

Abstract
Alcohol use disorder has been associated with the development of neurodegenerative diseases, including Alzheimer’s disease (AD). However, recent studies demonstrate that moderate alcohol consumption may be protective against dementia and cognitive decline. We examined astrocyte function, low-density lipoprotein (LDL) receptor-related protein 1 (LRP1), and the NF-κB p65 and IKK-α/β signaling pathways in modulating neuroinflammation and amyloid beta (Aβ) deposition. We assessed apolipoprotein E (ApoE) in the brain of APP/PS1 mice using IHC and ELISA in response to low to moderate ethanol exposure (MEE). First, to confirm the intracerebral distribution of ApoE, we co-stained with GFAP, a marker for astrocytes that biosynthesize ApoE. We sought to investigate whether the ethanol-induced upregulation of LRP1 could potentially inhibit the activity of IL-1β and TNF-α induced IKK-α/β towards NF-κB p65, resulting in a reduction of pro-inflammatory cytokines. To evaluate the actual Aβ load in the brains of APP/PS1 mice, we performed with a specific antibody Aβ (Thioflavin S) on both air- and ethanol-exposed groups, subsequently analyzing Aβ levels. We also measured glucose uptake using 18F- fluorodeoxyglucose (FDG)-positron emission tomography (PET). Finally, we investigated whether MEE induced cognitive and memory changes using the Y maze, noble object recognition test, and Morris water maze. Our findings demonstrate that MEE reduced astrocytic glial fibrillary acidic protein (GFAP) and ApoE levels in the cortex and hippocampus in presymptomatic APP/PS1 mice. Interestingly, increased LRP1 protein expression was accompanied by dampening the IKK-α/β-NF-κB p65 pathway, resulting in decreased IL-1β and TNF-α levels in male mice. Notably, female mice show reduced levels of anti-inflammatory cytokines IL-4, and IL-10 without altering IL-1β and TNF-α concentrations. In both males and females, Aβ plaques, a hallmark of AD, were reduced in the cortex and hippocampus of APP/PS1 mice exposed to ethanol starting at pre-symptomatic stage. Consistently, MEE increased FDG-PET-based brain activities and normalized cognitive and memory deficits in the APP/PS1 mice. Our findings suggest that MEE may benefit AD pathology via modulating LRP1 expression, potentially reducing neuroinflammation and attenuating Aβ deposition. Our study implies that reduced astrocyte-derived ApoE and LDL cholesterol levels are critical for attenuating AD pathology.
Subject terms: Alzheimer's disease, Cognitive ageing
Introduction
Alcohol use disorder (AUD) has been associated with the development of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD)1. Furthermore, chronic and excessive alcohol consumption is a crucial risk factor for cognitive decline and alcohol-related dementia (ARD)2,3. Recent studies have shown that excessive alcohol consumption reduces cognitive function in reversal learning4,5, indicating that excessive alcohol drinking dampens the ability to adapt to environmental changes6. On the other hand, it has been known that moderate alcohol consumption is possibly protective against cognitive decline7–10. However, it remains unclear how low to moderate ethanol exposure (MEE) prevents the accumulation of amyloid-beta (Aβ) peptides, reducing plaque formation in the brain11 and protecting hippocampal neurons from Aβ toxicity12.
APP/PS1 mice, a model of early-onset AD, have been used to study the molecular mechanisms of AD progression and therapeutic interventions13–17. We extensively characterized APP/PS1 mice, demonstrating that they recapitulate some aspects of human AD, including mitochondrial dysfunction, altered energy homeostasis15, progressive accumulation of amyloid plaques18, and cognitive dysfunction14,16,17. In addition, APP/PS1 mice are commonly used in studying ethanol-induced AD pathology19,20.
Cortical and hippocampal astrocyte reactivity is related to the development of AD21. Astrocytes synthesize ApoE and cholesterol, regulating cholesterol-dependent signaling in the brain22. In humans, the astrocytic ApoE4 allele, a genetic risk factor for AD, contributes to amyloidosis in neurons through increased ApoE4-derived cholesterol levels23. Moreover, astrocyte-derived cholesterol controls Aβ accumulation in vivo, linking ApoE, Aβ, and plaque formation, further underscoring astrocytes’ critical role in AD progression24.
Cholesterol is known to regulate amyloid deposition in AD pathology25,26. Apolipoproteins play a crucial role in cholesterol transport and metabolism, and any changes in their levels can result in dysregulation of lipid homeostasis in AD27,28. Astrocytes express several critical enzymes involved in cholesterol biosynthesis, such as 3-hydroxy-3-methyl glutaryl-CoA (HMG-CoA) reductase, and the uptake of these cholesterol-containing particles is dependent upon the low-density lipoprotein receptor (LDLR) in the plasma membrane29. Interestingly, LDLR-related protein (LRP) is involved in various cellular processes such as lipid metabolism, cell migration, and endocytosis30. Particularly, LRP1 activation inhibits Aβ aggregation31,32 and down-regulates NF-κB-dependent proinflammatory cytokines33,34, which are critical for AD pathology.
To determine a possible correlation between MEE and AD pathology, we examined how MEE alters cognitive function through LDL cholesterol, neuroinflammation, and Aβ deposition in age- and sex-dependent manner in APP/PS1 mice. Our data demonstrate the differential effect of MEE on AD pathology and suggest potential therapeutic targets.
Results
Low to moderate ethanol exposure reduces ApoE and LDL cholesterol levels in presymptomatic APP/PS1
To examine the impact of ethanol exposure in the age- and sex-dependent early onset AD mouse model, we used two different groups where ethanol exposure started at presymptomatic age, before the accumulation of Aβ plaques, between 12 and 24 weeks of age (molecular and behavior experiments were conducted between 24 and 26 weeks of age) and after the onset of the symptoms, between 36 and 48 weeks of age (molecular and behavior experiments were conducted between 48 and 50 weeks of age). Both male and female APP/PS1 mice were exposed to ethanol vapor or room air using vapor chambers for 4 h from 9 am to 1 pm, followed by 20 h of room air in their home cages to mimic alcohol drinking patterns in humans. This process was repeated for four consecutive days per week, followed by 3 days in their home cages with room air (withdrawal period) for 12 weeks to examine the long-term effect of alcohol exposure (Fig. S1A and B). After completing the 12-week cycle, we examined the effect of ethanol exposure as outlined in Fig. S1A and B. We found no weight loss due to the influence of ethanol in both male and female presymptomatic or symptomatic APP/PS1 mice (Fig. S1C and D). The average blood alcohol concentration (BAC) immediately after the vapor chamber was approximately 170 mg/dl (Fig. S1C and D).
In AD, astrocytes surrounding amyloid plaques become reactive, indicating neuroinflammation, a key feature of AD pathology24. Since astrocytes express ApoE, which is critical for Aβ clearance35, we first measured ApoE levels using immunohistochemistry (IHC) and enzyme-linked immunosorbent assay (ELISA) in response to MEE in presymptomatic APP/PS1 mice. To confirm the intracerebral distribution of ApoE, brain tissues were co-stained with GFAP, a marker for astrocytes that biosynthesize ApoE (Fig. 1A). Interestingly, we found a reduction of ApoE and astrocytes in the cortex (Fig. 1B and D) and hippocampus (Fig. 1C and E) of ethanol-exposed presymptomatic APP/PS1 mice compared to air-exposed counterparts. Consistently, we confirmed the reduced ApoE protein levels in ELISA experiments in the cortex (Fig. 1F) and hippocampus (Fig. 1J) of ethanol-exposed presymptomatic APP/PS1 mice. We found no sex-specific differences in GFAP and ApoE levels (Fig. S3A-C).
Fig. 1.

Impact of low to moderate ethanol exposure on ApoE and LDL cholesterol levels in the brains of presymptomatic APP/PS1 mice. (A) Representative immunohistochemistry images of ApoE (green) and GFAP (red) co-staining in the brains of air-exposed and ethanol-exposed APP/PS1 mice. (B and C) ApoE IHC evaluation revealed reduced ApoE levels in the cortex (B) and hippocampus (C) compared to the air exposure group. (D and E) GFAP IHC evaluation showed decreased astrocyte activation in the cortex (D) and hippocampus (E) in the ethanol exposure compared to the air group. (F–H) and (J–L) Analysis of ApoE, LDL-cholesterol, and Total-cholesterol levels in the cortex and hippocampus by ELISA after low to moderate ethanol exposure. (F) ApoE level in the cortex, G) LDL-cholesterol level in the cortex, (H) Total cholesterol level in the cortex, (I) Correlation in the cortex, (J) ApoE level in the hippocampus, (K) LDL cholesterol level in the hippocampus, (L) Total cholesterol level in the hippocampus. (M) Correlation in the hippocampus. Data represent mean ± SEM; n = 10 per group. *P < 0.05 comparing each group. (B-I and K-N) Two-tailed Mann–Whitney test. (I and M) Spearman correlation analysis. Linear regression (solid line) and 95% confidence bands (shaded are) are shown. See Table S1 for full statistical information.
Since ApoE is an essential protein involved in cholesterol transport and late-onset AD36,37, we examined whether MEE alters LDL-cholesterol levels in the brain. Interestingly, ethanol-exposed presymptomatic APP/PS1 mice have lower LDL-cholesterol levels than air-exposed counterparts (Fig. 1G, H, K, and L). However, the HDL-cholesterol levels did not differ between the air- and ethanol-exposed groups (Fig. S4A-H). A linear correlation of LDL cholesterol levels and ApoE (Fig. 1I and M) suggests that MEE reduces the biosynthesis of ApoE in astrocytes, which in turn causes a decrease in LDL cholesterol. On the other hand, we found no sex-specific difference (Fig. S5A-C).
