

Abstract
Apolipoprotein E3 (apoE) is a critical cholesterol transport protein in humans and is composed of two domains: a well characterized N-terminal (NT) domain that harbors the low-density lipoprotein LDL receptor, and a less understood C-terminal (CT) domain that is the site of protein oligomerization and initiation of lipid binding. To better understand the domain structure of apoE, the CT domain was fused to apolipophorin III (apoLp-III), a single-domain, monomeric apolipoprotein of insect origin, to yield a chimeric protein, apoLp-III/CT-apoE. Recombinant apoLp-III/CT-apoE maintained an overall helical content similar to that of the parent proteins, while chemical induced unfolding studies indicated that its structural integrity was not compromised. Analysis using 1-anilinonaphthalene-8-sulfonic acid (ANS), a sensitive fluorescent indicator of exposed hydrophobic sites and protein folding, demonstrated that whereas apoLp-III provided few ANS binding sites, the apoLp-III/CT-apoE harbored an abundance of ANS binding sites. Thus, this indicated tertiary structure formation in CT-apoE when part of the chimera. Size-exclusion chromatography and chemical crosslinking analysis demonstrated that while apoLp-III is monomeric, the chimeric protein formed large oligomeric complexes, similar to native apoE3. Compared to apoLp-III, the chimera showed a two-fold enhancement in phospholipid vesicle solubilization rates and a significantly improved ability to bind to lipolyzed low density lipoprotein, preventing the onset of lipoprotein aggregation at concentrations comparable to that of parent CT-apoE. These results confirm that high lipid binding and self-association sites are located in the CT domain of apoE, and that these properties can be transferred to an unrelated apolipoprotein, demonstrating that these properties operate independently from the NT domain.
Keywords: Apolipoprotein E3, apolipophorin III, lipoproteins, self-association, lipid binding, cross linking, fluorescence, chemical denaturation
Introduction
In the circulatory and the central nervous system, lipoproteins are the main vehicles that distribute lipids and other hydrophobic molecules between various cells and tissues. Due to their hydrophobic character, lipids are packaged with the aid of apolipoproteins into macromolecular complexes ranging in size from 10 to 1000 nm in diameter. Besides serving as a detergent to solubilize lipids, apolipoproteins provide sites for cell surface receptor recognition, and promote or support enzymatic reactions. All apolipoproteins are characterized by the presence of a series of α-amphipathic helical segments that provide a hydrophobic protein surface for lipid binding interactions. While some apolipoproteins are large and are an integral part of the lipoprotein complex (non-exchangeable), others superficially associate with the lipoprotein surface allowing them to switch between various lipoproteins [1].
The exchangeable apolipoproteins are an ensemble of amphipathic α-helices folded into a globular or slightly elongated conformation. Upon binding to the lipoprotein surface, α-helices rearrange, exposing their hydrophobic faces to anchor the protein onto the lipoprotein surface. They are typically small molecular mass proteins (7-34 kDa), with a remarkable structural flexibility that allows them to exist stably in lipid-free and different lipid-associated states. The focus of this study is on apolipoprotein (apo) E, a protein that plays a key role in lipid homeostasis in the plasma and central nervous system [2, 3]. In humans, it is a polymorphic protein with three major alleles APOE ε2, ε3 and ε4, coding the isoforms apoE2, apoE3 and apoE4, respectively. They differ by a single amino acid at position 112 or 158 (apoE2: C112 and C158; apoE3: C112 and R158; apoE4: R112 and R158). ApoE (299 residues) contains two independently folded structural and functional domains that are linked via a protease sensitive loop: the N-terminal (NT) domain (~24 kDa, residues 1-191) and the C-terminal (CT) domain (~10 kDa, 201-299). High resolution structural studies of the isolated NT domain of apoE2 [4], apoE3 [5, 6] and apoE4 [7] reveal a classic 4-helix bundle fold, with hydrophobic residues of the amphipathic α-helices buried in the protein interior and polar and charged residues residing on the protein surface. It contains the residues that recognize the LDL receptor, allowing uptake of apoE-containing lipoproteins [8, 9]. The helix bundle organization is amenable to structural characterization because of its high solubility, stability, and monomeric state [10, 11]. While much is known about the structural and functional role of apoE NT domain, the precise role of the CT domain, which has an identical sequence in all 3 isoforms, remains to be determined.
The CT domain is made of three amphipathic α-helices and bears high affinity lipid binding sites that govern metabolism of lipoproteins wherein apoE resides [12]. It is responsible for directing the protein to the lipoprotein surface and bringing the NT domain in close contact to the lipid environment [13, 14]. This allows anchoring of the entire protein to the lipid surface, potentially offering an option to the NT domain to exist in a lipid-free helix bundle state or a lipid-associated state; in the latter, the helix bundle has opened allowing interaction of the hydrophobic interior with the lipid surface. The lipid-bound conformation presents residues that form the LDL receptor recognition site in an optimal configuration [8, 15, 16]. In the lipid-free state, the CT domain promotes apoE tetramerization in solution, thereby protecting the hydrophobic face from exposure to the aqueous environment [17] [10, 18]. Incremental truncations of apoE3 [19] and apoE4 [20, 21] from the CT end revealed that the CT helix (267-299) promotes protein tetramerization, likely via dimerization mediated by residues 218-266 [17]. Consistent with these observations, mutation of select hydrophobic to polar residues (F257A, W264R, V269A, L279Q, and V287E) in isolated apoE CT domain [22] and full length apoE3 [23] resulted in a monomeric form of apoE3 that allowed high resolution structural analysis of apoE3 by NMR [24]. The NMR structure revealed that the CT domain makes extensive contact with the NT domain via ionic interactions and H-bond formation, in a configuration that exposes several hydrophobic sites. From these studies, it is not known if this arrangement of the CT domain is the result of the multiple mutations that were introduced to promote formation of monomeric protein or if it represents the native organization of the full-length tetrameric protein. The extensive protein-protein interactions observed between the NT and CT domain in the NMR solution structure of full-length apoE thus raises the question: is the CT domain affected by the presence of the NT domain?
In the present study we attempt to gain further insight into the structure and function of apoE CT domain in the context of an alternate helix bundle protein, apolipophorin III (apoLp-III), a single-domain, monomeric protein from the hemolymph of Locusta migratoria, with similar lipid transport properties as apoE. We report structural and functional findings when the apoE CT domain was appended to the CT end of apoLp-III to generate a novel chimera, apoLp-III/CT-apoE.
Materials and Methods
Protein expression and purification
DNA sequences for apoLp-III/CT-apoE and apoLp-III [25] bearing an N-terminal 6x poly-histidine tag (His6) and TEV protease site were commercially synthesized (Eurofins Genomics), and sub-cloned into a pET-20b(+) expression vector (Novagen) using NdeI and HindIII restriction sites. The DNA sequences were codon optimized to improve the production of recombinant proteins in E. coli [26]. ApoE3, CT-apoE (residues 201-299), and NT-apoE3 (residues 1-191) were purified as described previously [17, 27]. Briefly, the recombinant proteins were over-expressed in E. coli BL21(DE3) pLysS cells (Agilent Technologies, Santa Clara, CA) and purified by nickel affinity chromatography followed by size exclusion chromatography (Sepharose CL-6B, Sigma Aldrich) in 10 mM ammonium bicarbonate, pH 7.4, 1 mM EDTA, 0.002% sodium azide. The purified proteins were freeze-dried and stored at −20 °C until further use. Prior to experimentation, proteins were dissolved in 6 M guanidine hydrochloride (Gdn-HCl) and dialyzed extensively in the appropriate buffer. Protein concentrations were determined by bicinchoninic acid (BCA) assay (ThermoFisher Scientific) and by measuring absorbance at 280 nm. Purified proteins were electrophoresed by SDS-PAGE using NuPage 10% bis-tris polyacrylamide gel (ThermoFisher Scientific) and visualized by Amido black stain. The proteins were confirmed by Western blot analysis using the 3H1 monoclonal antibody against apoE-CT, or HRP-anti-His antibody (ThermoFisher Scientific) (1:1000) and IRDye 680RD goat anti-mouse IgG (Li-COR Biosciences) (1:5000).
