

Abstract
Malignant primary brain tumors are a group of highly aggressive and often infiltrating tumors that lack adequate therapeutic treatments to achieve long time survival. Complete tumor removal is one precondition to reach this goal. A promising approach to optimize resection margins and eliminate remaining infiltrative so-called guerilla cells is photodynamic therapy (PDT) using organic photosensitizers that can pass the disrupted blood–brain-barrier and selectively accumulate in tumor tissue. Hypericin fulfills these conditions and additionally offers outstanding photophysical properties, making it an excellent choice as a photosensitizing molecule for PDT. However, the actual hypericin-induced PDT cell death mechanism is still under debate. In this work, hypericin-induced PDT was investigated by employing the three distinct fluorescent probes hypericin, resorufin and propidium iodide (PI) in fluorescence-lifetime imaging microscopy (FLIM). This approach enables visualizing the PDT-induced photodamaging and dying of single, living glioma cells, as an in vitro tumor model for glioblastoma. Hypericin PDT and FLIM image acquisition were simultaneously induced by 405 nm laser irradiation and sequences of FLIM images and fluorescence spectra were recorded to analyze the PDT progression. The reproducibly observed cellular changes provide insight into the mechanism of cell death during PDT and suggest that apoptosis is the initial mechanism followed by necrosis after continued irradiation. These new insights into the mechanism of hypericin PDT of single glioma cells may help to adjust irradiation doses and improve the implementation as a therapy for primary brain tumors.
This study uses fluorescence lifetime imaging and spatially-resolved spectroscopy to explore morphological changes in single glioma cells during hypericin-induced photodynamic therapy, offering insights into the underlying cell death mechanisms.
Introduction
Among the deadliest and most challenging to treat cancers are malignant primary brain tumors, encompassing different tumor types depending on the cellular origin.1 They are known to grow aggressively, infiltrating the surrounding healthy brain tissue in the process.2,3 Many of the emerging primary brain tumors are astrocytomas, of which glioblastoma is the most common representative.4,5 The World Health Organization (WHO) classifies glioblastoma as a WHO grade IV astrocytoma due to its aggressive and fast growth and the high degree of tissue infiltration.6 To this day, there is no cure and treatment is limited to improving the patients’ quality of life until their inevitable death.7 The established treatment of primary brain tumors includes an initial tumor resection in a surgical procedure using fluorescence-supported tumor tissue visualization.8 For this purpose, the patient is administered a fluorescent dye, capable of passing the disrupted blood–brain-barrier that selectively accumulates in the cancerous tissue. Precise tumor removal is then aided by the use of intermittent dye-specific illumination, which allows better contrast between healthy and cancerous brain tissue.9 The application of 5-aminolevulinic acid (5-ALA), as a precursor dye, is currently considered the gold standard for primary brain tumor surgery.10,11 This molecule exhibits high water-solubility, which facilitates its implementation as pharmaceutical formulation, and thus its administration to the patient. After accumulating in the brain tumor, 5-ALA is enzymatically transformed into Protoporphyrin IX (PpIX),12 which is the fluorophore used for tumor visualization. The brain tumor surgery is followed by radiochemotherapy to reduce residual tissue and thus improve the overall tumor removal.13,14 However, these therapies do not selectively affect cancerous tissue and result in collateral damage of healthy, surrounding brain cells. Photodynamic therapy (PDT) is a recent therapeutic approach, with the goal of establishing a tumor specific therapeutic alternative to chemo- or radiotherapy. It focuses on enhancing the functionality of the already administered fluorescent dye used for tumor visualization to additionally act as a photosensitizing agent.15 In PDT, photosensitizers are optically excited followed by a transition to the triplet state, which further reacts with biological substrate or surrounding oxygen to form highly reactive oxygen species (ROS).16 These species induce a high oxidative stress and a pronounced level of photodamage inside the tumor cells, resulting in cell death.
As 5-ALA and thus PPIX are specifically enriched in brain tumors, their application in PDT was expected to be a promising approach. Several studies were performed to investigate the potential of 5-ALA-PDT for PDT applications.17,18 Eskiler et al. demonstrated the effectiveness of 5-ALA-PDT in the treatment of breast cancer,19 whereas its photodamaging capabilities on rhabdomyosarcoma were shown by Atif et al.20 Hypericin is a promising alternative photosensitizer for PDT. Like 5-ALA, it can pass the disturbed blood–brain barrier and additionally exhibits a high triplet quantum yield that allows for an effective ROS formation.21,22 A direct comparison between hypericin and 5-ALA revealed a significantly higher photo-cytotoxicity of hypericin.23 Hypericin uptake and distribution was further studied in tumor spheroid models24 followed by hypericin PDT.25 Although hypericin, like the photosensitive compound PpIX metabolized from 5-ALA, is mostly apolar, the two photosensitizers exhibit different accumulation behaviours in cancer cells. PpIX is synthesized from 5-ALA primarily in the mitochondria through the heme biosynthetic pathway, leading to its predominant accumulation in the mitochondria.26,27 In contrast, hypericin does not require metabolic conversion and accumulates based on its physicochemical properties and the cellular microenvironment. Hypericin has been shown to accumulate in the perinuclear region of various tumor cell lines, with primary enrichment in the endoplasmic reticulum (ER) and Golgi apparatus, as well as in mitochondria and lysosomes.28–30 The localization of the photosensitizer within the cell significantly influences PDT progression. In PpIX-induced PDT, mitochondrial damage is expected to play a central role. In contrast, hypericin-induced PDT may involve additional effects, such as endoplasmic reticulum (ER) stress or interference with protein processing, which could critically affect the cell death mechanism.
To further understand hypericin PDT, it is essential to uncover the mechanistic steps involved in the damage and cell death process it induces. Cellular damage can follow certain pathways until the cells are finally dead, which is very characteristic and can be assigned to concrete cell death mechanisms. The two most common programmed cell deaths are apoptosis and necrosis.31,32 Apoptosis can be triggered intrinsically via a mitochondrial pathway or by external stimuli.32 Depending on the pathway type, it undergoes a cascade of various protein or enzyme activations, including Bax, apoptotic protease, caspases or procaspase 8, that initiate the systematic progression of cellular degradation.32 Typical morphological features of apoptosis are cell and nucleus shrinkage, nuclear chromatin condensation, nucleus break up and membrane blebbing.33,34 Necrosis, however, is induced via caspases-independent pathways and shows a partly different morphological outcome, entailing bleb formation, cellular swelling, chromatin condensation and cellular lysis.35,36 Further cell death mechanisms are ferroptosis or paraptosis as well as several hybrid forms, which are all either morphologically or genetically distinguishable from one another.37,38 So far, hypericin PDT was tested on umbilical vein endothelial, HepG2 and A431 human epidermoid carcinoma cells, where apoptosis was observed to be the dominant cell death mechanism.39–41 Necrosis was demonstrated to be the type of cell death mechanism in human colon adenocarcinoma HT-29 cells,42 whereas paraptosis was identified to be the underlying cell death mechanism in OVCAR-5 cells.43 Glioma cell lines were (already) used as in vitro brain tumor models. Investigations upon hypericin PDT revealed either apoptosis or necrosis as the primary cell death mechanism.44–47 These studies were mainly performed with flow cytometry, biological assays, or different microscopy techniques, like confocal, conventional, and total internal reflection fluorescence microscopy. Distinguishing between different types of cell death is important for therapeutic applications due to their effect on the immune system. Necrosis can lead to inflammation through the release of intracellular contents. Apoptosis typically does not lead to inflammatory responses as the cell contents are contained and processed by phagocytes.48–51
This study deals with the investigation of hypericin PDT on single, living glioma cells by means of spatially resolved fluorescence spectroscopy and fluorescence lifetime imaging microscopy (FLIM). Three fluorescent markers: hypericin, resorufin and propidium iodide (PI), were chosen to initiate and monitor hypericin-induced PDT. FLIM is an ideal method for studying the effects of PDT on cell morphology as it enables intra-cellular differentiation between fluorescent dyes based on their distinct fluorescence lifetimes (FLTs), even when their emission spectra overlap significantly. Imaging the fluorescence lifetime ensures high image contrast in fully fluorescent samples and provides spatially-resolved, quantitative insights into the accumulation of the dyes and changes in their distribution and photophysical properties as PDT progresses.52–54 An introduction to the theory, acquisition, and analysis of fluorescence lifetime data is given in Section 1 of the ESI.† Combining FLIM with spatially resolved fluorescence spectroscopy allows imaging the photodamaging progression and identify the underlying cell death mechanism.