Ethanol exposure increases levels of LRP1 in APP/PS1 mice
Since reduced ApoE and LDL cholesterol levels38 are inversely associated with LRP1 expression, which regulates Aβ clearance in response to ApoE39–41, we examined LDLR-related protein 1 (LRP1) levels in air- and ethanol-exposed groups. We found a marked increase in LRP1 protein levels in the cortex (Fig. 2A and C) and hippocampus (Fig. 2B and D) of ethanol-exposed presymptomatic APP/PS1 mice compared to air-exposed counterparts. Consistent with the previous findings, these results confirm the increase of LRP1 in response to reduced ApoE42 or decreased LDL cholesterol levels36.
Fig. 2.

Effects of low to moderate ethanol exposure from IKK-α/β to IκB-α on LRP1 expression and NF-κB signaling in presymptomatic APP/PS1 mice. (A and B) Western blot quantification of LRP1/IKK-α/β/IκB α/NF-κB signaling. (C and D) Western blot results of LRP1/IKK-α/β/IκB-α/NF-κB in the cortex and hippocampus of the presymptomatic APP/PS1 mice brains. β-Actin was used as a loading control. (A and C) Cortex, (B and D) Hippocampus. Data represent mean ± SEM; n = 5 per group. *P < 0.05 comparing each group. Two-tailed Mann–Whitney test. See Fig. S17 for a full Western blot and see Table S1 for full statistical information.
Low to moderate ethanol exposure reduces IL-1β & TNF-α via LRP1 –IKKα/β – NF-κB p65 pathway
Next, we investigated whether the ethanol-induced upregulation of LRP1 reduces proinflammatory cytokines since LRP1 is known to down-regulate NF-κB-dependent cytokine expressions33,43. First, we examined ten different cytokines and found that levels of IL-1β and TNF-α were reduced in the serum of ethanol-exposed presymptomatic males but not in female APP/PS1 mice (Fig. S6A-J). IL-1β and TNF-α are cytokines that may promote inflammation by activating the transcription factor NF-κB p6542. Remarkably, we observed a reduction in p-IKK-α/β levels in ethanol-exposed presymptomatic APP/PS1 mice compared to air-exposed counterparts (Fig. 2A-D). This decrease in p-IKK-α/β led to a corresponding reduction in NF-κB p65 protein levels (Fig. 2A-D). We further assessed the IL-1β and TNF-α levels in the cortex and hippocampus using ELISA (Fig. 3). After 12 weeks of ethanol exposure, as expected, we found reduced levels of IL-1β and TNF-α in the cortex and hippocampus of ethanol-exposed presymptomatic APP/PS1 mice (Fig. 3A). Surprisingly, we found that IL-1β and TNF-α levels were reduced only in male but not in female mice (Fig. 3B and C). Instead, we found an upregulation of IL-10, an anti-inflammatory cytokine, in the hippocampus of ethanol-exposed presymptomatic APP/PS1 female mice (Fig. S7A-D).
Fig. 3.

Low to moderate ethanol exposure significantly reduces IL-1β and TNF-α levels in both brain regions compared to air-exposed presymptomatic APP/PS1 mice. (A-C) ELISA assay to detect IL-1β and TNF-α levels in the cortex and hippocampus of presymptomatic APP/PS1 mice. (A) Comparison of air and ethanol groups including male and female (B) Comparison of only male mice air and ethanol groups (C) Comparison of only female mice air and ethanol groups. Data represent mean ± SEM; n = 5 per group. *P < 0.05 comparing each group. Two-tailed Mann–Whitney test. See Table S1 for full statistical information.
Low to moderate ethanol exposure reduced Aβ plaques in presymptomatic APP/PS1 mice
We next examined whether MEE could potentially mitigate the deposition of Aβ plaques44,45. We utilized a simple and straightforward thioflavin S staining to evaluate the Aβ load in the brains of presymptomatic APP/PS1 mice46,47. We quantified the number of Aβ plaques, including intra-neuronal Aβ and extracellular plaques. We compared these in the cortex and dentate gyrus of the hippocampus across all experimental presymptomatic APP/PS1 mice (Fig. 4A-B). Our results revealed a notable reduction in Aβ plaques in the cortex (Fig. 4C) and hippocampus (Fig. 4D) in both presymptomatic APP/PS1 males and females (Fig. S8A-C) in response to ethanol exposure compared to air-exposed counterparts.
Fig. 4.
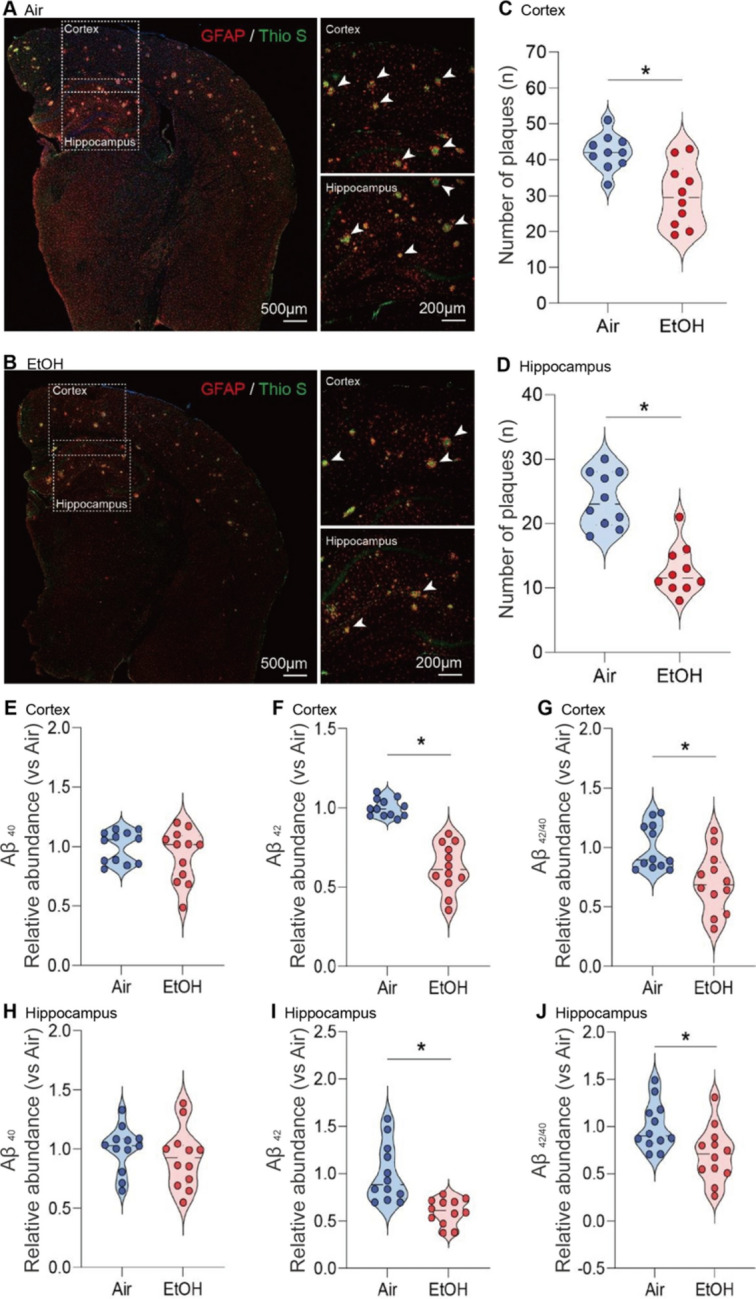
Reduction of Aβ plaque formation in the cortex and hippocampus of ethanol-exposed presymptomatic APP/PS1 mice. (A-D) Immunohistochemistry analysis shows the effects of MEE on Aβ plaque formation in the cortex and hippocampus of presymptomatic APP/PS1 mice. (A and B) Aβ plaques were visualized using Thioflavin S staining. (C and D) the number of plaques was significantly decreased in both cortex and hippocampus of ethanol-exposed presymptomatic APP/PS1 mice compared to air-exposed controls. (E-J) ELISA analysis shows the effects of chronic ethanol exposure on Aβ levels in the cortex and hippocampus of presymptomatic APP/PS1 mice. Quantification of amyloid Aβ1-40,42 levels using ELISA revealed a significant reduction in the cortex (E–G) and hippocampus (H-J) of ethanol-exposed presymptomatic APP/PS1 mice after 12 weeks of ethanol exposure compared to age-matched air-exposed presymptomatic APP/PS1 mice. Data represent mean ± SEM; n = 10 per group. *P < 0.05 comparing each group. Two-tailed Mann–Whitney test. See Table S1 for full statistical information.
We next performed an ELISA assay to detect Aβ42 in brain tissue supernatants collected from both experimental groups to quantify the level of amyloid protein in the brain. At 12 weeks of ethanol exposure, Aβ42 levels were markedly reduced in the cortex and hippocampus of ethanol-exposed presymptomatic APP/PS1 mice compared to air-exposed presymptomatic APP/PS1 mice (Fig. 4E-J). Consistently, we confirmed that a reduction of Aβ plaque formation and ApoE in the cortex and hippocampus of ethanol-exposed presymptomatic APP/PS1 mice (Fig. S8). Interestingly, we found no sex-specific differences in Aβ plaques (Fig. S9A-C), indicating that two different neuroinflammatory signaling pathways yield similar effects on the Aβ plaques in presymptomatic APP/PS1 mice. In addition, we observed no notable differences in Aβ levels in ethanol-exposed symptomatic APP/PS1 mice (Fig. S10A-D).
Ethanol exposure increases brain metabolism in presymptomatic APP/PS1 mice
The fluorodeoxyglucose-positron emission tomography (FDG-PET) measures glucose metabolism and brain activity48. Thus, we performed the micro-PET experiments using the18F-FDG radiotracer to examine whether MEE alters brain activity. We noted a higher uptake of18F-FDG in the ethanol-exposed presymptomatic APP/PS1 mice compared to the air-exposed presymptomatic APP/PS1 mice, especially in the cortex and hippocampus (Fig. 5A-C). The effect of alcohol was the same in male and female mice (Fig. S11A). However, in symptomatic APP/PS1 mice, ethanol exposure did not change FDG uptake (Fig. S12A-C), indicating that MEE increases brain activity only in presymptomatic APP/PS1 mice without apparent impact on symptomatic APP/PS1 mice.
