

Abstract
Ross River virus (RRV) and Barmah Forest virus (BFV) are arthritogenic arthropod-borne viruses (arboviruses) that exhibit generalist host associations and share distributions in Australia and Papua New Guinea (PNG). Using stochastic mapping and discrete-trait phylogenetic analyses, we profiled the independent evolution of RRV and BFV signature mutations. Analysis of 186 RRV and 88 BFV genomes demonstrated their viral evolution trajectories have involved repeated selection of mutations, particularly in the nonstructural protein 1 (nsP1) and envelope 3 (E3) genes suggesting convergent evolution. Convergent mutations in the nsP1 genes of RRV (residues 248 and 441) and BFV (residues 297 and 447) may be involved with catalytic enzyme mechanisms and host membrane interactions during viral RNA replication and capping. Convergent E3 mutations (RRV site 59 and BFV site 57) may be associated with enzymatic furin activity and cleavage of E3 from protein precursors assisting viral maturation and infectivity. Given their requirement to replicate in disparate insect and vertebrate hosts, convergent evolution in RRV and BFV may represent a dynamic link between their requirement to selectively ‘fine-tune’ intracellular host interactions and viral replicative enzymatic processes. Despite evidence of evolutionary convergence, selection pressure analyses did not reveal any RRV or BFV amino acid sites under strong positive selection and only weak positive selection for nonstructural protein sites. These findings may indicate that their alphavirus ancestors were subject to positive selection events which predisposed ongoing pervasive convergent evolution, and this largely supports continued purifying selection in RRV and BFV populations during their replication in mosquito and vertebrate hosts.
Keywords: Ross River virus, Barmah Forest virus, Alphavirus, Togaviridae, convergent evolution, discrete-trait analysis
1. Introduction
The alphaviruses Ross River virus (RRV) and Barmah Forest virus (BFV) are arthritogenic pathogens of medical importance with both epidemic potential and the capacity for convergent evolution. RRV and BFV share overlapping distributions in Australia and Papua New Guinea (PNG) (Harley et al. 2001, Caly et al. 2019). RRV was first isolated in 1959 from Aedes vigilax mosquitoes collected in Townsville, Queensland (QLD) Australia (Doherty et al. 1963), and BFV was first isolated in 1974 from Culex annulirostris mosquitoes in Victoria (VIC) and concurrently from other mosquito collections from QLD (Doherty et al. 1979, Marshall et al. 1982). Within Australia, they are the most commonly notified arboviruses associated with human disease and case numbers typically average about 5000 (RRV) and 400 (BFV) per year [National Notifiable Diseases Surveillance System (NNDSS) fortnightly reports (https://nindss.health.gov.au/pbi-dashboard/), Australian Government, Department of Health and Aged Care (DoHAC), National notifiable diseases surveillance system (NNDSS) 2024]. Symptomatic infection from either RRV or BFV often results in a debilitating arthritogenic syndrome, which is generally clinically indistinguishable (Flexman et al. 1998). Whilst a candidate vaccine for RRV has been previously under development (Aaskov et al. 1997, Wressnigg et al. 2015), there are currently no publicly available effective, licensed vaccines, or antiviral treatments for either virus (Aaskov et al. 1997, Caly et al. 2019).
RRV and BFV are maintained in zoonotic sylvatic transmission cycles involving multiple mosquito vectors (e.g. Aedes and Culex spp.) and reservoir vertebrate hosts. Native macropods, including kangaroos and wallabies are among the natural vertebrate host species for RRV, although possums, flying foxes, birds, horses, and humans may also contribute to enzootic and/or epizootic transmission dynamics (Russell 2002, Stephenson et al. 2018, Kain et al. 2021). The specific vertebrate host species for BFV are largely unknown (Lindsay et al. 1995), however, considering the homogeneity of geographically disparate isolates, they may include avian species (Poidinger et al. 1997). RRV and potentially BFV can cause epidemics in immunologically naïve human populations. Humans have been implicated as important primary hosts contributing to RRV transmission where other vertebrates were not notably recognized. For example, RRV caused an explosive outbreak in 1979–80 affecting several nations in the South Pacific, and with over 50 000 cases recorded, it remains the largest RRV epidemic to date (Aaskov et al. 1981; Rosen et al. 1981; Tesh et al. 1981; Fauran et al. 1984; Mackenzie et al. 1998). During 2014–15, RRV also caused the largest Australian alphavirus outbreak on record, affecting several states (Jansen et al. 2019).
The alphavirus positive-sense RNA genome (RRV ≈11.8 kb, BFV ≈11.5 kb) encodes four nonstructural proteins (nsP1 to nsP4) and six structural proteins [capsid (C), E3, E2, 6 K, TransFrame protein (TF), and E1] from two separate open-reading frames (Strauss and Strauss 1994, Firth et al. 2008). Alphavirus structure and replication are further summarized in the Supplementary data. RNA viruses, such as RRV and BFV, are more likely to be subjected to convergent evolution and utilize divergent and epistatic mutation interactions for fitness enhancement, as their smaller genome size potentially limits the number of adaptive/virulence determinants available for such mechanisms (Geoghegan and Holmes 2018). An example of the mutation potential of RNA viruses leading to host adaptation and consequent convergent evolution was demonstrated following the emergence of the highly transmissible chikungunya virus (CHIKV) Indian Ocean sublineages bearing the nonsynonymous E1 gene A226V mutation. This mutation and subsequent epistatic mutations in the E2 gene facilitated widespread persistence of CHIKV, despite ongoing host immune and other evolutionary selective pressures. Prompted by these recent events, and the propensity of RRV and BFV to acquire lineage-specific and potentially adaptive mutations (Michie et al. 2020a, 2020b), we investigated the occurrence and patterns of convergent evolution in RRV and BFV using whole-genome sequence (WGS) data and discrete-trait phylogenetic analyses. Ongoing analyses of these alphavirus genomes and assessment of potential adaptive phenomena, such as convergent evolution is needed for increased understanding of their transmission dynamics and identification of selected traits, which define their emergence and persistence, and potential for causing disease.
2. Results
We sequenced recent and historical isolates from human and mosquito sources. We further obtained available WGSs from the GenBank database (Sayers et al. 2022). For RRV, 186 sequences sampled between 1959 and 2018, representing six states and one territory within Australia and four Pacific Island countries and territories (PICTs), including PNG were analyzed. These sequences grouped within one of four previously identified RRV genotypes [G1 to G4, (Michie et al. 2020a)]. Of these, 79 new sequences were obtained from Australian states and territories, including Queensland (QLD), Tasmania (TAS), the Northern Territory (NT), New South Wales (NSW), and Victoria (VIC). Similarly, we analyzed 88 BFV sequences sampled over 44 years between 1974 and 2018, representing four Australian states and one territory [QLD, NSW, Western Australia (WA), VIC, and NT], and PNG, all of which grouped within one of three previously described BFV genotypes [G1 to G3, (Michie et al. 2020b)]. Details of all RRV and BFV isolates and sequences used in this study are summarized in Supplementary Table S1.
2.1. Discrete-trait reconstruction analyses
In Australia, both RRV and BFV have been responsible for seasonal epidemics and may share mosquito vector and vertebrate host species during transmission in overlying geographical regions. To investigate similarities in their evolutionary trajectories and uncover possible convergent transmission trends, we conducted discrete-trait phylogenetic analyses using the complete coding regions of the 186 RRV and 88 BFV sequence datasets.
To ascertain if independent coevolution of sites and convergence have contributed to RRV and BFV evolution, firstly, we manually scrutinized the protein alignments and investigated repetitive nonsynonymous mutations across or among lineages that potentially arose through convergent evolution (i.e. shared among isolates that did not arise from a common ancestor) and those shown to be subject to significant positive selection. These mutations were largely a result of single-nucleotide polymorphisms (SNPs) (Supplementary Table S2). We then selected a subset of such sites and reconstructed their evolution over time using phylogenetic ancestral-trait reconstruction, which allowed us to quantify statistical support for convergent evolution. To this end, we modeled these sites as a reversible Markovian process (Duchene et al. 2016, Anantharam 2022). By further measuring the degree of statistical uncertainty associated with each discrete trait, we were able to identify key converging mutations and additional diverging or epistatic amino acid (aa) transitions that likely support this convergence.
The discrete-trait analysis identified 17 (RRV) and 27 (BFV) aa transitions that had sufficient Bayes factor statistical support (>3.2; i.e. positive support; Kass and Raftery 1995) for convergent evolution. A full summary of these aa transitions is provided in Supplementary Table S3. Of these, we chose five RRV and seven BFV sites for further investigation, which had the highest mean rates of aa change and were considered to contribute to nonsynonymous, convergent mutations based on the appearance of homoplasy and evidence of independent, repeated evolution in the maximum clade credibility (MCC) trees. The trees were constructed to map the evolution of each of these aa sites independently assuming a Markovian process, also known as stochastic mapping (Huelsenbeck et al. 2003, Faria et al. 2011). Table 1 summarizes these convergent and RRV and BFV aa mutation sites and a schematic of their relative genomic positions and corresponding MCC trees derived from the discrete-trait analyses are shown in Fig. 1.
Table 1.
