

Abstract
Acute myeloid leukemia (AML) is the most common hematological cancer in the adult population worldwide. Approximately 35% of patients with AML present internal tandem duplication (ITD) mutations in the FMS-like tyrosine kinase 3 (FLT3) receptor associated with poor prognosis, and thus, this receptor is a relevant target for potential therapeutics. Tyrosine kinase inhibitors (TKIs) are used to treat AML; however, their molecular interactions and effects on leukemic cells are poorly understood. The present study aimed to gain insights into the molecular interactions and affinity forces of four TKI drugs (sorafenib, midostaurin, gilteritinib and quizartinib) with the wild-type (WT)-FLT3 and ITD-mutated (ITD-FLT3) structural models of FLT3, in its inactive aspartic acid-phenylalanine-glycine motif (DFG-out) and active aspartic acid-phenylalanine-glycine motif (DFG-in) conformations. Furthermore, the present study evaluated the effects of the second-generation TKIs gilteritinib and quizartinib on cancer cell viability, apoptosis and proliferation in the MV4-11 (ITD-FLT3) and HL60 (WT-FLT3) AML cell lines. Peripheral blood mononuclear cells (PBMCs) from a healthy volunteer were included as an FLT3-negative group. Molecular docking analysis indicated higher affinities of second-generation TKIs for WT-FLT3/DFG-out and WT-FLT3/DFG-in compared with those of the first-generation TKIs. However, the ITD mutation changed the affinity of all TKIs. The in vitro data supported the in silico predictions: MV4-11 cells presented high selective sensibility to gilteritinib and quizartinib compared with the HL60 cells, whereas the drugs had no effect on PBMCs. Thus, the current study presented novel information about molecular interactions between the FLT3 receptors (WT or ITD-mutated) and some of their inhibitors. It also paves the way for the search for novel inhibitory molecules with potential use against AML.
Keywords: acute myeloid leukemia, FMS-like tyrosine kinase 3, internal tandem duplication, homology modeling of proteins, molecular docking, tyrosine kinase inhibitors, cytotoxicity, MV4-11, HL60
Introduction
Acute myeloid leukemia (AML) is the most common hematopoietic tumor and mainly affects older adults (1). In 2020, worldwide statistics indicated that AML was the thirteenth most common type of cancer in terms of incidence and had the tenth highest mortality rate (2). Men are more susceptible to AML than women (6.4 vs. 4.3%) (2), and the overall survival rate is ~50% in young patients (18–50 years) but only 10% in adults >65 years old (3).
AML is characterized by accelerated and autonomous growth of immature myeloid cells, leading to the dysregulation of hematological components (4–6). Some mutations are relevant for cell survival and proliferation, such as tyrosine-protein kinase (c-Kit) and FMS-like tyrosine kinase 3 (Flt3) mutations, which are directly related to AML progression (7,8). Notably, 25–35% of patients with AML have mutations that result in the upregulation of expression of the FLT3 receptor, which is related to high malignancy and poor prognosis (9).
FLT3 is part of the class III receptor tyrosine kinase (RTK) family. It is typically expressed in early myeloid and lymphoid CD34+ precursors, and its expression gradually decreases during cell differentiation (10). The FLT3 receptor consists of five domains: An immunoglobulin-like extracellular domain, which is the site of ligand binding and FLT3 activation (which is activated in synergy with IL-3 and granulocyte-macrophage colony-stimulating factor), a transmembrane domain, a juxtamembrane domain (JMD) and two cytoplasmic tyrosine kinase-like domains (TKDs; TKD1 and TKD2), which constitute the catalytic domain of FLT3 and are critical for receptor activation (10–12). Upon ligand binding, FLT3 activity is mediated by the JMD through phosphorylation of Tyr589 and Tyr591 amino acid residues. This phosphorylation promotes conformational changes that free the activation loop (A-loop) present in TKD1 and the phenylalanine residue of the aspartic acid-phenylalanine-glycine motif (DFG motif) (13).
The DFG motif, which is part of the ATP-binding site, is highly conserved and essential for the function of RTKs such as FLT3 (14). Depending on the type of ligand, the DFG motif can change between its inactive DFG motif (DFG-out) or active DFG motif (DFG-in) form (13,15). The DFG-out conformation occurs when Asp flips the ATP binding site outward and Phe moves out of the hydrophobic site (14,16,17). Conversely, the DFG-in conformation occurs when Phe is packed in a hydrophobic pocket and Asp moves toward the ATP binding site to interact with Mg2+ (18). FLT3/DFG-in activation stimulates proliferation and survival signaling pathways via Janus kinase (JAK)/STAT5, AKT/ERK and PI3K (10–13,19).
In patients with AML, common activating mutations in FLT3 include the Asp835Tyr (D835Y) point mutation in TKD1 and the internal tandem duplication (ITD) in JMD or TKD1 (3,20). ITD mutations in JMD and TKD1 affect 5–16% of pediatric patients (<10 years old) and 25–35% of adult patients (3,20). Patients with ITD-FLT3 mutations exhibit poor prognosis associated with the upregulation of this receptor, which leads to high proliferation and survival of leukemic cells, resistance to treatment, increased risk of relapse and decreased survival (3,20). Therefore, the ITD-FLT3 receptor requires special consideration as a drug target (21).
The ITD insertion (3–400 base pairs) occurs between exons 14 and 15 of the Flt3 gene, corresponding to the JMD or TKD1 domains of FLT3. At the protein level, the new amino acids are commonly inserted within a sequence of one to seven amino acids [F(Y)DFREYE/YDLK], corresponding to codons 591–597 of the original open reading frame (3,8,22). The length of ITD insertion changes the size of FLT3 and may influence its activity and the effects of its inhibitors (3,8,22).
Drugs that inhibit tyrosine kinase activity are widely used as AML treatments, alone or in combination with chemotherapy (23,24). FLT3 inhibitors have improved the overall survival of patients with leukemia. Due to their efficacy and being associated with fewer adverse effects compared with chemotherapy, they represent a major improvement in AML treatment. However, first-generation tyrosine kinase inhibitors (TKIs; including midostaurin and sorafenib) lack specificity for FLT3 as they bind to several RTKs (C-KIT, platelet-derived growth factor receptor, RAS, RAF, JAK2 and VEGFR), contributing to strong side effects, such as diarrhea, fatigue, nausea, cardiovascular problems and myelosuppression (25). Additionally, some patients with AML may develop resistance to TKIs (24,26,27). Conversely, second-generation TKIs, including quizartinib and gilteritinib, have a more specific effect, both for wild-type (WT) and ITD-mutated forms of the FLT3 receptor. Therefore, they can be used in lower concentrations than first-generation TKIs, resulting in lower toxicity and fewer side effects (11,23,24). TKIs can also be classified based on their selective binding to the DFG-out or DFG-in conformations of the FLT3 receptor. Type I inhibitors (midostaurin and gilteritinib) bind preferably to the ATP pocket of the DFG-in conformation (active motif), while type II inhibitors (sorafenib and quizartinib) bind preferably to the hydrophobic region of FLT3 adjacent to the ATP pocket of the DFG-out conformation (inactive motif) (23).
Over the past 15 years, the efficiency of several TKIs has been evaluated in patients with AML. Midostaurin, gilteritinib and quizartinib have been approved by the Food and Drug Administration (FDA) for AML treatment (24). However, little is known about the effects of ITD mutations on the molecular interactions and affinity forces of these inhibitors. Therefore, it is crucial to carefully investigate these parameters and their impact on the inhibitory activity of TKIs in AML cells.
Bioinformatics provides valuable tools to assess molecular interactions and predict the theoretical potential of specific inhibitors for a particular target. Examples of these tools include homology modeling of protein structure (HMP) and molecular docking. HMP helps approximate the atomic structure of a specific protein based on the known structure of a related protein available from databases such as the Protein Data Base (PDB) (28–30). HMP is achieved using two phylogenetically close protein structures with a high percentage of similarity, one serving as a template and the other as a target (30). Once a good quality model of the protein of interest is obtained, molecular docking facilitates the prediction of the molecular interactions between a ligand and a protein, calculating their affinity force through polar (H-bonds) and non-polar bonds (31).
Several AML cell lines expressing WT or ITD-mutated FLT3 receptors have been established for experimental evaluation of inhibitors. These are useful for obtaining an in vitro model to study the disease and validate the predictions of in silico approaches. Previous studies have used WT-FLT3 homozygous cell lines (HL60, THP1, OCI-AML3 and MUTZ-2 cell lines), while other studies have used WT/ITD-FLT3 heterozygous cell lines (MOLM-13, MOLM-14 and PL21 cell lines) or the MV4-11 and MULT-11 ITD-FLT3 cell lines with loss of heterozygosity (LOH) of FLT3 (15,32–35).
The present study was designed to assess the molecular interactions and affinity forces of four TKIs (sorafenib, midostaurin, gilteritinib and quizartinib) with the WT-FLT3 and ITD-FLT3 receptors in their DFG-out and DFG-in conformations in silico. Furthermore, the present study evaluated the effect of the second-generation inhibitors gilteritinib and quizartinib on MV4-11 (ITD-FLT3) and HL60 (WT-FLT3) AML cell lines, and peripheral blood mononuclear cells (PBMCs; FLT3-negative) from a healthy volunteer in vitro.
Materials and methods
In silico assay
Homology modeling of proteins
The in silico models of WT-FLT3 and ITD-FLT3 in their DFG-in and DFG-out conformations were prepared using the modeling assay algorithm proposed by Ke et al (13), Zorn et al (36), and Todde and Friedman (37).
WT-FLT3 modeling
A four-step strategy was applied. For WT-FLT3 modeling, the NP_004110.2 National Center for Biotechnology Information (NCBI) reference sequence was chosen in addition to the 1RJB (autoinhibited; 2.1 Å resolution) and 6IL3 (inactive; 2.5 Å resolution) protein templates (38,39). Sequence identities of 1RJB and 6IL3 were calculated and compared with the reference sequence using the NCBI protein-Basic Local Alignment Search Tool v.2.15.0 (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (40). Paired global alignment of the sequences was also carried out to identify their extension and similarity. Sequence identity was calculated for the 1RJB and 6IL3 structures of WT-FLT3. The best quality WT-FLT3 model of the DFG-out (WT-FLT3/DFG-out) conformation was selected using the SWISS-MODEL tool (http://swissmodel.expasy.org) from the Expasy: Swiss Institute of Bioinformatics server (41). The WT-FLT3/DFG-in model was then obtained, considering the c-Kit protein in DFG-in conformation as a template (1PKG; 2.9 Å resolution) and our WT-FLT3/DFG-out model using the SWISS-MODEL server as the target (41,42). The identity between sequences was calculated using the Clustal W tool v.1.81 (43) from the MEGA X v.11.0.13 software (41).
Insertion of the ITD sequence in the FLT3 model
The ITD sequence [H(L)VDFREYEYD/LKWE], previously described by Reiter et al (44) in the Z region of JMD (JMD-Z), was incorporated using the MODELLER v. 9.25 program (45,46). The best ITD-FLT3 model was selected based on the normalized Discrete Optimized Protein Energy score (47). Finally, the 3Drefine (https://3drefine.mu.hekademeia.org/) server was used to refine the structures and relax or correct errors of interactions between amino acid residues (48).
Structure validation and obtention of quality scores
Structural evaluation of the 3D models of WT-FLT3 and ITD-FLT3 in DFG-out and DFG-in conformations was performed with the ERRAT (49), VERIFY3D (50) and PROCHECK (51) programs, available from the SAVE v.6.0 (https://saves.mbi.ucla.edu/) server (2020). The quality of the formed structures was assessed using the QMEAN (https://swissmodel.expasy.org/qmean/) (52), Molprobity v.4.5.2 (http://molprobity.biochem.duke.edu/) (53), ProSA (https://prosa.services.came.sbg.ac.at/prosa.php) (54) and ProQ v.1.2 (https://proq.bioinfo.se/cgi-bin/ProQ/ProQ.cgi) servers (55).
