

Abstract
The development of bimolecular homolytic substitution (SH2) catalysis has expanded cross-coupling logic by enabling the selective merger of any primary radical with any secondary or tertiary radical via a radical sorting mechanism1–8. Biomimetic9,10 SH2 catalysis can be used to merge common feedstock chemicals—such as alcohols, acids, and halides—in any permutation for the construction of a single C(sp3)–C(sp3) bond. The ability to sort these two distinct radicals across commercially available alkenes in a three-component manner would enable the simultaneous construction of two C(sp3)–C(sp3) bonds, greatly accelerating access to drug-like chemical space11. However, the simultaneous in situ formation of electrophilic and primary nucleophilic radicals in the presence of unactivated alkenes is problematic, typically leading to statistical radical recombination, hydrogen atom transfer, disproportionation, and other deleterious pathways12,13. Herein, we report the use of bimolecular homolytic substitution catalysis to sort an electrophilic radical and a nucleophilic radical across an unactivated alkene. This reaction involves the in situ formation of three distinct radical species, which are then differentiated by size and electronics, allowing for regioselective formation of desired dialkylated products. This work accelerates access to pharmaceutically relevant C(sp3)-rich molecules and defines a novel mechanistic paradigm for alkene dialkylation.
A key goal of organic chemistry is the development of new methods for the rapid synthesis of C(sp3)–C(sp3) bonds en route to three-dimensional drug-like molecules.14–16. Traditional cross-coupling paradigms rely on oxidative addition, transmetallation, and reductive elimination mechanistic steps that limit the pool of potential coupling partners. By contrast, bimolecular homolytic substitution (SH2) catalysis couples primary radicals with secondary or tertiary radicals through a radical sorting mechanism based on carbon–metal bond strength1–8,17. As this unique radical sorting mechanism is functional group-agnostic, abundant radical precursors, such as alcohols, acids, and halides, can be coupled in any desired combination to generate complex products from simple feedstock chemicals1–8 (Fig 1a). This novel approach greatly expands access to C(sp3)-rich chemical space and enables formation of otherwise elusive all-C(sp3) quaternary centers18. Although radical-sorting SH2 catalysis has been shown to construct a single C(sp3) bond from a variety of radical precursors, the more challenging three-component radical sorting mechanism has yet to be demonstrated.
Figure 1. Radical sorting-enabled alkene dialkylation.

a, Bimolecular homolytic substitution (SH2) radical sorting enables the use of any functional group in any combination for C(sp3)–C(sp3) bond formation. b, Three-component radical sorting enables alkene dialkylation. c, This work: alkene dialkylation of unactivated alkenes using primary alcohols and α-acyl chlorides as radical precursors. Boc, tert-butylcarbonyl; Me, methyl; Cbz, carbobenzyloxy; Ph, phenyl; Bn, Benzyl; NHC, N-heterocyclic carbene; Ni, nickel; PC, photocatalyst; Ar, aryl.
A one-step protocol for the regioselective dialkylation of unactivated alkenes is highly desirable11. Alkenes are widely available, and the simultaneous construction of two C(sp3)–C(sp3) bonds across an alkene would greatly accelerate access to therapeutically advantageous C(sp3)-rich small molecules14. Due to the propensity of alkyl-metal complexes to undergo β–H elimination19–21, existing methods for alkene dialkylation remain greatly limited, relying either on auxiliary functional groups to direct dialkylation22–24 or the presence of specific ground-state radical traps25. A general method in which two distinct radicals are formed and regioselectively added across any unactivated alkene represents an ideal approach.
