

Abstract
BCL-2 family members are known to be implicated in survival in numerous biological settings. Here, we provide evidence that in injury and repair processes in lungs, BCL-2 mainly acts to attenuate endoplasmic reticulum (ER) stress and limit extracellular matrix accumulation. Days after an intratracheal bleomycin challenge, mice lose a fraction of their alveolar type II epithelium from terminal ER stress driven by activation of the critical ER sensor and stress effector IRE1α. This fraction is dramatically increased by BCL-2 inhibition, because IRE1α activation is dependent on its physical association with the BCL-2–proapoptotic family member BAX, and we found BCL-2 to disrupt this association in vitro. In vivo, navitoclax (a BCL-2/BCL-xL inhibitor) given 15–21 days after bleomycin challenge evoked strong activation of IRE-1α in mesenchymal cells and markers of ER stress, but not apoptosis. Remarkably, after BCL-2 inhibition, bleomycin-exposed mice demonstrated persistent collagen accumulation at Day 42, compared with resolution in controls. Enhanced fibrosis proved to be due to the RNAase activity of IRE1α downregulating MRC2 mRNA and protein, a mediator of collagen turnover. The critical role of MRC2 was confirmed in precision-cut lung slice cultures of Day-42 lungs from bleomycin-exposed wild-type and MRC2 null mice. Soluble and tissue collagen accumulated in precision-cut lung slice cultures from navitoclax-treated, bleomycin-challenged mice compared with controls, in a manner nearly identical to that of challenged but untreated MRC2 null mice. Thus, apart from mitochondrial-based antiapoptosis, BCL-2 functions to attenuate ER stress responses, fostering tissue homeostasis and injury repair.
Keywords: Bcl-2, ER stress, fibrosis, fibroblast, alveolar epithelial cells
Clinical Relevance
This research sheds new light of the regulation of endoplasmic reticulum stress and it raises some caution of the use of BCL-2 inhibitors in fibrosis.
The BCL-2 protein family has been extensively studied in fibrotic lungs and as part of their well-known and established role as central regulator of the intrinsic mitochondria apoptosis pathway (1). Stress signals activate proapoptotic BH-3–only initiators (i.e., BIM, PUMA, and BID), which inhibit the antiapoptotic proteins (i.e., BCL-2, BCL-X, and BCL-W). This interaction between initiators and antiapoptotic proteins, in turn, allows the proapoptotic effectors (i.e., BAX, BAK, and BOK) that usually are maintained in an inactivated state by the antiapoptotic proteins to be activated, resulting in the mitochondrial outer membrane permeabilization initiating the caspase cascade. BCL-2/BCL-xL inhibitors such as navitoclax (ABT-263) have been used in multiple pulmonary fibrosis models to remove senescent cells and fibroblasts, resulting in the attenuation of fibrosis and improved lung function (2–7). The BCL-2 protein family has also additional lesser known functions that regulate other pathways; of interest to us, the adaptative endoplasmic reticulum (ER) stress response mediated by the ER transmembrane protein kinase and endoribonuclease inositol-requiring enzyme 1α (IRE1α) (8–10). It has been established that, in cells carrying the double knockout of BAX and BAK, IRE1α signaling was deficient (10). Subsequently, IRE1α activation was demonstrated to be dependent on the binding of BAX onto IRE1α in the cytoplasm. However, the potential role of BCL-2, known to bind BAX, in regulating IRE1α function is undefined.
ER stress has been implicated in the pathogenesis of pulmonary fibrosis in both animal models and humans with fibrosis, especially idiopathic pulmonary fibrosis (IPF) (11). Multiple mechanisms to activate an ER stress response in IPF have been invoked, including DNA damage responses to short telomeres, viral infections, hypoxia, and misfolded/mutated proteins accumulating in the ER (12). Genetic studies in animals and humans confirm a direct connection between ER stress and pulmonary fibrosis (13, 14). Accordingly, there is substantial evidence pointing to activation of the ER stress sensors PERK and IRE1α in the pathobiology of IPF (11, 15, 16). In IPF, the BCL-2 protein family has been studied mainly in relation to the sensitivity of alveolar epithelial cells (AECs) to apoptosis, thought at least in part to be due to unbalanced BCL-2 family member expression with relatively high levels of BAX and absent or low levels of BCL-2 (17, 18). However, no direct link between BCL-2 levels and IRE1α activity in human epithelial cells or fibrosis has been established. In mesenchymal cells, the expression of BCL-2 is reportedly higher than that of BAX, which is consistent with the concept of myofibroblasts being “resistant to apoptosis,” although whether this phenotype applies to all mesenchymal cells or only subpopulations remains to be defined (19). In this study, we elucidate a specific role for BCL-2 in ER stress by uncovering a BCL-2–dependent mechanism for defining the threshold of IRE1α activation and testing the functional impact in two phases of a fibrosis model, initial injury and subsequent repair.
Methods
Detailed methods are available elsewhere (see the data supplement).
In Vivo Studies
We used a mouse strain engineered by Shh-cre–mediated complete lung epithelial deletion of IRE1α (termed IRE1α, Shhcre), as described previously (20). Mrc2−/− mice that lack the collagen-binding domain were generated by T. H. Bugge and colleagues as previously described (21).
To induce pulmonary fibrosis, 8- to 10-week-old C57BL/6J, Mrc2−/−, and IRE1α−/−, Shhcre mice were given PBS or bleomycin (#B5507-15un; Millipore Sigma) at 2.4 U/kg body weight by oral aspiration. Injured mice were treated with navitoclax (ABT-263; catalog no. CT-A263; Chemietek) at 50 mg/kg in 10% EtOH, 20% polyethylene glycol 400, and 70% PHOSAL 40 (Sigma-Aldrich), or vehicle control. Animals were killed either 5, 21, or 42 days after the bleomycin challenge.
Animal experiments were conducted using a protocol approved by the University of California, San Francisco, Institutional Animal Care and Use Committee (Protocol AN109566).
TUNEL Staining
The TUNEL assay was used to detect apoptotic cells on 4-μm paraffin-embedded mouse lung sections according to the manufacturer’s protocol (catalog no. ab206386; Abcam).
Immunofluorescence
We fixed 7-μm cryosections in 4% paraformaldehyde and stained them with various antibodies and IgG isotype controls (see Table E1 in the data supplement). Sections were imaged for quantification on a Zeiss AxioImager.
Western Blot
Pulverized mouse lung or precision-cut lung slices (PCLSs) were lysed in RIPA buffer and analyzed by immunoblotting as previously described (22). (A list of antibodies is given in Table E1.) Densitometry was quantified using NIH ImageJ software. Concentrated conditioned media were blotted for collagen I.
XBP1 Splicing
XBP-1 primers (see Table E1) were used to amplify XBP-1 amplicons using 1/10 of the cDNA product. PCR fragments were resolved on 4% agarose gels, stained with SYBR Green and quantified by densitometry using NIH ImageJ.
Cell Culture
Human embryonic kidney 293 (HEK-293) and fetal CCL-171 fibroblast (MRC5) cells were purchased from ATCC and grown in Dulbecco’s modified Eagle medium supplemented with L-glutamine and 10% FBS (catalog no. SH30396.03; Hyclone Laboratories). HEK-293 cells were treated with DMSO (catalog no. D8418; Sigma), ABT-263 (navitoclax), or ABT-199 (venetoclax; catalog no. CT-A199; Chemietek). MRC5 cells were treated with bleomycin (0.1 U/ml) for 3 hours and then were exposed for 48 hours to navitoclax, venetoclax, and/or KIRA8 (1 μM; a gift from F.R.P., University of California, San Francisco).
Transfection of Plasmid or siRNA
HEK-293 cells with 80% confluency were transfected with IRE1α construct (23) and/or human Bcl-2/pcDNA3 (catalog no. 19279; Addgene,) plasmids using TurboFect transfection reagent (catalog no. R0531; Thermo Fisher Scientific) and were cultured in DMEM supplemented with L-glutamine and 10% FBS medium for 24 hours before lysis for coimmunoprecipitation. HEK-293 cells were transfected with siRNA using Lipofectamine RNAiMAX (catalog no. 13778-030; ThermoFisher). (For a list of siRNAs, see Table E3.)
