

Abstract
Background
Multidisciplinary discussion (MDD), in which physicians, radiologists, and pathologists communicate and diagnose together, has been reported to improve diagnostic accuracy compared to diagnoses made solely by physicians. However, even among experts, diagnostic concordance of MDD is not always good, and some patients may not receive a specific diagnosis due to insufficient findings. A provisional diagnosis based on the ontology with a diagnostic confidence level has recently been proposed. Additionally, we developed an artificial intelligence model to differentiate idiopathic pulmonary fibrosis (IPF) from other chronic interstitial lung diseases (ILD)s, which needs validation in a broader population.
Methods
This prospective nationwide ILD registry has recruited patients with newly diagnosed ILD at the referral respiratory hospitals in Japan and provides rapid MDD diagnoses and treatment recommendations through a central online MDD platform with a 3-year follow-up period. A modified diagnostic ontology is used. If no diagnosis reaches more than 50% certainty, the diagnosis is unclassifiable ILD. If multiple diseases are expected, the diagnosis with a high probability takes precedence. If the confidence levels for the top two possible diagnoses are equal, the diagnosis can be unclassifiable. The registry uses tentative diagnostic criteria for nonspecific interstitial pneumonia with organising pneumonia and smoking-related ILD not otherwise specified as possible new entities. Central MDD diagnosticians review the clinical data, test results, radiology images, and pathological specimens on a dedicated website and conduct MDD diagnoses using online meetings with a cloud-based reporting system. This study aims to (1) provide MDD diagnoses with treatment recommendations; (2) determine the overall ILD rates in Japan; (3) clarify the reasons for unclassifiable ILDs; (4) evaluate possible new disease entities; (5) identify progressive phenotypes and create a clinical prediction model; (6) measure the agreement rate between institutional and central diagnoses in ILD referral and non-referral centres; (7) identify key factors for each specific ILD diagnosis; and (8) create a new disease classification system based on treatment strategies, including the use of antifibrotic drugs.
Discussion
This study will provide ILD frequencies, including new entities, using central MDD on dedicated online systems, and develop a machine learning model for ILD diagnosis and prognosis prediction.
Trial registration
UMIN-CTR Clinical Trial Registry (UMIN000040678).
Supplementary Information
The online version contains supplementary material available at 10.1186/s12890-024-03232-1.
Keywords: Interstitial lung disease, Multidisciplinary discussion, Diagnosis, Diagnostic ontology, Unclassifiable interstitial lung disease, Hypersensitivity pneumonitis, Registry
Background
Interstitial lung disease (ILD) is a heterogeneous and challenging group of pulmonary disorders with varied prognoses and management options [1, 2]. Among ILDs, idiopathic pulmonary fibrosis (IPF) is a chronic, progressively worsening fibrotic lung disease of unknown aetiology with a devastating prognosis [1, 3, 4]. With the recently proven efficacy of two antifibrotic therapies, high accuracy in the diagnosis of IPF has become more important [3], and the guidelines for IPF diagnosis have been updated [5].
However, the correct diagnosis of fibrotic ILD is a challenge even for expert clinicians, radiologists, and pathologists [6, 7]. It requires multidisciplinary integration of clinical, radiological, and pathological features, which are then compared against a series of formal and informal diagnostic criteria for different conditions [7]. Considering this, the gold standard for diagnosing ILD is a dynamic integrated approach using multidisciplinary discussion (MDD), with close communication among clinicians, radiologists, and pathologists [1, 2]. In real-world settings, the number of facilities where specialists in the above three fields can discuss and diagnose using face-to-face MDD is limited. Thus, some patients with fibrotic ILD may not have been properly diagnosed or treated. One solution to this problem is the use of online MDD diagnosis, which has been proven to be effective only for patients with idiopathic interstitial pneumonia (IIP) using a retrospective dataset [8], and a trial in patients with IIPs has been initiated prospectively in Japan (JIPS registry). Another solution involves the use of artificial intelligence; however, we need to validate our artificial intelligence model to differentially diagnose IPF from other chronic ILDs, developed using data from a single facility [9], in the general population.
Recently, a provisional diagnostic approach based on ILD ontology with a diagnostic confidence level was proposed [7]. While this diagnostic method is useful for ILD specialists, respiratory physicians not specialised in ILD may have some difficulty in selecting possible diagnoses, assigning a level of confidence, and determining treatments. This is especially true for patients with a confidence level of less than 50% or those showing common clinical features associated with ILD but lacking a clearly defined clinical entity, such as smoking-related ILD (SR-ILD) and not otherwise specified or nonspecific interstitial pneumonia (NSIP) with organising pneumonia (OP), who are diagnosed with unclassifiable ILD at present. In addition, no report examined the impact of the recently published hypersensitivity pneumonitis (HP) guidelines on all types of ILDs [10]; thus, clarifying the nature of the current diagnostic ambiguity between IPF and HP is needed.
