Müller-Hinton or blood agar, as appropriate, and grown overnight at appropriate temperature. The following day, three to four isolated colonies were collected with a sterile cotton swab and resuspended in saline solution to obtain turbidity equal to McFarland Standard 0.5. Serial dilutions of the tested cystobactamids and reference antibiotics (0.03–64 μg/mL) were prepared in cation-adjusted Müller-Hinton broth or TSB, as appropriate, in sterile 96-well plates and the bacterial suspension was added. The growth inhibition was assessed after 16–20 h or after satisfactory growth of the controls was observed at appropriate temperature under static conditions. The determined MIC value represented the lowest concentration of antibiotic at which there was no visible growth. This determination method was applied for the testing of the compounds in Tables 1 and 2 as well as Tables S1–S4.
Determination of the Time-Kill Curve
Test articles were prepared in sterile dimethyl sulfoxide (DMSO) and used at final concentrations of 1, 2, 4, or 8× the minimum inhibitory concentration (MIC), with a final DMSO concentration of 1% (v/v). Killing kinetics assays were performed in triplicate on separate days, using separate preparations of growth medium and bacterial inocula. Starter cultures of each bacterial strain were inoculated by suspending a single colony from a Mueller-Hinton agar plate in 10 mL cation-adjusted Mueller-Hinton broth (caMHB) and incubating at 37 °C with shaking at 300 rpm. Following 18 h’ incubation, starter cultures were diluted into fresh prewarmed caMHB containing the appropriate concentration of CN-CC-861 (13) (or 1% DMSO for the vehicle-treated control) to achieve a target bacterial density of approximately 5 × 105 CFU/mL at the start of the assay. A sample (200 μL) was removed immediately from each tube for quantification of the total viable bacterial count, which was achieved by serial 10-fold dilution in sterile phosphate-buffered saline (PBS), followed by plating onto cystine–lactose–electrolyte-deficient (CLED) agar. Subsequently, at 1-, 2-, 4-, 6-, 8- and 24 h postinoculation, additional samples were taken for bacterial quantification as described above. Agar plates were incubated at 37 °C in air overnight prior to enumeration of colonies for each condition. The limit of detection (LoD) for the assay was 50 CFU/mL.
DNA Supercoiling Assay
The supercoiling assay for the determination if IC50 values on E. coli gyrase was carried out in two steps. The test compounds were diluted to a 0.75 mM for the assay. The method is described in the following.
DNA Relaxation
In two separate reactions 25 μL (1 μg/μL) purified circular pUC19 plasmid was mixed with 13.5 μL H2O, 0.5 μL Topoisomerase I (6.5 U) and 50 μL topoisomerase buffer (250 mM Tris [pH 7.5], 250 mM KCl, 50 mM MgCl2, 2.5 mM DTT, 0.5 mM EDTA and 150 μg/mL BSA). The mixture was incubated for 90 min at 37 °C. Both reactions were combined, and the relaxed plasmid DNA was purified by spin-columns according to the vendor’s manual. The concentration was determined by measurement of the optical density at 600 nm and adjusted to 50 ng/μL.
DNA Supercoiling Assay
The 0.75 mM test compound solution was diluted to obtain final concentrations of 25, 8.33, 2.78, 0.93, 0.31, 0.1, and 0.03 μM. For a singular determination 7.9 μL H2O, 3 μL DNA gyrase buffer (Inspiralis) and 0.1 μL E. coli gyrase (5 U/μL; Inspiralis) were added to a 0.2 mL test tube and a control tube. One μL of the compound dilution series was added to the test tube. Both tubes were vortexed. One μL relaxed plasmid DNA (50 ng) was diluted with 2 μL water, added to each tube and vortexed. The mixtures were incubated for 30 min at 37 °C and the reaction stopped by increased temperature at 60 °C for 10 min.
