

SUMMARY
Maladaptive plasticity is linked to the chronification of diseases such as pain, but the transition from acute to chronic pain is not well understood mechanistically. Neuroplasticity in the central nucleus of the amygdala (CeA) has emerged as a mechanism for sensory and emotional-affective aspects of injury-induced pain, although evidence comes from studies conducted almost exclusively in acute pain conditions and agnostic to cell type specificity. Here, we report time-dependent changes in genetically distinct and projection-specific CeA neurons in neuropathic pain. Hyperexcitability of CRF projection neurons and synaptic plasticity of parabrachial (PB) input at the acute stage shifted to hyperexcitability without synaptic plasticity in non-CRF neurons at the chronic phase. Accordingly, chemogenetic inhibition of the PB→CeA pathway mitigated pain-related behaviors in acute, but not chronic, neuropathic pain. Cell-type-specific temporal changes in neuroplasticity provide neurobiological evidence for the clinical observation that chronic pain is not simply the prolonged persistence of acute pain.
Graphical Abstract
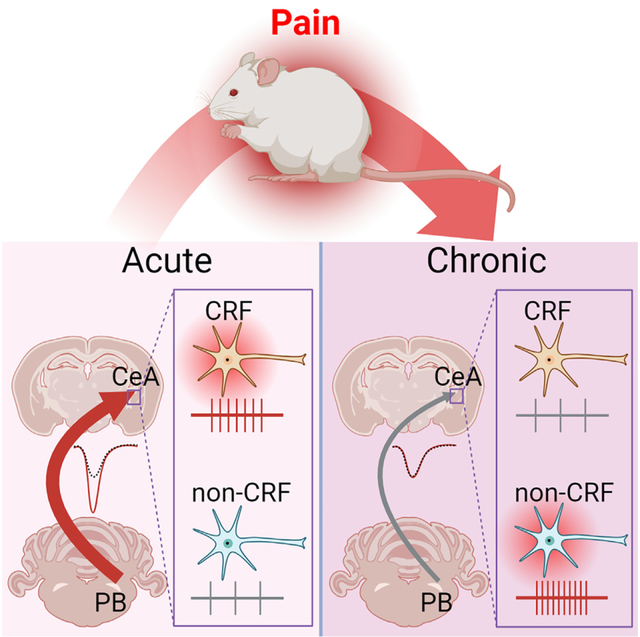
In brief
The transition from acute to chronic pain and the chronification of diseases in general are not well understood mechanistically. Kiritoshi et al. demonstrate time- and cell-type-specific forms of neuroplasticity in chronic neuropathic pain in the amygdala, a brain region that is linked to the emotional-affective aspects of pain and pain modulation.
INTRODUCTION
Neuroplasticity forms the basis for important functions such as learning and memory, but it is also a mechanism of neurological and psychiatric disorders,1,2 and structural and functional neuroplastic changes are observed in pain conditions.3,4 Chronic pain is a health problem that affects about 20% of the world’s population,5–7 impacting the quality of life of patients and caregivers as well as healthcare costs.8–10 Pain is a complex sensory and emotional experience that involves the encoding of nociceptive signals and higher integrated processing in brain circuits such as the corticolimbic system.11,12 Pain-related neuroplasticity in the brain, however, remains an understudied line of research despite an urgent need to identify mechanisms and targets for improved therapeutic approaches.
The amygdala, a key component of the corticolimbic system, is one of only a few brain regions that have been explored for neuroplastic changes in pain conditions.13 The central nucleus of the amygdala (CeA) receives nociceptive information via the spino-(trigemino-)parabrachio (PB)-amygdaloid pathway, and through integrative processing plays an important role in the somatosensory and emotional-affective aspects of pain.13–15 Synaptic plasticity in the PB→CeA pathway and changes in the excitability of CeA neurons have been linked to pain-related behaviors in different models.16–25 However, these studies were undertaken in acute pain conditions or at the early stages of persistent pain models.
Mechanisms of the transition to chronic pain are generally not well understood. Here, we addressed this important knowledge gap by evaluating amygdala neuroplasticity in chronic pain states in genetically distinct and projection-specific CeA cells. The CeA comprises highly heterogeneous populations of GABAergic neurons identified by their genetic, firing, and connectivity properties.24,26–32 Our recent studies using transgenic mice revealed the opposing role of CeA-somatostatin (SOM) and CeA-protein kinase C delta (PKCδ) neurons in pain-related behaviors as well as excitability changes in these neurons.23,24 Furthermore, we demonstrated important contributions of CeA-corticotropin-releasing factor (CRF) neurons to pain-related behaviors.33,34 Injury-induced CeA neuroplasticity is also firing type and projection specific.20,21 Importantly, a recent study provided evidence for synaptic changes in CeA-CRF neurons in brain slices at the acute phase of a neuropathic pain model.22 However, pain-related excitability changes in CeA-CRF neurons remain to be determined, and synaptic and cellular changes in different types of CeA neurons in chronic pain conditions are not known.
Here, we focused on CRF neurons in the CeA and sought to determine differences in excitability and PB-driven synaptic transmission between acute and chronic phases of neuropathic pain models using brain slice patch-clamp recordings. We also investigated anatomical and electrophysiological differences between periaqueductal gray (PAG)- and PB-projecting CeA-CRF neurons. Since we found a shift from plasticity at the PB→CeA synapse and hyperexcitability of CRF projection neurons at the acute stage to synaptic plasticity-independent hyperexcitability in non-CRF neurons at the chronic phase, we also explored the cell type of these non-CRF (SOM and PKCδ) neurons. Finally, we examined the contribution of the PB→CeA pathway to pain-related behaviors at the different pain stages using chemogenetics.
RESULTS
Loss of synaptic plasticity at the chronic stage of neuropathic pain
Electrophysiological studies were performed in brain slices from behaviorally tested animals. Paw withdrawal thresholds were measured 1 week (acute phase) or 4 weeks (chronic phase) after spinal nerve ligation surgery (SNL model) to confirm the development of neuropathic pain (Figure 1A). Consistent with previous studies on amygdala function in the SNL neuropathic pain model,18,34–37 neuropathic animals showed significant mechanical hypersensitivity at both acute and chronic phases of SNL compared to sham controls (F3, 80 = 39.55, p < 0.0001, one-way ANOVA with post hoc Bonferroni tests; Figure 1B).
Figure 1. Distinct types of CRF and non-CRF neurons in brain slices from acute and chronic sham/SNL rats.
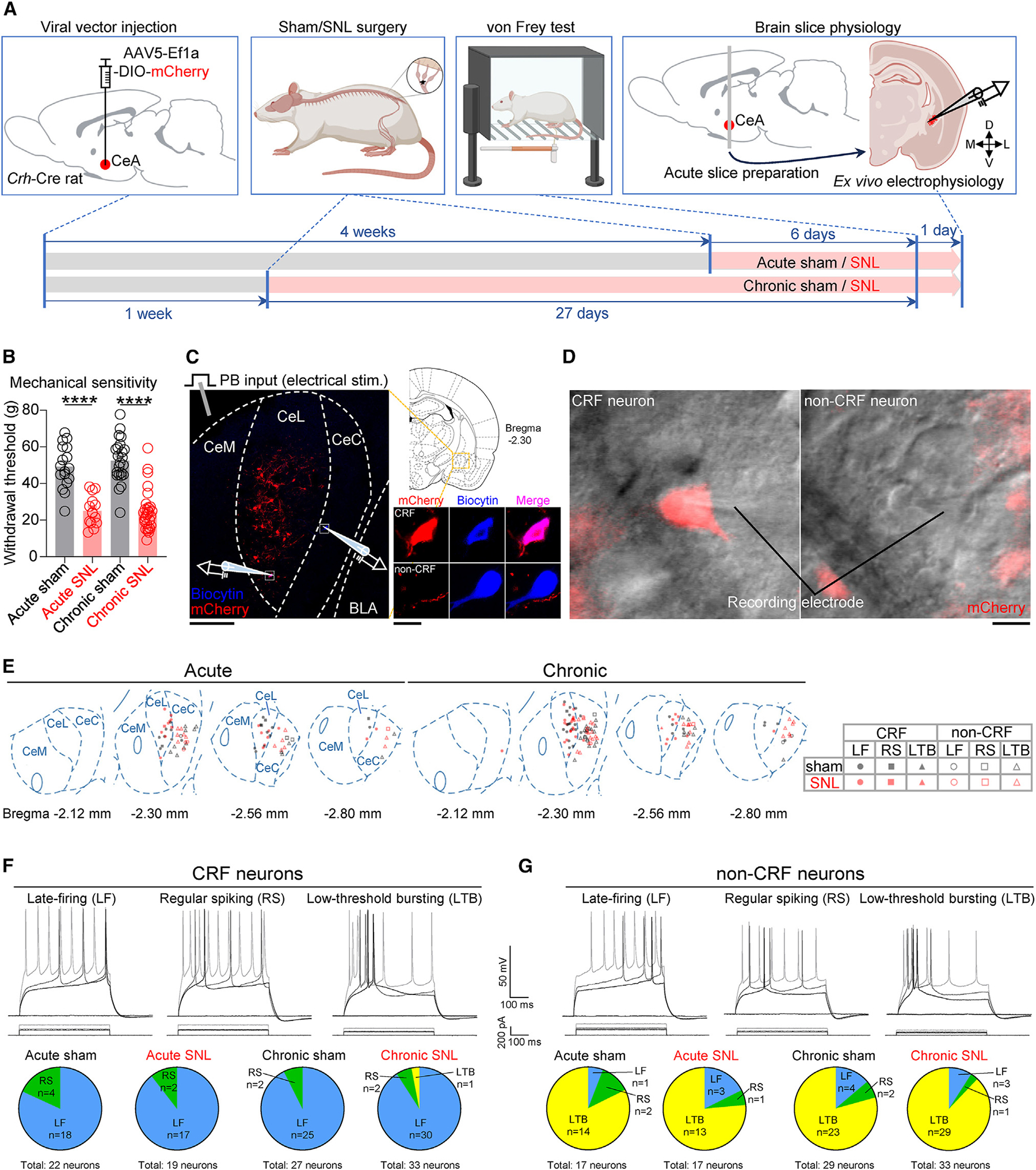
(A) Experimental design.
(B) Paw withdrawal thresholds. Acute sham: n = 17, acute SNL: n = 14, chronic sham: n = 23, chronic SNL: n = 30. ****p < 0.0001, one-way ANOVA with Bonferroni posttests compared with sham controls.
(C) Experimental setup. The area delineated by the white dotted square at left is shown at higher magnification at right. Scale bars, 200 μm (left) and 10 μm (right). BLA, basolateral amygdala; CeC, capsular divisions of the CeA; CeL, lateral; CeM, medial.
(D) Representative differential interference contrast (DIC) and fluorescent images of a patched mCherry-expressing CRF neuron and a patched mCherry− non-CRF neuron in the CeA. Scale bar, 10 μm.
(E) Distribution of recorded cells and their firing types.
(F) Representative recordings and proportions of CRF cell types based on action potential firing. Acute sham: 22 neurons from 14 rats, acute SNL: 19 neurons from 13 rats, chronic sham: 27 neurons from 18 rats, chronic SNL: 33 neurons from 20 rats.
(G) Same as in (F), but for non-CRF cell types. Acute sham: 17 neurons from 10 rats, acute SNL: 17 neurons from 9 rats, chronic sham: 29 neurons from 14 rats, chronic SNL: 33 neurons from 18 rats.
Data are represented as mean ± SEM.
To examine pain-related changes in CeA neurons at the acute and chronic phases of SNL, whole-cell patch-clamp recordings were made from fluorescently labeled CRF neurons in the lateral and capsular divisions of the CeA (CeL/C) or from unlabeled non-CRF neurons in the capsular division of the CeA (CeC) in brain slices collected from Crh-Cre rats injected into the amygdala with an adeno-associated virus (AAV) vector encoding the Cre-dependent fluorophore mCherry (Figures 1C and 1D). Consistent with previous reports using the same Crh-Cre rat line,38,39 the majority of CRF neurons was located in the lateral division of the CeA (CeL) and showed a late-firing (LF) pattern (Figures 1E and 1F). However, the majority of CeC non-CRF neurons showed a low-threshold bursting firing pattern (Figures 1E and 1G), which is one of the main firing types found in unidentified CeL/C neurons in rat brain slices.20,32,40,41 There was no significant difference in the ratio of cell types between the experimental groups (p > 0.05, Fisher’s exact test; Figures 1F and 1G), demonstrating that neither acute nor chronic neuropathic pain conditions affect the relative distribution of firing phenotypes of CRF or non-CRF neurons. Because of the very low incidence of regular spiking and low-threshold bursting types in CRF neurons, and regular spiking and LF types in non-CRF neurons, the remainder of this study focuses on LF CRF neurons and low-threshold bursting non-CRF neurons.
To investigate pain-related changes in synaptic transmission, we measured excitatory postsynaptic currents (EPSCs) in response to the electrical stimulation of PB inputs in the visually identified tract dorsomedial to the CeA and ventral to caudateputamen,16,42,43 where no other afferents to the CeA have been described.42,44 To validate the electrical stimulation of PB input, we used optogenetic activation of the terminals of PB afferents in the CeA and compared the properties of electrically and optogenetically evoked EPSCs in the same cell (Figure S1). We found substantial similarity in kinetics of the EPSCs evoked with both stimulation techniques (Figures S1D, S1E, S1H, and S1I), indicating that PB inputs were activated by electrical stimulation of these fibers. We further confirmed that the responses were purely glutamatergic by the application of glutamate receptor antagonists AP5 and CNQX (Figures S1F and S1J). Subsequent experiments to examine pain-related synaptic changes were conducted with electrical stimulation because of potentially confounding factors associated with optogenetics, such as variable opsin expression among animals.45
PB-driven EPSCs were significantly increased in both CRF (F1, 224 = 19.53, p < 0.0001, two-way ANOVA) and non-CRF (F1, 154 = 10.80, p < 0.01, two-way ANOVA) neurons in brain slices from acute, but not chronic, SNL rats (CRF neurons: F1, 308 = 1.013, p > 0.05; non-CRF neurons: F1, 294 = 3.568, p > 0.05; two-way ANOVA; Figures 2A–2E). There was no significant change in paired-pulse ratio (PPR) (p > 0.05, unpaired t test; Figures 2F–2I), arguing against a major role of changes in presynaptic release properties in pain-related synaptic plasticity. The loss of synaptic plasticity at the chronic stage of neuropathic pain represents an unexpected finding.
Figure 2. Loss of PB-driven synaptic plasticity and loss of beneficial behavioral effects of chemogenetic inhibition of CeA-projecting PB neurons in chronic neuropathic pain.
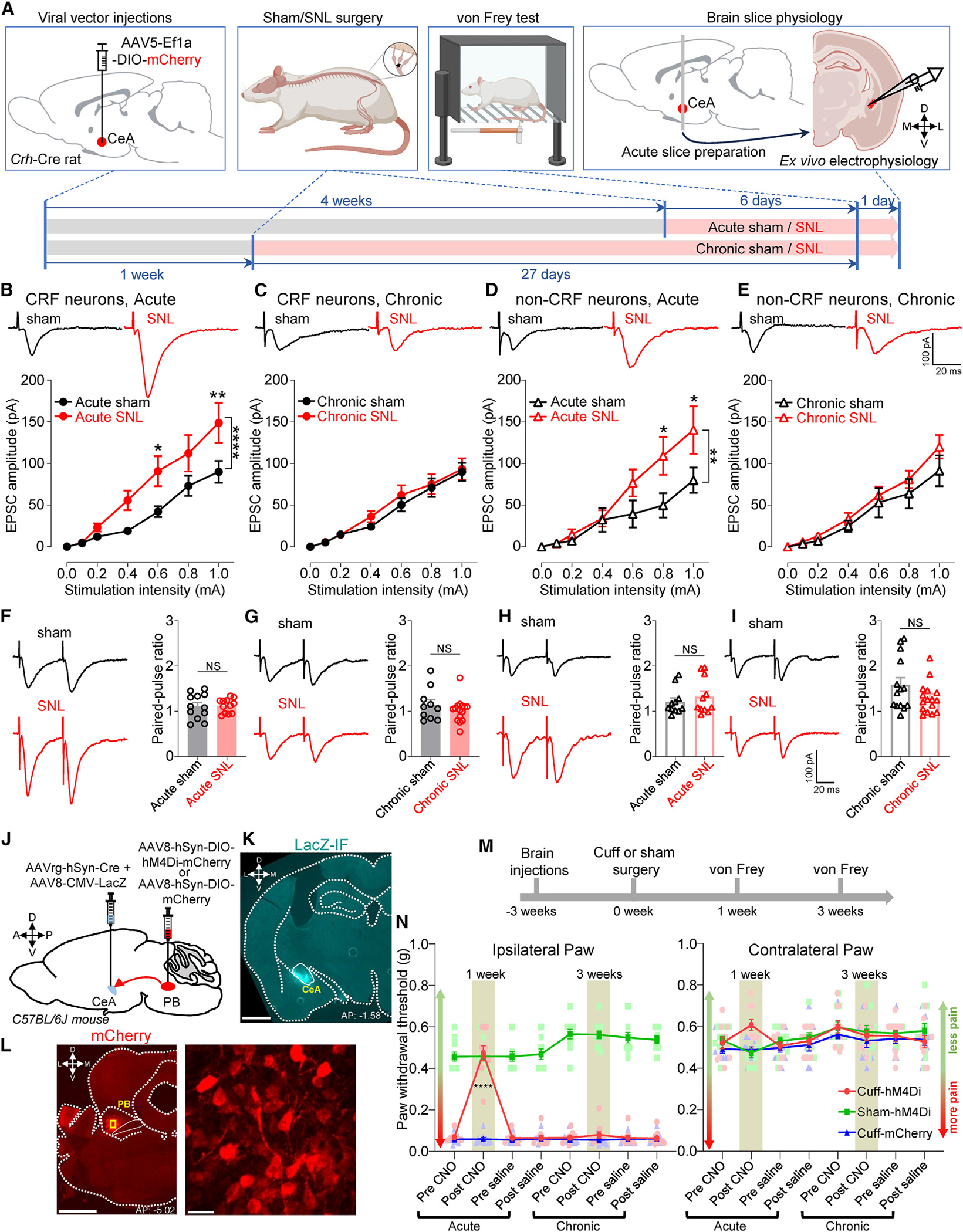
(A) Experimental design.