Materials and methods
Cell culture
Cell experiments were performed with the glioma cell line U-87 MG (ATCC HTB-14). Cells were grown in Eagle's Minimum essential medium (MEM, no glutamine and phenol red, Gibco™) supplemented with 10% fetal calve serum (FCS), 1% penicillin/streptomycin, 1% l-glutamine and 1% sodium pyruvate (all purchased from Gibco™). They were seeded with densities between 5000 cells per cm2 and 20 000 cells per cm2 and cultivated in glass-bottom cell culture dishes (Willco Wells, HBST-5040) for 24 h prior to further treatment. Cell culture dishes were equipped with glass coverslip bottoms of 170 μm thickness on average and coated with FCS to improve cell adherence. After 24 h, cells were mostly attached to the cell culture dish bottoms and used for hypericin incubation. Cultivation was accomplished at 37 °C and 5% CO2 atmosphere.
Hypericin incubation
Aliquots of hypericin (PhytoLab GmbH & Co. KG) stock solutions in dimethyl sulfoxide (DMSO; Molecular Probes™ D12345) with a concentration of 0.1 mM were freshly thawed for each cell incubation. Dilutions of this stock in complete cultivation medium and additional 10% FCS were prepared to achieve a final hypericin concentration of 1 μM. This procedure was adapted from Ritz et al.45 Dilutions of DMSO in complete cultivation medium and additional 10% FCS were used for controls, resulting in 1% DMSO within the final cultivation mixture. The hypericin and DMSO cultivation mixtures were added to the cells and incubated for 30 min at 37 °C and 5% CO2 conditions, respectively. Both incubations were disrupted by removing the cultivation mixtures and washing three times with phosphate buffered saline (PBS). Afterwards, fresh cultivation medium was added to the hypericin-incubated and control cells. All preparation steps and incubation experiments were performed in the dark. Cells were kept at 37 °C and 5% CO2 atmosphere until resorufin and PI treatments or FLIM measurements.
Resorufin conversion
U-87 MG cells were additionally incubated with a resazurin incubation mixture to convert resazurin to resorufin by metabolism in the cells. For this purpose, a resazurin stock solution with a concentration of 0.011 mg ml−1 in PBS was diluted 1 : 10 in complete cultivation medium and the final 4.3 μM resazurin incubation mixture was added to the cells overnight at 37 °C and 5% CO2 atmosphere. During this period, resazurin was quantitively metabolized to resorufin, which was mainly released into the cultivation medium by the cells. Resorufin conversion was completed after the incubation mixture turned from blue (resazurin) to pink (resorufin). No further exchange of cultivation medium was performed, since the released resorufin was needed for FLIM experiments. Hypericin-incubated-samples and controls were treated similarly according to this procedure. All preparation steps and incubation experiments were performed in the dark. Cells were kept at 37 °C and 5% CO2 atmosphere until PI supplementation or FLIM measurements.
PI supplementation
Aliquots of PI stock with a concentration of 500 μg ml−1 in PBS were freshly thawed for each cell treatment. To obtain a final PI concentration of 15 μg ml−1 (22.4 μM) in the cell culture dish, 100 μl PI stock was added to the cultivation medium after 100 μl cultivation medium was removed. This approach was carried out identically for hypericin-incubated cells and control cells. All preparation steps and incubation experiments were performed in the dark. Cells were kept at 37 °C and 5% CO2 atmosphere prior to the optical measurements.
Fluorescence lifetime imaging microscopy (FLIM)
FLIM experiments were performed with custom-built, inverted confocal laser stage scanning microscopes. A 405 nm laser diode (LDH P-C-405, PicoQuant GmbH, Germany) and a supercontinuum laser (SuperK Extreme EXB-4, NKT Photonics) equipped with an acousto-optic tunable filter (AOTF SuperK SELECT UV/VIS-nIR, NKT Photonics) were employed as excitation sources. Both sources supply picosecond pulses with adjustable repetition rates ranging from 2 to 78 MHz. Focusing of the excitation laser and collection of scattered and emitted light was achieved with high numerical aperture oil immersion objective lenses (Zeiss Plan-Apochromat, 100×, 1.4 Oil DIC and alpha Plan-Fluar 100×/1,49 Oil M27, Carl Zeiss AG, Germany). Two additional long-pass filters (458 nm: RazorEdge LP Edge Filter 458 RU, Semrock; 532 nm: LP EdgeBasic™ Filter 532, Semrock) were introduced into the detection beam paths to exclude the detection of the excitation wavelength. Single photon avalanche diodes (SPAD; PDM series, Micro Photon Devices, Italy) with an active area of 100 μm coupled with time-correlated single photon counting electronics (HydraHarp 400, PicoQuant GmbH, Germany) were used to acquire time-resolved photoluminescence data. The SymPhoTime64 software package (PicoQuant GmbH, Germany) was used to acquire and analyze time-correlated single photon counting (TCSPC) data. Further data analysis and plotting were carried out using the open-source general-purpose programming language Python.