Agarose Gel Electrophoresis
Three μL agarose gel loading buffer was added to each sample and the control. Fifteen μL of the sample was loaded to the 0.8% agarose gel. The gel was run at 25 min at 100 V and subsequently stained with ethidium bromide (1 μg/mL) for 5 min. The ethidium bromide fluorescence was documented under a UV lamp. The gel was divided into lanes. The intensity of the staining was assessed by Image Lab 5.0 (BioRad). By densitometric analysis, each lane was analyzed for bands corresponding to the coiled pUC19 DNA. The intensity of the bands was determined by relative intensity to the untreated control. A graph was generated with the concentration of the test sample at the X-axis and the relative intensity of the pUC19 DNA band at the Y-axis. The IC50 values were calculated by nonlinear regression by graph pad prism.27
E. coli Gyrase Supercoiling Inhibition Assay and Topoisomerase IV Relaxation Assay
Both assay kits were purchased from Inspiralis (Norwich, U.K.). The assays were performed according to the manufacturer’s instructions, with one modification. Prior to addition of either relaxed or supercoiled pBR322 plasmid, the enzyme was preincubated with the serial dilutions of investigated compound. In short, one unit of enzyme was pre incubated with the compound at room temperature for 15 min, final concentration of DMSO did not exceed 1%. After 15 min, 0.5 μg of either relaxed or supercoiled plasmid was added and the reaction was incubated for 30 min at 37 °C. Next, the reaction was stopped by addition of chloroform/isoamyl alcohol (v/v, 24.1) and GSTEB buffer. Samples were briefly vortexed and centrifuged for 1 min at 15,000 rpm. Upper aqueous phase was loaded onto a 1% TAE gel and ran at 90 V until the dye traveled 6–7 cm from the loading pocket to ensure sufficient separation of relaxed and supercoiled topoisomers. Next, the gels were stained in 1 μg/mL ethidium bromide bath for 15 min and destained in water for 5 min. In the final step, the gels were visualized with a gel documentation system. Gel band intensities were determined using ImageJ and normalized to the control. IC50 values were determined using nonlinear regression in graph pad prism.27
Determination of Cytotoxicity against HepG2 and CHO Cell Lines
The cytotoxicity assay was conducted as described previously.28 In brief, the epithelial cell line HepG2 (ATCC HB-8065TM) was cultivated in Dulbecco’s modified Eagle’s medium (DMEM) with 10% heat-inactivated fetal calf serum (FCS) at 37 °C and 5% CO2. Similarly, CHO cells (ATCC CCL-61) were cultivated in Gibco Ham’s F-12K (Kaighn’s) medium with 10% heat-inactivated fetal calf serum (FCS) at 37 °C and 5% CO2. Cells were tested for mycoplasma contamination via electron microscopy before use. HepG2 cells as well as CHO-cells were seeded into a 96-well plate (Nunc, Roskilde, Denmark) and grown to 75% confluency. CN-CC-861 (13) was tested in concentrations ranging from 0.01 to 300 μM for 24 h with a residual DMSO assay concentration of 1%. All assays were conducted in triplicates.
Determination of Plasma Stability
The plasma stability assay was conducted as described previously28 with procaine, procainamide and propoxycaine as controls. Plasma stability was determined for CN-CC-861 (13), 16, 24, 26, 27, 38, 39, 41 and 42. All samples were analyzed via HPLC-MS/MS.
Determination of Plasma Protein Binding
The plasma protein binding assay was conducted as described previously29 with naproxen as control. Plasma protein binding was determined for CN-CC-861 (13), 16, 24, 26, 27, 38, 39, 41 and 42. All samples were analyzed via HPLC-MS/MS.
