

Abstract
Clinical bleeding events are reported here from 773 patients with B‐cell malignancies receiving pirtobrutinib monotherapy from the phase 1/2 BRUIN study (ClinicalTrials.gov identifier: NCT03740529), either in the presence or absence of antithrombotic therapy (antithrombotic exposed [AT‐E], n = 216; antithrombotic nonexposed [AT‐NE], n = 557). Among the AT‐E cohort, 51.9% received platelet aggregation inhibitors, 36.6% received direct factor Xa inhibitors, 18.5% received heparins, 5.6% received salicylic acid for indications other than platelet aggregation inhibition, and 2.3% received thrombolytics. Warfarin was not permitted. Any‐grade bleeding/bruising events occurred in 97 patients (44.9%; 95% confidence interval [CI], 38.3–51.5) in the AT‐E cohort and 181 patients (32.5%; 95% CI, 28.6–36.4) in the AT‐NE cohort. Most bleeding/bruising events in both cohorts began within the first 6 months of treatment (AT‐E: 65.4%; AT‐NE: 72.5%). Contusion was the most common bleeding/bruising event in both cohorts (AT‐E: 22.7%; AT‐NE: 18.1%). Grade ≥3 bleeding/bruising events were reported in six patients (2.8%) in the AT‐E cohort and 11 patients (2.0%) in the AT‐NE cohort. Bleeding/bruising events requiring or prolonging hospitalization were reported in 2.3% and 1.6% of patients in the AT‐E and AT‐NE cohorts, respectively. No bleeding/bruising events led to pirtobrutinib dose reduction or permanent discontinuation in the AT‐E cohort, and one patient (0.2%) in the AT‐NE cohort experienced an event requiring dose reduction. These data support the safety of pirtobrutinib in patients requiring antithrombotic therapies.
Keywords: antithrombotic therapy, B‐cell cancers, bleeding, Bruton tyrosine kinase inhibitor, pirtobrutinib
1. INTRODUCTION
Covalent Bruton tyrosine kinase (BTK) inhibitors have demonstrated remarkable efficacy in the treatment of chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and other non‐Hodgkin lymphomas (NHL) [1, 2, 3, 4, 5]. Despite their efficacy, an increased risk of bleeding, including potentially serious events such as central nervous system bleeding and major gastrointestinal hemorrhage, has been associated with covalent BTK inhibitors and has caused patients to discontinue BTK inhibitor treatment [6, 7]. This may pose a significant dilemma in the clinical management of patients with hematologic malignancies who require concomitant antithrombotic therapy for a variety of comorbid conditions such as cardiovascular disease or stroke prevention [7, 8]. Bleeding risks with BTK inhibitors are higher when used alongside dual‐antiplatelet therapy or systemic anticoagulation [9]. A study of 111 patients with MCL treated with ibrutinib, wherein 61 patients (55%) had received concomitant treatment with anticoagulants or antiplatelet agents, noted that bleeding events occurred more frequently in patients receiving these agents (any grade, 69%; grade 3–4, 8%) compared with those not receiving these treatments (any grade, 28%; grade 3–4, 4%) [10]. Another retrospective analysis of 70 patients who received ibrutinib reported that 19% of patients experienced major bleeding (grade ≥3 by the Common Terminology Criteria for Adverse Events [CTCAE]), and a majority were taking a concurrent antiplatelet agent (70%) or an anticoagulant (17%). Furthermore, the combined use of both antiplatelet and anticoagulant therapy significantly increased bleeding risk (hazard ratio: 19.2, 95% confidence interval [CI]: 2.3–166.7, p < 0.01) [9]. The added bleeding risk posed by the combination of BTK inhibitors and antiplatelet agents, especially in an elderly population more vulnerable to excessive bleeding, may limit the use of standard‐of‐care BTK inhibitor therapy in multiple hematologic malignancies for many patients [6, 11].
The mechanism underlying BTK inhibitor‐related bleeding has been largely attributed to a combination of on‐target BTK inhibition (through interference with platelet glycoprotein [GP] VI signaling) [12] compounded by additional off‐target inhibition of related kinases (such as tyrosine‐protein kinase [TEC]) important for platelet function [6, 13]. Specifically, in the absence of BTK, TEC signaling has been shown to regulate platelet activation through GPVI, resulting in a deficit in collagen‐mediated activation [14]. Notably, patients taking a BTK inhibitor show reductions in collagen‐mediated platelet aggregation, correlating with the occurrence of clinical bleeding [15].
In a pooled analysis of phase 3 studies in 330 patients with CLL receiving the first‐generation covalent BTK inhibitor, ibrutinib, bleeding/bruising events were observed in 55% of patients, and 21 patients reported 25 major hemorrhage events.[16] Second‐generation covalent BTK inhibitors, acalabrutinib, and zanubrutinib, have higher specificity for BTK and fewer off‐target toxicities [17]; nonetheless, bleeding events have still been observed with these agents, albeit at a lower frequency than seen with ibrutinib therapy [18]. Specifically, a pooled analysis of 1040 patients with mature B‐cell malignancies treated with acalabrutinib monotherapy reported that 46% experienced hemorrhage, with 4% of these being major hemorrhage (grade ≥3, serious, or affecting the central nervous system) [19]. Similarly, in a pooled analysis of 779 patients who received zanubrutinib monotherapy, 55% reported hemorrhage, with 4% experiencing major hemorrhage [20].
Pirtobrutinib is a potent and highly selective noncovalent (reversible) BTK inhibitor designed to address some of the limitations of covalent BTK inhibitors. Among its unique attributes, pirtobrutinib was highly selective for BTK in >98% of the human kinome and retained greater than 100‐fold selectivity over other tested kinases, minimizing off‐target activity and potentially lowering the risk of toxicities, such as bleeding [21]. These properties suggest pirtobrutinib may represent the potential to treat certain hematologic malignancies with improved tolerability and provide patients with another opportunity to safely utilize BTK inhibitor therapy [21, 22, 23].
Based on results from the BRUIN study, pirtobrutinib has been approved in the United States for the treatment of relapsed or refractory (R/R) MCL after at least two lines of systemic therapy, including a BTK inhibitor [24], and in Europe for the treatment of adult patients with R/R MCL who have previously received a BTK inhibitor [25]. On December 1, 2023, the United States Food and Drug Administration granted accelerated approval to pirtobrutinib for adults with CLL/SLL who have received at least two prior lines of therapy, including a BTK inhibitor and a B‐cell lymphoma 2 inhibitor [26]. The BRUIN study was a first‐in‐human, global, multi‐center evaluation of pirtobrutinib in patients previously treated for MCL, CLL, or other NHL [22]. In this study, low rates of major bleeding events were reported with pirtobrutinib, despite enrolling patients with a history of these events and patients on concurrent antithrombotic therapy. Furthermore, prolonged treatment (≥12 months) with pirtobrutinib continued to exhibit a safety profile suitable for extended use in B‐cell malignancies [27]. Pirtobrutinib additionally demonstrated robust response rates in patients with B‐cell malignancies, including R/R MCL and CLL, previously treated with a covalent BTK inhibitor [22, 28]. Here, we report the bleeding risk in patients with B‐cell malignancies from the BRUIN study who received pirtobrutinib monotherapy in the presence or absence of concomitant antithrombotic therapy.