Evolving convergent RRV and BFV mutations with statistical support.
| Gene | Nucleotide positionsa | Amino acid position in genea | Convergentamino acid mutationb | Mean number of amino acid mutations at codon site | Mean rate of amino acid change | Bayes factor of transition ratec | Posterior probability | Median jumpsd | Lower (2.5%)–Upper (97.5%) credible intervale |
|---|---|---|---|---|---|---|---|---|---|
| RRV | |||||||||
| nsP1 | 821–823 | 248f | T to I | 37.100 | 0.999 | Inf | 1.0 | 21 | 14–26 |
| nsP1 | 1400–1402 | 441f | K to E | 7.014 | 1.759 | 4827.2 | 1.0 | 4 | 3–7 |
| E3 | 8554–8556 | 59 | G to R | 3.745 | 1.483 | 54.0 | 1.0 | 2 | 0–3 |
| E2 | 9736–9738 | 389 | A to T | 2.184 | 1.357 | Inf | 1.0 | 2 | 2–3 |
| E1 | 10 354–10 356 | 113 | I to V | 5.069 | 1.270 | Inf | 1.0 | 3 | 2–7 |
| BFV | |||||||||
| nsP1 | 951–953 | 297f | K to E | 5.964 | 1.930 | 2658.794 | 1.0 | 3 | 2–5 |
| nsP1 | 1400–1402 | 447f | S to G | 4.122 | 1.091 | Inf | 1.0 | 4 | 4–5 |
| nsP4 | 5772–5774 | 103 | V to A | 2.208 | 1.108 | Inf | 1.0 | 2 | 1–3 |
| E3 | 8146–8148 | 21 | G to D | 3.635 | 1.127 | Inf | 1.0 | 2 | 1–5 |
| E3 | 8254–8256 | 57 | T to M | 3.575 | 1.139 | 80.018 | 0.980 | 1 | 0–2 |
| E2 | 8359–8361 | 24 | T to A | 2.161 | 1.097 | Inf | 1.0 | 2 | 2–3 |
| E2 | 9129–9131 | 281 | A to V | 2.521 | 1.452 | Inf | 1.0 | 2 | 1–4 |
Nucleotide and aa sequence positions are numbered according to the prototype viruses, isolate T48 (GenBank accession number GQ433359) for RRV and isolate BH2193 (GenBank accession number U73745.1) for BFV.
Convergent aa mutations repeatedly observed in phylogenetic trees (Fig. 1).
Bayes factor statistical support (>3.2) for aa transition; Inf (infinity) was observed when the entire posterior included this rate as a parameter, such that the posterior odds were infinity and therefore also the Bayes factor.
Posterior median number of transition events (Markov jumps) for aa substitution.
Upper and lower bounds of the credible interval of the number of jumps.
Convergent mutations evolving under significant positive selection.
Figure 1.
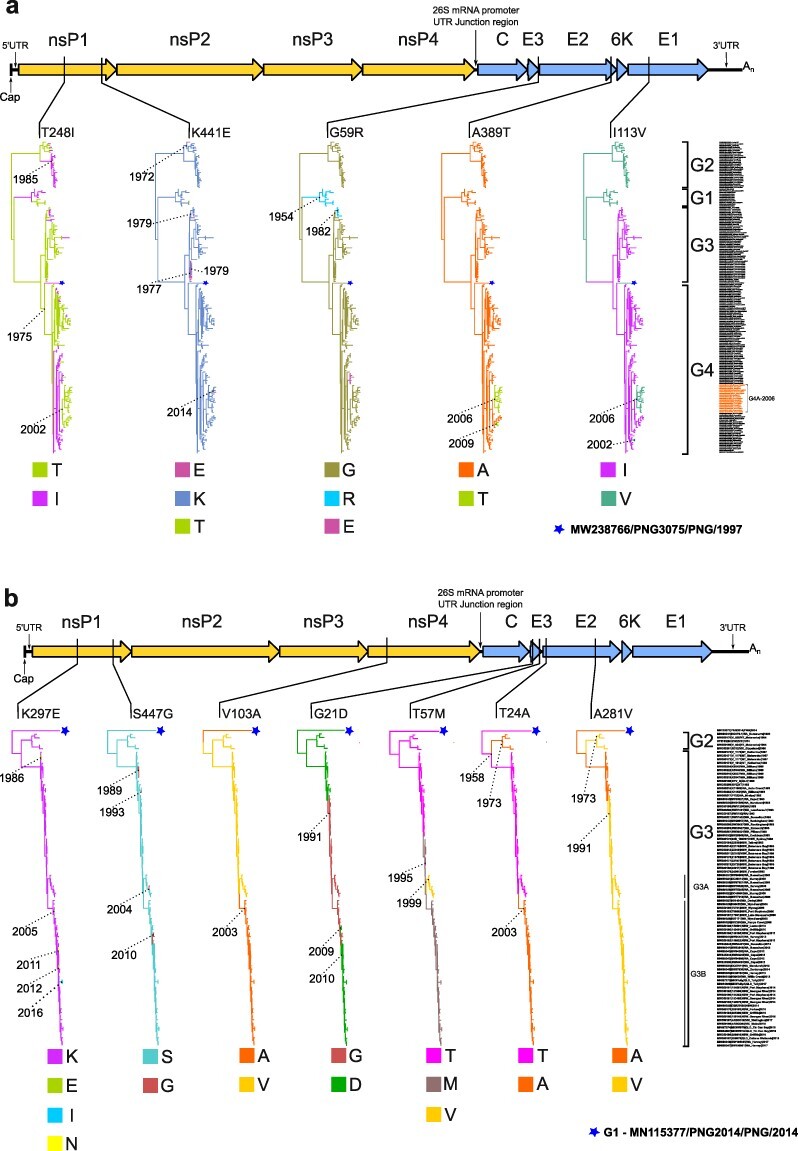
Alphavirus genome map and discrete-trait representation indicating genomic locations and lineage placement of Ross River virus and Barmah Forest virus convergent mutations. The schematic of the alphavirus genome at the top of each Fig. [A, Ross River virus (RRV) and B, Barmah Forest virus (BFV)] shows relative positions of convergent aa mutations within coding nonstructural (nsP1 to nsP4) or structural genes (C, E3, E2, 6 K, and E1). Each mutation is then further mapped below in individual maximum clade credibility (MCC) trees using their Markov rewards where branch color denotes the inferred ancestral state according to the amino acid named under each tree. Years are provided to denote the age of key nodes in the MCC trees. The taxa names and genotype groupings (RRV:G1 to G4, and BFV:G1 to G3) used to construct the MCC trees are also provided on the right side of each A and B sub-figure relative to their corresponding tree tip position. In Fig. 1a, the isolates belonging to the RRV G4A-2006 clade are highlighted on the figure (also summarized in Supplementary Table S8). The monophyletic PNG clade consisting of a single RRV PNG 1997 isolate, PNG3075 (MW238766) (Michie et al. 2021) in Fig. 1a, or BFV PNG 2014 isolate, PNG2014 (MN115377) (Caly et al. 2019) in Fig. 1b, are represented by stars.
Table 1 also summarizes the calculations for the mean number of aa mutations for each site, the mean rate at which each aa transition occurred along branches and the median Markov jumps, which define the posterior median number of transition events for each aa substitution. A mean number of mutations at an aa site corresponding to a value of >1 for a particular type of transition implies a higher chance of convergence. The most evident example of convergence was for RRV nsp1 site 248 where the median number of T to I changes (Markov jumps) was 21. As shown in the corresponding phylogeny (Fig. 1a), both T and I states occurred independently and, in some instances, their interchange involved whole clades.
2.2. Convergent mutations in nsP1 and E3 and analysis of selection pressure
Convergent mutations were particularly evident in the nsP1 and E3 genes of RRV and BFV suggesting that they confer a selective advantage. The majority of the nsP1 and E3 convergent mutations shared striking similarities in their respective RRV and BFV genomic placements. For nsP1, these were RRV T248I and BFV K297E, and further downstream, RRV K441E and BFV S447G. Within E3, the convergent mutations RRV G59R and BFV T57M were identified. The observed nsP1 and E3 viral mutations can be predominantly linked to either host intracellular membrane interactions and/or viral replicative enzymatic functions suggesting their role in fine-tuning these processes. This further supports the generalist host behaviour of RRV and BFV and potential sharing of mosquito and mammalian hosts (Lindsay et al. 1995, Claflin et al. 2015). Indeed, additional divergent mutations (occurring at the same genomic sites as the convergent mutations) were identified (RRV nsP1 K441T, E3 G59E; BFV nsP1 K297 I/N; Fig. 1) and are summarized in Supplementary data (Supplementary Tables S4, S5, and S6).
We also performed selection pressure analysis using measurements of the ratio of nonsynonymous nucleotide substitutions per non-synonymous site to synonymous nucleotide substitutions per synonymous site (dN/dS or ω) for the 186 RRV and 88 BFV strains in our datasets (Table 2). Results indicate that the two RRV nsP1 aa sites 248 and 441 were under significant positive selection pressure as reported previously (Michie et al. 2020a). Site 248, which is located in the recently defined β11/αg region of nsP1 (Jones et al. 2021) demonstrated positive selection under four methods of detection [fixed effect likelihood (FEL), mixed effects model of evolution (MEME), single-likelihood ancestor counting (SLAC), and fast, unconstrained Bayesian approximation (FUBAR)]. Similarly, two methods detected positive selection for nsP1 site 441 and three methods detected positive selection for nsP3 site 453. Whilst no RRV structural aa sites were shown to be under positive pressure selection by at least two methods, E3 site 59 was shown to demonstrate positive selection by one method [posterior probability value 0.910 (FUBAR)].
Table 2.
Selection pressure analyses of 186 RRV and 88 BFV strains collected in Australia and the Pacific between 1959 and 2018.
| Methodsa | |||||||
|---|---|---|---|---|---|---|---|
| Virus | Gene | Codon | Amino acid substitution | FEL | SLAC P value |
MEME | FUBAR posterior probability |
| RRV | nsP1 | 248 | T248I | 0.000 | 0.00006 | 0.000 | 1.000 |
| nsP1 | 441 | K441E/T | 0.064 | 0.102 | 0.030 | 0.986 | |
| nsP3 | 435 | A435I/G/T/Vb | 0.0498 | 0.198 | 0.000 | 0.967 | |
| BFV | nsP1 | 297 | K297E/I/N | 0.0862 | 0.233 | 0.010 | 0.980 |
| nsP1 | 447 | S447G | 0.0835 | 0.234 | 0.010 | 0.982 | |
The P-value thresholds used were <0.05 (FEL and MEME) and <0.1 (SLAC). The posterior probability (PP) threshold used for FUBAR was >0.900. Sites under significant positive selection by at least two methods are indicated by P-values and PPs shown in bold font.