(B–E) Representative traces and input-output functions of PB-driven EPSCs in CRF neurons from acute sham (n = 17 neurons from 11 rats), acute SNL (n = 17 neurons from 11 rats), chronic sham (n = 19 neurons from 15 rats), and chronic SNL (n = 27 neurons from 17 rats) rats (B and C), and in non-CRF neurons from acute sham (n = 13 neurons from 10 rats), acute SNL (n = 11 neurons from 7 rats), chronic sham (n = 20 neurons from 13 rats), and chronic SNL (n = 24 neurons from 16 rats) rats (D and E). *p < 0.05; **p < 0.01; ****p < 0.0001; two-way ANOVA with post hoc Bonferroni tests compared with sham controls.
(F) Representative traces of PB-driven EPSCs evoked by paired stimulation at 50-ms intervals and summary of PPR in CRF neurons from acute sham (n = 12 neurons from 9 rats) and SNL (n = 12 neurons from 8 rats) rats.
(G) Same as in (F), but for chronic sham (n = 10 neurons from 8 rats) and SNL (n = 15 neurons from 12 rats) rats.
(H and I) Same as in (F) and (G), but for non-CRF neurons in brain slices from acute sham (n = 11 neurons from 10 rats) and acute SNL (n = 11 neurons from 7 rats) rats (H), and from chronic sham (n = 14 neurons from 10 rats) and chronic SNL (n = 16 neurons from 12 rats) rats (I). NS, not significant, unpaired t test.
(J) Experimental approach.
(K) Representative image of the AAV8-CMV-LacZ-bGH injection into the right CeA shown in cyan. Scale bar, 1,000 μm.
(L) Representative images of the AAV8-hSyn-DIO-hM4Di-mCherry injection into the right PB shown in red. The area delineated by the yellow rectangle at left is shown at higher magnification at right. Scale bars, 1,000 μm (left) and 20 μm (right).
(M) Experimental design.
(N) Paw withdrawal thresholds in the ipsilateral (left) and contralateral (right) hindpaws before and 1 h after systemic (i.p.) CNO (10 mg/kg) or saline at 1 and 3 weeks after cuff or sham surgery (n = 10 for cuff-hM4Di and cuff-mCherry; n = 11 for sham-hM4Di). Scatter points represent individual mice. ****p < 0.0001; repeated measures two-way ANOVA followed by Dunnett’s multiple comparison test compared with pre-CNO or cuff-mCherry controls.
Data are represented as mean ± SEM.
PB input is the major if not exclusive source of calcitonin gene-related peptide (CGRP) in the CeA, where CGRP axon terminals from PB contact CRF and PKCδ but not SOM neurons13; and this system plays a crucial role in pain-related behaviors and synaptic plasticity.13,36,46–51 To better understand the loss of synaptic plasticity in the PB input, we tested the effect of a CGRP antagonist (BIBN4096BS) on PB-driven EPSCs in non-CRF neurons in the CeC at the chronic SNL phase when the pain-related enhancement of EPSCs was no longer detectable. In contrast to the significant inhibitory effect we had reported in an acute pain model,46 BIBN4096BS did not significantly decrease EPSCs in non-CRF CeC neurons at the chronic SNL phase (F1, 28 = 0.4856, p > 0.05, two-way repeated measures ANOVA; Figure S2).
Time-dependent contribution of PB-CeA inputs to neuropathic pain behaviors
To investigate behavioral consequences of the time-dependent change of PB input (Figures 2B–2E), we measured tactile hypersensitivity in a well-established sciatic nerve cuff mouse pain model52 and selectively inhibited the PB→CeA pathway at different phases using an intersectional chemogenetic approach (Figure 2J). We used the designer drug clozapine N-oxide (CNO) to selectively inhibit neurons expressing the inhibitory hM4Di designer receptor exclusively activated by designer drug (DREADD). This intersectional chemogenetic strategy allowed us to specifically inhibit CeA-projecting PB neurons. A virus expressing LacZ (AAV8-CMV-LacZ-bGH) was co-injected with the Cre-expressing retrograde tracer in the CeA to verify injection sites. LacZ immunostaining showed that all retrograde viral vector injections were restricted to the CeA (Figures 2K and S3A). Evaluation of mCherry+ cells further demonstrated robust transduction of inhibitory DREADDs (hM4Di) restricted to PB neurons (Figures 2L and S3B).
We measured stimulus-evoked tactile sensitivity 1 and 3 weeks after sciatic nerve injury (Figure 2M). Mice displayed hypersensitivity at both time points as demonstrated by lower mechanical thresholds in the ipsilateral hindpaw compared to the contralateral hindpaw and compared to sham-operated mice. As illustrated in Figure 2N, CNO (intraperitoneal [i.p.], 10 mg/kg body weight)-induced chemogenetic inhibition of CeA-projecting PB neurons at 1 week of nerve injury reversed tactile hypersensitivity in the ipsilateral paw compared to before CNO injection (F7, 392 = 59.73, p < 0.0001, two-way ANOVA) and mCherry control mice (F5, 56 = 450.4, p < 0.001, two-way ANOVA). No effect was seen after CNO treatment in mice injected with the control mCherry vector into PB, indicating that CNO by itself had no effect. Notably, tactile hypersensitivity after 3 weeks of nerve injury in the ipsilateral hindpaw was unaffected by CNO-mediated chemogenetic inhibition of CeA-projecting PB neurons (Figure 2N); thus, withdrawal thresholds in the ipsilateral paw before and after CNO or saline treatment were indistinguishable. Withdrawal thresholds in both paws of sham-operated mice and in the contralateral paw of cuff-implanted mice were comparable before and after chemogenetic inhibition of CeA-projecting PB neurons, indicating that baseline nociception was not affected by this pathway. Together, the loss of synaptic plasticity in the PB→CeA pathway and the lack of chemogenetic behavioral effects strongly suggest that CeA-projecting PB neurons contribute to the induction but not the maintenance of neuropathic pain.
Time-dependent changes in excitability of CeA CRF and non-CRF neurons in the transition from acute to chronic neuropathic pain
Since we discovered loss of synaptic plasticity in the PB→CeA pathway, we explored any changes in excitability of CeA neurons at the acute and chronic phases of SNL (Figure 3A). The excitability of CRF neurons in the CeL/C was significantly increased at the acute (F1, 576 = 5.174, p < 0.05, two-way ANOVA), but not the chronic (F1, 720 = 2.757, p > 0.05, two-way ANOVA), phase of SNL (Figures 3B and 3C). In contrast, the excitability of non-CRF neurons in the CeC was increased at the chronic (F1, 414 = 58.08, p < 0.0001, two-way ANOVA), but not the acute (F1, 396 = 1.421, p > 0.05, two-way ANOVA), phase of SNL (Figures 3D and 3E). There was no significant difference in resting membrane potential (RMP), rheobase, and input resistance (Rin) between neurons from sham and SNL rats (p > 0.05, unpaired t test; Figures S4A–S4D). The data suggest that hyperexcitability of CRF neurons at the acute pain stage may be linked to synaptic plasticity of PB input, but hyperexcitability of non-CRF neurons at the chronic stage is independent of increased excitatory drive from PB as synaptic plasticity is no longer observed in the PB→CeA pathway.
Figure 3. Neuronal excitability changes in CeA neurons in the transition from acute and chronic neuropathic pain.
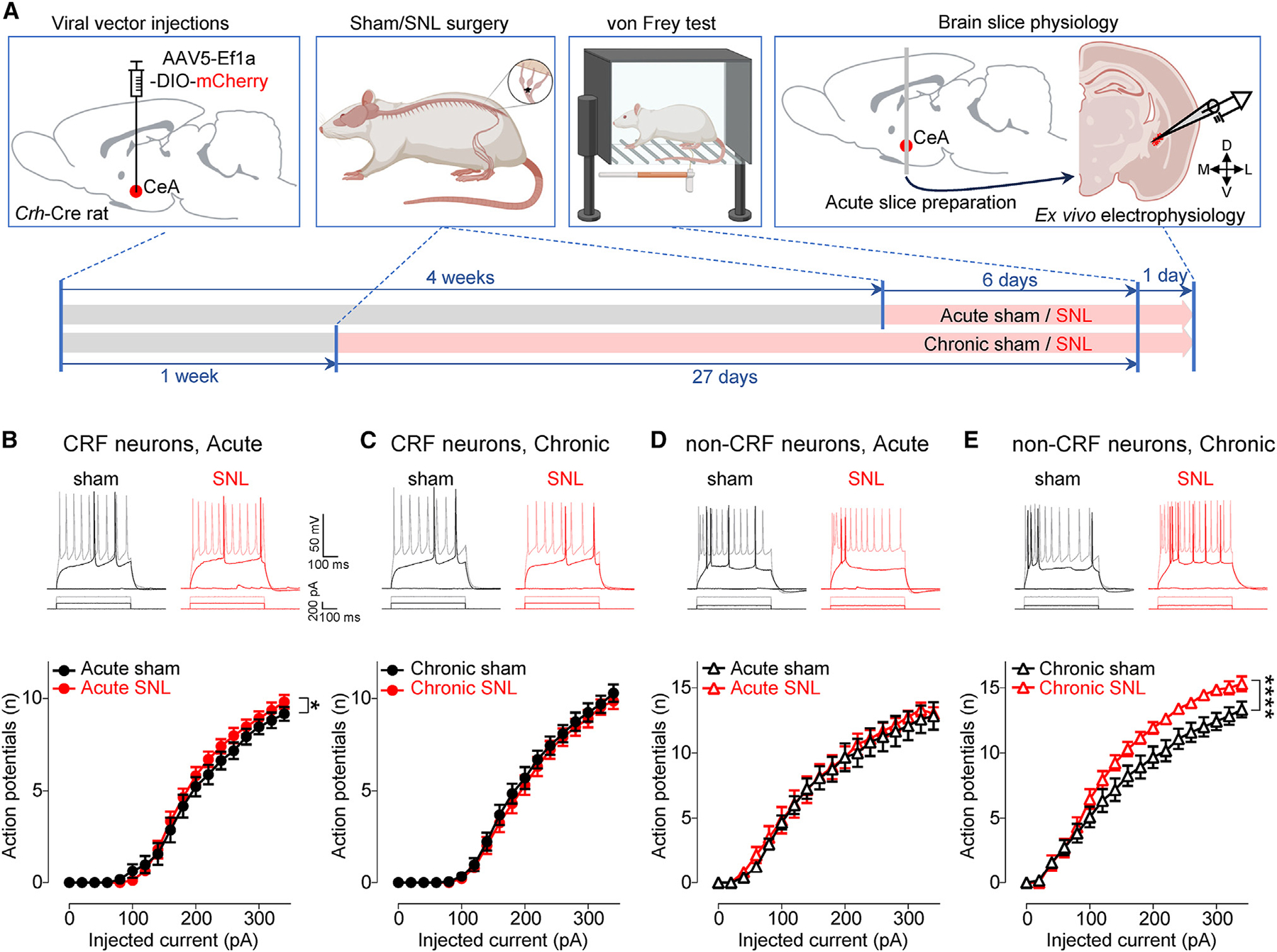
(A) Experimental design.
(B) Representative voltage responses to depolarizing current pulses and F-I relationship of CRF neurons from acute sham (n = 17 neurons from 11 rats) and acute SNL (n = 17 neurons from 11 rats) rats.
(C) Same as in (B), but for chronic sham (n = 20 neurons from 13 rats) and chronic SNL (n = 22 neurons from 10 rats) rats.
(D and E) Same as in (B) and (C), but for non-CRF neurons in brain slices from acute sham (n = 12 neurons from 9 rats) and acute SNL (n = 12 neurons from 8 rats) rats (D), and from chronic sham (n = 12 neurons from 8 rats) and chronic SNL (n = 13 neurons from 8 rats) rats (E).
*p < 0.05; ****p < 0.0001; two-way ANOVA with post hoc Bonferroni tests. Data are represented as mean ± SEM.
Cell-type-specific changes in excitability of CeA neurons at the chronic phase of neuropathic pain
We next sought to identify the cell type(s) of CeC non-CRF neurons that showed increased excitability at the chronic phase of neuropathic pain (Figure 3). We focused on CeL/C neurons expressing PKCδ, the CGRP receptor (CGRPR), or SOM given their previously shown contributions to nociceptive processing.22–24,51,53 To identify PKCδ, CGRPR, and SOM neurons in brain slices, we used transgenic Prkcd-Cre, Calcrl-Cre, and Sst-Cre mice crossed with homozygous Ai9 reporter mice (Figure 4A). The sciatic nerve cuff model of neuropathic pain52 was used for these experiments. Similar to our results with the SNL model of neuropathic pain (Figure 1B), robust hypersensitivity to tactile stimuli was found at both 1 week and 3 weeks postinduction (Figure 2N). Contrary to our expectations based on previous studies that showed minimal colocalization of PKCδ and CGRPR neurons with CRF neurons and their preferential localization in the CeC,23,38,47,54,55 neither CeA-PKCδ (F1, 30 = 0.1453, p > 0.05, two-way ANOVA) nor CeA-CGRPR (F1, 84 = 0.9114, p > 0.05, two-way ANOVA) neurons showed a significant increase in excitability at the chronic phase of neuropathic pain compared to sham controls (Figures 4B–4G). Instead, we found increased excitability of regular spiking (F1, 41 = 3.112, p < 0.05, two-way ANOVA), but not LF (F1, 62 = 1.645, p > 0.05, two-way ANOVA), SOM neurons at the chronic phase (Figures 4H–4K).
Figure 4. Cell-type-specific excitability changes at the chronic phase of neuropathic pain and cell-type- and time-specific effects of chemogenetic inhibition on neuropathic pain behaviors.
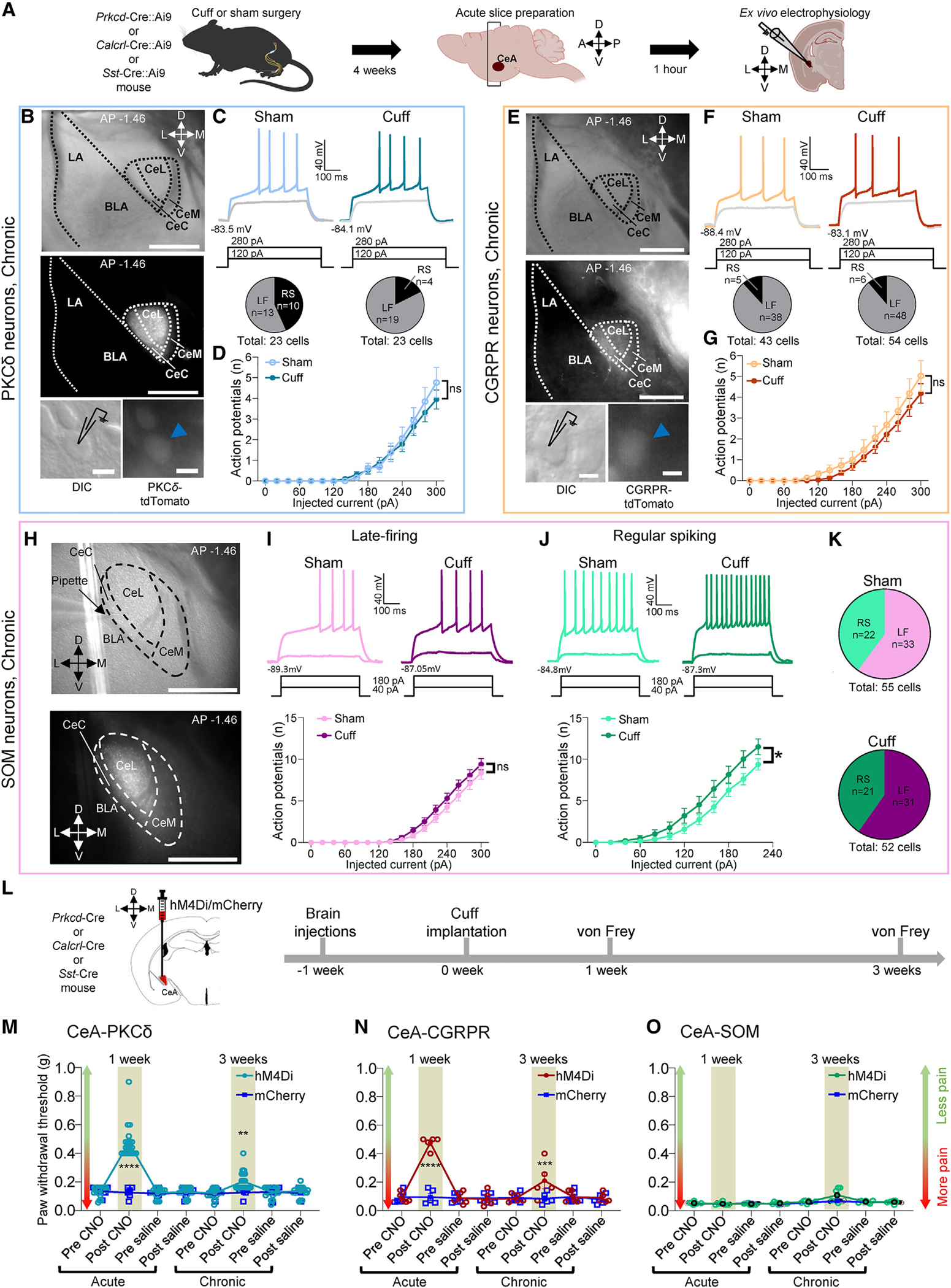
(A) Experimental design.
(B) Representative DIC and fluorescent images of the CeA (top and center). High-magnification DIC and fluorescent images of tdTomato-expressing PKCδ neurons (bottom). LA, lateral amygdala. Scale bars, 500 μm (top and center) and 10 μm (bottom).
(C) Representative voltage traces of LF PKCδ neurons in sham and neuropathic conditions (top). Proportions of LF and regular-spiking (RS) PKCδ neurons (bottom).
(D) F-I relationship in LF neurons (sham: n = 13 neurons from 6 mice, cuff: n = 19 neurons from 5 mice); ns, not significant, two-way ANOVA.
(E) Same as in (B), but for tdTomato-expressing CGRPR neurons.
(F and G) Same as in (C) and (D), but for CGRPR neurons (sham: n = 38 neurons from 9 mice, cuff: n = 48 neurons from 12 mice); ns, not significant, two-way ANOVA.
(H) Representative DIC (top) and fluorescent images of tdTomato-expressing SOM neurons (bottom) in the CeA. Scale bars in both images, 500 μm.