Immunoprecipitation
HEK-293 cells were transfected with IRE1α and/or BCL-2 constructs, and after 24 hours, the cells were lysed as described previously (24). Clarified lysates were immunoprecipitated with antibody to BAX. The immunoprecipitates were then blotted for BAX, IRE1α, or BCL-2 (Table E1).
Gene Expression Analysis
RNA was extracted from cells or tissues using commercially available kits (ReliaPrep RNA Miniprep Systems, catalog no. Z6011; Promega). cDNA synthesis was performed using the iScript Reverse Transcription Supermix for RT-PCR (catalog no. 1708841; BioRad Laboratories). We performed a qPCR on the CFX Connect Real-Time PCR detection system (BioRad) using SsoAdvanced Universal SYBR Green Supermix (catalog no. 1,725,271; BioRad). (A list of primers is given in Table E2.) All assays were performed according to the manufacturer’s instructions.
Lung Digestion and FACS
Mouse lung pieces were prepared as previously described, and sorted mesenchymal cells (live CD31− CD45− Epcam−) and epithelial cells (live CD31− CD45− Epcam+) were collected (25). (A list of antibodies is given in Table E1).
Single-Cell RNA Sequencing and Analysis
Single-cell sequencing was performed on a 10× Chromium instrument (10× Genomics) at the Institute of Human Genetics, University of California, San Francisco, as described previously (26). In the vehicle-treated group, 8,784 cells were sequenced at the average depth of 62,250 reads per cell to identify 2,924 median genes per cells. In the navitoclax-treated group, 8,064 cells were sequenced at the average depth of 59,153 reads per cell to identify 3,084 median genes per cell. All raw data generated in this article are hosted at the National Center for Biotechnology Information Gene Expression Omnibus database with the accession number GSE244568. Identification of canonical pathways was performed using Ingenuity Pathway Analysis (Qiagen). (For further details, see the Supplemental Methods in the data supplement.)
Collagen Deposition
Hydroxyproline measurements were performed as previously described using the mouse left lobe (27). Paraffin-embedded lung sections were stained with picrosirius red stain as previously described (4).
PCLS Culture
Fresh lung tissues were obtained from PBS-treated or bleomycin-injured wild-type (WT) and Mrc2−/− mouse lungs treated with vehicle or navitoclax collected at Day 42 postinjury. PCLSs were prepared as previously described (22).
Results
Inhibition of BCL-2 during the Injury Phase of the Bleomycin Mouse Model Leads to Terminal ER Stress of Type II AECs (AEC2s), Resulting in Enhanced Apoptosis and Subsequent Fibrosis
BCL-2 and BCL-xL are known to play a crucial role in cell survival and are upregulated when damaged cells attempt to avoid apoptosis. We confirmed a significant increase in the expression levels of Bcl-2 and Bcl-xL mRNA in whole lungs within the first week after bleomycin injury (see Figure E1A). To dissect the role of BCL-2 in early bleomycin injury, mice were immediately treated with the BCL-2/BCL-xL inhibitor navitoclax (50 mg/kg) by means of gavage for 5 consecutive days (Figure 1A). Navitoclax-treated mice displayed more severe lung injury, as evidenced by pronounced weight loss and increased protein concentration in BALs, compared with vehicle-treated mice that underwent bleomycin injury (Figures E1B and E1C). The accentuated injury level was associated with an increased number of TUNEL+ cells (Figures 1B and E1D) and depletion AEC2s (Figures 1C and 1D). To assess the mechanism of enhanced injury by BCL-2/BCL-xL inhibition, we measured whole-lung cleaved caspase-12, a caspase specifically activated during terminal ER stress (28), and found cleavage to be enhanced in navitoclax-treated mice (Figure 1E). Consistent with higher epithelial ER stress, we observed increased splicing of the transcription factor XBP1, a direct measure of activation of the ER sensor and ER stress effector IRE1α, suggesting that AEC2 apoptosis and enhanced injury were triggered by this mechanism (Figure 1F). XBP1 splicing was not observed in mesenchymal cells (Figure E1E).
Figure 1.
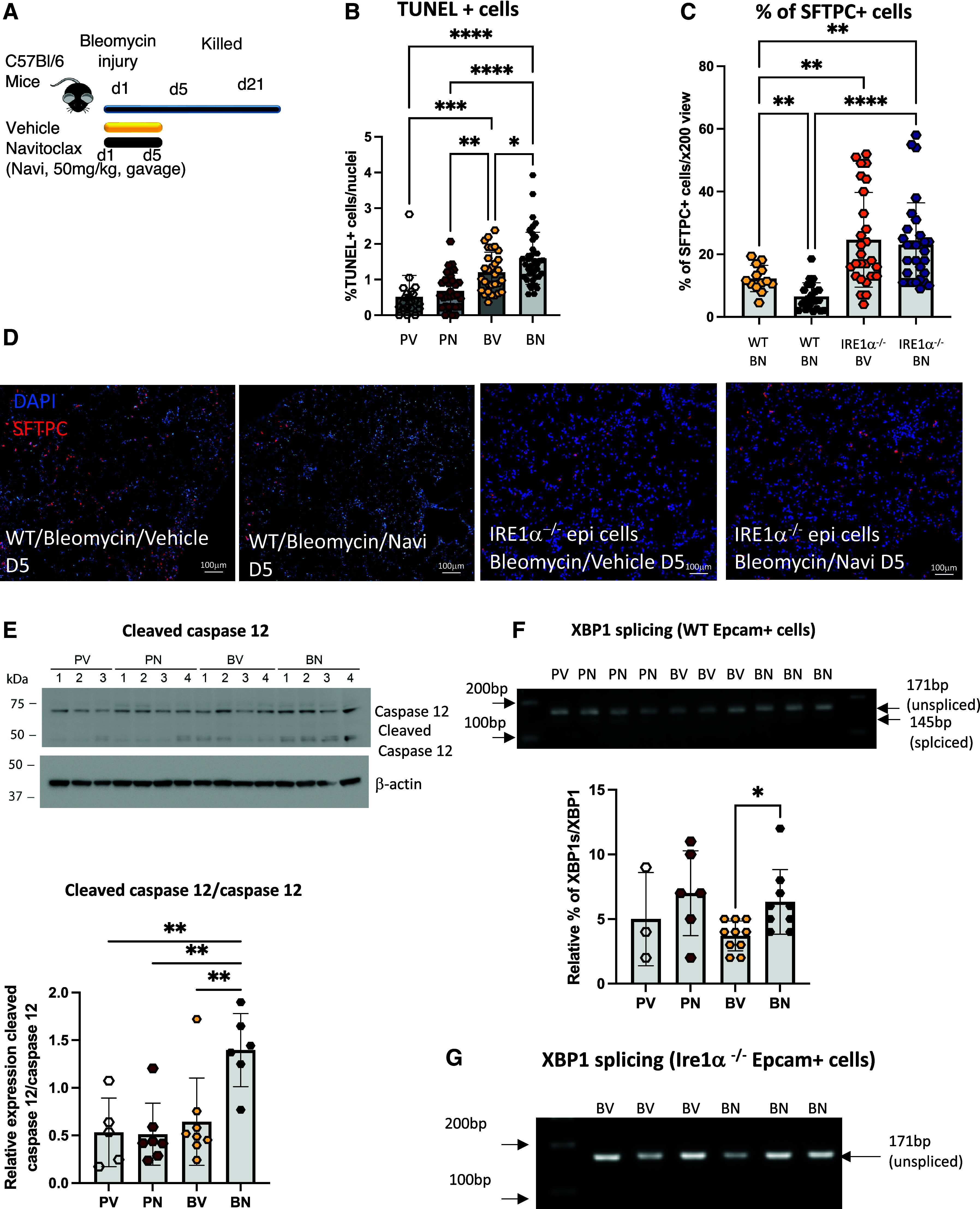
Inhibition of BCL-2/BCL-xL after bleomycin injury promotes an IRE1α-dependent loss of alveolar type II cells. (A) Schematic of bleomycin injury model and early navitoclax treatment. Navitoclax (Navi; 50 mg/kg) or vehicle was given by gavage for 5 consecutive days to the mice immediately after bleomycin injury. (B) Bar graph of the percentage of TUNEL-positive cells in PBS-treated (P) or bleomycin-injured (B) lungs after vehicle (V) or navitoclax (N) treatment. n = 3 mice per group. Bleomycin-injured (BN) wild-type (WT) C57BL/6J and Ire1α−/−Shhcre lungs after navitoclax treatment; BV = bleomycin-injured WT C57BL/6J and Ire1α−/−Shhcre lungs after vehicle treatment; PN = bleomycin-injured WT C57BL/6J and Ire1α−/−Shhcre lungs after navitoclax treatment; PV = bleomycin-injured WT C57BL/6J and Ire1α−/−Shhcre lungs after vehicle treatment. (C) Bar graph of the percentage of surfactant protein C–positive (SFTPC+) cells in BV and BN samples. n = 3 mice per group. (D) Representative images of SFTPC immunostaining of bleomycin-injured lungs treated with vehicle or navitoclax. Lungs were collected 5 days after bleomycin injury. Scale bars, 100 μm. (E) Western blot of caspase-12 and cleaved caspase-12 using whole WT mouse lungs collected 5 days after bleomycin injury and treatment with navitoclax. Bar graph representation of the relative level of expression of the ratio of cleaved caspase-12 to caspase-12. Each data point represents an individual mouse. (F) Representative ethidium bromide–stained agarose gel of XBP1 cDNA amplicons after vehicle or navitoclax treatment of PBS-treated or bleomycin-injured WT Epcam+ cells. Bar graph representation of the ratio of spliced amplicons to spliced plus unspliced amplicons. Each data point represents an individual mouse. (G) Representative ethidium bromide–stained agarose gel of XBP1 cDNA amplicons after navitoclax treatment of bleomycin-injured Ire1α−/−Shhcre Epcam+ cells. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 as determined by one-way ANOVA followed by Tukey multiple comparison test (B and E) or Dunnett’s multiple comparison (C and F). D = day; epi = epithelial; TUNEL = terminal deoxynucleotidyl transferase dUTP nick end labeling.
