

Microglia facilitate nociception; however, their molecular responses to varied pain types are uncertain. We show distinct adaptations during injury- and opioid-induced hypersensitivity, indicating a need for pain-type–specific interventions.
Keywords: Microglia, Opioids, Nerve injury, Analgesic tolerance, Hypersensitivity, Chronic neuropathic pain, RNA sequencing
Abstract
Microglia take on an altered morphology during chronic opioid treatment. This morphological change is broadly used to identify the activated microglial state associated with opioid side effects, including tolerance and opioid-induced hyperalgesia (OIH). Microglia display similar morphological responses in the spinal cord after peripheral nerve injury (PNI). Consistent with this observation, functional studies have suggested that microglia activated by opioids or PNI engage common molecular mechanisms to induce hypersensitivity. In this article, we conducted deep RNA sequencing (RNA-seq) and morphological analysis of spinal cord microglia in male mice to comprehensively interrogate transcriptional states and mechanistic commonality between multiple models of OIH and PNI. After PNI, we identify an early proliferative transcriptional event across models that precedes the upregulation of histological markers of microglial activation. However, we found no proliferative transcriptional response associated with opioid-induced microglial activation, consistent with histological data, indicating that the number of microglia remains stable during morphine treatment, whereas their morphological response differs from PNI models. Collectively, these results establish the diversity of pain-associated microglial transcriptomic responses and point towards the targeting of distinct insult-specific microglial responses to treat OIH, PNI, or other central nervous system pathologies.
1. Introduction
Microglia, the resident macrophages of the central nervous system (CNS), regulate a host of neural processes, such as synaptic pruning and programmed cell death.46,61 In the setting of injury or disease, microglia enter an activated state characterized by changes in number, morphology, and production of signaling molecules.1,59 After peripheral nerve injury (PNI), microglial morphology and distribution evolve in the spinal cord dorsal horn, exhibiting shortened processes and increased density where injured sensory neurons terminate.4,26,76 These observations have led to numerous functional studies that established the contribution of activated microglia to the development of chronic neuropathic pain.51,68
We and others have observed a similar activated morphology after chronic opioid treatment, corresponding with the onset of tolerance and opioid-induced hyperalgesia (OIH).13,19,75 Given these shared morphological alterations, common molecular mechanisms are believed to underlie the deleterious pronociceptive functions of microglia in both opioid- and injury-induced pain states. The ATP receptor P2X4 is upregulated in spinal microglia after either PNI or chronic morphine treatment, and P2X4 signaling is required for the development of tactile hypersensitivity in the context of both spinal nerve injury and morphine tolerance.34,71 Both neuropathic pain and OIH have been suggested to rely on P2X4-mediated release of brain-derived neurotrophic factor (BDNF) from microglia. BDNF could then act on TrkB expressed by spinal neurons to downregulate the chloride transporter KCC2 and disturb chloride homeostasis, resulting in pronociceptive neuronal hyperexcitability in the spinal cord dorsal horn.14,19,71 However, recent studies detected no BDNF mRNA expression in microglia,3,7,16,33,38 challenging this hypothesis. In addition, another microglial purinergic receptor, P2X7, is upregulated and implicated in both morphine tolerance43,70,78 and neuropathic pain.40 Although we detected no expression of the mu-opioid receptor (MOR) or its mRNA (Oprm1) in microglia,13 others have suggested that opioids can act directly on microglial toll-like receptor 4 (TLR4) and that this receptor may also contribute to neuropathic pain.36,48 Furthermore, we previously reported that global MOR knockout mice display microglial reactivity, but not OIH.13 These results suggested a mechanistic dissociation between microglial activation and the role of these cells in OIH, despite previous evidence that inhibiting microglial reactivity can reduce morphine side effects.58
Although many molecular mechanisms of microglial activation in pronociceptive states have been proposed, a complete understanding of microglial responses across pronociceptive conditions is required to determine whether these responses reflect a single transcriptional process. This clearer understanding of microglial responses to pain states could reveal selective interventions that precisely alter microglial function to reduce hyperalgesia. RNA sequencing has drastically expanded our knowledge of the transcriptomic and functional diversity of specific cell types. With this method, we can compare the transcriptomes of microglia to those of other CNS cell types77 and immune cells,22,42,52 including across disease states15,20,39,53 and across brain regions.9 To date, however, there has been no systematic comparison of microglial transcriptional responses across pain states and chronic opioid treatment to directly ask whether injuries and opioids engage similar microglial pronociceptive mechanisms.
Therefore, we used transcriptomics to resolve microglial response patterns in multiple settings of pain hypersensitivity. Our results command a reappraisal of the microglial mechanisms that underlie injury- and opioid-induced pain hypersensitivity and a redefinition of microglial activation based on both transcriptional and morphological profiles. Further interrogation of these transcriptional states, especially at the single-cell level, may reveal condition-specific interventions that precisely modulate microglial function to reduce hyperalgesia.
2. Methods
2.1. Animals
All procedures were approved by the Stanford University Administrative Panel on Laboratory Animal Care in accordance with the International Association for the Study of Pain. Male C57Bl6/J mice from The Jackson Laboratory, 8 to 12 weeks old, were housed 2 to 5 per cage on a 12-hour light/dark cycle with ad libitum access to food and water. Male Sprague-Dawley rats, used only for culture experiments, were obtained from Charles River Laboratories.
2.2. Drugs
Morphine sulfate (West-Ward NDC 0641-6070-01) was administered subcutaneously (s.c.) at 10 to 40 mg/kg (Fig. 1A). Dilutions and control injections were prepared with 0.9% sodium chloride (Hospira NDC 0409-4888-10).
Figure 1.
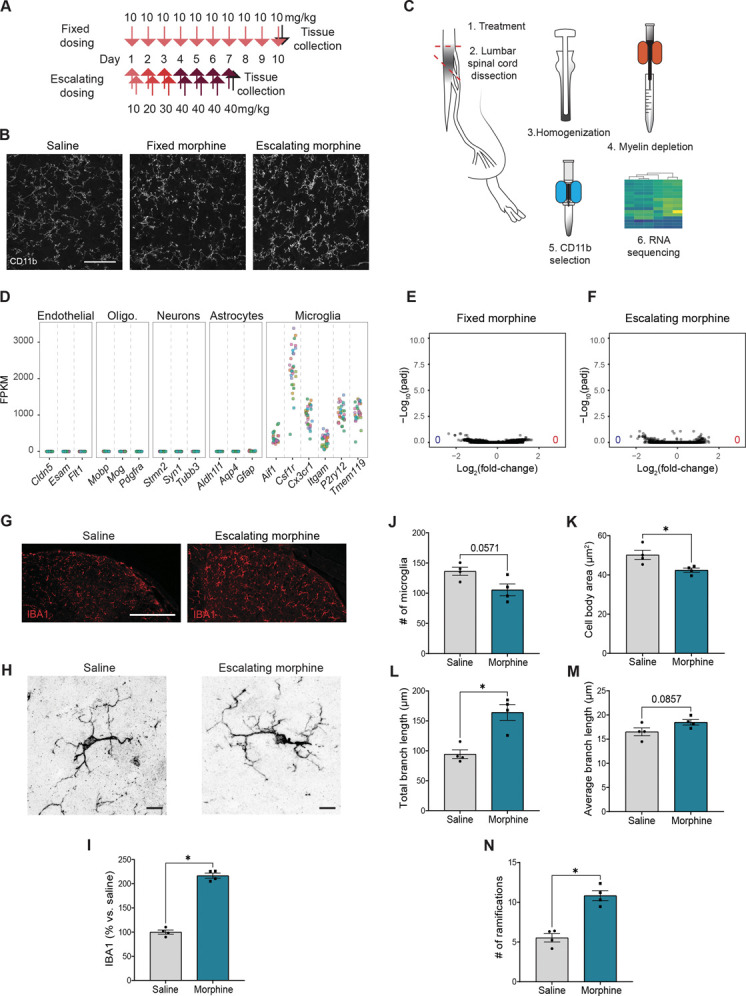
Minimal transcriptomic changes associated with chronic morphine–induced microglial activation despite strong morphological alterations. (A) Schematic illustrating the 2 opioid dosing paradigms. Fixed dosing was once daily 10 mg/kg (subcutaneous, s.c.) for 10 days, with tissue collection 1 hour after the final dose on day 10. Escalating dosing was twice daily 10, 20, or 30 mg/kg per day (s.c.) for the first 3 days, then twice daily 40 mg/kg for 3 days, and a final day of a single dose of 40 mg/kg 1 hour before perfusion. (B) Example images of CD11b immunoreactivity in the spinal cord dorsal horn after saline, fixed morphine, or escalating morphine treatment. Scale bar = 50 µm. (C) Schematic of tissue collection location and microglia isolation for RNA sequencing (RNA-seq). (D) Normalized reads for genes specifically expressed in possibly contaminating cell types and markers of microglia, from all conditions and replicates tested. FPKM, fragments per kilobase of transcript per million mapped reads. (E and F) Volcano plots summarizing gene expression changes in microglia after fixed (E) or escalating (F) morphine treatment. Blue and red dots indicate significantly downregulated or upregulated genes from DESeq2, respectively (Padj < 0.01). The numbers in the lower corners indicate the total numbers of downregulated (blue) and upregulated (red) genes (Padj < 0.01). (G) Example images of IBA1 immunoreactivity in the dorsal horn of lumbar spinal cords after saline or morphine treatment. Scale bars = 100 µm. (H) 63x example images of IBA1 immunoreactivity in the dorsal horn of lumbar spinal cords after saline or morphine treatment. Scale bars = 10 µm. (I) Quantification of IBA1 density (% area) in the dorsal horn, normalized as % vs control saline. (J) Quantification of the number of microglia in the dorsal horn in all conditions. (K-N) Quantification of microglial morphology, including number of ramifications, microglia body area, total branch length, average branch length, and number of microglia. Two-way ANOVA and Kruskal–Wallis test (I, N); * indicates P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001 vs sham or saline or morphine. Dots: n = 4 mice/condition.