Positioning, focusing, and determination of suitable acquisition parameters for each investigated cell was performed with low laser irradiance of 1.5–2.5 μW μm−2 to avoid damaging the cell prior to PDT induction. An irradiance between 5.5 μW μm−2 and 8 μW μm−2 was set for PDT initiation of hypericin-incubated cell, whereas control cells were measured at a minimum irradiance of 8 μW μm−2 up to 20 μW μm−2. The laser power was adjusted and regularly checked with a power meter (Optical power meter 1830-C Picowatt Digital, Newport) throughout the measurement session to ensure power stability. In total, 60 single cells were measured by FLIM for all incubation conditions to confirm all observations and ensure a high reproducibility (n = 60). Laser powers were measured in front of the 50/50 beam splitters in both setups. The cumulative losses through the beam splitter and the objective (the diameter of the collimated laser beam is approx. 20% larger than the back apertures of the objectives) are estimated to be approx. 70%. As the Airy disk contains 84% of the total excitation power, the irradiance I in the diffraction limited focus with a radius of approx. λ/4 is estimated to be: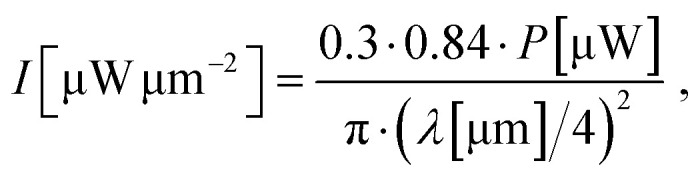 where P is the laser power measured in front of the objective and λ is the excitation wavelength. The total exposure time per pixel (exp/pix) in a FLIM image is calculated according to:
where P is the laser power measured in front of the objective and λ is the excitation wavelength. The total exposure time per pixel (exp/pix) in a FLIM image is calculated according to: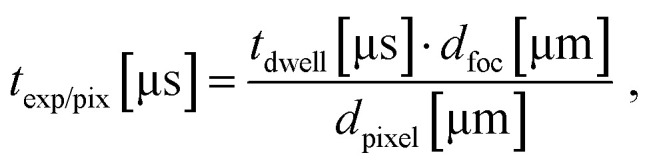 where tdwell is the dwell time per pixel and tfoc and dpixel are the diameters of the diffraction limited focal spot and the image pixel size, respectively.
where tdwell is the dwell time per pixel and tfoc and dpixel are the diameters of the diffraction limited focal spot and the image pixel size, respectively.
Widefield fluorescence imaging
The acquisition of widefield fluorescence images was performed with a custom-built, confocal microscope, equipped with a supercontinuum laser source (SuperK Extreme EXB-4, NKT Photonics) and a multi-channel tunable filter accessory (AOTF SuperK SELECT UV/VIS-nIR, NKT Photonics), controlled by the NKT Photonics CONTROL 1.2 software. Suitable long-pass filters were used to exclude the excitation laser wavelength from the detected signal. Widefield illumination was achieved by a lens (Ø1′′ UVFS Plano-Convex Lens, f = 150.0 mm, ARC: 350–700 nm, Thorlabs LA4874-A-ML), focused on the back focal plane of the objective (alpha Plan-Fluar 100×/1,49 Oil M27, Carl Zeiss AG, Germany). After light collection, a second lens (Biconvex lens; N-BK 7; D = 25.4; F = 100; mounted, Qioptiq G063854000) was used to focus the sample's fluorescence on the detection camera (ORCA-Flash4.0 V2 camera, Hamamatsu Photonics). Image acquisition was controlled by the camera software (HoKaWo 3.0). Each recording was manually stopped after cell death. The employed imaging camera can only record the total steady-state fluorescence signal; hence the localization of the different fluorescent markers and their individual contributions cannot be determined using this method. It should be noted that, in contrast to confocal scanning with a diffraction limited focal spot size, in widefield illumination the whole cell is exposed to laser irradiation for the entirety of the measurement. This, in conjunction with the large diameter of the collimated laser beam (several millimeters), leads to no direct comparability of the exposure times and the irradiation levels of the cells in widefield fluorescence image acquisition and the parameters texp/pix and I, defined for the acquisition of confocal scanning measurements.
Fluorescence spectra acquisition
Fluorescence spectra were acquired using an Acton SP300i spectrometer (Princeton Instruments, USA) with a grating of 300 grooves per mm, coupled to a thermoelectrically cooled CCD camera (PIXIS 100, Princeton Instruments, USA) at a detector temperature of −45 °C. Spectra acquisition was controlled by the Winspec software package (Princeton Instruments, USA).
Results
The focus of this manuscript is the investigation and visualization of cell damage and death caused by PDT on a subcellular level. For this purpose, three different dyes, the chemical structures of which are shown in Fig. 1(a), were employed. The first probe is hypericin, which takes on a dual function as both a fluorescent marker and the photosensitizer for PDT. The second dye, resorufin, is mainly released into the surrounding medium and only a small amount remains in the cell's cytoplasm. Thus, it enables drawing conclusions on the cell membrane integrity and indicates if the surrounding medium is entering the cell. The third fluorescent probe is propidium iodide (PI), which is only fluorescent when intercalated into DNA/RNA. Furthermore, it is unable to diffuse through intact cell membranes and can only enter the cell/nucleus in case of decreasing membrane integrity or even rupture. Therefore, PI mainly functions as an indicator for cellular damage and death. These three dyes have very distinct optical properties. The respective fluorescence spectra and lifetime histograms are presented in Fig. 1(b). All three dyes have different fluorescence emission maxima (584 nm resorufin, 600 nm hypericin and 622 nm PI), however, there is a significant overlap between the individual spectra making it difficult to distinguish the dyes only based on the fluorescence intensity. In contrast, the fluorescence lifetime strongly differs for these dyes, where resorufin has the shortest lifetime of 2.8 ns, followed by hypericin with 6.8 ns, and PI with a long lifetime of 12 ns in complete cell culture medium. The TCSPC histograms of the individual dyes and the fits thereof are displayed in Section 2 of the (ESI†). Fig. 1(c) shows an exemplary FLIM composite image, which is the superposition of a photoluminescence (PL) intensity image (gray in Fig. 1(c)) and a FLT image. In the final FLIM composite image, the FLT is represented by the color and the fluorescence intensity is given by the brightness. The black curve in Fig. 1(d) presents the histogram of the photon arrival times for all pixels in the FLT image showing three maxima at the average lifetimes of the three fluorescent probes. Additionally, the histograms of the regions of interest (ROI) marked in Fig. 1(c) are given in Fig. 1(d) in the respective colors. This data shows that the polar compound resorufin is mainly located outside the cell. As expected, we observe the highest accumulation of hypericin in the ER, but it is also distributed throughout the cytoplasm, likely associated with mitochondria, the Golgi apparatus, and lysosomes. A small amount of PI can already be detected inside the cell nucleus. The ability to detect this small amount of PI in the early stages of cell death, when membrane permeabilization is just beginning, is made possible by the single-molecule sensitivity of the employed confocal microscope. The accumulation behaviour of the dyes can be deduced from their individual contributions to the overall fluorescence of the cell, which are exemplarily imaged in Fig. S3.3 of the ESI.†
Fig. 1. Characterization of hypericin-induced PDT in single glioma cells. (a) The chemical structures of hypericin, resorufin and PI. (b) The fluorescence spectra and fluorescence decay curves for hypericin (green), resorufin (blue) and PI (red) in medium solution, acquired by excitation with a 405 nm laser. The respective fluorescence lifetimes τ are 2.8 ns for resorufin, 6.8 ns for hypericin and 13 ns for PI. (c) depicts a composite FLIM image of a single glioma cell in an early PDT stage. The composite FLIM image's color is given by the FLT recorded at each pixel, while the brightness is determined by the PL intensity. Three regions of interest (ROI) are indicated in (c), where the fluorescence signal is dominated by one specific dye, [ROI#1 (nucleoli): PI, ROI#2 (ER): hypericin, ROI#3 (medium): resorufin]. The occurrence histograms in (d) display the number of photons that are associated with a certain average FLT for the full image (black) and the ROIs in (c).