2. METHODS
2.1. Study design and participants
The detailed study design and treatment were previously described [22]. Excluding warfarin, all other ongoing treatment with select antithrombotic agents such as direct factor Xa inhibitors, heparin anticoagulants, and platelet aggregation inhibitors was permitted during the study. Patients with clinically significant uncontrolled cardiovascular disease were excluded as were patients receiving current treatment with strong CYP3A4 inhibitors or inducers.
Patients received pirtobrutinib monotherapy in either phase 1 (at doses ranging from 25 to 300 mg once daily in 28‐day cycles) or phase 2 (at the recommended dose of 200 mg once daily) portion of the study [22]. Treatment continued until disease progression, unacceptable toxicity, or patient withdrawal. The safety cohort in this analysis comprised patients who received at least one dose of pirtobrutinib monotherapy, regardless of the type of B‐cell malignancy, as of the data cutoff date of July 29, 2022.
This trial was registered with ClinicalTrials.gov, NCT03740529. The protocol was approved by the institutional review boards or independent ethics committees overseeing each site. The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local laws. All patients provided written informed consent.
2.2. Study endpoint and definitions
Safety was determined by the frequency and severity of adverse events (AE) graded according to the NCI CTCAE, version 5·0. Treatment‐emergent adverse events (TEAE) were defined as all AE reported from the date of the first dose through 30 days (+7‐day window) after the date of the last dose or the start of subsequent anticancer therapy, whichever occurred earlier. Serious AEs were further defined per the International Council for Harmonisation requirements as any untoward medical occurrence that resulted in death, was life‐threatening, required hospitalization or prolongation of existing hospitalization, resulted in disability or incapacity, was a congenital anomaly or birth defect, or was deemed an important medical event upon further determination by the investigator.
For this post hoc analysis, the bleeding/bruising event category included bruising and hemorrhage/hematoma subcategories, as well as preferred terms such as hematuria, gingival bleeding, hemoptysis, epistaxis, and other less common bleeding events. The bruising subcategory was defined as an aggregate of contusion, petechiae, ecchymosis, and an increased tendency to bruise. The hemorrhage/hematoma subcategory was defined as a combination of all preferred terms including hemorrhage or hematoma. Thrombocytopenia at baseline was defined as platelet counts of ≤100 × 109/L.
2.3. Statistical analysis
Descriptive statistics were used to summarize baseline demographic and clinical characteristics and AE rates. The rates of bleeding events that led to study drug dose interruption, reduction, or discontinuation were also determined and summarized. The cumulative incidence of bleeding events over time was estimated under the competing risks framework [29]. In these analyses, time to the first TEAE occurrence was measured from the date of the first dose to the TEAE onset date, regardless of when concomitant medication was administered. Patients still on treatment were censored at the data cutoff date. Going off treatment without a TEAE was considered a competing risk, with the date of the competing risk set to the earliest of the study exit date (death date, withdrawal of consent date, etc.), subsequent anti‐cancer therapy date, or the date corresponding to 30 days (+7‐day window) after the last dose date. All analyses were performed using SAS, version 9.4.
3. RESULTS
3.1. Patient characteristics
From March 21, 2019, through July 29, 2022, 773 patients with B‐cell malignancies were enrolled in the BRUIN study and treated with pirtobrutinib monotherapy. Of these, 216 patients received pirtobrutinib with concomitant antithrombotic therapy (antithrombotic exposed [AT‐E] cohort), and 557 patients received pirtobrutinib without concomitant antithrombotic therapy (antithrombotic nonexposed [AT‐NE] cohort).
Baseline demographic and disease characteristics are summarized in Table 1. The median age was 71.5 years (range: 43.0–95.0) for patients in the AT‐E cohort and 67.0 (range: 26.0–88.0) in the AT‐NE cohort; most patients in both cohorts were male (72.7% and 64.5%, respectively). CLL was the most common disease type, with 44.5% in the AT‐E cohort and 38.4% in the AT‐NE cohort, followed by MCL (19.9% and 22.1%, respectively) and Waldenström macroglobulinemia (9.7% and 10.6%, respectively). At baseline, thrombocytopenia was present in 34 patients (15.7%) in the AT‐E cohort and 95 patients (17.1%) in the AT‐NE cohort.
TABLE 1.
Patient characteristics at baseline.