2.3. Histology
2.3.1. Tissue collection and processing
Mice anesthetized with sodium pentobarbital were transcardially perfused with phosphate-buffered saline (PBS), followed by 10% formaldehyde in PBS. Spinal cords were dissected and postfixed in 10% formaldehyde for 4 hours and then cryoprotected overnight in 30% sucrose in PBS. Tissues were frozen in Tissue-Tek and sectioned using a cryostat (Leica, New York, NY). Tissue was sectioned at 40 μm and stored in PBS at 4°C and then transferred to glycerol-based cryoprotectant solution for long-term storage at −20°C.
2.3.2. Immunohistochemistry
Tissues were blocked for 1 hour in 0.1-M PBS with 0.3% Triton X-100 and 5% normal donkey serum (NDS). Primary and secondary antibodies were diluted in 0.1-M PBS with 0.3% Triton X-100 and 1% NDS. Sections were then incubated overnight at room temperature (RT) in primary antibody solution, washed in 0.1-M PBS with 0.3% Triton X-100 and 1% NDS for 3 × 10 minutes, incubated for 2 hours in secondary antibody (RT), and then washed in 0.1-M PBS for 3 × 10 minutes. Sections were then mounted using Fluoromount-G on Fisherbrand Superfrost Plus microscope slides. Images were acquired with a Leica TCS SPE confocal microscope.
Primary antibodies: anti-CD11b, Abd Serotec # MCA711G (rat, 1:1000); anti-IBA1, Fujifilm Wako # 019-19741 (rabbit, 1:1000). Alexa Fluor–conjugated secondary antibodies were acquired from Invitrogen and Jackson ImmunoResearch Labs.
2.3.3. Quantification of immunofluorescence and microglia number
For quantification of CD11b immunofluorescence, lumbar spinal cord tissue from all conditions was immunostained and imaged together. Single confocal plane images were taken of both dorsal horns. Using ImageJ, a region of interest was drawn around the dorsal horn to exclude image space without tissue. Ipsilateral and contralateral dorsal horns from each slice were processed together with the same threshold. CD11b integrated density was measured, and ipsilateral value was divided by contralateral for one measurement per slice. n = 5 to 17 slices per animal, 2 to 3 animals per condition. A similar quantification was performed for IBA1 density, using sham control mice as a reference, rather than the contralateral side.
For counts of microglial density after chronic opioid treatment, mice were treated with saline or morphine (fixed or escalating dose) and processed for CD11b immunohistochemistry as described above. Single confocal plane images of the dorsal horn were taken with DAPI labeling. Only clearly labeled microglia positive for both CD11b and DAPI were counted and normalized for tissue area. Because tissue was prepared and counted in 2 separate replicates by independent experimenters, results were normalized to the appropriate control tissue. Each treatment group and replicate had a minimum of 3 animals, with a minimum of 9 slices per animal.
For the analysis of microglial morphology, lumbar spinal cord tissue from all conditions analyzed was immunostained for IBA1 and imaged together. Z-stack confocal images of full spinal cords were taken. Using ImageJ, a region of interest was drawn around the dorsal horn on 63x images, and single microglial cells were subjected either to Sholl analysis (https://github.com/morphonets/SNT) or to manual quantification. Quantifications were performed for each slice: 4 slices per animal, 4 animals per condition.
2.4. In situ hybridization
2.4.1. Tissue collection and processing for RNAscope ISH
Mice anesthetized with isoflurane were decapitated. Brains were dissected and fresh frozen on dry ice for 30 minutes and then parafilmed overnight in a −80°C freezer. Tissues were then sectioned using a cryostat (Leica). Tissue was sectioned at 16 μm and directly mounted onto Superfrost Plus microscope slides. Slides were stored long term at −80°C.
2.4.2. RNAscope ISH
The RNAscope Multiplex Fluorescent v2 assay was used to simultaneously detect 3 RNA targets in a single sample using 3 target probes purchased from ACDBio designed for manual RNAscope assays in mouse species. Tissues were postfixed on-slide with 4% formalin for 1 hour and then washed twice with 0.1-M PBS. Tissue was then dehydrated with 50%, 70%, and then twice with 100% EtOH for 5 minutes each before a hydrophobic barrier was applied. Tissue was incubated in H2O2 for 10 minutes at RT and then washed twice with ddH20. The sample was then incubated with RNAscope Protease III for 20 minutes at RT and then washed with 0.1-M PBS. Target probes were warmed for 10 minutes in a 40°C incubator and then cooled to RT. Fifty volumes of C1 (6 drops per slide) and 1 volume of C2 and C3 (3 μL per slide) were mixed into the same tube. One-hundred fifty microliters of mixture were added onto each slide and then incubated for 2 hours at 40°C. Slides were then washed twice with 1X wash buffer. We then submerged slides in 5X saline sodium citrate overnight. The next day, slides were washed twice with a 1X wash buffer. Slides were then incubated with AMP1 for 30 minutes, AMP2 for 30 minutes, and then AMP3 for 15 minutes at 40°C. Slides were washed twice with 1X wash buffer after each AMP step. Next, HRP-C2 signal was developed by incubating slides in HRP-C2 for 15 minutes, 570 dye for 30 minutes, and then HRP blocker for 15 minutes, all at 40°C. Slides were washed twice with 1X wash buffer after each HRP signal development step. This step was repeated for HRP-C1 and C3. HRP-C1 was paired with 520 dye, whereas C3 was paired with 690 dye. Opal dyes were diluted with TSA buffer for staining. All washes were for 2 minutes with minimal agitation. Nuclei were then counterstained and visualized using DAPI fluorescent dye. Slides were mounted using Fluoromount-G. Images were acquired with a Leica SP8X Falcon confocal microscope.
Target probes: RNAscope Probe-Mm-ITGAM, ACDBio #311491; RNAscope Probe-Mm-Oprm1-O4-C2, ACDBio #544731-C2; and RNAscope Probe-Mm-Mki67-C3, ACDBio #416771-C3.
