

Abstract
The genetic landscape underlying the transformation of splenic diffuse red pulp small B‐cell lymphoma (SDRPL) is not well understood. The present study aimed to unravel the genomic alterations involved in the progression and transformation of SDRPL. We performed genetic studies on both SDRPL and subsequent or synchronous diffuse large B cell lymphoma (DLBCL) samples in three SDRPL patients who eventually developed DLBCL. Our findings revealed that SDRPL cases progressing to DLBCL acquired genomic alterations in genes related to the cell cycle (CDKN2A/B, TP53, MYC and CCND3) and B cell development (BCL6).
Keywords: B‐cell developement, cell cycle, clonal relationship, genetic alterations, histologic transformation, splenic diffuse red pulp small B‐cell lymphoma
Splenic diffuse red pulp small B‐cell lymphoma (SDRPL) is an uncommon splenomegalic lymphoma recognised in the International Consensus Classification (ICC) [1] and WHO Classification, 5th edition [2]. The diagnosis relies on spleen histology since SDRPL lacks recurrent chromosomal abnormalities and shows morphological and phenotypic features overlapping with splenic marginal zone lymphoma (SMZL) and hairy cell leukaemia variant (Table S1). Recurrent somatic variants in SDRPL include those involving BCOR, CCND3, MAP2K1 and NOTCH pathway [3, 4, 5]. Contrarily, alterations of TP53 and MYD88 genes are rare. The disease course is usually indolent, but a few patients pursue an aggressive course [3, 6]. Data on transformation to diffuse large B‐cell lymphoma (DLBCL) is scant with only few cases documented [7, 8, 9] and the impact of genetic aberrations in the progression or transformation of SDRPL is largely unknown. Therefore, we performed a detailed histopathological and molecular characterization of three patients with SDRPL and subsequent or synchronic DLBCL.
Histology with immunohistochemistry and flow cytometry were available in SDRPL and DLBCL samples (Table S2) and reviewed by expert pathologists. Tumour DNA was isolated from fresh frozen tissues (n = 2), formalin‐fixed paraffin‐embedded tissues (n = 4) and involved peripheral blood (PB)(n = 1) using standard protocols. Copy number alterations (CNA) were examined using Oncoscan CNV assay (ThermoFisher Scientific), single nucleotide variants (SNV) and small insertions and deletions (indels) were evaluated by a custom next‐generation sequencing panel as described elsewhere [10]. Cytogenetics were performed in SDRPL samples and fluorescent in situ hybridization (FISH) with dual fusion IGH::BCL2 and BCL2, BCL6, MYC, and IGH break‐apart probes (Metasystems) in DLBCL and SDRPL. The clonal relationship between SDRPL and DLBCL was confirmed in two patients (cases 1 and 2) by demonstrating identical IGHV gene rearrangement using consensus primers for the IGHV FR2 or FR3 region; the third patient had two clones in the DLBCL sample.
Case 1 was a 64‐year‐old woman with lymphocytosis, splenomegaly and bone marrow (BM) involvement (Figure S1A). Splenectomy was performed (Figure 1A–C and Table S2). Cytogenetics showed a complex karyotype (CK): 45–47, XX, t(2;5)(p11;p15), −8, −8, del(14)(q24), −17, i(17)(q10), +mar1, +mar2, +mar3, +mar4[cp10]/46, XX[10]. We identified losses of 14q24, 14q32 and 17p (TP53), gain of 17q (BIRC5) and variants in CCND3, SPIB, STAT6, B2M, CREBBP, SMARCA4 and STAT3 (Figure 2A and Tables S3 and S4). The two variants in CCND3 were in the PEST domain of the protein (Figure 2D). A diagnosis of SDRPL (SDRPL1) was made. The patient received chemoimmunotherapy achieving a complete response (CR). Nine years later, the patient developed urinary tract obstruction showing relapse of the disease with bladder infiltration and metabolic uptake in lymph nodes next to the bladder, uterus and iliac regions at the PET‐CT (Figure S1A). A bladder biopsy showed infiltration by large CD20+ cells. A diagnosis of SDRPL transformed to non‐germinal centre (non‐GCB) DLBCL (SDRPL1‐T1) was made (Figure 1D–H and Table S2). The tumour displayed the same CNA profile as SDRPL1, and acquired deletions of 1p36 (ARID1A), 6p22‐p21 (HLA), 8p21‐p12, 12p13‐p12 (ETV6) and 22q13, gain in Xp11, and trisomy 16 (Table S3). Shared variants were detected in CCND3 (frameshift variant) and SPIB, both with higher allele frequencies in the DLBCL compared to SDRPL1; one somatic variant in PIK3CD was acquired (Figure 2A and Table S4). BCL2, BCL6, MYC and IGH were not rearranged. The patient was treated with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R‐CHOP) achieving a CR. One year later, the patient was readmitted with respiratory failure. CT scan showed retroperitoneal and mediastinal lymphadenopathy and pleural involvement by tumour cells. The patient had blood circulating large cells (29%) with the immunophenotype of DLBCL (SDRPL1‐T2). The SDRPL1‐T2 had the same CNA than the SDRPL1‐T1 and acquired 1q42‐q44 and 9p21 (CDKN2A/B) deletions and a gain in 15q22‐q26, maintained the CCND3 and SPIB variants; however, the PIK3CD variant was not detected (Figure 2A). The patient was treated with salvage chemoimmunotherapy, achieving a short‐lasting response and progressing with aggressive disease (lost to follow‐up) (Figure S1A).