Secondary structure analysis
Far-UV circular dichroism (CD) was used to assess the secondary structure of apoLp-III/CT-apoE. The spectra of proteins (0.2 mg/mL in 20 mM sodium phosphate, pH 7.4) were recorded from 185 to 260 nm at a rate of 50 nm min−1 using a 1 mm pathlength cuvette (Hellma Cells) in a Jasco J-810 spectropolarimeter. An average of four scans was obtained for each acquisition and plotted as molar ellipticity using the equation below where [θ] is molar ellipticity at a given wavelength (deg.cm2 dmol−1), MRW is the mean residue weight (Da), θ is ellipticity (mdeg), l is cell pathlength (cm) and c is the protein concentration (g mL−1).
Molar ellipticity at 222 nm was used to calculate the % α-helical content:
Further, the resistance to Gdn-HCl-induced denaturation was used to assess the overall stability of the chimeras. Protein samples (0.2 mg/mL in 20 mM sodium phosphate pH 7.4) were incubated with increasing concentration of Gdn-HCl for 16 h and the ellipticity measured at 222 nm. The ellipticity values were converted into maximal change at a given denaturant concentration, which allowed the determination of the midpoint of Gdn-HCl-induced denaturation (the concentration of Gdn-HCl required to elicit 50 % decrease in molar ellipticity, [GdnHCl]1/2) [11].
Fluorescence spectroscopy
The tertiary fold of the chimeras was assessed by monitoring the exposure of hydrophobic surface as deduced by the fluorescence emission features of bound probe, 8-anilinonaphthalene-1-sulfonic acid (ANS, Sigma-Aldrich). Protein samples (2.5 μM) in 20 mM sodium phosphate, pH 7.4 containing 150 mM NaCl (phosphate buffered saline, PBS) were incubated with 125 μM ANS and 1 mM dithiothreitol (DTT) for 30 min at 24 °C. Fluorescence spectra were recorded using an LS 55 fluorescence spectrometer (PerkinElmer, Waltham, MA), recording the emission spectra from 400 to 600 nm with the excitation wavelength set at 395 nm, a scan speed of 50 nm min−1, excitation and emission slit width of 3 nm, and averaging three accumulations.
Self-association analysis
Self-association of the chimera proteins was assessed by FPLC system (Bio-Rad) using a Superdex 200 10/300 GL column (GE Healthcare) at a flow rate of 0.75 mL/min. Fifty μl of 0.5 mg/mL protein with 1 mM DTT in PBS was loaded and the eluted fractions were monitored at 280 nm. In addition, crosslinking analysis was used to determine the degree of self-association. Twenty μg of protein was preincubated in PBS with 1 mM DTT for 1 h, followed by addition of dimethylsuberimidate (DMS, 13 mM final concentration) in 63 mM triethanolamine for 2 h at 24 °C. The samples were electrophoresed as described above and stained with Amido black. Alternatively, proteins (300 ng) were visualized by western blotting using HRP-anti-His antibody.
Lipid binding interaction
The lipid binding of chimeras was assessed by measuring the rate of phospholipid vesicle solubilization using 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC). Large unilamellar vesicles (LUVs) were prepared by extrusion as described previously [25], and 250 μg of the vesicle suspension was equilibrated for 15 min at 24.1 °C in a 500 μL quartz cuvette (Starna cells). Protein (250 μg) was added to the vesicles, and the absorbance at 325 nm was monitored in a UV-2401PC spectrophotometer (Shimadzu) equipped with a Peltier unit. First order rate constants and time taken for initial absorbance to decrease by 50% (T1/2) were calculated as described previously [28]. The binding of the chimeras to lipoproteins was measured using human LDL treated with phospholipase-C (PL-C) as described previously [29]. Briefly, LDL (50 μg of protein) was incubated with apolipoprotein samples (150 μg) in Tris-HCl buffer (50 mM Tris HC1, 150 mM NaCl, 2 mM CaCl2, pH 7.4) in the presence or absence of 1 mM DTT. PL-C (Sigma Aldrich, 160 mU) was added to the mixture and the change in sample turbidity monitored at 340 nm using a Varioskan Lux 3020-514 plate reader (ThermoFisher Scientific).
Results
Chimera construct design and production
To generate the apoLp-III CT-apoE chimera, a DNA construct was designed harboring the coding sequence for L. migratoria apoLp-IIIb, the main isoform found in insect hemolymph corresponding to amino acid residues 3-164 [30], and residues 201 - 299 of apoE3, represented simply as CT-apoE without the isoform designation since the amino acid sequence is identical for the apoE isoforms in the CT domain (Fig. 1A). The construct also comprised a His6 tag at the NT end to facilitate affinity purification, and T20C and A149C mutations in the apoLp-III sequence to allow for formation of a disulfide bond. The disulfide bond tethers and limits the movement of helix-1 and -5, thereby severely hindering helix bundle opening and reducing apoLp-III lipid binding activity [25, 31]. This design allows us to attribute changes in functional properties to the additional apoE amino acids. The construct (henceforth referred to as apoLp-III/CT-apoE) was codon optimized for expression in E. coli and inserted into the pET-20b(+) vector to produce the recombinant chimeric protein. Purified apoLp-III/CT-apoE chimera was obtained in mg quantities from E. coli extracts. ApoLp-III harboring two cysteines at positions 20 and 149 as described for the chimera (henceforth referred to as apoLp-III), apoE3, NT-apoE3 (residues 1-191) and CT-apoE (residues 201-299) were also produced in E. coli and included for comparison. The observed and expected masses were 34.2 and 30.5 kDa for apoLp-III/CT-apoE, 20.1 and 19.1 kDa for apoLp-III, 15.1 and 13.3 kDa for CT-apoE, 43.6 and 36.3 kDa for apoE3, and 28.9 and 24.1 kDa for NT-apoE3, as shown in the SDS-PAGE (Fig. 1B). Additionally, a small amount of dimers was observed for each of the isolated apolipoproteins. Thus apoLp-III/CT-apoE showed the expected increase in mass of ~ 10 kDa compared to apoLp-III. Western blot analysis employing apoE antibody showed reactivity with the apoLp-III/CT-apoE chimera, apoE3, CT-apoE, but not with apoLp-III (Fig. 1C).
Figure 1.