| Characteristics |
AT‐E cohort (n = 216) |
AT‐NE cohort (n = 557) |
|---|---|---|
| Age, median (range), years | 71.5 (43.0–95.0) | 67.0 (26.0–88.0) |
| <50 | 2 (0.9) | 30 (5.4) |
| 50–64 | 46 (21.3) | 196 (35.2) |
| 65–74 | 95 (44.0) | 220 (39.5) |
| 75–84 | 60 (27.8) | 100 (18.0) |
| ≥85 | 13 (6.0) | 11 (2.0) |
| Sex, n (%) | ||
| Female | 59 (27.3) | 198 (35.5) |
| Male | 157 (72.7) | 359 (64.5) |
| ECOG PS, n (%) | ||
| 0 | 100 (46.3) | 285 (51.2) |
| 1 | 102 (47.2) | 241 (43.3) |
| 2 | 14 (6.5) | 31 (5.6) |
| Disease types, n (%) | ||
| CLL | 97 (44.9) | 214 (38.4) |
| MCL | 43 (19.9) | 123 (22.1) |
| WM | 21 (9.7) | 59 (10.6) |
| RT‐DLBCL | 20 (9.3) | 62 (11.1) |
| FL | 15 (6.9) | 33 (5.9) |
| DLBCL | 9 (4.2) | 20 (3.6) |
| MZL | 5 (2.3) | 26 (4.7) |
| PCNSL | 3 (1.4) | 1 (0.2) |
| SLL | 1 (0.5) | 5 (0.9) |
| B‐PLL | 1 (0.5) | 4 (0.7) |
| HCL | 1 (0.5) | 3 (0.5) |
| Low‐grade transformation | 0 | 5 (0.9) |
| LPL | 0 | 1 (0.2) |
| MCL‐RT | 0 | 1 (0.2) |
| Baseline thrombocytopenia, n (%) | ||
| Present | 34 (15.7) | 95 (17.1) |
| Absent | 182 (84.3) | 462 (82.9) |
| Number of previous lines of systemic therapy, median (range) | 3 (1–12) | 3 (0–13) |
| Distribution, n (%) | ||
| 0 | 0 | 1 (0.2) |
| 1 | 19 (8.8) | 36 (6.5) |
| 2 | 60 (27.8) | 145 (26.0) |
| 3 | 49 (22.7) | 115 (20.6) |
| ≥4 | 88 (40.7) | 260 (46.7) |
| Previous systemic therapy, n (%) | ||
| BTK inhibitor | 166 (76.9) | 431 (77.4) |
| Chemotherapy | 178 (82.4) | 490 (88.0) |
| Anti‐CD20 antibody | 196 (90.7) | 527 (94.6) |
| Anti‐CD20 antibody plus chemotherapy | 174 (80.6) | 482 (86.5) |
| BCL2 inhibitor | 59 (27.3) | 169 (30.3) |
| PI3K agent | 33 (15.3) | 93 (16.7) |
| Immunomodulator | 35 (16.2) | 65 (11.7) |
| CAR T‐cell therapy | 12 (5.6) | 43 (7.7) |
| Stem cell transplant | 15 (6.9) | 60 (10.8) |
| Autologous | 12 (5.6) | 47 (8.4) |
| Allogeneic | 3 (1.4) | 18 (3.2) |
| Other systemic therapy | 63 (29.2) | 150 (26.9) |
| Number of previous lines of BTK inhibitor therapy, median (range) | 1 (0–5.0) | 1 (0–7.0) |
| 0 | 50 (23.1) | 126 (22.6) |
| 1 | 137 (63.4) | 341 (61.2) |
| 2 | 23 (10.6) | 76 (13.6) |
| ≥3 | 6 (2.8) | 14 (2.5) |
| Reason for discontinuation of any previous BTK inhibitora | ||
| Disease progression | 120 (72.3) | 348 (80.7) |
| Toxicity | 34 (20.5) | 61 (14.2) |
| Other | 12 (7.2) | 16 (3.7) |
| Missing | 0 | 6 (1.4) |
Abbreviations: AT‐E, antithrombotic exposed; AT‐NE, antithrombotic nonexposed; B‐PLL, B‐cell prolymphocytic leukemia; BTK, Bruton tyrosine kinase; CAR‐T, chimeric antigen receptor – T‐cell therapy; CD20, cluster of differentiate 20; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B‐cell lymphoma; ECOG PS, Eastern Cooperative Oncology Group performance status; FL, follicular lymphoma; HCL, hairy cell leukemia; LPL, lymphoplasmacytic lymphoma; MCL, mantle cell lymphoma; MCL‐RT mantle cell lymphoma‐radiation therapy; MZL, marginal zone lymphoma; PCNSL, primary central nervous system lymphoma; PI3K, phosphoinositide 3‐kinase; RT‐DLBCL, Richter transformation‐diffuse large B‐cell lymphoma; SLL, small lymphocytic lymphoma; WM, Waldenström's macroglobulinemia.
aIn the event that more than one reason for discontinuation was noted, disease progression took priority, followed by toxicity, and then other reasons.
Among the 216 patients in the AT‐E cohort, the median number of previous lines of systemic therapy was 3 (range: 1–12) and 166 patients (76.9%) had received a previous BTK inhibitor, 196 (90.7%) an anti‐CD20 antibody, 178 (82.4%) chemotherapy, and 174 (80.6%) an anti‐CD20 antibody plus chemotherapy. Among the 557 patients in the AT‐NE cohort, the median number of previous lines of therapy was 3 (range: 0–13) and 431 patients (77.4%) had received a previous BTK inhibitor, 527 (94.6%) an anti‐CD20 antibody, 490 (88.0%) chemotherapy, and 482 (86.5%) an anti‐CD20 antibody plus chemotherapy. Most patients in both cohorts had discontinued a previous BTK inhibitor due to disease progression (AT‐E cohort: 120/166 [72.3%]; AT‐NE cohort: 348/431 [80.7%]), or due to toxicity (AT‐E cohort: 34/166 [20.5%]; AT‐NE cohort: 61/431 [14.2%]). There were five patients (2.3%) in the AT‐E cohort and four patients (0.7%) in the AT‐NE cohort who discontinued a prior BTK inhibitor therapy due to a bleeding‐related toxicity.
The majority of patients received the recommended phase 2 dose of pirtobrutinib, 200 mg once daily, as their starting dose (AT‐E cohort: 90.3%, n = 195; AT‐NE cohort: 88.5%, n = 493). In the AT‐E cohort, the median time on pirtobrutinib treatment was 10.6 months (range, 0.3–37.4), and 86 patients (39.8%) were still receiving pirtobrutinib at data cutoff. In the AT‐NE cohort, the median time on pirtobrutinib treatment was 9.3 months (range, <0.1–39.9), and 223 patients (40.0%) were still receiving pirtobrutinib at data cutoff. The primary reason for pirtobrutinib discontinuation was progressive disease in both cohorts (AT‐E cohort: 82/216 [38.0%]; AT‐NE cohort: 229/557 [41.1%]).
Patients in the AT‐E cohort received a wide range of antithrombotic agents (Table 2); 60.2% started antithrombotic therapy before pirtobrutinib, 30.1% on the same day as pirtobrutinib or after starting pirtobrutinib, and 9.7% were unknown. Among patients in the AT‐E cohort, 51.9% of patients (n = 112) received a platelet aggregation inhibitor, 36.6% (n = 79) a direct factor Xa inhibitor, 18.5% (n = 40) heparins, 5.6% (n = 12) salicylic acid or derivatives for indications besides platelet aggregation inhibition including pain management, fever, and other uses, and 2.3% (n = 5) thrombolytics. Among the five patients who received thrombolytics, three appeared to have been for the prevention or treatment of central venous catheter occlusions and two were for unknown reasons. The median duration of antithrombotic therapy with complete start and end dates at the time of data cutoff was 6.4 months (interquartile range [IQR]: 0.3–25.5). Some patients (19.0%) had received more than 1 type of antithrombotic agent during the study. Of these patients, 63.4% took 2 or more antithrombotic therapies simultaneously.
TABLE 2.
Concomitant antithrombotic therapy.
| Agents | Patients, n (%) a |
|---|---|
| Platelet aggregation inhibitors | 112 (51.9) |
| Acetylsalicylic acid | 105 (48.6) |
| Clopidogrel | 7 (3.2) |
| Ticagrelor | 2 (0.9) |
| Dipyridamole | 1 (0.5) |
| Prasugrel | 1 (0.5) |
| Direct factor Xa inhibitors | 79 (36.6) |
| Apixaban | 56 (25.9) |
| Rivaroxaban | 22 (10.2) |
| Edoxaban | 4 (1.9) |
| Heparins | 40 (18.5) |
| Enoxaparin | 29 (13.4) |
| Heparin | 15 (6.9) |
| Dalteparin | 1 (0.5) |
| Sulodexide | 1 (0.5) |
| Salicylic acid and derivatives | 12 (5.6) |
| Acetylsalicylic acid b | 12 (5.6) |
| Thrombolytics | 5 (2.3) |
| Alteplase c | 5 (2.3) |
Patients may have received more than one type/subtype of concomitant antithrombotic therapy.