FLIM composite image series were acquired to reveal regularities in the progression of cellular damage to determine a plausible mechanism of glioma cell death induced by hypericin PDT. FLIM image acquisition and hypericin PDT are simultaneously initiated by the excitation laser and image sequences are recorded for single glioma cells. The first step is to locate a cell of interest and to determine suitable acquisition parameters with a low irradiance of around 1 μW μm−2, which was found to not cause significant photodamage to the cells. For image acquisition, the irradiance is increased to initiate PDT while simultaneously acquiring time-resolved PL data. The irradiance was adjusted to 7–10 μW μm−2, which is low enough to cause a slowly proceeding cell damage and thus to enable the visualization of the PDT progression over the course of multiple images (which are labeled M1…M8). The acquisition and analysis principles of FLIM images presented in this work are given in Section 3 of the (ESI†). Fig. 2 shows the progression of PDT for a single glioma cell incubated with hypericin and resorufin displaying individual stages of cellular damage. In Fig. 2(a) M1, the cell is still intact, as indicated by its morphology with an elongated shape with narrow branches and the bulky body center containing the cell nucleus (dark due to extremely low fluorescence intensity). Resorufin with a FLT of 2.8 ns dominates the fluorescence of the medium surrounding the cell. Hypericin with a FLT of 6.8 ns is mainly accumulated in the ER around the nucleus but can also be found within the cytoplasm. In contrast to the fluorescence emitted from the outside medium and the cell's ER, the overall fluorescence of the cytoplasm is not dominated by a single dye but is a combination of both hypericin and resorufin fluorescence. As a result, the intensity weighted average fluorescence lifetime value (τAvInt) of the cytoplasm is approx. 5 ns, in between the values recorded for the two contributing dyes. With ongoing PDT progression (Fig. 2(a) M2–4), overall cell and nucleus contraction and rounding can be observed, which is accompanied by a formation of cell membrane blebs. Blebs mainly occur at membrane sites around the ER and expand to create large protrusions (M4, white arrows). While the cell membrane is already severely distorted by surface protrusions in M4, its integrity is still maintained as indicated by the low fluorescence intensity inside the protrusions and no significant increase of the amount of resorufin in the cell. Between Fig. 2(a) M5 and M6 rupture of the cell membrane occurs. During the final stages of PDT (Fig. 2(a) M6–M8) the cell membrane stability is compromised. The decreasing τAvInt values in the cell from approx. 5 ns in Fig. 2(a) M5 to approx. 3.5 ns in M8 indicate that the cell is progressively flooded with the surrounding medium, resulting in expansion and complete rounding of the cellular body. At this stage, cell organelles are hardly visible anymore and resorufin is the dominant fluorescence emitter within the entire glioma cell.
Fig. 2. (a) Composite FLIM image series of a single glioma cell after hypericin incubation and resorufin conversion. Cell contraction and rounding can be observed in M1 to M3, together with a decrease of hypericin PL intensity. Formation of protrusions of the cellular membrane close to the ER is visible in M4. Further swelling until rupture of the cell membrane occurs between M5 and M8. (b) Series of fluorescence spectra acquired in the cell's ER at the locations marked by the white cross in M1–M7. (c) Evolution of the overall fluorescence intensities of hypericin and resorufin recorded in each image of the series. The acquisition parameters of the imaging series are displayed on top of the figure.

With ongoing deformation and membrane blebbing the hypericin fluorescence in the overall image steadily decreases, as shown in Fig. 2(c), where the sum of all photons emitted by hypericin and resorufin are displayed for each image in the series (for information on how the total fluorescence emitted by the individual dyes in each image is extracted see Section 3.1 of the ESI†). This trend is also evident from the spatially resolved, steady state fluorescence spectra displayed in Fig. 2(b), acquired in the cell's ER after each FLIM acquisition. At time point M1, the fluorescence spectrum shows a strong signal of Hypericin at 600 nm with a small contribution of resorufin (shoulder at 584 nm). Over the course of the PDT, the fluorescence signal of hypericin steadily decreases, whereas the resorufin signal increases. Especially at Fig. 2(a) M7, a strong increase of the resorufin fluorescence intensity can be observed, which is consistent with the average FLTs observed in the FLIM images. At Fig. 2(a) M8, the cell is completely flooded with the surrounding medium and the overall fluorescence spectrum closely resembles the emission of resorufin with only a small contribution from hypericin. A second series of FLIM composite images for a hypericin-incubated and resorufin surrounded cell is displayed in Section 4 of the ESI.† The same mechanistic steps in the cell death are observed there, namely a contraction and rounding of the cell, a steadily decreasing hypericin contribution to the overall fluorescence, the formation of numerous small surface blebs that quickly merge to form larger surface protrusions, easing of the membrane stability and finally flooding of the cell with the surrounding medium.
To ensure that the reproducibly observed cell death is caused by hypericin-induced PDT and is not solely a result of photodamage caused by the laser irradiation, control measurements of glioma cells that were not incubated with hypericin were performed at laser powers larger than the ones used for hypericin PDT induction of 7–9 μW μm−2 at λexc = 405 nm. These control experiments of cells without hypericin incubation are displayed and discussed in Section 5 of the ESI.† In summary, without the presence of the PDT sensitizer hypericin, we observed no significant cell damage after irradiating the single glioma cells in a comparable manner.
Further experiments were carried out, where PI was supplemented to the cultivation to act as a fluorescence marker sensitive to cell stress and death in the lifetime imaging measurements. The investigation of hypericin PDT on a single glioma cell with resorufin and PI present in the culture medium is displayed in a series of FLIM composite images in Fig. 3.
Fig. 3. (a) Composite FLIM image series of a single glioma cell after hypericin incubation, PI supplementation, and resorufin conversion. (b) Evolution of the overall fluorescence intensities of hypericin, resorufin and PI recorded in each image of the series. The fluorescence spectra acquired at the position markers are displayed in Fig. 4. The acquisition parameters of the imaging series are displayed above.

Again, resorufin is mainly located in the surrounding medium, as demonstrated by average FLTs of 3 ns around the glioma cell. Immediately after PDT induction (Fig. 3(a) M1), the cell morphology is unchanged, and the glioma cell has an elongated, narrow shape. Hypericin is accumulated throughout the entire cell with the largest concentration inside the ER, as illustrated by the bright green color for hypericin FLTs of approximately 7 ns. Only miniscule amounts of PI are found inside the cell, as the selective permeability of the membrane is still intact. The contribution of the individual dyes to the overall fluorescence decay of the intact cell, and their evolution as the PDT progresses are depicted and discussed in Section 6 of the ESI.† In the early stages of PDT, contraction of the cellular body accompanied by membrane bleb formation is observed (Fig. 3(a) M2–4). At this early stage of cellular damage, small amounts of PI are found to already enter the cell and bind to DNA in the nucleoli in the nucleus. As a result, from Fig. 3(a) M2–4 the nucleoli become increasingly fluorescent with a long average FLT of 12–13 ns (red). The small membrane blebs on the bottom of the cell in Fig. 3(a) M3 merge and form a large protrusion in M4/M5. This is accompanied by a strong increase in PI fluorescence, as can be seen in Fig. 3(b) and the FLIM image itself. The final steps of PDT (Fig. 3(a) M5–8) are characterized by the glioma cell fully rounding and flooding with the surrounding medium. Large areas with average lifetimes above 9 ns show a strong increase of the amount of PI binding to DNA and/or RNA compared to earlier stages. This drastic increase of PI fluorescence is also evident from Fig. 3(b). However, remnants of the ER are still observed as spots in the cytoplasm. The FLT of 13 ns observed in the nucleus (red color) shows a strong accumulation of PI and indicates the final cell death. Overall, the fluorescence intensity is increased in the entire cell, which is illustrated by the brighter and more intense false colors of the FLTs. Fig. 3(b) illustrates this trend by showing the fluorescence intensity of the three fluorescent probes during PDT progression. Again, a decrease of the hypericin fluorescence intensity can be observed, which is accompanied by a strong increase of the resorufin and PI fluorescence intensity at later PDT stages. A comparable PDT progression of another single glioma cell with hypericin incubation and resorufin and PI supplementation is depicted in Section 7 of the ESI.† Within the composite FLIM images, steady state fluorescence spectra were recorded at the spatial positions marked in Fig. 3. These spectra are presented in Fig. 4.