RRV isolates containing nsP3 aa site 435 mutations in the intrinsically unstructured region of the hypervariable domain (HDV) included MN038278|SW72209|WA_Busselton|2003 (A435I), MN038284|SW94735|WA_Harvey|2013 (A435G), MW350155|19 502|QLD_Charleville|1976 (435 T), and MW321536|V-309|NT_ Jabiru|1983 (A435V).
For BFV, nsP1 aa sites 297 and 447 were shown to be under significant positive selection pressure according to the methods MEME and FUBAR. As reported previously, aa substitutions of K to E or I to N were detected at BFV site 297 (Michie et al. 2020b).
Overall, relatively weak significant positive selection pressure values were identified for some RRV nsP1/nsP3 and BFV nsP1 mutations in Table 1. However, all sites showed significant selection by analysis using MEME (including the RRV nsP3 mutations), indicating their possible subjection to positive pressure evolving under episodic directional selection (Engel et al. 2016), although false positives cannot be discounted. However, none of these mutations were selected at the same site for either RRV or BFV. Overall, only RRV T248I demonstrated significant positive selection by all four methods and each of these residues were convergently gained and lost frequently throughout the evolutionary history of RRV indicating pervasive selection (affecting all lineages) as highlighted in the MCC tree (Fig. 2).
Figure 2.
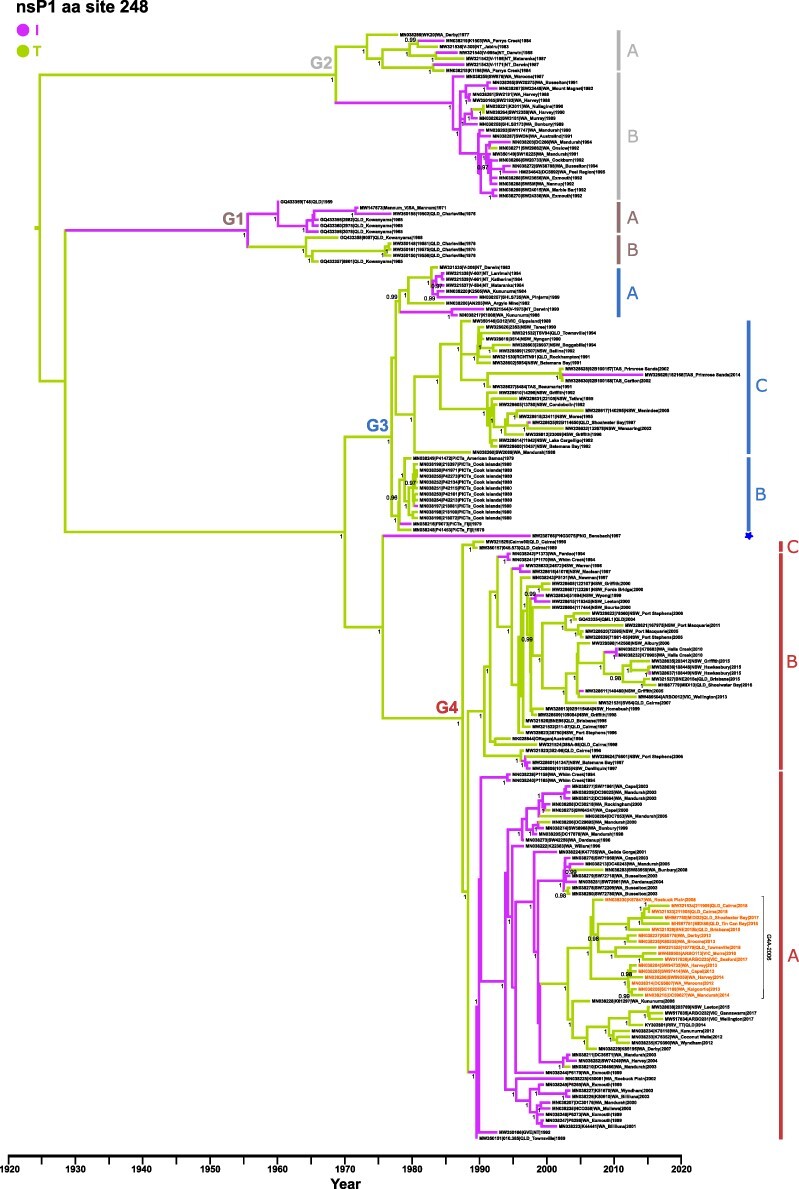
Ross River virus nsP1 maximum clade credibility tree. Time-measured (MCC tree of 186 RRV genomes following ancestral state reconstruction for aa transitions occurring at nsP1 site 248. The nucleotide substitution rate used to calibrate the tree was 3.21 × 10−4 substitutions/site/year. The four RRV genotypes (G1 to G4) and respective sublineages are indicated with Bayesian posterior probabilities of key nodes (>0.95) provided. Tree branches are colored according to the detected aa transition, namely purple (T to I) or green (I to T). Tree topology and taxa order are identical to each tree represented in main manuscript Fig. 1a. The 17 sequences belonging to the recently emerging G4A-2006 clade containing the signature mutations C T35A, E2 A389T, 6 K A58V, E1 I113V, and E1 V426L are highlighted in the figure.
Although the convergent RRV T248I mutation was not observed in BFV, a protein sequence alignment comparison (Fig. 3a) revealed evidence of alternating T or I residues at the corresponding 248 sites for other alphaviruses belonging to the Semliki Forest virus (SFV) complex, namely Getah virus (GETV) and Mayaro virus (MAYV) (Fig. 3a). Our dN/dS analyses above which was applied at the individual species level, may also be limited due to relatively small sequence datasets and lack of sequence divergences (Alvarez-Carretero et al. 2023). To further investigate the nature of selection pressures acting on RRV, BFV, GETV and MAYV nsP1 and E3 genes, we estimated the genus-wide and within-species variation in the (dN/dS) ratio using a phylogeny-free computational method, GenomegaMap (Wilson et al. 2020).
Figure 3.
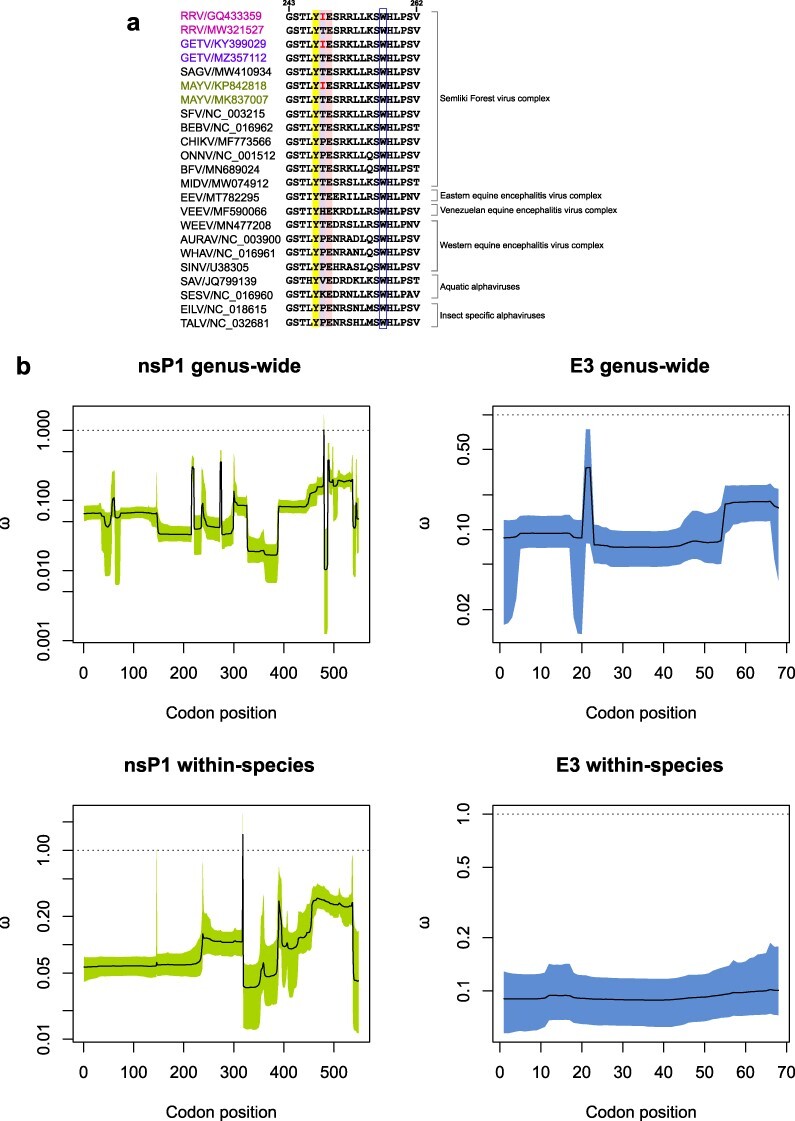
Protein sequence alignment of alphavirus nsP1 β11/αg region and GenomegaMap estimates of the nsP1 and E3 genus-wide and within-species dN/dS ratios ω. (A) Protein sequence alignment comparing RRV nsP1 residues G243 to V262 [previously reported as an amphipathic α-helix membrane binding peptide and encompassing the recently defined β11/αg region (Jones et al. 2021)] with cognate sequences from other representative Alphavirus species. The key residues, Y247 and E249, which are conserved in alphaviruses and have been identified as important for nsP1 methyltransferase activity, are highlighted in colour together with the RRV mutated residue at site 248. The conserved W257 site also considered an important membrane anchor for nsP1, is indicated in the open blue box. (B) GenomegaMap output plots. Solid lines (posterior medians) and shaded regions (95% credibility intervals) for the GenomegaMap point estimates for nsP1 and E3 genus-wide and within-species dN/dS ratios ω are shown. The dotted line (ω of 1) indicates the threshold for neutral selection.