HPLC-MS/MS Analysis of Cystobactamids
Samples were analyzed using an Agilent 1290 Infinity II HPLC system coupled to an AB Sciex QTrap 6500plus mass spectrometer. LC conditions were as follows: column: Agilent Zorbax Eclipse Plus C18, 50 mm × 2.1 mm, 1.8 μm; temperature: 30 °C; injection volume: 1 μL per sample; flow rate: 700 μL min–1. Samples were run under acidic conditions. Solvents: A: water +0.1% formic acid; solvent B: 95% acetonitrile/5% H2O + 0.1% formic acid. The following gradient was applied: 99% A at 0 min, 99% A until 0.1 min, 99–50% A from 0.1 to 3.5 min, 50–0% A from 3.5 min until 3.8 min, 0–99% A from 3.8 min until 4.7 min. The mass spectrometer was run in positive and negative mode with multiple reaction monitoring (MRM). Mass transitions for controls and compounds are depicted in SI Table 7. Samples were analyzed using MultiQuant 3.0 software (AB Sciex).
The HPLC traces were determined and analyzed using an Agilent 1100 HPLC system. LC conditions were as follows: column: Phenomenex Aeris PEPTIDE XB-C18, 50 mm × 2.1 mm, 3.6 μm; temperature: 25 °C; injection volume: 10 μL of a 1 mg/mL stock solution in DMSO; flow rate: 700 μL min–1. Samples were run under acidic conditions. Solvents A: water +0.1% trifluoroacetic acid; solvent B acetonitrile +0.1% trifluoroacetic acid. The following gradient was applied: 95% A at 0 min, 95% A until 3.0 min, 95–0% A from 3.0 to 23.0 min, 0% A from 23 to 28 min, 0–95% A from 28 to 28.1 min, 95% A from 28.1 to 33 min. The UV detection and quantification was carried out at 254 nm.
In Vivo Efficacy Study of CN-CC-861 (13) in an E. coli Thigh Infection Model
The inoculum of the E. coli ATCC 25922 was diluted from a frozen stock to 1.4 × 106 cfu/mL. N = 5 animals per group were used. N = 4 animals were used for the pretreatment group. Mice were rendered neutropenic by administration of 150 and 100 mg/kg cyclophosphamide intraperitoneally on day −4 and −1, respectively. On the day of infection (day 0), mice received 50 μL of the inoculum with E. coli into each lateral thigh under isoflurane aesthesia. While still under anesthesia, mice were administered a dose of Buprenorphine (0.03 mg/kg) subcutaneously for pain relief. Treatment started 1 h post infection. As vehicle hydroxyl-propyl-β cyclodextrin in Tris-buffer 1% at pH 9.0 (20:80 (w/v)) was used. For dissolution of cystobactamids vehicle was added to the respective amount of cystobactamid and sonicated for 10 min at 35 kHz. Then, the solution was stirred overnight with high speed at ambient temperature and protected from light. CN-DM-861 and CN-CC-861 were both dosed q6h starting from 1 h post infection. CN-DM-861 was dosed at 20, 40, and 50 mg/kg/day, whereas CN-CC-861 was dosed at 20, 40, and 80 mg/kg/day. Twenty-five hours after infection, mice were euthanized, blood was removed from the heart, thighs were aseptically removed. Twenty-five hours after infection the clinical score of every individual animal was assessed. When animals reached the humane end point, they were euthanized earlier and blood was removed from the heart and thighs were aseptically removed. Whole blood was collected into Eppendorf tubes coated with 0.5 M EDTA. Organs were homogenized in PBS. Thighs were plated onto agar plates in duplicates in serial dilutions and incubated at 37 °C for 24 h. The Evotec guidelines for animal testing are aligned with the 3Rs (Replacement, Reduction and Refinement) principles of Russel and Burch. These principles work toward good laboratory animal welfare and the principle of 3Rs is a holistic and integral part of R&D processes at Evotec. The responsible use of animals remains essential in research. Animals remain a small but an integral part of a comprehensive research that includes alternative methods and clinical research. Evotec animal facilities are AAALAC accredited or in the process of first accreditation and undergo regular visits by AAALAC. When animal experimentation is necessary, great care is taken to choose the most appropriate animal species for the research and optimize the study design to ensure that the results will be as meaningful as possible. All studies are designed to gain the maximum information from the fewest number of animals and the lowest burden possible. Each proposed use of animals is reviewed and approved by our veterinarians and scientists with an emphasis on eliminating or minimizing any potential pain or distress which may be experienced by the animals. Our standards of animal care and welfare meet or exceed those required by applicable local, national, or international laws and regulations. All Evotec personnel involved in the care, welfare and use of animals are trained to ensure that they are competent, aware of ethical issues and demonstrate respect and humane treatment toward the animals in their care within the procedures required to complete the proposed work.