We envisioned a catalytic alkene dialkylation platform commencing with addition of an electrophilic alkyl radical—such as trifluoromethyl or difluoroacyl radical—into an alkene. Subsequent radical–radical recombination of the resultant radical species with a nucleophilic alkyl radical would yield the dialkyl adduct in a single transformation (Fig. 1b). The proposed dialkylation would be expected to proceed with good regioselectivity and enable installation of electron-poor alkyl groups that are typically not compatible with nickel catalysis26. Through the course of the reaction, three distinct radicals would be formed and must be efficiently sorted. Unsurprisingly, the envisioned transformation does not proceed in the absence of any sorting catalysts; instead, deleterious pathways, including disproportionation, alkyl-alkyl dimerization, and hydrogen atom transfer (HAT) predominate12,13. We postulated that an appropriate SH2 catalyst, which has been shown to facilitate outer-sphere C(sp3)–C(sp3) bond formation, might be used to sort these three simultaneously generated radicals toward productive alkene dialkylation. For the hypothesized radical sorting to be operative, the catalyst used must be capable of preferentially binding primary alkyl radicals over high-energy electrophilic alkyl radicals and secondary or tertiary radicals, while still being capable of performing outer-sphere bimolecular homolytic substitution for C(sp3)–C(sp3) bond formation. Herein, we disclose a general strategy for the dialkylation of alkenes through the simultaneous generation of electrophilic alkyl radicals and primary nucleophilic radicals in the presence of unactivated alkenes (Fig. 1c). The functional group-agnostic nature of SH2 catalysis permits the use of commercially available primary alcohols and electron-poor alkyl chlorides as radical precursors, giving potential access to 2×1015 C(sp3)-rich dialkylated products27.
Mechanism
We envisioned that the alkene dialkylation would proceed via the mechanism outlined in Figure 2. Condensation of primary alcohol 1 onto a benzoxazolium salt (NHC) forms adduct 2 in situ28. Meanwhile, blue light excitation of photocatalyst 3 accesses a long-lived, triplet excited state 4 (E1/2red [*IrIII/IrII]=+1.21 vs. saturated calomel electrode (SCE) in MeCN)29. Stern-Volmer analysis (See SI pages S32–S33) suggests that 4 undergoes reductive quenching with 2. Subsequent deprotonation and facile β-scission, provides the desired primary alkyl radical (6) and a benign aromatized byproduct. The primary alkyl radical can then be captured by high valent nickel SH2 catalyst 7, producing nickel–alkyl complex 85. To close the photocatalytic cycle, reduced-state IrII (9) is capable of reducing an α-acyl alkyl chloride 10 (or dMesSCF3(OTf)) to produce an electrophilic carbon-centered radical (11) that can add into an unactivated alkene (12). Radical probes (See SI pages S28–S31) support the formation of tertiary radical (13) which is capable of being further functionalized30–32. This nucleophilic tertiary radical can undergo a SH2 reaction with 8, thereby regenerating 7 and forming the desired dialkylated product (14) (see SI for select mechanistic experiments). Key to the success of this reaction is the radical sorting of the many transient radicals, both electronically, through addition to the alkene, and sterically, through binding to a high valent nickel complex. We envisioned that this novel mechanistic paradigm might provide a general, modular strategy for the dialkylation of unactivated olefins.
Figure 2.