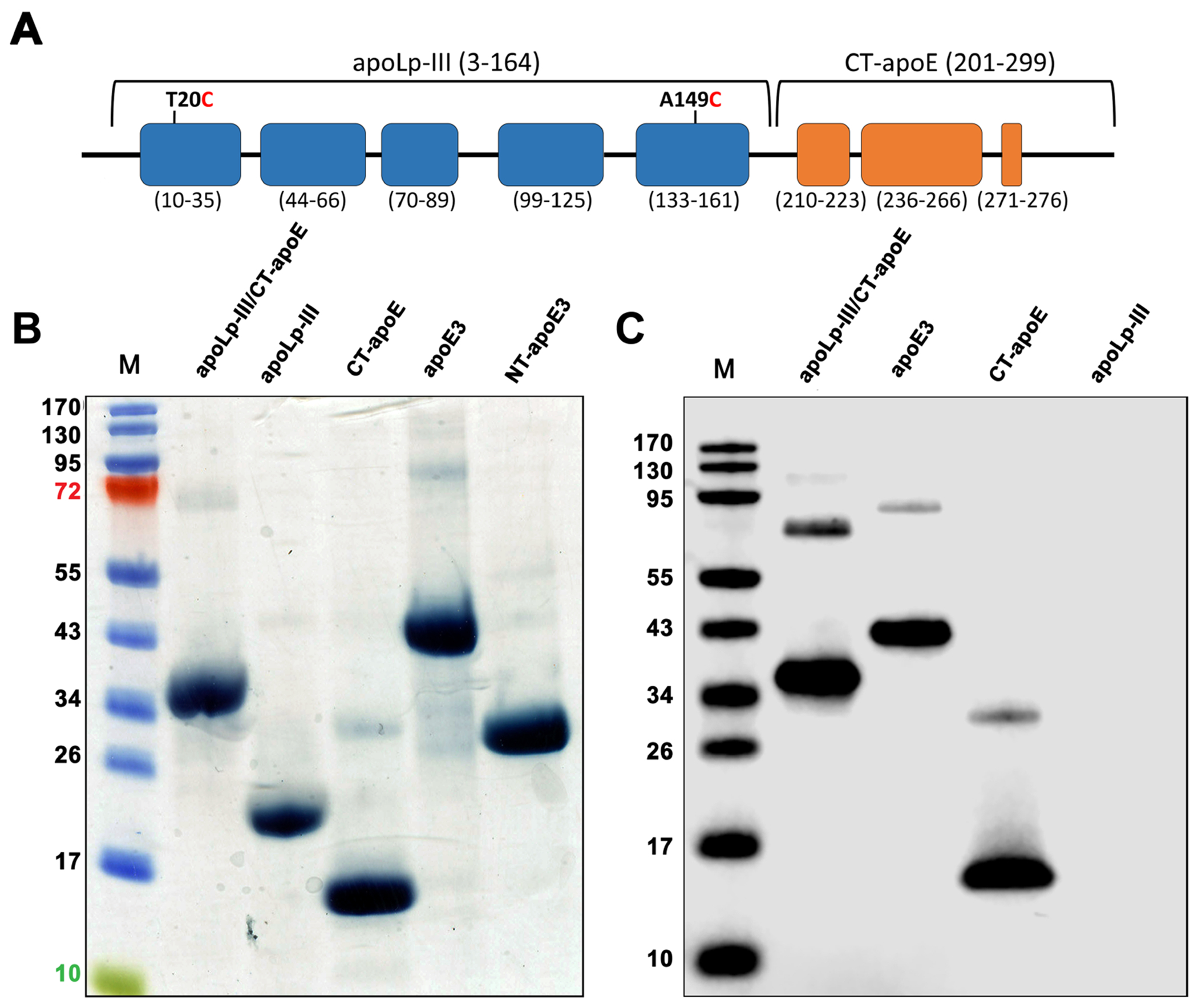
ApoLp-III/CT-apoE chimera design and purification. Panel A: Schematic illustration of apoLp-III/CT-apoE chimera, with apoLp-III helices in blue and CT-apoE helices in orange. The chimera comprises residues 3-164 of L. migratoria apoLp-III and residues 201-299 of human apoE (helix boundaries indicated below each helix). For sake of convenience and clarity, residue numbering of the parent proteins was retained for the chimeras. Panel B: SDS-PAGE analysis of apoLp-III/CT-apoE chimera and relevant parent proteins under reducing conditions. Shown are the apoLp-III/CT-apoE chimera, apoLp-III, CT-apoE, apoE3, and NT-apoE3. The far-left lane (M) contains the molecular weight standards. Panel C: Western blot of purified recombinant proteins using a monoclonal antibody (3H1) specific for the CT domain of apoE3.
Secondary structure and protein stability
To assess if the addition of CT-apoE to apoLp-III led to major alterations in protein secondary structure, CD analysis was caried out. The chimera and parent proteins displayed similar far-UV CD profiles with troughs at 208 and 222 nm, reminiscent of protein structures with a predominantly α-helical character (Fig. 2). Helical contents of all apolipoprotein constructs were between 59.5 ± 1.6 and 68.0 ± 1.9 % (Table 1), in good agreement with previously reported values for apoE3 and apoLp-III [11, 31]. Chemical-induced denaturation of apoLp-III/CT-apoE revealed a [GdnHCl]1/2 of 1.28 ± 0.01 M in the absence of DTT (Fig. 3), which is higher compared to the 0.63 M measured for apoLp-III or 1 M for CT-apoE as reported previously [11]. However, the disulfide bond in apoLp-III was responsible for the increased protein stability as reported earlier [25]. Indeed, addition of DTT to the apoLp-III containing proteins resulted in a substantial decrease in stability, and the [Gdn-HCl]1/2 changed from 1.40 ± 0.01 M to 0.63 ± 0.04 for apoLp-III, and from 1.28 ± 0.01 M to 0.81 ± 0.01 M for apoLp-III/CT-apoE. Therefore, CT-apoE has the higher contribution to the chimera’s protein stability in the reduced state, while the high protein stability of the chimera in the oxidized state can be attributed to apoLp-III. In agreement with previous studies, the stability of apoE3 and NT-apoE3, [GdnHCl]1/2 ~ 2.4 M, was substantially higher compared to apoLp-III and CT-apoE. Overall, the secondary structure and stability analyses indicated that the addition of CT-apoE to apoLp-III generated stable chimeric proteins that displayed characteristics of both parent proteins.
Figure 2:
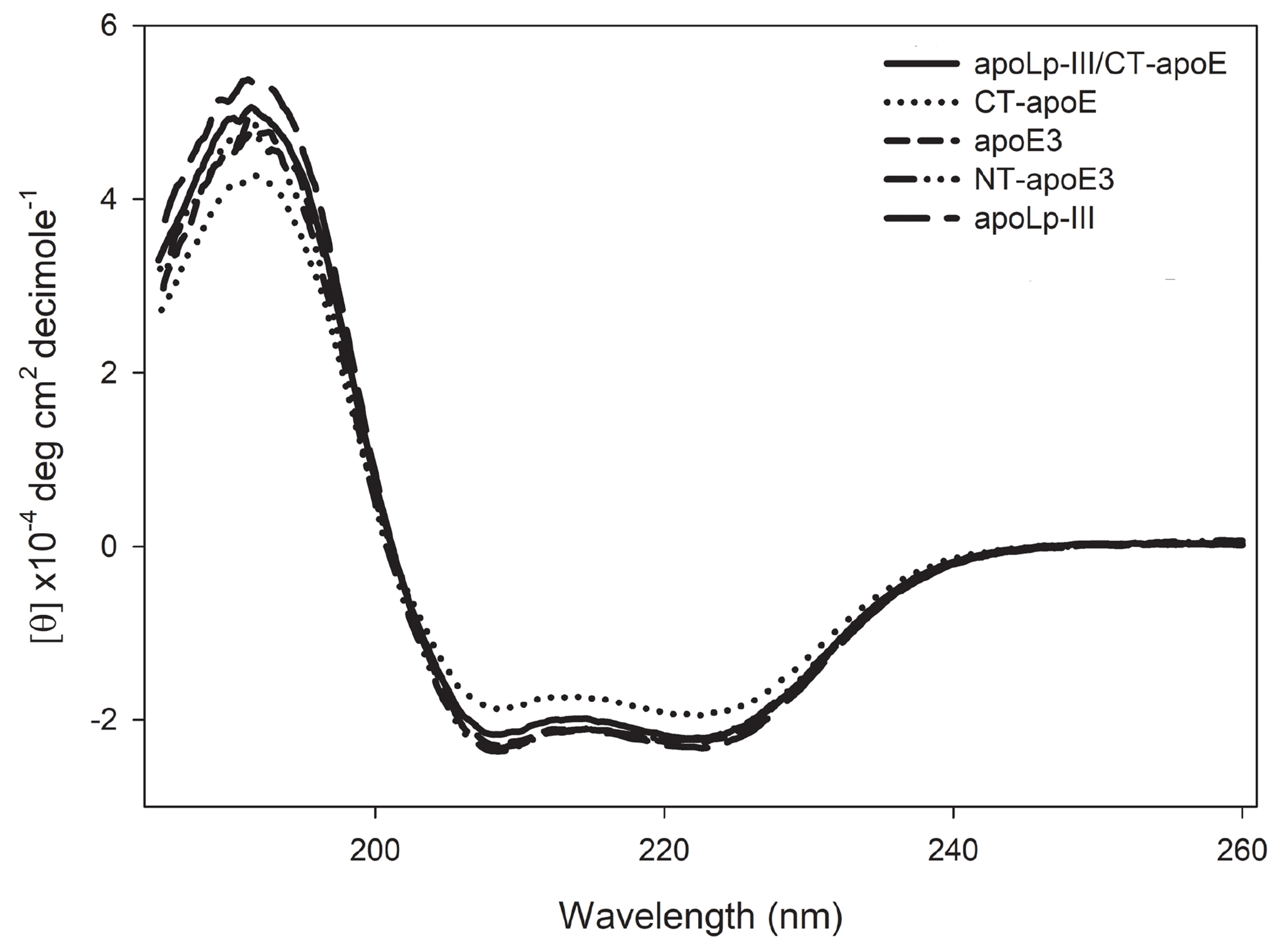
Circular dichroism analysis of apoLp-III/CT-apoE. Shown are the far-UV scans of apoLp-III/CT-apoE (solid line), apoLp-III (long dashed line), CT-apoE (dotted line), apoE3 (short dashed line), and NT-apoE3 (dashed-dotted line).