To solidify this point, we repeated the experiments using mice with epithelium-specific deletion of IRE1α (20). The deletion of epithelial IRE1α not only abrogated the hallmark features of IRE1α activation (Figure 1G) but also all measured elements of increased injury triggered by navitoclax exposure, preventing the loss of AEC2 numbers and the disruption of lung barrier integrity (Figures 1C, 1D, E1C, and E1F). These findings reveal a critical functional connection between BCL-2/BCL-xL, IRE1α, and ER stress, affecting not only AEC2 health but also alveolar barrier function in response to bleomycin.
As expected, exacerbating the injury through the loss of AECs, the activation of IRE1α promoted worsening of fibrosis 21 days after the bleomycin challenge (Figure E1G). In contrast to findings in the recovery phase after bleomycin injury (discussed later), there was no change by Day 21 in mRNA expression of Mrc2, one of the IRE1α–dependent proteins contributing to collagen resorption (Figure E1H).
BCL-2 Sequesters BAX to Modulate IRE1α Activation
The role of the BH3-protein family in the regulation of IRE1α activation has been documented (1). However, the contribution of BCL-2 in this mechanism is uncertain (9, 10, 29). To assess whether BCL-2 modulates IRE1α activation, HEK-293 cells were cotransfected with Ire1α and Bcl-2 constructs and treated with navitoclax, a potent BCL-2 family inhibitor that directly binds to BCL-2, preventing its interaction with proapoptotic BCL-2 family member partners such as BAX (30). Coimmunoprecipitation experiments using lysates from HEK-293 cells transfected with the Ire1α construct confirmed the previously established association of BCL-2 with BAX, as well as IRE1α with BAX, which was largely blocked by overexpression of BCL-2 (Figure 2A). In the presence of navitoclax, the reduced BCL-2 interaction with BAX is associated with an increase in the interaction of BAX with IRE1α. In HEK-293 cells cotransfected with IRE1α and BCL-2, 85% ± 10.2% of BAX protein was associated with BCL-2, resulting in a significant decrease of Xbp1 splicing (Figures 2A–2D). Blocking the BCL-2/BAX interaction with navitoclax (250 nM) resulted in only 15% ± 9% of BAX being associated with BCL-2 (Figures 2A and 2B) and dose-dependently increased Xbp1 splicing (Figure 2C). In addition, treatment with venetoclax, which specifically blocks BCL-2 activity, promoted IRE1α activation, as evidenced by increased XBP1 splicing (Figure 2D). Treatment with navitoclax or venetoclax did not promote apoptosis in HEK-293 cells (see Figure E2A).
Figure 2.
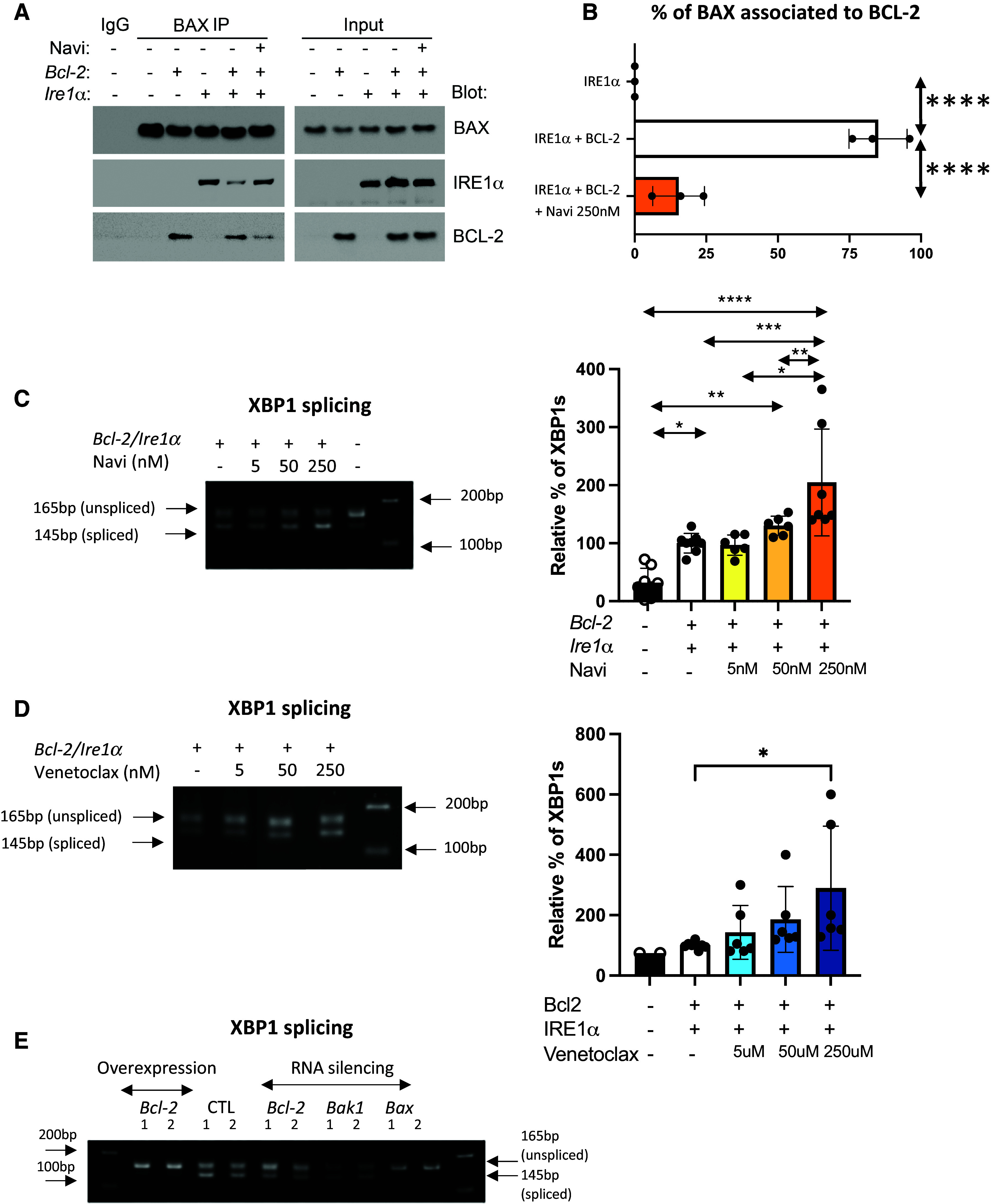
BCL-2 regulates IRE1α activation by sequestering BAX. (A) Coimmunoprecipitation of BAX with BCL-2 or IRE1α from human embryonic kidney 293 (HEK-293) cells cotransfected with BCL-2 and IRE1α plasmids followed by treatment with navitoclax (250 nM). BAX was immunoprecipitated (BAX IP), and BCL-2 and IRE1α were detected by western blot. n = 3 biological replicates. (B) Quantification of the intensity of bands, expressed as percentage of BAX associated with BCL-2. (C and D) Representative ethidium bromide–stained agarose gel of Xbp1 cDNA amplicons after transfection of Bcl-2 and pcDNA3 and Ire1α plasmid in HEK-293 cells followed by treatment with (C) navitoclax (5, 50, and 250 nM) or (D) venetoclax (5, 50, and 250 nM) for 24 hours. n = 6 (2 technical replicates in 3 biological replicates). Bar graphs indicate the percentage of spliced-form amplicons over spliced plus unspliced amplicons in samples transfected with Bcl-2 and pcDNA3 and Ire1α plasmid (control). The percentage of Xbp1 splicing in each control sample per replicate is the reference and equals 100. n = 6 (2 technical replicates in 3 biological replicates). (E) Representative ethidium bromide–stained agarose gel of Xbp1 cDNA amplicons after transfection of siRNA targeting Bcl-2, Bax, and Bak-1 in HEK-293 cells. n = 6 (2 technical replicates in 3 biological replicates). All data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, as determined by one-way ANOVA followed by Tukey multiple comparison test in (B–D).