Fig. 5.

[18F] Fluorodeoxyglucose uptake measured by in vivo microPET. (A) Representative FDG-PET images from air-exposed and ethanol-exposed presymptomatic APP/PS1 mice. The boundaries of ROIs were drawn on the coronal section. Glucose uptake increased in the cortex and hippocampus of ethanol exposed presymptomatic APP/PS1 mice. Quantification of glucose uptake by FDG-PET imaging in the cortex (B) and hippocampus (C). Data represent mean ± SEM; n = 10 per group. *P < 0.05 comparing each group. Two-tailed Mann–Whitney test. See Table S1 for full statistical information.
Low to moderate ethanol exposure restores cognitive, memory, and reversal learning deficits in presymptomatic APP/PS1 mice
Finally, we investigated whether MEE could rescue cognitive and memory functions in presymptomatic APP/PS1 mice. Our primary analysis involved evaluating the impact of ethanol exposure on spatial and procedural working memories in the Y-maze (Fig. 6A). Notably, we found an enhanced spontaneous alteration in the ethanol-exposed presymptomatic APP/PS1 compared to the air-exposed presymptomatic APP/PS1 mice (Fig. 6B) without changes in a total number of arm entries (Fig. 6C). In novel object recognition (NOR) task (Fig. 6D), the ethanol-exposed presymptomatic APP/PS1 mice exhibited a prolonged exploration time of the unfamiliar object (Fig. 6E). In contrast, air-exposed presymptomatic APP/PS1 mice displayed similar exploration times for both familiar and unfamiliar objects (Fig. 6F) without differences in total exploration time, speed, total distance, and frequency (Fig. S13). Importantly, as reported previously 49–51, we noted that presymptomatic APP/PS1 mice exhibited cognitive and memory decline in Y-maze and NOR compared to aged-matched non-transgenic (NTG) mice. Interestingly, we found that MEE does not affect cognitive performance in NTG mice (Fig. S14). Also, symptomatic APP/PS1 mice exhibited no difference in cognitive and memory function in response to MEE (Fig. S15). Our findings indicate that MEE rescues or normalizes cognitive function in presymptomatic APP/PS1 mice without affecting NTG or symptomatic APP/PS1 mice.
Fig. 6.

Effect of low to moderate ethanol exposure on cognitive performance in presymptomatic APP/PS1 mice. (A) Y-maze schematic diagram, (B) total arm entries and (C) Y-maze test showing spontaneous alteration for air-exposed and ethanol-exposed presymptomatic APP/PS1 mice. (D) Novel Object Recognition (NOR) task schematic diagram, (E and F) NOR task demonstrating time spent exploring familiar and unfamiliar objects for both groups. (G) Morris water maze (MWM) schematic diagram, (H and I) MWM acquisition training and probe test (J) reversal probe test. Data represent mean ± SEM; n = 12 ~ 28 per group. *P < 0.05 comparing each group. (B-C and E–F) Two-tailed Mann–Whitney test (H and J) Two-way ANOVA followed by Tukey’s multiple comparisons tests. See Table S1 for full statistical information.
Next, we tested spatial reference and working memory in the Morris water maze (MWM) (Fig. 6G). As previously published47, we measured spatial learning on the fourth day during the acquisition training. Consistently with Y-maze and NOR, the ethanol-exposed presymptomatic APP/PS1 mice showed a shorter latency to find a safe platform than the air-exposed counterparts (Fig. 6H). In a subsequent probe test, ethanol-exposed presymptomatic APP/PS1 mice exhibited an increased preference for the target quadrant relative to other quadrants (Fig. 6I). At the same time, the swimming distance remained similar between groups (Fig. S12E and F). We next assessed the reversal learning. Presymptomatic APP/PS1 mice showed enhanced cognitive function, indicating that the ethanol-exposed presymptomatic APP/PS1 mice are more flexibly adapted to the altered environment (Fig. 6J). We conducted an open-field test to ensure whether mice’s locomotor activity affects memory and cognitive abilities. We found no differences between the air- and ethanol-exposed groups regarding distance moved or velocity (Fig. S13). These results demonstrate that MEE only restores cognitive and memory function in alcohol-exposed male and female presymptomatic APP/PS1 mice (Fig. S16).
Discussion
This study provides a novel insight into how MEE contributes to cognitive improvement in the early-onset AD model, the APP/PS1 mice19,52. Our findings indicate that MEE reduces astrocyte activity and ApoE biosynthesis, possibly lowering the brain’s low-density lipoprotein (LDL) cholesterol levels. Also, consistent with a previously reported inverse correlation between ApoE and LRP1 expression53, we observed increased LRP1 protein levels in the brains of ethanol-exposed presymptomatic APP/PS1 mice.
It is worth noting that our initial hypothesis was that a short (binge-like) and naturalistic ethanol exposure might exacerbate AD pathology. However, our findings contradict the initial hypothesis. Although excessive drinking often refers to more than 4 drinks per day for women and more than 5 drinks per day for men54, low to moderate alcohol consumption has been reported to provide some health benefits, especially cardiovascular health55. Keeping these in mind (4–6), it is still an open question how to define “moderate” or "non-hazardous" drinking. In our study, contrasting to our previous56 or other studies57, we chose to expose mice to vaporized alcohol for 4 h per day without using pyrazole, an inhibitor of alcohol dehydrogenase (ADH), which naturally and slowly increases blood alcohol concentration to approximately 170 mg/dl. Although 170 mg/dl is not typically a low to moderate dose of alcohol, we consider this as a low to moderate dose because of relatively short alcohol exposure and ten times faster heart rate in mice compared to humans58. As we reported56, longer (16 h per day, 4 days per week, 4 weeks) daily ethanol exposures in the vapor chamber may worsen the AD-like pathology in APP/PS1 mice.
However, it is important to consider the method of alcohol administration when interpreting these results, as different routes of ethanol exposure may yield divergent outcomes. In our study, ethanol was administered via vapor exposure, which was chosen to maintain consistent blood alcohol concentrations (BAC) over an extended period, mimicking chronic alcohol consumption patterns. This method offers the advantage of reducing stress and variability that could arise from other methods, such as oral gavage. The method of alcohol administration is an important factor that may explain the differences between our results and those reported in other studies that administered alcohol using oral gavage19,59 Oral gavage introduces ethanol directly into the digestive system, where it undergoes first-pass metabolism in the liver, potentially leading to different metabolic and neurobiological effects compared to inhalation methods. This difference in ethanol metabolism and distribution may influence the extent and nature of its impact on amyloid plaque formation and cognitive function.
In the presymptomatic APP/PS1 group, MEE reduced Aβ plaque count and levels, with a corresponding improvement in cognitive function. This suggests that low to moderate alcohol consumption could mitigate neuronal loss and improve cognitive function in early disease stages. Importantly, we found that MEE has no effect on NTG mice nor symptomatic APP/PS1 mice (Figs. S14 and S15), suggesting that MEE delayed AD-like cognitive function in only presymptomatic APP/PS1 mice. Our results imply that possible neuroprotective effects of low to moderate alcohol consumption may not extend to the symptomatic group with established Aβ plaque burden and more advanced neurodegeneration60. Interestingly, a study on alcohol-induced autophagy in human hepatoma cells revealed that upregulation of PIASy promotes HCV replication through autophagy mechanisms. While the context of liver cells and HCV differs from brain cells and AD, the underlying principle that alcohol can induce autophagy through specific pathways is relevant. One way to remove these plaques is through autophagy activation, which helps clear Aβ. Studies have shown that alcohol can induce autophagy, facilitating Aβ removal61. This suggests that MEE’s neuroprotective effects might be partly due to its ability to promote autophagic Aβ clearance. In addition to the addictive nature of alcohol62, alcohol is known to damage multiple organs and cause many diseases, including cancers60. In particular, prolonged alcohol misuse causes alcohol liver disease63 and hepatitis64. The AST and ALT levels are hallmarks of liver function65. We found increased AST levels without altered ALT levels in presymptomatic APP/PS1 mice liver compared to aged-matched air-exposed APP/PS1 mice without differences in the cortex and hippocampus (Fig. S17). The AST/ALT ratio of over 1.5 is considered severe liver damage66,67. In this regard, a relatively high AST/ALT ratio (~ 2.3) in the liver of APP/PS1 mice indicates that the ethanol exposure paradigm has a detrimental effect on liver function (Fig. S17). Furthermore, it is essential to recognize that consuming even as little as one drink per day carries a spectrum of risks extending beyond liver damage, including the increased likelihood of developing breast cancer68.
Thus, despite some beneficial effects of alcohol in presymptomatic APP/PS1 mice, because only about 10% are early-onset AD patients and a few of them have APP/PS1 mutations69–71, our findings do not support alcohol drinking to prevent cognitive decline or AD pathology. Instead, we want to emphasize that even low- to moderate drinking could be harmful for those especially sensitive to the intoxication effects of alcohol.