(I) Representative voltage traces of LF SOM neurons recorded in sham and neuropathic conditions (top). F-I relationship in LF SOM neurons (sham: n = 33 neurons from 10 mice, cuff: n = 31 neurons from 11 mice); ns, two-way ANOVA (bottom).
(J) Same as in (I), but for RS SOM neurons (sham: n = 22 neurons from 11 mice, cuff: n = 21 neurons from 10 mice); *p < 0.05, two-way ANOVA.
(K) Proportions of RS and LF neurons in sham (top) or neuropathic (bottom) conditions.
(L) Experimental time line.
(M–O) Tactile hypersensitivity shown as the ipsilateral hindpaw withdrawal thresholds before and 1 h after systemic (i.p.) CNO (10 mg/kg) or saline at 1 and 3 weeks after cuff implantation. Scatter points represent individual mice. Repeated measures two-way ANOVA followed by Šídák’s multiple comparison test. CeA-PKCδ neurons (n = 20 for hM4Di and n = 6 for mCherry; 1 week, ****p < 0.0001; 3 weeks, **p < 0.01) (M), CeA-CGRPR neurons (n = 5 for hM4Di and mCherry; 1 week, ****p < 0.0001; 3 weeks, ***p < 0.001) (N), CeA-SOM neurons (n = 6 for hM4Di and n = 5 for mCherry) (O).
All values are presented as the mean ± SEM.
Time-dependent contribution of genetically distinct CeA neurons to neuropathic pain behaviors
The identification of time- and cell-type-specific changes in CeA neurons in neuropathic pain made us seek to determine the behavioral contributions of different CeA cell types. To do so, we chemogenetically inhibited PKCδ, CGRPR, or SOM neurons in the CeA at the acute and chronic phases of the cuff-induced neuropathic pain model (Figures 4L–4O). Inhibition of CeA-PKCδ or CeA-CGRPR neurons significantly mitigated tactile hypersensitivity at both the acute and chronic phases, but the effects were much more pronounced at the acute phase (CeA-PKCδ neurons: F1, 24 = 40.01, p < 0.0001; CeA-CGRPR neurons: F1, 8 = 8.801, p < 0.05; two-way ANOVA; Figures 4M and 4N), which may reflect synaptic plasticity in the PB→CeA pathway in acute but not chronic neuropathic pain and a lack of increased excitability at the chronic stage. Interestingly, chemogenetic inhibition of CeA-SOM neurons had no significant effect on tactile hypersensitivity at the acute and chronic phases (F1, 9 = 3.743, p > 0.05, two-way ANOVA; Figure 4O), which may be because only a subpopulation of SOM neurons showed hyperexcitability. These results suggest time- and cell-type-specific involvement of CeA neurons in pain-related behaviors.
Projection-specific pain-related changes in CeA-CRF neurons
Our experiments revealed that the mechanisms driving CeA modulation of the initiation and chronification of pain are distinct. To determine whether the changes in CRF neurons at the acute neuropathic pain stage (Figures 2 and 3) were projection specific, we focused on the efferent projections of CeA-CRF neurons to the PB and PAG, two key pain modulatory brainstem regions previously shown to receive inputs from CeA-CRF neurons.13,38,56 To identify PB- and PAG-projecting CeA-CRF neurons, we co-injected retrograde AAVs expressing the Cre-dependent fluorophores EGFP and tdTomato (AAVrg-hSyn-DIO-EGFP and AAVrg-FLEX-tdTomato) into the PB and PAG, respectively, of transgenic Crh-Cre rats (Figure 5A). After confirming the correct targeting (Figure 5B) and specificity of the viral vector strategy (Figure 5C), we examined the anatomical distribution of PB- and PAG-projecting CRF neurons within the CeA (Figure 5D). Quantification of retrogradely labeled neurons along the rostro-caudal levels of each CeA subdivision showed that the vast majority of retrogradely labeled neurons are within the CeL, with less than 48 neurons retrogradely labeled in the CeC or CeM. Additional analyses showed a significantly larger number of PB-projecting CRF neurons compared to PAG-projecting ones (p < 0.01, paired t test; Figure 5E). Evaluation of the percentage of retrogradely labeled cells further showed that the rostro-caudal distribution of CRF neurons projecting to PB or PAG was similar within the CeA (Figure 5F). Importantly, we found that PB- and PAG-projecting CRF neurons were largely non-overlapping populations (Figure 5G).
Figure 5. PAG- and PB-projecting CRF neurons.
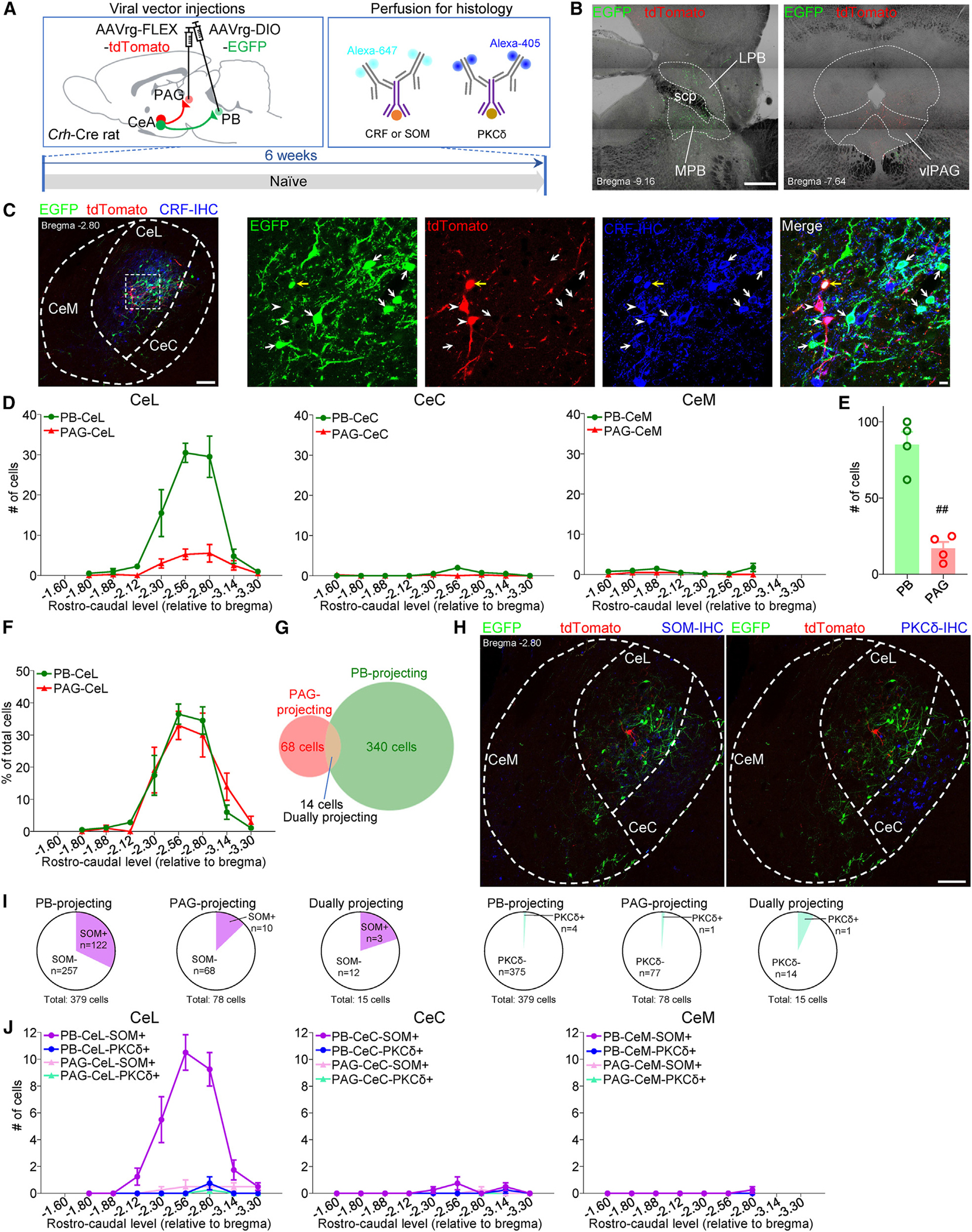
(A) Experimental time line.
(B) Representative images of injection sites in the PB and PAG. LPB, lateral parabrachial nucleus; MPB, medial parabrachial nucleus; scp, superior cerebellar peduncle; vlPAG, ventrolateral PAG. Scale bar, 500 μm.
(C) Immunohistochemical verification of the specificity of retrogradely labeled PAG- and PB-projecting CRF neurons in the CeA. The area delineated by the white dotted square at left is shown at higher magnification in the right panels. Scale bars, 100 μm (left) and 10 μm (right). White arrows indicate EGFP-labeled PB-projecting CRF neurons. White arrowheads indicate tdTomato-labeled PAG-projecting CRF neurons. Yellow arrow indicates dually projecting CRF neurons.
(D) Rostro-caudal distribution of PB- and PAG-projecting CRF neurons in CeL, CeC, and CeM (n = 4 rats).
(E) Total number of PB- and PAG-projecting CRF neurons in the CeL (n = 4 rats). ##p < 0.01, paired t test.
(F) Rostro-caudal distributions of PB- and PAG-projecting CRF neurons in the CeL expressed as percentage of total labeled cells (n = 4 rats).
(G) Venn diagram showing the number of overlapping and non-overlapping PB- and PAG-projecting CRF neurons in the CeL (n = 4 rats).
(H) Representative images of SOM (left) and PKCδ (right) IHC. Scale bar, 100 μm.
(I) Summary of colocalization analysis for SOM (left) and PKCδ (right) with CRF neurons (n = 4 rats).
(J) Rostro-caudal distributions of colocalization of PB- and PAG-projecting CRF neurons with SOM and PKCδ IHC in the CeL, CeC, and CeM (n = 4 rats).
Data are represented as mean ± SEM.
CeA-CRFneuronsexpressotherneurochemicalmarkers,38,54,55 but this is not known for CRF projection neurons. We conducted immunohistochemistry (IHC) to determine whether CeA-CRF neurons projecting to PB and PAG co-express neurochemical markers for SOM and PKCδ (Figure 5H). We found that 32.2% of PB-projecting, 12.8% of PAG-projecting, and 20.0% of dually projecting CeA-CRF neurons co-expressed SOM (Figure 5I). In contrast, less than 2% of PB-projecting or PAG-projecting and less than 7% of dually projecting CeA-CRF neurons co-expressed PKCδ (Figure 5I). Retrogradely labeled CeA neurons co-expressing SOM were also mostly localized to the CeL and were distributed throughout the rostro-caudal CeA (Figure 5J).
Next, we compared the electrophysiological properties between PB- and PAG-projecting CRF neurons (Figure 6). We found significantly higher excitability in PB-projecting CeA-CRF neurons compared to PAG-projecting ones in brain slices from naive rats (F1, 468 = 9.628, p < 0.01, two-way ANOVA; Figure 6F). There was no significant difference in other cellular properties (RMP, rheobase, Rin) (p > 0.05, unpaired t test, Figure S4E) and synaptic transmission (PB-driven EPSCs, F1, 154 = 0.4369, p > 0.05, two-way ANOVA; Figure 6G) and PPR (p > 0.05, unpaired t test; Figure 6H).
Figure 6. Baseline electrophysiological comparison of PAG- and PB-projecting CRF neurons.
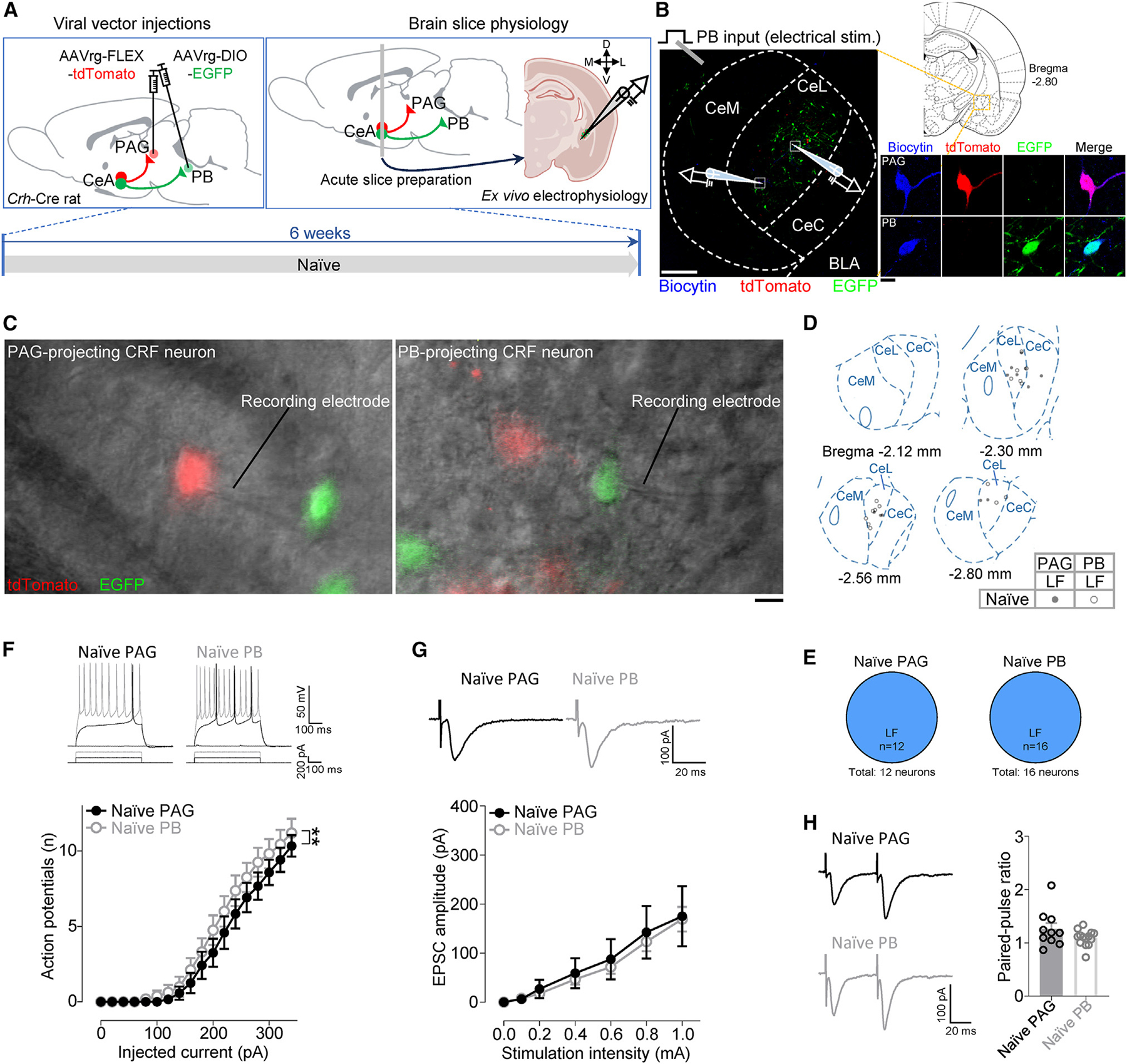
(A) Experimental time line.
(B) Experimental setup. The area delineated by the white dotted square at left is shown at higher magnification at right. Scale bars, 200 μm (left) and 10 μm (right).
(C) Representative DIC and fluorescent images of a patched tdTomato-expressing PAG-projecting CRF neuron (left) and a patched EGFP-expressing PB-projecting CRF neuron (right) in the CeA. Scale bar, 10 μm.
(D) Anatomical location of recorded cells and their firing types.
(E) Proportions of firing types of PAG- (12 neurons from 9 rats) and PB-projecting (16 neurons from 12 rats) CRF neurons in brain slices from naive animals.
(F) Representative voltage responses to depolarizing current pulses and excitability (F-I relationship) of PAG- (n = 12 neurons from 9 rats) and PB-projecting (n = 16 neurons from 12 rats) CRF neurons.
(G) Representative traces and input-output functions of PB-driven EPSCs in PAG- (n = 10 neurons from 8 rats) and PB-projecting (n = 14 neurons from 12 rats) CRF neurons.
(H) Representative traces and PPR (50-ms intervals) for PB-driven EPSCs in PAG- (n = 10 neurons from 8 rats) and PB-projecting (n = 14 neurons from 12 rats) CRF neurons.
**p < 0.01, two-way ANOVA. Data are represented as mean ± SEM.
Finally, we examined pain-related changes in these projection-defined CeA-CRF neurons in the acute phase of neuropathic pain (Figure 7), because at this time point, CeA-CRF neurons showed synaptic plasticity and hyperexcitability (Figures 2 and 3). We found increased excitability in PAG-projecting (F1, 450 = 17.47, p < 0.0001, two-way ANOVA; Figures 7F and 7G), but not PB-projecting (F1, 432 = 1.966, p > 0.05, two-way ANOVA; Figures 7J and 7K), CeA-CRF neurons in the SNL model compared to sham controls. We also found enhanced PB-driven EPSCs in both projection neurons, but the increase was greater in PAG-projecting CeA-CRF neurons (F1, 133 = 21.89, p < 0.0001, two-way ANOVA; Figure 7L) compared to that in PB-projecting ones (F1, 154 = 8.93, p < 0.01, two-way ANOVA; Figure 7N). There was no significant difference in PPR (p > 0.05, unpaired t tests; Figures 7M and 7O), arguing against presynaptic mechanisms. These results show that changes in synaptic transmission and excitability of CeA-CRF neurons at the acute stage of neuropathic pain are projection specific and more pronounced in CeA-CRF neurons projecting to the PAG.
Figure 7. Pain-related changes in PAG- and PB-projecting CRF neurons at the acute neuropathic pain stage.