Molecular docking
Identification of the binding site of FLT3
The identification of the parameters of the FLT3 binding site was based on the coordinates and co-crystallized ligands available from Protein Data Bank (PDB, http://www.rcsb.org/) (6IL3 and 1PGK) using the University of California, San Francisco (UCSF)-Chimera v.1.16 software, AutoDock Tools v.1.5.6 and ProQ v.1.2 servers (39,42,46,56). For the DFG-out conformation of FLT3, a box size of 24×24×24 Å, with a center localization in x=74.64, y=50.01 and z=24.46, was considered. For DFG-in, the box size was 24×24×24 Å, and the center was x=25, y=27.5 and z=26.8. The parameters of the binding site were validated by redocking assays using 6IL3 and 1PGK structures and their co-crystallized ligands (Fig. S1).
Molecular docking of sorafenib, midostaurin, gilteritinib and quizartinib with the FLT3 receptor models
The structures of the four TKIs were obtained from the ZINC 15 database (Table I) (57). Before docking, the ligands and FLT3 models were conditioned by removing water molecules and extra ligands from the protein and ligand surfaces, and by adding polar hydrogen atoms and atomic charges, such as semi-empirical with bond charge correction charges for FLT3 receptors and Gasteiger charges for ligands. Finally, all structures were minimized using the UCSF-Chimera v.1.16 and Autodock tools v.1.5.6 (46,58).
Table I.
Classification and ZINC ID of the four TKIs included in the study.
| TKI | Generation | Type | ZINC ID (ZINC15) |
|---|---|---|---|
| Sorafenib | First | II | ZINC100013130 |
| Midostaurin | I | ZINC1493878 | |
| Gilteritinib | Second | I | ZINC113476229 |
| Quizartinib | II | ZINC43204002 |
TKI, tyrosine kinase inhibitor.
Based on the established parameter sets, 100 models of molecular docking were run for each of the four FLT3 models and each of the four TKIs using Autodock-Vina software v.1.1.2 (59). The best ligand-protein complexes were selected based on the lowest ΔG free energy (kcal/mol), root-mean-square deviation ≤3.0, and the number of covalent and non-covalent bonds, considering true contact when polar bonds presented a length ≤5 Å (58). The most stable complexes were visualized with PyMOL v.2.5.4 (60) and Discovery Studio v.21.1.0.20298 (BIOVIA).
In vitro assays
Cell culture
Two AML cell lines were selected to study leukemia in vitro. The MV4-11 cell line (ITD-FLT3) was acquired from American Type Culture Collection (CRL-9591). According to Human Genome Variation Society nomenclature, the MV4-11 cells present the NP_004110.2:p.(Leu601His_Lys602insVDFREYEYD) variant (61). The HL60 cell line (WT-FLT3) was obtained from the Cell Bank of the Western Biomedical Research Center of Mexican Social Security Institute (Guadalajara, México). According to the French-American-British classification, which classifies AML cells into eight groups (M0 to M7) based on morphological and cytochemical characteristics, the MV4-11 cell line is classified as M5 and the HL60 cell line as M2 (32,62,63). The PBMCs (FLT3-negative) used as a negative control were obtained with the written informed consent of a healthy volunteer (30 years; female; Zapopan, Mexico; April-August 2023). The samples were collected at the Biomedicine and Ecology Molecular Markers Laboratory of Biological and Agricultural Sciences Campus, University of Guadalajara (Zapopan, Mexico) using ethical criteria for collecting biological samples. All cell cultures were performed at 37°C in a CO2 incubator (Binder CO2 incubator CB-S 170; BINDER GmbH) with 5% CO2 and 95% humidity in Iscove's Modified Dulbecco's medium (cat. no. I7633-10X1L; Sigma-Aldrich; Merck KGaA) supplemented with 10% FBS (cat. no. BIO-S1650-500; Biowest) and 1% penicillin/streptomycin (cat. no. P4333-100ML; Sigma-Aldrich; Merck KGaA).
Validation of the ITD mutation in FLT3 in the MV4-11 cell line
The presence/absence of the ITD mutation in FLT3 was validated by end-point PCR using genomic DNA (gDNA) isolated from the cell lines and PBMCs using the rapid isolation of mammalian DNA extraction protocol by Sambrook and Russell (64). The primer pair sequences were: FLT3 forward, 5′-GCAATTTAGGTATGAAAGCCAGC-3′ and reverse, 5′-CTTTCAGCATTTTGACGGCAACC-3′ (65). The primers were synthetized by Integrated DNA Technologies, Inc. The PCR reaction contained 50 ng gDNA, 1X PCR buffer, 1.5 mM MgCl2, 0.4 mM dNTPs, 0.4 µM forward and reverse primers, and 0.5 U Taq Polymerase (cat. no. 10342053; Invitrogen; Thermo Fisher Scientific, Inc.). The amplification program was as follows: Initial denaturation cycle at 94°C for 5 min, followed by 35 cycles of denaturation (94°C for 30 sec), hybridization (58°C for 40 sec) and elongation (72°C for 1 min), and a final elongation step at 72°C for 7 min. The amplification products were separated on a 1.2% agarose gel (cat. no. 16500-500; Invitrogen; Thermo Fisher Scientific, Inc.) stained with GelRed (1:10,000; cat. no. 41003; Biotium, Inc.) and images were captured with the D-DiGit image acquisition system (LI-COR Biosciences).
FLT3 expression in the in vitro AML model
To validate the presence of the FLT3 receptor, its expression was measured by flow cytometry after labeling cell surfaces with phycoerythrin (PE)-coupled anti-FLT3 (cat. no. 558996; BD Biosciences). The PE-coupled anti-IgG1 antibody (cat. no. 555749; BD Biosciences) was used as an isotype control. Parameter analysis was based on the intensity of the emitted fluorescence signal and the percentage of the cell population passing through the flow cell. A total of 10,000 events were acquired using the BD Accuri C6 cytometer (BD Biosciences) and analyzed using the statistical estimator super enhanced DMAX of the FlowJo v.10 data analysis software (BD Life Sciences) (66).
Evaluation of cell viability
The in vitro assays focused on the second-generation inhibitors gilteritinib (type I) and quizartinib (type II) because they showed the best molecular interactions and affinity forces with WT-FLT3 and ITD-FLT3 in both DFG-out and DFG-in conformations. Notably, the FDA has recently approved both inhibitors for clinical use in patients with AML (67,68).
Their cytotoxic effect on MV4-11 and HL60 cells, as wells as PBMCs, was evaluated using the MTT assay. The cells were seeded in 96-well plates (MV4-11 cells and PBMCs, 70,000 cells per well; HL60 cells, 20,000 cells per well), preincubated for 48 h at 37°C, and exposed to increasing concentrations of gilteritinib (cat. no. 21503; Cayman Chemical Company), or quizartinib (cat. no. 17986; Cayman Chemical Company) dissolved in DMSO (cat. no. D4540; Sigma-Aldrich; Merck KGaA). For MV4-11 cells the 20, 10, 5, 2.5 and 1.25 nM range was used, and for HL60 cells the 800, 400, 20, 10, 5, 2.5 and 1.25 nM concentrations were applied based on a previous report by Hu et al (69). PBMCs were treated with the calculated IC50 of both compounds in MV4-11 cells (7.99 nM gilteritinib or 4.76 nM quizartinib). In addition, 3% DMSO (cat. no. D4540; Sigma-Aldrich; Merck KGaA) was used as a death control, 100 nM etoposide (cat. no. E1383; Sigma-Aldrich; Merck KGaA) as an apoptosis control and 0.1% DMSO as a vehicle control. Cells without treatment were used as a negative control. The exposure time to each inhibitor was 48 h at 37°C, which coincides approximately with the half-life of these compounds (gilteritinib, 113 h; quizartinib, 73 h) (70,71). MTT (5 mg/ml in 1X phosphate buffered saline) was added at the end of the incubation period, and cell viability was evaluated according to the manufacturer's instructions (cat. no. M2003; Sigma-Aldrich; Merck KGaA). To dissolve the formazan crystals, half of the culture medium (100 µl) was removed and acidified-isopropanol was added (100 µl; 0.1 M HCl; cat. nos. 190764 and 320331; Sigma-Aldrich; Merck KGaA). Absorbance was measured at 570/690 nm on a plate reader (XMARK; Bio-Rad Laboratories, Inc.), and the absorbance values were converted to viability percentages for analysis. All assays were performed in triplicate and repeated three times. The IC50 (72) was calculated using the nonlinear regression method with Rstudio v.2022.12.0+353 (http://www.rstudio.com/) (73) and GraphPad Prism v. 9 (Dotmatics).
Evaluation of apoptosis
To determine the effect of drugs on the level of cell death, apoptosis was assessed by flow cytometry. First, the cells were placed in 24-well plates (100,000 MV4-11 cells or PBMCs per well; 40,000 HL60 cells per well) and incubated for 48 h at 37°C. The treatments (7.99 nM gilteritinib or 4.76 nM quizartinib) were applied for 48 h in the same conditions as aforementioned. These concentrations correspond to the IC50 of these compounds for MV4-11 cells as determined in the present study. Cells were also treated with the vehicle control (0.1% DMSO), death control (3% DMSO) and apoptosis control (100 nM etoposide). Finally, Annexin-V-FITC (cat. no. A13199; Invitrogen; Thermo Fisher Scientific, Inc.) and PI (cat. no. P4170; Sigma-Aldrich; Merck KGaA) were added to label the cells according to the manufacturer's instructions. Three independent experiments were carried out for each condition, and 10,000 events were acquired using the BD Accuri C6 Plus cytometer (BD Biosciences). The data were analyzed with FlowJo version 8 (BD Life Sciences) (66).
Evaluation of cell proliferation
The cells were plated in 24-well plates as aforementioned, preincubated for 48 h at 37°C and labeled with carboxyfluorescein succinimidyl ester (CFSE; 5 mM; cat. no. C1157; Invitrogen; Thermo Fisher Scientific, Inc.) for 5 min at room temperature. CFSE provides relevant information to monitor distinct cell generations across a given time-period. Subsequently, the cells were exposed to 7.99 nM gilteritinib or 4.76 nM quizartinib, as well as vehicle (0.1% DMSO), death (3% DMSO) and apoptosis (100 nM etoposide) controls for 48 h at 37°C and collected for flow cytometry (BD Accuri C6 Plus cytometer; BD Biosciences) analysis considering 10,000 events per sample. In addition, the basal proliferation indexes of untreated leukemia cells (MV4-11 and HL60) and PBMCs were evaluated at 0 and 48 h at 37°C and compared with the vehicle control. Three independent experiments were carried out for each condition. The data were analyzed with FCS Express 7 (De Novo Software).
Statistical analysis
One-way ANOVA was used to analyze the percentage of FLT3 expression positivity in cell lines and PBMCs and determine the effect of gilteritinib and quizartinib on viability. Tukey's and Dunnett's post hoc tests were then performed, respectively. Additionally, one-way ANOVA and two-way ANOVA, followed by Dunnett's and Bonferroni's post hoc tests, respectively, were used to determine the effect of gilteritinib and quizartinib on the proliferation index and the induction of apoptosis of each cell type and across cell types. Finally, the IC50 was determined by nonlinear regression analysis with transformed viability data. All data (triplicates repeated three times) are presented as the mean ± SD. All data analyzed passed the Shapiro-Wilk normality test and were analyzed with GraphPad Prism version 9 for Windows (Dotmatics) and R Studio v.2022.12.0+353 software (http://www.rstudio.com/) (73). P<0.05 was considered to indicate a statistically significant difference.
Results
In silico phase
Homology of the sequences used for WT-FLT3 and ITD-FLT3 model construction
The sequences used for HMP allowed the construction of high-quality models of WT-FLT3 and ITD-FLT3 in their DFG-out and DFG-in conformations. First, the three FLT3 sequences (NP_004110.2, 1RJB and 6IL3) used for the construction of the WT-FLT3/DFG-out model presented high amino acid homologies, reaching 96.1% between 1RJB and 6IL3, 87.09% between NP_004110.2 and 1RJB, and 86.90% between NP_004110.2 and 6IL3, indicating that these sequences were adequate for HMP (Fig. S2A; Table II). Second, the addition of the DFG-in motif of c-Kit (1PKG) was considered appropriate for WT-FLT3/DFG-in construction because its sequence presented 57.45 and 58.66% homology with 1RJB and 6IL3, respectively (Fig. S2B; Table II). Third, the ITD sequence VDFREYEYD (44) was successfully inserted into the JMD-Z region of WT-FLT3/DFG-out to obtain the ITD-mutated model of FLT3 (ITD-FLT3/DFG-out) (Fig. S2C). Fourth, the DFG-in motif of c-Kit (1PKG) was added to the ITD-FLT3/DFG-out model to obtain ITD-FLT3/DFG-in (Fig. S2D).