Despite the unclear mechanistic causality of altered LRP1 expression, our study establishes a compelling correlation between MEE and changes in ApoE, LDL cholesterol, and LRP1 levels. Additionally, our study revealed that ethanol exposure significantly mitigates the levels of proinflammatory cytokines, IL-1β and TNF-α, in the cortex and hippocampus of ethanol-exposed APP/PS1 mice compared to air-exposed counterparts, which is causally linked to the inhibition of the LRP1-IKK-α/β-NF-κB p65 pathway (Fig. 2 and Fig. S18). This finding is particularly significant as both LRP1 and Toll-like receptor 4 (TLR4) play influential roles in neuroinflammation and AD pathogenesis72,73. LRP1 suppresses microglial and astrocytic cell activation, critical contributors to neuroinflammation, by regulating the TLR4/NF-κB p65/MAPK signaling pathway72. This interaction regulates the release of proinflammatory cytokines and phagocytosis, maintaining brain homeostasis74. However, IκB-α levels responded to ethanol within 30 min in mixed hippocampal cell samples from wild-type mice but not in cells from TLR4- or MyD88-deficient mice75. Besides, insufficient LRP1 activation is associated with inflammation-induced tumor progression76,77, demonstrating the role of LRP1 in inflammation. Our findings illustrate alterations in sub-signals IKK-α/β and IKβ-α, notwithstanding the absence of an overall alteration in TLR4. In an additional experimental group, we assessed the impact of LRP1 inhibition on cognitive performance in ethanol-exposed presymptomatic APP/PS1 mice (Fig. S19). To inhibit the LRP1 function, we utilized Receptor-Associated Protein (RAP), a well-documented inhibitor, for the LRP178. Mice were divided into two groups: those receiving the LRP1 antagonist (RAP +) and those not treated with the antagonist (RAP-). We found a notable reduction in cognitive performance in Y-maze and NOR tasks in the RAP + group compared to the RAP- group without altering locomotor function (Fig. S19), indicating that LRP1 function is inversely correlated with cognitive abilities. Interestingly, a peptide ligand SP16 is known to activate LRP1, which decreases inflammation and increases cell survival in acute myocardial infarction79,80. A recent phase 1 clinical trial as a first-in-class anti-inflammatory LRP1 agonist shows a promising outcome in healthy volunteers81. Moreover, a new peptide agonist, COG1410, shows a similar anti-inflammatory effect in rats60. Thus, a future study will reveal a possible therapeutic effect of LRP1 agonist in neuroinflammation-related AD.
One intriguing finding is the reduced levels of proinflammatory cytokine IL-1β in male mice following MEE. IL-1β plays a significant role in inducing neuroinflammation, and its reduction suggests a decrease in the inflammatory response82. This reduction could potentially lead to alleviating the symptoms associated with neuroinflammation, such as cognitive dysfunction. Conversely, in female mice, MEE has been linked to the upregulation of IL-10, an anti-inflammatory cytokine. An increase in IL-10 suggests an enhanced anti-inflammatory response, potentially protecting the brain from the damaging effects of inflammation82,83. This upregulation might contribute to preserving cognitive function and neuronal health in female mice. Interestingly, we found no sex-specific changes in AD-like behaviors but only sex-specific changes in cytokine levels Table 1. Although the exact mechanisms of these sex-specific effects of ethanol are unclear, several plausible explanations underlie our findings. Hormonal differences between males and females, such as the influence of estrogen and testosterone, could potentially differentially respond to ethanol exposure84. Several recent studies revealed the sex-specific differences in the regulation of cytokines by astrocytes and microglia. In males, astrocytes primarily regulate proinflammatory cytokines85. Conversely, in females, microglia play a central role in regulating anti-inflammatory cytokines86. Furthermore, astrocytes are more active in anti-inflammatory phenotype in females87. These differences underscore the complex interplay of astrocytes, microglia, and cytokines and their roles in the brain. Similarly, recent studies have shown that astrocytes and microglia, essential central nervous system (CNS) components, respond differently to alcohol exposure and protracted withdrawal. Astrocytes primarily activate proinflammatory cytokines, proteins that heighten inflammation in response to alcohol consumption. This is part of their role in maintaining CNS homeostasis and their varying response to inflammatory stimuli83,88. On the other hand, microglia mainly activate anti-inflammatory cytokines, undermining the inflammation. Focusing on the role of TLR4 in alcohol-induced neuroinflammation and brain damage, alcohol consumption activates microglia through TLR4 to produce anti-inflammatory cytokines in the brain89. Thus, our findings imply that MEE preferentially reduces astrocyte-induced proinflammatory cytokines in males while increasing microglia-induced anti-proinflammatory cytokines; both yield similar MEE-induced behavior outcomes. Further research may reveal mechanisms underlying the sex difference in cytokine-mediated signaling and comprehensive AD pathology.
Table 1.
Summary of statistical analysis.
| Figure | Statistical Tests | Comparison | Value | P value | |
|---|---|---|---|---|---|
| Figure 1 | B | Mann–Whitney test | Air vs EtOH | U = 0 | P < 0.0001 |
| C | Mann–Whitney test | Air vs EtOH | U = 9 | P = 0.0011 | |
| D | Mann–Whitney test | Air vs EtOH | U = 1 | P < 0.0001 | |
| E | Mann–Whitney test | Air vs EtOH | U = 4 | P = 0.0001 | |
| F | Mann–Whitney test | Air vs EtOH | U = 1.5 | P < 0.0001 | |
| G | Mann–Whitney test | Air vs EtOH | U = 4 | P = 0.0001 | |
| H | Mann–Whitney test | Air vs EtOH | U = 37 | P = 0.3423 | |
| I | Correlation | ApolipoproteinE vs LDL-C | R = 0.6897 | P = 0.0008 | |
| J | Mann–Whitney test | Air vs EtOH | U = 13 | P = 0.0039 | |
| K | Mann–Whitney test | Air vs EtOH | U = 18 | P = 0.0138 | |
| L | Mann–Whitney test | Air vs EtOH | U = 47 | P = 0.8534 | |
| M | Correlation | ApolipoproteinE vs LDL-C | R = 0.5964 | P = 0.0021 | |
| Figure 2 | C (LRP1) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 |
| C (p-IKK-α/β) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| C (t-IKK-α/β) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| C (p-/t-IKK-α/β) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.0317 | |
| C (IκB-α) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| C (NF-κB) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| D (LRP1) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| D (p-IKK-α/β) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| D (t-IKK-α/β) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| D (p-/t-IKK-α/β) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| D (IκB-α) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.0317 | |
| D (NF-κB) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| Figure 3 | A (Total, IL-1β, Cortex) | Mann–Whitney test | Air vs EtOH | U = 10 | P = 0.0014 |
| A (Total, IL-1β, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 9.5 | P = 0.0011 | |
| A (Total, TNF-α, Cortex) | Mann–Whitney test | Air vs EtOH | U = 18 | P = 0.0147 | |
| A (Total, TNF-α, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 12 | P = 0.0018 | |
| B (Male, IL-1β, Cortex) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| B (Male, IL-1β, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| B (Male, TNF- α, Cortex) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.317 | |
| B (Male, TNF- α, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| C (Female, IL-1β, Cortex) | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.3905 | |
| C (Female, IL-1β, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 9 | P = 0.5476 | |
| C (Female, TNF- α, Cortex) | Mann–Whitney test | Air vs EtOH | U = 6 | P = 0.2222 | |
| C (Female, TNF- α, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 5 | P = 0.1508 | |
| Figure 4 | C | Mann–Whitney test | Air vs EtOH | U = 13 | P = 0.0038 |
| D | Mann–Whitney test | Air vs EtOH | U = 3.5 | P < 0.0001 | |
| E | Mann–Whitney test | Air vs EtOH | U = 56 | P = 0.3698 | |
| F | Mann–Whitney test | Air vs EtOH | U = 0 | P < 0.0001 | |
| G | Mann–Whitney test | Air vs EtOH | U = 23 | P = 0.0036 | |
| H | Mann–Whitney test | Air vs EtOH | U = 50 | P = 0.2133 | |
| I | Mann–Whitney test | Air vs EtOH | U = 10 | P = 0.0001 | |
| J | Mann–Whitney test | Air vs EtOH | U = 29 | P = 0.0121 | |
| Figure 5 | B | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.0005 |
| C | Mann–Whitney test | Air vs EtOH | U = 0 | P < 0.0001 | |
| Figure 6 | B | Mann–Whitney test | Air vs EtOH | U = 116.5 | P < 0.0001 |
| C | Mann–Whitney test | Air vs EtOH | U = 359.5 | P = 0.9623 | |
| E (Air) | Mann–Whitney test | Air vs EtOH | U = 256 | P = 0.8618 | |
| FD (EtOH) | Mann–Whitney test | Air vs EtOH | U = 108 | P < 0.0001 | |
| H | Two-way ANOVA | Air vs EtOH | F (3, 88) = 5.433 | P = 0.0018 | |
| I | Two-way ANOVA | (PF) Air vs (PF) EtOH | F (3, 88) = 6.786 | P = 0.0138 | |
| J | Two-way ANOVA | Air vs EtOH | F (3, 88) = 9.313 | P < 0.0001 | |
| Sup Fig. 1 | C (Male, Presymptomatic , Body weight) | Two-way ANOVA | Air vs EtOH | F (4, 62) = 0.3201 | P = 0.8635 |
| C (Female, Symptomatic, Body weight) | Two-way ANOVA | Air vs EtOH | F (4, 100) = 0.8585 | P = 0.4917 | |
| D (Male, Presymptomatic , Body weight) | Two-way ANOVA | Air vs EtOH | F (3, 32) = 0.3373 | P = 0.7984 | |