Table 1.
Biophysical and functional parameters of chimera and parent proteins.
| Protein | α-helical content (%) | Gdn-HCl midpoint a | ANS fluorescence intensity at 468 nm | LUV solubilization rate (s−1 x 10−3) |
|---|---|---|---|---|
| apoLp-III/CT-apoE1 | 66.0 ± 1.5 | 1.28 ± 0.01 | 119.2 ± 5.1 | 1.01 ± 0.25 |
| apoLp-III1 | 67.0 ± 1.2 | 1.40 ± 0.01 | 31.4 ± 1.3 | 0.59 ± 0.14 |
| CT-apoE | 59.5 ± 1.6 | 1.02 ± 0.01 | 198.7 ± 4.2 | 1.37 ± 0.21 |
| NT-apoE3 | 67.3 ± 2.1 | 2.19 ± 0.05 | 56.0 ± 1.7 | 0.85 ± 0.11 |
| apoE3 | 68.0 ± 1.9 | 1.72 ± 0.02 | 417.7 ± 18.4 | 1.73 ± 0.16 |
Analysis was done in the absence of DTT, which led to intramolecular disulfide bond formation, and resulted in an increased denaturation midpoint for apoLp-III.
Figure 3.
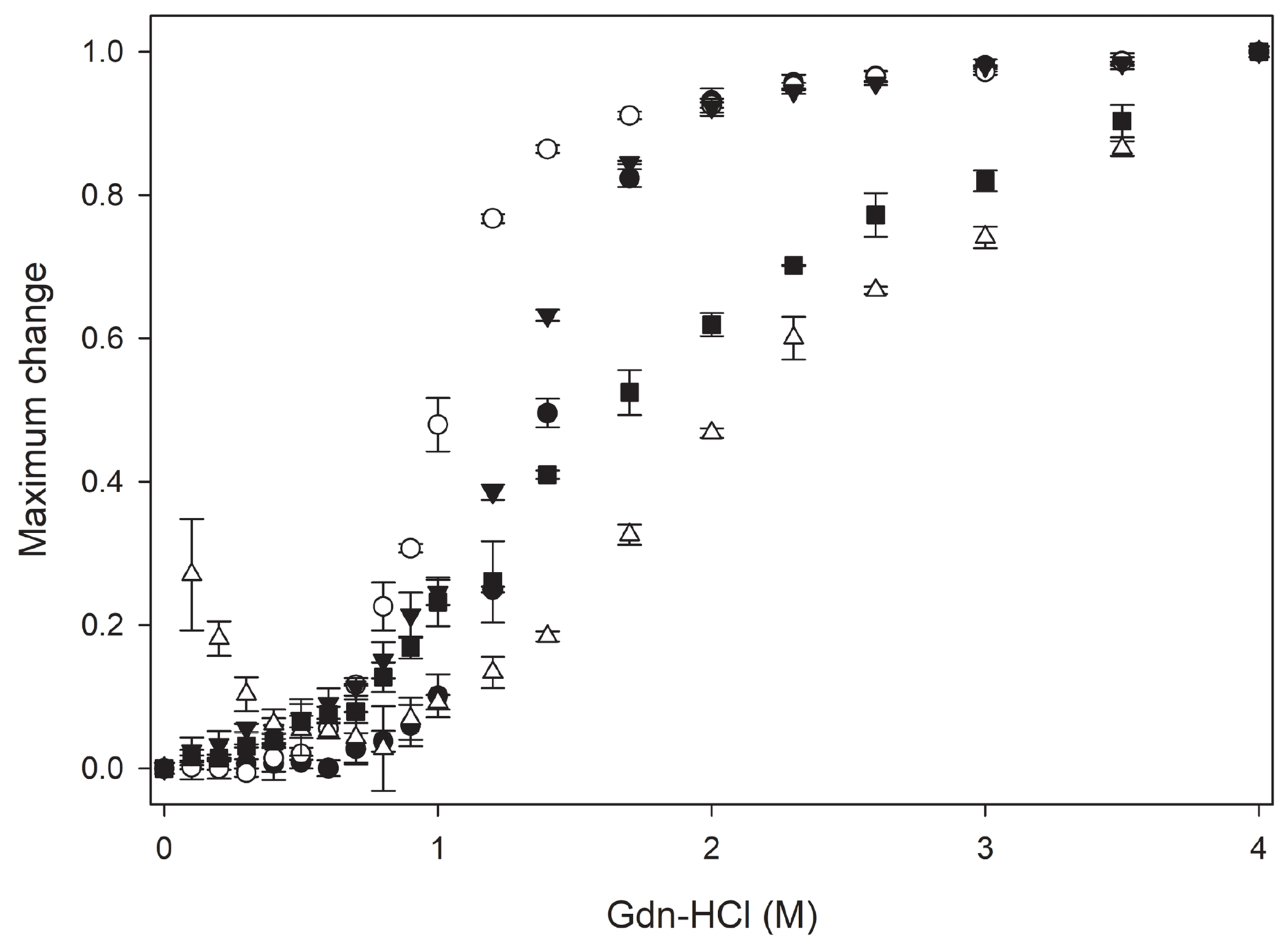
Gdn-HCl denaturation of apoLp-III/CT-apoE (closed triangle), apoLp-III (closed circle), CT-apoE (open circle), apoE3 (closed square), and NT-apoE3 (open triangle). Shown are average ± standard deviation (n=3).
Tertiary structure organization
ANS fluorescence was employed to gain insight into the chimera’s tertiary structure in comparison to the parent proteins. ANS fluorescence emission intensity in the absence of proteins was relatively low, with a wavelength of maximum emission (λmax) of ~ 525 nm. Addition of apoLp-III to the ANS solution resulted in a small increase in fluorescence intensity, indicating the 5-helix bundle of apoLp-III offers few ANS binding sites (Fig. 4). Similarly, ANS fluorescence intensity of NT-apoE3 was relatively low. On the other hand, the ANS emission spectrum of apoLp-III/CT-apoE chimera showed a 10-fold increase in intensity compared to apoLp-III or NT-apoE3, with a 57 nm blue-shift (λmax 468 nm) compared ANS in the absence of proteins. Similarly, strong ANS fluorescence emission intensities with blue-shifted λmax values were observed for apoE3 and CT-apoE. This result suggests that the majority of ANS sites in apoE3 are located in the CT domain, while the compact helix bundle conformation of NT-apoE3 provides few ANS binding sites, in agreement with previous analysis [21]. Thus, the helix bundles of NT-apoE3 and apoLp-III provide few exposed hydrophobic pockets to which ANS can bind.
Figure 4.