Table II.
Sequence homology presented as the percentage of identity of the three sequences used for the homology modeling of FMS-like tyrosine kinase 3.
| Sequence 1 | Sequence 2 | Identity (%) |
|---|---|---|
| (NP_004110.2) | 1RJB | 87.09 |
| (NP_004110.2) | 6IL3 | 86.90 |
| 1RJB | 6IL3 | 96.10 |
| 1RJB | 1PKG | 57.45 |
| 6IL3 | 1PKG | 58.66 |
The National center for biotechnology information protein basic local alignment search tool was used.
Quality evaluation of WT-FLT3 and ITD-FLT3 modeled structures in their DFG-out and DFG-in conformations
Several bioinformatics tools were used to confirm the quality of the four FLT3 models. These tools confirmed that the WT-FLT3 and ITD-FLT3 structural models were appropriate and of high quality in their DFG-out and DFG-in conformations (Table III).
Table III.
Quality scores of the in silico models of FLT3 evaluated using ERRAT, VERIFY3D, QMEAN, Molprobity, ProSA and ProQ.
| Receptor | Conformation | ERRAT, % | Verify 3D, % | QMEAN quality score | Molprobity | ProSA Z-score | ProQ LG-score |
|---|---|---|---|---|---|---|---|
| WT-FLT3 | DFG-out | 95.61 | 82.32 | 0.17 | 1.92 | −7.35 | 6.99 |
| DFG-in | 95.96 | 66.56 | −1.03 | 2.34 | −6.71 | 8.19 | |
| ITD-FLT3 | DFG-out | 90.27 | 75.67 | −0.48 | 2.63 | −7.40 | 8.62 |
| DFG-in | 95.75 | 71.60 | −1.01 | 2.42 | −6.37 | 8.39 |
DFG-in, active aspartic acid-phenylalanine-glycine motif; DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication; WT, wild-type.
The ERRAT program verifies the quality structure of proteins through the error function of the non-interactions of reported structures compared with the high-resolution structure database. Values of 95% or higher are considered to indicate good high-resolution structures (1.5–2 A), while lower values (91% or lower) correspond to lower resolution (2.5–3 A) (49). Thus, the WT-FLT3 in DFG-out and DFG-in conformations, as well as ITD-FLT3/DFG-in, modeled structures had high resolutions (95.61, 95.96 and 95.75%, respectively), while the resolution of ITD-FLT3/DFG-out was lower (90.27%) (Fig. S3; Table III).
The Verify 3D program determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) (50). The scores indicated that the WT-FLT3/DFG-out model presented the best quality, followed by the ITD-FLT3 models in DFG-out and DFG-in conformations and the WT-FLT3/DFG-in model (Table III).
The quality of the four models was confirmed using the QMEAN, Molprobity, Z-score, and LG-score (Table III). The QMEAN quantifies the accuracy of the model based on individual residual errors as well as at the overall structural level (52). Molprobity determines the quality of the structure based on the geometric scores of the Ramachandran plots and sidechain rotamer (53). The Z-score measures the deviation of the total energy of the structure with respect to an energy distribution derived from random conformations (54). The LG-score calculates the frequency of atom-atom contacts and predicts the quality of a model (55).
The Ramachandran plots showed that all four models presented >90% of their amino acid residues within the most favored regions according to the ψ and φ angles and none in non-favorable regions (Fig. 1; Table IV).
Figure 1.

Ramachandran plots of the FLT3 models. (A) WT-FLT3/DFG-out. (B) WT-FLT3/DFG-in. (C) ITD-FLT3/DFG-out. (D) ITD-FLT3/DFG-in. Red areas indicate the amino acid residues in most favored regions. Yellow areas show residues in allowed regions. Pale yellow areas correspond to residues in generously allowed regions. White areas show residues in disallowed regions (PROCHECK). DFG-in, active aspartic acid-phenylalanine-glycine motif; DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication; WT, wild-type.
Table IV.
Ramachandran and G-factor values of FLT3 in silico models determined using PROCHECK from the SAVE 6.0 server.
| PROCHECK | ||||||||
|---|---|---|---|---|---|---|---|---|
|
|
||||||||
| Ramachandran plot | ||||||||
|
|
G-factor | |||||||
| Most favored, % | Additional allowed, % | Generously allowed, % | Disallowed, % |
|
||||
| Receptor | Conformation | Dihedral | Covalent | Overall | ||||
| WT-FLT3 | DFG-out | 91.2 | 8.1 | 0.7 | 0.0 | 0.10 | −1.00 | −0.40 |
| DFG-in | 90.6 | 8.3 | 1.1 | 0.0 | 0.01 | −1.39 | −0.62 | |
| ITD-FLT3 | DFG-out | 91.8 | 7.6 | 0.7 | 0.0 | 0.11 | −1.16 | −0.47 |
| DFG-in | 92.6 | 6.3 | 1.1 | 0.0 | 0.04 | −1.26 | −0.55 | |
DFG-in, active aspartic acid-phenylalanine-glycine motif; DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication; WT, wild-type.
Finally, the G-factor values obtained with PROCHECK confirmed that each residue of the four structures lay in normal stereochemical distribution, resulting in ordered and favorable structures (Fig. 2; Table IV).
Figure 2.

In silico models of FLT3. (A) WT-FLT3/DFG-out. (B) WT-FLT3/DFG-in. (C) ITD-FLT3/DFG-out. (D) ITD-FLT3/DFG-in. The arrows show the JMD-Z, JMD-S and JMD-B regions of the JMD and the ITD mutation of the ITD-FLT3 models. The catalytic site (green β-sheets) and DFG motif (red β-sheets) are found within the yellow dotted circle. A-loop, activation loop; DFG-in, active aspartic acid-phenylalanine-glycine motif; DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication; JM-B, JM binding motif; JMD, juxtamembrane domain; JMD-Z, Z region of JMD; TKD, tyrosine kinase-like domain; WT, wild-type.
Altogether, these indexes confirmed the good quality scores applied using bioinformatics tools to all synthetic receptor 3D models of FLT3, guaranteeing that all were suitable for the following analysis.
Molecular docking of TKIs with WT-FLT3
Molecular docking with WT-FLT3/DFG-out showed that the second-generation inhibitors gilteritinib and quizartinib presented higher affinity forces (−9.6 and −9.3 kcal/mol, respectively) than the first-generation inhibitors sorafenib and midostaurin (−8.3 and −7.5 kcal/mol, respectively) (Fig. 3; Table V).
Figure 3.

3D and 2D representations of WT-FLT3/DFG-out/ligand complexes. (A) Overview of the overlay complexes. (B) Sorafenib. (C) Midostaurin. (D) Gilteritinib. (E) Quizartinib. PyMOL v. 2.5.4 and Discovery Studio v.21.1.0.20298 were used. The 3D representations (middle panel) show the conventional polar bonds. The 2D representations (right panel) show the conventional polar bonds (dark green) and the non-covalent interactions (other colors). DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; JM-B, JM binding motif; WT, wild-type.
Table V.
Characteristics of the affinity forces of the tyrosine kinase inhibitors towards the DFG-out and DFG-in conformations of WT-FLT3.
| Receptor | Inhibitor | Affinity, kcal/mol | Polar bonds | Non-covalent interactions |
|---|---|---|---|---|
| WT-FLT3 DFG-out | Sorafenib | −8.3 | Leu616-H; Leu616-H; | Tyr696, Glu724, Asp698, |
| Tyr693-O | Phe779, Arg783, Arg764 | |||
| Midostaurin | −7.5 | Arg764-H; Arg764-O; | Gly697, Leu616, Asp698, | |
| Asn701-O | Phe779 | |||
| Gilteritinib | −9.6 | Tyr696-H; Lys614-O; | Cys694, Phe691, Leu767, Asp698, | |
| Tyr693-H; Cys695-H | Phe779, Leu616, Gly697, Tyr696 | |||
| Quizartinib | −9.3 | Asp788-H; Asp788-H; | Arg704, Leu767, Leu616, | |
| Asp788-H; Asn790-N; | Asp698, Phe779 | |||
| Arg704-N; Arg704-O; | ||||
| Arg704-O | ||||
| WT-FLT3 DFG-in | Sorafenib | −9.4 | Asn701-O; Asp698-H | Phe691, Leu767, Val624, Cys777, |
| Leu700, Arg764, Ala642 | ||||
| Midostaurin | −7.4 | Asn836-H | Arg764, Trp803, Lys802, Pro800, | |
| Leu799 | ||||
| Gilteritinib | −9.2 | Cys694-H | Gly619, Val624, Leu767, Cys777, | |
| Asp698, Arg764 | ||||
| Quizartinib | −9.6 | Lys802-O; Val801-O | Phe691, Ala642, Leu767, Val624, | |
| Asp698, Arg764, Ala620 |
DFG-in, active aspartic acid-phenylalanine-glycine motif; DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; WT, wild-type.
Molecular docking with WT-FLT3/DFG-in showed that gilteritinib, quizartinib and sorafenib presented high affinity (−9.2, −9.6 and −9.4 kcal/mol, respectively) towards this receptor. By contrast, midostaurin presented the lowest affinity (−7.4 kcal/mol) (Fig. 4: Table V).
Figure 4.

3D and 2D representations of the WT-FLT3/DFG-in/ligand complexes. (A) Overview of the overlay complexes. (B) Sorafenib. (C) Midostaurin. (D) Gilteritinib. (E) Quizartinib. PyMOL v. 2.5.4 and Discovery Studio v.21.1.0.20298 were used. The 3D representations (middle panel) show the conventional polar bonds. The 2D representations (right panel) show the conventional polar bonds (dark green) and the non-covalent interactions (other colors). DFG-in, active aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; WT, wild-type.
The interactions of WT-FLT3/DFG-out with the first-generation inhibitors involved forming polar bonds with Leu616 and Tyr693 for sorafenib and Arg764 and Asn701 for midostaurin. Conversely, for WT-FLT3/DFG-in, Asn701 and Asp698 participated in the binding of sorafenib, and Asn836 was used for midostaurin. The interactions of WT-FLT3/DFG-out with the second-generation inhibitors involved Tyr696, Lys614, Tyr693 and Cys695 for gilteritinib, and Asp788, Asn790 and Arg704 for quizartinib. Furthermore, for WT-FLT3/DFG-in, Cys694 was required for the interaction with gilteritinib, and Lys802 and Val801 were required for the interaction with quizartinib (Fig. 3; Table V). Notably, the number of polar bonds of the four inhibitors tested was higher with the DFG-out conformation (3–7) than with the DFG-in conformation (1–2) of WT-FLT3 (Figs. 3 and 4).
Molecular docking of TKIs with ITD-FLT3
The affinity forces of ITD-FLT3/DFG-out with the first-generation inhibitors sorafenib (−9.5 kcal/mol) and midostaurin (−9.6 kcal/mol) were increased compared with those of WT-FLT3/DFG-out. By contrast, these forces were decreased for the second-generation inhibitors gilteritinib (−7.9 kcal/mol) and quizartinib (−9.0 kcal/mol) (Fig. 5; Table VI).
Figure 5.

3D and 2D representations of the ITD-FLT3/DFG-out/ligand complexes. (A) Overview of the overlay complexes. (B) Sorafenib. (C) Midostaurin. (D) Gilteritinib. (E) Quizartinib. PyMOL v. 2.5.4 and Discovery Studio v.21.1.0.20298 were used. The 3D representations (middle panel) show the conventional polar bonds. The 2D representations (right panel) show the conventional polar bonds (dark green) and the non-covalent interactions (other colors). DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication; JMD, juxtamembrane domain.
Table VI.