| D (Female, Symptomatic, Body weight) | Two-way ANOVA | Air vs EtOH | F (3, 31) = 0.6150 | P = 0.6105 | |
| Sup Fig. 3 | A (ApoE, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 |
| A (ApoE, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 1 | P = 0.0159 | |
| A (ApoE, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| A (ApoE, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.0317 | |
| B (GFAP, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 1 | P = 0.0159 | |
| B (GFAP, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| B (GFAP, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| B (GFAP, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| C (ApoE, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 1.5 | P = 0.0238 | |
| C (ApoE, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| C (ApoE, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 3 | P = 0.0478 | |
| C (ApoE, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 3 | P = 0.0456 | |
| Sup Fig. 4 | A | Mann–Whitney test | Air vs EtOH | U = 40.5 | P = 0.4926 |
| B | Mann–Whitney test | Air vs EtOH | U = 37 | P = 0.3423 | |
| C | Mann–Whitney test | Air vs EtOH | U = 26 | P = 0.0753 | |
| D | Mann–Whitney test | Air vs EtOH | U = 9 | P = 0.0011 | |
| E | Mann–Whitney test | Air vs EtOH | U = 31 | P = 0.1649 | |
| F | Mann–Whitney test | Air vs EtOH | U = 47 | P = 0.8534 | |
| G | Mann–Whitney test | Air vs EtOH | U = 38 | P = 0.3930 | |
| H | Mann–Whitney test | Air vs EtOH | U = 10 | P = 0.0278 | |
| Sup Fig. 5 | A (HDL-C, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.3095 |
| A (HDL-C, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 8.5 | P = 0.4603 | |
| A (HDL-C, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 12.5 | P > 0.9999 | |
| A (HDL-C, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.3095 | |
| B (LDL-C, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 1 | P = 0.0159 | |
| B (LDL-C, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 1 | P = 0.0159 | |
| B (LDL-C, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 2.5 | P = 0.0397 | |
| B (LDL-C, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.0317 | |
| C (Total-C, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.3095 | |
| C (Total-C, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 12 | P > 0.9999 | |
| C (Total-C, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 11 | P = 0.8413 | |
| C (Total-C, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 9 | P = 0.5476 | |
| Sup Fig. 6 | A | One-way ANOVA | Air vs EtOH (Male) | F (3, 9) = 1.031 | P = 0.9636 |
| Air vs EtOH (Female) | P = 0.5892 | ||||
| B | One-way ANOVA | Air vs EtOH (Male) | F (3, 9) = 0.9527 | P = 0.7369 | |
| Air vs EtOH (Female) | P = 0.5858 | ||||
| C | One-way ANOVA | Air vs EtOH (Male) | F (3, 11) = 8.388 | P = 0.0209 | |
| Air vs EtOH (Female) | P = 0.4809 | ||||
| D | One-way ANOVA | Air vs EtOH (Male) | F (3, 10) = 0.6277 | P = 0.8978 | |
| Air vs EtOH (Female) | P = 0.9988 | ||||
| E | One-way ANOVA | Air vs EtOH (Male) | F (3, 11) = 12.16 | P = 0.9998 | |
| Air vs EtOH (Female) | P = 0.0015 | ||||
| F | One-way ANOVA | Air vs EtOH (Male) | F (3, 10) = 1.222 | P = 0.9984 | |
| Air vs EtOH (Female) | P = 0.7469 | ||||
| G | One-way ANOVA | Air vs EtOH (Male) | F (3, 12) = 0.7291 | P = 0.9994 | |
| Air vs EtOH (Female) | P = 0.0454 | ||||
| H | One-way ANOVA | Air vs EtOH (Male) | F (3, 9) = 1.290 | P = 0.9815 | |
| Air vs EtOH (Female) | P = 0.4227 | ||||
| I | One-way ANOVA | Air vs EtOH (Male) | F (3, 12) = 1.549 | P = 0.2216 | |
| Air vs EtOH (Female) | P = 0.9233 | ||||
| J | One-way ANOVA | Air vs EtOH (Male) | F (3, 10) = 5.033 | P = 0.0097 | |
| Air vs EtOH (Female) | P = 0.9996 | ||||
| Sup Fig. 7 | A | Mann–Whitney test | Air vs EtOH | U = 12 | P > 0.9999 |
| B | Mann–Whitney test | Air vs EtOH | U = 10 | P = 0.6905 | |
| C | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.3095 | |
| D | Mann–Whitney test | Air vs EtOH | U = 1 | P = 0.0159 | |
| Sup Fig. 8 | A (Aβ plaque) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0006 |
| A (ApoE) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0006 | |
| Sup Fig. 9 | A (Aβ, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.0317 |
| A (Aβ, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 2 | P = 0.0269 | |
| A (Aβ, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 0.5 | P = 0.0159 | |
| A (Aβ, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0079 | |
| B (Aβ40, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 10 | P = 0.2251 | |
| B (Aβ40, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 16 | P = 0.7835 | |
| B (Aβ40, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 15.5 | P = 0.7338 | |
| B (Aβ40, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 7.5 | P = 0.1039 | |
| C (Aβ42, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0022 | |
| C (Aβ42, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0022 | |
| C (Aβ42, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0022 | |
| C (Aβ42, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0022 | |
| Sup Fig. 10 | C | Mann–Whitney test | Air vs EtOH | U = 38.5 | P = 0.9136 |
| D | Mann–Whitney test | Air vs EtOH | U = 32 | P = 0.4992 | |
| Sup Fig. 11 | A (FDG, Cortex, Male) | Mann–Whitney test | Air vs EtOH | U = 1.5 | P = 0.0203 |
| A (FDG, Cortex, Female) | Mann–Whitney test | Air vs EtOH | U = 3 | P = 0.0303 | |
| A (FDG, Hippocampus, Male) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0001 | |
| A (FDG, Hippocampus, Female) | Mann–Whitney test | Air vs EtOH | U = 3 | P = 0.0401 | |
| Sup Fig. 12 | B | Mann–Whitney test | Air vs EtOH | U = 8 | P > 0.9999 |
| C | Mann–Whitney test | Air vs EtOH | U = 7 | P = 0.8857 | |
| Sup Fig. 13 | A | Mann–Whitney test | Air vs EtOH | U = 101.5 | P = 0.1414 |
| B | Mann–Whitney test | Air vs EtOH | U = 171.5 | P = 0.8003 | |
| C | Mann–Whitney test | Air vs EtOH | U = 110 | P = 0.1626 | |
| D | Mann–Whitney test | Air vs EtOH | U = 109 | P = 0.1528 | |
| E | Mann–Whitney test | Air vs EtOH | U = 299 | P = 0.5061 | |
| F | Mann–Whitney test | Air vs EtOH | U = 326 | P = 0.7032 | |
| G | Mann–Whitney test | Air vs EtOH | U = 62 | P = 0.5899 | |
| H | Mann–Whitney test | Air vs EtOH | U = 70 | P = 0.9323 | |
| Sup Fig. 14 | A | Mann–Whitney test | Air vs EtOH | U = 122 | P = 0.2112 |
| B | Mann–Whitney test | Air vs EtOH | U = 132 | P = 0.3503 | |
| C | Mann–Whitney test | Air vs EtOH | U = 23 | P < 0.0001 | |
| D | Mann–Whitney test | Air vs EtOH | U = 22 | P < 0.0001 | |
| Sup Fig. 15 | A | Mann–Whitney test | Air vs EtOH | U = 26.5 | P = 0.2457 |
| B | Mann–Whitney test | Air vs EtOH | U = 19.5 | P = 0.0706 | |
| C | Mann–Whitney test | Air vs EtOH | U = 20 | P = 0.0831 | |
| D | Mann–Whitney test | Air vs EtOH | U = 40 | P > 0.9999 | |
| E | Mann–Whitney test | Air vs EtOH | U = 28 | P = 0.3154 | |
| F | Mann–Whitney test | Air vs EtOH | U = 38 | P = 0.8968 | |
| G | Mann–Whitney test | Air vs EtOH | U = 26 | P = 0.9546 | |
| H | Mann–Whitney test | Air vs EtOH | U = 17 | P = 0.1806 | |
| I | Two-way ANOVA | Air vs EtOH | F (3, 40) = 1.476 | P = 0.2356 | |
| J | Two-way ANOVA | Air vs EtOH | F (3, 88) = 1.716 | P = 0.1694 | |
| K | Two-way ANOVA | Air vs EtOH | F (3, 40) = 1.204 | P = 0.9961 | |
| Sup Fig. 16 | A | Mann–Whitney test | Air vs EtOH (Male) | U = 10.5 | P = 0.0004 |
| Air vs EtOH (Female) | U = 56 | P = 0.0113 | |||
| B | Mann–Whitney test | Air vs EtOH (Male) | U = 65.5 | P = 0.9878 | |
| Air vs EtOH (Female) | U = 118 | P = 0.9766 | |||
| C | Mann–Whitney test | Old vs New object (Air male) | U = 45 | P = 0.3653 | |
| Old vs New object (Air female) | U = 47 | P = 0.1600 | |||
| D | Mann–Whitney test | Old vs New object (EtOH male) | U = 15 | P = 0.0019 | |
| Air vs Old vs New object (EtOH female) | U = 42 | P = 0.0027 | |||
| E (Male) | Two-way ANOVA | Air vs EtOH | F (3, 32) = 5.215 | P = 0.0048 | |
| F (Male) | Two-way ANOVA | Air vs EtOH | F (3, 48) = 2.693 | P = 0.0465 | |
| G (Female) | Two-way ANOVA | Air vs EtOH | F (3, 32) = 6.761 | P = 0.0012 | |
| H (Female) | Two-way ANOVA | Air vs EtOH | F (3, 88) = 3.638 | P = 0.0191 | |
| Sup Fig. 17 | A (ALT, Cortex) | Mann–Whitney test | Air vs EtOH | U = 8 | P = 0.1320 |
| A (ALT, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 14 | P = 0.5887 | |
| A (ALT, Liver) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0022 | |
| B (AST, Cortex) | Mann–Whitney test | Air vs EtOH | U = 16 | P = 0.8182 | |
| B (AST, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 8.5 | P = 0.1450 | |
| B (AST, Liver) | Mann–Whitney test | Air vs EtOH | U = 15 | P = 0.6623 | |
| C (ALT/AST, Cortex) | Mann–Whitney test | Air vs EtOH | U = 12 | P = 0.3939 | |
| C (ALT/AST, Hippocampus) | Mann–Whitney test | Air vs EtOH | U = 11 | P = 0.3095 | |
| C (ALT/AST, Liver) | Mann–Whitney test | Air vs EtOH | U = 0 | P = 0.0022 | |
| Sup Fig. 19 | B (Number of arm entries) | Mann–Whitney test | RPA + vs RAP- | U = 1 | P = 0.0012 |
| B(Total arm entries) | Mann–Whitney test | RPA + vs RAP- | U = 37.5 | P = 0.8138 | |
| C(NOR) | Mann–Whitney test | RPA + vs RAP- | U = 2 | P = 0.0022 | |
| D(OFT) | Mann–Whitney test | RPA + vs RAP- | U = 20 | P = 0.0770 |
In this study, we have not explored a possible change of tau pathology. In 3 × TgAD (APPSwe, TauP301L, PSEN) mice, two weeks of 24 access to two bottles of home-cage alcohol consumption with 25% ethanol and 0.1% saccharin (in average 20 g/kg consumption) showed cognitive decline along with increased phosho-tau accumulation59. Future studies will address the effect of the ethanol exposure paradigm utilized in this report on tau pathology.