Additionally, there is growing interest in a subset of ILDs that exhibit progressive fibrosis. Studies on its pathology and criteria, as presented by clinical trials or international guidelines, have been published [5]. However, the disease behaviour can be significantly influenced by therapeutic interventions, and there is ongoing debate about the criteria and assessment timelines for progressive pulmonary fibrosis (PPF) [11]. In an era where antifibrotic drugs are already approved for PPF, a study examining the frequency and significance of PPF in a registry of all ILDs is important.
Therefore, we designed a prospective national cohort study of all types of ILDs in Japan in real-world settings.
Methods
Overview
This prospective nationwide registry study using centralised MDD was designed to identify Japanese patients with any type of newly diagnosed ILD. It aims to determine the prevalence and prognosis of each categorised ILD in real-world settings using web-based centralised MDD with diagnostic ontology. Additional main objectives of this study are:
To identify progressive phenotypes.
To determine the agreement rate between institutional and central diagnoses in patients with ILDs at ILD referral and non-referral centres. Moreover, the usefulness of ontology is examined using MDD diagnoses after three years of registration.
To identify the most important factors for each specific ILD diagnosis.
To create a new disease classification system based on treatment strategies, including antifibrotic drugs.
To evaluate the relationships between disease progression and baseline parameters, findings on high-resolution CT (HRCT) images, and patterns in patients with ILDs. To this end, we created a clinical prediction model.
To investigate the incidence of acute exacerbations according to ILD type and the prognosis after the onset of acute exacerbations.
To validate and improve a machine learning algorithm to differentiate IPF from other chronic ILDs or predict radiological and pathological patterns, prognosis, and disease progression [9].
To ensure data reliability, registration is performed via the Electronic Data Capture system (EDC). Clinical information and test results are stored in an online viewing system, and radiological images are stored in a dedicated viewing system. These multimodal data are provided to the central diagnostician via a seamless diagnostic system. The central MDD diagnostic team received requests from the study office via email and automated system notifications encouraging it to complete the MDD diagnosis within 2 weeks. Whole-slide images of pathology specimens from surgical lung biopsies or cryobiopsies are scanned and uploaded to a cloud-based viewer associated with a synoptic reporting system. The choice of treatment has to be based on regular medical practice and is at the discretion of the physician; no specific treatment is mandated or withheld from the patients. In addition, the robustness of the data collected by the EDC is ensured by conducting central monitoring.
Eligible patients and hospitals
An e-mail was sent to the 904 hospitals certified by the Japanese Respiratory Society for respiratory specialist training programs in Japan, and 223 hospitals were willing to participate in this study. The inclusion criteria are age > 20 years and suspected ILD within 24 months before enrolment at a participating hospital. The exclusion criteria are having undergone lobectomy or greater resection at the time of enrolment and an inadequate history and examination for diagnosis of ILD.
Data collection
Patient backgrounds
The following items are mainly acquired:
Date of ILD detection, date of diagnosis, diagnosis name at the registration facility, and assume disease progression within 24 months of registration.
Type of onset judged by respiratory symptoms (acute onset, within 1 month; subacute onset, within 1–3 months; chronic onset, > 3 months; asymptomatic).
Age, sex, life history, smoking history, and residence.
Comorbidities, medical history, and family history.
ILD drug treatment at the time of registration.
Physical findings and laboratory tests
Performance status, symptoms, and physical findings.
Laboratory tests, collagen-related autoantibodies, urine tests, arterial blood gas analysis, and oxygen saturation (SpO2).
Bronchoalveolar lavage fluid.
Pulmonary function test.
Six-minute walk test.
Chest images
Chest radiography (posterior-anterior view) and chest HRCT within 90 days of registration were acquired in the DICOM format. Sequential chest HRCTs in the supine position were obtained from the whole lungs with a slice thickness of 1.25 mm or less and reconstructed using a high-spatial-frequency algorithm. The MDD teams viewed these images using a web DICOM viewer (LOOKREC, MNES Inc., Hiroshima, Japan) for diagnosis.