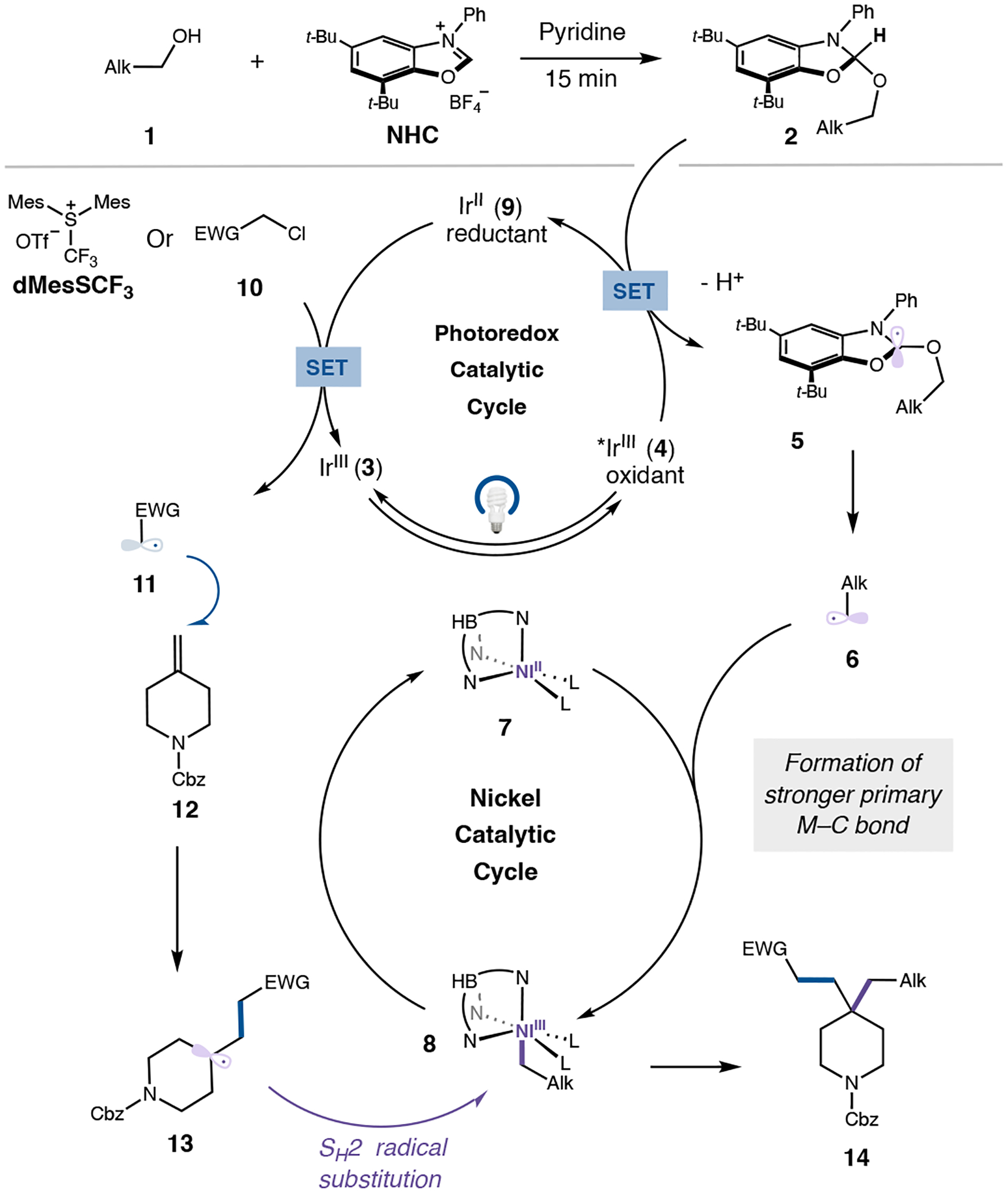
Proposed mechanism of alkene dialkylation Alk, Alkyl; t-Bu, tert-butyl; EWG, electron withdrawing group; dMesCF3(OTf), Dimesityl(trifluoromethyl)sulfonium trifluoromethanesulfonate; L, ligand.