FIGURE 1.
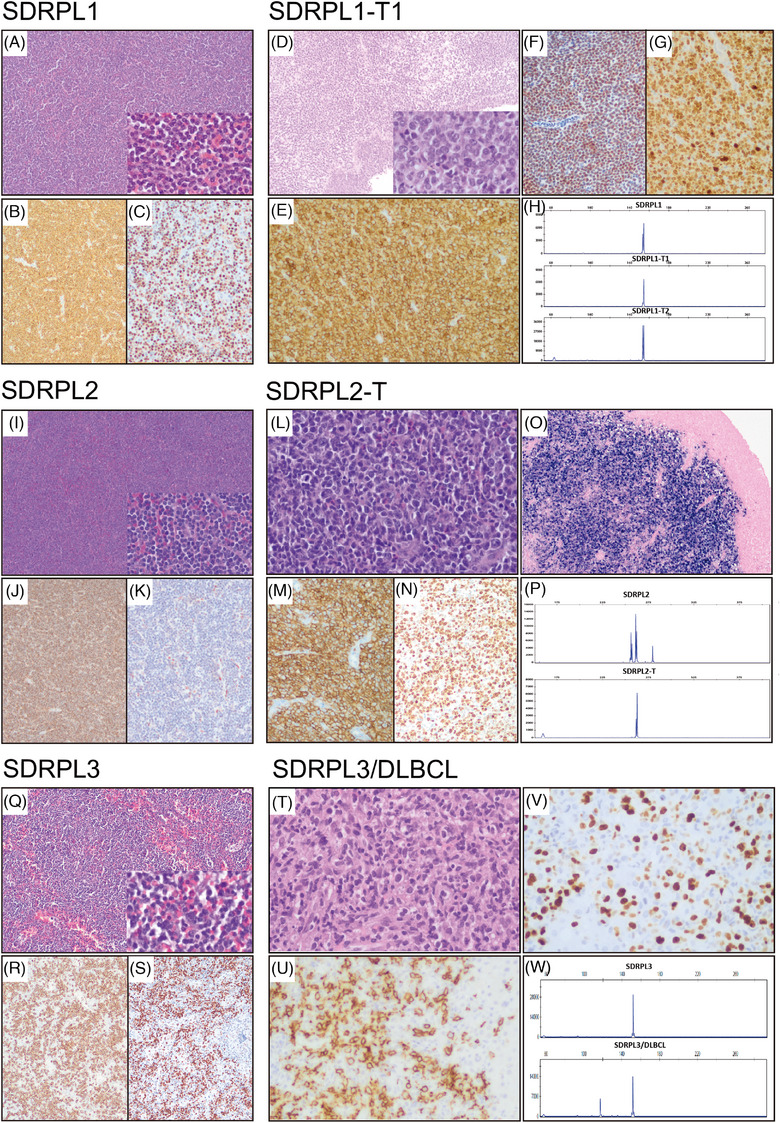
Morphologic and immunohistochemical features of splenic diffuse red pulp small B‐cell lymphoma (SDRPL) and transformed splenic diffuse red pulp lymphoma (SDRPL‐T). Histology of SDRPL1 (A–C): The panels show a proliferation of small lymphocytes diffusely infiltrating the red pulp of the spleen. They have a monomorphic appearance with round nuclei, clumped chromatin, and scant cytoplasm (A, hematoxylin and eosin (H&E) 10x; inset, 40x). On immunohistochemical analysis, they show positivity for CD20 (B, 10x), cyclin D3 (C, 20x), IgG and aberrant coexpression of CD2, and negativity for CD5, CD23, CD43, IgD, IgM, cyclin D1 and annexin A1. By flow cytometry, the cells are CD11c, FMC7 and kappa positive and negative for CD10, CD25 and CD103. Histology of SDRPL1‐T1 (D–G): There is a neoplastic proliferation of large cells invading the bladder muscular wall. The large cells have scant cytoplasm, irregular nuclei with coarse chromatin and nucleoli on higher power (D, H&E, 10x; inset, 40x). The cells express CD20 (E, 20x), BCL6, MUM1, MYC, cyclin D3 (F, 20x), IgG and aberrant CD2 and CD5. The Ki67 proliferation index is 95% (G, 20x). The cells are negative for CD10 and CD23. (H) IGH gene rearrangement studies in SDRPL1 (upper part) and SDRPL1‐T1 (lower part) showed same clonal peak in FR2. Histology of SDRPL2 (I–K): Histologic findings are similar to case 1 (I, H&E, 10x; inset, 40x). The cells are CD20 positive (J, 10x) with coexpression of IgG and DBA44, being negative for cyclin D1, annexin A1, cyclin D3 (K, 20x), CD25, LMO2, and BCL6. On flow cytometry the cells are positive for CD103, FMC7 and kappa, and negative for