Dyes: Opal 520, Opal 570, and Opal 690 from Akoya Biosciences, with recommended dilution range 1:750 for Opal 570 and Opal 690 and 1:1500 for Opal 520. Opal 690 was assigned to low-expression genes, Opal 570 to medium-expression genes, and Opal 520 to high-expression genes.
2.5. Injury models
Three peripheral nerve injuries were performed: complete transection of the sciatic nerve (SNT), chronic constriction injury of the sciatic nerve (CCI), and spared nerve injury (SNI).63,74 Adult mice were anesthetized with isoflurane, and the right sciatic nerve was exposed at midthigh level. For CCI, two 5-0 silk sutures were tied loosely around the nerve about 2 mm apart. For SNT, a 2-mm portion of the nerve was removed. For SNI, only the tibial and common peroneal branches were cut. The muscle was pinched closed, and the skin was sealed with tissue adhesive (Vetbond). For RNA sequencing, the procedure was repeated on the left side, and spinal cords were collected 1, 2, 4, and 7 days after SNT; 2 and 7 days after CCI; and 7 days after SNI. For histology, the injury was only performed on the right side.
2.6. Microglial RNA library construction and sequencing
Mice were perfused with ice-cold PBS, and caudal lumbar spinal cords were isolated. Caudal lumbar segments from 4 to 15 mice per group were pooled to obtain enough material to ensure tight correlation between replicates. Tissue was mechanically dissociated in HBSS (Gibco) containing 0.5% glucose, 15-mM HEPES (pH 7.5), and 125-U/mL DNaseI and then processed with MACS myelin removal (Myelin Removal Beads II, Miltenyi, Bergisch Gladbach, Germany) followed by CD11b selection (CD11b MicroBeads, Miltenyi) according to the manufacturer's instructions, except that all centrifugation steps were shortened to 30 seconds at 10,000 rpm.
Total RNA was extracted from CD11b-positive cells using the RNeasy Micro Kit (Qiagen, Venlo, Netherlands). Libraries were prepared using Smart-Seq256 and modified for sequencing using the Nextera XT DNA Sample Preparation Kit (Illumina, San Diego, CA) with 300 pg of cDNA as input material. Libraries were sequenced using the Illumina NextSeq or MiSeq to obtain 75-bp paired-end reads. At least 2 libraries for each condition were prepared and sequenced independently for an average of 10.1 million reads per group (range of 1.8-28.6 million).
Reads were mapped to the UCSC mouse reference genome mm10 through the Galaxy platform (http://usegalaxy.org) using HISAT2 65 version 2.0.5.1. Fragments per kilobase of transcript sequence per million mapped fragments (FPKM) values were obtained using Cufflinks 66 version 2.2.1. Reads were quantified using featureCounts version 1.4.6.p5.45 Differential analysis was conducted in R with DESeq2.49 A stringent significance cutoff was set using the Padj < 0.01 to account for low replicates in some conditions. No statistical analysis was performed on FPKM values; stars in figures illustrating FPKM values are derived from DESeq2 results. Gene ontogeny (GO) term analysis was conducted by creating individual lists of upregulated and downregulated genes in each condition with Padj < 0.01 from DESeq2 output and evaluated using DAVID.35 Initial analysis of Oprm1 expression from a subset of CCI, SNT, and morphine data sets was reported in our previous work.13 All sequencing data are available at GEO GSE117321 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117321).
For rank–rank hypergeometric overlap (RRHO) analysis, full threshold-free differential expression lists were ranked by the −log10(P value) multiplied by the sign of the fold change from the DESeq2 analysis. Rank–rank hypergeometric overlap analysis was used to evaluate the overlap of differential expression lists between our PNI and morphine conditions. Rank–rank hypergeometric overlap difference maps were produced for each comparison.
2.7. Microglia culture and stimulation
Rat microglia were isolated and cultured as described previously.8 Briefly, brains were extracted from perfused male 21-day-old rats, and the cerebellum of each brain was discarded. Brains were mechanically dissociated to a single-cell suspension and passed through a density gradient to remove myelin and cellular debris. Myeloid cells were isolated by CD11b immunopanning and plated in serum-free medium (DMEM/F12 containing 100-U/mL penicillin, 100-μg/mL streptomycin, 2-mM glutamine, 5-μg/mL N-acetyl cysteine, 5-μg/mL insulin, 100-μg/mL apo-transferrin, 100-ng/mL sodium selenite, 0.1-μg/mL oleic acid, 0.001-μg/mL gondoic acid, 1-μg/mL heparan sulfate, 1.5-μg/mL ovine wool cholesterol, 100-ng/mL murine IL-34, and 2-ng/mL human TGF-β2). Cells were maintained for 5 days with 50% medium change on day 3. After 5 days, cells were stimulated with TLR ligands LPS (1 ng/mL), PAM3CSK4 (1 ng/mL), zymosan (100 μg/mL), or morphine (1 μM or 10 μM) by introducing the drug in a 50% medium change.
2.8. Cultured cell gene expression analysis
For gene expression experiments, cells were incubated with morphine for 4 or 24 hours, or with TLR agonist for 24 hours, and then washed twice before lysis and RNA purification using the RNeasy Micro Kit (Qiagen, Venlo, Netherlands). For RNA-seq experiments, samples were prepared and analyzed as described above for freshly isolated mouse samples, except data were mapped using the rat genome annotation rn6. For qPCR experiments, RNA was reverse-transcribed using a High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Waltham, MA) and used as template for SYBR Green PCR Master Mix (Thermo Fisher Scientific) reactions (20-μL volume) with previously published primers.8 CT values were registered using noiseband thresholding, and melting curves were checked to ensure formation of a single product within the expected size range. ΔCT values were calculated relative to the housekeeping gene Rplp0. Consistent ΔCT values of a second housekeeping gene, Eef1a1, were measured across samples. All CT values were detected before completion of the 38th cycle.
3. Results
3.1. Morphine exposure induces no significant transcriptional response in microglia
We and others have previously shown histological evidence of microglial activation after chronic morphine treatment that coincides with the establishment of tolerance and OIH.13 Moreover, this activation is intact in mu-opioid receptor (MOR) knockout mice and is thus independent of MOR signaling.13 We, therefore, used transcriptomics to clarify the MOR-independent molecular mechanisms associated with opioid-induced microglial reactivity. First, we treated mice with 10-mg/kg morphine for 10 days, a fixed dosing paradigm (FIX) known to produce tolerance, OIH, and microglial activation (Fig. 1A, B). We then isolated the lumbar spinal cord because this region was particularly important for further comparisons to sciatic nerve injury conditions. We combined mechanical dissociation of tissue with magnetic-activated cell sorting (MACS) for myelin depletion and CD11b selection to isolate a highly enriched microglial population for transcriptional analyses (Fig. 1C). As we relied on CD11b surface expression to enrich for microglia, we also captured a small number of other myeloid cells such as neutrophils and perivascular macrophages. However, we saw strong enrichment of microglial genes compared with markers of other nonmyeloid CNS cell types including endothelial cells, oligodendrocytes, neurons, and astrocytes (Fig. 1D). Note that we found no evidence of infiltration or expansion of monocytes or neutrophils in any chronic pain model, consistent with previous studies.9,37,44
Strikingly, despite obvious morphological changes suggesting an activated state (Fig. 1B), chronic morphine treatment induced no significant change in microglial gene expression (Fig. 1E). In stark contrast, hundreds of genes showed differential expression in other chronic pain models when tested using the same methods (see below). We further interrogated this lack of transcriptional response using a more aggressive escalating dosing paradigm (ESC) that produced increased density of microglial labeling (Fig. 1B) but again detected no transcriptional difference compared with control (Fig. 1A, B, F-N).
We therefore closely re-examined the characteristics of microglia activation in tissues. We found that chronic morphine (ESC) resulted in a higher number of microglial ramifications, increased branch length, and decreased cell body area compared with controls (Fig. 1J-M). These characteristics notably contrast with the morphological changes commonly associated with microglial activation after PNI, which include decreased number of ramifications and decreased branch length.73 Importantly, morphine treatment caused no increase, and even a trend towards a decrease, in the number of microglia (Fig. 1N). Together, these observations challenge the notion of a shared mechanistic basis for PNI- and opioid-induced hypersensitivity.