Pathological tissue samples
Four unstained 3–4 μm thick sections of formalin-fixed paraffin-embedded tissue are prepared at each institution, and haematoxylin-eosin and Elastic van Gieson (EVG) stainings are performed at Nagasaki University. Whole-slide images scanned at 400× magnification are uploaded to a cloud-based viewer (PathPresenter, New York), and the pathologists evaluate the images using a synoptic reporting system (Porous, BonBon, Kyoto).
MDD diagnosis
Central MDD members recommended by the research committee of the Japanese Respiratory Society, which is independent of the study office, make the MDD diagnosis. Each central MDD team comprises one respiratory physician, one thoracic radiologist, and one pulmonary pathologist. In total, 13 MDD teams are considered. Central MDD will be assessed twice via the dedicated MDD diagnosis platform (Fig. 1), once at enrolment and again 3 years after enrolment. Central MDD members are not allowed to participate in central MDDs of cases registered at their institutions. If the registered data are insufficient to make an MDD diagnosis, the MDD team requests that the institution register the data and provide additional data through the study office.
Fig. 1.
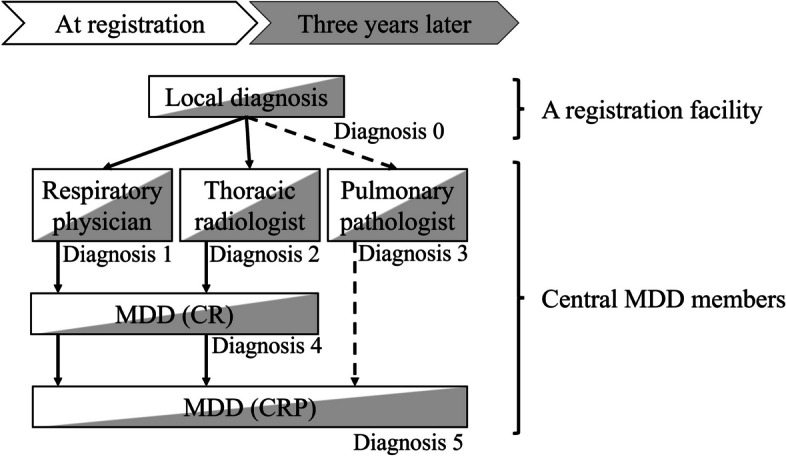
Diagnostic procedures. This shows the diagnostic process flow at two time points: at enrolment and 3 years later. Initially, local diagnoses are made by each registration facility (Diagnosis 0). Next, central MDD members’ diagnoses are made by individual respiratory physicians, thoracic radiologists, and pulmonary pathologists (if pathological samples are available), independently (Diagnosis 1 to 3). Next, the MDD is conducted in two stages: first, by a respiratory physician and a radiologist (shown as MDD (CR) and Diagnosis 4), and if pathology samples are available, by including a pulmonary pathologist in the second stage (shown as MDD (CRP) and Diagnosis 5). Abbreviations: MDD, multidisciplinary discussion.; CR, clinical and radiological discussion; clinical, radiological, and pathological discussion
The facility registering the case inputs the necessary information, image data, and digitised pathological slides if available. A diagnostic request is then emailed to one of the 13 MDD diagnostic teams. The team accesses a dedicated online diagnostic page to review the clinical information, image data using an online DICOM viewer, and digital pathology images. MDD members, including respiratory physicians, radiologists, and pathologists, access the required information, including patient interview data, physical findings, laboratory findings, pulmonary function testing results, and image findings from participating facilities. First, they independently generate a diagnostic report themselves in a preformatted form. Once individual diagnoses are complete, the system allows for MDD diagnosis input. The respiratory physician and radiologist conduct a remote diagnostic meeting via platforms like ZOOM to finalise the MDD diagnosis, which is then input into their forms. When pathology samples are available, a pathologist subsequently joins the team, and an MDD diagnosis is made based on the inputs of the respiratory physician, radiologist, and pathologist. After the diagnosis by the central MDD team, physicians of the participating facilities who register the patients are promptly notified of the diagnosis via automatic email distribution (Fig. 1).
The diagnostic process is classified into Steps 0–3 and is performed at both enrolment and 3 years later. The institutional diagnosis is mainly performed by a respiratory physician, possibly involving an MDD diagnostic procedure at the enrolment site. Central MDD diagnosis is performed via the dedicated online MDD diagnosis platform (Figs. 2 and 3), which provides access to clinical information and radiological images, except for the institutional diagnosis, to complete a standardised MDD diagnosis form. A pulmonologist and a radiologist independently complete the diagnostic report by referring to clinical information and radiological images (Diagnoses 1 and 2). If histopathologic images were available, a pathologist completes the diagnostic report by referring to the clinical information, radiologic images, and pathologic images (Diagnosis 3). Next, the pulmonologist and radiologist discuss the MDD diagnosis report via a web meeting (Diagnosis 4). If histopathological images were available, the pathologist re-creates the MDD diagnosis report (Diagnosis 5).