2.3. GenomegaMap analyses of RRV, BFV, GETV, and MAYV nsP1 and E3 genes
GenomegaMap analyses of RRV, BFV, GETV, and MAYV nsP1 and E3 genes demonstrated high pairwise diversity, both genus-wide [θnsP1 = 2.64, 95% credibility interval 2.41–2.89; θE3 = 3.24, 95% confidence interval (CI) 2.58–4.20] and within-species (θnsP1 = 0.266, 95% CI 0.247–0.287; θE3 = 0.372, 95% CI 0.312-0.451). However, we did not find strong evidence of positive selection, including the nsP1 site corresponding to RRV 248. These findings coincided with the dN/dS ratio and selection pressure analyses using FEL, SLAC, MEME, and FUBAR methods (Table 2), which also indicated weak positive selection for nsP1 RRV 248. In the genus-wide analyses, the highest probability of positive selection was 51.6% at nsP1 codon position 480 in the alignment (corresponding to RRV site 475), in which an I/L/M/Q/R/T/V amino acid polymorphism was observed. This site is situated in the crown region of nsP1 harbouring the RNA capping domains (Jones et al. 2021).
In the within-species analyses, the highest probability of positive selection was 82.8% at nsP1 codon position 318 in the alignment (corresponding to RRV site 313), in which L/M/V polymorphisms were observed in RRV, L/M/T/V polymorphisms were observed in GETV, while BFV and MAYV were both fixed for V. This nsP1 site is located in the ring-aperture membrane-binding and oligomerization (RAMBO) domain, which forms a platform on which the capping domain sits (Jones et al. 2021). The GenomegaMap output estimates of the nsP1 and E3 genus-wide and within-species dN/dS ratios ω are summarized in Fig. 3.
The overall low dN/dS ratios for RRV, BFV, GETV, and MAYV support stronger purifying selection for nsP1 and E3 genes, which is characteristic of arboviruses and is largely a result of alternate replication in mosquito and vertebrate hosts (Jones et al. 2010). However, the independent observance of the same T to I mutation within RRV, GETV, and MAYV populations at the corresponding RRV nsP1 site 248, together with evidence of convergent evolution in RRV (as determined by discrete-trait analysis), suggest this polymorphism is unlikely to have occurred by chance. Several factors may have contributed to the inability of demonstrating strong positive selection for the convergent site 248 via the employed dN/dS methods, which incidentally, are sensitive to the degree of nonsynonymous polymorphism and do not consider synonymous mutations that arise under positive selection. The discrepancy in dN/dS findings generated from individual virus populations compared to the GenomegaMap cross-species analyses is curious and could be owing to the differences in methodologies, sensitivities, or generation of false positives. It is also important to note that selective pressure analyses on the individual virus populations versus cross-species populations, may involve different proportions of sylvatic versus epidemic strains which can lead to alternate findings (Stica et al. 2022).
Convergent mutations may assist fine-tuning of intracellular host interactions and replicative enzymatic functions during viral replication
The convergent nsP1 mutations in RRV (sites 248 and 441) and BFV (297 and 447) were found at similar relative genome positions and may have fine-tuning implications during viral RNA capping and enzymatic processes. These mutations were mapped (Fig. 4) onto the predicted 3-dimensional cryogenic-electron microscopy (cryo-EM) crystal structure of the CHIKV nsP1 oligomer complex based on PDB code 7FGG (PDB DOI: 10.2210/pdb7FGG/pdb; Zhang et al. 2022). Alphavirus RNA methylation and capping are initiated by the catalytic enzymes S-adenosyl-l-methionine (SAM) dependent methyltransferase (MTase) and m7guanosine-5ʹ-triphosphate (GTP) transferase (GTase). Reviewed further in Supplementary data, these early infection processes, which promote viral RNA stability and limit degradation by host cell nucleases, are critically dependent on formation of the nsP1 oligomer RNA replication complex (spherule) and its anchorage in host intracellular cell membranes (Salonen et al. 2005, Jones et al. 2021, Zhang et al. 2021).
Figure 4.
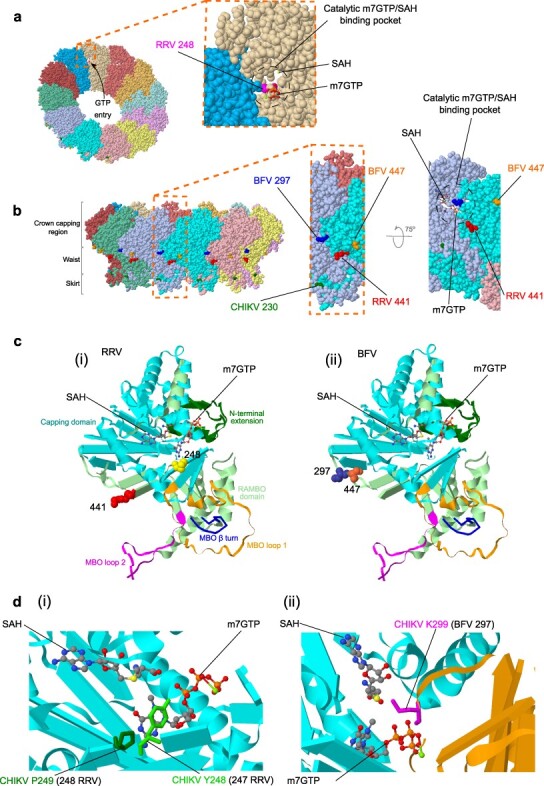
Mapping of convergent RRV/BFV nsP1 mutations to CHIKV cryo-EM structures. Cryo-EM crystal structure of the CHIKV nsP1 oligomer complex based on PDB code 7FGG (PDB DOI: 10.2210/pdb7FGG/pdb; Zhang et al. 2022) with 7-methylguanosine 5ʹ-triphosphate (m7GTP) and S-adenosyl homocysteine [SAH, byproduct produced during methylation of GTP requiring S-adenosyl-l-methionine (SAM)-dependent methyltransferase (MTase) and m7guanosine-5ʹ-triphosphate (GTP) transferase (GTase)] shown as sticks. (A) The 12 nsP1 monomers forming the ring structure are individually colored and the entrance of GTP into the catalytic m7GTP/SAH binding pocket is shown by an arrow. The inset shows how the catalytic pocket, which is involved during RNA capping, is formed from adjacent nsP1 molecules and the predicted position of mutated RRV site 248 is highlighted. (B) Lateral view of the nsP1 complex represented in (A) showing the three recently described regions (crown, waist, and skirt; Jones et al. 2021) and predicted positioning of RRV and BFV nsP1 mutations. The location of CHIKV site 230 implicated with site 299 (BFV site 297) in mutations, which confer resistance to the antiviral compound FHA (Kovacikova et al. 2021) is also highlighted. The inset further shows the relative proximity of the mutated RRV site 441 and BFV sites 297 and 447 within the nsP1 complex waist region involved with host cell membrane binding. Upward rotation of this magnified view allows visualization of the catalytic m7GTP/SAH binding pocket. (C) The nsP1 monomer structure showing the mapped positioning of (i) RRV and (ii) BFV nsP1 mutations relative to m7GTP and SAH bound molecules. (D) Close-up views showing key CHIKV residues (also represented as sticks) involved with m7GTP/SAH binding. The corresponding mutation site numbers for (i) RRV and (ii) BFV are shown in brackets.
Within the structural gene E3, the convergently evolving mutations at RRV G59R and BFV T57M were observed, and like the nsP1 RRV 248 and BFV 297 mutations, may be evidence of aa transitions, which assist fine-tuning of enzymatic processes. These E3 mutations are in the vicinity of the furin cleavage site located at the C-terminal end of the E3 gene (reviewed in Supplementary data). The host cell protease furin cleaves PE2 (E3–E2) protein precursors in the trans-Golgi compartment during viral assembly to activate the alphavirus PE2-E1 heterodimer spikes for host cell fusion and infectivity (Strauss and Strauss 1994, Smit et al. 2001).
Similar to the convergent nsP1 and E3 mutations, additional convergent mutations (RRV E2 A389T, BFV nsP4 V103A, BFV E3 G21D, and BFV E2 A281V), divergent mutations (RRV nsP1 K441T, RRV E3 G59E, RRV E2 T384A/I, BFV nsP1 K297I/N, BFV E3 M57V) and fixated signature mutations (RRV C T35A, RRV 6K A58V, RRV E1 V426L, BFV nsP2 V681A, BFV nsP3 T458M, BFV C P32L, and BFV E1 S171T) (summarized in Supplementary Table S7 and Supplementary Fig. S1), are highly likely to be associated with intracellular host cell membrane and/or enzymatic interactions. In contrast, we could only putatively associate one aa site from each virus with binding of a host cell receptor, namely the mammalian host receptor Mxra8 (Basore et al. 2019, Song et al. 2019). These sites were RRV E1 113 (Domain II, Supplementary Fig. S2A) and BFV E2 24 (Domain A, Supplementary Fig. S2B), which both demonstrated evidence of convergent mutations.