To further investigate these unexpected observations, we used an in vitro culture system. Microglial cultures have been used extensively to interrogate microglial response to opioids,9,43,44 with some studies suggesting that morphine may contribute to microglial activation through an unknown mechanism involving TLR4.37 To investigate the direct effects of morphine on microglia, we used a highly pure serum-free culture system8 to compare morphine treatment with TLR2 and TLR4 agonists (LPS, PAM3CSK4, and zymosan) known to induce robust transcriptional changes. As expected, all 3 TLR agonists produced extensive gene expression changes. Again, however, we observed no transcriptional changes associated with morphine treatment (Fig. S1A-D, http://links.lww.com/PAIN/C58). To confirm this finding, we conducted additional treatments with LPS or morphine (1 μM up to 10 μM) and performed qPCR for genes associated with TLR activation. We found that LPS induced significant upregulation of these genes, whereas morphine had no such effect at any concentration (Fig. S1E, http://links.lww.com/PAIN/C58). To ensure that the high variability between TLR agonism and control did not mask potential differential expression in morphine treatment, we again ran DESeq2 to compare morphine treatment to PBS alone and still identified no differences. Note that, consistent with our previous report,13 we detected no Oprm1 transcripts in serum-free cultured microglia, either before or after morphine or TLR agonist exposure. Even in the absence of MOR, it is possible that morphine could activate microglia by binding to one or both of the other opioid receptor types, delta (DOR) or kappa (KOR). However, we found no transcripts for the genes encoding DOR and KOR (Oprd1 and Oprk1, respectively). Taken together, this lack of transcripts encoding any of the 3 opioid receptors suggests that canonical opioid signaling may not occur in microglia.
Thus, from both our in vivo and in vitro experiments, we conclude that morphine exposure has no detectable effect on microglial gene expression profiles, even at doses that alter microglial morphology in vivo and produce behavioral hypersensitivity.
3.2. Distinct transcriptional signatures after peripheral nerve injury and morphine exposure
Given the reported mechanistic similarities of microglial activation and pronociceptive action after PNI, we next directly compared chronic opioid treatment against multiple broadly used models of PNI on a transcriptional level. First, we used the spared nerve injury (SNI) model,63 which consists of transecting 2 of the 3 branches of the sciatic nerve (Fig. 2A). As expected, SNI induced a strong increase in CD11b immunoreactivity 7 days postinjury (dpi) when comparing ipsilateral with contralateral spinal cord, indicating microglial activation (Fig. 2B). Remarkably, however, differential expression analysis with DESeq2 revealed only minimal gene expression changes in isolated microglia at 7 dpi (adjusted P value [Padj] < 0.01), with only 7 downregulated genes and 1 upregulated gene, the complement pathway component C4b (Fig. 2C).
Figure 2.

Diverse transcriptional responses of spinal microglia to chronic neuropathic pain conditions. (A) Schematic of peripheral nerve injuries: complete sciatic nerve transection (SNT), chronic constriction injury (CCI), and spared nerve injury (SNI). (B) Example images of CD11b immunostaining in lumbar spinal cords 7 days after unilateral SNI. dpi, days postinjury. Scale bar = 100 µm. (C) Volcano plots summarizing gene expression changes in microglia 7 days after bilateral SNI. Blue and red dots indicate significantly downregulated or upregulated genes, respectively, from DESeq2 (Padj < 0.01). The numbers in the lower corners indicate the total numbers of downregulated (blue) and upregulated (red) genes at Padj < 0.01. (D) Example images of CD11b immunoreactivity in lumbar spinal cords 7 days after unilateral SNT. dpi, days postinjury. Scale bar = 100 µm. (E) Volcano plots summarizing gene expression changes in microglia 7 days after bilateral SNT. Blue and red symbols indicate significantly downregulated or upregulated genes, respectively, from DESeq2 (Padj < 0.01). Triangles indicate values outside the axis scale. The numbers in the lower corners indicate the total numbers of down (blue) and upregulated (red) genes at Padj < 0.01. (F) Example images of CD11b immunoreactivity in the spinal cord 2 and 7 days after unilateral CCI. Scale bar = 100 µm. (G) Volcano plots summarizing gene expression changes in microglia 7 (D) days after bilateral CCI. Blue and red symbols indicate significantly downregulated or upregulated genes, respectively, from DESeq2 (Padj < 0.01). The numbers in the lower corners indicate the total numbers of downregulated (blue) and upregulated (red) genes at Padj < 0.01. (H) Top enriched GO terms associated with CCI at 7 dpi. FDR, false discovery rate. (I) Heatmap of genes differentially regulated (Padj < 0.01 in DESeq2) after fixed (FIX) or escalating (ESC) morphine treatment, and bilateral SNI, SNT, or CCI at 7 dpi, plotted as Log2(fold-change). Asterisks indicate significance in that condition (Padj < 0.05). GO, gene ontogeny.
Because SNI leaves a large proportion of somatosensory neurons intact,4 we hypothesized that the low detection of SNI-induced changes in RNA expression could result from a dilution effect associated with the presence of resting-state microglia. To test this possibility, we used a more severe injury, a complete bilateral sciatic nerve transection (SNT) (Fig. 2A). Immunohistological analysis of mice that underwent SNT confirmed previous reports of marked microglial activation (Fig. 2D).69 Despite this substantial change in the CD11b staining pattern, we found that after SNT, only 3 genes were significantly downregulated, whereas 13 were upregulated at 7 dpi (Padj < 0.01, Fig. 2E), supporting our previous results that nerve injury–induced microglial activation is accompanied by only minimal gene expression changes at 7 dpi.
Having observed minimal transcriptional changes 7 days after full or partial transection of the sciatic nerve, we next used a third allodynia-inducing PNI model: chronic constriction injury (CCI).6 For CCI, 2 ligatures are tied loosely around the sciatic nerve (Fig. 2A). We found that CCI induced a pattern of CD11b immunoreactivity similar to that of SNT and SNI, with increased CD11b expression at 7 dpi (Fig. 2F). However, specifically after CCI, we detected a larger number of differentially expressed genes, with 31 downregulated and 155 upregulated (Fig. 2G). To further characterize this response to CCI, we used GO term analysis and found that the microglial response 7 days after CCI was predominantly related to immune and inflammatory processes (Fig. 2H), including members of the complement pathway (C4b and Cfb) and cytokines (Ccl5 and Cxcl10). Among the 13 genes upregulated at 7 days after SNT, 9 were also upregulated at 7 days after CCI; most of these genes related to immune responses, including Cfb, Ifit1, and Ccl5 (Fig. 2I). Overlap between gene expression changes in CCI and SNI was much lower.
To identify transcriptome-wide overlap in a threshold-free manner across all our conditions, we performed RRHO RNA-seq analysis (Fig. S2A, http://links.lww.com/PAIN/C58). This analysis showed limited overlap for most comparisons, particularly between morphine treatments and PNI models, indicating different transcriptional signatures across conditions. We found some overlap between morphine treatment paradigms (FIX vs ESC) and some PNI models (SNT vs CCI and SNT vs SNI), and more surprisingly, SNI vs ESC and SNI vs FIX. Thus, the CCI and SNT models seem to have more similar transcriptional signatures compared with SNI and opioid treatments (Fig. S2A, http://links.lww.com/PAIN/C58).
To examine this finding further, we complemented this analysis with a more targeted hypergeometric gene overlap analysis to determine the P value and odds ratio relative to a genomic background. Consistent with the RRHO analysis, a comparison of the top 10 upregulated genes in each condition indicated no overlap for most comparisons (Fig. S2B, http://links.lww.com/PAIN/C58). Only FIX overlapped with ESC, and only SNT overlapped with CCI (Fig. S2B, http://links.lww.com/PAIN/C58), confirming the similarities in microglial transcriptional responses between our 2 morphine paradigms, and between SNT and CCI, and that the microglial transcriptional response after SNI is significantly different from that observed after the other 2 PNI models. Importantly, this test did not find a significant overlap between our morphine treatment conditions and SNI (Fig. S2B, http://links.lww.com/PAIN/C58). We also evaluated the distribution of overlapping genes between conditions for the top 10 upregulated genes and for the proliferation-associated genes among the top 100 upregulated genes (Fig. S2C-D, http://links.lww.com/PAIN/C58). Notably, we found more overlapping proliferation-associated genes in our PNI models at 2 dpi than at 7 dpi and overall more proliferation-associated genes in the top 100 upregulated genes in the PNI conditions than the morphine conditions (Fig. S2D, http://links.lww.com/PAIN/C58).