Conclusions
Our study demonstrates that reducing neuroinflammation and LDL cholesterol would have a therapeutic effect on cognitive impairment in AD, evidenced by the rescue of cognitive and memory deficits in the presymptomatic APP/PS1 mice. Considering alcohol’s harmful effect on liver function and addiction liability, our findings do not support even low- to moderate-level drinking. However, our findings offer a new insight that activation of LRP1 could be a therapeutic target for mitigating neuroinflammation and attenuating Aβ deposition, thereby ameliorating AD pathology.
Materials and methods
We confirm that our research complies with all pertinent ethical regulations, including the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guideline. The Committee on Animal Care and Use at Mayo Clinic (A00005502-20-R23) approved all experimental procedures.
Mice
The APP mice were heterozygous transgenic mice (C57B6/SJL, I.D. No. Tg2576) that expressed mutant human APP695 containing a double mutation (K670N, M671L). We purchased the APP mice (stock # 1349) from Taconic Biosciences (Germantown, NY, USA). The PS1 mice were homozygous transgenic mice (Swiss Webster/B6D2; I.D. No. M146L) that express mutant human PS1 containing a single mutation (M146L)17. Dr. Steven Younkin’s laboratory at Mayo Clinic generated PS117. The double transgenic mice, APP/PS1, were produced in house by crossbreeding of homozygous PS1 and heterozygous APP mice17. Based on previous studies13–17, we defined 12 ~ 24 weeks old mice as the presymptomatic group and 36 ~ 48 weeks old as the symptomatic group. Following the establishment of these groups, behavioral assessments were conducted on both the presymptomatic (24 ~ 26 weeks) and symptomatic (48 ~ 50 weeks) APP/PS1 mice to evaluate cognitive and motor functions, critical indicators of AD progression. After these behavioral tests, mice from the presymptomatic group at 26 weeks of age and the symptomatic group at 50 weeks of age were sacrificed to enable the molecular analyses (Fig. S1A and B). Mice were arbitrarily assigned to two distinct groups in a new cohort: one group was administered RAP (BML-SE552-0100; Enzo Life Science, Farmingdale, New York, USA) at a dosage of 3.5 mg/kg through intraperitoneal injections (80 μl), while the control group received a corresponding vehicle treatment consisting of 80 μl saline via intraperitoneal injection. These treatments were given once weekly, specifically on the fourth day of each week throughout the 12 weeks of ethanol exposure, ensuring a regular dosing schedule across the study duration (Fig. S19A). The group size was determined based on similar studies conducted by our labs and others. All animals were housed individually in standard mouse cages under a 12-h artificial light–dark cycle with ad libitum access to food and water. Room temperature and humidity were kept constant (22 ± 1 °C; relative humidity: 55 ± 5%). Standard laboratory rodent chow (LabDiet 5P00 Prolab RMH 3000 rodent chow) and tap water were provided ad libitum throughout the experimental period. Mice underwent a battery of behavioral tests at baseline and various stages during ethanol exposure.
Low to moderate ethanol exposure
As previously described56, air or ethanol vapor was delivered in Plexiglas inhalation chambers (Fig. S2). We have previously used the vapor chamber system to successfully maintain stable blood ethanol concentration (BAC) in mice. Mice were exposed to ethanol vapor or room air using vapor administration chambers for 4 h from 09:00 to 13:00, followed by 20 h of room air in their home cages. This process was repeated for four consecutive days, followed by 3 days in their home cages with room air (withdrawal period). After the last ethanol exposure, the mice’s tail blood was collected immediately and centrifuged to extract the serum. BACs were measured by Analox GL5 multi metabolite analyzer (Analox Instruments, Stourbridge, United Kingdom) with the accompanying kits.
FDG-PET
The micro-PET scanning was done using a Siemens Inveon MicroPET/CT Scanner (Siemens Preclinical Solutions Inc., Erlangen, Germany) with the 30 min list mode acquisition protocol. Mice were fasted one hour before the IP injection of 200 ~ 270 μCi of fludeoxyglucose F18(18F-FDG) in 200 μl injection volume prepared the same day at the Mayo Clinic Nuclear Medicine Animal Imaging Resource90. CT-based attenuation corrections were applied. During the scan, mice were anesthetized by inhalation of ~ 2% isoflurane supplemented with oxygen. PET images were spatially normalized to the mouse brain PET template91 using PMOD v4.3 (PMOD technologies, Zurich, Switzerland). Then, brain18F-FDG uptake was calculated as standard uptake value ratio (SUVR) with the cerebellum as referencing tissue. For group-wise comparisons, regional SUVRs were calculated as the average uptake over the total voxels in the region of interest (ROI).
ELISA assay
The Aβ1-40 (Thermo Fisher Scientific, #KMB3481), Aβ1-42 (Thermo Fisher Scientific, KMB3441), Apolipoprotein E (Abcam, #ab215086), HDL and LDL / total cholesterol levels (Abcam, #ab65390), IL-1β (Abcam, #ab197742), and TNF-α (Abcam, #ab208348) in the soluble fraction of frozen brain tissues were quantified using an ELISA assay. All assays were performed according to the manufacturer’s instructions. Levels of these proteins were calculated from a standard curve developed with specific optical density versus serial dilutions of a known concentration. Each standard and experimental sample was run in duplicate, and the results were averaged.
Immunohistochemistry
After deeply anesthetizing the mouse by inhaling isoflurane, perfuse it with PBS and quickly remove the brain. For immunohistochemistry on frozen sections, the 35 μm-thick brain sections were washed three times in PBS containing 0.2% Triton X-100 and were then incubated in a blocking solution (0.5% bovine serum albumin and 3% normal goat serum in PBS with 0.4% Tween 20) for 1 h at RT21. The sections were incubated with the primary GFAP (Abcam, CAMB, UK, #ab4674), Aβ42 (Thermo Fisher Scientific, Walthan, MA, USA#700,254), ApoE (Abcam, #ab183596) and Thioflavin S overnight at 4 °C. Following this, the sections were washed three times and were then incubated with an Alexa Fluor 488 or Alexa Fluor 555 donkey anti-rabbit IgG antibody (Invitrogen, Carlsbad, CA, USA) for 1 h at RT. The brain slices were then washed three times and mounted onto slides using Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA, USA). Tissue specimens were taken using a Nikon TS2-S-SM microscope equipped with a Nikon DS-Qi2 camera (Nikon Microscopy, Tokyo, Japan). Serial images of the cortex and hippocampus were captured on 4 consecutive 30 μm sections with × 100 magnification. Once the ROIs were defined, we quantified the fluorescence intensity and percentage area of a red signal representing Alexa Fluro 555 within each ROI per Sect. (5 mice per group).
Western blotting
Western blot (WB) analysis was conducted using cortex and hippocampus homogenates from both alcohol (n = 5) and air groups (n = 5), including male and female samples. Protein concentrations were determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA), and 20 μg of protein from each sample was loaded. Proteins were separated on a 4–12% Nu-Page Bis–Tris gel (MOPS buffer, Invitrogen, Carlsbad, CA, USA) at 140 V for 2 h and subsequently transferred to a PVDF membrane (Invitrogen) at 30 V for 1 h. The membrane was blocked with 5% non-fat milk for 1.5 h. Primary antibody incubation was performed overnight at 4 °C in 5% BSA in 1 × TBST with antibodies against LRP-1 (1:2000; Abcam, #ab92544), p-IKK-α/β (1:500; Abcam, #ab194528), t-IKK-α/β (1:500; Abcam, #ab178870), IKB-α (1:500; Abcam, #ab76429), NF-κB p65 (1:500; Cell Signaling, #8284), and β-actin (1:1000; Abcam, #ab8226). After three 10-min washes in 1 × TBST, membranes were incubated with anti-rabbit and anti-mouse secondary antibodies (1:1000, Millipore) in 5% BSA for 1 h at room temperature. Blots were visualized using the Radiance Q Chemiluminescent Substrate (Azure Biosystems, Dublin, CA, USA) and images were captured using an Azure 300 Chemiluminescent Western Blot Imaging System (Azure Biosystems, Dublin, CA, USA). Band optical density was quantified using NIH ImageJ software (ij153-win-java8). Uncropped, full-length images of the Western blot membranes can be found in Fig. S17.
Y-maze spontaneous alternation
In the presymptomatic and symptomatic groups, mice were tested for spontaneous alternation in a three-armed Y-maze constructed from white polyvinyl plastic. Each arm (A, B, and C) was 40 cm long, 6.8 cm wide, and 15.5 cm high, and the three-folding angle was 120°. Mice were placed in the equipment and allowed to explore freely. During each 8-min period, the number of times that the tail of each animal fully entered each arm was counted for each arm, and then, the number of times each animal entered the arm one after another (in A, B, or C sequence) was also counted, which was assigned one point (actual alternation). The inside of the Y-maze was wiped with 70% ethanol between different animal trials. Alternation behavior was defined as no overlap into all three arms and was calculated by the following equation: Rate of spontaneous alterations (%) = (number of alternations) / (the total number of arm entries − 2) × 100. All tests were recorded by a technician blind to the genotype of the animals.
Novel objective recognition task
We conducted a novel objective recognition (NOR) task with the young and symptomatic groups to assess the changes in cognition and memory. The setup comprised a black-walled square box measuring 35 × 35 × 30 cm. Mice were placed in the center of the open field box and allowed to acclimate for 30 min. Then, two of the same objects were placed in the box, and the mice were habituated for 10 min. After 1 h, one object was replaced with a novel object, and the exploration time of the new or familiar object was recorded for 10 min using the Ethovision XT 9 system (Noldus Information Technology, Wageningen, Netherlands). The following parameters were measured: the total exploration time, frequency, objective recognition time, and memory index. The memory index is calculated by the exploration time for each object divided by the total exploration time.