Characteristics of the affinity forces of tyrosine kinase inhibitors towards the DFG-out and DFG-in conformations of ITD-FLT3.
| Receptor | Inhibitor | Affinity, kcal/mol | Polar bonds | Non-covalent interactions |
|---|---|---|---|---|
| ITD-FLT3 DFG-out | Sorafenib | −9.5 | Arg 792-O; Asn 710-O; | Arg773, Asp797, Phe788, |
| Asp 707-O | Glu733, Cys704, Tyr705 | |||
| Midostaurin | −9.6 | Arg 773-H | Gly706, Asn710, Phe788, Leu625 | |
| Gilteritinib | −7.9 | Ser 796-H; Cys 703-H | Phe 788 | |
| Quizartinib | −9.0 | Asn 799-N; Asp 797-H; | Asp707, Phe788, Arg773, | |
| Cys 623-O | Asp797, Leu625, Glu733 | |||
| ITD-FLT3 DFG-in | Sorafenib | −9.4 | Ala 629-O; Arg 773-N | Gly628, Val633, Cys786, |
| Leu776, Phe700, Ala651 | ||||
| Midostaurin | −7.5 | Arg 773-O; Asn 774-H; | Asp707, Gly626, Arg773 | |
| Asp 787-H | ||||
| Gilteritinib | −7.5 | Arg 773-H | Asp707, Ser627, Ala629, | |
| Trp812, Lys811, Pro846 | ||||
| Quizartinib | −9.7 | Val 810-O; Lys 811-O | Phe700, Val684, Ala651, Cys786, | |
| Leu776, Val633, Asp707, Arg773 |
DFG-in, active aspartic acid-phenylalanine-glycine motif; DFG-out, inactive aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication.
Compared with WT-FLT3/DFG-in, the ITD mutation in FLT3/DFG-in did not affect the affinity force of sorafenib (−9.4 kcal/mol), midostaurin (−7.5 kcal/mol) or quizartinib (−9.7 kcal/mol); however, it decreased the affinity force of gilteritinib (−7.5 kcal/mol) (Fig. 6; Table VI). The ITD mutation in FLT3 modified the amino acid residues and the number of polar bonds involved in the interaction of this receptor with its ligands. The main amino acid residues involved in the formation of the ITD-FLT3/DFG-out/ligand complexes were Arg792, Asn710 and Asp707 for sorafenib, Arg773 for midostaurin, Ser796 and Cys703 for gilteritinib, and Asn799, Asp797 and Cys623 for quizartinib. The complexes between ITD-FLT3/DFG-in and its ligands involved Ala629 and Arg773 for sorafenib, Arg773, Asn774 and Asp787 for midostaurin, Arg773 for gilteritinib, and Val810 and Lys811 for quizartinib (Figs. 5 and 6; Table VI). The number of polar bonds between ITD-FLT3 and its ligands was higher for its DFG-out than its DFG-in conformations, except for midostaurin.
Figure 6.

3D and 2D representations of the ITD-FLT3/DFG-in/ligand complexes. (A) Overview of the overlay complexes. (B) Sorafenib. (C) Midostaurin. (D) Gilteritinib. (E) Quizartinib. PyMOL v. 2.5.4 and Discovery Studio v.21.1.0.20298 were used. The 3D representations (middle panel) show the conventional polar bonds. The 2D representations (right panel) also show the conventional polar bonds (dark green) and the non-covalent interactions (other colors). DFG-in, active aspartic acid-phenylalanine-glycine motif; FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication.
As aforementioned, the non-covalent molecular interactions of ITD-FLT3 were more abundant with the ITD-FLT3/DFG-out conformation than with the ITD-FLT3/DFG-in conformation (Figs. 5 and 6).
In vitro evaluation
FLT3 expression and identification of the ITD mutation in AML cells
The FLT3 receptor was expressed in 60.03±3.5% of MV4-11 cells and 14.23±3.4% of HL60 cells (Fig. 7A). The amount of FLT3 receptor (mean fluorescence index) was 1.7 times higher on the surface of MV4-11 cells than on that of HL60 cells (Fig. 7B). The PBMC samples obtained from the clinically healthy volunteer showed no FLT3-positive cells and the mean fluorescence intensity value of these cells was similar to that of the unstained control (Fig. 7A and B). Fig. S4 shows the dot plots and histograms of the flow cytometric analysis of FLT3 expression on MV4-11 and HL60 cells, and PBMCs.
Figure 7.

Validation of FLT3 protein expression and evaluation of the presence of the ITD mutation in the MV4-11 (ITD-FLT3) and HL60 (WT-FLT3) cell lines and PBMCs from a healthy volunteer (FLT3-negative). (A) Cell positivity for FLT3. (B) MFI of FLT3. (C) Amplification of the ITD fragment between exons 14 and 15 of FLT3 in MV4-11 cells. Data were analyzed using one-way ANOVA (% positivity) and two-way ANOVA (MFI) with post hoc Tukey's and Bonferroni's tests, respectively. *P<0.05; **P<0.01. FLT3, FMS-like tyrosine kinase 3; ITD, internal tandem duplication; M, marker; MFI, mean fluorescence index; PBMCs, peripheral blood mononuclear cells; WT, wild-type.
The FLT3 amplicon, which corresponds to the sequence between exons 14 and 15, showed a larger PCR product for the MV4-11 cell line than the HL60 cell line and PBMCs (WT-FLT3) (Fig. 7C). The increase in the PCR product size (354 vs. 329 bp) corresponds to the presence of the ITD mutation in MV4-11 cells as described by Quentmeier et al (32), Huang et al (65) and Jilani et al (74).
Effect of gilteritinib and quizartinib on the viability of MV4-11 cells, HL60 cells and PBMCs
The IC50 determined for gilteritinib in MV4-11 and HL60 cells was 7.99±1.07 and 57.54±3.99 nM, respectively. For quizartinib, these values were 4.76±0.55 and 38.75±2.4 nM, respectively (Fig. 8A and B). Thus, gilteritinib and quizartinib presented a significantly higher cytotoxic effect (lower viability %) against MV4-11 cells than HL60 cells. This effect was especially marked for gilteritinib since its IC50 was seven times lower in MV4-11 cells (7.99±1.07 nM) than in HL60 cells (57.54±3.99 nM), while for quizartinib the IC50 was eight times lower in MV4-11 cells (4.76±0.55 nM) compared with HL60 cells (38.75±2.4 nM) (Fig. 8A and B). The exposure of PBMCs to gilteritinib or quizartinib (using the IC50 value calculated for MV4-11 cells) did not affect their viability (Fig. 8C; Table VII).
Figure 8.
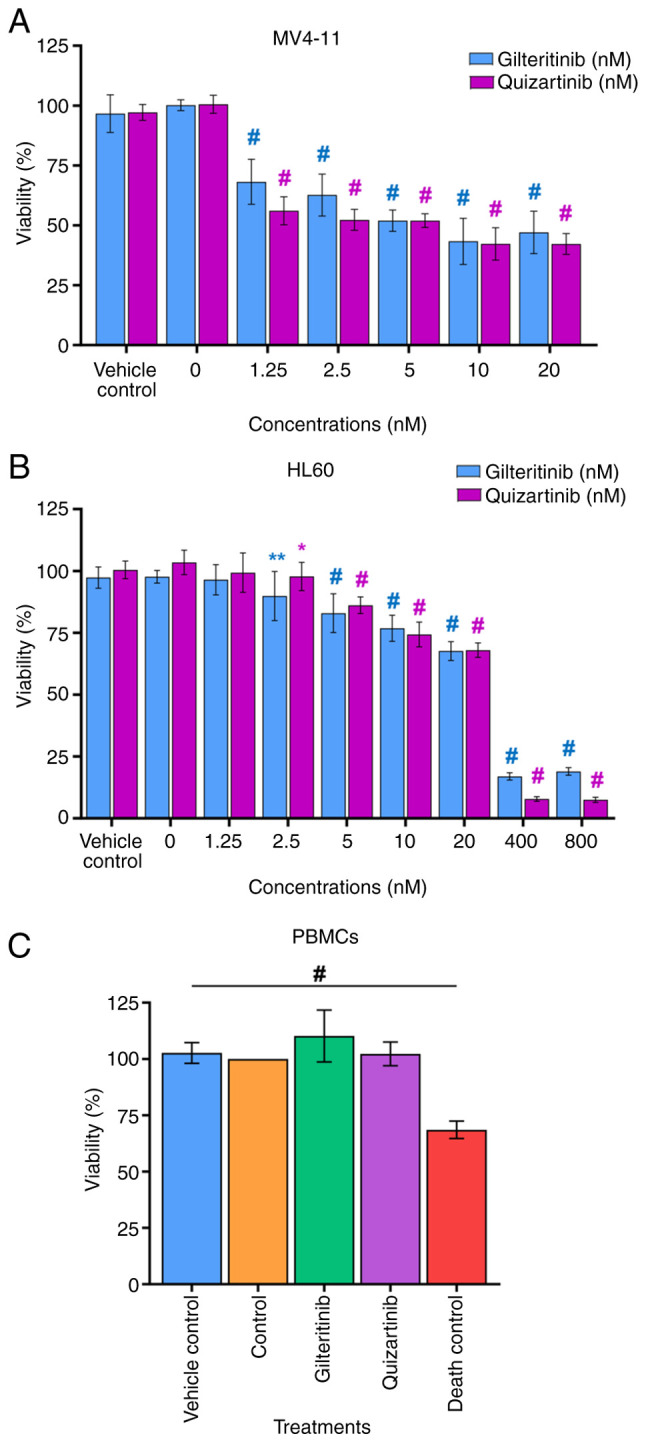
Effect of gilteritinib and quizartinib on the viability of acute myeloid leukemia cell lines and PBMCs. (A) MV4-11 cells. (B) HL60 cells. (C) PBMCs from a healthy volunteer. Vehicle control, 0.1% DMSO; gilteritinib, 7.99 nM; quizartinib, 4.76 nM; death control, 3% DMSO. Data are presented as the percentage of viability. One-way ANOVA with Dunnett's post hoc test comparing the viability of the vehicle control with each inhibitor concentration and untreated cells (0 nM). Blue symbols indicate significative differences between the concentrations of gilteritinib. Pink symbols indicate significative differences between the concentrations of quizartinib. *P<0.05; **P<0.01; #P<0.0001 vs. vehicle control. PBMCs, peripheral blood mononuclear cells.
Table VII.
IC50 of gilteritinib and quizartinib in in vitro models of acute myeloid leukemia.
| Cell line | IC50 of gilteritinib, nM | IC50 of quizartinib, nM |
|---|---|---|
| MV4-11 | 7.99±1.07 | 4.76±0.55 |
| HL60 | 57.54±3.99 | 38.75±2.40 |
| PBMCs | NA | NA |
NA, not applicable; PBMCs, peripheral blood mononuclear cells.
Effects of gilteritinib and quizartinib on the apoptosis and proliferation of MV4-11 cells, HL60 cells and PBMCs
The effects of gilteritinib and quizartinib on apoptosis induction and cell proliferation inhibition of MV4-11 cells, HL60 cells and PBMCs were evaluated by flow cytometry. For this purpose, the cells were exposed to 7.99 nM gilteritinib or 4.76 nM quizartinib, corresponding to the IC50 of these compounds, as calculated for the MV4-11 cell line.
Gilteritinib appeared to induce apoptosis in MV4-11 cells (6.4±8.0% of cells in early apoptosis and 2.3±1.2% in late apoptosis), although this effect was not statistically significant compared with the untreated control. Quizartinib induced irreversible necrotic damage to MV4-11 cells: 23±2.8% of the cell population showed high PI staining, 7.6±2.3% were in late apoptosis and 1.78±0.9% were in early apoptosis (Fig. 9A). The exposure to gilteritinib or quizartinib did not induce apoptosis in HL60 cells or PBMCs, and the apoptosis levels were similar compared with the vehicle control (Fig. 9B and C). Fig. S5 shows the dot plot representation of the effect of gilteritinib and quizartinib on the induction of apoptosis in AML cell lines and PBMCs.
Figure 9.

Effect of gilteritinib and quizartinib on the induction of apoptosis in acute myeloid leukemia cell lines and PBMCs. (A) MV4-11 cells. (B) HL60 cells. (C) PBMCs from a clinically healthy volunteer. Annexin V-FITC and PI staining was performed. Vehicle control (0.1% DMSO), IC50 gilteritinib (7.99 nM), IC50 quizartinib (4.76 nM), death control (3% DMSO) and etoposide (100 nM). Two-way ANOVA with Bonferroni's post hoc test was performed. *P<0.05; **P<0.01; #P<0.0001. PBMCs, peripheral blood mononuclear cells.