Finally, we pooled our data from all control, all morphine, and all injury conditions and ran these 3 groups through DESeq2 analysis (Fig. S3, http://links.lww.com/PAIN/C58). Compared with our unpooled DESeq2 analysis, this approach revealed a greater number of differentially expressed genes after PNI or morphine exposure. Indeed, we identified 113 downregulated genes and 91 upregulated genes after PNI (Fig. S3A, http://links.lww.com/PAIN/C58) vs 65 downregulated genes and 25 upregulated genes after morphine exposure (Fig. S3B; Table S1, http://links.lww.com/PAIN/C58 for the injury group and Table S2, http://links.lww.com/PAIN/C58 for the morphine group). Subsequent RRHO analysis revealed little transcriptomic overlap between injury and morphine conditions (Fig. S3C, http://links.lww.com/PAIN/C58). Among the top 50 upregulated (Fig. S3D, http://links.lww.com/PAIN/C58) or downregulated genes (Fig. S3E, http://links.lww.com/PAIN/C58), we found only 4 overlapping upregulated genes and no overlapping downregulated genes. Importantly, in comparison with unpooled SNT, SNI, or CCI, morphine treatment produced a distinct transcriptional signature (Fig. 2I). The immune-response genes detected after CCI showed no expression changes in either morphine dosing paradigm. Together, these results further confirm that microglia undergo distinct transcriptional responses after PNI or morphine treatment, despite similar hyperalgesic phenotypes. Similar to previous transcriptomic studies,3,7,16,33,38 we detected no transcripts for BDNF, which is believed to be a key mediator for the contribution of microglia to both neuropathic pain and OIH.14,19
Our differential transcriptomic analysis therefore posits that although both opioids and PNI elicit histological indicators of microglial activation (Fig. 1B, G-I; Fig. 3A-E), proliferation is observed only after PNI (Fig. 3F). This observation challenges the notion of a shared mechanistic basis for PNI- and opioid-induced hypersensitivity. To test this concept further, we examined the morphological characteristics of activated microglia using IBA1 immunostaining in our PNI models (Fig. 3C-E). We found that, across all our PNI conditions, microglia display similar increases in both IBA1 expression and microglia number, no change in cell body area or number of ramifications, and decreases in total and average branch length (Fig. 3D-J). Microglia from all PNI conditions displayed an amoeboid shape, suggesting that microglia present similar activation profiles regardless of the type of nerve injury. By contrast, morphine increased IBA1 expression without affecting microglia number, increased both the number of ramifications and the total branch length, and decreased cell body area (Fig. S4, http://links.lww.com/PAIN/C58).
Figure 3.

PNI models induce proliferative and morphological features of microglial activation. (A) Example images of IBA1 immunoreactivity in the ipsilateral dorsal horn of lumbar spinal cords at 7 dpi after sham, CCI, SNI, or SNT. Scale bar = 100 µm. (B) Similar to (A), but for contralateral dorsal horn. (C) Example 63x images of IBA1 immunoreactivity in the ipsilateral dorsal horn of lumbar spinal cords at 7 dpi after sham, CCI, SNI, or SNT. Scale bars = 10 µm. (D) Quantification of IBA1 density (% area) in the ipsilateral (left) and contralateral (right) dorsal horn. Normalized as % vs control sham. (E) Statistical comparisons of IBA1 density (% area) between ipsilateral and contralateral sides after sham, CCI, SNI, or SNT. (F) Quantification of the number of microglia in the dorsal horn in all conditions. (G–J) Quantification of microglial morphology, including number of ramifications, microglia body area, total branch length, average branch length, and number of microglia. One-way ANOVA and Holm-Sidak's test (I, N); * indicates P < 0.05; ***, P < 0.001; ****, P < 0.0001 vs sham or saline or morphine. n = 4 mice. CCI, chronic constriction injury; PNI, peripheral nerve injury; SNI, spared nerve injury; SNT, transection of the sciatic nerve.
3.3. Proliferation drives transcriptional changes after peripheral nerve injury but not opioid exposure
Given the unexpected contrast between the dramatic histological changes and the limited transcriptional changes after PNI, we next conducted a time-course study of microglial responses. To maximize detection of differentially expressed genes across time, we used the SNT model. We performed bilateral SNT and RNA-seq as described above at 7 dpi, but also collected tissue at 1, 2, and 4 dpi. Differential expression analysis revealed extensive transcriptomic changes soon after injury that were not detected at later time points (Fig. 4A, B). We identified 316 differentially expressed genes 24 hours after injury (Fig. 4A) and detected the greatest number of differentially expressed genes, 550, at 2 dpi (Fig. 4B). By contrast, at 4 dpi, only 15 genes were differentially expressed (Fig. 4C). We selected the time point with the greatest differential expression, 2 dpi, for comparison with the CCI model. Two days after CCI, we detected 711 upregulated genes and 301 downregulated genes (Fig. 4D), the largest response from all groups. In addition, we found extensive overlap between differentially expressed genes 2 days after SNT or CCI, suggesting similar early microglial responses between these conditions (Fig. 4E).
Figure 4.
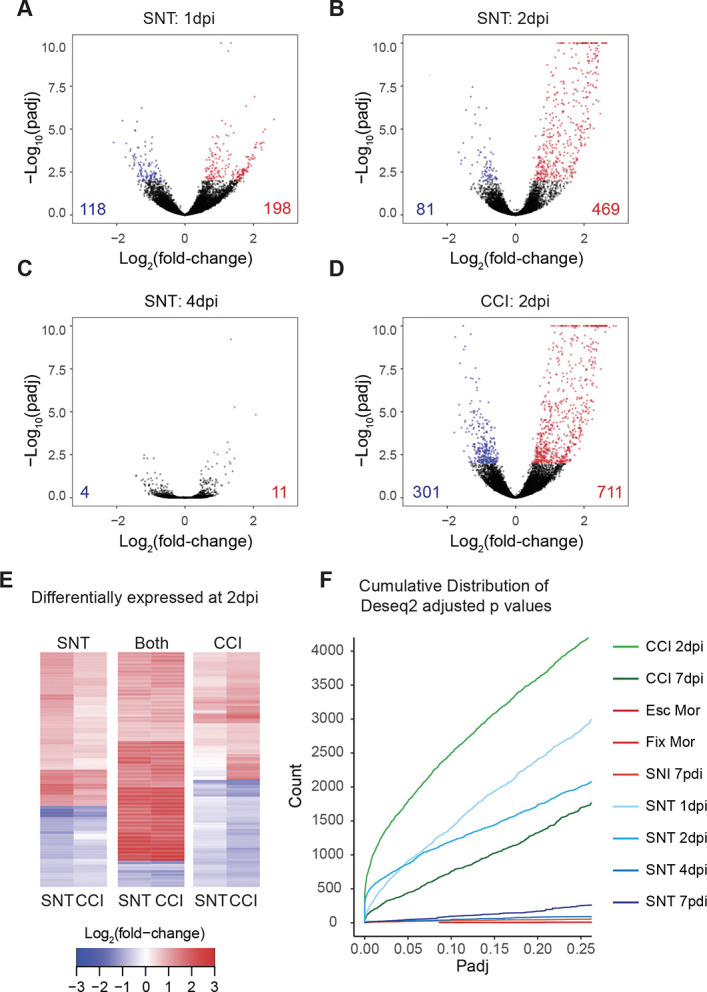
The greatest transcriptional variability occurs early after injury. (A–D) Volcano plots summarizing gene expression changes in microglia at 1 (A), 2 (B), or 4 (C) days after complete bilateral transection or 2 (D) days after bilateral CCI. Blue and red symbols indicate significantly downregulated or upregulated genes, respectively, from DESeq2 (Padj < 0.01). The numbers in the lower corners indicate the total numbers of downregulated (blue) and upregulated (red) genes at Padj < 0.01. (E) Heatmaps of genes differentially expressed 2 days after SNI only, both SNT and CCI or CCI only. Values are plotted as Log2(fold-change). (F) Cumulative distribution of read counts at Padj values (≤0.25) from DESeq2 for all injury and treatment conditions. CCI, chronic constriction injury; SNI, spared nerve injury; SNT, transection of the sciatic nerve.
