

ABSTRACT
Syndromes characterized by congenital diarrhea, hearing loss, and intrahepatic cholestasis are uncommon and often misdiagnosed as progressive familial intrahepatic cholestasis (PFIC). Recent genetic studies have widened the array of genes linked with cholestatic disorders. Among these, UNC45A has recently been implicated in microvillous inclusion disease (MVID), although only a few cases exist. This case highlights a 20-year-old woman initially diagnosed clinically with PFIC type 1 during childhood. After ileal bypass at age 4 years, she had a resolution of intractable pruritus and cholestasis. Despite remaining symptom-free for over a decade, she returned in adulthood with recurrent cholestatic pruritus. Odevixibat was initiated for presumed PFIC while awaiting additional testing with symptomatic improvement and laboratory normalization. Whole genome sequencing identified novel compound heterozygous mutations in UNC45A and small bowel biopsies confirmed villous atrophy. Odevixibat, currently approved for cholestatic pruritus in PFIC and Alagille syndrome, demonstrates efficacy in managing cholestatic pruritus in MVID.
KEYWORDS: congenital diarrhea, intrahepatic cholestasis, UNC45A mutations, microvillous inclusion disease, MVID, odevixibat
INTRODUCTION
Syndromes consisting of congenital diarrhea, hearing loss, and intrahepatic cholestasis in infancy and early childhood are rare. Patients with these clinical symptoms are often diagnosed with progressive familial intrahepatic cholestasis (PFIC) based on clinical presentations. However, studies have reported that many patients have a negative mutation in the areas classically responsible for PFIC including ATP8B1, ABCB11, and ABCB4.1 Recently, because of the advancement in genetic testing technologies, newer studies have demonstrated that other mutations in areas such as TJP2, NR1H4, SLC51A, and MYO5B are also responsible for PFIC-like clinical pictures.2
Microvillous inclusion disease (MVID) is an autosomal recessive disorder classically associated with mutations in the MYO5B, STX3, and STX2BP genes responsible for encoding proteins involved in the apical membrane trafficking in enterocytes.3 This congenital condition typically manifests as intrahepatic cholestasis and diarrhea. In 2018, a novel mutation in UNC45A was identified to be responsible for a new variant of MVID similar to those with MYO5B mutations.4 UNC45A is responsible for encoding a cochaperone that promotes the proper folding of myosin. UNC45A mutations manifest as congenital diarrhea, intrahepatic cholestasis, hearing loss, and bone fragility. A comprehensive investigation of 4 children with this condition demonstrated varying phenotypic expressions of the condition and severity.4 These findings were then confirmed by a second study on an additional 6 individuals showing UNC45A mutations result in MVID.3
We present a case of a young female with a novel compound heterozygous UNC45A mutation causing MVID, successfully treated with odevixibat for cholestasis and pruritus. Initially diagnosed with presumptive PFIC type 1 at the age of 4 years, genetic testing was delayed until recurrent symptoms prompted reevaluation over a decade later.
CASE REPORT
A 20-year-old woman presented to hepatology clinic with episodic pruritus, loose clay-colored stools, and jaundice. She was diagnosed with PFIC type 1 during infancy now status-post ileal bypass surgery. She had performed well throughout childhood and early adulthood until she developed these episodic symptoms that occurred in association with starting combined oral contraceptive pills for various gynecologic disorders. Examination was notable for jaundice. Laboratory test results demonstrated high total bilirubin of 8.2 (reference range 0.3–1.0 mg/dL), conjugated bilirubin of 4.3 mg/dL (reference range 0.1–0.5 mg/dL), alanine transaminase of 127 U/L (reference range 7–52 U/L), aspartate aminotransferase of 55 U/L (reference range 15–41 U/L), alkaline phosphatase of 423 (reference range 32–91 U/L), and total bile acids >1,350 μmol/L (reference range 0–10 μmol/L). Gamma-glutamyl transferase ranged between 6 and 33 U/L (reference range 7–26 U/L). The liver and gallbladder appearance were normal on ultrasound. A colonoscopy with biopsy revealed villous atrophy of her terminal small bowel. Hormone therapy was believed to be contributing to cholestasis, so she was instructed to avoid these medications. Despite stopping hormone therapy and trials of various medications, including cholestyramine, colestipol, rifampin, ursodeoxycholic acid, and phenobarbital, relief was not achieved.
History included the development of clay-colored stools and intractable pruritus by 12 months old and sensorineural hearing loss requiring cochlear implants in infancy. She had 3 fractures during childhood. By age 3 years, she had undergone 3 liver biopsies showing intrahepatic cholestasis and mild fibrosis. Recurrent episodes of intractable pruritis led to hospitalizations, during which various therapies, including naloxone, ursodeoxycholic acid, and laxatives, were attempted with some success until age 4 years when they became largely ineffective. At this time, she was diagnosed clinically with PFIC type I given her cholestasis, diarrhea, and hearing loss. Because of refractory pruritus requiring frequent hospitalizations, ileal bypass surgery was performed at age 4 years.
Given symptom recurrence in adulthood despite ileal bypass surgery at age 4 years, additional diagnostic evaluation was warranted. A cholestatic genetic panel (Invitae) was negative. Given refractory symptoms, odevixibat was initiated for presumptive PFIC type 1. Significant improvement was noted after treatment, with normalization of laboratory values (total bilirubin, aspartate aminotransferase, alanine transaminase, and total bile acids) and resolution of episodic pruritus and cholestasis. Bile acid levels remained normal since odevixibat initiation.