It is important to note that the baseline activation of endogenous IRE1α as indicated by splicing of Xbp1 mRNA in HEK-293 (control) was significantly prevented by the overexpression of Bcl-2 as measured by the increased expression of Bcl-2 mRNA (Figures E2B–E2D). Using siRNA targeting Bax in HEK-293 cells limited the activation of IRE1α as measured by reduced level of spliced Xbp1 mRNA (Figure 2E). Silencing the expression of Bcl-2 or Bak-1 did not affect IRE1α activation compared with baseline, perhaps because of low baseline expression in HEK-293 cells (https://www.proteinatlas.org/ENSG00000087088-BAX/cell+line) (Figure 1E). Our data confirmed that BAX is necessary for IRE1α activation and that BCL-2 attenuates IRE1α activation by functionally sequestering BAX.
Inhibition of BCL-2 during the Late Fibrotic Phase of the Bleomycin Model Promotes ER Stress in Fibroblasts
Fibrobasts in fibrotic environment are known to evade apoptosis due to high level of BCL-2 family protein expression (31). To explore the potential interaction between ER stress and the BCL-2 protein family in fibroblasts, injured mice were treated with navitoclax for 5 consecutive days from Day 15 to Day 20 post–bleomycin challenge (Figure 3A). At Day 15, before the first dose of navitoclax, Bcl-2, but not Bcl-xl, mRNA levels were significantly elevated in the mesenchymal cells isolated from bleomycin-injured lung compared with the mesenchymal cells from naive mice (Figures 3B and E3A). In contrast, Bcl-2 mRNA levels were similar in the epithelial cells isolated from naive and bleomycin-injured mice (Figure 3B). Surprisingly, no increase in cleaved caspase-3–positive cells were detected, implying that no significant number of cells underwent apoptosis (Figure E3B). Furthermore, AEC2 numbers were unaffected by navitoclax treatment (Figure E3C). In addition, no change in the BAL protein concentration was measured between bleomycin-injured mice treated with navitoclax and those treated with vehicle (Figure E3D).
Figure 3.
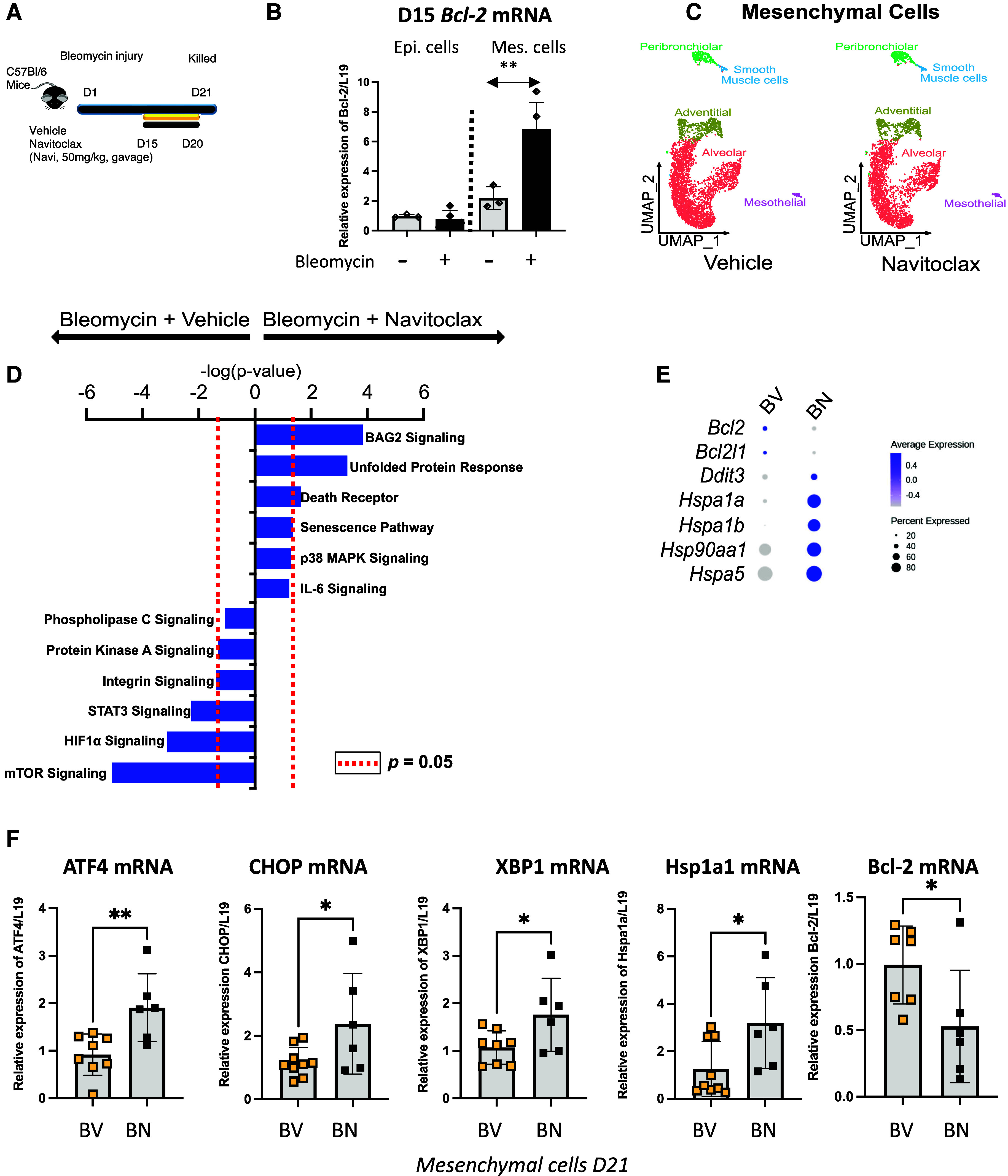
Inhibition of BCL-2 and BCL-xL during the late fibrotic and resolution phase of the bleomycin mouse model activates the unfolded protein response (UPR) in the mesenchymal cells. (A) Schematic of bleomycin injury model and late treatment with navitoclax. Vehicle or navitoclax (50 mg/kg) was given by gavage to the mice 15 days after bleomycin injury and for 5 consecutive days. (B) Bcl-2 mRNA expression 15 days after bleomycin injury measured in flow-sorted epithelial and mesenchymal cells. n = 3 mice. (C) UMAP of mesenchymal cells isolated 21 days post–bleomycin injury and treated with vehicle or navitoclax. (D) Top enriched canonical pathways identified using ingenuity pathway analysis on the differentially expressed genes in the alveolar fibroblast cluster between vehicle- and navitoclax-treated samples. (E) Dot plot of differentially expressed genes in mesenchymal cells isolated from BV or BN samples selected for their relevance to UPR. The expression levels of genes were indicated by the color and size of the circle. (F) Validation in an independent cohort of navitoclax-treated bleomycin-injured mice of the reduced expression of Bcl-2 mRNA after treatment and activation of expression of genes encoding key players of UPR in flow-sorted mesenchymal cells. n = 6–9 mice per group. Data represent mean ± SD. *P < 0.05, **P < 0.01, and NS = not statistically significant, as determined by unpaired two-tailed t test. Each data point represents one mouse sample. Epi. = Epcam+cells; Mes. = mesenchymal cells; UMAP = uniform manifold approximation and projection.