Overall, we found that the major transcriptional changes at 2 dpi had largely resolved at 4 days after SNT and had evolved into an inflammatory response at 7 days after CCI. We also confirmed that the increased number of differentially expressed gene calls at early time points after SNT or CCI was not a result of selecting a particular P value cutoff because the difference in the extent of changes was observed regardless of the significance cutoff selected (Fig. 4F).
To elucidate the classes of genes upregulated during early stages, we used GO term analysis. Markers associated with cell cycle and cell division emerged as the primary drivers of differential gene expression between control and injury conditions at 2 dpi (Fig. S5A, http://links.lww.com/PAIN/C58). Interestingly, the patterns were remarkably consistent between SNT and CCI. This early cellular response is exemplified by the expression of canonical cell proliferation markers such as Mki67, Top2a, Ccn2, and Cdk1 predominantly at 2 dpi in both conditions (Fig. S5B, http://links.lww.com/PAIN/C58). These findings suggest a conserved temporal sequence in microglial responses across different nerve injury models, characterized by an initial increase and subsequent decrease in proliferation markers. Notably, after this initial proliferative phase, persistent alterations in the microglial transcriptome are relatively limited across various injury models. By contrast, chronic morphine treatment caused no increase in proliferation markers (Fig. S5B, http://links.lww.com/PAIN/C58). To validate these disparities, we used in situ hybridization (ISH) to examine Oprm1, Cd11b, and Mki67 expression in the spinal cord dorsal horn at 2 dpi after either injury or morphine treatment (Fig. S5C, http://links.lww.com/PAIN/C58). We found that after morphine treatment, in contrast to CCI, neither Cd11b nor Mki67 RNA expression was upregulated. Furthermore, the number of Mki67+ cells remained stable, including at early time points, confirming the absence of microglial proliferation after morphine treatment (Fig. S5C-E, http://links.lww.com/PAIN/C58).
3.4. Diversity of microglial transcriptional responses to insult and disease
Given that our analysis revealed a diversity of microglial transcriptional responses associated with hyperalgesic conditions, we next sought to compare these pain-associated microglial transcriptional responses to those of other disease states. A meta-analysis across 336 purified myeloid transcription data sets identified modules of coregulated genes characteristic of diverse disease or injury states.20 We used these modules to compare the transcriptomic responses of microglia with PNI or morphine treatment to those associated with proliferation, interferon stimulation, LPS treatment, and neurodegenerative disease models (Fig. 5A-D). Importantly, and in agreement with our initial analysis, we found that genes implicated in microglial proliferation response such as Gal-3 are upregulated 2 days after SNT or CCI, but remain relatively unchanged at other time points or after morphine treatment (Fig. 5D). Further supporting our interpretation that microglia take on an immune-reactive phenotype at late time points, we also found that, after SNT or CCI, microglia exhibit increased expression of genes associated with interferon stimulation, such as the interferon-stimulated gene Ifit1 (Fig. 5B). The stronger immune response induced by CCI was also reflected in the distribution of the LPS treatment module, as illustrated by the increase of complement component C3 expression (Fig. 5C). In stark contrast, morphine treatment did not induce LPS-like changes in gene expression. Although a minority of genes related to neurodegeneration were significantly upregulated in one or more of our manipulations, the proportion of genes from this module and the magnitude of these transcriptional changes were very limited compared with the changes associated with neurodegeneration models (Fig. 5D). These observations strongly support the idea that microglial activation as detected by histological examination cannot be assumed to correspond to any stereotyped transcriptional state, but may instead reflect widely varying conditional responses.
Figure 5.

Diversity of microglial transcriptional responses. (A-D) Differential expression, displayed as Log2(fold-change) from DESeq2 output compared with control, for each gene in 4 modules, Proliferation (A), Interferon-related (B), LPS (C), and Neurodegeneration related (D), selected from Friedman et al., 2018 across conditions tested, with FPKM values plotted, eg, genes. Asterisks indicate significance from DESeq2 output. * indicates P < 0.05. FPKM, fragments per kilobase of transcript per million mapped reads.
Together, the transcriptional data presented here and the comparison with published data sets establish the need for multimodal descriptions of activation states to resolve the molecular mechanisms of microglial function, including after PNI- and opioid-induced hyperalgesia.
4. Discussion
In this study, we provide a full transcriptional characterization of acutely isolated spinal cord microglia across multiple hyperalgesic states (chronic opioid exposure, SNI, SNT, and CCI), including temporal resolution of the progression of microglial activation. Microglial depletion after chronic morphine treatment is known to reverse hypersensitivity, supporting the importance of microglial function at this time point.19 Unexpectedly, we found no significant changes in gene expression after chronic opioid treatment. Previous studies suggested that microglial activation after morphine treatment occurs because of binding of morphine to MORs expressed by microglia.19 However, we detected no MOR expression in spinal microglia using immunohistochemistry, ISH, or RNA-seq and previously found that opioid-induced microglial activation is intact in MOR-null mice.13 Furthermore, we found no evidence of 2 other opioid receptors, KOR (encoded by Oprk1) and DOR (encoded by Oprd1), ruling out morphine-binding microglial DORs or KORs as a potential mechanism of microglial activation. Another proposed mechanism implicates opioid engagement of microglial TLR4.18,23,37 Because TLR4 activation by LPS produces significant transcriptomic changes in microglia both in vivo10 and in culture (Fig. 2), we expected to detect similar transcriptional changes if morphine was indeed acting as a TLR4 agonist. However, note that the transcript for the TLR4 cosignaling molecule MD-2, implicated in the interaction of morphine and TLR4,37 was not detected in our cultures. Nevertheless, the lack of transcriptional evidence of LPS-like signaling in response to morphine both in vivo and in vitro argues against direct TLR4 activation, consistent with reports showing intact microglial activation or morphine tolerance in TLR4-null mice.21,50 Together, these data suggest that receptors other than opioid receptors and TLRs may act as sensors for microglia to detect either opioids or secondary mediators released after opioid treatments.
To address the possibility that some biologically meaningful gene expression changes may exist below our statistical threshold of detection, we further compared the trends in our data set against the transcriptional response of spinal microglia with PNI. We found that, after PNI, microglia undergo a transient, proliferation-driven transcriptional response. This signature is consistent with previous studies that used histological and imaging methods to quantify microglial proliferation after PNI.17,25,27,60 We found the greatest transcriptional variability 24 to 48 hours after injury, preceding maximum changes in histological markers of activation. Interestingly, depletion of microglia or their inhibition with minocycline effectively diminishes pain hypersensitivity only when introduced at these early proliferative time points.55,57 However, the minimal transcriptional changes detected at later time points may also be important, given that we detected divergence between injury types at 7 dpi, with a strong inflammatory immune response after only CCI. Interestingly, CCI also triggers an epineural inflammation not seen in SNI, suggesting a commonality between peripheral and microglial immune responses to pronociceptive insults.11
By combining our data sets with those of Friedman et al.,20 we performed a meta-analysis that further revealed the diversity of microglial responses. For example, even at time points with the greatest transcriptional variability, PNI- and OIH-associated microglial transcriptional signatures differed from those associated with neurodegenerative conditions. In addition, we see transcriptional changes similar to those induced by LPS after CCI, but not after morphine treatment, as would be predicted by opioid activation of TLR4. Although these and other sequencing studies have aided the search for selective markers for microglia and their activated state, our findings suggest there may be no universal marker or signature across conditions. Note, however, that recent studies using single-cell RNA sequencing2,28,47 have focused on the discovery of different subtypes of microglia, as illustrated by the Cd11c population,41 a transcriptionally and functionally distinct subset of the Cd11b population. This subset, enriched in insulin receptor IGF1, was shown to be involved in remission and relapse of neuropathic pain. Unlike neurons, microglia undergo different states of activation during aging, and their intrinsic properties (eg, highly proliferative) make it unclear whether a unique transcriptional and functional signature suffices to define a new microglia subtype.5 However, future single-cell sequencing and morphological studies will likely further unravel the heterogeneity of microglia.