Alkene scope
We first sought to interrogate the scope of the alkene coupling partner (Fig 3). We selected trifluoromethyl and difluoroacetamide radicals as the electrophilic alkyl radical partners, as both groups are important in drug discovery33, 34. For the primary radical partner, we opted to use the methyl radical, both for its ability to favorably influence the properties of drugs (termed the magic methyl effect) and due to the challenge often posed by its incorporation into complex molecules35. Gratifyingly, the coupling reaction proceeded efficiently across a wide range of alkenes. Unactivated terminal alkenes with relatively low π-nucleophilicity36 were dialkylated in good yields, tolerating protic functionality (15, 16) as well as homolytically labile allyl–benzylic C–H bonds (17, 18). The coordinatively saturated SH2 catalyst is incapable of oxidative addition; as such, aryl and alkyl halide-containing vinyl ethers and enamides were dialkylated in good yield (19–21). The generation of quaternary centers, a longstanding challenge in organic synthesis, can be achieved from 4, 5, 6, and 7-member rings as well as from acyclic 1,1-disubstituted alkenes (22–27). Notably, tertiary boronic esters (28), ethers (29), and ureas (30) were effectively formed under our reaction conditions. Moreover, a range of 1,2-disubstituted and trisubstituted alkenes were competent substrates (31–34). As a demonstration of the mild and robust nature of the reaction conditions, we successfully dialkylated several complex bioactive molecules. Specifically, efinconazole (35), paroxetine derivative (36), vinclozolin (37), retapamulin (38), quinine (39), and ataluren derivative (40) were dialkylated in good yields, showcasing the ability of the reaction to tolerate tertiary amines, alcohols, sulfides, quinuclidine, and oxidative addition-prone oxadiazole functionality. These results suggest that the protocol should be applicable to the late-stage dialkylation of alkenes.
Figure 3. Alkene scope.

aMeOH (2 equiv.), NHC-1 (2.1 equiv.), pyridine (2.1 equiv.), Ni(acac)2 (15 mol%), KTp* (15 mol%), (Ir[dF(CF3)ppy]2(dtbbpy))PF6 (1 mol%), dMesSCF3(OTf) (2 equiv.), alkene (0.50 mmol), CsOAc (2.5 equiv.), TBME/tAmOH (1:1, 0.05 M), IPR(2% light intensity, 12 h). bMeOH (2 equiv.), NHC-1 (2.1 equiv.), Pyridine (2.1 equiv.), Ni(acac)2 (25 mol%), 4CzPN (1 mol%), difluorochloroacetamide (2 equiv.), alkene (0.50 mmol), CsOAc (2.5 equiv.), TBME/MeCN (1:1, 0.05 M), IPR (50% light intensity, 12 h). Isolated yields. Ir, iridium; Ni(acac)(Tp*), Nickel(acetylacetone)tris(3,5-dimetyl-1-pyrazolyl)borate; Ir-1, Ir(dFCF3ppy)2(dtbbpy)PF6; TBS, tert-butyldimethylsilyl; Ln, ligand; Ni(acac)2, nickel(II) bis(acetylacetone); KTp*, potassium tris(3,5-dimetyl-1-pyrazolyl)borate; 4CzPN, 3,4,5,6-tetra(9H-carbazol-9-yl)phthalonitrile; IPR, integrated photoreactor.
Alkyl chloride scope
We next turned our attention to exploring the scope of the electrophilic radical partner (Fig 4). Alkyl chlorides were selected as radical precursors for their ease of synthesis, commercial availability, and enhanced stability over their bromide counterparts. We found that α-acyl radicals spanning a range of electrophilicity profiles, from α-ester to α-difluoroester radicals (41–43), reacted in good yields. Both acetamide (44), a ubiquitous moiety in drugs, and substituted difluoroacetamides (45), which are particularly important in fragment-based drug discovery, could be incorporated through the dialkylation protocol. Synthetically useful Horner-Wadsworth-Emmons reagents (46) and α-acyl chlorides (47) were prepared in good yields and offer the potential for further elaboration. In addition to α-acyl radicals, we found numerous other electrophilic carbon-centered radicals to be viable electrophilic radical partners, including α-difluorosulfonyl (48), difluorobenzyl (49), and α-nitrile (50) radicals.
Figure 4. Scope of electrophilic and nucleophilic radicals.