Open field test
The open field task is a simple sensorimotor test used to determine general activity levels, gross locomotor activity, and exploration habits in rodent models of CNS disorders. Presymptomatic and symptomatic mice were tested in a black-walled square box measuring 35 × 35 × 30 cm. The animal is placed in the arena and allowed to freely move about for 10 min while being recorded by an overhead camera. The footage is then analyzed by an automated tracking system for the following parameters: distance moved, velocity, and time spent in pre-defined zones.
Morris water maze (MWM) test
Spatial learning and memory were assessed using the MWM test using the animal cognitive functions assessment meter (Ethovision Maze task system, Noldus Information Technology, Wageningen, Netherlands). Mice were tested in a circular pool (100 cm diameter, 45 cm high, and outer height 61.5 cm from the ground floor) filled with opaque water equilibrated to room temperature (22 °C). The tank was divided into four quadrants with different navigation marks (cues) for each quadrant. Mice were continuously trained with 4 trials per mouse daily (once per navigation mark) for 4 days to search for the escape platform within a maximum of 60 s 47. The platform location stayed constant during the test, and the time taken to reach the platform was recorded as the escape latency. After the mouse found the hidden platform, it was kept on the platform for 2 s. If mice could not find the platform within 60 s, they were placed on the platform for 20 s to encode the location of the escape platform; for these trials, the escape latency was recorded as 60 s. Mice removed from the pool were dried and returned to the home cage. The MWM probe test was performed within 48 h of the final trial. The platform was removed from the pool, and the mice were placed in the water and allowed to swim for 60 s. The time spent in the quadrant that previously contained the platform indicates long-term memory maintenance. Swim distance, velocity, and frequency were recorded as measures of motor function. All the tests were performed by a technician who was blind to the genotype of the animals.
Supplementary Information
Acknowledgements
We thank all the laboratory members for their helpful discussion and comments.
Abbreviations
- AUD
Alcohol use disorder
- AD
Alzheimer’s disease
- MEE
Low to moderate ethanol exposure
- ApoE
Apolipoprotein E
- LDL- cholesterol
Low-density lipoprotein cholesterol
Author contributions
SK, ET, and DSC designed the study; SK and DA performed all the behavioral and molecular experiments; JL and PM carried out the FDG-PET experiment; SC did the Western blot experiment. JN and SC bred and genotyped mice; SK and DSC analyzed data; SK and DSC wrote the paper.
Funding
This research was supported by the Samuel C. Johnson for Genomics of Addiction Program at Mayo Clinic, the Soonchunhyang University, Ulm Foundation, and the National Institute of Health (AG072898, AA029258, and AA028968).
Data availability
All data generated or analyzed during this study are included in this published article and its additional files. Data analysis results are included in the table file.
Declarations
Competing interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-75202-w.
References
- 1.Kamal, H. et al. Alcohol use disorder, neurodegeneration, Alzheimer’s and Parkinson’s disease: interplay between oxidative stress, neuroimmune response and excitotoxicity. Front. Cell Neurosci.14, 282. 10.3389/fncel.2020.00282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ridley, N. J., Draper, B. & Withall, A. Alcohol-related dementia: an update of the evidence. Alzheimers Res. Ther.5, 3. 10.1186/alzrt157 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyas, S. L. Alcohol use and the risk of developing Alzheimer’s disease. Alcohol Res. Health25, 299–306 (2001). [PMC free article] [PubMed] [Google Scholar]
- 4.Barnett, A. et al. Adolescent binge alcohol enhances early Alzheimer’s disease pathology in adulthood through proinflammatory neuroimmune activation. Front. Pharmacol.13, 884170. 10.3389/fphar.2022.884170 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tucker, A. E., Alicea Pauneto, C. D. M., Barnett, A. M. & Coleman, L. G. Jr. Chronic ethanol causes persistent increases in Alzheimer’s tau pathology in female 3xTg-AD mice: a potential role for lysosomal impairment. Front. Behav. Neurosci.16, 886634. 10.3389/fnbeh.2022.886634 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho, A. M. et al. Chronic intermittent ethanol exposure alters behavioral flexibility in aged rats compared to adult rats and modifies protein and protein pathways related to Alzheimer’s disease. ACS Omega7, 46260–46276. 10.1021/acsomega.2c04528 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch, M. et al. Alcohol consumption and risk of dementia and cognitive decline among older adults with or without mild cognitive impairment. JAMA Netw. Open2, e1910319. 10.1001/jamanetworkopen.2019.10319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neafsey, E. J. & Collins, M. A. Moderate alcohol consumption and cognitive risk. Neuropsychiatr. Dis. Treat.7, 465–484. 10.2147/NDT.S23159 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabia, S. et al. Alcohol consumption and risk of dementia: 23 year follow-up of Whitehall II cohort study. BMJ362, k2927. 10.1136/bmj.k2927 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeon, K. H. et al. Changes in alcohol consumption and risk of dementia in a nationwide cohort in South Korea. JAMA Netw. Open6, e2254771. 10.1001/jamanetworkopen.2022.54771 (2023). [DOI] [PubMed] [Google Scholar]
- 11.Ormeno, D. et al. Ethanol reduces amyloid aggregation in vitro and prevents toxicity in cell lines. Arch. Med. Res.44, 1–7. 10.1016/j.arcmed.2012.12.004 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Munoz, G. et al. Low concentrations of ethanol protect against synaptotoxicity induced by Abeta in hippocampal neurons. Neurobiol. Aging36, 845–856. 10.1016/j.neurobiolaging.2014.10.017 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Zhang, L. et al. Differential effect of amyloid beta peptides on mitochondrial axonal trafficking depends on their state of aggregation and binding to the plasma membrane. Neurobiol. Dis.114, 1–16. 10.1016/j.nbd.2018.02.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang, L. et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. EBioMedicine2, 294–305. 10.1016/j.ebiom.2015.03.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trushina, E. et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE7, e32737. 10.1371/journal.pone.0032737 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holcomb, L. A. et al. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav. Genet.29, 177–185. 10.1023/a:1021691918517 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Holcomb, L. et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med.4, 97–100. 10.1038/nm0198-097 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Wengenack, T. M., Whelan, S., Curran, G. L., Duff, K. E. & Poduslo, J. F. Quantitative histological analysis of amyloid deposition in Alzheimer’s double transgenic mouse brain. Neuroscience101, 939–944. 10.1016/s0306-4522(00)00388-2 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Day, S. M. et al. Ethanol exposure alters Alzheimer’s-related pathology, behavior, and metabolism in APP/PS1 mice. Neurobiol. Dis.177, 105967. 10.1016/j.nbd.2022.105967 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong, Y. S., Hou, F. L., Guo, J., Lin, L. & Zhu, F. Y. Effects of alcohol intake on cognitive function and beta-amyloid protein in APP/PS1 transgenic mice. Food Chem. Toxicol.151, 112105. 10.1016/j.fct.2021.112105 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Carter, S. F. et al. Astrocyte biomarkers in Alzheimer’s disease. Trends Mol. Med.25, 77–95. 10.1016/j.molmed.2018.11.006 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Staurenghi, E. et al. Cholesterol dysmetabolism in Alzheimer’s disease: a starring role for astrocytes?. Antioxidants (Basel)10.3390/antiox10121890 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, S. I. et al. APOE4-carrying human astrocytes oversupply cholesterol to promote neuronal lipid raft expansion and Abeta generation. Stem Cell Reports16, 2128–2137. 10.1016/j.stemcr.2021.07.017 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang, H. et al. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA10.1073/pnas.2102191118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudajev, V. & Novotny, J. Cholesterol as a key player in amyloid beta-mediated toxicity in Alzheimer’s disease. Front. Mol. Neurosci.15, 937056. 10.3389/fnmol.2022.937056 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raulin, A. C. et al. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol. Neurodegener.17, 72. 10.1186/s13024-022-00574-4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahley, R. W. & Huang, Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron76, 871–885. 10.1016/j.neuron.2012.11.020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen, K. L. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: A review. Atherosclerosis255, 145–155. 10.1016/j.atherosclerosis.2016.10.037 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Dietschy, J. M. & Turley, S. D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol.12, 105–112. 10.1097/00041433-200104000-00003 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Feingold, K. R. in Endotext (eds K. R. Feingold et al.) (2000).