Discussion
The alphaviruses RRV and BFV are medically important pathogens because of their potential to cause debilitating arthralgia, particularly in Australia, where both viruses are widespread and responsible for periodic epidemics and annual activity. Herein, we have demonstrated that there is underlying evidence for convergent evolution in independent RRV and BFV populations, despite being largely under the influence of overall purifying selection that is commonly observed among arboviruses (Weaver et al. 2012, Di Giallonardo et al. 2016, Pollett et al. 2018). In the absence of evidence of strong positive selection, convergent mutations in these alphaviruses may indicate that their ancestral progenitors were subject to adapative positive selection resulting in favorable polymorphisms. We infer that RRV and BFV are subject to ongoing pervasive selection (affecting all lineages) related to the continued requirement to replicate in disparate insect and vertebrate hosts, and are periodically subject to episodic selection (affecting some lineages) imposed during significant environmental or host changes. While these selection phenomena essentially act as purifying forces, there is repeated evidence of convergent mutations that are likely to have originated in the ancestral genomes.
In general, most of the converging and signature mutations we investigated could be putatively associated with fine-tuning of intracellular host interactions and/or viral replicative enzymatic functions (Supplementary Table S7). Convergent evolution is innately pervasive in nature and may be a result of pervasive selection related to host cellular constraints. Indeed, there was little evidence that host immune selection pressure increased RRV or BFV fitness. Given very few human or other vertebrate sequences were available (RRV, n = 38; BFV, n = 4), our findings cannot confirm a direct link between any of the observed convergent or other fixed RRV and BFV mutations with viral fitness for specific mammalian or mosquito species. Only the converging mutations RRV E1 113 and BFV E2 24 could be putatively associated with binding of the mammalian host cell receptor Mxra8 (Basore et al. 2019, Song et al. 2019) and further phenotypic studies will be required to investigate if these mutations are associated with viral fitness and/or host immune mechanisms.
The lack of evidence for host immune selection pressure was also indicated by overall increased patterns of weak episodic positive selection in nonstructural genes compared to structural genes, a phenomena reported for other arboviruses, including MAYV (Mavian et al. 2017), Usutu virus (Engel et al. 2016), West Nile virus (Di Giallonardo et al. 2016), Zika virus (Sironi et al. 2016), and dengue virus (Pollett et al. 2018). Episodic selection may have occurred during notable environmental changes or other significant events, including spillover from natural sylvatic transmission cycles to enzootic transmission cycles involving incidental or dead-end hosts.
The most impressive demonstration of pervasive/episodic selection among the studied RRV and BFV populations was the nsP1 T/I polymorphism at the corresponding RRV site 248. Our dN/dS selection analyses for RRV using FUBAR and SLAC indicated that site 248 may be subject to weak pervasive positive selection and results using MEME indicated possible episodic selection (Table 2). Indeed, the T/I polymorphism involved all four RRV genotypes (Fig. 2) and was further observed in GETV and MAYV species, also belonging to the SFV complex (Fig. 3). However, both the above individual species and additional cross-species (GenomegaMap) dN/dS positive selection analyses did not demonstrate evidence of positive selection for any RRV, BFV, GETV, or MAYV nsP1 or E3 gene sites. Interestingly, inspection of representative alphavirus sequences revealed several residues (T/I/P/H/V/K) have been coded at nsP1 site 248, which is flanked by Y and E residues that are highly conserved among alphavirus species (Fig. 3). Collectively, this may indicate that mutations at site 248 may have resulted from past positive selection, possibly during adaptation of species to new environments or when there were shifts in different vector-host species interactions. Specifically, for RRV, GETV, and MAYV, the T/I polymorphism at site 248 further supports their previously reported phylogenetic clustering within the SFV complex and could be reminiscent of a shared historical geographical distribution and divergence from an emerging ancestor in the tropics (Forrester et al. 2012). Similar evolutionary events have been reported for other arboviruses, including the orthobunyavirus Bangui virus, where studies indicate that only distant common ancestors of this arbovirus have undergone positive selection and that ongoing pervasive selection may drive mutational constraint further limiting viral transmission to incidental human hosts (Orf et al. 2023).
We further considered if geographical distribution could be linked to convergence patterns in RRV and BFV populations. Previous RRV phylogenetic studies on partial E2 gene sequences have indicated that there is a clear spatial structure between eastern and western Australia RRV populations (Jones et al. 2010), but this has not been supported by more recent analyses using near whole genome sequences (Michie et al. 2020a). No such phylogenetic inferences have yet been reported for BFV and we also found little evidence of spatial structure in our near whole-genome data. However, RRV nsP1 I248T convergence patterns were associated with some geographically distinct clustering of viral sublineages. For example, predominance for I coding in WA isolates within G2B, and predominance of T coding in eastern Australian isolates belonging to G3C/B and G4B (Fig. 2). This further supports a historical predisposition for eastern and western lineages, at least for evolution of some RRV genes. Collectively, there was a higher incidence of T (80%) coded at site 248 from a total of 91 isolates sourced from eastern Australian states (QLD, NSW, VIC, and TAS) and during the 1979–80 human epidemic in the Pacific (Cook Islands, Fiji, and American Samoa) compared with 34 of 92 isolates (37%) coding T from the NT and WA (Supplementary Fig. S3).
The prevalence of T at site 248 in isolates from more densely populated eastern Australian states and in isolates from the explosive 1979–80 Pacific human epidemic, is curious and may also indicate episodic selection involving spillover hosts such as humans. Interestingly, between 2014 and 2015, eastern Australia was affected by the largest Australian RRV epidemic on record and 63% of human case notifications were from QLD (Jansen et al. 2019). Of the 10 RRV isolates in our dataset from 2014 to 2015, 80% (including two 2015 outbreak QLD human isolates, MW321527 and MW321528), contained a T coded at nsP1 248. Similarly, 97% of GETV sequences and 90% of MAYV sequences used in our GenomegaMap positive selection analyses contained a T residue at this corresponding nsP1 site. Many of these sequences were obtained from what are usually considered incidental vertebrate hosts. For MAYV, a large proportion of sequences were from humans (Mavian et al. 2017), and in the case of GETV, domesticated horses and pigs were common hosts (Li et al. 2022).
While geographical constraint may have historically contributed to RRV nsP1 convergence and be related to specific host cell tropisms, in contrast, the recently emerging RRV G4A-2006 clade (highlighted in Fig. 2, and reviewed in detail in Supplementary data) demonstrates a broad, diverse geographical range, incorporating eastern Australian (QLD, NSW, and VIC) and WA isolates. It is plausible that its widespread emergence reflects involvement of highly mobile hosts such as humans, horses (Azuolas et al. 2003, Barton and Bielefeldt-Ohmann 2017, Yuen et al. 2022) or potentially avian species. Indeed, the G4A-2006 clade contained a QLD human isolate MW325128 obtained during the 2014–15 Australian outbreak. In addition to coding of T at nsP1 site 248, members of the G4A-2006 clade carried E2 A389T and E1 I113V convergent mutations as well as fixed C T35A, 6K A58V, and E1 V426L mutations, which are a signature for G4. These additional mutations may suggest a significant environmental and/or host-related evolutionary event and possible episodic selection. Interestingly, most could be putatively associated with hydrophobic adjustment and viral protein stabilization at interactive sites with host cell intracellular membranes during viral replication (Supplementary Table S7).
The specific selection pressures acting on alphaviruses such as RRV and BFV and the implications for viral evolution, emergence, and pathogenesis remain largely understudied. More robust selection pressure analyses of alphavirus species than is presented herein are warranted, including assessment of the diversification of ancestral progenitors and identification of adaptive selective events that have shaped alphavirus populations and influenced their subsequent evolutionary landscapes. This may involve convergent, divergent, or other mutations, which become fixed over time as demonstrated by our discrete-trait analysis of RRV and BFV populations. The identification of pervasive selection at sites such as nsP1 248 in which the T/I polymorphism was observed for RRV, GETV, and MAYV, could assist future therapeutic discoveries, development of vaccines, or improved vector control strategies. Interestingly, the CHIKV nsP1 site 299, which corresponds to the convergent mutating BFV site 297 (Fig. 4d (ii)), has also been implicated with mutations at CHIKV site 230, which confer resistance to the potent antiviral compound 6ʹ-β-fluoro-homoaristeromycin (FHA) (Kovacikova et al. 2021). Similarly, our data identifying convergent mutations in RRV and BFV E3 genes may also help guide the development of specific furin inhibitors like dec-RVKR-cmk (Ozden et al. 2008) or identify resistance markers.
Limitations of the study
Of note, our phylogenomic assessment of RRV and BFV convergent evolution is based on consensus genome sequences from coding regions and does not include a detailed analysis of the 5ʹ and 3ʹ untranslated regions (UTRs), which could also contain adaptive mutations. In addition, we have not fully investigated viral subpopulations within isolates that could differ in mutation frequency for a given site and lead to potentially altered phenotypes or fitness traits. Sampling of isolate sequences was limited by availability, and some regions such as the NT, could only be represented by historical strains. Hence, the interpretation of all our phylogenetic and comparative evolutionary analyses is partly biased by sampling, which may have constrained temporal and spatial data comparisons. However, the stochastic nature of these observed convergent mutations and the fixture of other nonsynonymous mutations within genotypes or sublineages, suggest that artifactual adaptation and selection via cell culture passage is unlikely.
In the absence of phenotypic assessment of the observed RRV and BFV mutations, our bioinformatics-based dN/dS selection pressure analyses may also be limited as these methods do not consider synonymous mutations that arise under positive selection pressure. Further scrutiny and phenotypic assessment are therefore required to characterize the RRV and BFV signature mutations highlighted in this study more extensively.
The absence of evidence for strong positive selection in any RRV or BFV gene, including nsP1 or E3 where convergent mutations were demonstrated, is curious and may further reflect limitations in our methodologies and their sensitivities. Here, we have applied dN/dS based analyses to RRV and BFV proteins, which consider excessive nonsynonymous substitutions as a measure of positive selection. While evidence of episodic positive selection was inferred for nonstructural proteins of both viruses (in particular, nsP1) using MEME, this evolutionary phenomenon may not have heightened the nonsynonymous substitution rate sufficiently for its detection during cross-species analyses using GenomegaMap.