Microglia contribute to hyperalgesic states through other known nontranscriptional mechanisms. Epigenetic changes in microglia have been identified after PNI, contributing to the long-term maintenance of neuropathic pain.12,16,24,62 C1q and complement signaling are associated with synaptic pruning during development66; microglia may use similar complement-mediated mechanisms to remodel synapses in the dorsal horn after PNI or opioid treatment. Notably, despite its proposed involvement in both neuropathic pain and OIH, we detected no Bndf transcripts in the majority of our acutely isolated or cultured microglia samples, consistent with recently published microglial sequencing data sets.16,32 This adds complexity to the interpretation of studies in which genetic deletion of BDNF from microglia impacted not only pain but also learning and memory14,19,54 and warrants further study to examine the potential disconnect between mRNA and protein expression. Alternatively, the presence of BDNF transcripts in astrocytes,77 dorsal root ganglia,72 and spinal cord dorsal horn neurons31 suggests that BDNF of nonmicroglial origin could alter the excitability of dorsal horn neurons during neuropathic pain and/or OIH. Future proteomic studies may be required to fully resolve the mechanisms of BDNF production and action in the dorsal horn.
This study was conducted using only male mice. This decision was informed by an extensive body of existing literature demonstrating the critical role of microglial activation specifically in males.29,30 Recent findings indicate that there are sex differences in how non-neuronal cells, particularly microglia, contribute to pain. Studies have shown that in male mice, but not female mice, spinal microglial toll-like receptor 4 (TLR4) plays a role in controlling inflammatory and neuropathic pain, suggesting that microglia in the spinal cord may predominantly affect pain in males.64 In addition, interventions aiming to inhibit or ablate microglia using drugs such as minocycline, inhibitors of microglial BDNF signaling, or toxins reduced pain in rodent assays in males but not females.65,67 Furthermore, nerve damage was found to increase P2X4 receptor expression in the spinal cords of male but not female mice.65 This sex-specific response of microglia is influenced by testosterone, as evidenced by the lack of pain reduction by minocycline in castrated males and its effectiveness in reducing pain in females treated with testosterone.65 Conversely, in females but not males, T cells are the critical non-neuronal cells driving insult-induced hypersensitivity.65 Given this extensive literature that established the crucial contribution of microglia to pain in males but not females, combined with our ethical obligation to use the smallest number of animals possible to advance scientific knowledge, we were obligated to use only male mice for this study.
It is also important to recognize the complexities associated with the study of microglia, given their highly dynamic and intricately ramified nature. Our methodologies, although grounded in previous research, necessitate a selection of specific time points, which may overlook other significant periods in microglial function. Even with z-stack confocal imaging, our ability to evaluate the full three-dimensional morphology of these cells is limited. Future three-dimensional longitudinal studies may provide a more comprehensive analysis of the morphological phenotypes observed here, as well as the intricate responses of microglia during activation, migration, and phagocytosis.
Collectively, our data reveal dynamic transcriptional responses in microglia that vary across disease models and hyperalgesic states. Despite similarities in microglial marker expression after PNI or opioid exposure, microglia engage insult-specific transcriptional mechanisms. We conclude that, in the effort to combat the ongoing opioid epidemic74 by limiting opioid side effects and discovering alternative treatments for pain, therapeutics targeting microglia must address the specific pronociceptive mechanisms associated with each injury, drug, or disease, rather than general canonical inflammatory mediators.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/C58.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health grants DA031777 and DA044481 (G.S.), the New York Stem Cell Foundation (G.S.), a Department of Defense Neurosensory Award MR130053 (G.S.), a Rita Allen Foundation and American Pain Society Award in Pain (G.S.), a Department of Defense National Defense Science and Engineering Graduate (NDSEG) Fellowship (E.I.S.), NIH grant R37DA15043 (B.A.B.), and a Damon Runyon Cancer Research Foundation postdoctoral fellowship (C.J.B.). G.S. is a New York Stem Cell Foundation—Robertson Investigator. The authors thank Amaury François for his contributions to the development of this manuscript, Joshua Blair for manuscript editing, and Jesse Niehaus for his input regarding RNA sequencing analysis. This article was prepared while E.I.S. was employed at Stanford University. The opinions expressed in this article are the authors' own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the US Government.
Data availability statement: All data from original data sets are available upon request to Dr. Grégory Scherrer.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
C. J. Bohlen and G. Scherrer are co-senior authors.
E. I. Sypek and A. Tassou contributed equally.
Contributor Information
Elizabeth I. Sypek, Email: elizabeth.sypek@nih.gov.
Adrien Tassou, Email: adrtas@email.unc.edu.
Hannah Y. Collins, Email: collinha@ohsu.edu.
Karen Huang, Email: khuang3@email.unc.edu.
William M. McCallum, Email: will8mccallum@gmail.com.
Alexandra T. Bourdillon, Email: Alexandra.bourdillon@ucsf.edu.
Ben A. Barres, Email: barres@stanford.edu.
Christopher J. Bohlen, Email: cjbohlen@gmail.com.
References
- [1].Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science 2013;339:156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Anderson SR, Roberts JM, Zhang J, Steele MR, Romero CO, Bosco A, Vetter ML. Developmental apoptosis promotes a disease-related gene signature and independence from CSF1R signaling in retinal microglia. Cell Rep 2019;27:2002–13.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh Y-HE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, Sullivan JM, Purushothaman I, Scarpa JR, Goate AM, Busslinger M, Shen L, Losic B, Schaefer A. Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci 2018;21:1049–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain Behav Immun 2007;21:624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Benmamar-Badel A, Owens T, Wlodarczyk A. Protective microglial subset in development, aging, and disease: lessons from transcriptomic studies. Front Immunol 2020;11:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bennett GJ, Xie Y-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. PAIN 1988;33:87–107. [DOI] [PubMed] [Google Scholar]
- [7].Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 2016;113:E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron 2017;94:759–73.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, Mousseau M, Shankara JV, Stemkowski PL, Baimoukhametova D, Bains JS, Antle MC, Zamponi GW, Cahill CM, Borgland SL, DeKoninck Y, Trang T. Erratum: blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat Med 2017;23:788. [DOI] [PubMed] [Google Scholar]
- [10].Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 2012;32:11706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Colleoni M, Sacerdote P. Murine models of human neuropathic pain. Biochim Biophys Acta 2010;1802:924–33. [DOI] [PubMed] [Google Scholar]
- [12].Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol 2018;39:240–55. [DOI] [PubMed] [Google Scholar]
- [13].Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, Scherrer G. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med 2017;23:164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005;438:1017–21. [DOI] [PubMed] [Google Scholar]
- [15].Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, Cattaneo E, Gage FH, Cleveland DW, Glass CK. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci 2014;17:513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Denk F, Crow M, Didangelos A, Lopes DM, McMahon SB. Persistent alterations in microglial enhancers in a model of chronic pain. Cell Rep 2016;15:1771–81. [DOI] [PubMed] [Google Scholar]
- [17].Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. PAIN 2008;135:37–47. [DOI] [PubMed] [Google Scholar]
- [18].Eidson LN, Murphy AZ. Inflammatory mediators of opioid tolerance: implications for dependency and addiction. Peptides 2019;115:51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ferrini F, Trang T, Mattioli T-AM, Laffray S, Del'Guidice T, Lorenzo L-E, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu J-M, Cahill CM, Salter MW, De Koninck Y. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl−homeostasis. Nat Neurosci 2013;16:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee S-H, Haddick PCG, Ngu H, Modrusan Z, Larson JL, Kaminker JS, van der Brug MP, Hansen DV. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer's disease not evident in mouse models. Cell Rep 2018;22:832–47. [DOI] [PubMed] [Google Scholar]