CD43, CD23, IgM, CD10 and CD5. Histology of SDRPL2‐T (L–O): Ileum with extensive ulceration and transmural infiltration by large, atypical cells (L, H&E, 40x). Mitotic figures are present. The large cells are positive for CD20 (M, 20x), BCL6 and MUM1, and negative for cyclin D1, IgD, CD10, CD5 and CD23. The Ki67 proliferation index is 90% (N, 20x). in situ hybridization for Epstein–Barr virus is positive (O, 10x). (P) IGH gene rearrangement studies in SDRPL2 (upper part) and SDRPL2‐T (lower part) showed the same clonal peak in FR2 displaying IGHJ4*02–IGHD1‐14*01–IGHV1‐18*04 rearrangement in both samples. Histology of SDRPL3 (Q–S): The splenic histology is similar to cases 1 and 2 (Q, H&E, 10x; inset, 40x). On immunohistochemistry the cells are CD20 (R, 10x), BCL2, IgD, IgM and cyclin D3 (S, 10x) positive and are negative for cyclin D1, CD23, CD10 and CD5. On flow cytometry, cells are FMC7 and Lambda positive and CD5, CD23, CD43 and CD103 negative. Histology of SDRPL3/DLBCL (T–V): Liver biopsy with a patchy lymphoid infiltration composed predominantly of large, atypical cells and some small lymphocytes (T, H&E, 40x). The neoplastic cells are CD20 (U, 40x), BCL2 and IgD positive, being negative for CD5 and CD10. Ki67 immunostaining shows that most of the large cells are proliferating in the context of an inflammatory background (V, 40x). (W) IGH gene rearrangement studies in SDRPL3 (upper part) and SDRPL3/DLBCL (lower part) showing the same clonal peak in FR3 shared between both samples, corresponding to IGHJ4*02‐IGHD3‐3*01 ‐ IGHV3‐21*01 rearrangements and an additional peak in SDRPL3/DLBCL showing IGHJ4*02 ‐ IGHD4‐23*01 ‐ IGHV3‐11*04 rearrangement.
FIGURE 2.