Fig. 4. Spatially resolved fluorescence spectra recorded on the locations of the corresponding markers indicated in Fig. 3. All spectra were recorded at the same wavelength and laser power used for the FLIM image acquisition (λexc = 405 nm, I = 7.8 μW μm−2) with an integration time of one second.

After the recording of each FLIM image presented in Fig. 3, fluorescence spectra were acquired at different spatial positions corresponding to certain cellular features; namely ER, nucleus, protrusion and outside. The exact positions are indicated in Fig. 3. Each subplot of Fig. 4 shows a 2D surface representation (left), the individual spectra (upper right) and the spectral composition (lower right) determined by spectral unmixing. In this context, the spectral composition refers to the relative contribution of the individual components to the sum spectrum and is not a direct measure of the concentrations of the dyes (see ESI,† Section 8 for more information). The spectra acquired in the ER are shown in Fig. 4(a). At time point M1, the fluorescence spectrum shows a strong signal of hypericin at 600 nm with a small contribution of resorufin (shoulder at 584 nm). In the first steps of the PDT (M1–4), the hypericin fluorescence intensity in the ER decreases, which can be attributed to a combination of direct photobleaching of hypericin due to the irradiation and bleaching due to the formation of ROS created in the immediate surrounding of the hypericin during the PDT. The triplet transition and the quenching of the triplet absorption in the presence of oxygen was confirmed with nanosecond transient absorption measurements of hypericin in DMSO and in aqueous solution which are displayed and discussed in Section 9 of the ESI.† The fluorescence intensity of resorufin and PI are hardly altered since the selective permeability of the still intact cell membrane prevents them from entering the cell. At later stages, the contributions of resorufin and PI to the spectra significantly increase, indicating the influx of the surrounding medium into the cell (as reflected by the increased resorufin PL intensity) and the progression of cellular damage (as reflected by the increased PI PL intensity). In Fig. 4(b), the corresponding spectra acquired in the cell's nucleus are shown. For the early measurements (M1–M4), the fluorescence intensity in this region is only approx. 20% of the PL intensity of the spots measured in the ER (see inset in the top right plot in Fig. 4(b)) and only small amounts of hypericin, resorufin and PI can be found. The spectral composition suggests that the main fluorescence component is hypericin, followed by resorufin. After M5, the PI fluorescence intensity strongly increases, until hardly any signal of hypericin and resorufin are visible, suggesting strong damage to the cell and ultimately cell death. Fluorescence spectra acquired in the large protrusion after M4 (Fig. 3) are shown in Fig. 4(c). Immediately after bleb formation in M4, the spectra closely resemble the spectrum of a combination of resorufin and hypericin, which is validated by the spectral composition plot. This implies that the initial protrusion is filled with the cell's cytoplasm, within which both dyes are found to contribute approximately equally to the overall PL decay. In the next measurement (M5), however, the membrane stability appears to be compromised as the composition changes towards higher contributions of resorufin and PI, which are entering the damaged cell. Over the course of PDT, the hypericin fluorescence intensity decreases, whereas resorufin only shows small changes. The main indicator of the overall cell damage is again PI, which shows a strong increase of its fluorescence intensity at the later PDT stages. Finally, the fluorescence spectra acquired from the medium outside the cell are shown in Fig. 4(d) and are mainly dominated by resorufin. Only at the last PDT stages, a small amount of hypericin and PI are observed here, since they are released into the medium due to the damaged cell membrane.
A typical acquisition time for a FLIM image is 5–10 min, hence fast dynamics of the PDT process cannot be resolved. By contrast, morphogenic changes can be visualized much faster with widefield fluorescence images. This enables insights into cellular processes that are happening too fast to be captured by the slow, pixel-by-pixel-scanning based FLIM measurements. Fig. 5 shows selected frames of a video recorded at 0.33 frames per s to visualize fast dynamics of hypericin-induced PDT in single glioma cells. More widefield fluorescence image series are shown in Section 10 of the ESI.† The stages of cell death observed in the widefield images are in good agreement with those observed by means of confocal scanning measurements. In the beginning, the cell has an intact, elongated shape and the strongest total fluorescence signal is observed in the ER which we know from the FLIM experiments to be caused by hypericin accumulation. With ongoing PDT progression, we observe cell contraction, rounding, and a decrease of the hypericin fluorescence, especially in the ER. After approx. 120 s, the cell surface exhibits blebs that merge to form larger protrusions. This is followed by a significant increase of the overall PL throughout the whole cell body, with the most pronounced increase observed in the nucleolus. This results from an increasing PI intercalation into RNA and DNA in the nucleus and fragments in the cytoplasm due to a loss of cell membrane stability. The rapid acquisition time of the widefield fluorescence measurements allows us to capture the explosive rupture of the cell membrane (Fig. 5(g)) occurring after 237 seconds of irradiation. The final stage of PDT is illustrated in Fig. 5(h), where the rounded cell exhibits a high total fluorescence intensity signal, which from the FLIM experiments we know to be nearly exclusively PI emission, indicating cell death.
Fig. 5. (a) Series of widefield fluorescence images showing the effect of hypericin-induced PDT on a single glioma cell incubated with hypericin and PI. Different stages of cell damage are deductible, including rounding of the cell body (b)–(d), membrane bleb formation (d)–(f), explosive membrane rupture ((g), yellow arrow), and finally cell death (h). The membrane rupture is accompanied by the release of cytoplasm content into the extracellular environment. The overall recorded fluorescence intensity is continuously increasing, which results from more PI entering the damaged cellular body and binding to RNA/DNA.

Discussion
Spatially resolved fluorescence lifetime measurements and spectroscopic examination of hypericin-induced PDT on glioma cells revealed a sequence of cell damaging steps that include cell shrinkage and rounding, membrane bleb formation, degradation of the ER and finally breakdown of membrane stability resulting in flooding of the cell with the surrounding medium (Fig. 2 and 3).