We performed single-cell RNA sequencing of CD45−/CD31−/EpCAM− (mesenchymal) cells and CD45−/CD31−/EpCAM+ (epithelial) cells isolated from mouse lungs treated with vehicle or navitoclax 21 days after bleomycin injury (see Figures E4A and E4B in the online supplement). We analyzed mesenchymal cells separately to identify heterogenous cell populations on the basis of the markers described by Tuskui and colleagues (32) (Figure E4C). Likewise, we also analyzed the epithelial cells separately and identified different cell populations on the basis of canonical markers (33) (Figure E4D). The distribution of the cells in each subset clearly indicates that navitoclax treatment had no effect on the proportion of different mesenchymal and epithelial cell types (Figures E4E and E4F), in line with our cleaved caspase-3 staining (Figures E3B and E3C). We focused our analysis on the mesenchymal cells because of their elevated Bcl-2 mRNA expression (Figures 3B and 3C) and performed an ingenuity pathway analysis (Figure 3D). Navitoclax-treated mesenchymal cells had significant upregulation of several signaling pathways, including Bag2, Unfolded Protein Response (UPR), and Death Receptor signaling, and downregulation of mTOR signaling, suggesting that BCL-2 inhibition after bleomycin injury contributed to increased pathologic stress signaling (Figure 3D). Consistent with this observation, we found that Bcl-2 and Bcl2-l1 transcripts were downregulated and that transcripts of heat shock proteins involved in UPR were upregulated across all mesenchymal cell clusters after BCL-2/BCL-xL inhibition (Figure 3E). We confirmed in flow-sorted mesenchymal cells from bleomycin-injured and navitoclax-treated mice that BCL-2/BCL-xL inhibition upregulated transcripts of UPR-associated genes such as Atf4, Hspa5 (CHOP), Xbp1, and Hspa1a and downregulated Bcl-2 mRNA (Figure 3F). Expression of Atf4, Hspa5 (CHOP), Ddit3 (BIP), Xbp1, and Bcl-2 was not affected in epithelial cells (see Figure E5). Collectively, these results indicate that the inhibition of BCL-2/BCL-xL in the late phase of the bleomycin mouse model did not affect cell survival but, instead, promoted stress signaling, including the UPR/ER stress response specifically in the mesenchymal cells.
Increased ER Stress during the Late Fibrotic Phase of Bleomycin Leads to Lack of Fibrosis Resolution
To determine whether the activation of ER stress responses in mesenchymal cells affected the fibrotic process, we collected lung samples at the peak of collagen deposition (21 days after bleomycin injury) or at the time when some resolution should have occurred (42 days after bleomycin injury) (see Figure E6A). The upregulation of collagen content assessed by hydroxyproline measurement was similar in vehicle- and navitoclax-treated mice 21 days after bleomycin injury (Figure 4A). However, 42 days after bleomycin injury, we measured a reduction in collagen content only in the vehicle-treated injured mice. Using picrosirius red staining, we confirmed that inhibition of BCL-2/BCL-xL led to persistent excess collagen accumulation (Figure 4B). We did not measure a statistically significant difference in the expression of Col1a2 mRNA in the samples collected 42 days after bleomycin injury (Figure E6B), indicating that the excess of collagen deposition was not associated with a difference in collagen expression at that time. We next examined whether Mrc2 mRNA expression, which encodes a receptor (also known as uPARAP, or Endo180) that binds collagen, is critical to cell-mediated collagen internalization during fibrosis resolution (34). Mrc2 mRNA expression was significantly upregulated in the bleomycin-injured, vehicle-treated mice, consistent with its role in collagen internalization during normal fibrosis resolution (Figure 4C). This upregulation was abolished after navitoclax treatment (Figure 4C), which corresponded to our observed lack of fibrosis resolution (Figures 4A and 4B). Furthermore, in PCLSs of WT lungs treated with vehicle or navitoclax collected 42 days after bleomycin injury, we measured, by western blot, a significant reduction in MRC2 expression and increased Collagen 1 expression in the navitoclax-treated samples (Figures E6C and E6D). Large amounts of soluble Collagen 1, presumably partially degraded, were only observed in the supernatant of the WT bleomycin-injured, navitoclax-treated PCLS cultures when we controlled for total amount of protein (Figures 4D and 4F). When we used MRC2 null mice lungs for PCLS, the amount of soluble collagen measured was increased after bleomycin injury but not further affected by navitoclax treatment. The total amount of protein in the concentrated condition media was comparable among all samples (Figure E6E). It is interesting that the amount of soluble collagen accumulated in PCLS cultures from bleomycin-injured, navitoclax-treated WT mice was nearly identical to that of bleomycin-injured, vehicle-treated MRC2 null mice (Figures 4E and 4F).
Figure 4.
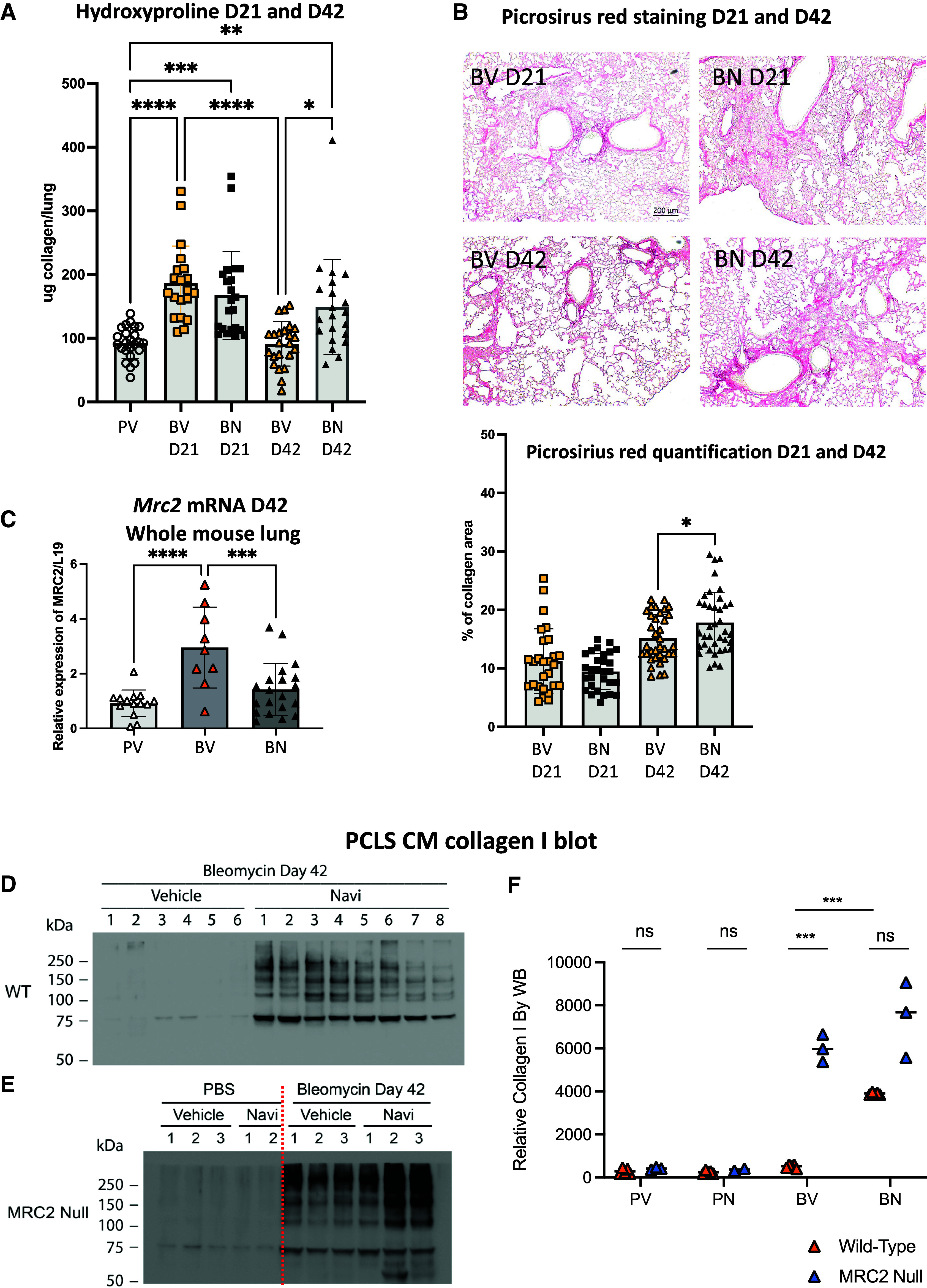
Inhibition of BCL-2 and BCL-xL during the late fibrotic/resolution phase of the bleomycin mouse model averts collagen resolution. (A) Hydroxyproline analysis of lung tissues from naive and injured mice treated with vehicle or navitoclax collected 21 and 42 days post–bleomycin injury. Samples were collected from three independent cohorts. (B) Representative images of picrosirus red–stained tissue sections collected 21 and 42 days after bleomycin injury and treated with vehicle or navitoclax from Day 15 to Day 20. Bar graph represents the quantification of collagen deposition performed using ImageJ software. Scale bars, 200 μm. (C) Level of expression of Mrc2 mRNA measured by qPCR in mesenchymal cells isolated from naive or injured lungs 42 days post–bleomycin injury treated with vehicle or navitoclax from Day 15 to Day 20. (D–F) Representative of western blot of collagen I obtained from conditioned media of precision-cut lung slices (PCLS CM) from bleomycin-injured (D) WT and (E) Mrc2−/− samples. n = 2–8 mice per group. (F) Dot plot representation of the relative level of expression of collagen I. Data represent mean ± SD. *P < 0.05, **P < 0.01, ****P < 0.0001, and ns significant as determined by one-way ANOVA followed by Tukey multiple comparison test in (A) and (B) or unpaired two-tailed t test in (F). Each data point represents one mouse sample. Because of a limitation in the availability of Mrc2−/− mice, only 2 mice were included in the PBS treated with Navi group (PV) to favor n = 3 in the BV and BN groups.