Patients were taking salicylic acid and derivatives for indications besides platelet aggregation inhibition including pain management, fever, and other uses.
Among the five patients who received thrombolytics, three appeared to have been for prevention or treatment of central venous catheter occlusions and two were for unknown reasons.
3.2. Overview of bleeding/bruising events
Any grade bleeding/bruising TEAE was reported in 97 patients (44.9%; 95% CI, 38.3–51.5) and 181 patients (32.5%; 95% CI, 28.6–36.4) in the AT‐E and AT‐NE cohorts, respectively (Table 3). The majority of bleeding/bruising events in both cohorts began within the first 6 months of treatment (AT‐E: 65.4%; AT‐NE: 72.5%). Most (>95%) bleeding/bruising events in both cohorts were grade ≤2. In the AT‐E cohort, six patients (2.8%) had a grade 3 bleeding/bruising event. Of these, two patients had a grade 3 bleeding/bruising event deemed related to pirtobrutinib by investigators: upper gastrointestinal bleeding and a hemarthrosis after a knee injury (1 each). No patients had a grade 4 or 5 bleeding/bruising event in the AT‐E cohort. In the AT‐NE cohort, grade ≥3 bleeding/bruising events occurred in 11 patients (2%), including 10 patients with grade 3 events, and 1 patient with a grade 5 bleeding/bruising event (hemorrhage due to a fall at home and considered unrelated to pirtobrutinib by investigators). Of these, four patients had a grade 3 bleeding/bruising event deemed related to pirtobrutinib by investigators: hematoma (n = 2), upper gastrointestinal hemorrhage (n = 1), and pulmonary hemorrhage (n = 1).
TABLE 3.
Overview of bleeding/bruising treatment‐emergent adverse events.
| Patient‐level summary |
AT‐E cohort (n = 216) |
AT‐NE cohort (n = 557) |
|---|---|---|
| Bleeding/bruising, n (%) | ||
| Any grade | 97 (44.9) | 181 (32.5) |
| Grade ≥3 | 6 (2.8) | 11 (2.0) |
| Serious bleeding/bruising, n (%) | 6 (2.8) | 10 (1.8) |
| Bleeding/bruising requiring | ||
| Dose interruption, n (%) | 8 (3.7) | 14 (2.5) |
| Dose reduction, n (%) | 0 | 1 (0.2) |
| Dose discontinuation, n (%) | 0 | 0 |
| Hospitalization a , n (%) | 5 (2.3) | 9 (1.6) |
| Median time to first onset of bleeding/bruising, weeks (IQR) | 8.1 (2.6–24.0) | 4.1 (1.3–16.1) |
| Event‐level summary | ||
|---|---|---|
| Total number of bleeding/bruising events | 157 | 296 |
| Recovered/resolved, n (%) | 90 (57.3) | 164 (55.4) |
| With treatment | 10 (6.4) | 17 (5.7) |
| Without treatment | 80 (51.0) | 147 (49.7) |
| Median duration b , weeks (IQR) | 2.1 (0.6–4.3) | 4.0 (1.1–7.9) |
Abbreviations: AT‐E, antithrombotic exposed; AT‐NE, antithrombotic nonexposed; IQR, interquartile range.
Including prolonged hospitalization.
Duration was calculated for 88 and 159 recovered/resolved adverse events with nonmissing end dates for the AT‐E cohort and AT‐NE cohorts, respectively.
Specific to the bruising subcategory, 60 patients (27.8%; 95% CI: 21.8–33.8) in the AT‐E cohort and 123 patients (22.1%; 95% CI: 18.6–25.5) in the AT‐NE cohort reported bruising events (all events were grade 1 and 2). Specific to the hemorrhage/hematoma subcategory, 34 patients (15.7%; 95% CI: 10.9–20.6) and 54 patients (9.7%; 95% CI: 7.2–12.2) in the AT‐E and AT‐NE cohorts reported events, respectively. Four patients (1.9%) in the AT‐E cohort and 10 patients (1.8%) in the AT‐NE cohort experienced grade ≥3 hemorrhage/hematoma.
Treatment‐emergent all‐cause and pirtobrutinib treatment‐related bleeding/bruising events are summarized in Table 4. In the AT‐E cohort, the most common bleeding/bruising events, regardless of attribution included contusion (22.7%), hematuria (5.6%), epistaxis (5.1%), petechiae (3.7%), and hematoma (3.2%). Similarly, in the AT‐NE cohort, the most common bleeding/bruising events, regardless of attribution, were contusion (18.1%), epistaxis (3.4%), and petechiae (4.8%). Notably, contusion was also the most frequent treatment‐related bleeding/bruising event in both cohorts (AT‐E: 14.4%; AT‐NE: 12.2%).
TABLE 4.