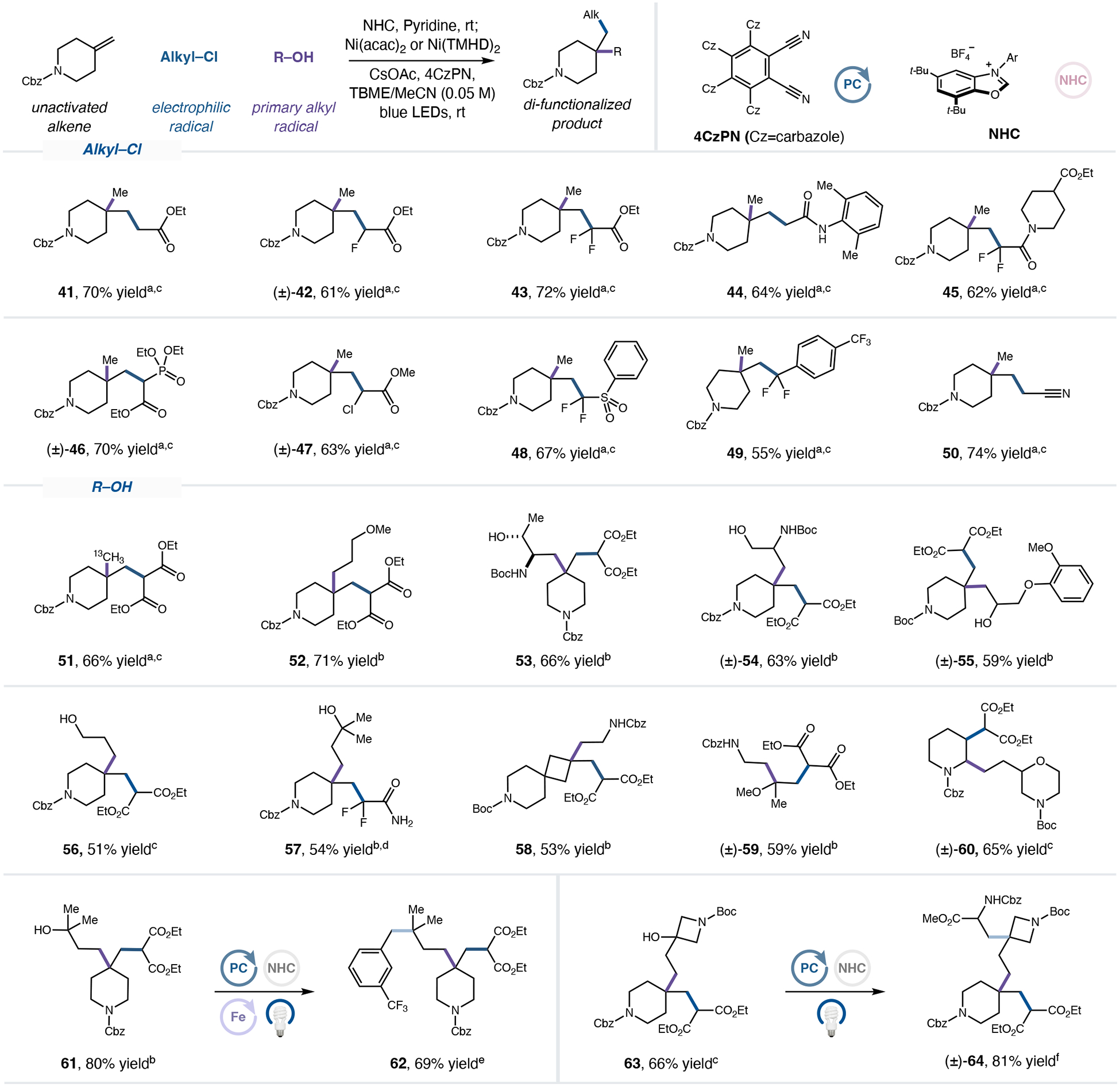
aMeOH (2.0 equiv.), NHC-1 (2.1 equiv.), pyridine (2.1 equiv.), Ni(acac)2 (25 mol%), 4CzPN (1 mol%), alkyl chloride (2 equiv.), alkene (0.50 mmol), CsOAc (2.5 equiv.), TBME/MeCN (1:1, 0.05 M). bprimary alcohol (2.5 equiv.), NHC-pCF3 (2.6 equiv.), pyridine (2.6 equiv.), Ni(TMHD)2 (25 mol%), 4CzPN (1 mol%), alkyl chloride (2.5 equiv.), alkene (0.50 mmol), CsOAc (3.0 equiv.), TBACl (0.6 equiv.) TBME/tAmOH (1:1, 0.05 M), IPR (5% intensity, 16 h). cSee supplementary information for experimental details. dYield determined by 19F NMR analysis with 1,4-difluorobenzene as an internal standard. eUtilizing 3-(trifluoromethyl)benzyl bromide as a cross-coupling partner; see supplementary information for experimental details. fUtilizing methyl 2-(((benzyloxy)carbonyl)amino)acrylate as a cross-coupling partner; see supplementary information for experimental details. All yields are isolated unless otherwise noted. Et, ethyl, Ni(TMHD)2, nickel(II) bis(2,2,6,6-tetramethyl-3,5-heptanedionate).
Primary alcohol scope
Finally, we explored the scope of the primary alcohol coupling partner. As shown in Figure 4, a broad scope of pharmaceutically relevant alkyl fragments were incorporated through our protocol. Coupling with commercially available 13C methanol served to install an isotopically labelled methyl group at the quaternary center (51). Moreover, a range of alcohols were found to be effective alkyl coupling partners, including ether alcohol (52), threonine (53), serine (54), guaifenesin (55), and other diols (56, 57). Reaction of diol substrates proceeds with full regioselectivity for the primary alcohol. These complex, coupled products (53-57) bear a free hydroxyl group that can be subjected to further elaboration via NHC activation. Moreover, 1,1-disubstituted alkenes in 4-membered rings (58) or acyclic substrates (59) were observed to undergo efficient alkylation with Cbz-glycinol. Notably, 1,2-disubstituted alkenes (60) were also readily dialkylated, providing an orthogonally protected morpholine scaffold. The complexity-building potential of this