Genetic landscape of splenic diffuse red pulp small B‐cell lymphoma (SDRPL) and transformed splenic diffuse red pulp lymphoma (SDRPL‐T). Oncoprint displaying the genomic alterations: copy number alterations (CNA), copy neutral loss of heterozygosity (CN‐LOH), single nucleotide variants (SNVs) and small insertions and deletions (indels) found in paired SDRPL and SDRPL‐T samples of case 1(A), case 2(B) and case 3(C). SNVs and indels are depicted in the upper part, and CNA and CN‐LOH in the lower part. Each column represents an individual sample. Rows correspond to individual genetic alterations. The decreasing intensity colour squares represent lower variant allele frequency in comparison to other samples of the same patient. Some genetic data of SDRPL2 and SDRPL3 have been previously published [3]. (D) Distribution of CCND3 SNVs and indels across the cyclin D3 protein sequence found in cases 1 and 3. Different colours indicate different types of variants scattered within the functional domains of cyclin D3 protein.aa, amino acids.
Case 2 was a 45‐year‐old woman presenting with lymphocytosis, hypogammaglobulinemia and splenomegaly. Two years later, the patient progressed and was treated with one cycle of fludarabine achieving a partial response. Five years later, the patient developed progressive lymphocytosis and splenomegaly (Figure S1B). Splenectomy was performed and a diagnosis of SDRPL (SDRPL2) was made (Figure 1I–K and Table S2). This tumour carried deletions of 2p24, 19q13 and Xp21‐p11, a copy‐number neutral loss of heterozygosity (CN‐LOH) at 1p and variants in KMT2D, CREBBP, OSBPL10 and NFKBIE (Figure 2B and Tables S3 and S4). An IGH::BCL2 rearrangement was detected. A diagnosis of follicular lymphoma was ruled out by the histological splenic features with massive red pulp infiltration, positivity of IgG, DBA44 and CD103, and the lack of CD10, BCL6 and LMO2 expression. Four years later, a CT scan showed ileal thickening, and a PET‐CT revealed intense metabolic uptake in the ileum, Waldeyer's ring, oesophagus and small volume lymphadenopathy (Figure S1B). A biopsy of the ileum showed an infiltration of sheets of large cells that expressed the Epstein‐Barr virus (EBV), RNAs (EBERs) and LMP‐1, whilst EBNA‐2 was negative (EBV latency II). A diagnosis of non‐GCB, EBV‐positive DLBCL (SDRPL2‐T) was made (Figure 1L–P). In SDRPL2‐T, we observed the same alterations present in SDRPL2, except for the small deletions of 2p24 and 19q13, which were not present. A BCL6 rearrangement was acquired. The IGH::BCL2 rearrangement was also detected in SDRPL2‐T, whereas MYC was not rearranged. No somatic variants were acquired (Figure 2B and Table S4). The patient received R‐CHOP, achieving a CR with residual blood circulating small lymphocytes and was alive after 29 months of follow‐up (Figure S1B).
Case 3 was a 43‐year‐old man with a history of hepatitis C who presented with splenomegaly, BM lymphoid infiltration and increased serum lactate dehydrogenase and B2 microglobulin. A splenectomy was performed, and the histology was consistent with the diagnosis of SDRPL (SDRPL3) (Figure 1Q–S). There were blood circulating lymphoma cells. Following splenectomy, the patient developed systemic symptoms (Figure S1C). BM and liver biopsies were performed and both tissues, unlike the spleen, showed infiltration by large cells with residual SDRPL3 (SDRPL3/DLBCL) (Figure 1T–W and Table S2). In this case the IGHV gene rearrangement showed IGHV3‐21*01 in both samples, and an additional rearrangement (IGHV3‐11*04) in SDRPL3/DLBCL. The patient received R‐CHOP, achieving a CR, and was alive nine years after diagnosis without evidence of lymphoma (Figure S1C). In the SDRPL3 sample we detected losses in 1p36 (TNFRSF14), 9p21 (CDKN2A/B) and 19q13, gains in 5p (TERT), 17q22‐q25 (BIRC5), Xq26‐q28, and CN‐LOH in 19q12‐q13, and variants in CCND3, ARID1A, TNFAIP3, NOTCH1, POU2AF1, TET2, HIST1H1E, PTPRD, PAX5, KRAS, BTG1, DTX1, ZFP36L1 and KMT2D (Tables S3 and S4). SDRPL3 displayed a CK 47, XY, add(1)(p36), t(9;14)(p13;q32), i(17)(q10), +mar[3]/50, XY, idem, +4, +16, +21[8]/46, XY[10]. The translocation t(9;14)(p13;q32) involved IGH::PAX5, as confirmed by FISH[3]. Regarding SDRPL3/DLBCL, only 5p and Xq26‐q28 gains were detected subclonally. All variants in SDRPL3 were maintained in SDRPL3/DLBCL with a lower frequency, whilst new variants in ID3, SGK1, TP53, SMARCA4, ARID1B and MYC emerged (Figure 2C and Tables S3 and S4), and SDRPL3/DLBCL had an additional CCND3 alteration that truncated the PEST domain (Figure 2D and Table S4). BCL6 and MYC translocations were not detected.