Gilteritinib and quizartinib significantly decreased the proliferation (20.36 and 24.00%, respectively) of MV4-11 cells compared with the vehicle control (70.29%). This effect was comparable to the 3% DMSO death control (23.7%) (Fig. 10A and D). However, these inhibitors did not affect HL60 cell proliferation (51 and 61% proliferation index, respectively) or PBMC proliferation (11 and 12% proliferation index, respectively) (Fig. 10B-D). Fig. S6, Fig. S7, Fig. S8 show the dot plots and histograms of the flow cytometry analysis of cell division of MV4-11 cells, HL60 cells and PBMCs using CFSE.
Figure 10.

Effects of gilteritinib (7.99 nM) and quizartinib (4.76 nM) on the proliferation of acute myeloid leukemia cells and PBMCs. (A) MV4-11 cells. (B) HL60 cells. (C) PBMCs from a clinically healthy volunteer. (D) Comparison of the effects of the inhibitors on the proliferation index of MV4-11 and HL60 cells, and PBMCs. Vehicle control (0.1% DMSO), IC50 gilteritinib (7.99 nM), IC50 quizartinib (4.76 nM), death control (3% DMSO) and etoposide (100 nM). The data were analyzed using one-way ANOVA and two-way ANOVA followed by Dunnett's and Bonferroni's post hoc tests, respectively. *P<0.05; **P<0.01; ***P<0.001; #P<0.0001. PBMCs, peripheral blood mononuclear cells.
Discussion
The upregulation of FLT3 expression leads to the excessive proliferation and death evasion of leukemic cells, as well as the rapid progression of the disease. FLT3 participates in cell proliferation, motility and differentiation. Its activation is induced by autophosphorylation of Tyr589, Tyr591 and Tyr599, which promotes conformational changes in the conserved DFG motif of this receptor and makes the ATP-binding site available (75,76). Additionally, ITD insertions in the JMD of FLT3 are present in 69.5% of patients with AML and are associated with higher cell proliferation and malignancy (13,77,78). Therefore, WT-FLT3 and ITD-FLT3 are essential targets for AML therapy.
The conformation of the FLT3 receptor is dynamic in biological systems, alternating between DFG-out (inactive) and DFG-in (active) forms depending on cellular processes and signaling (79). Thus, it was essential to obtain the structural models of the DFG-out and DFG-in forms of WT-FLT3 and ITD-FLT3 receptors. This contributes to understanding the impact of the ITD in JMD on the conformational changes of FLT3, and the molecular interactions, affinity and effectiveness of TKIs.
The first homology model of WT-FLT3/DFG-in was constructed by Ke et al (13), who used the FLT3/DFG-out (1RJB) and the colony stimulating factor 1 (3LCD) (DFG-in) structures. This model helped define the molecular interaction parameters of type I TKIs and allowed the identification of novel inhibitors through structure-based virtual screening (13). Furthermore, Mashkani et al (15) described two potential models of WT-FLT3/DFG-in based on the structures of c-Kit (1PKG) and Abelson kinases (2GQG), and their molecular interactions with adenosine diphosphate and a type I TKI (dasatinib). Lee et al (80) also constructed a model of WT-FLT3/DFG-in using dual specificity tyrosine phosphorylation regulated kinase 1A (4NCT) to study the effect of the G697R point mutation on the molecular interactions and drug responses to sorafenib and midostaurin. The WT-FLT3/DFG-out structure obtained in the present study complements these previous models and the ones reported by Griffith et al (38) (1RJB) and Thomas (6IL3) (39).
Based on this structural model, WT-FLT3/DFG-in was then constructed using the DFG-in portion of the crystalized c-Kit structure (1PGK) as described in previous works employing c-Kit (42) and other RTKs as templates (13,15,42). Despite the relevance of the ITD-FLT3 mutation in AML progression, few structural models have been reported so far. Most of them focus on elucidating the effect of ITD length on conformational changes of the receptor (37,81). Therefore, to the best of our knowledge, the present study was the first to report the ITD-FLT3 structural models in DFG-out and DFG-in conformations.
To evaluate the quality scores of the four models, seven programs (ERRAT, Profile 3D Verify, QMEAN, Molprobity, ProSA, ProQ and PROCHECK) were used, providing a set of parameters indicating their high structural quality. This strategy demonstrated that they could be used to assess the molecular interactions and affinity scores of sorafenib, midostaurin, gilteritinib and quizartinib.
The molecular interactions of TKIs with the FLT3 receptor are determined by the biochemical characteristics of each inhibitor and the conformation of the receptor to which they bind (76,82). First-generation TKIs were initially designed to target RTKs, but they are not specific for FLT3. For example, sorafenib and midostaurin can efficiently treat solid tumors, such as hepatocarcinoma, advanced renal carcinoma and non-small cell lung cancer, through the inhibition of Raf serine/threonine kinase (83) and protein kinase C (84), respectively. However, the lack of specificity of these inhibitors leads to strong cytotoxic effects at therapeutic doses, limiting their therapeutic use (77,85). Conversely, gilteritinib and quizartinib (second-generation inhibitors) were explicitly designed against FLT3 to treat patients with AML bearing ITD mutations or the D835 mutation in the TKD (86–88). The molecular docking presented in the present study confirmed that gilteritinib and quizartinib (second-generation TKIs) have higher affinities for the WT-FLT3/DFG-out and WT-FLT3/DFG-in models compared with sorafenib and midostaurin (first-generation TKIs).
The molecular docking of WT-FLT3 and the use of PyMOL2 and Discovery Studio visualizer programs indicated that Leu616 and Lys614 [from the phosphate-binding loop (P-loop)], Tyr693, Cys695, and Tyr696 [from the amino terminal lobe (N-lobe)] were the main residues participating in the interactions of WT-FLT3/DFG-out with its inhibitors, whereas Asp698 and Cys694 (from the N-lobe) were involved in the interaction with WT-FLT3/DFG-in. These data partially coincide (Leu616, Tyr693, Cys694, Cys695 and Tyr696) with those of Pandurang et al (89), who also reported the participation of Gly617, Val624, Ala642, Phe830 and Gly697 during the formation of inhibitor/WT-FLT3 complexes. The present study also indicated the participation of other residues, including Arg764, Asp788, Asn790 (from the DFG-motif), Arg704 (from the N-lobe) for WT-FLT3/DFG-out, and Asn701 (from the N-lobe), Asn836 (from the A-loop), Lys802 and Val801 (from the catalytic loop) for WT-FLT3/DFG-in. These sites are associated with the backbone and hinge region (JMD-TKD) of FLT3 and are crucial for the inhibitory activity of the drugs (13,85). Therefore, the ITD insertion in JMD, known to affect the conformation of this region of FLT3, may modify the molecular interactions with its inhibitors and modulate their activity.
Regarding the ITD-FLT3 models, it should be noted that the amino acid position (number) increased downstream of the ITD insertion site (9 aa long). The dockings showed that Asn710 (WT, Asn701), Asp707 (WT, Asp698), Arg773 (WT, Arg764), Cys703 (WT, Cys694), Asn799 (WT, Asn790), Asp797 (WT, Asp788), Val810 (WT, Val801) and Lys811 (WT, Lys802) were involved for both ITD-FLT3 and WT-FLT3 receptors (common amino acids). Most amino acids participating in the formation of the inhibitor/ITD-FLT3 complexes belonged to the hinge region, P-loop, A-loop and N-lobe of the receptor, as observed for WT-FLT3. The ITD insertion in FLT3 also led to the participation of new amino acids from the interacting region of the receptor. In particular, the sorafenib/ITD-FLT3/DFG-out complex involved Arg792 (WT, Arg783), the gilteritinib/ITD-FLT3/DFG-out complex involved Ser796 (WT, Ser787), the quizartinib/ITD-FLT3/DFG-out complex involved Cys623 (WT, Cys614), and the midostaurin/ITD-FLT3/DFG-in complex involved Arg773, Asn774 and Asp787 (WT, Arg764, Asn765 and Asp778, respectively). The different residues forming these complexes may change the affinity of the inhibitors.
The ITD mutation in FLT3 modified the affinity forces of the TKIs included in the present study. This observation supports previous reports by Zorn et al (36) and Friedman (76), who showed that the ITD mutation increased receptor flexibility and facilitated the interaction between ATP or TKIs (drugs) with their binding site, leading to constitutive activation (through ATP binding) or modulation of the activity of the receptors (through TKI binding). Sorafenib and midostaurin exhibited increased affinity towards ITD-FLT3/DFG-out compared with WT-FLT3/DFG-out. Conversely, sorafenib, midostaurin and quizartinib exhibited no marked changes in their affinities towards ITD-FLT3/DFG-in and WT-FLT3/DFG-in. Furthermore, gilteritinib exhibited decreased affinity towards ITD-FLT3 compared with WT-FLT3 (both DFG-out and DFG-in). Therefore, this inhibitor was more sensitive to the effect of the ITD mutation than the rest. Despite this in silico prediction, Perl et al (90) and Smith et al (91) reported that gilteritinib was associated with a good survival outcome in patients with refractory AML, 70% of which presented the ITD mutation.
Regarding the DFG motif, changes in its conformation may modulate the affinity of its inhibitors and their efficacy in AML treatment. Type I TKIs (midostaurin and gilteritinib) preferentially bind to the ATP-binding pocket of the DFG-in conformation, which is highly conserved between RTKs. By contrast, type II TKIs (sorafenib and quizartinib) preferentially bind to a less conserved region adjacent to the ATP binding pocket of the DFG-out conformation (23,92). The present data on type I TKIs showed that gilteritinib exhibited improved affinity compared with midostaurin for WT-FLT3 (DFG-out and DFG-in). These data agree with those of Egbuna et al (93) and Bultum et al (94), who reported high affinity of gilteritinib for WT-FLT3/DFG-out. Mashkani et al (15) reported that midostaurin had a similar affinity for WT-FLT3, in DFG-out and DFG-in conformations. The present results on type II TKIs also showed that sorafenib and quizartinib had high affinity for the four FLT3 models, which coincides with reports by Egbuna et al (93) for sorafenib and Mirza et al (95) for quizartinib using a WT-FLT3/DFG-out structural model of the receptor.
The differences in the molecular interactions and affinities of the inhibitors may be related to their biochemical characteristics. Both type I and type II inhibitors included in the present study are heterocyclic aromatic compounds; however, they have different characteristic structural groups; the type I inhibitors are indoles and the type II inhibitors are benzimidazoles. This may favor the binding of type I TKIs (WT-FLT3/DFG-in) to the ATP-binding site and the binding of type II TKIs (WT-FLT3/DFG-out) adjacent to the ATP-binding site (96). The changes in affinity forces may also be due to the differences in polar and non-polar bonds, which modulate the stability of the ligand/protein complexes (80).
Gilteritinib (type I TKIs) exhibited low affinity for the active ITD-FLT3/DFG-in conformation compared with sorafenib and quizartinib (type II TKIs) in both conformations (DFG-out and DFG-in), while midostaurin showed an increase in affinity for DFG-out. This difference may be related to the impact of the ITD insertion on the availability of the binding site of FLT3 (97,98).
Although both inhibitors (type I and type II) suppress FLT3, Wodicka et al (99) and Ke et al (13) reported that type I TKIs bind more strongly to the WT-FLT3/DFG-in model. Contrarily, Vijayan et al (18) reported that type II TKIs have an improved selectivity for the inactive conformations of FLT3 (DFG-out) because they are specific for each RTK.
In summary, the HMP and molecular docking tools applied revealed that the four models were suitable for studying the molecular interactions of TKIs with FLT3. They also highlighted the affinity of the second-generation drugs gilteritinib and quizartinib for WT and mutated FLT3 (DFG-out and DFG-in). To corroborate the accuracy of these predictions, their effect was evaluated in an in vitro cellular model of AML.
Several AML cell lines are available to evaluate the cytotoxicity and specificity of TKIs, including the HL60 (WT-FLT3) and MV4-11 (ITD-FLT3-LOH) homozygote cell lines (32). It has been reported that LOH of FLT3 in MV4-11 leukemia cells and patients with AML leads to the constitutive phosphorylation of FLT3, the activation of its related pathways and high malignancy (15). Commonly, ITD-FLT3-LOH is present in 35% of patients with newly diagnosed AML but in 70–80% of refractory patients (32,100); thus, it is particularly important to characterize inhibitors of this receptor.