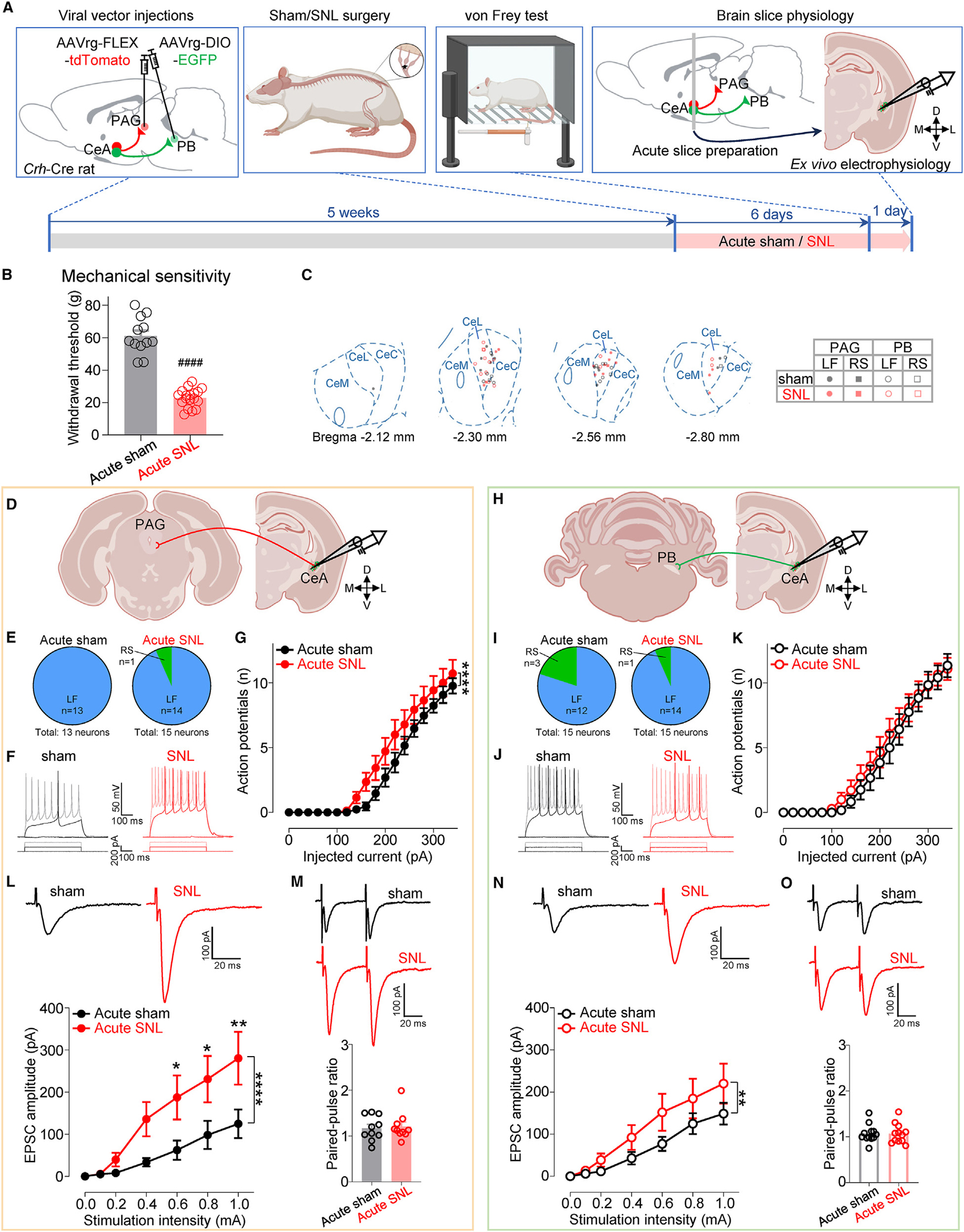
(A) Experimental design.
(B) Paw withdrawal thresholds (sham: n = 12, SNL: n = 18). ####p < 0.0001, unpaired t test.
(C) Location of recorded cells and their firing types.
(D) Recordings from PAG-projecting CeA-CRF neurons.
(E) Proportions of firing types of PAG-projecting CRF neurons in brain slices from acute sham (13 neurons from 10 rats) and acute SNL (15 neurons from 11 rats) rats.
(F) Representative voltage responses of PAG-projecting CRF neurons from acute sham/SNL rats to depolarizing current pulses.
(G) Excitability (F-I relationship) of PAG-projecting LF-CRF neurons from acute sham (n = 13 neurons from 10 rats) and acute SNL (n = 14 neurons from 11 rats) rats.
(H–K) Same as in (D)–(G), but for PB-projecting CeA-CRF neurons from acute sham (15 neurons from 10 rats) and acute SNL (15 neurons from 13 rats) rats (I), and from acute sham (n = 12 neurons from 9 rats) and acute SNL (n = 14 neurons from 12 rats) rats (K).
(L) Representative traces and input-output functions of PB-driven EPSCs in PAG-projecting CRF neurons from acute sham (n = 11 neurons from 8 rats) and acute SNL (n = 10 neurons from 10 rats) rats.
(M) Representative traces of PB-driven EPSCs evoked by paired stimulation at 50-ms intervals and summary of PPR in PAG-projecting CRF neurons from acute sham (n = 10 neurons from 8 rats) and acute SNL (n = 10 neurons from 10 rats) rats.
(N and O) Same as in (L) and (M) but for PB-projecting CRF neurons from acute sham (n = 12 neurons from 9 rats) and acute SNL (n = 12 neurons from 11 rats) rats (N), and from acute sham (n = 12 neurons from 9 rats) and acute SNL (n = 12 neurons from 11 rats) rats (O).
*p < 0.05, **p < 0.01, ****p < 0.0001, two-way ANOVA with post hoc Bonferroni tests compared with sham controls. Data are represented as mean ± SEM.
DISCUSSION
This study addressed an important knowledge gap in the field of pain research and neuroplasticity by identifying cell-type-specific synaptic and cellular mechanisms of the chronification of pain. This is important because the pathophysiology of chronic diseases is generally not well understood. This is also true for chronic pain, where emerging evidence points to a reorganization of brain activity and networks, including in limbic regions,57,58 but brain mechanisms of pain chronification are an understudied area of research. Here, we show fundamental changes in neuroplasticity driving the transition from acute to chronic pain in the amygdala, a limbic brain region concerned with pain modulation.59 Specifically, the present study demonstrates that the PB→CeA-CRF pathway and CRF neuronal hyperexcitability are critical for the initiation but not the maintenance of chronic neuropathic pain and that this pain-related plasticity is projection specific.
Critical contribution of the PB→CeA pathway to acute but not chronic neuropathic-pain-related behaviors
The CeA receives nociceptive input through the PB pathway.14 Although previous studies showed enhanced synaptic transmission at the PB-CeA synapse in different pain models,16–19,35,60–62 they focused on acute/subacute pain (hours to 2 weeks), and synaptic plasticity at the chronic phase was not shown. Here, we compared PB-driven synaptic transmissions in the CeA between chronic neuropathic pain and sham control, and found no significant enhancement of PB-CeA synaptic transmission. This represents a dramatic change in the role of PB→CeA pathway and the mechanism of neuroplasticity in chronic pain, which may involve structural synaptic reorganization, as we observed previously.63 The change in synaptic plasticity translated into behavioral consequences, because the inhibition of CeA-projecting PB neurons mitigated hypersensitivity at the acute phase of the neuropathic pain model, which is consistent with our recent study.15 However, inhibition of CeA-projecting PB neurons was no longer effective against the hypersensitivity at the chronic phase, further supporting the idea of a time-dependent contribution of the PB→CeA pathway to pain-related neuroplasticity and behaviors. Therefore, inhibition of the PB-CeA pathway at the acute phase may have translational implications to prevent the transition to chronic pain. Our findings align well with a recent study that found activation of PB Calca neurons to be necessary and sufficient to induce chronic pain, but their inhibition was ineffective against already-established allodynia.64 Their results suggest that neuroplasticity downstream of the PB had become independent from PB plasticity under chronic pain conditions, and here we identified the CeA as a key brain area for the maintenance of so-called nociplastic pain.65,66
Although we did not extensively study pre- versus postsynaptic mechanisms here, it is important to note that a previous study showed increased PPR in CeA-CRF neurons at the acute phase of a neuropathic pain model using optogenetic stimulation of PB inputs,22 which would reflect decreased transmitter release probability. We found increased transmission but no change in PPR using electrical stimulation (Figure 2F), which could be related to previously reported differences between optogenetic and electrical stimulations when they were used to evoke synaptic transmission at high frequency.67 While we report comparable data for electrically and optogenetically evoked synaptic responses (Figure S1), differences could exist when it comes to studying synaptic plasticity. It is also possible that differences in time points after the surgery (10 days vs. 7 days) or the pain models (spared nerve injury [SNI] vs. SNL) could account for the discrepancy.
Time- and cell-type-specific excitability changes in CeA neurons in neuropathic pain
Increased excitability of CeA neurons has been reported in different pain models and is thought to be critical for pain-related behaviors.16,17,19,25,63,68 Recent studies focused on molecularly defined CeA neurons revealed cell-type-specific differential excitability and synaptic changes in pain models.22,23,69 Specifically, the excitability of PKCδ and SOM neurons was increased and decreased, respectively, in the sciatic cuff neuropathic pain model.23 In contrast, increased excitatory synaptic transmission and excitability were found in SOM neurons and the opposite changes were found in PKCδ neurons in a muscle pain model.69 Another study with more focus on the subdivisions of the CeA suggests decreased excitatory input to CeC-SOM and CeL-CRF neurons in the SNI model.22 While these findings highlight the importance of identifying cell types to understand complex processing in the CeA, the studies were conducted in acute pain models or at the acute/subacute phase, and information is lacking about the situation at the chronic stage of pain conditions.
This knowledge gap was addressed here. Using transgenic Crh-Cre rats, we found time-dependent differential synaptic and excitability changes in CRF and non-CRF neurons in neuropathic pain (SNL model). Our results suggest an important role for CRF neurons in the initiation of neuropathic pain, whereas non-CRF cell types contribute to the chronic phase. This may explain the failure of CRF-based interventions in clinical studies70 and may have therapeutic implications for chronic pain. The persistent hyperexcitability of non-CRF neurons at the chronic phase of SNL is consistent with those from unidentified CeC neurons in our previous studies, which have been linked to pain-related behaviors.63,71 Using Prkcd-Cre, Calcrl-Cre, and Sst-Cre mice, we further show that it is not PKCδ and CGRPR neurons but a subset of CeA-SOM neurons that develop increased excitability at the chronic stage of neuropathic pain. These results are consistent with previous evidence that SOM neurons do not overlap substantially with CRF, PKCδ, or CGRPR neurons,23,47,54 and SOM neurons showed increased excitability at the chronic phase of a neuropathic pain model.72 While that study did not distinguish firing types, their example traces and frequency-current (F-I) relationship were similar to our regular spiking rather than LF neurons.72
Interestingly, we found that chemogenetic inhibition of CeA-SOM neurons did not measurably affect tactile hypersensitivity at either acute or chronic neuropathic stages, whereas the chemogenetic inhibition of CeA-PKCδ or CeA-CGRPR neurons reversed tactile hypersensitivity at the acute phase, with only a small partial reversal at the chronic phase. The reason why chemogenetic inhibition of CeA-SOM neurons had no significant behavioral effect at the chronic phase remains to be determined. Our data suggest that only a subpopulation of CeA-SOM neurons contributes to pain chronification, and the beneficial effect of their inhibition is masked by the nonselective inhibition of different subpopulations of CeA-SOM neurons, including those that might retain their antinociceptive function we observed at the acute phase.23 Supporting this view, recent evidence suggests differential roles of CeA-SOM neurons in pain depending on their projection targets53,73–75 or anatomical location within the CeA.22 Importantly, different firing patterns have also been implicated in the functional heterogeneity of CeA-SOM neurons.76 Together, our results suggest that CeA neurons contributing to pain chronification most likely involve a subpopulation of SOM-expressing non-CRF neurons in the CeA.
Differential pain-related changes in PAG- and PB-projecting CeA-CRF neurons in neuropathic pain
The CeA is well positioned to modulate pain behaviors through widespread projections to forebrain and brainstem areas involved in behaviors and pain modulation.13 Their downstream targets contribute to distinct behaviors.77–81 Anatomically, consistent with previous studies,27,28,56 our histological experiments revealed that subsets of PB- and PAG-projecting CRF neurons co-express SOM but very rarely co-express PKCδ. Functionally, a recent study revealed the important role of CeA-CRF projections to the PB under acute pain conditions.56 However, pain-related synaptic and cellular changes in projection-defined CeA-CRF neurons remained to be determined. Here, we examined baseline differences between PB- and PAG-projecting CRF neurons, and pain-related changes in these neurons. We found higher excitability in PB-projecting CeA-CRF neurons compared to PAG-projecting ones at baseline, but more pronounced pain-related changes in PAG-projecting ones. While the effects of CRF projections on the PAG with regard to pain modulation are not clear, this change could be a mechanism of the well-documented switch from pain inhibition to pain facilitation.82 Because both PB and PAG form reciprocal connections with CeA-CRF neurons,38,83 it will be interesting to see how these circuits contribute to pain processing in each area and participate in the transition from acute to chronic pain. Also unknown is whether other CeA-CRF projections differentially modulate components of pain and how different CeA-CRF projections interact with one another during the development of chronic pain.
Potential mechanisms of pain chronification and future directions
The key finding of our study is that synaptic plasticity develops initially in the PB→CeA pathway but no longer contributes to hypersensitivity at the chronic phase. Since PB activity has been shown to remain increased at the chronic phase of neuropathic pain models,64,84 PB neurons may contribute to pain processing at the chronic phase via projections to other brain areas,85,86 possibly in a firing type-specific manner.87 It is also possible that CeA hyperexcitability is driven by input from other brain areas implicated in chronic neuropathic pain conditions such as the basolateral amygdala (BLA),88,89 insular cortex,90 and the paraventricular nucleus of the thalamus,91 also known to provide direct inputs to the CeA.92,93 In fact, we previously showed increased BLA-driven excitatory synaptic transmission in CeL/C neurons at the chronic phase of neuropathic pain.71 These other inputs could contribute to mechanisms underlying PB-independent hyperexcitability of CeC neurons at the chronic phase and would indicate a shift in network neuroplasticity.
Reciprocal inhibitory synaptic connections and their shift to disinhibition between these different types of GABAergic neurons94–97 might also be an important mechanism underlying the cell-type-specific transition in excitability from acute to chronic pain. In addition, when interpreting these interactions, it is important to note that activation of CeA-CRF neurons has been shown to activate, rather than inhibit, non-CRF neurons in the CeA, likely through the activation of CRF1 receptors by locally released CRF.38
In addition to neuronal interactions, non-neuronal components could contribute to neuronal plasticity at the chronic phase and to the transition from acute to chronic pain by driving synaptic plasticity-independent hyperexcitability identified in this study. Accumulating evidence suggests a critical role of neuroimmune signaling in the pathogenesis of chronic pain.98,99 However, the primary focus of previous studies was on the periphery and spinal cord, and the role of neuroimmune signaling in pain processing in the brain is largely unknown. Recent evidence has implicated neuroimmune signaling in the CeA in sensory and emotional aspects of pain.37,100–108 It remains to be determined whether and how neuroimmune signaling in the CeA contributes to neuroplasticity and to the transition from acute to chronic pain. One recent study revealed critical interactions between CeA neurons and microglia in acute stress-induced anxiety-like behaviors in mice, where a cytokine protein (CX3CL1) released from hyperexcitable CeA neurons activated microglia.109 Activated microglia in turn inhibited hyperexcitability of CeA neurons by the engulfment of their dendritic spines.109 These neuron-microglia interactions may serve as a negative feedback mechanism that could explain the loss of hyperexcitability in CRF neurons and the absence of enhanced synaptic transmission at the chronic pain stage. The mechanism by which microglia could modify CeA neurons in a cell-type-specific manner remains to be determined, but there is evidence that microglia interact with neurons differently, depending on the presence or absence of their contacts with CRF neurons,110,111 possibly through microglial CRF receptors.112,113 Additionally, since the activation of μ-opioid receptors has been shown to decrease PB-CeL/C EPSCs presynaptically,114,115 it is possible that the release of endogenous opioids from neighboring CeA enkephalinergic neurons116 caused the loss of enhanced PB-CeA synaptic transmission at the chronic phase. Interestingly, a recent study implicated systemic inflammation in the modulation of the endogenous opioid system in the PB→CeA pathway.115
Understanding the temporal regulation of neuroplastic changes is conceptually and perhaps therapeutically significant. One important question is whether the neuroplastic changes observed here are pain model specific or are linked to the different stages of the neuropathic pain condition. While similar neuroplastic changes have been reported for acute pain conditions in different models,16–25 a comparison for chronic pain conditions is lacking. Interestingly, recent studies using two different types of pain models (acid-induced muscle pain vs. cuff-induced neuropathic pain) showed opposite changes in CeA-SOM and CeA-PKCδ neurons at similar (sub)acute time points (2 weeks vs. 1–2 weeks) after pain induction.23,69 Here, we compared the 1-week and 3- to 4-week time points as acute and chronic phases, respectively, based on our recent studies that showed significant molecular and behavioral differences between these time points.34,36,37,108 Neuropathic-pain-related synaptic changes have been consistently reported at the 1-week time point,18,35 and therefore, we selected this time point for the acute pain stage to allow the comparison of results. The 3- to 4-week time point is well established as the chronic stage of neuropathic pain in different models.52,117,118 Recent evidence suggests a shift in the activity and role of PB neurons at this time point in a neuropathic pain model,64 further justifying the 4-week time point for our study of the PB→CeA pathway here. No difference in the effects of inhibition of CeA-SOM neurons on hypersensitivity was found between 4 h and 2 weeks after muscle pain induction,69 which suggests that these time points still reflect the acute phase and also justifies our choice of the 1-week (earlier than 2 weeks) and the 3- to 4-week (later than 2 weeks) time points to distinguish acute and chronic pain mechanisms. Hyperexcitability of CeA-SOM neurons at the 3-week time point in our study matches the data from another study at the 6-week time point.72 The cell-type-specific69,119,120 longitudinal monitoring of neuronal activity88 could provide a more fine-grained analysis of the time-dependent contribution of each CeA cell type to the transition to chronic pain.
Our previous study showed beneficial effects of inhibition of CeA-CRF neurons,34 while activation of CeA-CRF terminals in the PB also was antinociceptive at the acute phase.56 These contrasting results suggest differential, potentially opposing, roles of CeA-CRF neurons in pain processing, depending on their projection targets. Based on the more pronounced pain-related changes we found in PAG-projecting neurons and recent evidence suggesting a pain-facilitatory role of the CeA-PAG pathway,121 it is possible that hyperexcitable CeA-CRF neurons at the acute phase function as part of a pain-facilitatory circuit through their synaptic outputs to specific target regions such as PAG. However, a hyperexcitable subpopulation of CeA-SOM neurons at the chronic phase could mediate chronic pain and comorbidities through their synaptic outputs to the lateral habenula72 or to the zona incerta.53,73
Summary and conclusion
This study identified a potential cellular mechanism of neuroplasticity in the amygdala underlying the transition from acute to chronic pain, which suggests that chronic pain is not simply an extension of acute pain. The loss of synaptic plasticity in the PB→CeA pathway is intriguing, and underlying mechanisms remain to be determined. Synaptic plasticity at the acute and chronic stages of pain needs to be explored in different pathways such as the corticolimbic system as there could be a relative shift of the role of ascending pathways to reverberating brain circuits in amygdala neuroplasticity in chronic pain.