The cellular contraction and rounding we observed shortly after initiating the PDT with the excitation laser is reported to be a typical feature of early apoptotic cell death.55–58 In the early stages of PDT, following and accompanying the contraction and rounding, we reproducibly observed membrane blebbing, which is morphologically characterized by the formation of small bubble-like bulges on the cell surface that can expand and retract over time.59 During bleb expansion, the pressure inside the cell temporarily increases and cytoplasm from the cell body flows into the bleb.60 A prominent example of this membrane blebbing can be seen in FLIM image M3 of Fig. 3(a), where the cellular membrane towards the bottom of the image shows small bubble-like bulges. This type of membrane blebbing is also reported to be a typical feature of apoptotic cell death.55,58
As the PDT progresses, the formation of one or multiple large membrane protrusions was observed. These are caused by a detachment of the membrane from the cytoskeleton (actomyosin cortex).61,62 Such protrusions are characteristic for necrosis, and are described to be larger, more transparent and non-retractable in comparison to apoptosis blebs.61,63 The formation of such large surface protrusions, which we most commonly observed forming close to the ER, being the site of highest hypericin concentration, can be seen in panel M4 of Fig. 2 and 3. The extent of this phenomenon is perhaps most dramatic in the panels following M5 of Fig. S4.1 in the ESI.† The fact that initially only very little fluorescence is recorded from within the protrusions, despite using an extremely sensitive avalanche photodiode for detection, indicates two key points. Firstly, the stability of the cell membrane, at this stage, is still largely intact. If the membrane had been compromised, the surrounding medium would have flooded the cell, and the photoluminescence of resorufin would have been detected inside. Secondly, the cytoskeleton of the cell initially remains mostly intact. Otherwise, the fluorescence of cellular components from within the cytoskeleton would have been detected within the protrusion. This tells us that the protrusion is a result of an active cellular process set in motion by the PDT, rather than a passive event caused by membrane rupture or cell lysis. As the PDT progresses, the cell membrane stability eases and the surrounding medium floods the cell. This results in resorufin and/or PI dominating the photoluminescence (PL) of the cell, as seen in the final panels of the FLIM series.
We quantified the fluorescence emitted by the individual dyes to show that the absolute amount of hypericin fluorescence from the cells decreases drastically as PDT progresses (Fig. 1(c), 2(b) and 4(a)–(c) of the main manuscript and Fig. S4.1 of the ESI†). Given that very low powered, pulsed excitation was used, this decrease in hypericin fluorescence is unlikely to be solely due to photobleaching. Instead, we attribute it to the decomposition of cellular structures where hypericin accumulates and the degradation of hypericin itself caused by PDT-generated reactive oxygen species. This hypothesis is supported by the loss of distinct ER structure observed in the FLIM images, ultimately leaving behind only fragmented remnants of the ER. This is to be expected, as hypericin accumulates in the ER, making it the primary site of PDT-induced ROS production, resulting in a significant level of ER stress during PDT. The ER as an important reaction site in hypericin PDT is highly linked to mitochondria via mitochondria-associated membranes (MAMs) that are stated to play a role in various cell death mechanisms in cancer, including apoptosis and necrosis.64 The activated cell death mechanism is supposed to be strongly dependent on the stressors, availability of certain compounds in the ER and mitochondria, and on cellular, as well as environmental conditions.65–67 ER stress is reported to primarily induce apoptosis,68–70 but was in some cases also shown to cause necrosis.71–74
The observed initial contraction and rounding of the cell and the observation of small, retracting membrane blebs imply that hypericin-induced PDT of glioma cells initially triggers apoptosis. The formation of the large surface protrusions indicates that this initially apoptotic cell death is followed by necrosis as the PDT progresses. The assumption of an initial apoptotic and continued necrotic cell death mechanism is also supported by the observed breakdown of membrane stability and flooding of the cells with the surrounding medium (Fig. 2: between M6 and M7, Fig. 3: between M5 and M6). Widefield fluorescence images revealed that the rupture of the membrane resulting in the release of cytosolic content occurs in an explosive manner (Fig. 5(g)). The abrupt pressure release due to membrane rupture is defined to be a necrotic attribute, which further emphasizes the previously proposed mechanism of cell death.75 After membrane disruption, large amounts of PI diffuse into the cell at once intercalating into DNA/RNA fragments in the cytoplasm and nucleus, marking the final stage of necrotic cell death, where no apoptotic bodies are observed. In conclusion, based on the analysis of FLIM composite images, spatially resolved spectroscopy and widefield fluorescence microscopy, apoptotic and necrotic features were observed for hypericin-induced PDT on glioma cells, suggesting apoptosis to be the initial mechanism followed by necrosis after sustained irradiation.
Conclusions
In this work, the mechanism of hypericin-induced PDT was investigated in single glioma cells. For this purpose, glioma cells were incubated with hypericin and supplemented with the two additional fluorescence markers resorufin and PI. These three fluorescence markers were chosen as they display distinct FLTs, which allows us to easily distinguish and locate them within a glioma cell. Furthermore, all three markers serve a specific purpose, i.e., hypericin is deployed as photosensitizer for PDT, resorufin is used to label the surrounding medium and PI indicates cellular stress/damage. The combination of all three fluorescence compounds enables us to investigate the individual steps of hypericin-induced PDT on glioma cells and to deduce specific damaging patterns that are indicative of the different programmed cell death mechanisms. We attribute the observed initial cell body shrinkage and bleb formation to be typical features of apoptosis, while the formation of large, non-retracting protrusions, the absence of apoptotic bodies and the explosive membrane rupture were identified as necrotic cell death characteristics. In accordance with control cells, all observed cellular changes during hypericin PDT were proven to be treatment-specific and not to be triggered by laser irradiation. As a result, the cell death occurring in hypericin-induced PDT on glioma cells was found to be primarily apoptosis followed by necrosis. Our study generated new insights and perspectives on hypericin PDT that should contribute to its successful implementation and improvement as an application in brain tumor treatments.
Author contributions
J. Hiller and M. Bassler performed and analyzed the spatially resolved optical measurements. P. Frech performed and analyzed the transient absorption measurements. M. Bassler cultivated the glioblastoma cells. C. Kertzscher and F. Schmidt helped with the data analysis. T. Rammler assisted in building and maintaining the confocal setup. F. Wackenhut, S. z. Oven-Krockhaus, K. Braun, M. Scheele, A. J. Meixner and M. Brecht conceived and supervised the project. R. Ritz and M. Brecht developed the research idea. M. Bassler, J. Hiller and F. Wackenhut wrote the manuscript with input from all authors. All authors have given approval to the final version of the manuscript.