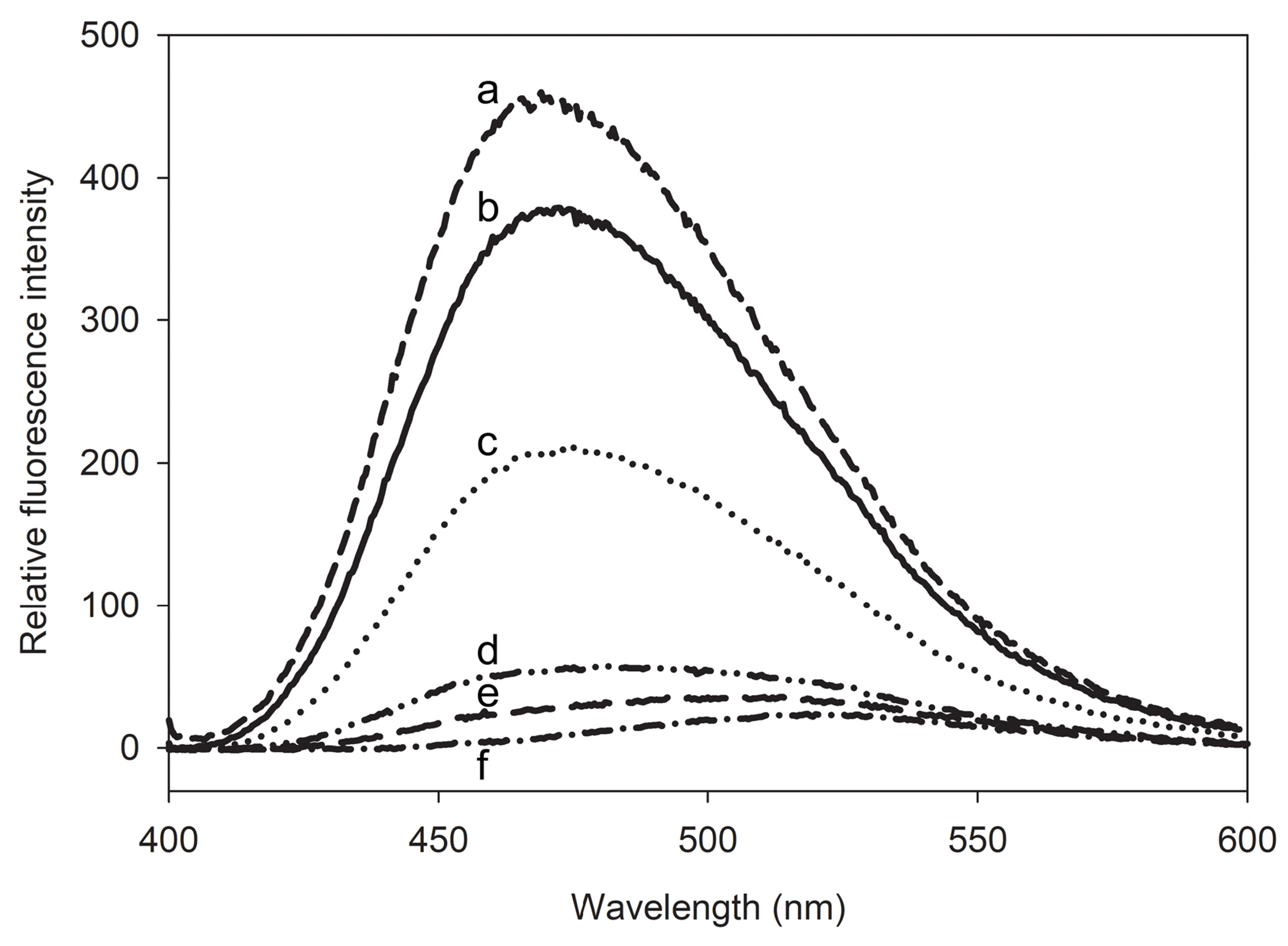
ANS fluorescence emission spectra of apoLp-III/CT-apoE. Following excitation at 395 nm, the fluorescence emission spectra of 125 μM ANS were recorded in the presence of 2.5 μM apoE3 (a), apoLp-III/CT-apoE (b), CT-apoE (c), NT-apoE3 (d), and apoLp-III (e). The ANS fluorescence profile in the absence of protein is shown in plot f.
Self-association properties
While apoE3 self-associates through its CT domain, NT-apoE3 and apoLp-III are predominantly monomeric proteins [21, 32]. Size-exclusion chromatography analysis showed single distinct peaks for apoLp-III (~18 kDa, elution at 16.5 mL), and NT-apoE3 (~24 kDa, 15.7 mL), in agreement with the 6 kDa size difference (Fig. 5). The much smaller CT-apoE (~10 kDa) eluted at 13.6 mL, prior to apoLp-III and NT-apoE3, indicative that the isolated CT domain forms oligomers. Further, the apoLp-III/CT-apoE chimera (30.5 kDa) displayed a prominent peak at 11.3 mL, indicative of self-association caused by the inclusion of CT-apoE. The elution profile of apoE3 showed a similar peak at 11.1 mL and included an additional broader peak at ~8 mL, suggesting the protein is present in different oligomerization states. Both apoLp-III/CT-apoE and CT-apoE showed similar additional elution peaks at ~ 8 ml, but these were much smaller in intensity.
Figure 5.
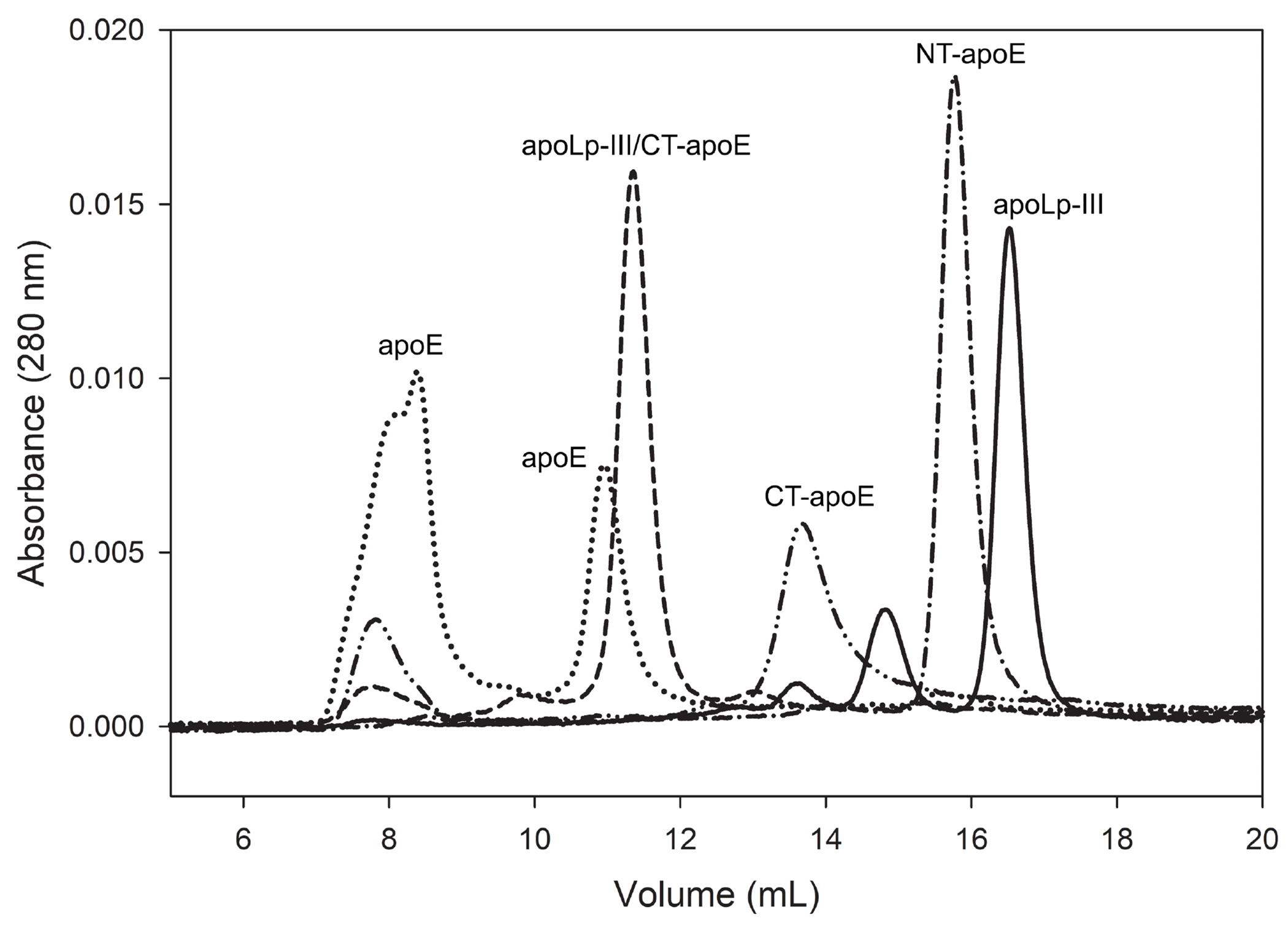
Apolipoprotein size-exclusion chromatography profiles. ApoLp-III (solid line), NT-apoE3 (dash-dotted line), CT-apoE (dash-double dotted line), apoLp-III/CT-apoE (dashed), and apoE3 (dotted) were injected (50 μl of 0.5 mg/mL protein with 1 mM DTT) on a Superdex 200 10/300 GL column operating at a flow rate of 0.75 mL/min in PBS.