Acknowledgments
We thank Janine Schreiber for excellent technical assistance with ADME experiments, and Ulrike Beutling and Kirsten Harmrolfs for mass spectrometry and NMR support, respectively (all HZI). We also thank Jennifer Williams, Andy Wise, and Andrew Sharp for TKC experiments and Thomas Taillier for chemistry support.
Glossary
Abbreviations Used
- cfu
colony-forming unit
- CIP
ciprofloxacin
- Cryo-EM
cryogenic electron microscopy
- EEDQ
ethyl 2-ethoxyquinoline-1(2H)-carboxylate
- FoR
frequency of resistance
- LPS
lipopolysaccharide
- PABA
para-aminobenzoic acid
- T3P
propanephosphonic acid anhydride
- TI IV
topoisomerase IV
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00927.
MIC tables including 13, 21-27, 32, 33, 37–43; antibiotic activities of CN-DM-861 (4), CN-CC-861 (13) and ciprofloxacin (CIP) against susceptible and multiresistant bacteria; docking study with Albi-1, 17 and 18; mass transitions of the internal standard (caffeine), controls and cystobactamids; synthetic schemes; 1H and 13C NMR spectra; HPLC chromatograms for CN-CC 861 (13) and exemplified for 17, 21, 38 and 42 (PDF)
Molecular formula strings (CSV)
The studies were cofunded by the German Centre for Infection Research (DZIF; Grant no TTU09.710) and the BMBF Project “Wirkstoffentwicklung auf Basis von Naturstoffen zur Bekämpfung von Infektionskrankheiten” (no GGNATM27).
The authors declare the following competing financial interest(s): A part of the authors have filed patent applications (published: WO2015003816, WO2016082934, WO2019038405) on cystobactamids.
Special Issue
Published as part of Journal of Medicinal Chemistryspecial issue “Natural Products Driven Medicinal Chemistry”.
Supplementary Material
References
- Murray C. J. L.; Ikuta K. S.; Sharara F.; Swetschinski L.; Aguilar G. R.; Gray A.; Naghavi M.; et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399, 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacconelli E.; Carrara E.; Savoldi A.; Harbarth S.; Mendelson M.; Monnet D. L.; Pulcini C.; Kahlmeter G.; Kluytmans J.; Carmeli Y.; et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18 (3), 318–327. 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Adamiak J. W.; Bonifay V.; Mehla J.; Zgurskaya H. I.; Tan D. S. Defining new chemical space for drug penetration into Gram-negative bacteria. Nat. Chem. Biol. 2020, 16 (12), 1293–1302. 10.1038/s41589-020-00674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes E. J.; Gugger M. K.; Garcia A.; Chavez M. G.; Lee M. R.; Perlmutter S. J.; Bieniossek C.; Guasch L.; Hergenrother P. J. Porin-independent accumulation in Pseudomonas enables antibiotic discovery. Nature 2023, 624 (7990), 145–153. 10.1038/s41586-023-06760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. S.; Gigante V.; Sati H.; Paulin S.; Al-Sulaiman L.; Rex J. H.; Fernandes P.; Arias C. A.; Paul M.; Thwaites G. E.; et al. Analysis of the clinical pipeline of treatments for drug resistant bacterial infections: despite progress, more action is needed. Antimicrob. Agents Chemother. 