3. Materials and methods
3.1. Human ethics statement
This work has received ethical clearance from the Forensic and Scientific Services Human Ethics Committee, approval reference HEC 24-15.
3.2. Virus isolate processing and WGS
A total of 186 near whole-genome RRV sequences were analyzed encompassing sampling over a 59-year period (1959–2018). Along with 107 sequences obtained from GenBank, 79 new sequences were obtained, which included one from a recent isolate from human patient sera (19 779) sourced specifically for this study, and other sequences from historical human and mosquito isolates sampled from 45 sites in 6 states and 1 territory within Australia.
Similarly, 88 near whole-genome BFV sequences were analyzed, including 36 sequences retrieved from GenBank and 52 new sequences which were collectively sampled over a 44-year period (1974–2018) from 39 sites in 4 states and 1 territory within Australia. Details of all RRV and BFV sequences including their original isolation source and GenBank accession numbers are summarized in Supplementary Table S2. To ensure adequate amounts of viral RNA prior to sequencing, isolates were passaged once in Ae. albopictus C6/36 cells (ATTC, CRL-1660TM) as previously described (Pyke et al. 2018) and assessed using an immunofluorescence assay and RRV monoclonal antibody 83/8 (Queensland Health, unpublished data), or if obtained as an historical culture with no known viral titer, were further subcultured once in these cells. Total RNA extraction, first and second strand cDNA synthesis, and massive parallel sequencing using the Nextera XT kit for library construction were performed as previously described (Pyke et al. 2020), with sequencing conducted on NextSeq 500 (Illumina) using a mid-output V2 reagent kit v2.5 (250 cycles). Raw sequence reads were processed by conducting quality control and read trimming analyses, and near WGSs (most had only partially resolved 5ʹ and 3ʹ terminal untranslated regions) were assembled within Geneious R10 v10.2.6 (Kearse et al. 2012) using the SPAdes v3.10.0 plugin (Bankevich et al. 2012).
3.3. Sequence alignment and manual concatenation
Multiple sequence RRV (186 taxa) and BFV (88 taxa) alignments were performed using MAFFT v7.450 (Katoh and Standley 2013) in Geneious R10 v10.2.6 (Kearse et al. 2012). The nt alignments were trimmed to remove 5ʹ and 3ʹ-terminal untranslated regions before screening for evidence of molecular recombination using the Recombination Detection Program (RDP v.4.97) employing the RDP, Bootscan, 3Seq, GENECONV, MaxChi, Chimaera, and SiScan methods to detect and characterize distinct recombination signals (Martin et al. 2015). The nt alignments were then further manually refined to remove the nsP4 3ʹ-terminal stop codons and noncoding 26S RNA promoter junction regions (RRV 47 nt and BFV 31 nt). For each of the individual RRV and BFV nt alignments, the remaining coding nonstructural and structural ORFs were then concatenated to create final RRV and BFV multiple sequence alignments.
3.4. Discrete-trait reconstruction analyses
To reconstruct the evolution of each of the signature convergent and single site aa mutations, we analyzed each independently assuming a Markovian process, otherwise known as discrete-trait reconstruction, which is also commonly used to model geographic movement (Huelsenbeck et al. 2003, Lemey et al. 2009, Faria et al. 2011). Under this method, we assume a nonsymmetric rate matrix to describe the probability of state changes for each site. The realization of the process produces the number of changes between states (aas), known as Markov jumps. To do this, we analyzed the data in BEAST1.10 (Suchard et al. 2018) under a fixed strict clock for the nt sequence alignment and an independent relaxed clock with an underlying lognormal distribution for each aa site in question. We fixed the nt molecular clock rate at 3.21 × 10−4 (RRV) and 2.11 × 10−4 (BFV) substitutions/site/year, as estimated previously (Michie et al. 2020a, 2020a) and used a constant-size coalescent tree prior, for statistical convenience. To sample from the posterior distribution, we used a Markov chain Monte Carlo of length 5 × 108 steps and ensured that the effective sample size for all parameters was at least 200 in Beastiary (Wirth et al. 2022). To determine statistical support for particular stage changes (i.e. changes from one aa to another), we determined the corresponding Bayes factor, the ratio of the posterior and prior odds for the inclusion of the corresponding transition rates (Lemey et al. 2009). For each convergent aa site, the individual MCC trees were constructed and visualized using FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) using their Markov rewards.
3.5. Analysis of selection pressures
The concatenated RRV and BFV multiple nt sequence alignments were further manually refined to remove the in-frame opal termination codon in the nsP3 gene. The nonstructural and structural ORFs were then analyzed for evidence of positive pervasive diversifying selection using the DataMonkey webserver (www.datamonkey.org/; Delport et al. 2010). DataMonkey employs the MEME (Murrell et al. 2012), the FEL, SLAC, and the FUBAR methods (Kosakovsky Pond and Frost 2005, Murrell et al. 2012). Codons were considered under positive selection if at least two methods provided support, namely, a P-value of < 0.05 with MEME and FEL methods, a P value of < 0.1 with the SLAC method, and a posterior probability of > 90% with the FUBAR method.
3.5. Cross-species selection pressure analyses of nsP1 and E3
3.5.1. ClonalFrameML analyses
Available complete coding sequences of GETV and MAYV were obtained from GenBank. The nsP1 and E3 sequences from BFV (n = 88), GETV (n = 76), MAYV (n = 71), and RRV (n = 186) were aligned using the codon-aware package pagan. We estimated a maximum likelihood phylogeny using phyML with arguments -d nt -p -b 0 -m HKY85 -f e -t e -v 0 -c 1 -a 2 -s BEST -o tlr—r_seed 0. We searched for evidence of homoplasy and recombination tracts using ClonalFrameML, assuming the tree topology and transition:transversion ratio estimated by phyML. For the homoplasy analysis, we used the -imputation only option. While there was widespread homoplasy, visual assessment did not reveal convincing evidence of recombination tracts, and the species were monophyletic in the phyML tree.
3.5.2. GenomegaMap genus-wide analyses
In the genus-wide analyses, all sequences of the nsP1 and E3 genes were analyzed together, regardless of their species-of-origin. We estimated variation in the dN/dS ratio (ω) assuming a sliding window model with mean window size 30 codons and an exponential prior distribution on ω with mean 1. We co-estimated the pairwise diversity (θ) and transition:transversion ratio (κ), assuming equal codon usage and improper log uniform priors on θ and κ. For each analysis, we ran two chains of Markov Chain Monte Carlo (MCMC) for 500 000 iterations. Chains were compared to assess convergence and merged after removing 250 000 iterations burn-in from each chain. We summarized the posterior distributions of θ and κ using the posterior median and (2.5%, 97.5%) quantiles. We summarized the posterior distribution of ω along the sequences using the posterior median and (10%, 90%) quantiles.
3.5.3. GenomegaMap within-species analyses
To assess evidence of convergent evolution across the four species, we repeated the GenomegaMap analysis of nsP1 and E3 genes, this time ignoring substitutions between species, while estimating common parameters (θ, κ, and ω) across the species using the same priors, MCMC strategy and posterior summaries as before.
3.6. Mapping of convergent RRV/BFV nsP1 mutations to CHIKV cryo-EM structures
The convergent nsP1 mutations in RRV (sites 248 and 441) and BFV (297 and 447) were mapped onto the predicted 3-dimensional cryo-EM crystal structure of the CHIKV nsP1 oligomer complex (Fig. 4) based on PDB code 7FGG (PDB DOI: 10.2210/pdb7FGG/pdb; Zhang et al. 2022), using Geneious Prime® 2022.1 software (http://www.geneious.com/).
Supplementary Material
Acknowledgements
We are sincerely grateful to Professor Edward Holmes, University of Sydney, for reading and providing insightful comments and suggestions for the manuscript. We also thank laboratory staff from Public Health Virology, Public and Environmental Health Reference Laboratories (PEHRL) Queensland Health, Berrimah Veterinary Labs, Department of Primary Industries and Fisheries, Darwin, NT, Department of Microbiology and Molecular Pathology, Sullivan and Nicolaides Pathology and Serology, Pathology Queensland Central Laboratory, Royal Brisbane and Women’s Hospital, QLD for assistance with specimen processing and transport.
Contributor Information
Alyssa T Pyke, Public Health Virology Laboratory, Public and Environmental Health Reference Laboratories, Department of Health, Queensland Government, P.O. Box 594, Archerfield, Coopers Plains, Queensland, Australia.
Daniel J Wilson, Big Data Institute, Oxford Population Health, University of Oxford, Li Ka Shing Centre for Health Information and Discovery, Old Road Campus, Oxford OX3 7LF, United Kingdom; Department for Continuing Education, University of Oxford, 1 Wellington Square, Oxford OX1 2JA, United Kingdom.
Alice Michie, School of Biomedical Sciences, University of Western Australia, 35 Stirling Highway, Perth, Western Australia 6009, Australia.
John S Mackenzie, Faculty of Health Sciences, Curtin University, G.P.O. Box U1987, Bentley, Western Australia 6845, Australia.
Allison Imrie, School of Biomedical Sciences, University of Western Australia, 35 Stirling Highway, Perth, Western Australia 6009, Australia.
Jane Cameron, Public Health Virology Laboratory, Public and Environmental Health Reference Laboratories, Department of Health, Queensland Government, P.O. Box 594, Archerfield, Coopers Plains, Queensland, Australia.
Stephen L Doggett, NSW Health Pathology, Westmead Hospital, 166-174 Hawkesbury Road Westmead, Sydney, New South Wales 2145, Australia.
John Haniotis, NSW Health Pathology, Westmead Hospital, 166-174 Hawkesbury Road Westmead, Sydney, New South Wales 2145, Australia.