In an independent approach, the self-association behavior of apoLp-III/CT-apoE was assessed by DMS cross-linking analysis. ApoLp-III/CT-apoE or the parent proteins were treated with molar excess of DMS, separated by SDS-PAGE, and visualized by western blot using HRP-anti-His antibody (Fig. 6). The immunoblot showed the unmodified protein as the smallest protein in each lane. For apoE3 and apoLp-III/CT-apoE multiple high-molecular weight bands caused by extensive crosslinking corresponding to dimers, trimers, and tetramers were visible in the DMS-treated samples. This indicated that these proteins were in close proximity and confirmed the results of the size-exclusion chromatography analysis. It is of note that CT-apoE lacked the extensive crosslinking pattern seen for the other two proteins. It is possible that the four lysine residues that reside in CT-apoE were not in close proximity to each other to form a covalent bond with each end of the crosslinker. Alternatively, it is possible that the CT domain brings the NT domains into closer proximity to allow more efficient cross-linking.
Figure 6.
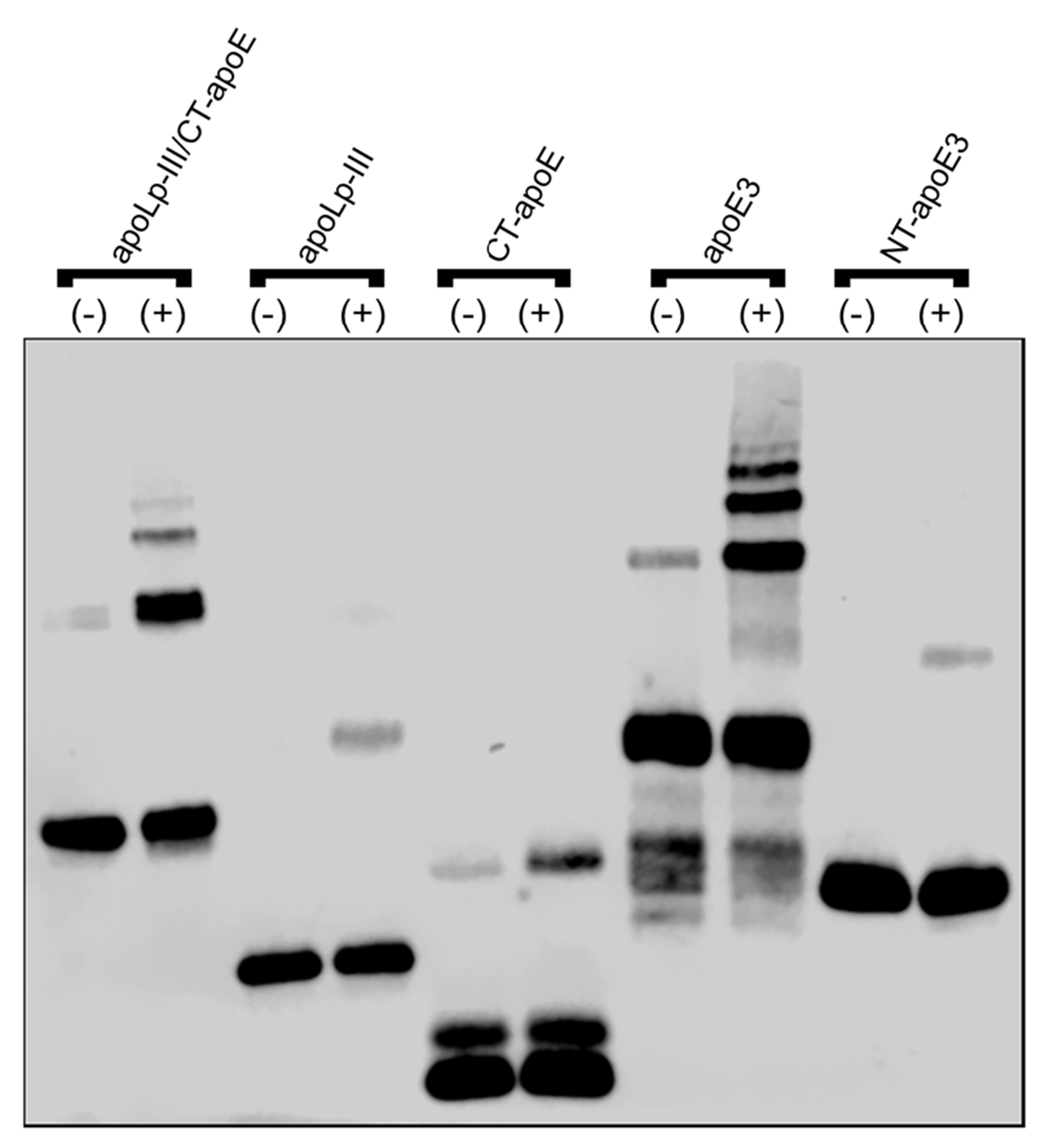
DMS crosslinking of apoLp-III/CT-apoE. Proteins were incubated in the absence (−) or presence (+) of DMS crosslinker, were separated by SDS-PAGE, and visualized by western blot using HRP-anti-His antibody.
Lipid binding
The effect of apoE-CT on the lipid binding properties of the chimera was determined using two distinct lipid-binding assays: measurement of the rate of phospholipid vesicle solubilization and the association with lipoproteins. Protein induced phospholipid vesicle solubilization was carried out using DMPC LUVs with a diameter of ~200 nm at the phospholipid-transition temperature (Tm) of 24.1 °C. Apolipoproteins typically convert the vesicles into small (10-15 nm diameter) discoidal lipid-protein complexes upon mixing at the Tm. This is evident by a decrease in sample turbidity when DMPC vesicles are solubilized by the apolipoprotein yielding smaller discoidal particles. The experiment was carried out in the absence of DTT, thus apoLp-III, both in the free protein and when part of the chimera, were present in the oxidized state which allows for disulfide bond formation between the two Cys residues. As shown in Fig. 7. addition of apoLp-III to the LUV suspension induced a 43 % decrease in turbidity by the end of the experiment (30 min), while the apoLp-III/CT-apoE chimera reduced the sample turbidity by 85%. Similarly, NT-apoE3 was less potent in solubilizing LUVs (50% decrease), while apoE3 caused a decrease in sample turbidity by 80%. Calculation of the initial rate constant showed a two-fold increase for the proteins in which CT-apoE was present (apoLp-III/CT-apoE and apoE) compared to the proteins that lacked CT-apoE (apoLp-III, NT-apoE3) (Table 1).
Figure 7.