From a clinical perspective, it is of interest that in all three cases the DLBCL was predominantly extranodal, involving bowel, urinary tract or liver. In patient two, the clonally related DLBCL was EBV‐driven, raising the hypothesis that this patient had underlying immune deficiency related to hypogammaglobulinaemia and fludarabine treatment. The outcome was favourable for patients 2 and 3 whilst patient 1 had multiple relapses and refractory disease. The DLBCL had a non‐GCB phenotype in the two cases evaluated. Noteworthy, all three DLBCL had a very high proliferation rate.
All cases showed aberrations that were exclusive to the diagnosis, as it has been described in other lymphomas [11, 12, 13]. Two patients had a CK, a risk factor for SMZL transformation [14]. Both patients had CCND3 alterations, an event documented in SDRPL [5], that resulted in a truncated cyclin D3 PEST domain, impairing protein degradation and leading to protein overexpression. Supporting this, we observed cyclin D3 overexpression by immunohistochemistry in both CCND3 mutated cases (Figure 1). Many alterations detected in our cases (i.e. TP53, CDKN2A/B, NOTCH1, CREBBP and TNFRSF14) had been reported in few SDRPL prior to transformation [7, 8, 9], suggesting that they could play a role in this event. All three cases in the present study acquired additional alterations in DLBCL samples, including a CDKN2A/B deletion and genomic complexity in Case 1, BCL6 rearrangement in Case 2, and TP53, MYC, SGK1, SMARCA4, ARID1B and CCND3 variants in Case 3. Of the three cases documented in the literature [8, 9], only one acquired an additional IGH::MYC rearrangement. The other two cases already harboured high risk alterations at diagnosis (NOTCH1 and RB1 variants and a CDKN2A/B homozygous deletion). Our Case 3 also acquired MYC mutations and had a frameshift NOTCH1 variant, together with CDKN2A/B deletion at diagnosis. Of interest, our Case 2, an EBV‐driven DLBCL, had very low genomic complexity, and did not acquire further somatic variants or CNA, except for a BCL6 rearrangement, which has been rarely described in transformed marginal zone lymphomas [11, 15].
The standard of care in clinical practice for transformed lymphomas, irrespective of their origin, is immunochemotherapy, with good results in patients not previously treated, but limited in the remainder. More recently, other “agnostic” approaches, including immunotherapies (i.e., bispecific antibodies) and cellular therapy (CAR‐T therapy) have shown high activity in relapsed/refractory aggressive lymphomas [16]. There is less evidence for small molecules with targeted effect, either alone or in combination with conventional immunochemotherapy. In this context, some of the genetic alterations herein found merit to be tested in vitro and eventually in vivo, since they might improve the treatment opportunities in some cases. There is evidence for antitumor activity of drugs targeting TP53 mutations [17] and different MYC alterations [18] in the lymphoma setting. Besides, drugs targeting SMARCB1/SMARCA4 or EZH2 alterations in paediatric tumours, such as Tazemetostat, seem to have a potential effect on disease stabilization [19]. Nevertheless, more data are needed in the setting of transformed lymphomas.
In summary, we document three SDRPL that underwent DLBCL transformation, which acquired genomic aberrations related to cell cycle (CDKN2A/B, TP53, MYC and CCND3) and B cell development (BCL6) that might have driven the progression of low‐grade B‐cell lymphoma to DLBCL.