- [21].Fukagawa H, Koyama T, Kakuyama M, Fukuda K. Microglial activation involved in morphine tolerance is not mediated by toll-like receptor 4. J Anesth 2013;27:93–7. [DOI] [PubMed] [Google Scholar]
- [22].Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014;159:1327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Fabisiak TJ, Anderson ND, Cheng K, Greene LI, Berkelhammer D, Zhang Y, Ellis AL, Yin HH, Campeau S, Rice KC, Roth BL, Maier SF, Watkins LR. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A 2016;113:E3441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Griffin RS, Costigan M, Brenner GJ, Ma CHE, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci 2007;27:8699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI. Injured sensory neuron–derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 2016;19:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gu N, Eyo UB, Murugan M, Peng J, Matta S, Dong H, Wu LJ. Microglial P2Y12 receptors regulate microglial activation and surveillance during neuropathic pain. Brain Behav Immun 2016;55:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H, Wu L-J. Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Rep 2016;16:605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, Marsh SE, Saunders A, Macosko E, Ginhoux F, Chen J, Franklin RJM, Piao X, McCarroll SA, Stevens B. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019;50:253–71.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hanamsagar R, Alter MD, Block CS, Sullivan H, Bolton JL, Bilbo SD. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia 2018;66:460. [DOI] [PubMed] [Google Scholar]
- [30].Hanamsagar R, Bilbo SD. Sex differences in neurodevelopmental and neurodegenerative disorders: focus on microglial function and neuroinflammation during development. J Steroid Biochem Mol Biol 2016;160:127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Häring M, Zeisel A, Hochgerner H, Rinwa P, Jakobsson JET, Lönnerberg P, La Manno G, Sharma N, Borgius L, Kiehn O, Lagerström MC, Linnarsson S, Ernfors P. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat Neurosci 2018;21:869–80. [DOI] [PubMed] [Google Scholar]
- [32].Hirbec H, Marmai C, Duroux-Richard I, Roubert C, Esclangon A, Croze S, Lachuer J, Peyroutou R, Rassendren F. The microglial reaction signature revealed by RNAseq from individual mice. Glia 2018;66:971–86. [DOI] [PubMed] [Google Scholar]
- [33].Honey D, Wosnitzka E, Klann E, Weinhard L. Analysis of microglial BDNF function and expression in the motor cortex. Front Cell Neurosci 2022;16:961276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Horvath RJ, Romero-Sandoval AE, De Leo JA. Inhibition of microglial P2X4 receptors attenuates morphine tolerance, Iba1, GFAP and mu opioid receptor protein expression while enhancing perivascular microglial ED2. PAIN 2010;150:401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, Somogyi AA, Yin H, Maier SF, Rice KC, Watkins LR. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience 2010;167:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 2010;24:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, Kocher J-P, Petrucelli L, Fryer JD. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med 2018;215:2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A unique microglia type associated with restricting development of alzheimer's disease. Cell 2017;169:1276–90.e17. [DOI] [PubMed] [Google Scholar]
- [40].Kobayashi K, Takahashi E, Miyagawa Y, Yamanaka H, Noguchi K. Induction of the P2X7 receptor in spinal microglia in a neuropathic pain model. Neurosci Lett 2011;504:57–61. [DOI] [PubMed] [Google Scholar]
- [41].Kohno K, Shirasaka R, Yoshihara K, Mikuriya S, Tanaka K, Takanami K, Inoue K, Sakamoto H, Ohkawa Y, Masuda T, Tsuda M. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science 2022;376:86–90. [DOI] [PubMed] [Google Scholar]
- [42].Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014;159:1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Leduc-Pessah H, Weilinger NL, Fan CY, Burma NE, Thompson RJ, Trang T. Site-specific regulation of P2X7 receptor function in microglia gates morphine analgesic tolerance. J Neurosci 2017;37:10154–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liang Y, Chu H, Jiang Y, Yuan L. Morphine enhances IL-1β release through toll-like receptor 4-mediated endocytic pathway in microglia. Purinergic Signal 2016;12:637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- [46].Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 2018;18:225–42. [DOI] [PubMed] [Google Scholar]
- [47].Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, Marschallinger J, Yu G, Quake SR, Wyss-Coray T, Barres BA. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 2019;101:207–23.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu X-J, Liu T, Chen G, Wang B, Yu X-L, Yin C, Ji R-R. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep 2016;6:28188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mattioli TA, Leduc-Pessah H, Skelhorne-Gross G, Nicol CJB, Milne B, Trang T, Cahill CM. Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One 2014;9:e97361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 2009;10:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Niehaus JK, Taylor-Blake B, Loo L, Simon JM, Zylka MJ. Spinal macrophages resolve nociceptive hypersensitivity after peripheral injury. Neuron 2021;109:1274–82.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Noristani HN, Gerber YN, Sabourin J-C, Le Corre M, Lonjon N, Mestre-Frances N, Hirbec HE, Perrin FE. RNA-seq analysis of microglia reveals time-dependent activation of specific genetic programs following spinal cord injury. Front Mol Neurosci 2017;10:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan W-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013;155:1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Peng J, Gu N, Zhou L, B Eyo U, Murugan M, Gan W-B, Wu L-J. Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat Commun 2016;7:12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 2013;10:1096–8. [DOI] [PubMed] [Google Scholar]
- [57].Raghavendra V, Tanga F, Deleo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003;306:624–30. [DOI] [PubMed] [Google Scholar]
- [58].Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology 2004;29:327–34. [DOI] [PubMed] [Google Scholar]
- [59].Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 2016;19:987–91. [DOI] [PubMed] [Google Scholar]
- [60].Rotterman TM, Alvarez FJ. Microglia dynamics and interactions with motoneurons axotomized after nerve injuries revealed by two-photon imaging. Sci Rep 2020;10:8648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med 2017;23:1018–27. [DOI] [PubMed] [Google Scholar]
- [62].Schwarz JM, Smith SH, Bilbo SD. FACS analysis of neuronal–glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology 2013;230:525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shields SD, Eckert WA, III, Basbaum AI. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain 2003;4:465–70. [DOI] [PubMed] [Google Scholar]
- [64].Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin J-S, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci 2011;31:15450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sorge RE, Mapplebeck JCS, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 2015;18:1081–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell 2007;131:1164–78. [DOI] [PubMed] [Google Scholar]
- [67].Taves S, Berta T, Liu D-L, Gan S, Chen G, Kim YH, Van de Ven T, Laufer S, Ji R-R. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: sex-dependent microglial signaling in the spinal cord. Brain Behav Immun 2016;55:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tsuda M, Beggs S, Salter MW, Inoue K. Microglia and intractable chronic pain. Glia 2013;61:55–61. [DOI] [PubMed] [Google Scholar]
- [69].Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci 2005;28:101–7. [DOI] [PubMed] [Google Scholar]
- [70].Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K. Microglial regulation of neuropathic pain. J Pharmacol Sci 2013;121:89–94. [DOI] [PubMed] [Google Scholar]
- [71].Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003;424:778–83. [DOI] [PubMed] [Google Scholar]
- [72].Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015;18:145–53. [DOI] [PubMed] [Google Scholar]
- [73].Vidal-Itriago A, Radford RAW, Aramideh JA, Maurel C, Scherer NM, Don EK, Lee A, Chung RS, Graeber MB, Morsch M. Microglia morphophysiological diversity and its implications for the CNS. Front Immunol 2022;13:997786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Volkow ND, Collins FS. The role of science in addressing the opioid crisis. N Engl J Med 2017;377:391–4. [DOI] [PubMed] [Google Scholar]
- [75].Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci 2005;28:661–9. [DOI] [PubMed] [Google Scholar]
- [76].Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem 2006;97:772–83. [DOI] [PubMed] [Google Scholar]
- [77].Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014;34:11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhou D, Chen M-L, Zhang Y-Q, Zhao Z-Q. Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. J Neurosci 2010;30:8042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/C58.