Summary of treatment‐emergent bleeding/bruising events. a
| All‐cause | Treatment‐related | |||||||
|---|---|---|---|---|---|---|---|---|
|
AT‐E cohort (n = 216) |
AT‐NE cohort (n = 557) |
AT‐E cohort (n = 216) |
AT‐NE cohort (n = 557) |
|||||
| Events |
All grades n (%) |
Grade ≥3 b n (%) |
All grades n (%) |
Grade ≥3 n (%) |
All grades n (%) |
Grade ≥3 b n (%) |
All grades n (%) |
Grade ≥3 n (%) |
| Bleeding/bruising | 97 (44.9) | 6 (2.8) | 181 (32.5) | 11 (2.0) | 46 (21.3) | 2 (0.9) | 103 (18.5) | 4 (0.7) |
| Hematuria | 12 (5.6) | 0 | 15 (2.7) | 0 | 1 (0.5) | 0 | 3 (0.5) | 0 |
| Epistaxis | 11 (5.1) | 0 | 19 (3.4) | 0 | 1 (0.5) | 0 | 12 (2.2) | 0 |
| Gingival bleeding | 3 (1.4) | 0 | 2 (0.4) | 0 | 1 (0.5) | 0 | 1 (0.2) | 0 |
| Hemoptysis | 3 (1.4) | 0 | 1 (0.2) | 0 | 1 (0.5) | 0 | 0 | 0 |
| Hemarthrosis | 2 (0.9) | 1 (0.5) | 2 (0.4) | 0 | 1 (0.5) | 1 (0.5) | 0 | 0 |
| Hematochezia | 2 (0.9) | 1 (0.5) | 1 (0.2) | 0 | 0 | 0 | 0 | 0 |
| Blood loss anemia | 1 (0.5) | 0 | 1 (0.2) | 1 (0.2) | 0 | 0 | 0 | 0 |
| Bleeding/bruising subcategories | ||||||||
|---|---|---|---|---|---|---|---|---|
| Bruising c | 60 (27.8) | 0 | 123 (22.1) | 0 | 34 (15.7) | 0 | 83 (14.9) | 0 |
| Contusion | 49 (22.7) | 0 | 101 (18.1) | 0 | 31 (14.4) | 0 | 68 (12.2) | 0 |
| Petechiae | 8 (3.7) | 0 | 27 (4.8) | 0 | 3 (1.4) | 0 | 19 (3.4) | 0 |
| Ecchymosis | 4 (1.9) | 0 | 6 (1.1) | 0 | 1 (0.5) | 0 | 2 (0.4) | 0 |
| Increased tendency to bruise | 3 (1.4) | 0 | 4 (0.7) | 0 | 0 | 0 | 3 (0.5) | 0 |
| Hemorrhage/hematoma c | 34 (15.7) | 4 (1.9) | 54 (9.7) | 10 (1.8) | 13 (6.0) | 1 (0.5) | 18 (3.2) | 4 (0.7) |
| Hematoma | 7 (3.2) | 0 | 10 (1.8) | 2 (0.4) | 3 (1.4) | 0 | 5 (0.9) | 2 (0.4) |
| Conjunctival hemorrhage | 4 (1.9) | 0 | 6 (1.1) | 0 | 2 (0.9) | 0 | 2 (0.4) | 0 |
| Upper gastrointestinal hemorrhage | 3 (1.4) | 2 (0.9) | 3 (0.5) | 2 (0.4) | 2 (0.9) | 1 (0.5) | 1 (0.2) | 1 (0.2) |
| Vitreous hemorrhage | 3 (1.4) | 0 | 1 (0.2) | 0 | 2 (0.9) | 0 | 1 (0.2) | 0 |
| Hemorrhage | 1 (0.5) | 0 | 5 (0.9) | 1 (0.2) | 1 (0.5) | 0 | 3 (0.5) | 0 |
| Gastrointestinal hemorrhage | 1 (0.5) | 0 | 2 (0.4) | 2 (0.4) | 0 | 0 | 0 | 0 |
| Postprocedural hemorrhage | 1 (0.5) | 1 (0.5) | 1 (0.2) | 1 (0.2) | 0 | 0 | 0 | 0 |
| Subdural hematoma | 2 (0.9) | 1 (0.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Pulmonary hemorrhage | 0 | 0 | 1 (0.2) | 1 (0.2) | 0 | 0 | 1 (0.2) | 1 (0.2) |
| Subarachnoid hemorrhage | 0 | 0 | 1 (0.2) | 1 (0.2) | 0 | 0 | 0 | 0 |
Abbreviations: AT‐E, antithrombotic exposed; AT‐NE, antithrombotic nonexposed.
Events occurring in ≥3% of patients and events of interest occurring at a lower percentage are presented.
No grade 4–5 bleeding/bruising events occurred in patients in the AT‐E cohort.
Bruising and hemorrhage/hematoma are subcategories of bleeding/bruising.
In the AT‐E cohort, the first onset of most bleeding/bruising events occurred in the first 6 months of pirtobrutinib treatment, with the estimated cumulative incidence at 6, 12, 18, and 24 months being 34.9%, 41.2%, 44.3%, and 47.1%, respectively. Similarly, in the AT‐NE cohort, the estimated cumulative incidence at 6, 12, 18, and 24 months was 28.2%, 31.0%, 32.2%, and 33.0%, respectively. Cumulative incidence of bleeding/bruising events by AT‐E and AT‐NE cohorts is presented in Figure 1.
FIGURE 1.
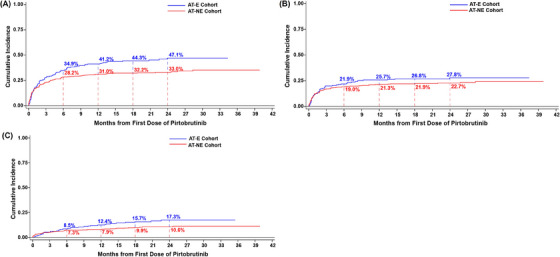
Cumulative incidence curves for bleeding/bruising events (A), and subcategories of bruising (B) and hemorrhage/hematoma (C). AT‐E, antithrombotic exposed; AT‐NE, antithrombotic nonexposed.
In the AT‐E cohort (n = 216), 157 individual bleeding/bruising events were reported in 97 patients (Table 3). Among those with events, the median time to first onset was 8.1 weeks (IQR: 2.6–24.0). Of the 157 individual bleeding/bruising events, 90 (57.3%) had recovered or resolved by the data cutoff, and the median duration of a recovered or resolved event was 2.1 weeks (IQR: 0.6–4.3). Eight patients (3.7%) experienced bleeding/bruising events requiring a temporary pirtobrutinib dose interruption, and 5 patients (2.3%) had events requiring or prolonging hospitalization. A summary of patients with pirtobrutinib treatment‐related bleeding/bruising events can be found in Table S1. No bleeding/bruising events led to dose reduction or permanent discontinuation of pirtobrutinib.
In the AT‐NE cohort (n = 557), a total of 296 individual bleeding/bruising events were reported in 181 patients with a median time to first onset of 4.1 weeks (IQR: 1.3–16.1). Of the 296 individual bleeding/bruising events, 164 (55.4%) had recovered or resolved by the data cutoff, and the median duration of a recovered or resolved event was 4.0 weeks (IQR: 1.1–7.9). Bleeding/bruising events requiring pirtobrutinib dose interruption occurred in 14 patients (2.5%), 1 (0.2%) patient had an event leading to pirtobrutinib dose reduction, and 9 patients (1.6%) had events requiring or prolonging hospitalization. Table S1 presents an overview of pirtobrutinib treatment‐related bleeding/bruising events.
3.3. Bleeding/bruising events by antithrombotic therapy
The distribution of bleeding/bruising events by individual antithrombotic agents is presented in Table 5. Bleeding/bruising events of any grade were observed in 46.8% of patients who received direct factor Xa inhibitors, 55.0% who received heparins, 43.8% who received platelet aggregation inhibitors, 33.3% who received salicylic acid and its derivatives (for indications besides platelet aggregation inhibition including pain management, fever, and other uses), and 60% who were administered thrombolytics.
TABLE 5.