Fig. 2.

An example of the dedicated online MDD platform. MDD diagnosis is performed by accessing a dedicated page for each case. Each MDD diagnosis team can only view cases assigned by the Research Office, and the system is restricted so that each MDD diagnosis team can only view its own cases. 2A) A page displaying clinical information for each case. Detailed medical information can be viewed for each case. 2B) Chest radiographic images can also be viewed from a dedicated web page. 2C) Pathological whole slide images can be viewed from a dedicated web page with an unlimited range of magnification. Abbreviations: MDD, multidisciplinary discussion
Fig. 3.

A page to be filled in the MDD diagnosis system. In the left column, the MDD diagnosis at the time of registration is entered, and in the right column, the MDD diagnosis 3 years after registration.The institutional diagnosis is undisclosed to MDD members. This image has been edited to replace the Japanese text with English text. Abbreviations: MDD, multidisciplinary discussion
Diagnosis definitions
Previous registries and cohort studies indicated that unclassifiable ILD is more common in Japan, whereas HP is considerably less common. The epidemiology of all types of ILDs has not been examined since the establishment of the international HP guidelines [10]. The concept of unclassifiable ILD may also differ among physicians and countries, which in turn may affect the frequency of each disease. In Japan, the following disease classifications are commonly used. We redefined each classification and established a diagnostic manual for this study:
IIPs
Classifications are based on the 2018 ATS/ERS/JRS/ALAT Guidelines for Idiopathic Pulmonary Fibrosis Diagnosis [1] and the 2013 ATS/ERS statement for IIP classification [2]. Note that only “NSIP with OP” and “SR-ILD” are tentatively used as diagnostic classifications for possible new classifications not specified in current guidelines (Table 1, online supplement).
Table 1.
Diagnostic criteria arrangement for MDD in the PROMISE study
| Confidence level of diagnosis |
| ■ Definite (≥ 90%), high-confidence (70 − 89%), low-confidence (51 − 69%), very low-confidence (30 − 50%). |
| Definition of unclassifiable interstitial lung disease |
|
■ No diagnosis exceeds 50% certainty. ■ Combined PPFE and IPF lesions that are nearly equivalent. ■ Possible new entities as listed below. |
| Tentative diagnostic criteria for possible new entities |
|
■ NSIP with OP is defined morphologically as containing features of both NSIP and OP. The clinical course is typically subacute and usually observed in cases of anti-synthetase syndrome. HRCT images show bilateral areas of airspace consolidation and reticulation with or without traction bronchiectasis, usually showing both peribronchovascular and lower lung predominance, often with loss of volume. ■ SR-ILD on HRCT is defined as cases with ground-glass opacity and mild reticulation in the periphery of bilateral lower lobes with relative subpleural sparing, typically accompanied by cystic formations connecting to the bronchi or emphysema. Cases with prominent pleural surface irregularity or upper irregular lines perpendicular to the pleura were excluded. Pathologic findings show airspace enlargement with fibrosis and cystic lesions in the central airway with fibrosis. SR-ILD is distinct from known disease entities such as IPF, DIP, RB-ILD, and PLCH. |
| Handling of IPAF cases |
| ■ IPAF is diagnosed based on the classification of IIP and established disease concepts. |
Abbreviations: DIP Desquamative interstitial pneumonia, IIP Idiopathic interstitial pneumonia, IPAF Interstitial pneumonia with autoimmune features, IPF Idiopathic pulmonary fibrosis, HRCT High-resolution CT, MDD Multidisciplinary discussion, NSIP Nonspecific interstitial pneumonia, OP Organising pneumonia, PLCH Pulmonary Langerhans cell histiocytosis, PPFE Pleuroparenchymal fibroelastosis, RB-ILD Respiratory bronchiolitis, SR-ILD Smoking-related interstitial lung disease, IPAF Interstitial pneumonia with autoimmune features
In this study, the definition of unclassifiable ILD is as follows: (1) No diagnosis with more than 50% certainty. (2) Combined idiopathic pleuroparenchymal fibroelastosis (PPFE; Table 2, online supplement) and IPF lesions that are nearly equivalent. (3) Possible new entities of “NSIP with OP” and “SR-ILD” (Table 1, online supplement).