AUTHOR CONTRIBUTIONS
Marta Grau and Melina Pol analysed and interpreted data and wrote the manuscript. Anna Montaner, Ferran Nadeu and Ian Márquez‐López performed bioinformatic analyses. Melina Pol, Gerard Frigola, Olga Balagué, Elias Campo and Estella Matutes reviewed the pathology material. Anna Montaner, Daniel Martinez, Jose Ramon Álamo, Dolors Colomer and Mónica Lopez‐Guerra provided and analysed data. Melika Bashiri and Silvia Ruiz‐Gaspà provided technical and logistic assistance, respectively. Pablo Mozas, Juan Correa, Eva Giné and Armando López‐Guillermo provided clinical data. Cristina López, Estella Matutes and Sílvia Beà designed the study, supervised the research, interpreted data and wrote the manuscript. All authors read, commented on and approved the manuscript.
CONFLICT OF INTEREST STATEMENT
Elias Campo has been a consultant for GenMab, and Takeda; has received research support from AstraZeneca; received honoraria from Janssen, EUSPharma, Takeda and Roche for speaking at educational activities; and is an inventor on a Lymphoma and Leukemia Molecular Profiling Project patent ‘Method for subtyping lymphoma subtypes by means of expression profiling’ (PCT/US2014/64161) and a bioinformatic tool (IgCaller) licenced to Diagnostic Longwood. Armando López‐Guillermo. served on the advisory board of Roche, Celgene, Novartis and Gilead/Kite, and received grants from Celgene and Gilead/Kite. Ferran Nadeu has received honoraria from Janssen, AbbVie, AstraZeneca and SOPHiA GENETICS for speaking at educational activities; has received research support from Gilead; and has licensed the use of the protected IgCaller algorithm to Diagnóstica Longwood. The remaining authors declare no competing financial interests.
ETHICS STATEMENT
The study was approved by the Ethics Committee of Hospital Clinic de Barcelona.
PATIENT CONSENT STATEMENT
The authors have confirmed patient consent statement is not needed for this submission.
CLINICAL TRIAL REGISTRATION
The authors have confirmed clinical trial registration is not needed for this submission.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
This study was supported by Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III, and Fondos FEDER: European Regional Development Fund “Una manera de fer Europa”: PI22/00203 (E.G. and S.B.), INT23/00037 (S.B.) and PI23/01500 (A.L‐G. and P.M.); Ministry of Science and Innovation (MCIN) MCIN/AEI/10.13039/501100011033; FEDER European Regional Development Fund “Una manera de fer Europa”: PID2021‐123054OB‐I00 (E.C.); Marató TV3 TV3‐Cancer (201904–30, S.B.); and Generalitat de Catalunya Suport Grups de Recerca AGAUR [2021‐SGR‐01293 (S.B.), 2021‐SGR‐01172 (E.C.)]. M.G. was funded by FPI pre‐doctoral fellowship: PRE2019‐088443. M.P. was funded by the 2021‐Estella Matutes fellowship. C.L. was supported by postdoctoral Beatriu de Pinós grant from Secretaria d'Universitats i Recerca del Departament d'Empresa i Coneixement de la Generalitat de Catalunya and by Marie Sklodowska‐Curie COFUND program from H2020 (2018‐BP‐00055). E.C. is an Academia Researcher of the “Institució Catalana de Recerca i Estudis Avançats” of the Generalitat de Catalunya. This work was developed at the Centre Esther Koplowitz (CEK), Barcelona, Spain. The authors thank the Hematopathology Collection registered at the Biobank of Hospital Clínic‐IDIBAPS for sample procurement and to the IDIBAPS Functional Genomics core facility.
Grau M, Pol M, Montaner A, Mozas P, Nadeu F, Márquez‐López I, et al. The genomic landscape of transformed splenic diffuse red pulp small B‐cell lymphoma. eJHaem. 2024;5:1014–1020. 10.1002/jha2.1018
Marta Grau and Melina Pol contributed equally to this work.
Cristina López, Estella Matutes and Sílvia Beà jointly supervised this work.
DATA AVAILABILITY STATEMENT
Our genomic data set, including, NGS and CNA arrays, will be deposited at the European Genome‐phenome Archive (link will be available at the moment of publication).