Lara J Herrero, Gold Coast Campus, Institute for Glycomics, Griffith University, 1 Parklands Drive, Southport, Queensland 4215, Australia.
Leon Caly, Victorian Infectious Diseases Reference Laboratory, Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, 792 Elizabeth Street, Melbourne, Victoria 3000, Australia.
Stacey E Lynch, Agriculture Victoria Research, AgriBio, Centre for AgriBioscience, 5 Ring Road, Bundoora, Victoria 3083, Australia.
Peter T Mee, Agriculture Victoria Research, AgriBio, Centre for AgriBioscience, 5 Ring Road, Bundoora, Victoria 3083, Australia.
Eugene T Madzokere, Gold Coast Campus, Institute for Glycomics, Griffith University, 1 Parklands Drive, Southport, Queensland 4215, Australia.
Ana L Ramirez, College of Public Health, Medical and Veterinary Sciences, James Cook University, P.O. Box 6811, Cairns, Queensland 4870, Australia; Australian Institute of Tropical Health and Medicine, James Cook University, P.O. Box 6811, Cairns, Queensland 4870, Australia; The Jackson Laboratory, 10 Discovery Drive Connecticut, Farmington, CT 06032, United States of America.
Devina Paramitha, School of Chemistry and Molecular Biosciences, The University of Queensland, Bdg 68 Cooper Road, St. Lucia, Queensland 4072, Australia.
Jody Hobson-Peters, School of Chemistry and Molecular Biosciences, The University of Queensland, Bdg 68 Cooper Road, St. Lucia, Queensland 4072, Australia.
David W Smith, NSW Health Pathology, Westmead Hospital, 166-174 Hawkesbury Road Westmead, Sydney, New South Wales 2145, Australia; School of Medicine, University of Western Australia, 35 Stirling Highway, Perth, Western Australia 6009, Australia.
Richard Weir, Department of Primary Industries and Fisheries, Berrimah Veterinary Laboratory, P.O. Box 3000, Darwin, Northern Territory 0801, Australia.
Mitchell Sullivan, Public and Environmental Health Reference Laboratories, Department of Health, Queensland Government, P.O Box 594 Archerfield, Coopers Plains, Queensland 4108, Australia.
Julian Druce, Victorian Infectious Diseases Reference Laboratory, Royal Melbourne Hospital at the Peter Doherty Institute for Infection and Immunity, 792 Elizabeth Street, Melbourne, Victoria 3000, Australia.
Lorna Melville, Department of Primary Industries and Fisheries, Berrimah Veterinary Laboratory, P.O. Box 3000, Darwin, Northern Territory 0801, Australia.
Jennifer Robson, Department of Microbiology and Molecular Pathology, Sullivan Nicolaides Pathology, P.O. Box 2014 Fortitude Valley, Brisbane, Queensland 4006, Australia.
Robert Gibb, Serology, Pathology Queensland Central Laboratory, Royal Brisbane and Women’s Hospital, 40 Butterfield Street Herston, Brisbane, Queensland 4029, Australia.
Andrew F van den Hurk, Public Health Virology Laboratory, Public and Environmental Health Reference Laboratories, Department of Health, Queensland Government, P.O. Box 594, Archerfield, Coopers Plains, Queensland, Australia.
Sebastian Duchene, Department of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, 792 Elizabeth Street, Melbourne, Victoria 3000, Australia; Evolutionary Dynamics of Infectious Diseases, Department of Computational Biology, Institut Pasteur, 28 Rue du Dr Roux, Paris 75015, France.
Author contributions
Project administration: A.T.P.; conceptualization and methodology: A.T.P, S.D., and D.J.W.; sample procurement: A.T.P., A.F.v.d.H., S.L.D., J.H., S.E.L., P.T.M., A.L.R., D.P., J.H.P., R.W., L.M., J.R., and R.G.; sample analysis and data curation, A.T.P., S.D, D.J.W., and J.C.; initial draft writing and interpretation, A.T.P., S.L.D., and D.J.W.; manuscript review and input from A.F.v.d.H., J.S.M., A.L., A.M., S.L.D., S.E.L., M.S., L.C., L.J.H., E.T.M., and D.W.S.
Supplementary data
Supplementary data is available at VEVOLU Journal online.
Conflict of interest:
The authors declare that there are no conflicts of interest.
Funding
This work was supported by Queensland Health, Griffith University, and the Peter Doherty Institute for Infection and Immunity, Agriculture Victoria, the Victorian Department of Health, AgriBio, Centre for AgriBioscience, Bundoora, Victoria. D.J.W. is supported by a Big Data Institute Robertson Fellowship. L.J.H. is the recipient of the Australian National Health and Medical Research Council Career Development Award (ID: 105760). S.D. is supported by the Australian Research Council (FT220100629) and the Australian National 324Health and Medical Research Council (grant number 2017284).
Data availability
Virus sequences are available on the National Center for Biotechnology Information (NCBI) GenBank database at https://www.ncbi.nlm.nih.gov/genbank/.
References
- Aaskov JG, Mataika JU, Lawrence GW. et al. An epidemic of Ross River virus infection in Fiji, 1979. Am J Trop Med Hyg 1981;30:1053–9. doi: 10.4269/ajtmh.1981.30.1053 [DOI] [PubMed] [Google Scholar]
- Aaskov J, Williams L, Yu S. A candidate Ross River virus vaccine: preclinical evaluation. Vaccine 1997;15:1396–404. doi: 10.1016/S0264-410X(97)00051-0 [DOI] [PubMed] [Google Scholar]
- Alvarez-Carretero S, Kapli P, Yang Z. Beginner’s guide on the use of PAML to detect positive selection. Mol Biol Evol 2023:40: msad041. doi: 10.1093/molbev/msad041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam V. ReversibleMarkov decision processes and the Gaussian free field. arXiv 2022. doi: 10.48550/arXiv.2207.05217 [DOI] [Google Scholar]
- Azuolas JK, Knight PK, Evans DL. et al. Isolation of Ross River virus from mosquitoes and from horses with signs of musculo-skeletal disease. Aust Vet J 2003;81:344–47. doi: 10.1111/j.1751-0813.2003.tb11511.x [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. doi: 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton AJ, Bielefeldt-Ohmann H. Clinical presentation, progression, and management of five cases of Ross River virus infection in performance horses located in southeast Queensland: A longitudinal case series. J Equine Vet Sci 2017;51:34–40. doi: 10.1016/j.jevs.2016.12.010 [DOI] [Google Scholar]
- Basore K, Kim AS, Nelson CA. et al. Cryo-EM structure of chikungunya virus in complex with the Mxra8 receptor. Cell 2019;177:1725–1737e1716. doi: 10.1016/j.cell.2019.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly L, Horwood PF, Vijaykrishna D. et al. Divergent Barmah Forest virus from Papua New Guinea. Emerg Infect Dis 2019;25:2266–69. doi: 10.3201/eid2512.191070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claflin SB, Webb CE, Dutch RE. Ross River Virus: many vectors and unusual hosts make for an unpredictable pathogen. PLoS Pathog 2015;11:e1005070. doi: 10.1371/journal.ppat.1005070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delport W, Poon AF, Frost SD. et al. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010;26:2455–57. doi: 10.1093/bioinformatics/btq429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giallonardo F, Geoghegan JL, Docherty DE. et al. Fluid spatial dynamics of West Nile virus in the United States: rapid spread in a permissive host environment. J Virol 2016;90:862–72. doi: 10.1128/JVI.02305-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty RL, Carley JG, Kay BH. et al. Isolation of virus strains from mosquitoes collected in Queensland, 1972-1976. Aust J Exp Biol Med Sci 1979;57:509–20. doi: 10.1038/icb.1979.52 [DOI] [PubMed] [Google Scholar]
- Doherty RL, Whitehead RH, Gorman BM. et al. The isolation of a third group A arbovirus in Australia, with preliminary observations on its relationships to epidemic polyarthritis. Aust J Sci 1963;26:183–84. [Google Scholar]
- Duchene S, Di Giallonardo F, Holmes EC. Substitution model adequacy and assessing the reliability of estimates of virus evolutionary rates and time scales. Mol Biol Evol 2016;33:255–67. doi: 10.1093/molbev/msv207 [DOI] [PubMed] [Google Scholar]
- Engel D, Jost H, Wink M. et al. Reconstruction of the evolutionary history and dispersal of Usutu virus, a neglected emerging arbovirus in Europe and Africa. mBio 2016;7:e01938–01915. doi: 10.1128/mBio.01938-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria NR, Suchard MA, Rambaut A. et al. Toward a quantitative understanding of viral phylogeography. Curr Opin Virol 2011;1:423–29. doi: 10.1016/j.coviro.2011.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauran P, Donaldson M, Harper J. et al. Characterization of Ross River viruses isolated from patients with polyarthritis in New Caledonia and Wallis and Futuna Islands. Am J Trop Med Hyg 1984;33:1228–31. doi: 10.4269/ajtmh.1984.33.1228 [DOI] [PubMed] [Google Scholar]
- Firth AE, Chung BY, Fleeton MN. et al. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol J 2008;5:108. doi: 10.1186/1743-422X-5-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexman JP, Smith DW, Mackenzie JS. et al. A comparison of the diseases caused by Ross River virus and Barmah Forest virus. Med J Aust 1998;169:159–63. doi: 10.5694/j.1326-5377.1998.tb116019.x [DOI] [PubMed] [Google Scholar]
- Forrester NL, Palacios G, Tesh RB. et al. Genome-scale phylogeny of the Alphavirus genus suggests a marine origin. J Virol 2012;86:2729–38. doi: 10.1128/JVI.05591-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan JL, Holmes EC. The phylogenomics of evolving virus virulence. Nat Rev Genet 2018;19:756–69. doi: 10.1038/s41576-018-0055-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley D, Sleigh A, Ritchie S. Ross River virus transmission, infection, and disease: a cross- disciplinary review. Clin Microbiol Rev 2001;14:909–32. doi: 10.1128/CMR.14.4.909-932.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Nielsen R, Bollback JP. Stochastic mapping of morphological characters. Syst Biol 2003;52:131–58. doi: 10.1080/10635150390192780 [DOI] [PubMed] [Google Scholar]