2022, 66, e01991-21 10.1128/aac.01991-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke M.; Pieroni M.; Weber T.; Brönstrup M.; Hammann P.; Halby L.; Arimondo P. B.; Glaser P.; Aigle B.; Bode H. B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5 (10), 726–749. 10.1038/s41570-021-00313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S.; Herrmann J.; Raju R.; Steinmetz H.; Mohr K. I.; Hüttel S.; Harmrolfs K.; Stadler M.; Müller R. Cystobactamids: myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew. Chem., Int. Ed. 2014, 53 (52), 14605–14609. 10.1002/anie.201409964. [DOI] [PubMed] [Google Scholar]
- Cociancich S.; Pesic A.; Petras D.; Uhlmann S.; Kretz J.; Schubert V.; Vieweg L.; Duplan S.; Marguerettaz M.; Noëll J.; et al. The gyrase inhibitor albicidin consists of p-aminobenzoic acids and cyanoalanine. Nat. Chem. Biol. 2015, 11 (3), 195–197. 10.1038/nchembio.1734. [DOI] [PubMed] [Google Scholar]
- Seedorf T.; Kirschning A.; Solga D. Natural and Synthetic Oligoarylamides: Privileged Structures for Medical Applications. Chem. - Eur. J. 2021, 27 (26), 7321–7339. 10.1002/chem.202005086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalczyk E.; Hommernick K.; Behroz I.; Kulike M.; Pakosz-Stepien Z.; Mazurek L.; Seidel M.; Kunert M.; Santos K.; von Moeller H.; et al. Molecular mechanism of topoisomerase poisoning by the peptide antibiotic albicidin. Nat. Catal. 2023, 6 (1), 52–67. 10.1038/s41929-022-00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttel S.; Testolin G.; Herrmann J.; Planke T.; Gille F.; Moreno M.; Stadler M.; Bronstrup M.; Kirschning A.; Muller R. Discovery and Total Synthesis of Natural Cystobactamid Derivatives with Superior Activity against Gram-Negative Pathogens. Angew. Chem., Int. Ed. 2017, 56 (41), 12760–12764. 10.1002/anie.201705913. [DOI] [PubMed] [Google Scholar]
- Trauner D.; Cheng B.; Muller R. Total Syntheses of Cystobactamids and Structural Confirmation of Cystobactamid 919–2. Angew. Chem., Int. Ed. 2017, 56, 12755–12759. 10.1002/anie.201705387. [DOI] [PubMed] [Google Scholar]
- Kretz J.; Kerwat D.; Schubert V.; Gratz S.; Pesic A.; Semsary S.; Cociancich S.; Royer M.; Sussmuth R. D. Total synthesis of albicidin: a lead structure from Xanthomonas albilineans for potent antibacterial gyrase inhibitors. Angew. Chem., Int. Ed. 2015, 54 (6), 1969–1973. 10.1002/anie.201409584. [DOI] [PubMed] [Google Scholar]
- Kleebauer L.; Zborovsky L.; Hommernick K.; Seidel M.; Weston J. B.; Süssmuth R. D. Overcoming AlbD Protease Resistance and Improving Potency: Synthesis and Bioactivity of Antibacterial Albicidin Analogues with Amide Bond Isosteres. Org. Lett. 2021, 23 (18), 7023–7027. 10.1021/acs.orglett.1c02312. [DOI] [PubMed] [Google Scholar]
- Testolin G.; Cirnski K.; Rox K.; Prochnow H.; Fetz V.; Grandclaudon C.; Mollner T.; Baiyoumy A.; Ritter A.; Leitner C.; et al. Synthetic studies of cystobactamids as antibiotics and bacterial imaging carriers lead to compounds with high in vivo efficacy. Chem. Sci. 2020, 11 (5), 1316–1334. 10.1039/C9SC04769G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behroz I.; Durkin P.; Grätz S.; Seidel M.; Rostock L.; Spinczyk M.; Weston J. B.; Süssmuth R. D. Extensive Structure–Activity Relationship Study of Albicidin’s C-Terminal Dipeptidic p-Aminobenzoic Acid Moiety. Chem. - Eur. J. 2019, 25 (72), 16538–16543. 10.1002/chem.201904752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grätz S.; Kerwat D.; Kretz J.; von Eckardstein L.; Semsary S.; Seidel M.; Kunert M.; Weston J. B.; Süssmuth R. D. Synthesis and Antimicrobial Activity of Albicidin Derivatives with Variations of the Central Cyanoalanine Building Block. ChemMedChem 2016, 11 (14), 1499–1502. 10.1002/cmdc.201600163. [DOI] [PubMed] [Google Scholar]