Data availability
The data that support the findings of this study are available at Zenodo.org at 10.5281/zenodo.13760690.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This study was funded by the doctoral program of the University of Tübingen and Reutlingen University “Intelligente Prozess- und Materialentwicklung in der Biomateriomics (IPMB)” which is supported by the MWK Baden-Württemberg (PhD student fellowship to MCB). Additionally, this work was supported by a MINT-funded project from Vector Stiftung Stuttgart and by the German Research Foundation (CRC 1101-Z02). Additional financial support for this work has been provided by the DFG under grant SCHE1905/9-1 (project no. 426008387). The TA measurements were funded by the DFG under contract INST 37/1160-1 FUGG (project no. 458406921).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00107a
Notes and references
- Chandana S. R. Movva S. Arora M. Singh T. Am. Fam. Physician. 2008;77:1423–1430. [PubMed] [Google Scholar]
- Oliver T. G. Wechsler-Reya R. J. Neuron. 2004;42:885–888. doi: 10.1016/j.neuron.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Castro M. G. Cowen R. Williamson I. K. David A. Jimenez-Dalmaroni M. J. Yuan X. Bigliari A. Williams J. C. Hu J. Lowenstein P. R. Pharmacol. Ther. 2003;98:71–108. doi: 10.1016/S0163-7258(03)00014-7. [DOI] [PubMed] [Google Scholar]
- Tan A. C. Ashley D. M. López G. Y. Malinzak M. Friedman H. S. Khasraw M. Ca-Cancer J. Clin. 2020;70:299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- Behin A. Hoang-Xuan K. Carpentier A. F. Delattre J.-Y. Lancet. 2003;361:323–331. doi: 10.1016/S0140-6736(03)12328-8. [DOI] [PubMed] [Google Scholar]
- Louis D. N. Perry A. Reifenberger G. von Deimling A. Figarella-Branger D. Cavenee W. K. Ohgaki H. Wiestler O. D. Kleihues P. Ellison D. W. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- Tran B. Rosenthal M. A. J. Clin. Neurosci. 2010;17:417–421. doi: 10.1016/j.jocn.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Eljamel M. S. Photodiagn. Photodyn. Ther. 2008;5:260–263. doi: 10.1016/j.pdpdt.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Nagaya T. Nakamura Y. A. Choyke P. L. Kobayashi H. Front. Oncol. 2017;7:314. doi: 10.3389/fonc.2017.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L. Roberts D. W. Sanai N. Liu J. T. C. J. Neuro-Oncol. 2019;141:495–505. doi: 10.1007/s11060-018-03077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens L. C. Krabbenhøft M. G. Hansen R. W. Mikic N. Pedersen C. B. Poulsen F. R. Korshoej A. R. Cancers. 2022;14:617. doi: 10.3390/cancers14030617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari K. M. Eggleston I. M. Photochem. Photobiol. Sci. 2018;17:1553–1572. doi: 10.1039/c8pp00362a. [DOI] [PubMed] [Google Scholar]
- Matsutani M. Int. J. Clin. Oncol. 2004;9:471–474. doi: 10.1007/s10147-004-0456-7. [DOI] [PubMed] [Google Scholar]
- Graham C. A. Cloughesy T. F. Semin. Oncol. Nurs. 2004;20:260–272. doi: 10.1016/S0749-2081(04)00090-7. [DOI] [PubMed] [Google Scholar]
- Stepp H. Stummer W. Lasers Surg. Med. 2018;50:399–419. doi: 10.1002/lsm.22933. [DOI] [PubMed] [Google Scholar]
- Castano A. P. Demidova T. N. Hamblin M. R. Photodiagn. Photodyn. Ther. 2004;1:279–293. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi K. Garvey K. L. Bouras A. Cramer G. Stepp H. Jesu Raj J. G. Bozec D. Busch T. M. Hadjipanayis C. G. J. Neuro-Oncol. 2019;141:595–607. doi: 10.1007/s11060-019-03103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius J. F. Slotty P. J. El Khatib M. Giannakis A. Senger B. Steiger H. J. Photodiagn. Photodyn. Ther. 2014;11:1–6. doi: 10.1016/j.pdpdt.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Guney Eskiler G. Deveci Ozkan A. Sozen Kucukkara E. Kamanlı A. F. Gunoğlu B. Yıldız M. Z. Photodiagn. Photodyn. Ther. 2020;31:101854. doi: 10.1016/j.pdpdt.2020.101854. [DOI] [PubMed] [Google Scholar]
- Atif M. Fakhar-E-Alam M. Firdous S. Zaidi S. S. Z. Suleman R. Ikram M. Laser Phys. Lett. 2010;7:757–764. doi: 10.1002/lapl.201010061. [DOI] [Google Scholar]
- Ehrenberg B. Anderson J. L. Foote C. S. Photochem. Photobiol. 1998;68:135–140. doi: 10.1111/j.1751-1097.1998.tb02479.x. [DOI] [PubMed] [Google Scholar]
- Noell S. Mayer D. Strauss W. S. L. Tatagiba M. S. Ritz R. Int. J. Oncol. 2011;38:1343–1348. doi: 10.3892/ijo.2011.968. [DOI] [PubMed] [Google Scholar]
- Ritz R. Scheidle C. Noell S. Roser F. Schenk M. Dietz K. Strauss W. S. L. PLoS One. 2012;7:e51974. doi: 10.1371/journal.pone.0051974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler M. C. Rammler T. Wackenhut F. zur Oven-Krockhaus S. Secic I. Ritz R. Meixner A. J. Brecht M. Anal. Bioanal. Chem. 2022;414:4849–4860. doi: 10.1007/s00216-022-04107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamuhabwa A. A. R. Huygens A. De Witte P. A. M. Int. J. Oncol. 2003;23:1445–1450. [PubMed] [Google Scholar]
- Lai H. W. Nakayama T. Ogura S. Int. J. Clin. Oncol. 2021;26:26–33. doi: 10.1007/s10147-020-01766-y. [DOI] [PubMed] [Google Scholar]
- De Rosa F. S. Bentley M. V. L. B. Pharm. Res. 2000;17:1447–1455. doi: 10.1023/A:1007612905378. [DOI] [PubMed] [Google Scholar]
- Ritz R. Roser F. Radomski N. Strauss W. S. L. Tatagiba M. Gharabaghi A. Anticancer Res. 2008;28:2033–2038. [PubMed] [Google Scholar]
- Delaey E. M. Obermuëller R. Zupkó I. De Vos D. Falk H. De Witte P. A. M. Photochem. Photobiol. 2001;74:164–171. doi: 10.1562/0031-8655(2001)074<0164:IVSOTP>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Huntosova V. Nadova Z. Dzurova L. Jakusova V. Sureau F. Miskovsky P. Photochem. Photobiol. Sci. 2012;11:1428–1436. doi: 10.1039/c2pp05409d. [DOI] [PubMed] [Google Scholar]
- Kanduc D. Mittelman A. Serpico R. Sinigaglia E. Sinha A. A. Natale C. Santacroce R. Di Corcia M. G. Lucchese A. Dini L. Pani P. Santacroce S. Simone S. Bucci R. Farber E. Int. J. Oncol. 2002;21:165–170. [PubMed] [Google Scholar]
- D’Arcy M. S. Cell Biol. Int. 2019;43:582–592. doi: 10.1002/cbin.11137. [DOI] [PubMed] [Google Scholar]
- Saraste A. Pulkki K. Cardiovasc. Res. 2000;45:528–537. doi: 10.1016/S0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- Coleman M. L. Sahai E. A. Yeo M. Bosch M. Dewar A. Olson M. F. Nat. Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Van Cruchten S. Van den Broeck W. Anat., Histol., Embryol. 2002;31:214–223. doi: 10.1046/j.1439-0264.2002.00398.x. [DOI] [PubMed] [Google Scholar]