Table 2.
Idiopathic PPFE diagnostic criteria
| Idiopathic PPFE diagnostic criteria |
|---|
| 1. Slowly progressive dry cough or exertional dyspnoea. |
| 2. Alveolar consolidation just below the pleura with traction bronchiectasis, predominantly in the bilateral upper lobes on HRCT. |
| 3. Upward shift of bilateral pulmonary hilum on chest x-ray or volume reduction of the upper lobes on HRCT. |
| 4. Exclusion of other diseases with known causes, such as fibrotic hypersensitivity pneumonitis, connective tissue disease, occupational lung disease, hematopoietic stem cells, or lung transplant-related lung disease, were excluded. |
Radiologically possible idiopathic PPFE criteria described in [12], partially modified
Abbreviations: HRCT High-resolution CT, PPFE Pleuroparenchymal fibroelastosis
Idiopathic pleuroparenchymal fibroelastosis
Idiopathic PPFE is an ILD confined to or predominant in the upper lobe. Idiopathic PPFE is defined as a disease that meets all the revised diagnostic criteria listed in Table 2. A surgical lung biopsy is optional.
If the cause is unknown and interstitial pneumonia lesions are found in the lower lung fields, a diagnosis is made based on the predominant lesion. If the upper and lower lung fields are comparable, the disease is considered unclassifiable.
Connective tissue disease-related ILD
The diagnosis of CTD is based on the published diagnostic or classification criteria ([13–20]; online supplement). CTD is possibly, but not necessarily diagnosed by rheumatologists or physicians specialised in CTD diagnosis. ILD with autoimmune features (IPAF) that do not fulfil any of the CTD criteria diagnosed under the category of the current IIP statement [21].
If guidelines exist at the time of study initiation, a diagnosis is made using the guidelines at the time of study initiation, even if they are revised during the course of the study. If no corresponding guidelines exist at the time of study initiation, the criteria used in daily clinical practice at the time of study initiation are employed without further changes, even if a new guideline is developed.
Confidence level
Although confidence levels are used in each ILD guideline, the confidence classification varies among guidelines [1, 10]. In addition, even if the confidence level is equal to or less than 50%, the diagnosis is determined unclassifiable ILD [7], while the treatment strategy depends on the clinically assumed disease. Therefore, this study defined and used the following confidence levels:
Confident diagnosis: The diagnostic likelihood was believed to be 90% or more.
High confidence: The diagnostic likelihood was believed to be 70–89%.
Low confidence: The diagnostic likelihood was believed to be 51–69%.
Very low confidence: The diagnostic likelihood was believed to be 30–50%.
Definition of disease progression
Disease progression was defined in this study as at least one of the following criteria being met within 24 months:
Relative decline in forced vital capacity (FVC) of 10% or more (% predicted).
Relative FVC decrease by more than 5% but less than 10% (% predicted), and the site investigator judged that the respiratory symptoms worsened.
Relative FVC decrease by more than 5% but less than 10% (% predicted), and the site investigator determine that the extent of fibrotic changes on chest imaging (HRCT scan) increased by more than 10% from the image at enrolment.
The site investigator judged whether the respiratory symptoms worsened independently of changes in FVC and that the extent of fibrotic changes on chest imaging (HRCT scan) increased by more than 10%.
A definition of progressive pulmonary fibrosis (PPF) was recently proposed [5], but evidence of its real-world clinical significance is lacking. Therefore, this study will also examine the significance of the definition of PPF by examining the proportion of cases and characteristics that meet the criteria in years one, two, and three.
Sample size and recruitment
This prospective cohort study has no previous studies to base the sample size calculation on. Descriptive statistical methods are used to analyse the results. The sample size of INSIGHT-IPF was at least 500 patients and that of IPF-PRO was 300 patients [22, 23]. Therefore, at least 200 patients with IPF who fulfil the diagnostic criteria for IPF will be enrolled in this prospective study, as this is an adequate number to obtain meaningful outcomes and allow for comparison with other cohorts. A prior registry study in Japan, the JIPS registry, included 868 patients with IIP over 16 months in 85 Japanese facilities [24]. Patients with IPF comprised approximately 30–40% of IIPs at each facility, providing an expected IPF population of at least 260 patients. Based on the historical cohort of Tosei General Hospital in Japan, secondary ILD accounted for approximately 25–40% of all ILDs. Moreover, based on a prior survey, a maximum of 30% of the 706 Japanese Respiratory Society-certified facilities will participate in this study. Considering that many respiratory referral centres not specialising in ILD will participate in this study, it is assumed that 50% of registered patients with ILDs per participating center will be registered.