REFERENCES
- 1. Campo E, Jaffe ES, Cook JR, Quintanilla‐Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140(11):1229–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBDeO, Berti E, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martinez D, Navarro A, Martinez‐Trillos A, Molina‐Urra R, Gonzalez‐Farre B, Salaverria I, et al. NOTCH1, TP53, and MAP2K1 mutations in splenic diffuse red pulp small B‐cell lymphoma are associated with progressive disease. Am J Surg Pathol. 2016;40(2):192–201. [DOI] [PubMed] [Google Scholar]
- 4. Jallades L, Baseggio L, Sujobert P, Huet S, Chabane K, Callet‐Bauchu E, et al. Exome sequencing identifies recurrent BCOR alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B‐cell lymphoma. Haematologica. 2017;102(10):1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Curiel‐Olmo S, Mondéjar R, Almaraz C, Mollejo M, Cereceda L, Marès R, et al. Splenic diffuse red pulp small B‐cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood. 2017;129(8):1042–1045. [DOI] [PubMed] [Google Scholar]
- 6. Kanellis G, Mollejo M, Montes‐Moreno S, Rodriguez‐Pinilla S‐M, Cigudosa JC, et al. Splenic diffuse red pulp small B‐cell lymphoma: revision of a series of cases reveals characteristic clinico‐pathological features. Haematologica. 2010;95(7):1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lopedote P, Al NA, Malek A, Faller G, Hattar M, Dow E, et al. Early progression and transformation of a splenic diffuse red pulp small B‐cell lymphoma with NOTCH1, ARID2, CREBBP, and TNFRSF14 gene mutations. Leuk Res Rep. 2023;20:100384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christensen L‐M, Severinsen MT, Katoch P, Ovlisen AK, Hoyer T, Jensen P, et al. An aggressive course of transformed splenic diffuse red pulp small B‐cell lymphoma with novel somatic loss‐of‐function mutation in RB1 . J Hematol. 2023;12(3):118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cook JR, Amador C, Czader M, Duffield A, Goodlad J, Ott G, et al. Transformations of marginal zone lymphomas and lymphoplasmacytic lymphomas: report from the 2021 SH/EAHP workshop. Am J Clin Pathol. 2023;159(6):614–625. [DOI] [PubMed] [Google Scholar]
- 10. Mozas P, López C, Grau M, Nadeu F, Clot G, Valle S, et al. Genomic landscape of follicular lymphoma across a wide spectrum of clinical behaviors. Hematol Oncol. 2023;41(4):631–643. [DOI] [PubMed] [Google Scholar]
- 11. Grau M, López C, Navarro A, Frigola G, Nadeu F, Clot G, et al. Unraveling the genetics of transformed splenic marginal zone lymphoma. Blood Adv. 2023;7(14):3695–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nadeu F, Royo R, Massoni‐Badosa R, Playa‐Albinyana H, Garcia‐Torre B, Duran‐Ferrer M, et al. Detection of early seeding of Richter transformation in chronic lymphocytic leukemia. Nat Med. 2022;28(8):1662–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bastidas‐Mora G, Beà S, Navarro A, Gine E, Costa D, Delgado J, et al. Clinico‐biological features and outcome of patients with splenic marginal zone lymphoma with histological transformation. Br J Haematol. 2022;196(1):146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Flossbach L, Antoneag E, Buck M, Siebert R, Mattfeldt T, Möller P, et al. BCL6 gene rearrangement and protein expression are associated with large cell presentation of extranodal marginal zone B‐cell lymphoma of mucosa‐associated lymphoid tissue. Int J Cancer. 2011;129(1):70–77. [DOI] [PubMed] [Google Scholar]
- 16. Vodicka P, Klener P, Trneny M. Diffuse large B‐cell lymphoma (DLBCL): early patient management and emerging treatment options. Onco Targets Ther. 2022;151481–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H, Guo M, Wei H, Chen Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduction Targeted Ther. 2023;8(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Witte HM, Riedl J, Künstner A, Fähnrich A, Ketzer J, Fliedner SMJ, et al. Molecularly stratified treatment options in primary refractory DLBCL/HGBL with MYC and BCL2 or BCL6 rearrangements (HGBL, NOS with MYC/BCL6). Target Oncol. 2023;18(5):749–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chi SN, Yi JS, Williams PM, Roy‐Chowdhuri S, Patton DR, Coffey BD, et al. Tazemetostat for tumors harboring SMARCB1/SMARCA4 or EZH2 alterations: results from NCI‐COG pediatric MATCH APEC1621C. J Natl Cancer Inst. 2023;115(11):1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Our genomic data set, including, NGS and CNA arrays, will be deposited at the European Genome‐phenome Archive (link will be available at the moment of publication).