- Zborovsky L.; Kleebauer L.; Seidel M.; Kostenko A.; von Eckardstein L.; Gombert F. O.; Weston J.; Süssmuth R. D. Improvement of the antimicrobial potency, pharmacokinetic and pharmacodynamic properties of albicidin by incorporation of nitrogen atoms. Chem. Sci. 2021, 12 (43), 14606–14617. 10.1039/D1SC04019G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller M.; Norris M. D.; Planke T.; Cirnski K.; Herrmann J.; Müller R.; Kirschning A. Scalable Syntheses of Methoxyaspartate and Preparation of the Antibiotic Cystobactamid 861–2 and Highly Potent Derivatives. Org. Lett. 2019, 21 (20), 8369–8372. 10.1021/acs.orglett.9b03143. [DOI] [PubMed] [Google Scholar]
- Carpino L. A.; Cohen B. J.; Stephens K. E.; Sadat-Aalaee S. Y.; Tien J. H.; Langridge D. C. (Fluoren-9-ylmethoxy)carbonyl (Fmoc) amino acid chlorides. Synthesis, characterization, and application to the rapid synthesis of short peptide segments. J. Org. Chem. 1986, 51 (19), 3732–3734. 10.1021/jo00369a042. [DOI] [Google Scholar]
- Michalczyk E.; Hommernick K.; Behroz I.; Kulike M.; Pakosz-Stępień Z.; Mazurek L.; Seidel M.; Kunert M.; Santos K.; von Moeller H.; et al. Molecular mechanism of topoisomerase poisoning by the peptide antibiotic albicidin. Nat. Catal. 2023, 6 (1), 52–67. 10.1038/s41929-022-00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51 (10), 2871–2878. 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- Krass J. D.; Jastorff B.; Genieser H.-G. Determination of Lipophilicity by Gradient Elution High-Performance Liquid Chromatography. Anal. Chem. 1997, 69 (13), 2575–2581. 10.1021/ac961246i. [DOI] [PubMed] [Google Scholar]
- Wiegand I.; Hilpert K.; Hancock R. E. W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3 (2), 163–175. 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- GraphPad Prism Version. Regression analysis and IC50 determination was performed using GraphPad Prism Version 8.4.3 for Windows, GraphPad Software. www.graphpad.com.
- Sommer R.; Wagner S.; Rox K.; Varrot A.; Hauck D.; Wamhoff E.-C.; Schreiber J.; Ryckmans T.; Brunner T.; Rademacher C.; Hartmann R. W.; Brönstrup M.; Imberty A.; Titz A. Glycomimetic, Orally Bioavailable LecB Inhibitors Block Biofilm Formation of Pseudomonas aeruginosa. J. Am. Chem. Soc. 2018, 140 (7), 2537–2545. 10.1021/jacs.7b11133. [DOI] [PubMed] [Google Scholar]
- Sommer R.; Rox K.; Wagner S.; Hauck D.; Henrikus S. S.; Newsad S.; Arnold T.; Ryckmans T.; Brönstrup M.; Imberty A.; Varrot A.; Hartmann R. W.; Titz A. Anti-biofilm Agents against Pseudomonas aeruginosa: A Structure–Activity Relationship Study of C-Glycosidic LecB Inhibitors. J. Med. Chem. 2019, 62 (20), 9201–9216. 10.1021/acs.jmedchem.9b01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.