protocol was demonstrated through elaboration of dialkylated products into complex C(sp3) rich frameworks (61, 63). The tertiary alcohol of 61 was activated by NHC, and subsequent benzylation8 proceeded efficiently to generate a second quaternary center (62). In the case of 63, the tertiary alcohol served as a radical precursor en route to alkylation with dehydroalanine37 to yield complex scaffold, 64.
Conclusion
Herein, we describe a general procedure for the undirected dialkylation of unactivated alkenes. Key to the strategy is an outer-sphere C(sp3)–C(sp3) bond formation capable of forming quaternary centers. A wide range of unactivated alkenes were dialkylated, including tertiary amines, alcohols, aryl halides, and other reactive functionalities. Several examples of both the electrophilic radical and primary radical partners containing sites for further elaboration were demonstrated in good yield. Modulation of all three reaction components should allow for the rapid synthesis of C(sp3)-rich small molecule libraries. Furthermore, the described mechanistic paradigm provides a framework for future efforts in the development of C(sp3)–C(sp3) bond-forming alkene difunctionalization.
Supplementary Material
Acknowledgements
We thank Nathan W. Dow and Colin A. Gould for helpful scientific discussion. Research reported in this work was supported by the NIH National Institute of General Medical Sciences (R35 GM134897-04) and kind gifts from Merck, Pfizer, Janssen, Bristol-Myers Squibb, Genentech, and Genmab. J.Z.W. and W.L.L. acknowledge Princeton University, E. Taylor, and the Taylor family for an Edward C. Taylor Fellowship. W.L.L. acknowledges the National Science Foundation for a predoctoral fellowship (Award DGE-2039656). The authors thank Rebecca Lambert for assistance in preparing this manuscript.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Competing Interests D.W.C.M. declares a competing financial interest with respect to the Penn PhD Integrated Photoreactor, which is used to irradiate reactions in this work. The remaining authors declare no competing interests.