Treatment‐emergent bleeding/bruising events by antithrombotic therapy class.
| Antithrombotic agents a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Direct factor Xa inhibitors (n = 79) |
Heparins (n = 40) |
Platelet aggregation inhibitors (n = 112) |
Salicylic acid and derivatives b (n = 12) |
Thrombolytics c (n = 5) |
||||||
| Events | All grades n (%) |
Grade ≥3 n (%) |
All grades n (%) |
Grade ≥3 n (%) |
All grades n (%) |
Grade ≥3 n (%) |
All grades n (%) |
Grade ≥3 n (%) |
All grades n (%) |
Grade ≥3 n (%) |
| Bleeding/bruising | 37 (46.8) | 2 (2.5) | 22 (55.0) | 1 (2.5) | 49 (43.8) | 4 (3.6) | 4 (33.3) | 1 (8.3) | 3 (60) | 0 |
| Hematuria | 4 (5.1) | 0 | 2 (5) | 0 | 7 (6.3) | 0 | 0 | 0 | 0 | 0 |
| Epistaxis | 6 (7.6) | 0 | 2 (5) | 0 | 4 (3.6) | 0 | 2 (16.7) | 0 | 0 | 0 |
| Gingival bleeding | 3 (3.8) | 0 | 1 (2.5) | 0 | 2 (1.8) | 0 | 0 | 0 | 0 | 0 |
| Hemoptysis | 1 (1.3) | 0 | 2 (5) | 0 | 2 (1.8) | 0 | 0 | 0 | 1 (20) | 0 |
| Hemarthrosis | 1 (1.3) | 1 (1.3) | 1 (2.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hematochezia | 2 (2.5) | 1 (1.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Blood Loss anemia | 0 | 0 | 0 | 0 | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
| Bleeding/bruising subcategories | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bruising d | 22 (27.8) | 0 | 12 (30) | 0 | 32 (28.6) | 0 | 3 (25.0) | 0 | 2 (40) | 0 |
| Contusion | 18 (22.8) | 0 | 10 (25) | 0 | 25 (22.3) | 0 | 2 (16.7) | 0 | 1 (20) | 0 |
| Petechiae | 4 (5.1) | 0 | 1 (2.5) | 0 | 5 (4.5) | 0 | 1 (8.3) | 0 | 0 | 0 |
| Ecchymosis | 1 (1.3) | 0 | 1 (2.5) | 0 | 2 (1.8) | 0 | 0 | 0 | 1 (20) | 0 |
| Increased tendency to bruise | 1 (1.3) | 0 | 0 | 0 | 2 (1.8) | 0 | 0 | 0 | 0 | 0 |
| Hemorrhage/hematoma d | 13 (16.5) | 0 | 10 (25) | 1 (2.5) | 18 (16.1) | 4 (3.6) | 2 (16.7) | 1 (8.3) | 1 (20) | 0 |
| Hematoma | 2 (2.5) | 0 | 5 (12.5) | 0 | 3 (2.7) | 0 | 1 (8.3) | 0 | 0 | 0 |
| Conjunctival hemorrhage | 4 (5.1) | 0 | 0 | 0 | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
|
Upper gastrointestinal hemorrhage |
0 | 0 | 0 | 0 | 3 (2.7) | 2 (1.8) | 0 | 0 | 0 | 0 |
| Vitreous hemorrhage | 1 (1.3) | 0 | 1 (2.5) | 0 | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
| Hemorrhage | 1 (1.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal hemorrhage | 0 | 0 | 0 | 0 | 1 (0.9) | 0 | 0 | 0 | 0 | 0 |
| Postprocedural hemorrhage | 0 | 0 | 1 (2.5) | 1 (2.5) | 1 (0.9) | 1 (0.9) | 1 (8.3) | 1 (8.3) | 0 | 0 |
| Subdural hematoma | 0 | 0 | 1 (2.5) | 0 | 2 (1.8) | 1 (0.9) | 0 | 0 | 0 | 0 |
| Pulmonary hemorrhage | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Subarachnoid hemorrhage | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Patients may have received more than one type/subtype of concomitant antithrombotic therapy.
Patients were taking salicylic acid and derivatives for indications besides platelet aggregation inhibition including pain management, fever, and other uses.
Among the five patients who received thrombolytics, three appeared to have been for prevention or treatment of central venous catheter occlusions, and two were for unknown reasons.
Bruising and hemorrhage/hematoma are subcategories of bleeding/bruising.
In the subcategory of bruising, contusion was the most frequent AE, reported in 25 (22.3%), 18 (22.8%), 10 (25%), and 2 (16.7%) of the patients who received platelet aggregation inhibitors, direct factor Xa inhibitors, heparins, and salicylic acid and derivatives, respectively.
Any‐grade hemorrhage/hematoma occurred in 13 patients (16.5%) who received direct factor Xa inhibitors, 10 patients (25%) who received heparins, and 18 patients (16.1%) who received platelet aggregation inhibitors. Upper gastrointestinal hemorrhage was observed in three patients who received platelet aggregation inhibitors, including two patients who had grade ≥3 events.
Pirtobrutinib treatment‐related bleeding/bruising events by antithrombotic therapy are summarized in Table S2. Bleeding/bruising events by baseline thrombocytopenia status are summarized in Table S3.
4. DISCUSSION
The current post hoc analysis assessed the safety profile of pirtobrutinib with respect to bleeding risk in patients with B‐cell malignancies in the presence or absence of concomitant antithrombotic therapy. Overall, bleeding/bruising events were mostly low‐grade, with grade 3 events occurring in fewer than 3% of patients treated with pirtobrutinib. As expected, bleeding/bruising events were higher (44.9% vs. 32.5%) among patients who received concomitant antithrombotic therapy; however, no grade 4–5 bleeding/bruising events were reported in patients who received concomitant antithrombotic therapy. Bleeding/bruising events that required or prolonged hospitalization were uncommon in both cohorts, occurring in 2.3% and 1.6% of patients in the AT‐E and AT‐NE cohorts, respectively. In contrast, a similar study assessing the safety of ibrutinib when co‐administered with anticoagulants reported a 2.5‐fold higher risk of bleeding among patients treated with ibrutinib and concurrent anticoagulant use compared with those treated with ibrutinib alone [30].
The differential impact of noncovalent versus covalent BTK inhibitors on hemostasis is noteworthy as another potential mechanism behind bleeding with different BTK inhibitors. A recent study suggests that the reversible nature of pirtobrutinib can facilitate subsequent recovery of platelet function that might contribute to the low rates of hemorrhagic AEs [31]. Covalent BTK inhibitors irreversibly block BTK by modifying a cysteine in its ATP‐binding pocket. Recovery of platelet function from inhibition by covalent BTK inhibitors is dependent on platelet turnover, which has a half‐life of approximately 7 to 10 days [31]. Pirtobrutinib also targets the ATP binding pocket, but, in contrast, binds noncovalently to BTK and is reversible, so restoration of normal platelet function may be more likely determined by drug washout rather than platelet turnover [21, 22]. Although these observations generate a hypothesis with some evidence related to the differential effect of pirtobrutinib on platelet function as compared with covalent BTK inhibitors and associated bleeding risks, further studies are still warranted.