Therefore, 870–2,700 patients will be registered in 24 months at approximately 85–210 facilities in Japan.
Diagnostic concordance
To assess the diagnostic concordance among the MDD teams, 65 cases will be randomly selected from the registered cases, and all teams will make separate MDD diagnoses for the same selected cases, blinded to the fact that the cases are being diagnosed by all teams. This procedure will be notified before the trial begins.
Statistical analysis methods
Continuous data are presented as mean ± standard deviation. Categorical variables are reported as frequencies (%). Between-group differences are assessed using a two-sided t-test or chi-squared test, as appropriate. Cohen’s kappa coefficient (κ) is used to evaluate the agreement between institutional and MDD ILD diagnoses. Temporal changes in sensitivity and specificity of IPF diagnosis likelihood are analysed using generalised estimating equations. The results of the Cox proportional hazards analyses are presented as estimated values used to investigate the ability of each Cox proportional hazards regression model to predict mortality. Cumulative probabilities of survival are plotted using the Kaplan–Meier method and are compared using the log-rank test. Additionally, the diagnostic concordance between teams will be evaluated using the distribution of kappa values based on this blinded data. P < 0.05 is considered statistically significant.
Management system
This study is mainly funded by Nippon Boehringer Ingelheim, and Nagoya University Hospital is the study administrator with Mebix Corporation as the contract research organisation.
Discussion
This article provides an overview of the Providing Multidisciplinary ILD Diagnoses (PROMISE) study, one of the largest prospective national registries of all types of ILDs. A diverse group of respiratory centres in Japan participated in the study and possible new diagnostic entities were proposed.
This study is unique in its approach to identifying and validating new disease entities within the spectrum of ILD. Unlike previous studies where diagnostic classifications were often determined by multidisciplinary experts through consensus discussions, our study employs a large-scale, prospective registry to systematically extract candidate new disease entities based on prespecified diagnostic criteria. By evaluating these candidates through multiple diagnostic teams, we aim to establish their distinctiveness as independent diseases. This includes a thorough analysis of clinical presentation, imaging findings, pathological features, treatment responses, and prognostic outcomes. This methodology not only enhances the robustness of the diagnostic process but also provides a comprehensive framework for understanding the heterogeneity of ILD.
This study also aims to improve the efficiency of MDD diagnoses by validating and refining the developed diagnostic artificial intelligence [9]. The gold standard for the diagnosis of ILD is MDD [2]. However, few facilities have specialists in all three MDD areas to discuss and perform an MDD. As the number of patients with IPF increases annually, with a reported prevalence of 725 cases per 100,000 [25], rapid and convenient MDD diagnosis for many patients with ILDs, including those with IPF, may benefit from artificial intelligence assistance. In addition, a correct prediction of ILD prognosis is currently impossible. Thus, we aim to develop and validate an artificial intelligence system to predict the prognosis with high accuracy, thereby revolutionising clinical practice for patients with ILDs.
Furthermore, the PROMISE study differs from previous registries for ILD because it aims:
-
i.
To determine the relative prevalence and nature of all types of ILDs based on the latest guidelines, including the HP guidelines [10], in real-world settings.
Although differentiation between IPF and HP is crucial [10], the diagnostic concordance rate for HP is low, even in MDD diagnosis [26] because HP shares characteristics with other acute and chronic ILDs. Recently, guidelines for HP have been established; therefore, standardisation for diagnosing HP is expected. With this registry, the true nature of IPF and HP under the new guidelines can be clarified through centralised MDD.
-
ii.
To perform centralised MDD at enrolment and at the end of the 3-year follow-up period to evaluate any changes in MDD diagnosis.
The diagnosis of ILD may change over the disease course [7]. This study clarifies the transition from diagnosis at enrolment to diagnosis 3 years after enrolment using the diagnostic ontology of confidence levels. This approach enables clarification of the certainty of the diagnosis at enrolment and the significance of the diagnostic ontology.
-
iii.
To facilitate quick patient enrolment, MDD diagnosis, and treatment recommendations based on the newly developed web platform. This MDD diagnosis approach will overcome the temporal and geographic limitations of ILD practices.
According to the guidelines, MDD-based diagnosis is the gold standard for patients with ILDs and an important diagnostic step [1, 2]. However, few facilities, even in Japan, have all the necessary MDD specialists in the same facility. Therefore, in this study, we developed a standardised diagnostic platform that can quickly perform MDD diagnoses by interactively connecting registered facilities and central MDD specialists via a web platform. This rapid and precise MDD diagnostic system will also improve data quality, a frequent problem of disease registries [27], by enabling them to register precise information necessary for MDD diagnosis.