The presence of the ITD insertion (~30 bp) in the FLT3 sequence of MV4-11 cells was validated by end-point PCR, confirming other reports (32,43,67). In addition, the expression of FLT3 in the MV4-11 and HL60 cells was confirmed by flow cytometry, which also showed that MV4-11 cells expressed more FLT3 receptors on their surface than HL60 cells. Quentmeier et al (32) also reported high expression levels of ITD-FLT3 in MV4-11 cells, which enhanced their proliferation compared with that of MULT-11 or MOLM-13 heterozygote cells, which express WT-FLT3 and ITD-FLT3.
The upregulation of WT-FLT3 expression is commonly observed in patients with newly diagnosed AML who respond satisfactorily to conventional therapy (induction treatments with daunorubicin and cytarabine, and consolidation treatment by bone marrow transplant) (101). However, if the patient (new or refractory) is positive for ITD-FLT3 (homozygous or heterozygous), the only viable alternative is TKI therapy (11). At present, the only FDA-approved TKIs for the treatment of AML are midostaurin (Rydapt®; 2017), gilteritinib (Xosapta®; 2018) and quizartinib (Vanflyta®; 2023) (25,68,90,102). Midostaurin induces adverse effects and presents high toxicity (103). Additionally, this molecule may induce drug resistance, leading to a progressive loss of effectiveness and the need to increase the therapeutic dose during the illness (76). Therefore, it is mainly used in chemotherapy as a complement to cytostatic drugs (daunorubicin and cytarabine) in newly diagnosed patients (86). Sorafenib has not yet been approved for AML because of its strong side effects (104). Gilteritinib and quizartinib are considered competitive inhibitors of WT- and ITD-FLT3 because of their transitory binding to the receptor at the ATP-binding site (type I) or adjacent to it (type II) (76,96). Although both molecules were explicitly designed against WT- and ITD-FLT3, quizartinib has a more potent inhibitory activity (22 mg/day) than gilteritinib (120 mg/day) (68,105,106), Gilteritinib is better tolerated by patients than quizartinib (76). Thus, at present, quizartinib is approved to be used in combination with chemotherapy in newly diagnosed patients, and gilteritinib is recommended for monotherapy in patients with refractory or relapsed AML (68,106).
In the MTT assay, both compounds strongly affected AML cell viability, with IC50 values of 7.99 nM (gilteritinib) and 4.76 nM (quizartinib) against MV4-11 cells, and 57.54 nM (gilteritinib) and 38.75 nM (quizartinib) against HL60 cells. These IC50 values were consistent with previous reports in MV4-11 cells. Kawase et al (34) reported an IC50 of 3.8 nM for gilteritinib and 0.7 nM for quizartinib; Wang and Baron (103) reported an IC50 of 3.34 nM for gilteritinib and 1.07 nM for quizartinib; Hu et al (69) reported an IC50 of 3.02 nM for gilteritinib; and Reiter et al (44) reported an IC50 of 1.8 nM for quizartinib. Furthermore, Mori et al (25) reported that, in Ba/F3 cells (murine interleukin-3-dependent pro-B cell line) transfected with the ITD mutation, gilteritinib (IC50, 1.8 nM) and quizartinib (IC50, 0.46 nM) showed a similar effect to that observed in the present study. Kawase et al (34) also reported an IC50 of 15.5 nM for gilteritinib and 1.6 nM for quizartinib in MOLM-13 cells expressing the WT and ITD FLT3 receptor. These data highlight the strong in vitro effect of gilteritinib (type I TKI) and quizartinib (type II TKI) against leukemic cells and support the clinical evidence showing that they are currently the best TKIs for the treatment of patients with refractory AML.
The discrepancy between the high cytotoxic effect of gilteritinib against MV4-11 cells that express ITD-FLT3 and the prediction using molecular docking of a low affinity for ITD-FLT3 may be related to the Cys703 residue of ITD-FLT3 (WT, Cys694). Ke et al (13), Mashkani et al (15) and Pandurang et al (89) pointed out the importance of this amino acid for the formation of the gilteritinib/FLT3 complexes because it helps maintain the inhibitory effect. This may also explain why gilteritinib is useful (as monotherapy) in AML in refractory patients that present a high percentage of ITD-positive cells (68,90,100,106). Furthermore, the IC50 values revealed that the MV4-11 cells (ITD-FLT3) were more sensitive to both inhibitors than the HL60 cells (WT-FLT3), reflecting the fact that these drugs were designed to inhibit ITD-FLT3.
Cell death by apoptosis and inhibition of cell proliferation are among the main mechanisms that may explain the effect of gilteritinib and quizartinib on MV4-11 and HL60 cells through inhibition of the FLT3 receptor (69,104). Thus, apoptosis was examined by flow cytometry using annexin V (a marker of phosphatidylserine implicated in the induction of apoptosis) and PI (a DNA marker related to loss of membrane integrity) labeling, and cell proliferation was examined using CFSE labeling (a marker of cell generation).
Under the present experimental conditions, both gilteritinib (7.99 nM) and quizartinib (4.76 nM) slightly induced early and late apoptosis in MV4-11 cells after 48 h of exposure, and significantly induced necrosis compared with the vehicle control. These results differ from those of Hu et al (69), who reported a higher percentage (37.2%) of annexin V-positive MV4-11 cells exposed to 2.5 nM gilteritinib for 48 h. However, the present results align with those of other authors, such as Ueno et al (107), who observed that doses of 3–30 nM only induced 20% apoptosis. Similarly, Qiao et al (108) tested gilteritinib concentrations of 12.5, 25 and 50 nM, and observed that the highest concentration promoted apoptosis in only 25% of MV4-11 cells after 24 h. Quizartinib was recently evaluated by Qiu et al (109), who showed that 48 h of exposure to a high concentration of this molecule (50 nM) induced apoptosis in 65.8% of MV4-11 cells. In addition, the present study demonstrated that 48 h of exposure to gilteritinib or quizartinib did not induce apoptosis in HL60 cells or PBMCs. These data agreed with Qiao et al (108) and Darici et al (110) for THP1 cells, HL60 cells and PBMCs.
The present study also demonstrated that the 48-h exposure to the inhibitors significantly decreased MV4-11 cell proliferation from 80.68% (vehicle control) to 20.36% (gilteritinib) and 24.48% (quizartinib). However, this effect was not observed in HL60 cells which showed a similar percentage of proliferation after exposure to gilteritinib or quizartinib compared with the vehicle control, suggesting that one of the mechanisms responsible for the activity of these drugs is through their specific and competitive binding to ITD-FLT3 in MV4-11 cells, which inhibits its activity and delays the generation of new cells. This observation confirmed previous studies with other methodologies (Cell Counting Kit-8 and MTT assays) reporting that both inhibitors (used at nM ranges) have a higher specificity for ITD-FLT3 than for WT-FLT3 (70,111,112).
The viability of PBMCs was not affected following 48 h of exposure to gilteritinib or quizartinib. This result agrees with reports by Qiao et al (108) and Darici et al (110) on PBMCs exposed to high concentrations of gilteritinib (8,000 nM) and quizartinib (500 nM). These cells exhibited no sign of apoptosis or impact on their proliferation. These results are supported by the fact that the main target of both inhibitors is the FLT3 protein. This observation is of importance to avoid unwanted toxic effects in non-tumor blood cells, which are crucial in the functioning and defense of the organism. AML treatments have progressed in the last decade, fostering the development of precision TKIs targeting WT and ITD-mutated FLT3 receptors (76,85,89). The present in silico study showed a strong affinity of second-generation TKIs for WT-FLT3 (DFG-out and DFG-in) compared with the first-generation drugs. Additionally, the ITD mutation in FLT3 impacted the binding of the TKIs tested, and the effects were different for each inhibitor. The high affinities of gilteritinib (for WT-FLT3/DFG-out and WT-FLT3/DFG-in) and quizartinib (for all four FLT3 models) predicted in silico were tested in vitro by evaluating their IC50 in MV4-11 (ITD-FLT3) and HL60 (WT-FLT3) cell lines. The MV4-11 cells showed a slight induction of apoptosis and were more sensitive to both inhibitors than the HL60 cells. A significant decrease in proliferation in MV4-11 cells and a milder effect on HL60 cells were observed. No cytotoxic effect against non-tumoral blood cells (PBMCs from a healthy volunteer with null FLT3 expression) was observed, suggesting a specific effect of these TKIs against the FLT3 receptor expressed in leukemic cells.
As aforementioned, in the early stages of AML, patients express low levels of WT-FLT3 (such as HL60 cells). By contrast, 70–80% of patients with refractory or advanced-stage AML present the ITD-FLT3 mutation due to LOH (such as MV4-11 cells), which favors leukemic cell proliferation because of the constitutive expression of FLT3 (12,100). Thus, the rationale for using MV4-11 cells that possess the homozygous ITD-FLT3 mutation and HL60 cells that only express WT-FLT3 was to compare the effect of gilteritinib and quizartinib in a progressive cellular model of AML. Based on the present data and to further explore the contribution of FLT3 in leukemic cell proliferation and the inhibitory mechanism of TKIs, it would be interesting to complement the present in vitro model with more AML cell lines that express WT-FLT3 (THP1 cells) and WT/ITD-FLT3 (MOLM-13 and PL21 cells), and with null expression of FLT3 (U937 and HEL cells) (32,63). Additionally, more work is needed to include an MV4-11 cell line with equal expression of WT-FLT3 and ITD-FLT3 receptor, as well as an FLT3-free control system, which could be achieved by transfection and knockout of expression of FLT3 using interfering RNA (short hairpin RNA or small interfering RNA).
The present study provides a valuable starting point for further research. The construction of the structural models of WT-FLT3 and ITD-FLT3 (DFG-out and DFG-in), the descriptions of the molecular interactions and the calculations of the affinity forces of TKIs are relevant as they contribute to the knowledge of their mechanisms of action and provide high-quality data to search for novel, more specific and potent FLT3 inhibitory molecules of synthetic or natural origins, which are urgently needed for patients with AML. In addition, the present study provides the basis of an in vitro progressive model of AML to test their efficiency.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Abel Gutiérrez Ortega and Dr Rodolfo Hernández Gutiérrez for facilitating access to the Medical Biotechnology and Pharmaceutical Unit of the Center for Research and Assistance in Technology and Design of the State of Jalisco, Guadalajara, Mexico, Dr Susana del Toro Arreola (Health Science Campus of the University of Guadalajara, Guadalajara, Mexico) and Dr Jesse Haramati (Biological and Agricultural Sciences Campus of the University of Guadalajara, Zapopan, Mexico) for their expert scientific guidelines, and Dr Luis Javier Reséndiz Castillo (Health Science Campus of the University of Guadalajara, Guadalajara, Mexico) for revising the manuscript. The authors would also like to thank Dr Rebeca Mendez-Hernandez (Monell Chemical Senses Center, Philadelphia, PA, USA) for editing the manuscript in English.
Funding Statement
The present study was supported by the Research Promoting Programs of the University of Guadalajara, Mexico (grant no. 271980) and a PhD degree grant (grant no. 842471) from the CONAHCYT, Mexico.