Regardless of concomitant antithrombotic therapy type and increased risk of bleeding/bruising with their use, patients experienced few bleeding/bruising events that resulted in hospitalization, treatment modification (dose reduction or interruption), or permanent discontinuation. These data represent the potential to improve tolerability and adherence for patients with risk factors associated with excessive bleeding and/or who require antithrombotic therapy to utilize BTK inhibitor therapy in the treatment of B‐cell cancers.
This analysis has some important limitations. Although the study population received a wide array of concomitant antithrombotic agents while on pirtobrutinib treatment, not all agents used in everyday clinical practice were represented among patient data and the number of patients was small for some subgroups. Information regarding whether the antithrombotic drugs received by patients were considered prophylactic or therapeutic was not fully available. Missing dates prevented the complete evaluation of bleeding/bruising risk during intervals of concomitant antithrombotic therapy, so analyses could only be performed in patients who received antithrombotic therapy at any time, regardless of when the bleeding/bruising events occurred before, during, or after antithrombotic therapy. Furthermore, determining the association of bleeding/bruising events with the use of antithrombotic agents is challenging especially if multiple concomitant medications were used. It is also worth noting that while some patients may have had disease‐associated bleeding disorders, these were not documented or systematically collected except for 1 patient with Waldenström's macroglobulinemia who had a documented bleeding disorder in this study. Additional follow‐up is required to assess the long‐term safety of patients and the interactions between BTK inhibitors and specific antithrombotic agent classes and their potential impact on bleeding risks.
Available covalent BTK inhibitors may increase the risk of bleeding/bruising events due to on and off‐target effects. Pirtobrutinib is a potent and highly selective noncovalent (reversible) BTK inhibitor, and the findings presented here support the safety and manageability of the risk of bleeding in patients with B‐cell malignancies who require concomitant antithrombotic therapy.
AUTHOR CONTRIBUTIONS
Donald E. Tsai conceptualized and designed the study. Arrin Kontos, Heiko Konig, Amy S. Ruppert, Anindya Chatterjee, Richard Sizelove, Livia Compte, and Donald E. Tsai verified and interpreted the acquired study data and performed the analysis. Amy S. Ruppert and Richard Sizelove conducted the statistical analyses. Nicole Lamanna, Constantine S. Tam, Jennifer A. Woyach, Alvaro J. Alencar, M. Lia Palomba, Pier Luigi Zinzani, Ian W. Flinn, Bita Fakhri, Jonathon B. Cohen, and Wojciech Jurczak acquired and interpreted the study data. All authors had access to the clinical data. All authors participated in the interpretation of the study results and the drafting, critical revision, and approval of the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
NL reports research funding from Genentech, TG Therapeutics, BeiGene, AstraZeneca, AbbVie, MingSight, Eli Lilly and Company/Loxo@Lilly, Oncternal Therapeutics, and Octapharma, and consulting fees/honoraria from Genentech, Pharmacyclics, BeiGene, AstraZeneca, AbbVie, Adaptive Biotechnologies, Eli Lilly and Company/Loxo@Lilly, and Janssen. CST reports honoraria from Loxo@Lilly. JAW reports grants from the National Cancer Institute, Leukemia and Lymphoma Society, and CLL Global Society; consulting fees from AbbVie, AstraZeneca, BeiGene, Genentech, Janssen, Loxo@Lilly, Merck, Newave, and Pharmacyclics; and is on the advisory board of Gilead. AJA reports honoraria from Dr. Reddy's; is on advisory boards for Genentech, Eli Lilly and Company, Amgen, TG Therapeutics, Incyte, BeiGene, Janssen, Epizyme, and SeaGen; and reports research funding from Eli Lilly and Company, Incyte, and BeiGene. MLP reports participation on a data safety monitoring board or advisory board at Bristol‐Myers Squibb, Cellectar Biosciences, MustangBio, and Synthekine. PLZ reports membership on an entity's board of directors or advisory committee at AstraZeneca, Sandoz, Celltrion Healthcare, MSD, Secura Bio, Gilead, Janssen‐Cilag, Bristol‐Myers Squibb, Takeda, Roche, Servier Pharma, Kyowa Kirin, EUSA Pharma, Novartis, ADC Therapeutics, Incyte, and BeiGene; is on the speakers bureau of AstraZeneca, Celltrion Healthcare, MSD, Gilead, Janssen‐Cilag, Bristol‐Myers Squibb, Takeda, Roche, Servier Pharma, Kyowa Kirin, EUSA Pharma, Novartis, Incyte, and BeiGene; and has consultancy roles at MSD, EUSA Pharma, and Novartis. IWF reports consultancy roles at Genmab, BeiGene, Genentech, Secura Bio, Hutchison MediPharma, Kite Pharma, InnoCare Pharma, AbbVie, Novartis, Myeloid Therapeutics, Servier Pharma, Century Therapeutics, TG Therapeutics, and Vincerx Pharma. BF reports consultancy roles and membership on an entity's board of directors or advisory committee at BeiGene, AstraZeneca, ADC Therapeutics, AbbVie, Bristol‐Myers Squibb/Juno, Genentech, Genmab/AbbVie, Loxo@Lilly, and Pharmacyclics; and research funding from Genentech, Genmab/AbbVie, Loxo@Lilly, and Pharmacyclics. JBC reports research funding from Bristol‐Myers Squibb/Celgene, Novartis, Genentech, BioInvent, LAM Therapeutics, Takeda, ADC Therapeutics, AbbVie, and Loxo/Lilly; and consultancy roles at AstraZeneca, Janssen, BeiGene, and Loxo/Lilly. WJ reports research funding from Eli Lilly and Company; grants from AstraZeneca and BeiGene; and is on the advisory board for Eli Lilly and Company, AstraZeneca, and BeiGene. AK, HK, AC, and DET report full‐time employment with Loxo Oncology during the conduct of the study. ASR, RS, and LC report full‐time employment with Eli Lilly and Company during the conduct of the study.
ETHICS STATEMENT
The protocol was approved by the institutional review boards or independent ethics committees overseeing each site. The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local laws.
PATIENT CONSENT STATEMENT
Written informed consent was obtained from all the patients.
CLINICAL TRIAL REGISTRATION
ClinicalTrials.gov (NCT03740529).
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
The authors thank all the patients and their caregivers for participating in this trial. The authors also thank all the investigators and their support staff who participated in this work. Medical writing assistance and editing assistance, funded by Eli Lilly and Company, were provided by Anchal Sood, Ph.D., and Antonia Baldo, of Syneos Health. This work was funded by Loxo Oncology.