- 31.Chandrashekar, D. V., Steinberg, R. A., Han, D. & Sumbria, R. K. Alcohol as a modifiable risk factor for Alzheimer’s disease-evidence from experimental studies. Int. J. Mol. Sci.10.3390/ijms24119492 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gan, M., Jiang, P., McLean, P., Kanekiyo, T. & Bu, G. Low-density lipoprotein receptor-related protein 1 (LRP1) regulates the stability and function of GluA1 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor in neurons. PLoS ONE9, e113237. 10.1371/journal.pone.0113237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Frutos Lucas, J. et al. How does apolipoprotein E genotype influence the relationship between physical activity and Alzheimer’s disease risk? A novel integrative model. Alzheimers Res. Ther.15, 22. 10.1186/s13195-023-01170-4 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cramer, P. E. et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science335, 1503–1506. 10.1126/science.1217697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feringa, F. M. & van der Kant, R. Cholesterol and Alzheimer’s disease; from risk genes to pathological effects. Front. Aging Neurosci.13, 690372. 10.3389/fnagi.2021.690372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson, L. A. et al. Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol. Dis.64, 150–162. 10.1016/j.nbd.2013.12.016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin, M. G., Pfrieger, F. & Dotti, C. G. Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep.15, 1036–1052. 10.15252/embr.201439225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanekiyo, T., Liu, C. C., Shinohara, M., Li, J. & Bu, G. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J. Neurosci.32, 16458–16465. 10.1523/JNEUROSCI.3987-12.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu, Q. et al. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci.30, 17068–17078. 10.1523/JNEUROSCI.4067-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deane, R. et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron43, 333–344. 10.1016/j.neuron.2004.07.017 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Chen, K. et al. LRP1 is a neuronal receptor for alpha-synuclein uptake and spread. Mol. Neurodegener.17, 57. 10.1186/s13024-022-00560-w (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rex, J. et al. IL-1beta and TNFalpha differentially influence NF-kappaB activity and FasL-induced apoptosis in primary murine hepatocytes during LPS-induced inflammation. Front. Physiol.10, 117. 10.3389/fphys.2019.00117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corrao, G., Bagnardi, V., Zambon, A. & La Vecchia, C. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev. Med.38, 613–619. 10.1016/j.ypmed.2003.11.027 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Patel, N. S. et al. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm.2, 9. 10.1186/1742-2094-2-9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rea, I. M. et al. Age and age-related diseases: role of inflammation triggers and cytokines. Front. Immunol.9, 586. 10.3389/fimmu.2018.00586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi, D. S. et al. PKCepsilon increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. USA103, 8215–8220. 10.1073/pnas.0509725103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang, S., Kim, J. & Chang, K. A. Spatial memory deficiency early in 6xTg Alzheimer’s disease mouse model. Sci. Rep.11, 1334. 10.1038/s41598-020-79344-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen, B. et al. PET imaging in animal models of Alzheimer’s disease. Front. Neurosci.16, 872509. 10.3389/fnins.2022.872509 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang, H. et al. Characterization of AD-like phenotype in aged APPSwe/PS1dE9 mice. Age (Dordr)38, 303–322. 10.1007/s11357-016-9929-7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lonnemann, N., Korte, M. & Hosseini, S. Repeated performance of spatial memory tasks ameliorates cognitive decline in APP/PS1 mice. Behav. Brain Res.438, 114218. 10.1016/j.bbr.2022.114218 (2023). [DOI] [PubMed] [Google Scholar]
- 51.Zhu, S. et al. The role of neuroinflammation and amyloid in cognitive impairment in an APP/PS1 transgenic mouse model of Alzheimer’s disease. CNS Neurosci. Ther.23, 310–320. 10.1111/cns.12677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uchoa, M. F., Moser, V. A. & Pike, C. J. Interactions between inflammation, sex steroids, and Alzheimer’s disease risk factors. Front. Neuroendocrinol.43, 60–82. 10.1016/j.yfrne.2016.09.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peng, J. et al. LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats. Redox Biol.21, 101121. 10.1016/j.redox.2019.101121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips, J. A. Dietary guidelines for Americans, 2020–2025. Workplace Health Saf.69, 395. 10.1177/21650799211026980 (2021). [DOI] [PubMed] [Google Scholar]
- 55.Osna, N. A., Donohue, T. M. Jr. & Kharbanda, K. K. Alcoholic liver disease: pathogenesis and current management. Alcohol. Res.38, 147–161 (2017). [PMC free article] [PubMed] [Google Scholar]
- 56.Shang, P. et al. Chronic alcohol exposure induces aberrant mitochondrial morphology and inhibits respiratory capacity in the medial prefrontal cortex of mice. Front. Neurosci.14, 561173. 10.3389/fnins.2020.561173 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morton, R. A., Diaz, M. R., Topper, L. A. & Valenzuela, C. F. Construction of vapor chambers used to expose mice to alcohol during the equivalent of all three trimesters of human development. J. Vis. Exp.10.3791/51839 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Janssen, P. M., Biesiadecki, B. J., Ziolo, M. T. & Davis, J. P. The need for speed: mice, men, and myocardial kinetic reserve. Circ. Res.119, 418–421. 10.1161/CIRCRESAHA.116.309126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoffman, J. L. et al. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Int. Rev. Neurobiol.148, 169–230. 10.1016/bs.irn.2019.10.017 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peng, B. et al. Role of alcohol drinking in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Int. J. Mol. Sci.10.3390/ijms21072316 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ran, M. et al. Alcohol-induced autophagy via upregulation of PIASy promotes HCV replication in human hepatoma cells. Cell Death Dis.9, 898. 10.1038/s41419-018-0845-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koob, G. F. & Le Moal, M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology24, 97–129. 10.1016/S0893-133X(00)00195-0 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Seitz, H. K. et al. Alcoholic liver disease. Nat. Rev. Dis. Primers4, 16. 10.1038/s41572-018-0014-7 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Bataller, R., Arab, J. P. & Shah, V. H. Alcohol-associated hepatitis. N Engl. J. Med.387, 2436–2448. 10.1056/NEJMra2207599 (2022). [DOI] [PubMed] [Google Scholar]
- 65.Kim, H. R. & Han, M. A. Association between Serum liver enzymes and metabolic syndrome in Korean adults. Int. J. Environ. Res. Public Health10.3390/ijerph15081658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alves, P. S., Camilo, E. A. & Correia, J. P. The SGOT/SGPT ratio in alcoholic liver disease. Acta Med. Port3, 255–260 (1981). [PubMed] [Google Scholar]
- 67.Salaspuro, M. Use of enzymes for the diagnosis of alcohol-related organ damage. Enzyme37, 87–107. 10.1159/000469243 (1987). [DOI] [PubMed] [Google Scholar]
- 68.Topiwala, A., Ebmeier, K. P., Maullin-Sapey, T. & Nichols, T. E. Alcohol consumption and MRI markers of brain structure and function: cohort study of 25,378 UK Biobank participants. Neuroimage Clin.35, 103066. 10.1016/j.nicl.2022.103066 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cacace, R., Sleegers, K. & Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement12, 733–748. 10.1016/j.jalz.2016.01.012 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Hoogmartens, J., Cacace, R. & Van Broeckhoven, C. Insight into the genetic etiology of Alzheimer’s disease: a comprehensive review of the role of rare variants. Alzheimers Dement (Amst)13, e12155. 10.1002/dad2.12155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wingo, T. S., Lah, J. J., Levey, A. I. & Cutler, D. J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol.69, 59–64. 10.1001/archneurol.2011.221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He, Y. et al. Silencing of LRP1 exacerbates inflammatory response via TLR4/NF-kappaB/MAPKs signaling pathways in APP/PS1 transgenic mice. Mol. Neurobiol.57, 3727–3743. 10.1007/s12035-020-01982-7 (2020). [DOI] [PubMed] [Google Scholar]
- 73.Wu, L. et al. Toll-like receptor 4: a promising therapeutic target for Alzheimer’s disease. Mediators Inflamm.2022, 7924199. 10.1155/2022/7924199 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanekiyo, T. & Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer’s disease. Front. Aging Neurosci.6, 93. 10.3389/fnagi.2014.00093 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu, Y. et al. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br. J. Pharmacol.165, 1319–1329. 10.1111/j.1476-5381.2011.01572.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song, H., Li, Y., Lee, J., Schwartz, A. L. & Bu, G. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res.69, 879–886. 10.1158/0008-5472.CAN-08-3379 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xing, P. et al. Roles of low-density lipoprotein receptor-related protein 1 in tumors. Chin J. Cancer35, 6. 10.1186/s40880-015-0064-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu, G. et al. Receptor-associated protein (RAP) plays a central role in modulating Abeta deposition in APP/PS1 transgenic mice. PLoS ONE3, e3159. 10.1371/journal.pone.0003159 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Potere, N. et al. Developing LRP1 agonists into a therapeutic strategy in acute myocardial infarction. Int. J. Mol. Sci.10.3390/ijms20030544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toldo, S. et al. Low-density lipoprotein receptor-related protein-1 is a therapeutic target in acute myocardial infarction. JACC Basic Transl. Sci.2, 561–574. 10.1016/j.jacbts.2017.05.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wohlford, G. F. et al. A phase 1 clinical trial of SP16, a first-in-class anti-inflammatory LRP1 agonist, in healthy volunteers. PLoS ONE16, e0247357. 10.1371/journal.pone.0247357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Varodayan, F. P. et al. Chronic ethanol induces a pro-inflammatory switch in interleukin-1beta regulation of GABAergic signaling in the medial prefrontal cortex of male mice. Brain Behav. Immun.110, 125–139. 10.1016/j.bbi.2023.02.020 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dukay, B. et al. Neuroinflammatory processes are augmented in mice overexpressing human heat-shock protein B1 following ethanol-induced brain injury. J. Neuroinflamm.18, 22. 10.1186/s12974-020-02070-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marsland, P. et al. Sex-specific effects of ethanol consumption in older Fischer 344 rats on microglial dynamics and Abeta((1–42)) accumulation. Alcohol107, 108–118. 10.1016/j.alcohol.2022.08.013 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Crespo-Castrillo, A. & Arevalo, M. A. Microglial and astrocytic function in physiological and pathological conditions: estrogenic modulation. Int. J. Mol. Sci.10.3390/ijms21093219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han, J., Fan, Y., Zhou, K., Blomgren, K. & Harris, R. A. Uncovering sex differences of rodent microglia. J. Neuroinflamm.18, 74. 10.1186/s12974-021-02124-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niu, P. et al. Immune regulation based on sex differences in ischemic stroke pathology. Front. Immunol.14, 1087815. 10.3389/fimmu.2023.1087815 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Erickson, E. K., Grantham, E. K., Warden, A. S. & Harris, R. A. Neuroimmune signaling in alcohol use disorder. Pharmacol. Biochem. Behav.177, 34–60. 10.1016/j.pbb.2018.12.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alfonso-Loeches, S., Pascual-Lucas, M., Blanco, A. M., Sanchez-Vera, I. & Guerri, C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci.30, 8285–8295. 10.1523/JNEUROSCI.0976-10.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stojakovic, A. et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun. Biol.4, 61. 10.1038/s42003-020-01584-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mirrione, M. M. et al. A novel approach for imaging brain-behavior relationships in mice reveals unexpected metabolic patterns during seizures in the absence of tissue plasminogen activator. Neuroimage38, 34–42. 10.1016/j.neuroimage.2007.06.032 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional files. Data analysis results are included in the table file.