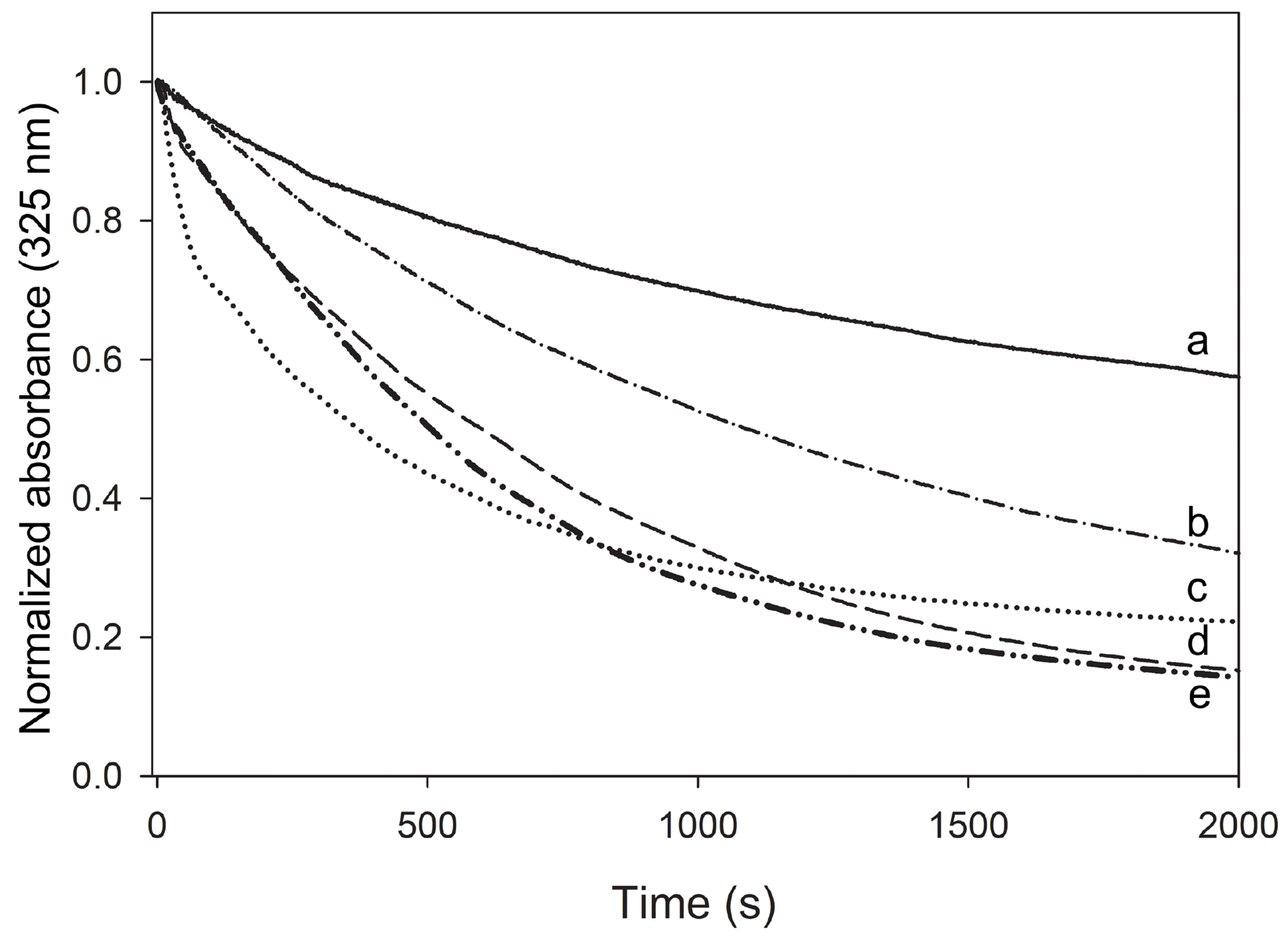
Protein-induced solubilization of DMPC LUVs. Proteins and LUVs were mixed at a 1:1 weight ratio and incubated at 24.1 °C, during which LUVs were converted into small discoidal particles. Changes in LUV light scatter intensity were measured by following the absorbance at 325 nm. Shown are apoLp-III (a, solid line), NT-apoE3 (b, dash-dotted line), apoE3 (c, dotted line), apoLp-III/CT-apoE (d, dashed line), and CT-apoE (e, dash-double-dotted line). The samples did not contain DTT, therefore apoLp-III was mainly present in the oxidized state. When vesicles were monitored in the absence of protein, the absorbance did not change during the incubation time (not shown).
The ability to associate with lipoproteins was measured with surface-modified LDL. Enzymatic removal of the phosphocholine headgroup from phosphatidylcholine by PL-C results in the appearance of hydrophobic spots comprised of diacylglycerol on the LDL surface and recruitment of apolipoproteins [29]. In the absence of apolipoproteins, the increased hydrophobicity on the lipoprotein surface created by PL-C prompted LDL to aggregate following an initial 10 min lag period, which was recorded as an increase in sample turbidity (Fig. 8). However, when apoLp-III/CT-apoE was included in the incubation mixture, the solution remained optically clear and thus prevented the onset of aggregation, indicating that its association with the lipolyzed LDL surface provided full protection against lipoprotein aggregation. Similar results were obtained for CT-apoE and apoE3. On the other hand, neither apoLp-III nor NT-apoE3 afforded protection against PL-C-mediated LDL aggregation, though they delayed the onset of aggregation by ~ 20 min. Thus, both the helix bundle proteins were not efficient in providing protection against aggregation in the absence of the CT domain or by themselves. The high protein stability of NT-apoE3 may have prevented an effective response to the appearance of hydrophobic patches; while in the case of apoLp-III, the disulfide bond that tethered the apoLp-III helix bundle (since DTT was omitted in the reaction mixture) may have precluded the helix bundle opening to bind the hydrophobic patches. Previously it was shown that when DTT was included, it resulted in the reduction of the apoLp-III disulfide bond, restoring lipoprotein binding evident by an effective protection against PLC-induced LDL aggregation [25, 31].
Figure 8.
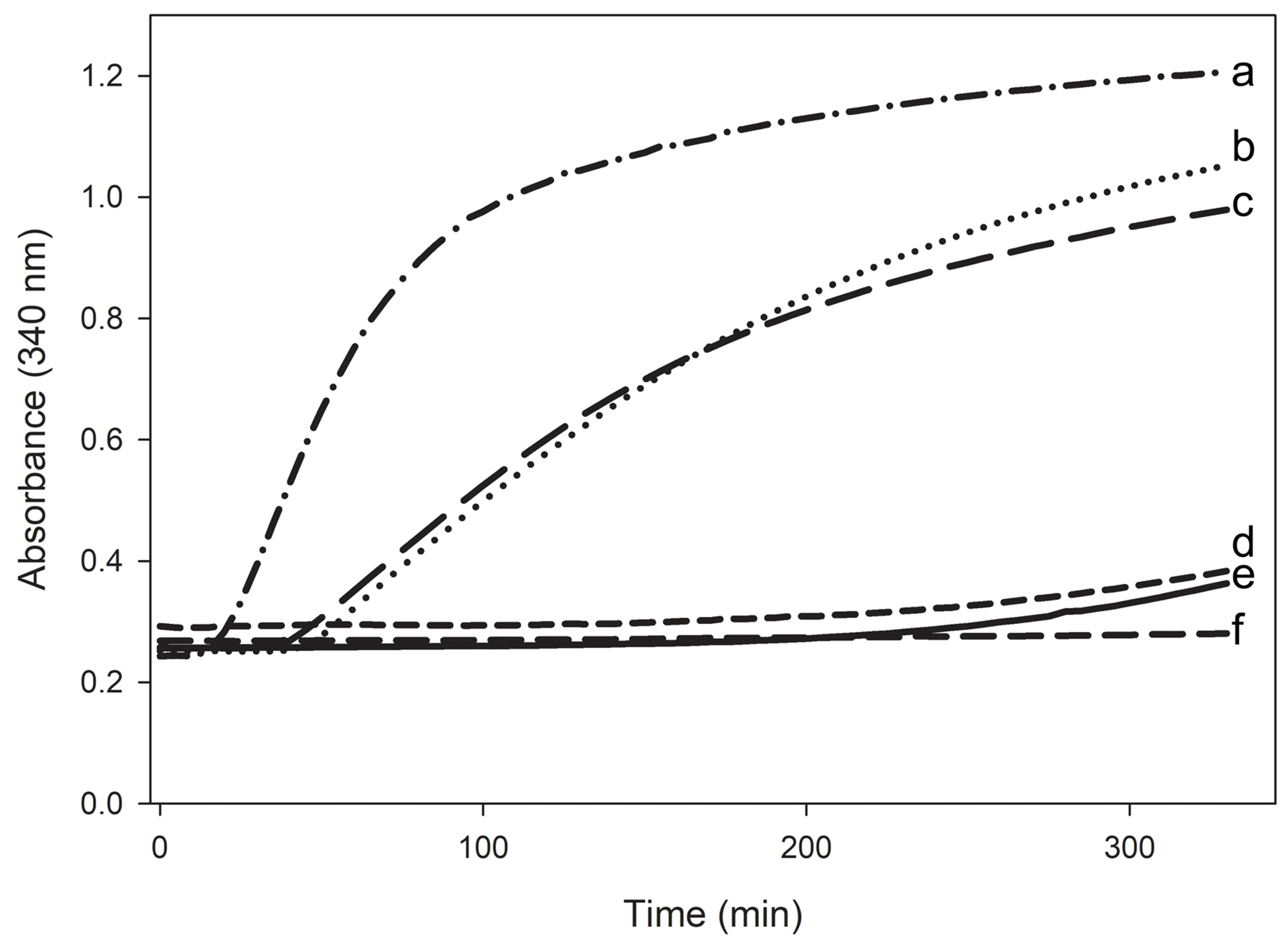
PL-C induced LDL aggregation and rescue by the apoLp-III/CT-apoE chimera. PL-C (160 mU) caused a rapid aggregation of LDL (50 μg protein) as seen by the increase in absorbance at 340 nm (a). PL-C induced LDL aggregation process was monitored in the presence of 150 μg of NT apoE3 (b); apoLp-III (c); apoE3 (d); apoLp-III/CT-apoE (e); or CT-apoE (f). All reactions were carried out in 50 mM Tris HCl, 150 mM NaCl, 2 mM CaCl2, pH 7.4 in the absence of DTT.