Collectively, these data suggest that activation of UPR/ER stress in the mesenchymal cells results in the lack of fibrosis resolution measured in the bleomycin-injured lungs after navitoclax because of the failed upregulation of Mrc2 expression compromising fibroblast uptake of partially degraded Collagen 1. Presumably, the persistence of Collagen 1 in the Day-42 PCLS analysis is reflected in the higher hydroxyproline levels in whole lungs at Day 42 of navitoclax-treated mice.
In Vitro Studies to Determine the Relationship between BCL-2 Inhibition, ER Stress, and Fibrosis Resolution
We next sought to confirm that UPR activation directly leads to Mrc2 expression regulation in mesenchymal cells. Under remedial ER stress levels, IRE1α catalyzes the cleavage and splicing of the mRNA encoding XBP1, a transcription factor that upregulates factors that promote a return to protein-folding homeostasis. In addition, activated IRE1α induces rapid degradation of mRNAs encoding secretory proteins and microRNAs, called “regulated IRE1α-dependent decay” (RIDD). Previous studies identified Mrc2 as a RIDD target of IRE1α (23, 35).
Therefore, we tested whether UPR downregulates Mrc2 in mesenchymal cells. We treated MRC5 fibroblasts with bleomycin to induce an injury level without inducing apoptosis and subsequently treated them with navitoclax to inhibit BCL-2 interactions with BAX. We first ascertained the activation of IRE1α by measuring the presence of spliced Xbp1 mRNA form in our MRC5 in vitro model and then measured the expression of Mrc2 mRNA (Figure 5A). In cells with documented IRE1α activation, the level of Mrc2 mRNA was reduced (Figures 5B and 5C). We also confirmed the role of Bcl-2 in the activation of UPR by exposing MRC5 cells to venetoclax after bleomycin injury and measuring the upregulation of transcripts of UPR-associated genes such as Atf4, Hspa5 (CHOP), and Xbp1 (Figure 5D). We detected that Mrc2 downregulation was IRE1α dependent, using KIRA8, an imidazopyrazine compound that binds the IRE1α kinase to allosterically inhibit its ribonuclease activity (36, 37). KIRA8 abolished Xbp1 splicing and rescued Mrc2 mRNA expression to levels similar to those of untreated, uninjured MRC5 cells (Figures 5B and 5C). These data confirm that the inhibition of BCL-2 and BCL-xL leads to the activation of IRE1α, resulting in the downregulation of Mrc2 mRNA (Figure 5E).
Figure 5.
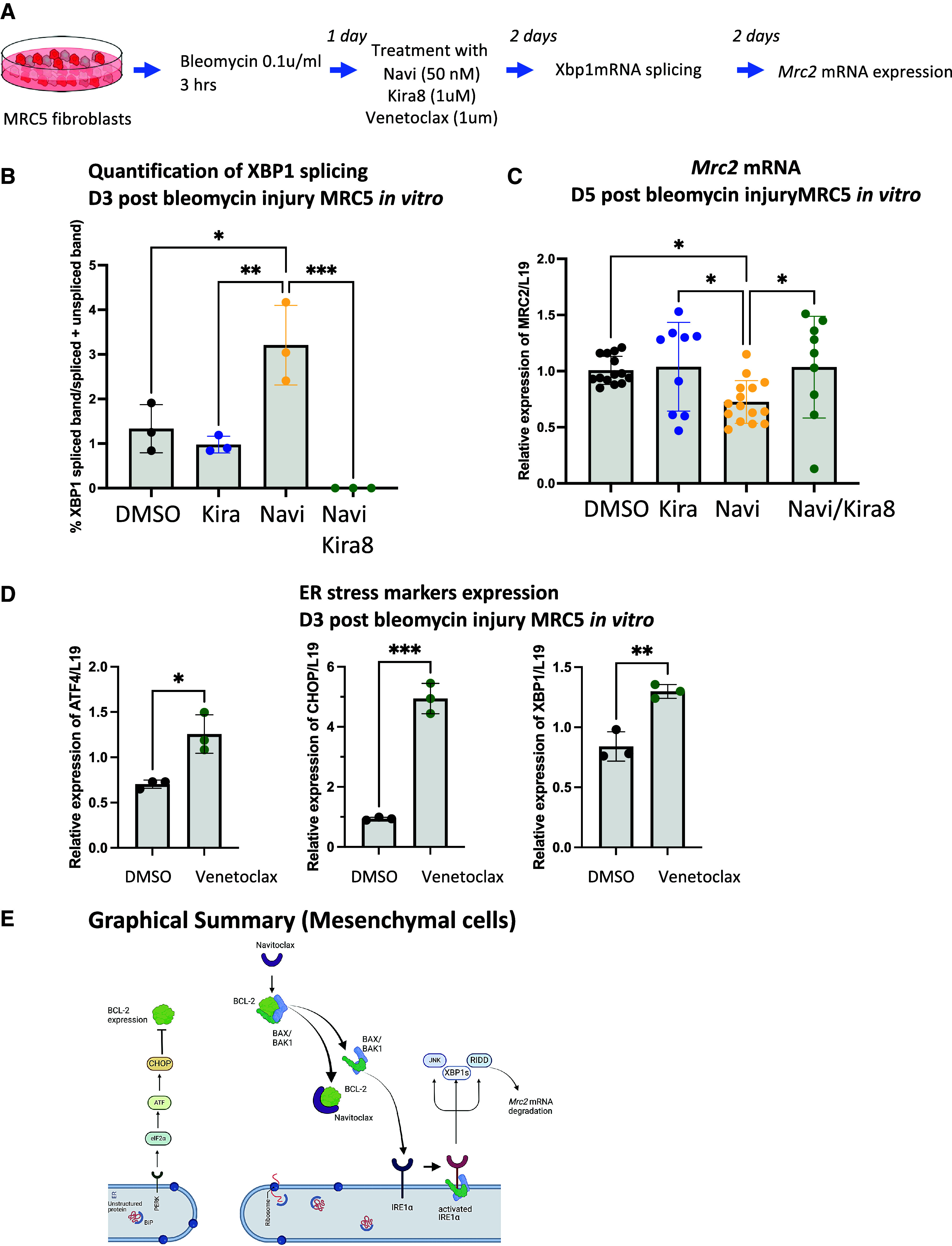
Activation of IRE1α by inhibition of BCL-2 results in MRC2 expression in vitro. (A) Schematic of in vitro model developed to test BCL-2 inhibition, IRE1α activation, and expression of the Mrc2 gene. (B) Bar graph representation of the ratio of spliced amplicons to (spliced plus unspliced amplicons measured in bleomycin-injured MRC5 fibroblasts treated with navitoclax (50 nM) and/or Kira8 (1 μM). N = 3 biological replicates. (C) Level of expression of Mrc2 mRNA in bleomycin-treated MRC5 fibroblasts treated with navitoclax (50 nM) and/or Kira8 (1 μM). n = at least 3 technical triplicates in 3 biological replicates. (D) Bar graph representation of the level of expression of the transcript of UPR-associated genes measured in bleomycin-injured MRC5 fibroblasts treated with venetoclax (1 μM). n = 3 biological replicates. (E) Schematic summary of the sequestering role of BCL-2 of BAX, resulting in the activation of IRE1α activation and lack of collagen resolution was prepared using Biorender. Data represent mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, as determined by one-way ANOVA followed by Tukey multiple comparison test (B), Holm-Šídák’s multiple comparison test (C), or unpaired two-tailed t test (D). ER = endoplasmic reticulum; RIDD = regulated IRE1α-dependent decay.