Limitations of the study
In the present study, we focused on the right CeA because of lateralized pain processing in the CeA,122,123 which was suggested to be determined by the PB-CeA circuit.51 It would be important to examine both hemispheres to gain a better understanding of neuroplasticity and mechanisms underlying the transition from acute to chronic pain. However, a previous study demonstrated a shift of CeA neuronal activity increases from left to right during the development of neuropathic pain,124 which would justify our focus on the role of the right in chronic neuropathic pain.
Another important aspect to consider is sex differences. Accumulating clinical and preclinical evidence suggests sex differences in pain processing along the neuraxis.125 We recently found sexually dimorphic, potentially time-dependent, functions of CGRP in the CeA in a neuropathic pain model,36 perhaps suggesting a change in the of source of CGRP from PB to the thalamus126 in females at the chronic stage. Sex differences in amygdala pain processing and underlying mechanisms remain a largely understudied area of research. Here, we found no differences between male and female mice in the electrophysiology of CeA-PKCδ, CeA-CGRPR, and CeA-SOM neurons and in the behavioral effects of chemogenetic modulations of these CeA neurons, and therefore data were pooled. While the rat experiments included only males, the consistencies between the results from experiments in male rats and those in mice of both sexes suggest that these findings apply to both males and females. However, given the growing evidence for sex differences in the development of chronic pain and chronic pain syndromes,127,128 their better understanding is important to gain insight into mechanisms of pain chronification.
Another caveat is that only sensory but not affective behavioral aspects were assessed here. However, we previously reported affective pain behaviors such as anxiety-like behaviors and vocalizations in neuropathic pain and showed that they were dependent on amygdala neuronal activity.34,68,71 Any mechanistic differences in the modulation of sensory vs. affective behaviors by different types of CeA neurons remain to be determined. Finally, we identified a subpopulation of CeA-SOM neurons as a potential key player in chronic neuroplasticity, but it is not clear whether different subpopulations are distinct with regard to their specific projection targets and how that might affect behavioral modulation; studies are needed to determine the behavioral effects of the specific manipulation of individual subpopulations of CeA-SOM neurons.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Volker Neugebauer (volker.neugebauer@ttuhsc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
In this collaborative study between two laboratories, we used both rats and mice to address specific experimental questions. Because of the expertise of each laboratory, all rat experiments were conducted in Dr. Neugebauer’s laboratory at Texas Tech University Health Sciences Center (TTUHSC) and all mouse experiments were conducted in Dr. Carrasquillo’s laboratory at the National Institutes of Health (NIH).
For electrophysiological, behavioral and immunohistochemical experiments at TTUHSC, male hemizygous transgenic Crh-Cre rats on Wistar background38 (initial breeding pairs kindly provided by Dr. Robert Messing, UT Austin) were housed in a temperature-controlled room under a 12-h light/dark cycle. The offspring was genotyped to identify the presence of Cre-recombinase from tail biopsies with the following primers: GAGTGAACGAACCTGGTCGAAATCAGTGCG (reverse) and GCATTACCGGTCGATGCAACGAGTGATGAG (forward) (Integrated DNA Technologies (IDT)). On the day of the experiment, rats were transferred from the animal facility and allowed to acclimate to the laboratory for at least 1 h. They were 4 weeks of age at the start of the surgical procedure.
For behavioral experiment with chemogenetic manipulations, male and female C57BL/6J mice were used at NIH. They were 8–17 weeks of age at the start of the surgical procedure. For electrophysiology experiments in brain slices from mouse, adult male and female transgenic mice (12–18 weeks) were used at NIH. Heterozygote Prkcd-Cre female and male mice (GENSAT-founder line 011559-UCD) were obtained from the Mutant Mouse Resource and Research Center (MMRRC) and backcrossed with C57BL/6J mice (The Jackson Laboratory, stock number 000664) for over 10 generations. Calcrl-Cre mice were generated and kindly provided by Dr. Richard Palmiter (University of Washington). Prkcd-Cre heterozygote, Calcrl-Cre homozygote, or Sst-Cre heterozygote mice (The Jackson Laboratory, founder line 018973) were crossed with homozygous Ai9 mice (The Jackson Laboratory, founder line 007909). The generation of Prkcd-Cre, Calcrl-Cre, and Sst-Cre mice has been previously described.47,134 The offspring were genotyped to identify a presence of Cre-recombinase from tail biopsies and PCR (Transnetyx) with the following primers: CAGGCTAAGTGCCTTCTCTACA (reverse) and TTAATCCATATTGGCAGAACGAAAACG (forward). Mice were housed in a vivarium with controlled humidity and temperature under a reversed 12-h light/dark cycle (9 a.m.–9 p.m. dark) and separated by a perforated Plexiglass divider. All procedures and behavioral experiments were performed during the dark phase (9 a.m.–6 p.m.).
All experimental procedures were approved by the Institutional Animal Care and Use Committees (IACUC) at TTUHSC or the National Institute of Neurological Disorders and Stroke and the National Institute of Deafness and other Communication Disorders, and conform to the guidelines of the International Association for the Study of Pain (IASP) and of the NIH. Water and food were available ad libitum, and all efforts were made to minimize animal suffering.
METHOD DETAILS
Viral vector injections
Brain slice electrophysiology
Four-week-old (60–100 g) rats were used for microinjections for brain slice electrophysiology or IHC. A Cre-dependent adeno-associated viral vector (AAV5-Ef1a-DIO-mCherry, 0.5 μL, Dr. Karl Deisseroth, packaged by the Vector Core at the University of North Carolina, Chapel Hill) was stereotaxically injected into the CeA of transgenic Crh-Cre rats using a 5 μL Neuros Syringe (33 gauge, Hamilton) to identify CeA-CRF neurons in brain slices. For the selective activation of PB input to CeA neurons in amygdala brain slices, an AAV encoding channelrhodopsin 2 (ChR2) under the control of the CaMKII promoter (AAV5-CaMKII-hChR2(134R)-EYFP, 0.3 μL, Dr. Karl Deisseroth, packaged by the Vector Core at the University of North Carolina, Chapel Hill) was injected into the PB for optical activation of PB fiber terminals in the CeA. In a separate group of animals, retrograde Cre-dependent adeno-associated viral vectors AAVrg-hSyn-DIO-EGFP (0.5 μL, 50457-AAVrg, Addgene) and AAVrg-FLEX-tdTomato (0.5 μL, 28306-AAVrg, Addgene) were injected into the PB and PAG, respectively, of transgenic Crh-Cre rats to identify PB- and PAG-projecting CeA-CRF neurons in brain slices. The following coordinates135 were used: CeA, 2.0–2.5 mm caudal to bregma, 4.0–4.2 mm lateral to midline, 7.5–7.6 mm deep; PB, 15° anteroposterior angle,20 6.3–6.9 mm caudal to bregma, 1.6–1.9 mm lateral to midline, 6.8–7.2 mm deep; PAG, 6.9–7.2 mm caudal to bregma, 0.6–0.8 mm lateral to midline, 5.7–5.8 mm deep. Animals were allowed to recover 5 (AAV5) or 6 (AAVrg) weeks for viral expression before brain slices were obtained for the electrophysiology or before transcardial perfusion for IHC.
Chemogenetics
Mice were initially anesthetized with 5% isoflurane and head fixed in a stereotaxic frame (David Kopf Instruments). Surgery was performed with 1.5–2% isoflurane at a flow rate of 0.5 L/min. A hand warmer (Hot Rods Hand Warmers) was used for thermal maintenance during the procedure.
For intersectional PB→CeA pathway analysis, AAVrg-hSyn-HI-eGFP-Cre-WPRE-SV40 (Addgene 105540-AAVrg) in combination with AAV8-CMV-LacZ-bGH (Addgene 105531-AAV8) were unilaterally microinjected (1:1, 70 nL) into the right CeA and simultaneously microinjected 100 nL of AAV8-hSyn-DIO-hM4D(Gi)-mCherry (Addgene 44362-AAV8) or AAV8-hSyn-DIO-mCherry (Addgene 50459-AAV8) in the right PB.
For CeA-PKCδ, CeA-CGRPR and CeA-SOM neurons inhibition, 0.3 μL, 0.05 μL and 0.03 μL of AAV8-hSyn-DIO-mCherry (Addgene 50459-AAV8), or AAV8-hSyn-DIO-hM4Di-mCherry (Addgene 44362-AAV8) virus was microinjected into the right CeA of Prkcd-Cre, Calcrl-Cre, or Sst-Cre mice respectively using a 32-gauge, 0.5 μL Hamilton Neuros syringe. The injections were performed at a flow rate of 0.1 μL/min, and the injector was left in place for an additional 5 min at the end of the injection to allow for virus diffusion.
The stereotaxic coordinates were as follows: for right CeA: 1.25 mm posterior from bregma, 3.0 mm lateral to midline, 4.5 mm ventral to skull surface, and for right PB: 4.9 mm posterior from bregma, 1.2 mm lateral to midline, 3.78 mm ventral to skull surface. At the end of the experiments, mice were transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer, pH 7.4. Post-experiment, histological confirmation of injections sites was performed and only animals with both correct injection sites were included in the analyses.
Spinal nerve ligation neuropathic pain model in rats
The well-established spinal nerve ligation (SNL) model117 was used to study acute (1 week) and chronic (4 weeks) phases of neuropathic pain. Rats were randomly assigned to the SNL and sham groups. As described in our previous studies,34,68,71 the left L5 spinal nerve was exposed and tightly ligated with 6–0 silk thread under isoflurane anesthesia (3–5% induction and 2% maintenance). Shamoperated animals underwent the same surgical procedure except for ligation of the nerve and were used as controls. Behavioral tests began after a one- or four-week recovery period. Electrophysiological recordings were performed 1 or 4 weeks after the SNL or sham procedure. Rats were handled to minimize stress as described previously.136
Sciatic cuff neuropathic pain model in mice
In order to avoid the reported variability or inconsistency of the SNL model in mice,137,138 we used the sciatic cuff model of neuropathic pain in the mouse experiments, which is a more suitable neuropathic pain model in mice with low interindividual variability because of the standardized cuffs and procedures.52,118 Validation across different neuropathic pain models also enhances scientific rigor. Mice were randomly assigned to receive a cuff implantation or a sham procedure on the left sciatic nerve as described previously.52,118 Briefly, mice were initially anesthetized with 5% isoflurane (flow rate of 0.5 L/min) and maintained at 1.5–2% for the duration of the surgery. An incision (1 cm) was made along the proximal one-third of the lateral left thigh. The sciatic nerve was gently stretched using forceps, and a non-toxic, sterile polyethylene tube (PE-20, 2 mm length, 0.38 mm ID, 1.09 mm OD; Daigger Scientific) was inserted onto the sciatic nerve for cuff animals. Sham control mice underwent the same process of sciatic nerve exposure and stretching but without tube implantation. Following the procedure, the incision was closed using wound clips. Mice were returned to a housing cage and placed on top of a hand warmer (Kobayashi Consumer Products, LLC) during postoperative recovery. Behavioral tests began after a one-week recovery period. Electrophysiological recordings were performed 28–32 days after the cuff or sham procedure. Mice were handled to minimize stress for at least 5 days prior to the experiment as described previously.139 Mice also received mock intraperitoneal (i.p.) injections of sterile saline (70 μL) during handling to minimize stress of anesthesia injection prior to transcardial perfusion and acute slice preparation.
Mechanosensitivity
In rats, the paw withdrawal thresholds to mechanical stimuli were measured using an electronic von Frey anesthesiometer with the rigid tip (IITC Life Science Inc., Woodland Hills, CA) as described previously.34,37,106 The tip was applied perpendicular to the plantar surface of the left hind paw with increasing force until the paw was withdrawn, and the maximum force (in grams) applied was automatically recorded. The measurement was repeated 3 times with an interval of at least 10 min and the values were averaged to determine the withdrawal thresholds.
In mice, mechanical hypersensitivity was determined with von Frey filaments (North Coast Medical, Inc. San Jose, CA) as previously described.140 Briefly, mice were handled 4–5 days for habituation. On testing day, Mice were placed individually in ventilated opaque white Plexiglas testing chambers (11 × 11 × 13 cm) on an elevated mesh platform for 2–3 h before stimulus application. Each calibrated filament was applied five times to the plantar surface of the hind paw until they bowed slightly. If no paw withdrawal response was observed, next filament with large force was used. The smallest filament that evoked a paw withdrawal response in at least three of five measurements was taken as the mechanical threshold for that trial. The average of five trials was calculated and used as the threshold value per hindpaw. Mechanical hypersensitivity was evaluated 1 week and 3 weeks after cuff or sham surgery in the same mice. On each testing day, paw withdrawal threshold was assessed at pre and 30–45 min post i.p. injection of either saline or Clozapine-N-oxide (CNO, BML-NS105, Enzo Life Sciences, Farmingdale, NY; 10 mg/kg body weight). Mice were randomly assigned to the saline or CNO group on the first day of each test and were tested on the opposite treatment on the second day of the same test.
In all behavioral experiments, cohorts were counterbalanced, animals were randomly assigned to different experimental groups, and experimenters performing injections and behavioral procedures were blinded to the experimental groups.
Acute brain slice preparation
Rat brain slices containing the amygdala were obtained from naive, sham, and SNL rats (9–10 weeks old, 280–400 g) as described before.34,68,71 Brains were quickly removed and immersed in oxygenated ice-cold sucrose-based physiological solution containing (in mM): 87 NaCl, 75 sucrose, 25 glucose, 5 KCl, 21 MgCl2, 0.5 CaCl2 and 1.25 NaH2PO4. Coronal brain slices (400 μm) were prepared using a vibratome (VT1200 S, Leica Biosystems, Nussloch, Germany). The amygdala slices were then incubated in oxygenated artificial cerebrospinal fluid (ACSF) at 32°C for 30 min before being allowed to equilibrate at room temperature (21°C) for at least an additional 30 min prior to recording. ACSF contained the following (in mM): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3 and 11 glucose. A single brain slice was transferred to the recording chamber and submerged in ACSF (31 ± 1°C) superfusing the slice at ~2 mL/min. Only one or two brain slices per animal were used.
Mouse brain slices containing the amygdala were obtained from sham and cuff implanted mice (12–18 week old) as described before.23,24 Mice were deeply anesthetized following an i.p. injection of Avertin (1.25%, 0.025 mL/g body weight), and perfused transcardially with a choline chloride-based cutting solution (110.0 mM choline chloride, 12.7 mM L-ascorbic acid, 3.1 mM pyruvic acid, 25.0 mM D-Glucose, 7.2 mM MgCl2, 0.5 mM CaCl2, 25.0 NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl). The brains were immediately extracted, submerged in ice-cold cutting solution, and cut into 250 μm-thick coronal brain slices using a VT1200 S vibratome. Coronal slices including the CeA from the right hemisphere were recovered in a 33°C heating chamber including ACSF (25.0 mM D-glucose, 1.0 mM MgCl2, 2.0 mM CaCl2, 25.0 NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 125.0 mM NaCl) 30 min after cutting. Following incubation, slices recovered for an additional 20 min at room temperature prior to electrophysiological recordings. During slice preparation and recovery, the cutting solution and ACSF were continuously oxygenated with 95%/5% O2/CO2.
Electrophysiology in brain slices
Rat amygdala slices
Whole-cell patch-clamp recordings were obtained from visually identified mCherry-expressing CRF neurons in the CeL/C, mCherry-negative non-CRF neurons in the CeC, and tdTomato- or EGFP-expressing CRF neurons in the CeL/C of the right hemisphere, using LED illumination system (X-Cite 120 LED, Excelitas Technologies Corp.) with ET-DSRed (TRITC/Cy3) (49005, Chroma Technology Corp.) (for mCherry and tdTomato) and ET-EGFP (FITC/Cy2) (49002, Chroma Technology Corp.) (for EGFP) filter sets, and infrared DIC videomicroscopy as described previously.34,39 Recording electrodes (tip resistance 5–8 MΩ) were made from borosilicate glass and filled with intracellular solution containing (in mM): 122 K-gluconate, 5 NaCl, 0.3 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, and 0.4 Na3-GTP; pH was adjusted to 7.2–7.3 with KOH and osmolarity to 280 mOsm/kg with sucrose. On the day of recording, 0.1% biocytin was included in the intracellular solution. Data acquisition and analysis were done using a dual 4-pole Bessel filter (Warner Instr., Hamden, CT), low-noise Digidata 1322 interface (Axon Instr., Molecular Devices, Sunnyvale, CA), Axoclamp-2B amplifier (Axon Instr., Molecular Devices, Sunnyvale, CA), Pentium PC, and pCLAMP 8.2 software (Axon Instr., Molecular Devices).
In current-clamp mode, depolarizing current steps (500 ms) were applied from resting membrane potential (RMP) to characterize the electroresponsive properties (firing types and rheobase, 10 pA steps) or to measure frequency-current (F-I) relationships (20 pA steps) of recorded neurons. Neuronal excitability was assessed from F-I relationships. Neurons were voltage-clamped at −70 mV for the study of excitatory postsynaptic currents (EPSCs). To evoke PB-driven EPSCs in amygdala neurons by focal stimulation (150 μs square-wave pulses), a concentric bipolar electrode (David Kopf Instruments) was placed onto the visually identified fiber tract dorsomedial to the CeA and ventral to but outside of the caudate-putamen as previously described.16,42,43 For paired-pulse ratio (PPR) analysis, two stimuli of equal intensity were applied with an inter-stimulus interval of 50 ms and the resulting EPSCs were recorded. Peak amplitudes of the first EPSC (EPSC1) and the second EPSC (EPSC2) were measured, and PPR was calculated as the ratio of EPSC2 over EPSC1. Any alterations in PPR suggest a presynaptic site of action.42,141 For the definitive analysis of PB synaptic input, ChR2 expressing afferent fibers from the PB were activated optically by blue light pulses (29.7 mW/mm2, 5 ms, 0.05 Hz) generated by a LED illumination system with a blue filter (ET470/40x, Chroma Technology Corp.) and a 40× objective of the microscope as described previously.39,142 Illumination area of the 40× objective (0.24 mm2) was centered on the soma of the patched cell. Light power density was measured using an optical power meter (PM200, Thorlabs) placed under the objective.