-
iv.
To demonstrate the problem of diagnosing unclassifiable ILDs such as ILDs with low diagnostic confidence, PPFE spectrum disorders including issues on the relationship of “PPFE+IPF” with “PPFE” and “IPF” [28], and possible new entities like "NSIP with OP" and "SR-ILD" which are not established but are assumed to be present in a certain percentage of patients.
This study will clarify current problems associated with ILD diagnosis and develop a clinical model that will innovate medical practice for patients with ILDs.
Supplementary Information
Acknowledgements
All authors contributed to the design of the PROMISE study and writing of this manuscript. TF is responsible for the overall content as the guarantor.
Abbreviations
- ANCA
Antineutrophil cytoplasmic antibody
- CTD
Connective tissue disease
- DIP
Desquamative interstitial pneumonia
- EDC
Electronic Data Capture system
- HP
Hypersensitivity pneumonitis
- HRCT
High-resolution CT
- IIP
Idiopathic interstitial pneumonia
- ILD
Interstitial lung disease
- IPAF
Interstitial pneumonia with autoimmune features
- IPF
Idiopathic pulmonary fibrosis
- MDD
Multidisciplinary discussion
- NSIP
Nonspecific interstitial pneumonia
- OP
Organising pneumonia
- PLCH
Pulmonary Langerhans cell histiocytosis
- PPF
Progressive pulmonary fibrosis
- PPFE
Pleuroparenchymal fibroelastosis
- RB-ILD
Respiratory bronchiolitis interstitial lung disease
- SR-ILD
Smoking-related interstitial lung disease
Authors’ contributions
YK and TF accepted full responsibility for the completed work and conduct of the study, had access to the data, and made the decision to publish. All authors designed the study. TF, SO, and JF created the web-based central MDD system. TH, AN, and ST performed data analysis. All authors contributed to the data interpretation. YK, TF, and TH wrote the manuscript. All authors contributed to the revision of the manuscript and approved the final version to be published.
Funding
The PROMISE study is funded by Boehringer Ingelheim Pharmaceuticals, Inc., JSPS KAKENHI (grant number JP19110253), The Hori Science and Arts Foundation, and a grant from the Japanese Respiratory Foundation. None of the sponsors participated in the study design, data collection, data analysis, or manuscript preparation. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as it relates to Boehringer Ingelheim substances, as well as intellectual property considerations.
Availability of data and materials
Data availability is permissible when approved by the appropriate ethics review committee for each study and by the specialised committee of this research. Not applicable because this manuscript does not report data generation or analysis.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
This study was approved by the Nagoya University Ethics Committee and the Ethics Committee of each participating institution. All enrolled patients provided written informed consent prior to enrolment in the study. This study was registered with the UMIN Clinical Trials Registry (https://www.umin.ac.jp/ctr/index.htm, UMIN000040678) on July 1, 2020.
Consent for publication
Not applicable.
Competing interests
YK reports personal fees from Asahi Kasei Pharma Corp.; Boehringer Ingelheim Co., Ltd.; Eisai Inc.; KYORIN Pharmaceutical Co., Ltd.; Novartis Pharma K.K.; and Shionogi & Co. outside of the submitted work. KS reports grants from the Pulmonary Fibrosis Foundation and Novartis Pharma outside the submitted work. JF holds stocks in PathPresenter and serves on an advisory board at N Lab Co., Ltd. MI reports advisory board participation and lecture fees from GlaxoSmithKline plc. and Moderna Japan Co., Ltd., and lecture fees from Shionogi & Co., Ltd., AstraZeneca K.K., KYORIN Pharmaceutical Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., MSD K.K., Insmed, Inc., Daiichi Sankyo Co., Ltd., Pfizer Japan Inc., Asahi Kasei Pharma Corporation, Chugai Pharmaceutical Co., Ltd., Eli Lilly Japan K.K., Abbott Diagnostics Medical Co., Ltd., TAIHO PHARMACEUTICAL Co., LTD., Amgen K.K., and Novartis Pharma K.K., outside the submitted work. All other authors report no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yasuhiro Kondoh and Taiki Furukawa contributed equally to this work.