- Rana P. S. Mudrak N. J. Lopez R. Lynn M. Kershner L. Model M. A. Biochem. Biophys. Res. Commun. 2017;492:300–303. doi: 10.1016/j.bbrc.2017.08.123. [DOI] [PubMed] [Google Scholar]
- Yu P. Zhang X. Liu N. Tang L. Peng C. Chen X. Signal Transduction Targeted Ther. 2021;6:128. doi: 10.1038/s41392-021-00507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnus W. Meyer C. Paliege A. Belavgeni A. von Mässenhausen A. Bornstein S. R. Hugo C. Becker J. U. Linkermann A. Journal of Pathology. 2019;247:697–707. doi: 10.1002/path.5248. [DOI] [PubMed] [Google Scholar]
- Zhang Q. Li Z.-h. Li Y.-y. Shi S.-j. Zhou S.-w. Fu Y.-y. Zhang Q. Yang X. Fu R.-q. Lu L.-c. Sci. Rep. 2015;5:18398. doi: 10.1038/srep18398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barathan M. Mariappan V. Shankar E. M. Abdullah B. J. Goh K. L. Vadivelu J. Cell Death Dis. 2013;4:e697–e697. doi: 10.1038/cddis.2013.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlanda J. Kiesslich T. Oberdanner C. B. Obermair F. J. Krammer B. Plaetzer K. J. Photochem. Photobiol., B. 2006;25:173–188. doi: 10.1615/jenvironpatholtoxicoloncol.v25.i1-2.100. [DOI] [PubMed] [Google Scholar]
- Mikeš J. Kleban J. Sačková V. Horváth V. Jamborová E. Vaculová A. Kozubík A. Hofmanová J. Fedoročko P. Photochem. Photobiol. Sci. 2007;6:758–766. doi: 10.1039/b700350a. [DOI] [PubMed] [Google Scholar]
- Kessel D. Photochem. Photobiol. 2020;96:1101–1104. doi: 10.1111/php.13275. [DOI] [PubMed] [Google Scholar]
- Pfaffel-Schubart G. Scalfi-Happ C. Rück A. Med. Laser Appl. 2008;23:25–30. doi: 10.1016/j.mla.2007.11.003. [DOI] [Google Scholar]
- Ritz R. Wein H. T. Dietz K. Schenk M. Roser F. Tatagiba M. Strauss W. S. L. Int. J. Oncol. 2007;30:659–667. [PubMed] [Google Scholar]
- Pevna V. Wagnières G. Huntosova V. Biomedicines. 2021;9:1703. doi: 10.3390/biomedicines9111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesslich T. Krammer B. Plaetzer K. Curr. Med. Chem. 2006;13:2189–2204. doi: 10.2174/092986706777935267. [DOI] [PubMed] [Google Scholar]
- Golstein P. Kroemer G. Trends Biochem. Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Edinger A. L. Thompson C. B. Curr. Opin. Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Festjens N. Vanden Berghe T. Vandenabeele P. Bba-Bioenergetics. 2006;1757:1371–1387. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Zitvogel L. Casares N. Péquignot M. O. Chaput N. Albert M. L. Kroemer G. Adv. Immunol. 2004;84:131–179. doi: 10.1016/S0065-2776(04)84004-5. [DOI] [PubMed] [Google Scholar]
- Rück A. Dolp F. Hülshoff C. Hauser C. Scalfi-Happ C. Proc. SPIE. 2005;5700:182–187. doi: 10.1117/12.590439. [DOI] [Google Scholar]
- Li M. Chen Y. He W. Guo Z. Chemistry. 2024;30:e202401285. doi: 10.1002/chem.202401285. [DOI] [PubMed] [Google Scholar]
- Dai J. Zhang X. Chem. Biomed. Imaging. 2023;1:796–816. doi: 10.1021/cbmi.3c00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndozangue-Touriguine O. Hamelin J. Bréard J. Biochem. Pharmacol. 2008;76:11–18. doi: 10.1016/j.bcp.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Povea-Cabello S. Oropesa-Avila M. de la Cruz-Ojeda P. Villanueva-Paz M. de la Mata M. Suárez-Rivero J. M. Alvarez-Córdoba M. Villalón-García I. Cotán D. Ybot-González P. Sánchez-Alcázar J. A. Int. J. Mol. Sci. 2017;18:2393. doi: 10.3390/ijms18112393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson R. S. Heer S. Dive C. Watson A. J. Am. J. Physiol.: Cell Physiol. 1996;270:C1190–C1203. doi: 10.1152/ajpcell.1996.270.4.C1190. [DOI] [PubMed] [Google Scholar]
- Doonan F. Cotter T. G. Methods. 2008;44:200–204. doi: 10.1016/j.ymeth.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Gores G. J. Herman B. Lemasters J. J. Hepatology. 1990;11:690–698. doi: 10.1002/hep.1840110425. [DOI] [PubMed] [Google Scholar]
- Bovellan M. Fritzsche M. Stevens C. Charras G. FEBS J. 2010;277:58–65. doi: 10.1111/j.1742-4658.2009.07412.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Chen X. Gueydan C. Han J. Cell Res. 2018;28:9–21. doi: 10.1038/cr.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackler O. T. Grosse R. J. Cell Biol. 2008;181:879–884. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. Maeno E. Shimizu T. Dezaki K. Wang J. Morishima S. J. Physiol. 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danese A. Patergnani S. Bonora M. Wieckowski M. R. Previati M. Giorgi C. Pinton P. Biochim. Biophys. Acta. 2017;1858:615–627. doi: 10.1016/j.bbabio.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Kim J.-S. He L. Lemasters J. J. Biochem. Biophys. Res. Commun. 2003;304:463–470. doi: 10.1016/S0006-291X(03)00618-1. [DOI] [PubMed] [Google Scholar]
- Iurlaro R. Muñoz-Pinedo C. FEBS J. 2016;283:2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- Orrenius S. Gogvadze V. Zhivotovsky B. Biochem. Biophys. Res. Commun. 2015;460:72–81. doi: 10.1016/j.bbrc.2015.01.137. [DOI] [PubMed] [Google Scholar]
- Tabas I. Ron D. Nat. Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman A. M. Healy S. J. M. Jäger R. Samali A. Pharmacol. Ther. 2012;134:306–316. doi: 10.1016/j.pharmthera.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Logue S. E. Cleary P. Saveljeva S. Samali A. Apoptosis. 2013;18:537–546. doi: 10.1007/s10495-013-0818-6. [DOI] [PubMed] [Google Scholar]
- Ullman E. Fan Y. Stawowczyk M. Chen H. M. Yue Z. Zong W. X. Cell Death Differ. 2008;15:422–425. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore G. C. Papa F. R. Oakes S. A. Curr. Opin. Cell Biol. 2011;23:143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redza-Dutordoir M. Averill-Bates D. A. Biochim. Biophys. Acta. 2016;1863:2977–2992. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- Lin H. Peng Y. Li J. Wang Z. Chen S. Qing X. Pu F. Lei M. Shao Z. Oxid. Med. Cell. Longevity. 2021;2021:8810698. doi: 10.1155/2021/8810698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rello S. Stockert J. C. Moreno V. Gámez A. Pacheco M. Juarranz A. Cañete M. Villanueva A. Apoptosis. 2005;10:201–208. doi: 10.1007/s10495-005-6075-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available at Zenodo.org at 10.5281/zenodo.13760690.