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
ASCA, AS and SEHR conceptualized the study. ASCA, LFJS and FYFH were involved in designing the methodology. ASCA performed the experiments. ASCA and MDRHL curated the data and performed formal analyses. ASCA, AS and SEHR wrote the draft and final version of the manuscript. ASCA, AS, SEHR, LFJS, MDRHL and FYFH reviewed and edited the manuscript. AS and SEHR acquired the funding, were involved in project administration and confirm the authenticity of all the raw data. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
The peripheral blood mononuclear cells used in the present study were obtained with the written informed consent of one healthy volunteer for participation in this project based on ethical criteria for collecting biological samples and in accordance with relevant guidelines and regulations of the Declaration of Helsinki. The present study was approved by the Bioethical Committee of the Biological and Agricultural Sciences Campus of the University of Guadalajara (approval no. 2019-2024; Zapopan, Jalisco, Mexico).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Shimony S, Stahl M, Stone RM. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023;98:502–526. doi: 10.1002/ajh.26822. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Ervik M, Lam F, Laversanne M, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F. International agency for research on cancer; Lyon: 2024. [ May 06; 2024 ]. Global Cancer Observatory: Cancer Today. Available from: [Google Scholar]
- 3.Gokhale P, Chauhan APS, Arora A, Khandekar N, Nayarisseri A, Singh SK. FLT3 inhibitor design using molecular docking based virtual screening for acute myeloid leukemia. Bioinformation. 2019;15:104–115. doi: 10.6026/97320630015104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azizidoost S, Babashah S, Rahim F, Shahjahani M, Saki N. Bone marrow neoplastic niche in leukemia. Hematology. 2014;19:232–238. doi: 10.1179/1607845413Y.0000000111. [DOI] [PubMed] [Google Scholar]
- 5.Ding L, Ley T, Larson D, Miller C, Koboldt D, Welch J, Ritchey J, Young M, Lamprecht T, McLellan M, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santoyo-Sánchez A, Ramos-Peñafiel CO, Saavedra-González A, González-Almanza L, Martínez-Tovar A, Olarte-Carrillo I, Collazo-Jaloma J. The age and sex frequencies of patients with leukemia seen in two reference centers in the metropolitan area of Mexico City. Gac Med Mex. 2016;152:186–189. [PubMed] [Google Scholar]
- 7.Gilliland D, Griffin J. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 8.Lagunas-Rangel FA, Chávez-Valencia V. FLT3-ITD and its current role in acute myeloid leukaemia. Med Oncol. 2017;34:114. doi: 10.1007/s12032-017-0970-x. [DOI] [PubMed] [Google Scholar]
- 9.Arber D, Orazi A, Hasserjia R, Thiele J, Borowitz M, Beau M, Bloomfield C, Cazzola M, Vardiman J. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 10.Kazi J, Rönnstrand L. FMS-like tyrosine kinase 3/FLT3: From basic science to clinical implications. Physiol Rev. 2019;99:1433–1466. doi: 10.1152/physrev.00029.2018. [DOI] [PubMed] [Google Scholar]
- 11.Fernández S, Desplat V, Villacreces A, Guitart A, Milpied N, Rigneux A, Vigo I, Pasquet J, Dumas P. Targeting tyrosine kinases in acute myeloid leukemia: Why, who and how? Int J Mol Sci. 2019;20:3429. doi: 10.3390/ijms20143429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marensi V, Keeshan KR, MacEwan DJ. Pharmacological impact of FLT3 mutations on receptor activity and responsiveness to tyrosine kinase inhibitors. Biochem Pharmacol. 2021;183:114348. doi: 10.1016/j.bcp.2020.114348. [DOI] [PubMed] [Google Scholar]
- 13.Ke YY, Singh VK, Coumar MS, Hsu YC, Wang WC, Song JS, Chen CH, Lin WH, Wu SH, Hsu JT, et al. Homology modeling if DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci Rep. 2015;5:11702. doi: 10.1038/srep11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng YH, Shiao HY, Tu CH, Liu PM, Hsu JT, Amancha PK, Wu JS, Coumar MS, Chen CH, Wang SY, et al. Protein kinase inhibitor design by targeting the Asp-Phe-Gly (DFG) motif: The role of the DFG motif in the design of epidermal growth factor receptor inhibitors. J Med Chem. 2013;56:3889–3903. doi: 10.1021/jm400072p. [DOI] [PubMed] [Google Scholar]
- 15.Mashkani B, Tanipour MH, Saadatmandzadeh M, Ashman LK, Griffith R. FMS-like tyrosine kinase 3 (FLT3) inhibitors: Molecular docking and experimental studies. Eur J Pharmacol. 2016;776:156–166. doi: 10.1016/j.ejphar.2016.02.048. [DOI] [PubMed] [Google Scholar]
- 16.Fabbro D, Cowan-Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR review 14. Br J Pharmacol. 2015;172:2675–2700. doi: 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ung PM, Schlessinger A. DFGmodel: Predicting protein kinase structures in inactive states for structure-based discovery of type-II inhibitors. ACS Chem Biol. 2015;10:269–278. doi: 10.1021/cb500696t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vijayan RS, He P, Modi V, Duong-Ly KC, Ma H, Peterson JR, Dunbrack RL, Levy RM. Conformational analysis of the DFG-out kinase motif and biochemical profiling of structurally validated type II inhibitors. Med Chem. 2015;58:466–479. doi: 10.1021/jm501603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uras I, Maurer B, Nebenfuehr S, Zujer M, Valent P, Sexl V. Therapeutic vulnerabilities in FLT3-mutant AML unmasked by Palbociclib. Int J Mol Sci. 2018;19:3987. doi: 10.3390/ijms19123987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rücker FG, Du L, Luck TJ, Benner A, Krzykalla J, Gathmann I, Voso MT, Amadori S, Prior TW, Brandwein JM, et al. Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia. 2022;36:90–99. doi: 10.1038/s41375-021-01323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohl SR, Bullinger L, Rücker FG. New targeted agents in acute myeloid leukemia: New hope on the rise. Int J Mol Sci. 2019;20:1983. doi: 10.3390/ijms20081983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, Groner S, Späth D, Krauter J, Ganser A, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–2392. doi: 10.1182/blood-2009-03-209999. [DOI] [PubMed] [Google Scholar]
- 23.Sexauer A, Tasian S. Targeting FLT3 signaling in childhood acute myeloid leukemia. Front pediatr. 2017;5:248. doi: 10.3389/fped.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, Shimada I, Mori K, Kuromitsu S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs. 2017;35:556–565. doi: 10.1007/s10637-017-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loschi M, Sammut R, Chiche E, Cluzeau T. FLT3 tyrosine kinase inhibitors for the treatment of fit and unfit patients with FLT3-mutated AML: A systematic review. Int J Mol Sci. 2021;11:5873. doi: 10.3390/ijms22115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun J, Hu R, Han M, Tan Y, Xie M, Gao S, Hu JF. Mechanisms underlying therapeutic resistance of tyrosine kinase inhibitors in chronic myeloid leukemia. Int J Biol Sci. 2024;1:175–181. doi: 10.7150/ijbs.86305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barton GJ. Protein sequence alignment techniques. Acta Crystallogr D Biol. 1998;54:1139–1146. doi: 10.1107/S0907444998008324. [DOI] [PubMed] [Google Scholar]
- 29.Contreras-Moreira B, Fitzjohn P, Bates P. Comparative modelling: And essential methodology for protein structure prediction in the post-genomic era. Appl Bioinformatics. 2002;1:177–190. [PubMed] [Google Scholar]
- 30.Saxena A, Sangwan RS, Mishra S. Fundamentals of homology modeling steps and comparison among important bioinformatics tools: An overview. Sci Int. 2013;1:237–252. doi: 10.17311/sciintl.2013.237.252. [DOI] [Google Scholar]
- 31.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: A powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17:120–124. doi: 10.1038/sj.leu.2402911. [DOI] [PubMed] [Google Scholar]
- 33.Xu B, Zhao Y, Wang X, Gong P, Ge W. MZH29 is a novel potent inhibitor that overcomes drug resistance FLT3 mutations in acute myeloid leukemia. Leukemia. 2017;31:913–921. doi: 10.1038/leu.2016.297. [DOI] [PubMed] [Google Scholar]
- 34.Kawase T, Nakazawa T, Eguchi T, Tsuzuki H, Ueno Y, Amano Y, Suzuki T, Mori M, Yoshida T. Effect of Fms-like tyrosine kinase 3 (FLT3) ligand (FL) on antitumor activity of gilteritinib, a FLT3 inhibitor, in mice xenografted with FL-overexpressing cells. Oncotarget. 2019;101:6111–6123. doi: 10.18632/oncotarget.27222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akwata D, Kempen AL, Sintim HO. Identification of a selective FLT3 inhibitor with low activity against VEGFR, FGFR, PDGFR, c-KIT, and RET anti-targets. ChemMedChem. 2023;19:e202300442. doi: 10.1002/cmdc.202300442. [DOI] [PubMed] [Google Scholar]
- 36.Zorn JA, Wang Q, Fujimura E, Barros T, Kuriyan J. Crystal structure of the FLT3 kinase domain bound to the inhibitor Quizartinib (AC220) PLoS One. 2015;10:e0121177. doi: 10.1371/journal.pone.0121177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Todde G, Friedman R. Conformational modifications induced by internal tandem duplication on the FLT3 kinase and juxtamembrane domains. Phys Chem Chem Phys. 2019;21:18467–18476. doi: 10.1039/C9CP02938A. [DOI] [PubMed] [Google Scholar]
- 38.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, Lippke J, Kumkum Saxena K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–178. doi: 10.1016/S1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 39.Thomas C. Crystal structure of the FLT3 kinase bound to a small molecule inhibitor. 2018 doi: 10.2210/pdb6il3/pdb. Available from: [DOI] [Google Scholar]
- 40.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 42.Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-kit product complex reveals the basis for kinase transactivation. JBC. 2003;278:31461–31464. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- 43.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reiter K, Polzer H, Krupka C, Maiser A, Vick B, Rothenberg-Thurley M, Metzeler KH, Dörfel D, Salih HR, Jung G, et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia. 2018;32:313–322. doi: 10.1038/leu.2017.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webb B, Sali A. Comparative protein structure modeling using modeller. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E, Ferriny T. UCSF chimera visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 47.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharya D, Nowotny J, Cao R, Cheng J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016;44:W406–W409. doi: 10.1093/nar/gkw336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colvos C, Yeates T. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eisenberg D, Luthy R, Bowie J. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/S0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 51.Laskowski R, Moss D, Thornton J. Main-chain bond length and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 52.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiederstein M, Sippl M. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cristobal S, Zemla A, Fischer D, Rychlewski L, Elofsson A. A study of quality measures for protein threading models. BMC Bioinformatics. 2001;2:5. doi: 10.1186/1471-2105-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris G, Huey R, Lindstrom W, Sanner M, Belew R, Goodsell D, Olson A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Irwin J, Shoichet B. ZINC-A Free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goodsell D, Morris G, Olson A. Automated docking of flexible ligands: Applications of autodock. JMR. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 59.Trott O, Olson A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schrödinger L, DeLano W. http://www.pymol.org/pymol PyMOL. 2020 Available from: [Google Scholar]
- 61.Den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PEM. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- 62.Schiffer CA, Stone RM. Morphologic Classification and Clinical and Laboratory Correlates. In: Kufe DW, Pollock RE, Weichselbaum RR, et al., editors. Holland-Frei Cancer Medicine. 6th edition. BC Decker; Hamilton, ON: 2003. Available from: [Google Scholar]