Data Availability:
The data supporting the findings of this study are available within the paper and its Supplementary Information.
References
- 1.Liu W, Lavagnino MN, Gould CA, Alcazar J & MacMillan DWC A biomimetic SH2 cross-coupling mechanism for quaternary sp3-carbon formation. Science 6572, 1258–1263 (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsymbal AV, Bizzini LD & MacMillan DWC Nickel catalysis vis SH2 homolytic substitution: the double decarboxylative cross-coupling of aliphatic acids. J. Am. Chem. Soc 144, 21278–21286 (2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mao E & MacMillan DWC Late-stage C(sp3)-H methylation of drug molecules. J. Am. Chem. Soc 145, 2787–2793 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakai HA & MacMillan DWC, Nontraditional fragment coupling of alcohol and carboxylic acids: C(sp3)–C(sp3) cross-coupling via radical sorting. J. Am. Chem. Soc 144, 6185–6192, (2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gould CA, Pace AL & MacMillan DWC, Rapid and modular access to quaternary carbons from tertiary alcohols via bimolecular homolytic substitution. J. Am. Chem. Soc 145, 16330–16336 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gan X, Kotesova S, Castanedo A, Green SA, Moller SLB & Shenvi RA, Iron-catalyzed hydrobenzylation: Stereoselective synthesis of (−)-Eugenial C. J. Am. Chem. Soc 145, 15714–15720 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ga. X, et al. Carbon quaternization of redox active esters and olefins via decarboxylative coupling. ChemRxiv November 17, 2023. DOI: 10.26434/chemrxiv-2023-7vb8x (Accessed 2023-11-19) [DOI] [Google Scholar]
- 8.Lovett VP, Kong L, Gan X & Shenvi RA Single catalyst double outer-sphere alkene cross-coupling. ChemRxiv November 17, 2023. DOI: 10.26434/chemrxiv-2023-zw9c2 (Accessed 2023-11-19) [DOI] [Google Scholar]
- 9.Zhang Q, van der Donk WA & Liu W Radical-mediated enzymatic methylation: A tale of two SAMS. Acc. Chem. Res 45, 555–564 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauerle MR, Schwalm EL & Booker SJ Mechanistic diversity of radical S-adenomethionine (SAM)-dependent methylation. J. Biol. Chem 290, 3995–4002 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qi X & Diao T Nickel-catalyzed dicarbofunctionalization of alkenes. ACS Catal 10, 8542–8556 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibian MJ & Corley RC Organic radical-radical reactions. disproportionations vs. combination. Chem. Rev 73, 441–465 (1973) [Google Scholar]
- 13.Leifert D & Studer A The persistent radical effect in organic synthesis. Angew. Chem. Int. Ed 59, 74–108 (2020) [DOI] [PubMed] [Google Scholar]
- 14.Lovering F Escape from flatland 2: complexity and promiscuity. Med. Chem. Commun 4, 515–519 (2013) [Google Scholar]
- 15.Choi J & Fu GC, Transition metal-catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dombrowski AW, et al. Expanding the medicinal chemist toolbox: Comparing seven C(sp2)–C(sp3) cross-coupling methods by library synthesis. ACS Med. Chem. Lett 11, 597–604 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simoes JAM & Beauchamp JL Transition metal-hydrogen and metal-carbon bond strengths: the keys to catalysis. Chem. Rev 90, 629–688 (1990) [Google Scholar]