Discussion
The studies reported here, as well as previous research, indicate that suppressing BCL-2 and BCL-xL activity in mice through the injection of the BCL-2 and BCL-xL binding agent ABT-263 (navitoclax) leads to stress responses in mouse lungs. However, these responses depend on the timing of exposure after bleomycin injury and can be separated into three distinct phases: an early phase, the early fibrotic phase (Days 7–14), and the later fibrotic phase (Days 14–28).
In an early phase, the genotoxic stress caused by bleomycin predominantly promotes epithelial apoptosis and cell loss, which is partially offset by the rise in BCL-2 levels. However, the exposure to the BCL-2 and BCL-xL binding agent navitoclax at this stage exacerbates IRE1α–dependent terminal ER stress, resulting in worse epithelial cell death. This is evidenced by the activation of caspase-12; aggravated lung injury; and, ultimately, increased fibrosis (Figures 1 and E1). The significance of the interplay between BCL-2 levels and IRE1α activation, even in the absence of BCL-2 and BCL-xL inhibition, is underscored by the reduction in AEC2 numbers during this early phase, a phenomenon that is completely reversed by the deletion of epithelial IRE1α (Figure 1C).
In the early fibrotic phase (Days 7–14), epithelial cells appear to have largely recovered or been replenished. Mesenchymal and senescent cells are expanding during this phase but are now susceptible to proapoptotic signaling and apoptosis unless relatively high BCL-2 levels sustain their survival and permit the typical progression of the fibrotic responses. Apoptosis induced by BCL-2 and BCL-xL inhibition at this stage is commonly thought to be due to mitochondrial proapoptotic signaling (38). Because mitochondrial UPR activation and dysfunction are reported as a potential consequence of ER stress, it remains to be determined whether the BCL-2–regulated terminal ER stress pathway identified here contributes to mitochondrial dysfunction, further promoting apoptosis (16, 39).
In the later fibrotic phase (Days 14–28), progressive collagen deposition and loss of lung function are measured. During this stage, neither epithelial nor mesenchymal cells are susceptible to BCL-mediated apoptosis. Instead, a decrease in BCL-2 and BCL-xL activity from navitoclax exposure and a potential downregulation of Bcl-2 expression promote the activation of the unfolded protein response (UPR) and, in particular, IRE1α in mesenchymal cells (Figures 3, E3, and E4) (40). This, in turn, induces the downregulation of Mrc2 gene expression, a target of RIDD (23, 35), and persistence of matrix collagen during what is normally a time of slow collagen resorption and return of homeostasis, as detailed in our findings here (Figures 4 and E6). The microenvironment and outcome at this stage parallel the low levels of lung MRC2, slow collagen resorption, and prolonged fibrosis previously reported in aged lungs with or without injury (41).
The significance of ER stress and the UPR in the development of fibrotic diseases, particularly in IPF, is well documented (12, 16). In familial forms of IPF, genetic mutations in surfactant proteins and increased apoptosis in AEC2s have been identified as contributing factors. The ER stress or UPR signaling pathway consists of three arms: PERK/ATF4/CHOP, ATF6, and IRE1α. These arms are activated in an attempt to restore cellular homeostasis and promote cell survival (42). However, when these efforts fail, terminal ER stress and apoptosis become evident. The determinants of whether activation of IRE1α leads primarily to apoptosis or a transcriptional response intended to rescue the cells from stress and a return to normal are complex, involving both ER stress signaling and crosstalk with mitochondrial function (16, 39, 43). A critical aspect of IRE1α activation is its graded response to activating signals, which seems, at a molecular level, at least partly dependent on the degree of oligomerization among active IRE1α monomers that, in turn, is a function of the strength of activating signals on the cell surface (44, 45). At some point that is not completely defined, extensive oligomerization leads to the assembly of proapoptotic complexes in the cytoplasm, caspase-12 cleavage, and terminal ER stress–dependent apoptosis (45). It is thought that a sudden or prolonged increase in IRE1α and PERK activation favors apoptosis (43). However, short of such oligomerization, activated IRE1α dimers suppress the translation of proteins that could further stress the cells through activation of the XBP1 transcriptional factor and RIDD-dependent mRNA degradation. Each of the ER stress arms is intricately linked to mitochondrial biology and the BCL-2 protein family, which can enhance susceptibility to apoptosis (46). Indeed, it has been established that bcl-2 mRNA expression is dependent on CHOP and that, on activation, JNK (IRE1α pathway) phosphorylates BCL-2, leading to its inhibition (40, 47, 48). ER stress alone can initiate proapoptotic signaling, with caspase-12 serving as a specific marker (28). However, the activation of these pathways is highly dependent on the cell type and the context in which they occur. Previous data have suggested that ER stress can promote apoptosis in epithelial cells, wherea in fibroblasts, it alters differentiation, promoting fibrosis rather than apoptosis as the primary outcome, as observed in our study (48, 49).
The beneficial effects on fibrosis observed by several groups—including ours, using senolytic agents in mice after lung injury to remove senescent mesenchymal cells—have led to the concept of similar agents as therapeutics in human fibrotic disorders, especially IPF (2–6, 50). The most common preclinical approach has been a BCL-2 binding agent used during the early fibrotic phase after injury (the second phase, described earlier), although other apoptosis-promoting agents have also been reported with similar results (3, 7). The findings reported here suggest some caution in this approach. Human lung structural cells subjected to repetitive injury or activation could potentially transit through the phases highlighted here by the distinct responses to BCL-2 and BCL-xL in lungs of bleomycin-treated mice. As reported here, the exact vulnerability of mesenchymal cells to apoptosis is uncertain and likely dynamic. Apoptosis of human alveolar epithelial lining cells or induction of IRE1α activation and the UPR, instead of apoptosis, could also worsen rather than improve lung function.
In summary, these studies reveal—for the first time, to our knowledge—the modulatory role of BCL-2 in the UPR by its interaction with BAX to suppress IRE1α activation in both mesenchymal and epithelial cells in vivo. In vitro studies confirmed that the suppression of IRE1α activation in fibroblasts is associated with reduced collagen accumulation through the enhanced expression of the Mrc2 gene. Thus, apart from its mitochondrial antiapoptotic role, BCL-2 functions in the cytoplasm to attenuate ER stress responses, fostering tissue homeostasis and injury repair.
Footnotes
Supported by NIH grants R01HL44712, U01HL134766, and R35HL150767 (to H.A.C.), K99HL155785 (to J.J.K.), K08HL157654 (to V.C.A.), and R01HL145037 (to F.R.P.).