- 63.Skopek R, Palusińsk M, Kaczor-Keller K, Pingwara R, Papierniak-Wyglądała A, Schenk T, Lewicki S, Zelent A, Szymański Ł. Choosing the right cell line for acute myeloid leukemia (AML) research. Int J Mol Sci. 2023;24:5377. doi: 10.3390/ijms24065377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sambrook J, Russell DW. 3rd edition. Vol. 1. Cold Spring Harbor Laboratory Press; New York, NY: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 65.Huang Y, Hu J, Lu T, Luo Y, Shi J, Wu W, Han X, Zheng W, He J, Cai Z, et al. Acute myeloid leukemia patient with FLT3-ITD and NPM1 double mutation should undergo allogeneic hematopoietic stem cell transplantation in CR1 for better prognosis. Cancer Treat Res Commun. 2019;11:4129–4142. doi: 10.2147/CMAR.S194523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Becton-Dickinson and Company; Ashland, OR: 2023. FlowJo™ Software (Windows) [software application] Version 10.6.2. [Google Scholar]
- 67.Pulte ED, Norsworthy KJ, Wang Y, Xu Q, Qosa H, Gudi R, Przepiorka D, Fu W, Okusanya OO, Goldberg KB, et al. FDA approval summary: Gilteritinib for relapsed or refractory acute myeloid leukemia with a FLT3 mutation. Clin Cancer Re. 2021;27:3515–3521. doi: 10.1158/1078-0432.CCR-20-4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Erba HP, Montesinos P, Kim HJ, Patkowska E, Vrhovac R, Žák P, Wang PN, Mitov T, Hanyok J, Kamel YM, et al. Quizartinib plus chemotherapy in newly diagnosed patients with FLT3-internal-tandem-duplication-positive acute myeloid leukaemia (QuANTUM-First): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Glob Health. 2023;401:1571–1583. doi: 10.1016/S0140-6736(23)00464-6. [DOI] [PubMed] [Google Scholar]
- 69.Hu X, Cai J, Zhu J, Lang W, Zhong J, Zhong H, Chen F. Arsenic trioxide potentiates Gilteritinib-induced apoptosis in FLT3-ITD positive leukemic cells via IRE1a-JNK-mediated endoplasmic reticulum stress. Cancer Cell Int. 2020;20:250. doi: 10.1186/s12935-020-01341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.James AJ, Smith CC, Litzow M, Perl AE, Altman JK, Shepard D, Kadokura T, Souda K, Patton M, Lu Z, et al. Pharmacokinetic profile of Gilteritinib: A novel FLT-3 tyrosine kinase inhibitor. Clin Pharmacokinet. 2020;59:1273–1290. doi: 10.1007/s40262-020-00888-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Trone D, Mendell J, O'Donnel P, Cook N. A drug-drug interaction study to assess the potential effect of acid-reducing agent, lansoprazole, on quizartinib pharmacokinetics. Cancer Chemother Pharmacol. 2019;84:799–807. doi: 10.1007/s00280-019-03915-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Motulsky H, Christopoulos A. Oxford Press; Oxford, UK: 2004. Fitting models to biological data using linear and nonlinear regression; pp. 211–316. in press. [Google Scholar]
- 73.RStudio Team. PBC; Boston, MA: 2020. RStudio: Integrated Development for R. RStudio. Available from: [Google Scholar]
- 74.Jilani I, Estey E, Manshuri T, Caligiuri M, Keating M, Giles F, Thomas D, Kantarjian H, Albitar M. Better detection of FLT3 internal tandem duplication using peripheral blood plasma DNA. Leukemia. 2003;17:114–119. doi: 10.1038/sj.leu.2402743. [DOI] [PubMed] [Google Scholar]
- 75.Razumovskaya E, Masson K, Khan R, Bengtsson S, Rönnstrand L. Oncogenic Flt3 receptors display different specificity and kinetics of autophosphorylation. Exp Hematol. 2009;37:979–989. doi: 10.1016/j.exphem.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 76.Friedman R. The molecular mechanisms behind activation of FLT3 in acute myeloid leukemia and resistance to therapy by selective inhibitors. Biochim Biophys Acta Rev Cancer. 2022;1877:188666. doi: 10.1016/j.bbcan.2021.188666. [DOI] [PubMed] [Google Scholar]
- 77.Patnaik MM. The importance of FLT3 mutational analysis in acute myeloid leukemia. Leuk Lymphoma. 2018;59:2273–2286. doi: 10.1080/10428194.2017.1399312. [DOI] [PubMed] [Google Scholar]
- 78.Castaño-Bonilla T, Alonso-Dominguez JM, Barragán E, Rodríguez-Veiga R, Sargas C, Gil C, Chillón C, Vidriales MB, García R, Martínez-López J, et al. Prognostic significance of FLT3-ITD length in AML patients treated with intensive regimens. Sci Rep. 2021;11:20745. doi: 10.1038/s41598-021-00050-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie T, Saleh T, Rossi P, Kalodimos CG. Conformational states dynamically populated by a kinase determine its function. Science. 2020;370:eabc2754. doi: 10.1126/science.abc2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee CC, Chuang YC, Liu YL, Yang CN. A molecular dynamics simulation study for variant drug responses due to FMS-like tyrosine kinase 3 G697R mutation. RSC Adv. 2017;7:29871–29881. doi: 10.1039/C7RA04099G. [DOI] [Google Scholar]
- 81.Todde G, Friedman R. Pattern and dynamics of FLT3 duplications. J Chem Inf Model. 2020;60:4005–4020. doi: 10.1021/acs.jcim.0c00544. [DOI] [PubMed] [Google Scholar]
- 82.Zhao J, Song Y, Liu D. Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark Res. 2019;7:19. doi: 10.1186/s40364-019-0170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iyer R, Fetterly G, Lugade A, Thanavala Y. Sorafenib: A clinical and pharmacologic review. Expert Opin Pharmacother. 2010;11:1943–1955. doi: 10.1517/14656566.2010.496453. [DOI] [PubMed] [Google Scholar]
- 84.Kawano T, Inokuchi J, Eto M, Murata M, Kang JH. Activators and inhibitors of protein kinase C (PKC): Their applications in clinical trials. Pharmaceutics. 2021;13:1748. doi: 10.3390/pharmaceutics13111748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: Choosing the best when the optimal does not exist. Am J Hematol. 2018;93:553–563. doi: 10.1002/ajh.25027. [DOI] [PubMed] [Google Scholar]
- 86.Tarver TC, Hill JE, Rahmat L, Perl AE, Bahceci E, Mori K, Smith CC. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Blood Adv. 2020;4:514–524. doi: 10.1182/bloodadvances.2019000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou F, Ge Z, Chen B. Quizartinib (AC220) a promising option for acute myeloid leukemia. Drug Des Devel Ther. 2019;13:1117–1125. doi: 10.2147/DDDT.S198950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kennedy VE, Smith CC. FLT3 mutations in acute myeloid leukemia: Key concepts and emerging controversies. Front Oncol. 2020;10:612880. doi: 10.3389/fonc.2020.612880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pandurang S, Keretsu S, Joo S. Design of new therapeutic agents targeting FLT3 receptor tyrosine kinase using molecular docking and 3D-QSAR approach. Lett Drug Des Discov. 2020;17:585–596. doi: 10.2174/1570180816666190618104632. [DOI] [Google Scholar]
- 90.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, Montesinos P, Baer MR, Larson RA, Ustun C, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381:1728–1740. doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 91.Smith CC, Levis MJ, Perl AE, Hill JE, Rosales M, Bahceci E. Molecular profile of FLT3-mutated relapsed/refractory patients with AML in the phase 3 ADMIRAL study of gilteritinib. Blood Adv. 2022;6:2144–2155. doi: 10.1182/bloodadvances.2021006489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: Current status and future directions. Mol Cancer Ther. 2017;16:991–1001. doi: 10.1158/1535-7163.MCT-16-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Egbuna C, Patrick-Iwuanyanwu KC, Onyeike EN, Khan J, Alshehri B. FMS-like tyrosine kinase-3 (FLT3) inhibitors with better binding affinity and ADMET properties than sorafenib and gilteritinib against acute myeloid leukemia: In silico studies. J Biomol Struct Dyn. 2022;40:12248–12259. doi: 10.1080/07391102.2021.1969286. [DOI] [PubMed] [Google Scholar]
- 94.Bultum LE, Tolossa GB, Lee D. Combining empirical knowledge, in silico molecular docking and ADMET profiling to identify therapeutic phytochemicals from Brucea antidysentrica for acute myeloid leukemia. PLoS One. 2022;17:e0270050. doi: 10.1371/journal.pone.0270050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mirza Z, Al-Saedi DA, Alganmi N, Karim S. Landscape of FLT3 variations associated with structural and functional impact on acute myeloid leukemia: A computational study. Int J Mol Sci. 2024;25:3419. doi: 10.3390/ijms25063419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ezelarab HAA, Ali TFS, Abbas SH, Hassan HA, Beshr EAM. Indole-based FLT3 inhibitors and related scaffolds as potential therapeutic agents for acute myeloid leukemia. BMC Chem. 2023;17:73. doi: 10.1186/s13065-023-00981-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen D, Oezguen N, Urvil P, Ferguson C, Dann SM, Savidge TC. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci Adv. 2016;2:e1501240. doi: 10.1126/sciadv.1501240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Madushanka A, Moura Jr RT, Verma N, Kraka E. Quantum mechanical assessment of protein-ligand hydrogen bond strength patterns: Insights from semiempirical tight-binding and local vibrational mode theory. Int J Mol Med Sci. 2023;24:6311. doi: 10.3390/ijms24076311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wodicka LM, Ciceri P, Davis MI, Hunt JP, Floyd M, Salerno S, Hua XH, Ford JM, Armstrong RC, Zarrinkar PP, Treiber DK. Activation state-dependent binding of small molecule kinase inhibitors: structural insights from biochemistry. Chem Biol. 2010;17:1241–1249. doi: 10.1016/j.chembiol.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 100.Stirewalt DL, Pogosova-Agadjanyan EL, Tsuchiya K, Joaquin J, Meshinchi S. Copy-neutral loss of heterozygosity is prevalent and a late event in the pathogenesis of FLT3/ITD AML. Blood Cancer J. 2014;4:e208. doi: 10.1038/bcj.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Daver N, Venugopal S, Ravandi F. FLT3 mutated acute myeloid leukemia: 2021 treatment algorithm. Blood Cancer J. 2021;11:104. doi: 10.1038/s41408-021-00495-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim ES. Midostaurin: First global approval. Drugs. 2017;77:1251–1259. doi: 10.1007/s40265-017-0779-0. [DOI] [PubMed] [Google Scholar]
- 103.Wang ES, Baron J. Management of toxicities associated with targeted therapies for acute myeloid leukemia: When to push through and when to stop. Hematology Am Soc Hematol Educ Program. 2020;1:57–66. doi: 10.1182/hematology.2020000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Scholl S, Fleischmann M, Schnetzke U, Heidel FH. Molecular mechanisms of resistance to FLT3 inhibitors in acute myeloid leukemia: Ongoing challenges and future treatments. Cells. 2020;9:2493. doi: 10.3390/cells9112493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Levis M. Quizartinib for the treatment of FLT3/ITD acute myeloid leukemia. Future Oncol. 2014;10:1571–1579. doi: 10.2217/fon.14.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perl AE, Hosono N, Montesinos P, Podoltsev N, Martinelli G, Panoskaltsis N, Recher C, Smith CC, Levis MJ, Strickland S, et al. Clinical outcomes in patients with relapsed/refractory FLT3-mutated acute myeloid leukemia treated with gilteritinib who received prior midostaurin or sorafenib. Blood Cancer J. 2022;12:84. doi: 10.1038/s41408-022-00677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ueno Y, Mori M, Kamiyama Y, Saito R, Kaneko N, Isshiki E, Kuromitsu S, Takeuchi M. Evaluation of gilteritinib in combination with chemotherapy in preclinical models of FLT3-ITD+ acute myeloid leukemia. Oncotarget. 2019;10:2530–2545. doi: 10.18632/oncotarget.26811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qiao X, Ma J, Knight T, Su Y, Edwards H, Polin L, Li J, Kushner J, Dzinic SH, White K, et al. The combination of CUDC-907 and gilteritinib shows promising in vitro and in vivo antileukemic activity against FLT3-ITD AML. Blood Cancer J. 2021;11:111. doi: 10.1038/s41408-021-00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Qiu Y, Li Y, Chai M, Hua H, Wang R, Waxman S, Jing Y. The GSK3β/Mcl-1 axis is regulated by both FLT3-ITD and Axl and determines the apoptosis induction abilities of FLT3-ITD inhibitors. Cell Death Discov. 2023;9:44. doi: 10.1038/s41420-023-01317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Darici S, Jørgensen HG, Huang X, Serafin V, Antolini L, Barozzi P, Luppi M, Forghieri F, Marmiroli S, Zavatti M. Improved efficacy of quizartinib in combination therapy with PI3K inhibition in primary FLT3-ITD AML cells. Adv Biol Regul. 2023;89:100974. doi: 10.1016/j.jbior.2023.100974. [DOI] [PubMed] [Google Scholar]
- 111.Wang Y, Zhang L, Tang X, Luo J, Tu Z, Jiang K, Ren X, Xu F, Chan S, Li Y, et al. GZD824 as a FLT3, FGFR1 and PDGFRα inhibitor against leukemia in vitro and in vivo. Transl Oncol. 2020;13 doi: 10.1016/j.tranon.2020.100766. 100766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhu R, Li L, Nguyen B, Seo J, Min Wu M, Seale T, Levis M, Duffield A, Hu Y, Small D. FLT3 tyrosine kinase inhibitors synergize with BCL-2 inhibition to eliminate FLT3/ITD acute leukemia cells through BIM activation. Signal Transduct Target Ther. 2021;6:186. doi: 10.1038/s41392-021-00578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in the present study may be requested from the corresponding author.