After an initial negative genetic panel, the patient underwent whole exome sequencing, revealing compound heterozygous mutations in UNC45A with c.2225 T>C; p.Leu742Pro, c.2297 A>T; p.Glu766Val, and c.2348 G>A; p.Cys783Tyr (Figure 1). Her clinical symptoms are consistent with MVID including cholestasis, hearing loss, diarrhea, and multiple fractures. Since diagnosis, odevixibat treatment has maintained an exceptional clinical response for nearly a year and a half, with normal laboratory values and absence of symptoms.
Figure 1.
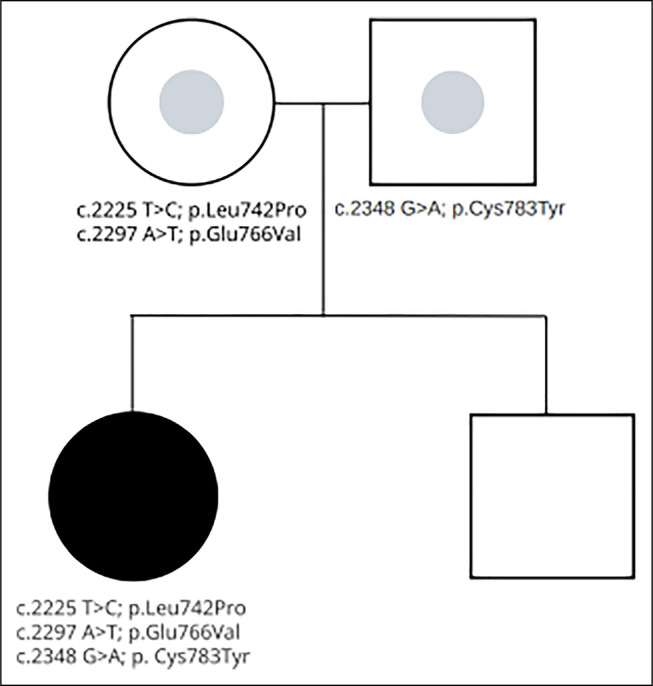
Whole exome sequencing results demonstrating inheritance of mutations at UNC45A.
DISCUSSION
This patient displayed clinical features resembling PFIC including intractable pruritis, cholestasis, and hearing loss. However, inconsistent findings such as villous atrophy on intestinal biopsy suggested MVID. Genetic analysis revealed a compound heterozygote mutation in the UNC45A gene, implicated in encoding a myosin chaperone expressed in intestinal and liver epithelial cells. This chaperone is used in apical trafficking and microvillus differentiation in enterocytes. UNC45A mutations have been linked to MVID variants characterized by congenital diarrhea, cholestasis, hearing loss, and bone fragility. The possible association with varying levels of intellectual disability has also been reported.4 Overall, genetic testing, small bowel biopsy, and the patient's clinical presentation align with MVID. Despite having 3 fractures during childhood, it is unknown whether any of these fractures would have been classified as fragility fractures. No intellectual disability has been identified in this patient. Her compound heterozygote mutation in UNC45A represents the first documented case of MVID with these specific mutations.
Most importantly, in this case, we demonstrated that odevixibat can be effective in the treatment of cholestasis and pruritus associated with MVID. There has been no change in this patient's issues with malabsorption or chronic diarrhea since starting odevixibat. Another patient in Germany with MY05B-related MVID also had a resolution of cholestasis and pruritus after starting odevixibat.5 Odevixibat is currently approved by the Food and Drug Administration for the treatment of PFIC in patients ≥3 months of age and Alagille syndrome in patients ≥12 months of age. These 2 cases support that although it is not currently Food and Drug Administration–approved for MVID, odevixibat effectively treats cholestatic pruritus in patients with MVID.
In conclusion, we report a case of MVID, an exceedingly rare disorder, and highlight novel compound heterozygous mutations of UNC45A. Moreover, this case introduces odevixibat as an effective therapeutic option for pruritus in patients with MVID demonstrating clinical remission of symptoms in our patient. This suggests its use could be considered in the management of future cases of MVID that are poorly responding to other therapies.
DISCLOSURES
Author contributions: A. Fiedler is the article guarantor. A. Fiedler and K. Brittan are responsible for the overall composition, edits, and submission. W. Manatsathit critically revised the manuscript for important intellectual content, was directly involved in the patient's care, and helped manage the clinical case. The authors reviewed and approved the final version of the manuscript.
Financial disclosure: None to report.
Informed consent was obtained for this case report.
Footnotes
Alexandra Fiedler and Kevin Brittan contributed equally to this work and are considered co-first authors.
Contributor Information
Kevin Brittan, Email: kbrittan@unmc.edu.
Wuttiporn Manatsathit, Email: ShaneManatsathit@creighton.edu.
REFERENCES
- 1.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie S, Wei S, Ma X, et al. Genetic alterations and molecular mechanisms underlying hereditary intrahepatic cholestasis. Front Pharmacol. 2023;14:1173542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duclaux-Loras R, Lebreton C, Berthelet J, et al. UNC45A deficiency causes microvillus inclusion disease–like phenotype by impairing myosin VB–dependent apical trafficking. J Clin Invest. 2022;132(10):e154997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esteve C, Francescatto L, Tan PL, et al. Loss-of-function mutations in UNC45A cause a syndrome associating cholestasis, diarrhea, impaired hearing, and bone fragility. Am J Hum Genet. 2018;102(3):364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roquelaure B Sciveres M Grammatikopoulos T, et al. . Odevixibat therapy in patients with progressive familial intrahepatic cholestasis (PFIC) associated with MYO5B mutations: A retrospective case series. Poster presented at EASL Congress June 21-24: Vienna. (www.postersessiononline.eu/173580348_eu/congresos/ILC2023/aula/-THU_336_ILC2023.pdf) (2023). [Google Scholar]