Author Contributions: C.J.L.S. codesigned and performed some experiments, analyzed the data, and wrote the manuscript. T.C.H. and A.M.B. performed most of the experiments. J.J.K. aided in the bioinformatics analysis and interpretation of results. Y.W. performed and analyzed all the biochemistry experiments. K.G. performed the picrosirius staining and quantification. J.-W.B.H. assisted with the effect of navitoclax on fibroblasts. V.C.A., F.R.P., and K.A. provided important reagents and conceptual inputs. H.A.C. aided in design of the study, analysis of the data, and writing of the manuscript. All authors contributed to editing the manuscript.
This article has a data supplement, which is accessible at the Supplements tab.
Originally Published in Press as DOI: 10.1165/rcmb.2023-0109OC on December 20, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ . 2018;25:65–80. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun . 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys . 2017;99:353–361. doi: 10.1016/j.ijrobp.2017.02.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiley CD, Brumwell AN, Davis SS, Jackson JR, Valdovinos A, Calhoun C, et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight . 2019;4:e130056. doi: 10.1172/jci.insight.130056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J . 2017;50:1602367. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He Y, Li F, Zhang C, Geng X, Syeda MZ, Du X, et al. Therapeutic effects of the Bcl-2 inhibitor on bleomycin-induced pulmonary fibrosis in mice. Front Mol Biosci . 2021;8:645846. doi: 10.3389/fmolb.2021.645846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cooley JC, Javkhlan N, Wilson JA, Foster DG, Edelman BL, Ortiz LA, et al. Inhibition of antiapoptotic BCL-2 proteins with ABT-263 induces fibroblast apoptosis, reversing persistent pulmonary fibrosis. JCI Insight . 2023;8:e163762. doi: 10.1172/jci.insight.163762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szegezdi E, Macdonald DC, Ní Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol . 2009;296:C941–C953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 9. Rodriguez DA, Zamorano S, Lisbona F, Rojas-Rivera D, Urra H, Cubillos-Ruiz JR, et al. BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1α. EMBO J . 2012;31:2322–2335. doi: 10.1038/emboj.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science . 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 11. Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med . 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2012;302:L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA . 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thamsen M, Ghosh R, Auyeung VC, Brumwell A, Chapman HA, Backes BJ, et al. Small molecule inhibition of IRE1α kinase/RNase has anti-fibrotic effects in the lung. PLoS One . 2019;14:e0209824. doi: 10.1371/journal.pone.0209824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe S, Markov NS, Lu Z, Piseaux Aillon R, Soberanes S, Runyan CE, et al. Resetting proteostasis with ISRIB promotes epithelial differentiation to attenuate pulmonary fibrosis. Proc Natl Acad Sci USA . 2021;118:118. doi: 10.1073/pnas.2101100118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest . 2018;128:64–73. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuwano K, Hagimoto N, Tanaka T, Kawasaki M, Kunitake R, Miyazaki H, et al. Expression of apoptosis-regulatory genes in epithelial cells in pulmonary fibrosis in mice. J Pathol . 2000;190:221–229. doi: 10.1002/(SICI)1096-9896(200002)190:2<221::AID-PATH495>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18. Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest . 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 19. Kazufumi M, Sonoko N, Masanori K, Takateru I, Akira O. Expression of bcl-2 protein and APO-1 (Fas antigen) in the lung tissue from patients with idiopathic pulmonary fibrosis. Microsc Res Tech . 1997;38:480–487. doi: 10.1002/(SICI)1097-0029(19970901)38:5<480::AID-JEMT4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 20. Auyeung VC, Downey MS, Thamsen M, Wenger TA, Backes BJ, Sheppard D, et al. IRE1α drives lung epithelial progenitor dysfunction to establish a niche for pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2022;322:L564–L580. doi: 10.1152/ajplung.00408.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Engelholm LH, List K, Netzel-Arnett S, Cukierman E, Mitola DJ, Aaronson H, et al. uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J Cell Biol . 2003;160:1009–1015. doi: 10.1083/jcb.200211091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei Y, Dong W, Jackson J, Ho TC, Le Saux CJ, Brumwell A, et al. Blocking LOXL2 and TGFβ1 signalling induces collagen I turnover in precision-cut lung slices derived from patients with idiopathic pulmonary fibrosis. Thorax . 2021;76:729–732. doi: 10.1136/thoraxjnl-2020-215745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell . 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei Y, Tang CH, Kim Y, Robillard L, Zhang F, Kugler MC, et al. Urokinase receptors are required for alpha 5 beta 1 integrin-mediated signaling in tumor cells. J Biol Chem . 2007;282:3929–3939. doi: 10.1074/jbc.M607989200. [DOI] [PubMed] [Google Scholar]
- 25. Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature . 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell . 2020;26:346–358.e4. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med . 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature . 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 29. Chonghaile TN, Gupta S, John M, Szegezdi E, Logue SE, Samali A. BCL-2 modulates the unfolded protein response by enhancing splicing of X-box binding protein-1. Biochem Biophys Res Commun . 2015;466:40–45. doi: 10.1016/j.bbrc.2015.08.100. [DOI] [PubMed] [Google Scholar]
- 30. Chen J, Jin S, Abraham V, Huang X, Liu B, Mitten MJ, et al. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol Cancer Ther . 2011;10:2340–2349. doi: 10.1158/1535-7163.MCT-11-0415. [DOI] [PubMed] [Google Scholar]
- 31. Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol . 2020;16:11–31. doi: 10.1038/s41584-019-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun . 2020;11:1920. doi: 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature . 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atabai K, Yang CD, Podolsky MJ. You say you want a resolution (of fibrosis) Am J Respir Cell Mol Biol . 2020;63:424–435. doi: 10.1165/rcmb.2020-0182TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol . 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, et al. Unfolded protein response in cancer: IRE1α inhibition by selective kinase ligands does not impair tumor cell viability. ACS Med Chem Lett . 2014;6:68–72. doi: 10.1021/ml500315b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab . 2017;25:1207. doi: 10.1016/j.cmet.2017.04.026. [DOI] [PubMed] [Google Scholar]
- 38. Kuehl T, Lagares D. BH3 mimetics as anti-fibrotic therapy: unleashing the mitochondrial pathway of apoptosis in myofibroblasts. Matrix Biol . 2018;68-69:94–105. doi: 10.1016/j.matbio.2018.01.020. [DOI] [PubMed] [Google Scholar]
- 39. Jiang D, Cui H, Xie N, Banerjee S, Liu RM, Dai H, et al. ATF4 mediates mitochondrial unfolded protein response in alveolar epithelial cells. Am J Respir Cell Mol Biol . 2020;63:478–489. doi: 10.1165/rcmb.2020-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol . 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Podolsky MJ, Yang CD, Valenzuela CL, Datta R, Huang SK, Nishimura SL, et al. Age-dependent regulation of cell-mediated collagen turnover. JCI Insight . 2020;5:e137519. doi: 10.1172/jci.insight.137519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci . 2019;6:11. doi: 10.3389/fmolb.2019.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Siwecka N, Rozpędek-Kamińska W, Wawrzynkiewicz A, Pytel D, Diehl JA, Majsterek I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines . 2021;9:156. doi: 10.3390/biomedicines9020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell . 2018;69:169–181. doi: 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 45. Belyy V, Zuazo-Gaztelu I, Alamban A, Ashkenazi A, Walter P. Endoplasmic reticulum stress activates human IRE1α through reversible assembly of inactive dimers into small oligomers. eLife . 2022;11:e74342. doi: 10.7554/eLife.74342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pihán P, Carreras-Sureda A, Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ . 2017;24:1478–1487. doi: 10.1038/cdd.2017.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gross A, Katz SG. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ . 2017;24:1348–1358. doi: 10.1038/cdd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett . 1996;395:143–147. doi: 10.1016/0014-5793(96)01016-2. [DOI] [PubMed] [Google Scholar]
- 49. Matsuzaki S, Hiratsuka T, Taniguchi M, Shingaki K, Kubo T, Kiya K, et al. Physiological ER stress mediates the differentiation of fibroblasts. PLoS One . 2015;10:e0123578. doi: 10.1371/journal.pone.0123578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med . 2017;9:eaal3765. doi: 10.1126/scitranslmed.aal3765. [DOI] [PMC free article] [PubMed] [Google Scholar]