Mouse amygdala slices
Whole-cell current-clamp recordings were collected from visually identified tdTomato-expressing neurons located in the CeL/C. The CeL/C regions were identified using distinctive fiber bundles and anatomical landmarks described in the mouse brain atlas.143 Cells were identified using differential interference contrast optics with infrared illumination and fluorescence microscopy (Nikon Eclipse FN1). An in-line solution heater and recording chamber heater (Warner Instruments) maintained the bath temperature at 33 ± 1°C throughout the experiment. Additionally, ACSF oxygenated with 95%/5% O2/CO2 was perfused into the recording chamber at a rate of 1 mL/min. Patch pipettes (2–5 MΩ resistance) were filled with a potassium methylsulfate internal solution (120.0 mM KMeSO4, 20.0 mM KCl, 10.0 mM HEPES, 0.2 mM EGTA, 8.0 mM NaCl, 4.0 mM Mg-ATP, 0.3 mM Tris-GTP, 14.0 mM phosphocreatine, solution pH adjusted to 7.3 with KOH, ~300 mosmol−1). Recordings were performed using a Multiclamp 700B patch-clamp amplifier with a Digidata 1500 acquisition system and pCLAMP 11.2 software (Molecular Devices). Pipette tip potentials were zeroed before forming a gigaseal with the cell membrane. During current clamp recordings, a 500 ms depolarizing current (0–280 pA) was injected to assess repetitive action potential firing of fluorescently labeled neurons from resting membrane potential in slices from Prkcd-CreAi9, Calcrl-CreAi9, or Sst-CreAi9 mice. Recordings were acquired at 100 kHz and low-pass filtered at 5 kHz. Pipette capacitances and series resistances were monitored during each recording by holding the cells at −70 mV, followed by ± 10 mV voltage steps of 25 ms duration. All neurons analyzed had series resistances lower than 20 MΩ. Liquid junction potentials were not corrected during recordings.
Drugs
An NMDA receptor antagonist (DL-2-amino-5-phosphonopentanoic acid, Cat# 0105, AP5) and a non-NMDA receptor antagonist (6-cyano-7-nitroquinoxaline-2,3-dione disodium, Cat# 1045, CNQX) were purchased from Tocris Bioscience (R&D Systems, Minneapolis, MN). A CGRP receptor antagonist (BIBN4096BS, Cat# SML2426) was purchased from MilliporeSigma (Burlington, MA). All drugs were applied by gravity-driven superfusion of the brain slice in ACSF (~2 mL/min). Solution flow into the recording chamber (1 mL volume) was controlled with a three-way stopcock. Drugs were applied for at least 15 min to establish equilibrium in the tissue. Clozapine-N-oxide (CNO, Cat# BML-NS105) was purchased from Enzo Life Sciences (Farming-dale, NY).
Verification of viral vector injection
Rats
Targeting of viral vectors into the PB and PAG in rats was verified histologically. During acute brain slice preparation, the removed brainstem was dipped in 4% PFA in 0.1 M phosphate buffer and fixed overnight at 4°C, and then cryoprotected in a 30% sucrose in 0.1 M phosphate buffer for 48 h at 4°C. Coronal sections (40 μm) containing the PB and PAG were collected using a cryostat (HM525 NX, epredia), mounted on slides, and coverslipped for later image acquisition. Only data from animals with correct virus injection sites were included in the analysis.
Mice
To evaluate injection sites of chemogenetic manipulations, mice were deeply anesthetized with 1.25% Avertin anesthesia (2,2,2-tribromoethanol and tert-amyl alcohol in 0.9% NaCl; 0.025 mL/g body weight) at the end of the experiments, then perfused transcardially with 0.9% NaCl (37°C), followed by 100 mL of ice-cold 4% PFA in 0.1 M phosphate buffer. The brain was dissected and post fixed in 4% PFA in 0.1 M phosphate buffer overnight followed by 30% sucrose in 0.1 M phosphate buffer for 48 h at 4°C. For injection site verification of intersectional chemogenetic experiment, coronal slices (30–45 μm) containing the CeA and PB were collected using a freezing sliding microtome and stored in 0.1 M phosphate buffered saline (PBS), pH 7.4 containing 0.01% sodium azide (Sigma) at 4°C until immunostaining. Sections were rinsed in PBS, incubated in 0.1% Triton X-100 in PBS for 10 min at room temperature and blocked in 5% normal goat serum (NGS) (Vector Labs, Burlingame, CA) with 0.1% Triton X-100, 0.05% Tween 20 and 1% bovine serum albumin (BSA) for 30 min at room temperature. The slices were incubated in rat anti-mCherry (1:125, M11217, Invitrogen) and rabbit anti-β-gal (1:500, ab986, Millipore Sigma) primary antibodies in 1.5% NGS blocking solution with 0.1% Triton X-100, 0.05% Tween 20 and 1% BSA overnight at 4°C. Slices were then rinsed in PBS and incubated in goat anti-rat Cy3 (1:250, A10522, Invitrogen) and Alexa Fluor 647-conjugated goat anti-rabbit (1:500, A21244, Invitrogen) secondary antibodies in 1.5% NGS blocking solution with 0.1% Triton X-100, 0.05% Tween 20 and 1% BSA, protected from light, for 2 h at room temperature. Sections were then rinsed in PBS, mounted on positively charged glass slides, air-dried and coverslips were placed using Fluoromount-G (Southern Biotech).
Post-hoc staining and identification of recorded neurons
In a subset of experiments, the recorded slices were fixed in 4% PFA in 0.1 M phosphate buffer overnight at 4°C. After fixation, slices were washed in PBS (3 × 10 min) and permeabilized in PBS containing 0.4% Triton X-100 for 1 h at room temperature. Slices were then incubated in fluorescently conjugated streptavidin (1:1000, streptavidin, Alexa Fluor 405 conjugate, S32351, Invitrogen) for 12–24 h at 4°C. Finally, the slices were washed in PBS (3 × 10 min), mounted on slides with ProLong Glass antifade mounting media (P36984, Invitrogen).
Immunohistochemistry
Rats were deeply anesthetized with isoflurane and transcardially perfused with 0.9% NaCl followed by 4% PFA in 0.1 M phosphate buffer. The brain was postfixed in 4% PFA in 0.1 M phosphate buffer overnight at 4°C and then cryoprotected in a 30% sucrose in 0.1 M phosphate buffer for 48 h at 4°C. Coronal sections (40 μm) containing the CeA were collected using a cryostat and stored in 0.01 M PBS at 4°C until immunostaining. Sections were washed in PBS (3 × 10 min) and blocked for 2 h at room temperature in blocking solution (0.1% Triton X-100, 1.5% BSA, and 5 or 10% NGS) followed by 48 h incubation at 4°C in rat anti-somatostatin (1:100, MAB354, Millipore Sigma) and mouse anti-PKCδ (1:1000, 610398, BD Biosciences), or rabbit anti-human/rat CRF (1:20000, PBL rC68, kindly provided by Dr. Paul Sawchenko, The Salk Institute, La Jolla, CA) in blocking solution. Sections were then washed in PBS (4 × 10 min) and incubated in Alexa Fluor 647-conjugated goat anti-rat (1:500, A-21247, Invitrogen) and Alexa Fluor Plus 405-conjugated goat anti-mouse (1:1000, A48255, Invitrogen), or Alexa Fluor 647-conjugated goat anti-rabbit (1:1000, A-21244, Invitrogen) in blocking solution for 24 h at 4°C. Finally, sections were washed in PBS (4 × 10 min), mounted on slides, and coverslipped with ProLong Glass antifade mounting media.
Image acquisition and analysis
Images of rat brain sections were acquired using a confocal microscope equipped with 2×, 20×, and 60× objectives (FV3000, Olympus, Center Valley, PA). The 2× or 20× objectives were used to capture wide field images to verify viral vector injection sites in the brainstem, to determine rostro-caudal level of sections, and to delineate CeA subregions.135 The 20× objective was used to acquire stitched (3× 3) z stack (3 μm steps) images for quantification. The 60× objective was used to acquire representative high magnification images. The same imaging parameters were used for all sections. Image stacks were converted into maximum intensity z-projections using ImageJ (Fiji, NIH, Bethesda, MD) and the number of labeled cells was counted manually. Quantification was performed using 1 section per rostro-caudal level per rat.
Images of mouse brain sections were acquired using a Nikon A1R laser scanning confocal microscope. 2× (for low magnification) and 10× (for high magnification) objectives were used for analyses. High magnification mCherry representative image was taken using a 20× objective. LacZ expression in the CeA and mCherry expression in PB were evaluated using 647 and Cy3 channels respectively. Once images were acquired, rostro-caudal level was determined, and anatomical location of positive cells was delineated based on the mouse brain atlas.143 Correct injection sites were defined as brains that showed localized LacZ transduced cells in the CeA, mCherry transduced cell bodies in the PB and mCherry-expressing terminals in the CeA.
QUANTIFICATION AND STATISTICAL ANALYSIS
The following data analysis methods (including neuronal classification) and statistics were used.
Rat experiments
Electrophysiological data were analyzed with Clampfit 11.2 (Molecular Devices) and GraphPad Prism (version 10.0.2, GraphPad Software, San Diego, CA). The Venn diagram was plotted using the venn2 function in matplotlib-venn (matplotlib version 3.8.0, matplotlib-venn version 0.11.9) package in Python (Anaconda Distribution version 2023.09–0, Python version 3.11.5, Spyder version 5.4.3). Firing types of CRF and non-CRF neurons from rats were determined according to methods described previously,20,32,40 Action potential (s) in response to depolarizing current (500 ms, 10 pA steps) that only just exceed the spike threshold were used for classification. Neurons with latencies of more than 250 ms for the first action potential generation were classified as late-firing (LF) neurons. Neurons with bursting action potentials were classified as low-threshold bursting (LTB) neurons. The remaining neurons that did not meet either criterion were classified as regular spiking (RS) neurons. Student’s t test was used to compare two sets of data that had Gaussian distribution and similar variances. For multiple comparisons, ANOVA was used with Bonferroni posttests as indicated in the text and figure legends.
Mouse experiments
Electrophysiological data were obtained, compiled and analyzed using Clampfit 11.2 (Molecular Devices), Microsoft Excel, and GraphPad Prism (version 9.5.1, GraphPad Software, San Diego, CA). At least 5 mice were used as biological replicates per treatment and genotype. To determine the firing phenotype of PKCδ neurons, the latency to the first action potential was calculated as previously categorized.24 LF PKCδ and SOM neurons had latencies longer than 100 ms in response to a 280 pA depolarizing current injection (sham conditions) and 90 ms following a 220 pA current injection (cuff conditions). LF CGRPR neurons were characterized at a current injection step that elicited between 4 and 5 action potentials and had latencies longer than 100 ms (sham conditions) or 90 ms (cuff conditions). The range of current injection that elicited 4 to 5 action potentials was 140–300 pA in LF CGRPR neurons. RS PKCδ and SOM neurons had latencies shorter than 100 ms following a 280 pA current injection (sham conditions) or 90 ms following a 220 pA current injection (cuff conditions). RS CGRPR neurons were characterized at a current injection step that elicited between 4 and 5 action potentials and had latencies shorter than 100 ms (sham conditions) or 90 ms (cuff conditions). Action potentials elicited in response to prolonged (500 ms) depolarizing current injections of increasing amplitudes (0–300 pA) were tallied at each respective current step and compiled in Prism according to cell type, treatment and sex in order to generate input-output curves as described previously.16,23 Statistical analyses were performed using two-way ANOVA in Prism.
All averaged values are given as the mean ± standard error of the mean (SEM). Statistical significance was accepted at the level p < 0.05.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rat anti-mCherry | Invitrogen | Cat# M11217; RRID: AB_2536611 |
| Rabbit anti-β-gal | Millipore Sigma | Cat# ab986; RRID: AB_92401 |
| Rat anti-somatostatin | Millipore Sigma | Cat# MAB354; RRID: AB_2255365 |
| Mouse anti-PKCδ | BD Biosciences | Cat# 610398; RRID: AB_397781 |
| Rabbit anti-human/rat CRF | Joan Vaughan, Paul Sawchenko | Cat# PBL rC68; RRID: AB_2650435 |
| Goat anti-rat Cy3 | Invitrogen | Cat# A10522; RRID: AB_2534031 |
| Alexa Fluor 647-conjugated goat anti-rabbit | Invitrogen | Cat# A-21244; RRID: AB_2535812 |
| Alexa Fluor 647-conjugated goat anti-rat | Invitrogen | Cat# A-21247; RRID: AB_141778 |
| Alexa Fluor Plus 405-conjugated goat anti-mouse | Invitrogen | Cat# A48255; RRID: AB_2890536 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV5-Ef1a-DIO-mCherry | University of North Carolina Vector Core | Lot# AV4311D, AV4311E, AV4311F, AV4311G |
| AAV5-CaMKII-hChR2(H134R)-EYFP-WPRE-PA | University of North Carolina Vector Core | Lot# AV4316P, AV4316Q |
| AAVrg-hSyn-DIO-EGFP | Bryan Roth | Addgene viral prep # 50457-AAVrg |
| AAVrg-FLEX-tdTomato | Edward Boyden | Addgene viral prep # 28306-AAVrg |
| AAVrg-hSyn-HI-eGFP-Cre-WPRE-SV40 | James M. Wilson | Addgene viral prep # 105540-AAVrg |
| AAV8-CMV-LacZ-bGH | James M. Wilson | Addgene viral prep # 105531-AAV8 |
| AAV8-hSyn-DIO-hM4D(Gi)-mCherry | Krashes et al.129 | Addgene viral prep # 44362-AAV8 |
| AAV8-hSyn-DIO-mCherry | Bryan Roth | Addgene viral prep # 50459-AAV8 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DL-AP5 | Tocris | Cat# 0105; CAS# 76326–31-3 |
| CNQX disodium salt | Tocris | Cat# 1045; CAS# 479347–85-8 |
| BIBN4096BS | Millipore Sigma | Cat# SML2426; CAS# 204697–65-4 |
| Clozapine N-oxide | Enzo Life Sciences | Cat# BML-NS105; CAS# 34233–69-7 |
| Streptavidin, Alexa Fluor 405 conjugate | Invitrogen | Cat# S32351 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Rat: Crh-Cre | Robert Messing (Pomrenze et al.38) | N/A |
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Prkcd-Cre | GENSAT | RRID:MMRRC_011559-UCD |
| Mouse: Calcrl-Cre | Richard Palmiter (Han et al.47) | N/A |
| Mouse: Sst-Cre | Jackson Laboratory | RRID:IMSR_JAX:018973 |
| Mouse: Ai9 | Jackson Laboratory | RRID:IMSR_JAX:007909 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Forward primer to genotype for the presence of Cre-recombinase in rats: GCATTACCGGTCGATGCAACGAGTGATGAG | Integrated DNA Technologies (IDT) | www.idtdna.com |
| Reverse primer to genotype for the presence of Cre-recombinase in rats: GAGTGAACGAACCTGGTCGAAATCAGTGCG | Integrated DNA Technologies (IDT) | www.idtdna.com |
| Forward primer to genotype for the presence of Cre-recombinase in mice: TTAATCCATATTGGCAGAACGAAAACG | Transnetyx | https://www.transnetyx.com/ |
| Reverse primer to genotype for the presence of Cre-recombinase in mice: CAGGCTAAGTGCCTTCTCTACA | Transnetyx | https://www.transnetyx.com/ |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al.130 | RRID:SCR_003070;https://imagej.net/ |
| Fiji | Schindelin et al.131 | RRID:SCR_002285;https://fiji.sc/ |
| pCLAMP 8.2/pCLAMP 11.2/Clampfit 11.2 | Molecular Devices, Axon Instruments | RRID:SCR_011323;https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite |
| GraphPad Prism (version 9.5.1 or 10.0.2) | GraphPad Software | RRID:SCR_002798;https://www.graphpad.com/ |
| Anaconda Distribution version 2023. 09–0 (Python Version 3.11.5) | Anaconda, Inc. | RRID:SCR_008394;https://anaconda.com |
| Spyder version 5.4.3 | Spyder Project Contributors | RRID:SCR_017585;https://www.spyder-ide.org/ |
| matplotlib version 3.8.0 | Hunter et al.132 | RRID:SCR_008624;https://matplotlib.org/ |
| matplotlib-venn version 0.11.9 | Tretyakov133 | https://pypi.org/project/matplotlib-venn/0.11.9/ |
Highlights.
Synaptic plasticity at the PB→CeA pathway is lost in chronic neuropathic pain
Chemogenetic inhibition of the PB→CeA pathway inhibits acute but not chronic pain behaviors
CeA hyperexcitability shifts from CRF to non-CRF neurons at the chronic pain stage
CeA hyperexcitability no longer depends on PB→CeA synaptic plasticity in chronic pain
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 NS038261 and NS118731 (to V.N.) and the Intramural Research Program of the NIH National Center for Complementary and Integrative Health (to Y.C.). We thank Dr. Robert Messing (The University of Texas at Austin) for kindly providing the initial Crh-Cre transgenic rat breeding pairs. We wish to thank Dr. Richard Palmiter (University of Washington) for kindly providing the Calcrl-Cre mice. We also thank the National Institute of Neurological Disorders and Stroke mouse facility staff for their work in animal husbandry, the NIH Section on Instrumentation for their help with the electrophysiology equipment, and Dr. Jose-Luis Redondo for genotyping all the rats used in this study. Schematic illustrations were created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114669.