- 18.Quasdorf KW & Overman LE Catalytic enantioselective synthesis of quaternary carbon stereocenters. Nature, 516, 181–191 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhungana RK, Sapkota RR, Wickham LM, Niroula D & Giri R Ni-Catalyzed regioselective 1,2-dialkylation of alkenes enabled by the formation of two C(sp3)–C(sp3) bonds. J. Am. Chem. Soc 142, 20930–20936 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jana R, Pathak TP & Sigman MS Advances in transition metal (Pd, Ni, Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev 111, 1417–1492 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisch AC & Beller M Catalysts for cross-coupling reactions with non-activated alkyl halides. Angew. Chem. Int. Ed 44, 674–688 (2005) [DOI] [PubMed] [Google Scholar]
- 22.Zhang JX & Shu W Ni-Catalyzed reductive 1,2-cross-dialkylation of unactivated alkenes with two alkyl bromides. Org. Lett 24, 3844–3849 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Dorsea J, Puyl VA, Tran VA, Liu M & Engle KM, Directed nickel-catalyzed 1,2-dialkylation of alkenyl carbonyl compounds. Chem. Sci 9, 5278–5283 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang T, Jiang Y, Luo Y, Lim JH, Lan Y & Koh MJ Chemoselective union of olefins, organohalides, and redox-active esters enables regioselective alkene dialkylation. J. Am. Chem. Soc 142, 21410–21419 (2020) [DOI] [PubMed] [Google Scholar]
- 25.Rao CQ, Zhang TZ, Liu HC & Huan HM Double alkyl–alkyl bond construction across alkenes enabled by nickel electron-shuttle catalysis, Nat. Catal (2023) [Google Scholar]
- 26.Le C, Chen TQ, Liang Y, Zhang P MacMillan D. W. C. A radical approach to the copper oxidative addition problem: trifluoromethylation of bromoarenes. Science, 360, 1010–1014 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reaxys search from October 2023 of commercially abailable fragments: 1° alcohols (261,696), α-acyl chlorides (29,000), alkenes (268553)
- 28.Dong Z & MacMillan DWC, Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 598, 451–456 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowry MS, et al. Single-layer electroluminescent devices and phtoinduced hydrogen production form an ionic iridium(III) complex. Chem. Mater 17, 5712–5719, (2005) [Google Scholar]
- 30.Kharasch MS, Skell PS & Fisher P, Reactions of atoms and free radicals in solution. XII. The addition of bromo esters to olefins. J. Am. Chem. Soc 70, 1055–1059 (1948) [Google Scholar]
- 31.Bian KJ, Nemoto D Jr, Kao SC, He Y, Li Y, Wang XS & West JG Modular difunctionalization of unactivated alkenes through bio-inspired radical ligand transfer catalysis. J. Am. Chem. Soc 144, 11810–11821 (2022) [DOI] [PubMed] [Google Scholar]
- 32.Patra S, Giri R & Katayev D Nitrative difunctionalization of alkenes via cobalt-mediated radical ligand transfer and radical-polar crossover photoredox catalysis. ACS Catal 13, 16136–16147 (2023) [Google Scholar]
- 33.Nair AS, et al. FDA-approved trifluoromethyl group-containing drugs: A review of 20 years. Processes 10, 2054–2078 (2022) [Google Scholar]
- 34.Vaas S, et al. Principles and applications of CF2X moieties as unconventional halogen bond donors in medicinal chemistry, chemical biology, and drug discovery. J. Med. Chem 66, 10202–10225 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schönherr H & Cernak T Profound methyl effects in drug discovery and a call for new C–H methylation reactions. Angew. Chem. Int. Ed 52, 12256–12267 (2013) [DOI] [PubMed] [Google Scholar]
- 36.Mayr H, Kempf B & Ofial AR, π-Nucleophilicity in carbon–carbon bond-forming reactions. Acc. Chem. Res 36, 66–77 (2003) [DOI] [PubMed] [Google Scholar]
- 37.Wang JZ, Sakai HA & MacMillan DWC, Alcohols as alkylating agents: Photoredox-catalyzed conjugate alkylation via in situ deoxygenation. Angew. Chem. Int. Ed 61, e202207150 (2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the paper and its Supplementary Information.