References
- 1.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–68. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Costabel U, Hansell DM, King TE Jr., Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3–19. [DOI] [PubMed] [Google Scholar]
- 4.Salton F, Ruaro B, Confalonieri P, Confalonieri M. Epithelial-mesenchymal transition: a major pathogenic driver in idiopathic pulmonary fibrosis? Med (Kaunas). 2020;56:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh SLF, Maher TM, Kolb M, Poletti V, Nusser R, Richeldi L, et al. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J. 2017;50:1700936. [DOI] [PubMed] [Google Scholar]
- 7.Ryerson CJ, Corte TJ, Lee JS, Richeldi L, Walsh SLF, Myers JL, et al. A standardized diagnostic ontology for fibrotic interstitial lung disease. An international working group perspective. Am J Respir Crit Care Med. 2017;196:1249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujisawa T, Mori K, Mikamo M, Ohno T, Kataoka K, Sugimoto C et al. Nationwide cloud-based integrated database of idiopathic interstitial pneumonias for multidisciplinary discussion. Eur Respir J. 2019;53(5):1802243. [DOI] [PMC free article] [PubMed]
- 9.Furukawa T, Oyama S, Yokota H, Kondoh Y, Kataoka K, Johkoh T, et al. A comprehensible machine learning tool to differentially diagnose idiopathic pulmonary fibrosis from other chronic interstitial lung diseases. Respirology. 2022;27:739–46. [DOI] [PubMed] [Google Scholar]
- 10.Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2020;202:e36–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cottin V, Brown KK, Flaherty KR, Wells AU. Progressive pulmonary fibrosis: should the timelines be taken out of the definition? Am J Respir Crit Care Med. 2022;206:1293–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe K, Ishii H, Kiyomi F, Terasaki Y, Hebisawa A, Kawabata Y, et al. Criteria for the diagnosis of idiopathic pleuroparenchymal fibroelastosis: a proposal. Respir Investig. 2019;57:312–20. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka Y, Kuwana M, Fujii T, Kameda H, Muro Y, Fujio K, et al. 2019 diagnostic criteria for mixed connective tissue disease (MCTD): from the Japan research committee of the ministry of health, labor, and welfare for systemic autoimmune diseases. Mod Rheumatol. 2021;31:29–33. [DOI] [PubMed] [Google Scholar]
- 14.Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69:2271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2016;69:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–8. [DOI] [PubMed] [Google Scholar]
- 18.Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic Lupus International collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robson JC, Grayson PC, Ponte C, Suppiah R, Craven A, Judge A, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for granulomatosis with polyangiitis. Ann Rheum Dis Am Coll Rheumatol. 2022;81:315–20. [DOI] [PubMed] [Google Scholar]
- 20.Suppiah R, Robson JC, Grayson PC, Ponte C, Craven A, Khalid S, et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann Rheum Dis Am Coll Rheumatol. 2022;81:321–6. [DOI] [PubMed] [Google Scholar]
- 21.Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–87. [DOI] [PubMed] [Google Scholar]
- 22.Behr J, Kreuter M, Hoeper MM, Wirtz H, Klotsche J, Koschel D, et al. Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J. 2015;46:186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien EC, Durheim MT, Gamerman V, Garfinkel S, Anstrom KJ, Palmer SM, et al. Rationale for and design of the idiopathic pulmonary Fibrosis-PRospective outcomes (IPF-PRO) registry. BMJ Open Respir Res. 2016;3:e000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okuda R, Ogura T, Hisata S, Baba T, Kondoh Y, Suda T, et al. Design and rationale of the Japanese idiopathic interstitial pneumonias (JIPS) Registry. Respir Investig. 2023;61:95–102. [DOI] [PubMed] [Google Scholar]
- 25.Kaul B, Lee JS, Zhang N, Vittinghoff E, Sarmiento K, Collard HR, et al. Epidemiology of idiopathic pulmonary fibrosis among U.S. veterans, 2010–2019. Ann Am Thorac Soc. 2022;19:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh SLF, Wells AU, Desai SR, Poletti V, Piciucchi S, Dubini A, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4:557–65. [DOI] [PubMed] [Google Scholar]
- 27.Moor CC, Kreuter M, Luppi F, Wuyts WA. The world is not enough – the value of increasing registry data in idiopathic pulmonary fibrosis. Respir Res. 2020;21:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oda T, Ogura T, Kitamura H, Hagiwara E, Baba T, Enomoto Y, et al. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared with idiopathic pulmonary fibrosis. Chest. 2014;146:1248–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data availability is permissible when approved by the appropriate ethics review committee for each study and by the specialised committee of this research. Not applicable because this manuscript does not report data generation or analysis.
No datasets were generated or analysed during the current study.