REFERENCES
- 1.Cramer SC, Sur M, Dobkin BH, O’Brien C, Sanger TD, Trojanowski JQ, Rumsey JM, Hicks R, Cameron J, Chen D, et al. (2011). Harnessing neuroplasticity for clinical applications. Brain 134, 1591–1609. 10.1093/brain/awr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhuriya YK, and Sharma D (2020). Neuronal Plasticity: Neuronal Organization is Associated with Neurological Disorders. J. Mol. Neurosci. 70, 1684–1701. 10.1007/s12031-020-01555-2. [DOI] [PubMed] [Google Scholar]
- 3.Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, Freeman R, Truini A, Attal N, Finnerup NB, et al. (2017). Neuropathic pain. Nat. Rev. Dis. Primers 3, 1–20. 10.1038/nrdp.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farmer MA, Baliki MN, and Apkarian AV (2012). A dynamic network perspective of chronic pain. Neurosci. Lett. 520, 197–203. 10.1016/j.neulet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breivik H, Eisenberg E, and O’Brien T; OPENMinds (2013). The individual and societal burden of chronic pain in Europe: The case for strategic prioritisation and action to improve knowledge and availability of appropriate care. BMC Publ. Health 13, 1229. 10.1186/1471-2458-13-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sá KN, Moreira L, Baptista AF, Yeng LT, Teixeira MJ, Galhardoni R, and de Andrade DC (2019). Prevalence of chronic pain in developing countries: systematic review and meta-analysis. Pain Rep. 4, e779. 10.1097/PR9.0000000000000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yong RJ, Mullins PM, and Bhattacharyya N (2022). Prevalence of chronic pain among adults in the United States. Pain 163, e328–e332. 10.1097/j.pain.0000000000002291. [DOI] [PubMed] [Google Scholar]
- 8.Phillips CJ (2006). Economic burden of chronic pain. Expert Rev. Pharmacoecon. Outcomes Res. 6, 591–601. 10.1586/14737167.6.5.591. [DOI] [PubMed] [Google Scholar]
- 9.Dueñas M, Ojeda B, Salazar A, Mico JA, and Failde I (2016). A review of chronic pain impact on patients, their social environment and the health care system. J. Pain Res. 9, 457–467. 10.2147/JPR.S105892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mutubuki EN, Beljon Y, Maas ET, Huygen FJPM, Ostelo RWJG, van Tulder MW, and van Dongen JM (2020). The longitudinal relationships between pain severity and disability versus health-related quality of life and costs among chronic low back pain patients. Qual. Life Res. 29, 275–287. 10.1007/s11136-019-02302-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apkarian VA, Hashmi JA, and Baliki MN (2011). Pain and the brain: Specificity and plasticity of the brain in clinical chronic pain. Pain 152, S49–S64. 10.1016/j.pain.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bushnell MC, Čeko M, and Low LA (2013). Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci. 14, 502–511. 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neugebauer V, Mazzitelli M, Cragg B, Ji G, Navratilova E, and Porreca F (2020). Amygdala, neuropeptides, and chronic pain-related affective behaviors. Neuropharmacology 170, 108052. 10.1016/j.neuropharm.2020.108052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato F, Sugimura YK, and Takahashi Y (2018). Pain-associated neural plasticity in the parabrachial to central amygdala circuit: Pain changes the brain, and the brain changes the pain. Adv. Exp. Med. Biol. 1099, 157–166. 10.1007/978-981-13-1756-9_14. [DOI] [PubMed] [Google Scholar]
- 15.Torres-Rodriguez JM, Wilson TD, Singh S, Torruella-Suárez ML, Chaudhry S, Adke AP, Becker JJ, Neugebauer B, Lin JL, Martinez Gonzalez S, et al. (2024). The parabrachial to central amygdala pathway is critical to injury-induced pain sensitization in mice. Neuropsy-chopharmacology 49, 508–520. 10.1038/s41386-023-01673-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neugebauer V, Li W, Bird GC, Bhave G, and Gereau RW 4th. (2003). Synaptic plasticity in the amygdala in a model of arthritic pain: Differential roles of metabotropic glutamate receptors 1 and 5. J. Neurosci. 23, 52–63. 10.1523/jneurosci.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han JS, and Neugebauer V (2004). Synaptic plasticity in the amygdala in a visceral pain model in rats. Neurosci. Lett. 361, 254–257. 10.1016/j.neulet.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 18.Ikeda R, Takahashi Y, Inoue K, and Kato F (2007). NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain 127, 161–172. 10.1016/j.pain.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Adedoyin MO, Vicini S, and Neale JH (2010). Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol. Pain 6, 60. 10.1186/1744-8069-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugimura YK, Takahashi Y, Watabe AM, and Kato F (2016). Synaptic and network consequences of monosynaptic nociceptive inputs of parabrachial nucleus origin in the central amygdala. J. Neurophysiol. 115, 2721–2739. 10.1152/jn.00946.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li JN, and Sheets PL (2018). The central amygdala to periaqueductal gray pathway comprises intrinsically distinct neurons differentially affected in a model of inflammatory pain. J. Physiol. 596, 6289–6305. 10.1113/JP276935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JN, and Sheets PL (2020). Spared nerve injury differentially alters parabrachial monosynaptic excitatory inputs to molecularly specific neurons in distinct subregions of the central amygdala. Pain 161, 166–176. 10.1097/j.pain.0000000000001691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson TD, Valdivia S, Khan A, Ahn HS, Adke AP, Martinez Gonzalez S, Sugimura YK, and Carrasquillo Y (2019). Dual and Opposing Functions of the Central Amygdala in the Modulation of Pain. Cell Rep. 29, 332–346.e5. 10.1016/j.celrep.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adke AP, Khan A, Ahn HS, Becker JJ, Wilson TD, Valdivia S, Sugimura YK, Martinez Gonzalez S, and Carrasquillo Y (2021). Cell-type specificity of neuronal excitability and morphology in the central Amygdala. eNeuro 8, 1–28. 10.1523/ENEURO.0402-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang H, Fang D, Kong LY, Jin ZR, Cai J, Kang XJ, Wan Y, and Xing GG (2014). Sensitization of neurons in the central nucleus of the amygdala via the decreased GABAergic inhibition contributes to the development of neuropathic pain-related anxiety-like behaviors in rats. Mol. Brain 7, 1–22. 10.1186/s13041-014-0072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samineni VK, Grajales-Reyes JG, Grajales-Reyes GE, Tycksen E, Copits BA, Pedersen C, Ankudey ES, Sackey JN, Sewell SB, Bruchas MR, and Gereau RW (2021). Cellular, circuit and transcriptional framework for modulation of itch in the central Amygdala. Elife 10, e68130. 10.7554/eLife.68130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Krabbe S, Eddison M, Henry FE, Fleishman G, Lemire AL, Wang L, Korff W, Tillberg PW, Lüthi A, and Sternson SM (2023). Multimodal mapping of cell types and projections in the central nucleus of the amygdala. Elife 12, e84262. 10.7554/eLife.84262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Leary TP, Kendrick RM, Bristow BN, Sullivan KE, Wang L, Clements J, Lemire AL, and Cembrowski MS (2022). Neuronal cell types, projections, and spatial organization of the central amygdala. iScience 25, 105497. 10.1016/j.isci.2022.105497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumont ÉC, Martina M, Samson RD, Drolet G, and Paré D (2002). Physiological properties of central amygdala neurons: Species differences. Eur. J. Neurosci. 15, 545–552. 10.1046/j.0953-816x.2001.01879.x. [DOI] [PubMed] [Google Scholar]
- 30.Lopez de Armentia M, and Sah P (2004). Firing properties and connectivity of neurons in the rat lateral central nucleus of the amygdala. J. Neurophysiol. 92, 1285–1294. 10.1152/jn.00211.2004. [DOI] [PubMed] [Google Scholar]
- 31.Schiess MC, Callahan PM, and Zheng H (1999). Characterization of the electrophysiological and morphological properties of rat central amygdala neurons in vitro. J. Neurosci. Res. 58, 663–673. . [DOI] [PubMed] [Google Scholar]
- 32.Chieng BCH, Christie MJ, and Osborne PB (2006). Characterization of neurons in the rat central nucleus of the amygdala: Cellular physiology, morphology, and opioid sensitivity. J. Comp. Neurol. 497, 910–927. 10.1002/cne.21025. [DOI] [PubMed] [Google Scholar]
- 33.Mazzitelli M, Marshall K, Pham A, Ji G, and Neugebauer V (2021). Optogenetic Manipulations of Amygdala Neurons Modulate Spinal Nociceptive Processing and Behavior Under Normal Conditions and in an Arthritis Pain Model. Front. Pharmacol. 12, 1–18. 10.3389/fphar.2021.668337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazzitelli M, Yakhnitsa V, Neugebauer B, and Neugebauer V (2022). Optogenetic manipulations of CeA-CRF neurons modulate pain- and anxiety-like behaviors in neuropathic pain and control rats. Neuropharmacology 210, 109031. 10.1016/j.neuropharm.2022.109031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakao A, Takahashi Y, Nagase M, Ikeda R, and Kato F (2012). Role of capsaicin-sensitive C-fiber afferents in neuropathic pain-induced synaptic potentiation in the nociceptive amygdala. Mol. Pain 8, 1–13. 10.1186/1744-8069-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Presto P, and Neugebauer V (2022). Sex Differences in CGRP Regulation and Function in the Amygdala in a Rat Model of Neuropathic Pain. Front. Mol. Neurosci. 15, 1–14. 10.3389/fnmol.2022.928587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Presto P, Ji G, Ponomareva O, Ponomarev I, and Neugebauer V (2023). Hmgb1 Silencing in the Amygdala Inhibits Pain-Related Behaviors in a Rat Model of Neuropathic Pain. Int. J. Mol. Sci. 24, 11944. 10.3390/ijms241511944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pomrenze MB, Millan EZ, Hopf FW, Keiflin R, Maiya R, Blasio A, Dadgar J, Kharazia V, De Guglielmo G, Crawford E, et al. (2015). A transgenic rat for investigating the anatomy and function of corticotrophin releasing factor circuits. Front. Neurosci. 9, 487. 10.3389/fnins.2015.00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yakhnitsa V, Ji G, Hein M, Presto P, Griffin Z, Ponomareva O, Navratilova E, Porreca F, and Neugebauer V (2022). Kappa Opioid Receptor Blockade in the Amygdala Mitigates Pain Like-Behaviors by Inhibiting Corticotropin Releasing Factor Neurons in a Rat Model of Functional Pain. Front. Pharmacol. 13, 903978. 10.3389/fphar.2022.903978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amano T, Amir A, Goswami S, and Paré D (2012). Morphology, PKCδ expression, and synaptic responsiveness of different types of rat central lateral amygdala neurons. J. Neurophysiol. 108, 3196–3205. 10.1152/jn.00514.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiritoshi T, and Neugebauer V (2018). Pathway-Specific Alterations of Cortico-Amygdala Transmission in an Arthritis Pain Model. ACS Chem. Neurosci. 9, 2252–2261. 10.1021/acschemneuro.8b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y, and Neugebauer V (2008). Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J. Neurosci. 28, 3861–3876. 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto S, Takahashi Y, and Kato F (2021). Input-dependent synaptic suppression by pregabalin in the central amygdala in male mice with inflammatory pain. Neurobiol. Pain 10, 100078. 10.1016/j.ynpai.2021.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwaber JS, Sternini C, Brecha NC, Rogers WT, and Card JP (1988). Neurons containing calcitonin gene-related peptide in the parabrachial nucleus project to the central nucleus of the amygdala. J. Comp. Neurol. 270, 416–426. 10.1002/cne.902700310. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Z, and Zamponi GW (2021). Protocol for detecting plastic changes in defined neuronal populations in neuropathic mice. STAR Protoc. 2, 100698. 10.1016/j.xpro.2021.100698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han JS, Li W, and Neugebauer V (2005). Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J. Neurosci. 25, 10717–10728. 10.1523/JNEUROSCI.4112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han S, Soleiman MT, Soden ME, Zweifel LS, and Palmiter RD (2015). Elucidating an Affective Pain Circuit that Creates a Threat Memory. Cell 162, 363–374. 10.1016/j.cell.2015.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu YC, Chen YZ, Wei YY, He XT, Li X, Hu W, Yanagawa Y, Wang W, Wu SX, and Dong YL (2015). Neurochemical properties of the synapses between the parabrachial nucleus-derived CGRP-positive axonal terminals and the GABAergic neurons in the lateral capsular division of central nucleus of amygdala. Mol. Neurobiol. 51, 105–118. 10.1007/s12035-014-8713-x. [DOI] [PubMed] [Google Scholar]
- 49.Shinohara K, Watabe AM, Nagase M, Okutsu Y, Takahashi Y, Kurihara H, and Kato F (2017). Essential role of endogenous calcitonin gene-related peptide in pain-associated plasticity in the central amygdala. Eur. J. Neurosci. 46, 2149–2160. 10.1111/ejn.13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugimoto M, Takahashi Y, Sugimura YK, Tokunaga R, Yajima M, and Kato F (2021). Active role of the central amygdala in widespread mechanical sensitization in rats with facial inflammatory pain. Pain 162, 2273–2286. 10.1097/j.pain.0000000000002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allen HN, Chaudhry S, Hong VM, Lewter LA, Sinha GP, Carrasquillo Y, Taylor BK, and Kolber BJ (2023). A Parabrachial-to-Amygdala Circuit That Determines Hemispheric Lateralization of Somatosensory Processing. Biol. Psychiatry 93, 370–381. 10.1016/j.biopsych.2022.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benbouzid M, Pallage V, Rajalu M, Waltisperger E, Doridot S, Poisbeau P, Freund-Mercier MJ, and Barrot M (2008). Sciatic nerve cuffing in mice: A model of sustained neuropathic pain. Eur. J. Pain 12, 591–599. 10.1016/j.ejpain.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Singh S, Wilson TD, Valdivia S, Benowitz B, Chaudhry S, Ma J, Adke AP, Soler-Cedeño O, Velasquez D, Penzo MA, and Carrasquillo Y (2022). An inhibitory circuit from central amygdala to zona incerta drives pain-related behaviors in mice. Elife 11, e68760. 10.7554/eLife.68760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim J, Zhang X, Muralidhar S, LeBlanc SA, and Tonegawa S (2017). Basolateral to Central Amygdala Neural Circuits for Appetitive Behaviors. Neuron 93, 1464–1479.e5. 10.1016/j.neuron.2017.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCullough KM, Morrison FG, Hartmann J, Carlezon WA, and Ressler KJ (2018). Quantified coexpression analysis of central amygdala subpopulations. eNeuro 5, 1–12. 10.1523/ENEURO.0010-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raver C, Uddin O, Ji Y, Li Y, Cramer N, Jenne C, Morales M, Masri R, and Keller A (2020). An amygdalo-parabrachial pathway regulates pain perception and chronic pain. J. Neurosci. 40, 3424–3442. 10.1523/JNEUROSCI.0075-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barroso J, Branco P, and Apkarian AV (2021). Brain mechanisms of chronic pain: critical role of translational approach. Transl. Res. 238, 76–89. 10.1016/j.trsl.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vachon-Presseau E, Centeno MV, Ren W, Berger SE, Tétreault P, Ghantous M, Baria A, Farmer M, Baliki MN, Schnitzer TJ, and Apkarian AV (2016). The emotional brain as a predictor and amplifier of chronic pain. J. Dent. Res. 95, 605–612. 10.1177/0022034516638027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neugebauer V (2020). Amygdala physiology in pain. In Handb BehavNeurosci, pp. 101–113. 10.1016/B978-0-12-815134-1.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu Y, Han J, Ishola T, Scerbo M, Adwanikar H, Ramsey C, and Neugebauer V (2008). PKA and ERK, but not PKC, in the amygdala contribute to pain-related synaptic plasticity and behavior. Mol. Pain 4, 26. 10.1186/1744-8069-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi Y, Ikeda R, and Kato F (2009). Synaptic potentiation in the central amygdala involves different mechanisms depending on pain model. Pain Res. 24, 137–146. [Google Scholar]
- 62.Ji G, Li Z, and Neugebauer V (2015). Reactive oxygen species mediate visceral pain-related amygdala plasticity and behaviors. Pain 156, 825–836. 10.1097/j.pain.0000000000000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandhi PJ, Gawande DY, Shelkar GP, Gakare SG, Kiritoshi T, and Dravid SM (2021). Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-synaptic Signaling in the Central Amygdala in Chronic Pain. Cells 10, 2644. 10.3390/cells10102644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Condon LF, Yu Y, Park S, Cao F, Pauli JL, Nelson TS, and Palmiter RD (2024). Parabrachial Calca neurons drive nociplasticity. Cell Rep. 43, 114057. 10.1016/j.celrep.2024.114057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fitzcharles MA, Cohen SP, Clauw DJ, Littlejohn G, Usui C, and Häuser W (2021). Nociplastic pain: towards an understanding of prevalent pain conditions. Lancet 397, 2098–2110. 10.1016/S0140-6736(21)00392-5. [DOI] [PubMed] [Google Scholar]
- 66.Nijs J, Lahousse A, Kapreli E, Bilika P, Saraçoğlu İ, Malfliet A, Coppieters I, De Baets L, Leysen L, Roose E, et al. (2021). Nociplastic pain criteria or recognition of central sensitization? Pain phenotyping in the past, present and future. J. Clin. Med. 10, 3203. 10.3390/jcm10153203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jackman SL, Beneduce BM, Drew IR, and Regehr WG (2014). Achieving high-frequency optical control of synaptic transmission. J. Neurosci. 34, 7704–7714. 10.1523/JNEUROSCI.4694-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson JM, Yakhnitsa V, Ji G, and Neugebauer V (2018). Small conductance calcium activated potassium (SK) channel dependent and independent effects of riluzole on neuropathic pain-related amygdala activity and behaviors in rats. Neuropharmacology 138, 219–231. 10.1016/j.neuropharm.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin YL, Yang ZS, Wong WY, Lin SC, Wang SJ, Chen SP, Cheng JK, Lu H, and Lien CC (2022). Cellular mechanisms underlying central sensitization in a mouse model of chronic muscle pain. Elife 11, e78610. 10.7554/eLife.78610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spierling SR, and Zorrilla EP (2017). Don’t stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology (Berl) 234, 1467–1481. 10.1007/s00213-017-4556-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ji G, Zhang W, Mahimainathan L, Narasimhan M, Kiritoshi T, Fan X, Wang J, Green TA, and Neugebauer V (2017). 5-HT2c receptor knockdown in the amygdala inhibits neuropathic-pain-related plasticity and behaviors. J. Neurosci. 37, 1378–1393. 10.1523/JNEUROSCI.2468-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou W, Jin Y, Meng Q, Zhu X, Bai T, Tian Y, Mao Y, Wang L, Xie W, Zhong H, et al. (2019). A neural circuit for comorbid depressive symptoms in chronic pain. Nat. Neurosci. 22, 1649–1658. 10.1038/s41593-019-0468-2. [DOI] [PubMed] [Google Scholar]