- Jansen CC, Shivas MA, May FJ. et al. Epidemiologic, entomologic, and virologic factors of the 2014-15 Ross River virus outbreak, Queensland, Australia. Emerg Infect Dis 2019;25:2243–52. doi: 10.3201/eid2512.181810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A, Lowry K, Aaskov J. et al. Molecular evolutionary dynamics of Ross River virus and implications for vaccine efficacy. J Gen Virol 2010;91:182–88. doi: 10.1099/vir.0.014209-0 [DOI] [PubMed] [Google Scholar]
- Jones R, Bragagnolo G, Arranz R. et al. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2021;589:615–19. doi: 10.1038/s41586-020-3036-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain M, Skinner E, van den Hurk A. et al. Physiology and ecology combine to determine host and vector importance for Ross River virus. eLife 2021;10:e67018. doi: 10.7554/eLife.67018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. Bayes factors. J Amer Stat Assoc 1995;90:773–95. doi: 10.1080/01621459.1995.10476572 [DOI] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30:772–80. doi: 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A. et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012;28:1647–49. doi: 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005;21:2531–33. doi: 10.1093/bioinformatics/bti320 [DOI] [PubMed] [Google Scholar]
- Kovacikova K, Gorostiola GM, Jones R. et al. Structural insights into the mechanisms of action of functionally distinct classes of chikungunya virus nonstructural protein 1 inhibitors. Antimicrob Agents Chemother 2021;65:e0256620. doi: 10.1128/AAC.02566-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Drummond AJ. et al. Bayesian phylogeography finds its roots. PLoS Comput Biol 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Wang H, Liang G. Getah virus (Alphavirus): an emerging, spreading zoonotic virus. Pathogens 2022;11:945. doi: 10.3390/pathogens11080945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay M, Johansen C, Broom AK. et al. Emergence of Barmah Forest virus in Western Australia. Emerg Infect Dis 1995;1:22–26. doi: 10.3201/eid0101.950104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JS, Broom AK, Hall RA. et al. Arboviruses in the Australian region, 1990 to 1998. Commun Dis Intell 1998;22:93–100. [PubMed] [Google Scholar]
- Marshall ID, Woodroofe GM, Hirsch S. Viruses recovered from mosquitoes and wildlife serum collected in the Murray Valley of southeastern Australia, February 1974, during an epidemic of encephalitis. Aust J Exp Biol Med Sci 1982;60:457–70. doi: 10.1038/icb.1982.51 [DOI] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M. et al. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 2015;1:vev003. doi: 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavian C, Rife BD, Dollar JJ. et al. Emergence of recombinant Mayaro virus strains from the Amazon basin. Sci Rep 2017;7:8718. doi: 10.1038/s41598-017-07152-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie A, Dhanasekaran V, Lindsay MDA. et al. Genome-scale phylogeny and evolutionary analysis of Ross River virus reveals periodic sweeps of lineage dominance in Western Australia, 1977-2014. J Virol 2020a;94:e01234–19. doi: 10.1128/JVI.01234-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie A, Ernst T, Chua IJ. et al. Phylogenetic and timescale analysis of Barmah Forest virus as inferred from genome sequence analysis. Viruses 2020b;12:732. doi: 10.3390/v12070732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie A, Mackenzie JS, Smith DW. et al. Genome sequence analysis of first Ross River Virus isolate from Papua New Guinea indicates long-term, local evolution. Viruses 2021;13:482. doi: 10.3390/v13030482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Wertheim JO, Moola S. et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 2012;8:e1002764. doi: 10.1371/journal.pgen.1002764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- National notifiable diseases surveillance system (NNDSS) . Australian Government, Department of Health and Aged Care (DoHaAC). https://www.health.gov.au/resources/collections/nndss-fortnightly-reports(24 May 2024, date last accessed).
- Orf GS, Perez LJ, Meyer TV. et al. Purifying selection decreases the potential for Bangui orthobunyavirus outbreaks in humans. Virus Evol 2023;9:vead018. doi: 10.1093/ve/vead018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozden S, Lucas-Hourani M, Ceccaldi PE. et al. Inhibition of chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J Biol Chem 2008;283:21899–908. doi: 10.1074/jbc.M802444200 [DOI] [PubMed] [Google Scholar]
- Poidinger M, Roy S, Hall RA. et al. Genetic stability among temporally and geographically diverse isolates of Barmah Forest virus. Am J Trop Med Hyg 1997;57:230–34. doi: 10.4269/ajtmh.1997.57.230 [DOI] [PubMed] [Google Scholar]
- Pollett S, Melendrez MC, Maljkovic BI. et al. Understanding dengue virus evolution to support epidemic surveillance and counter-measure development. Infect Genet Evol 2018;62:279–95. doi: 10.1016/j.meegid.2018.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyke AT, Gunn W, Taylor C. et al. On the Home Front: Specialised reference testing for dengue in the Australasian region. Trop Med Infect Dis 2018;3:75. doi: 10.3390/tropicalmed3030075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyke AT, McMahon J, Burtonclay P. et al. Genome Sequences of chikungunya virus strains from Bangladesh and Thailand. Microbiol Resour Announc 2020;9:e01452–19. doi: 10.1128/MRA.01452-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen L, Gubler DJ, Bennett PH. Epidemic polyarthritis (Ross River) virus infection in the Cook Islands. Am J Trop Med Hyg 1981;30:1294–302. doi: 10.4269/ajtmh.1981.30.1294 [DOI] [PubMed] [Google Scholar]
- Russell RC. Ross River virus: ecology and distribution. Annul Rev Entomol 2002;47:1–31. doi: 10.1146/annurev.ento.47.091201.145100 [DOI] [PubMed] [Google Scholar]
- Salonen A, Ahola T, Kaariainen L. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 2005;285:139–73. doi: 10.1007/3-540-26764-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Bolton EE, Brister JR. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2022;50:D20–D26. doi: 10.1093/nar/gkab1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi M, Forni D, Clerici M. et al. Nonstructural proteins are preferential positive selection targets in Zika virus and related flaviviruses. PLoS Negl Trop Dis 2016;10:e0004978. doi: 10.1371/journal.pntd.0004978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit JM, Klimstra WB, Ryman KD. et al. PE2 cleavage mutants of Sindbis virus: correlation between viral infectivity and pH-dependent membrane fusion activation of the spike heterodimer. J Virol 2001;75:11196–204. doi: 10.1128/JVI.75.22.11196-11204.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Zhao Z, Chai Y. et al. Molecular basis of arthritogenic alphavirus receptor MXRA8 binding to chikungunya virus envelope protein. Cell 2019;177:1714–24. doi: 10.1016/j.cell.2019.04.008 [DOI] [PubMed] [Google Scholar]
- Stephenson EB, Peel AJ, Reid SA. et al. The non-human reservoirs of Ross River virus: a systematic review of the evidence. Parasit Vectors 2018;11:188. doi: 10.1186/s13071-018-2733-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stica CJ, Barrero RA, Murray RZ. et al. Global evolutionary history and dynamics of dengue viruses inferred from whole genome sequences. Viruses 2022;14:703. doi: 10.3390/v14040703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution [published erratum appears in Microbiol Rev 1994 Dec; 58:806]. Microbiol Rev 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard MA, Lemey P, Baele G. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 2018;4:vey016. doi: 10.1093/ve/vey016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesh RB, McLean RG, Shroyer DA. et al. Ross River virus (Togaviridae: Alphavirus) infection (epidemic polyarthritis) in American Samoa. Trans R Soc Trop Med Hyg 1981;75:426–31. doi: 10.1016/0035-9203(81)90112-7 [DOI] [PubMed] [Google Scholar]
- Weaver SC, Winegar R, Manger ID. et al. Alphaviruses: population genetics and determinants of emergence. Antiviral Res 2012;94:242–57. doi: 10.1016/j.antiviral.2012.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DJ, Consortium CR, Peto TEA. GenomegaMap: Within-species genome-wide dN/dS estimation from over 10,000 genomes. Mol Biol Evol 2020;37:2450–60. doi: 10.1093/molbev/msaa069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth W, Duchene S, Falush D. Real-time and remote MCMC trace inspection with Beastiary. Mol Biol Evol 2022;39:msac095. doi: 10.1093/molbev/msac095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wressnigg N, van der Velden MV, Portsmouth D. et al. An inactivated Ross River virus vaccine is well tolerated and immunogenic in an adult population in a randomized phase 3 trial. Clin Vaccine Immunol 2015;22:267–73. doi: 10.1128/CVI.00546-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen KY, Henning J, Eng MD. et al. Epidemiological study of multiple zoonotic mosquito-borne alphaviruses in horses in Queensland, Australia (2018-2020). Viruses 2022;14:1846. doi: 10.3390/v14091846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Law MCY, Nguyen TM. et al. Molecular basis of specific viral RNA recognition and 5ʹ-end capping by the chikungunya virus nsP1. Cell Rep 2022;40:111133. doi: 10.1016/j.celrep.2022.111133 [DOI] [PubMed] [Google Scholar]
- Zhang K, Law YS, Law MCY. et al. Structural insights into viral RNA capping and plasma membrane targeting by chikungunya virus nonstructural protein 1. Cell Host Microbe 2021;29:757–764.e3. doi: 10.1016/j.chom.2021.02.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Virus sequences are available on the National Center for Biotechnology Information (NCBI) GenBank database at https://www.ncbi.nlm.nih.gov/genbank/.