Lamanna N, Tam CS, Woyach JA, Alencar AJ, Palomba ML, Zinzani PL, et al. Evaluation of bleeding risk in patients who received pirtobrutinib in the presence or absence of antithrombotic therapy. eJHaem. 2024;5:929–939. 10.1002/jha2.1013
DATA AVAILABILITY STATEMENT
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date for data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data‐sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data‐sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
REFERENCES
- 1. Barr PM, Owen C, Robak T, et al. Up to 8‐year follow‐up from RESONATE‐2: first‐line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022;6(11):3440–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown JR, Eichhorst B, Hillmen P, et al. Zanubrutinib or ibrutinib in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2023;388(4):319–332. [DOI] [PubMed] [Google Scholar]
- 3. Song Y, Zhou K, Zou D, et al. Zanubrutinib in relapsed/refractory mantle cell lymphoma: long‐term efficacy and safety results from a phase 2 study. Blood. 2022;139(21):3148–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharman JP, Egyed M, Jurczak W, et al. Efficacy and safety in a 4‐year follow‐up of the ELEVATE‐TN study comparing acalabrutinib with or without obinutuzumab versus obinutuzumab plus chlorambucil in treatment‐naïve chronic lymphocytic leukemia. Leukemia. 2022;36(4):1171–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Montoya S, Thompson MC. Non‐covalent Bruton's tyrosine kinase inhibitors in the treatment of chronic lymphocytic leukemia. Cancers (Basel). 2023;15(14):3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. von Hundelshausen P, Siess W. Bleeding by Bruton tyrosine kinase‐inhibitors: dependency on drug type and disease. Cancers (Basel). 2021;13(5):1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fleming MR, Xiao L, Jackson KD, Beckman JA, Barac A, Moslehi JJ. Vascular impact of cancer therapies: the case of BTK (Bruton tyrosine kinase) inhibitors. Circ Res. 2021;128(12):1973–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pellegrini L, Novak U, Andres M, Suter T, Nagler M. Risk of bleeding complications and atrial fibrillation associated with ibrutinib treatment: a systematic review and meta‐analysis. Crit Rev Oncol Hematol. 2021;159:103238. [DOI] [PubMed] [Google Scholar]
- 9. Mock J, Kunk PR, Palkimas S, et al. Risk of major bleeding with ibrutinib. Clin Lymphoma Myeloma Leuk. 2018;18(11):755–61. [DOI] [PubMed] [Google Scholar]
- 10. Wang ML, Blum KA, Martin P, et al. Long‐term follow‐up of MCL patients treated with single‐agent ibrutinib: updated safety and efficacy results. Blood. 2015;126(6):739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mendez‐Ruiz A, Lossos IS, Cohen MG. Bleeding risk with antiplatelets and Bruton's tyrosine kinase inhibitors in patients with percutaneous coronary intervention. J Soc Cardiovasc Angiogr Interv. 2023; 2(3):100608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quek LS, Bolen J, Watson SP. A role for Bruton's tyrosine kinase (BTK) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–1140. [DOI] [PubMed] [Google Scholar]
- 13. Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor‐dependent platelet functions. Blood. 2014;124(26):3991–3995. [DOI] [PubMed] [Google Scholar]
- 14. Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592–3599. [DOI] [PubMed] [Google Scholar]
- 15. Lipsky AH, Farooqui MZ, Tian X, et al. Incidence and risk factors of bleeding‐related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015;100(12):1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coutre SE, Byrd JC, Hillmen P, et al. Long‐term safety of single‐agent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv. 2019;3(12):1799–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. St‐Pierre F, Ma S. Use of BTK inhibitors in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): a practical guidance. Blood Lymphat Cancer. 2022;12:81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frustaci AM, Deodato M, Zamprogna G, Cairoli R, Montillo M, Tedeschi A. Next generation BTK inhibitors in CLL: evolving challenges and new opportunities. Cancers (Basel). 2023;15(5):1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Furman RR, Byrd JC, Owen RG, et al. Pooled analysis of safety data from clinical trials evaluating acalabrutinib monotherapy in mature B‐cell malignancies. Leukemia. 2021;35:3201–3211. [DOI] [PubMed] [Google Scholar]
- 20. Tam CS, Dimopoulos M, Garcia‐Sanz R, et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B‐cell malignancies. Blood Adv. 2022;6(4):1296–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gomez EB, Ebata K, Randeria HS, et al. Preclinical characterization of pirtobrutinib, a highly selective, noncovalent (reversible) BTK inhibitor. Blood. 2023;142(1):62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B‐cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892–901. [DOI] [PubMed] [Google Scholar]
- 23. Thompson PA, Tam CS. Pirtobrutinib: a new hope for patients with BTK inhibitor‐refractory lymphoproliferative disorders. Blood. 2023;141(26):3137–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. U.S. Food and Drug Administration . FDA grants accelerated approval to pirtobrutinib for relapsed or refractory mantle cell lymphoma. 27 January 2023. Accessed 7 December 2023 https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐grants‐accelerated‐approval‐pirtobrutinib‐relapsed‐or‐refractory‐mantle‐cell‐lymphoma
- 25. European Medicines Agency . Jaypirca – EPAR medicine overview. 2023. Accessed 7 December 2023. https://www.ema.europa.eu/en/medicines/human/EPAR/jaypirca
- 26. The ASCO Post . FDA grants accelerated approval to pirtobrutnib for CLL/SLL. 2023. Accessed 29 December 2023. https://ascopost.com/news/december‐2023/fda‐grants‐accelerated‐approval‐to‐pirtobrutinib‐for‐cllsll
- 27. Coombs CC, Shah NN, Jurczak W, et al. Long‐term safety with ≥12 months of pirtobrutinib in relapsed/refractory (R/R) B‐cell malignancies. J Clin Oncol. 2023;41(16_suppl):7513. [Google Scholar]
- 28. Mato AR, Woyach JA, Brown JR, et al. Pirtobrutinib after a covalent BTK inhibitor in chronic lymphocytic leukemia. N Engl J Med. 2023;389(1):33–44. [DOI] [PubMed] [Google Scholar]
- 29. Gray RJ. A class of k‐sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16(3):1141–1154. [Google Scholar]
- 30. Allouchery M, Tomowiak C, Singier A, et al. Bleeding risk with concurrent use of anticoagulants and ibrutinib: a population‐based nested case‐control study. Br J Haematol. 2023;203(2):311–318. [DOI] [PubMed] [Google Scholar]
- 31. Bye AP, Kriek N, Sage T, et al. Pirtobrutinib results in reversible platelet dysfunction compared to ibrutinib and acalabrutinib. Haematologica. 2023;108(5):1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date for data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data‐sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data‐sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.