- 73.Zhou M, Liu Z, Melin MD, Ng YH, Xu W, and Südhof TC (2018). A central amygdala to zona incerta projection is required for acquisition and remote recall of conditioned fear memory. Nat. Neurosci. 21, 1515–1519. 10.1038/s41593-018-0248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hogri R, Teuchmann HL, Heinke B, Holzinger R, Trofimova L, and Sandkühler J (2022). GABAergic CaMKIIa1 Amygdala Output Attenuates Pain and Modulates Emotional-Motivational Behavior via Parabrachial Inhibition. J. Neurosci. 42, 5373–5388. 10.1523/JNEUROSCI.2067-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wank I, Pliota P, Badurek S, Kraitsy K, Kaczanowska J, Griessner J, Kreitz S, Hess A, and Haubensak W (2021). Central amygdala circuitry modulates nociceptive processing through differential hierarchical interaction with affective network dynamics. Commun. Biol. 4, 732. 10.1038/s42003-021-02262-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mork BE, Lamerand SR, Zhou S, Taylor BK, and Sheets PL (2022). Sphingosine-1-phosphate receptor 1 agonist SEW2871 alters membrane properties of late-firing somatostatin expressing neurons in the central lateral amygdala. Neuropharmacology 203, 108885. 10.1016/j.neuropharm.2021.108885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, and Bruchas MR (2015). CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 87, 605–620. 10.1016/j.neuron.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Asok A, Draper A, Hoffman AF, Schulkin J, Lupica CR, and Rosen JB (2018). Optogenetic silencing of a corticotropin-releasing factor pathway from the central amygdala to the bed nucleus of the stria terminalis disrupts sustained fear. Mol. Psychiatry 23, 914–922. 10.1038/mp.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Guglielmo G, Kallupi M, Pomrenze MB, Crawford E, Simpson S, Schweitzer P, Koob GF, Messing RO, and George O (2019). Inactivation of a CRF-dependent amygdalofugal pathway reverses addiction-like behaviors in alcohol-dependent rats. Nat. Commun. 10, 1238. 10.1038/s41467-019-09183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pomrenze MB, Tovar-Diaz J, Blasio A, Maiya R, Giovanetti SM, Lei K, Morikawa H, Hopf FW, and Messing RO (2019). A corticotropin releasing factor network in the extended amygdala for anxiety. J. Neurosci. 39, 1030–1043. 10.1523/JNEUROSCI.2143-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang C, Yang X, He G, Wang F, Wang Z, Xu W, Mao Y, Ma L, and Wang F (2021). CRHCeA→VTA inputs inhibit the positive ensembles to induce negative effect of opiate withdrawal. Mol. Psychiatry 26, 6170–6186. 10.1038/s41380-021-01321-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heinricher MM, Tavares I, Leith JL, and Lumb BM (2009). Descending control of nociception: Specificity, recruitment and plasticity. Brain Res. Rev. 60, 214–225. 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang C, Wang Y, Chen P, Shan QH, Wang H, Ding LF, Bi GQ, and Zhou JN (2022). Single-cell reconstruction reveals input patterns and pathways into corticotropin-releasing factor neurons in the central amygdala in mice. Commun. Biol. 5, 322. 10.1038/s42003-022-03260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith JA, Ji Y, Lorsung R, Breault MS, Koenig J, Cramer N, Masri R, and Keller A (2023). Parabrachial Nucleus Activity in Nociception and Pain in Awake Mice. J. Neurosci. 43, 5656–5667. 10.1523/JNEUROSCI.0587-23.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chiang MC, Nguyen EK, Canto-Bustos M, Papale AE, Oswald AMM, and Ross SE (2020). Divergent Neural Pathways Emanating from the Lateral Parabrachial Nucleus Mediate Distinct Components of the Pain Response. Neuron 106, 927–939.e5. 10.1016/j.neuron.2020.03.014. [DOI] [PubMed] [Google Scholar]
- 86.Pauli JL, Chen JY, Basiri ML, Park S, Carter ME, Sanz E, McKnight GS, Stuber GD, and Palmiter RD (2022). Molecular and anatomical characterization of parabrachial neurons and their axonal projections. Elife 11, e81868. 10.7554/eLife.81868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Torruella-Suárez ML, Neugebauer B, Flores-Felix K, Keller A, Carrasquillo Y, and Cramer N (2024). Divergent changes in PBN excitability in a mouse model of neuropathic pain. eNeuro 11, ENEURO.0416–23.2024. 10.1523/ENEURO.0416-23.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Corder G, Ahanonu B, Grewe BF, Wang D, Schnitzer MJ, and Scherrer G (2019). An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281. 10.1126/science.aap8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li Z, Wang J, Chen L, Zhang M, and Wan Y (2013). Basolateral Amygdala Lesion Inhibits the Development of Pain Chronicity in Neuropathic Pain Rats. PLoS One 8, e70921. 10.1371/journal.pone.0070921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qiu S, Chen T, Koga K, Guo YY, Xu H, Song Q, Wang JJ, Descalzi G, Kaang BK, Luo JH, et al. (2013). An increase in synaptic NMDA receptors in the insular cortex contributes to neuropathic pain. Sci. Signal. 6, ra34. 10.1126/scisignal.2003778. [DOI] [PubMed] [Google Scholar]
- 91.Liang SH, Zhao WJ, Yin JB, Chen YB, Li JN, Feng B, Lu YC, Wang J, Dong YL, and Li YQ (2020). A neural circuit from thalamic paraventricular nucleus to central amygdala for the facilitation of neuropathic pain. J. Neurosci. 40, 7837–7854. 10.1523/JNEUROSCI.2487-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, Darvas M, Parada LF, Palmiter RD, He M, et al. (2015). The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459. 10.1038/nature13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hartley ND, Gaulden AD, Báldi R, Winters ND, Salimando GJ, Rosas-Vidal LE, Jameson A, Winder DG, and Patel S (2019). Dynamic remodeling of a basolateral-to-central amygdala glutamatergic circuit across fear states. Nat. Neurosci. 22, 2000–2012. 10.1038/s41593-019-0528-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hunt S, Sun Y, Kucukdereli H, Klein R, and Sah P (2017). Intrinsic circuits in the lateral central amygdala. eNeuro 4, 1–18. 10.1523/ENEURO.0367-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, et al. (2010). Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468, 270–276. 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fadok JP, Krabbe S, Markovic M, Courtin J, Xu C, Massi L, Botta P, Bylund K, Müller C, Kovacevic A, et al. (2017). A competitive inhibitory circuit for selection of active and passive fear responses. Nature 542, 96–100. 10.1038/nature21047. [DOI] [PubMed] [Google Scholar]
- 97.Chen WH, Lien CC, and Chen CC (2022). Neuronal basis for pain-like and anxiety-like behaviors in the central nucleus of the amygdala. Pain 163, E463–E475. 10.1097/j.pain.0000000000002389. [DOI] [PubMed] [Google Scholar]
- 98.Donnelly CR, Andriessen AS, Chen G, Wang K, Jiang C, Maixner W, and Ji RR (2020). Central Nervous System Targets: Glial Cell Mechanisms in Chronic Pain. Neurotherapeutics 17, 846–860. 10.1007/s13311-020-00905-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shen CL, Castro L, Fang CY, Castro M, Sherali S, White S, Wang R, and Neugebauer V (2022). Bioactive compounds for neuropathic pain: An update on preclinical studies and future perspectives. J. Nutr. Biochem. 104, 108979. 10.1016/j.jnutbio.2022.108979. [DOI] [PubMed] [Google Scholar]
- 100.Yuan T, Orock A, and Greenwood-Van Meerveld B (2021). Amygdala microglia modify neuronal plasticity via complement C1q/C3-CR3 signaling and contribute to visceral pain in a rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 320, G1081–G1092. 10.1152/AJPGI.00123.2021. [DOI] [PubMed] [Google Scholar]
- 101.Yuan T, Orock A, and Greenwood-VanMeerveld B (2022). An enriched environment reduces chronic stress-induced visceral pain through modulating microglial activity in the central nucleus of the amygdala. Am. J. Physiol. Gastrointest. Liver Physiol. 322, G223–G233. 10.1152/ajpgi.00307.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yuan T, Manohar K, Latorre R, Orock A, and Greenwood-Van Meerveld B (2020). Inhibition of Microglial Activation in the Amygdala Reverses Stress-Induced Abdominal Pain in the Male Rat. Cell. Mol. Gastroenterol. Hepatol. 10, 527–543. 10.1016/j.jcmgh.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wahis J, Baudon A, Althammer F, Kerspern D, Goyon S, Hagiwara D, Lefevre A, Barteczko L, Boury-Jamot B, Bellanger B, et al. (2021). Astrocytes mediate the effect of oxytocin in the central amygdala on neuronal activity and affective states in rodents. Nat. Neurosci. 24, 529–541. 10.1038/s41593-021-00800-0. [DOI] [PubMed] [Google Scholar]
- 104.Dworsky-Fried Z, Faig CA, Vogel HA, Kerr BJ, and Taylor AMW (2022). Central amygdala inflammation drives pain hypersensitivity and attenuates morphine analgesia in experimental autoimmune encephalomyelitis. Pain 163, E49–E61. 10.1097/j.pain.0000000000002307. [DOI] [PubMed] [Google Scholar]
- 105.Nascimento GC, Lucas G, and Leite-Panissi CRA (2023). Emerging role of microglia and astrocyte in the affective-motivational response induced by a rat model of persistent orofacial pain. Brain Res. Bull. 195, 86–98. 10.1016/j.brainresbull.2023.02.005. [DOI] [PubMed] [Google Scholar]
- 106.Santos JM, Wang R, Bhakta V, Driver Z, Vadim Y, Kiritoshi T, Ji G, Neugebauer V, and Shen CL (2023). Turmeric Bioactive Compounds Alleviate Spinal Nerve Ligation-Induced Neuropathic Pain by Suppressing Glial Activation and Improving Mitochondrial Function in Spinal Cord and Amygdala. Nutrients 15, 4403. 10.3390/nu15204403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shen CL, Wang R, Yakhnitsa V, Santos JM, Watson C, Kiritoshi T, Ji G, Hamood AN, and Neugebauer V (2022). Gingerol-Enriched Ginger Supplementation Mitigates Neuropathic Pain via Mitigating Intestinal Permeability and Neuroinflammation: Gut-Brain Connection. Front. Pharmacol. 13, 912609. 10.3389/fphar.2022.912609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mazzitelli M, Ponomareva O, Presto P, John J, and Neugebauer V (2024). Impaired amygdala astrocytic signaling worsens neuropathic pain-associated neuronal functions and behaviors. Front. Pharmacol. 15, 1368634. 10.3389/fphar.2024.1368634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen D, Lou Q, Song XJ, Kang F, Liu A, Zheng C, Li Y, Wang D, Qun S, Zhang Z, et al. (2024). Microglia govern the extinction of acute stress-induced anxiety-like behaviors in male mice. Nat. Commun. 15, 449. 10.1038/s41467-024-44704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bolton JL, Short AK, Othy S, Kooiker CL, Shao M, Gunn BG, Beck J, Bai X, Law SM, Savage JC, et al. (2022). Early stress-induced impaired microglial pruning of excitatory synapses on immature CRH-expressing neurons provokes aberrant adult stress responses. Cell Rep. 38, 110600. 10.1016/j.celrep.2022.110600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ngozi Z, and Bolton JL (2022). Microglia Don’t Treat All Neurons the Same: The Importance of Neuronal Subtype in Microglia-Neuron Interactions in the Developing Hypothalamus. Front. Cell. Neurosci. 16, 867217. 10.3389/fncel.2022.867217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stevens SL, Shaw TE, Dykhuizen E, Lessov NS, Hill JK, Wurst W, and Stenzel-Poore MP (2003). Reduced cerebral injury in CRH-R1 deficient mice after focal ischemia: A potential link to microglia and atrocytes that express CRH-R1. J. Cereb. Blood Flow Metab. 23, 1151–1159. 10.1097/01.WCB.0000086957.72078.D4. [DOI] [PubMed] [Google Scholar]
- 113.Wang W, Ji P, Riopelle RJ, and Dow KE (2002). Functional expression of corticotropin-releasing hormone (CRH) receptor 1 in cultured rat microglia. J. Neurochem. 80, 287–294. 10.1046/j.0022-3042.2001.00687.x. [DOI] [PubMed] [Google Scholar]
- 114.Kissiwaa SA, Patel SD, Winters BL, and Bagley EE (2020). Opioids differentially modulate two synapses important for pain processing in the amygdala. Br. J. Pharmacol. 177, 420–431. 10.1111/bph.14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sato N, Takahashi Y, Sugimura YK, and Kato F (2024). Presynaptic inhibition of excitatory synaptic transmission from the calcitonin gene-related peptide-containing parabrachial neurons to the central amygdala in mice – unexpected influence of systemic inflammation thereon. J. Pharmacol. Sci. 154, 264–273. 10.1016/j.jphs.2024.02.004. [DOI] [PubMed] [Google Scholar]
- 116.Paretkar T, and Dimitrov E (2019). Activation of enkephalinergic (Enk) interneurons in the central amygdala (CeA) buffers the behavioral effects of persistent pain. Neurobiol. Dis. 124, 364–372. 10.1016/j.nbd.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ho Kim S, and Mo Chung J (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363. 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 118.Yalcin I, Megat S, Barthas F, Waltisperger E, Kremer M, Salvat E, and Barrot M (2014). The sciatic nerve cuffing model of neuropathic pain in mice. J. Vis. Exp, 51608. 10.3791/51608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jo YS, Namboodiri VMK, Stuber GD, and Zweifel LS (2020). Persistent activation of central amygdala CRF neurons helps drive the immediate fear extinction deficit. Nat. Commun. 11, 422. 10.1038/s41467-020-14393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Griessner J, Pasieka M, Böhm V, Grössl F, Kaczanowska J, Pliota P, Kargl D, Werner B, Kaouane N, Strobelt S, et al. (2021). Central amygdala circuit dynamics underlying the benzodiazepine anxiolytic effect. Mol. Psychiatry 26, 534–544. 10.1038/s41380-018-0310-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cantu DJ, Kaur S, Murphy AZ, and Averitt DL (2022). Sex differences in the amygdaloid projections to the ventrolateral periaqueductal gray and their activation during inflammatory pain in the rat. J. Chem. Neuroanat. 124, 102123. 10.1016/j.jchemneu.2022.102123. [DOI] [PubMed] [Google Scholar]
- 122.Allen HN, Bobnar HJ, and Kolber BJ (2021). Left and right hemispheric lateralization of the amygdala in pain. Prog. Neurobiol. 196, 101891. 10.1016/j.pneurobio.2020.101891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Barroso J, Branco P, Pinto-Ramos J, Vigotsky AD, Reis AM, Schnitzer TJ, Galhardo V, and Apkarian AV (2023). Subcortical brain anatomy as a potential biomarker of persistent pain after total knee replacement in osteoarthritis. Pain 164, 2306–2315. 10.1097/j.pain.0000000000002932. [DOI] [PubMed] [Google Scholar]
- 124.Gonçalves L, and Dickenson AH (2012). Asymmetric time-dependent activation of right central amygdala neurones in rats with peripheral neuropathy and pregabalin modulation. Eur. J. Neurosci. 36, 3204–3213. 10.1111/j.1460-9568.2012.08235.x. [DOI] [PubMed] [Google Scholar]
- 125.Presto P, Mazzitelli M, Junell R, Griffin Z, and Neugebauer V (2022). Sex differences in pain along the neuraxis. Neuropharmacology 210, 109030. 10.1016/j.neuropharm.2022.109030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kang SJ, Liu S, Ye M, Kim DI, Pao GM, Copits BA, Roberts BZ, Lee KF, Bruchas MR, and Han S (2022). A central alarm system that gates multi-sensory innate threat cues to the amygdala. Cell Rep. 40, 111222. 10.1016/j.celrep.2022.111222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mogil JS (2012). Sex differences in pain and pain inhibition: Multiple explanations of a controversial phenomenon. Nat. Rev. Neurosci. 13, 859–866. 10.1038/nrn3360. [DOI] [PubMed] [Google Scholar]
- 128.Melchior M, Poisbeau P, Gaumond I, and Marchand S (2016). Insights into the mechanisms and the emergence of sex-differences in pain. Neuroscience 338, 63–80. 10.1016/j.neuroscience.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 129.Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest. 121, 1424–1428. 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 133.Tretyakov K (2017). matplotlib-venn: Functions for plotting area-proportional two-and three-way venn diagrams in matplotlib. https://pypi.org/project/matplotlib-venn/0.11.9/.
- 134.Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, and Heintz N (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925. 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 135.Paxinos G, and Watson C (1998). The Rat Brain in Stereotaxic Coordinates (Academic Press). [DOI] [PubMed] [Google Scholar]
- 136.Bigelow LJ, Pope EK, MacDonald DS, Rock JE, and Bernard PB (2023). Getting a handle on rat familiarization: The impact of handling protocols on classic tests of stress in Rattus norvegicus. Lab. Anim. 57, 259–269. 10.1177/00236772221142687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rigaud M, Gemes G, Barabas ME, Chernoff DI, Abram SE, Stucky CL, and Hogan QH (2008). Species and strain differences in rodent sciatic nerve anatomy: Implications for studies of neuropathic pain. Pain 136, 188–201. 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ye GL, Savelieva KV, Vogel P, Baker KB, Mason S, Lanthorn TH, and Rajan I (2015). Ligation of mouse L4 and L5 spinal nerves produces robust allodynia without major motor function deficit. Behav. Brain Res. 276, 99–110. 10.1016/j.bbr.2014.04.039. [DOI] [PubMed] [Google Scholar]
- 139.Hurst JL, and West RS (2010). Taming anxiety in laboratory mice. Nat. Methods 7, 825–826. 10.1038/nmeth.1500. [DOI] [PubMed] [Google Scholar]
- 140.Carrasquillo Y, and Gereau RW 4th. (2007). Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J. Neurosci. 27, 1543–1551. 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McKernan MG, and Shinnick-Gallagher P (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611. 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 142.Hein M, Ji G, Tidwell D, D’Souza P, Kiritoshi T, Yakhnitsa V, Navratilova E, Porreca F, and Neugebauer V (2021). Kappa opioid receptor activation in the amygdala disinhibits CRF neurons to generate pain-like behaviors. Neuropharmacology 185, 108456. 10.1016/j.neuropharm.2021.108456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Paxinos G, and Franklin K (2001). The Mouse Brain in Stereotaxic Coordinates (Academic Press; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.