Discussion
Human apoE is a two-domain protein, comprised of a 4-helix bundle NT domain and three additional short α-helices that form the CT-domain. On the other hand, the smaller insect apoLp-III is a one-domain helix bundle protein formed by five α-helices. The α-helices of these two proteins are amphipathic in nature, providing a hydrophobic surface for lipid binding interaction, which is a signature feature of exchangeable apolipoproteins [33]. In the present study, the CT domain of apoE was appended to apoLp-III in a chimera protein construct creating a two-domain protein, to better understand the structure and function of the apoE CT domain. The additional domain significantly altered the behavior of apoLp-III by conferring two properties of CT-apoE. First, the chimera adopted self-association properties similar to apoE3 as deduced from cross-linking and size-exclusion chromatographic analyses. While apoLp-III is monomeric in solution [32], CT-apoE provided additional α-helices with a propensity to form a coiled-coil helix, thus facilitating formation of a multimeric protein. Second, the chimera displayed improved lipid binding properties compared to apoLp-III as inferred by the protection afforded against aggregation of lipolyzed LDL and a 2-fold increase in the rate of phospholipid vesicle solubilization. Thus, self-association and high lipid binding sites that are located in the CT domain of apoE, were transferred to an unrelated albeit a helix bundle containing apolipoprotein, suggesting it operates independently from the apoE NT domain. Previously, a similar observation was made when the CT domain of apoAI, a major exchangeable apolipoprotein of HDL that plays a critical role in reverse cholesterol transport, was tacked on to apoLp-III [25], illustrating the protein architectural similarity between apoE3 and apoAI. Further, the CT domains of apoE and apoAI were swapped to yield apoE3-NT/apoAI-CT and apoAI-NT/apoE-CT, both of which were stable chimeras [34]. While CT-apoAI enhanced the lipid binding of apoE, CT-apoE improved cholesterol efflux ability of apoAI.
NT-apoE3 and apoLp-III are helix bundle proteins with helices arranged in an up-and-down topology. The NT-apoE3 fold is a classic four-helix bundle, with a free energy change of ~10 kcal mol−1 needed for unfolding [10]. It is four times more stable compared to apoLp-III, which has a similar fold, with the exception that the helix bundle has an uncommon 5-helix bundle architecture [32, 35]. The extra helix may lower the protein stability perhaps by decreasing helix-helix interactions; in addition, polar residues residing in the helix interface may also contribute to destabilize the protein [36, 37]. NT-apoE3 and apoLp-III share more similarities. This study showed that there are few ANS binding sites on NT-apoE3 and apoLp-III, indicating a helix packing with almost no exposed hydrophobic surface. Previous studies showed that ANS binding sites can be generated in both apolipoproteins by lowering the pH, reminiscent of a molten globule with ruptured tertiary structure and intact secondary structure [38, 39]. This pH induced loosening of tertiary contacts creates an apolipoprotein with enhanced lipid binding activity [38–40]. Indeed, studies with apoE isoforms and apoLp-III mutants have demonstrated an inverse correlation between helix bundle stability and phospholipid vesicle solubilization [38, 41]. During this process, apoE and apoLp-III undergo a dramatic rearrangement of their helical segments when helix-helix contacts are replaced by helix-lipid contacts, resulting in formation of highly stable discoidal complexes. In case of apoE, the CT domain offers exposed hydrophobic surfaces that can readily engage with lipid packaging defects to promote protein-lipid interactions. These CT residues can firmly anchor the protein onto a suitable lipid surface [42], positioning the NT helix bundle in close proximity to the lipid, and triggering the opening of the NT helix domain through several helix rearrangements [43] to adopt the lipid bound conformation. The initial anchoring has been proposed to function as a regulatory feature that allows the NT domain to switch between a LDLr-active open state and LDLr-inactive closed helix bundle state [1, 14, 44, 45]. It is envisaged that the switch is triggered by the lipid components that signal a particle to be competent for LDLr binding and be internalized by the cells.
In addition to harboring sites for lipid binding, the CT domain of apoE contains sites for protein-protein interactions causing the proteins to form a tetramer above a critical protein concentration. ApoE-CT self-association is likely driven by formation of a coiled-coil helix forming a dimer, which dimerizes with another coiled-coil dimer into a tetramer [17, 46]. The CT-domain mediated self-association may shield hydrophobic sides from the aqueous environment, which could potentially prevent the protein to engage in lipid binding. Therefore, upon encountering a suitable lipid surface, the tetrameric protein may dissociate into a monomer, activating the now exposed amphipathic helices, becoming available to bind to lipid surfaces. While the current study is in agreement that apoE-CT is an independently folded domain, the NMR solution structure of full-length apoE3 shows the CT α-helices wrapped around the NT 4-helix bundle and thus may not be considered a separate domain [24]. However, it is possible that the five mutations required to shift the protein from a tetrameric state into a monomer altered the organization of the CT α-helices. While the mutations may have obstructed formation of a coiled-coil dimer, the CT α-helices may have docked onto the NT-apoE3 surface to prevent exposure of the hydrophobic faces of the helices.
A comparative analysis of the apoLp-III/CT-apoAI generated previously with residues 190-243 of apoAI appended to apoLp-III [25] with apoLp-III/CT-apoE in the current study is instructive, revealing notable differences and yielding valuable insights into the molecular organization of apolipoproteins in general. Although the CT domains of both apoAI and apoE bear high affinity lipid binding and self-associations sites, the phospholipid solubilization rate increased 10-fold when the CT-apoAI was tacked to apoLp-III, and 2-fold when the CT-apoE was used. This indicates a higher lipid binding affinity for CT-apoAI compared to CT-apoE, confirming earlier studies [44]. Further, DMS crosslinking showed a higher degree of oligomers when CT-apoA-I was part of the chimera construct (Fig. S1, Supplementary Information), suggesting that CT-apoAI is better capable to promote self-association compared to CT-apoE. ANS fluorescence intensity was six times higher for CT-apoE compared to CT-apoAI (Table S1, Supplementary Information), which manifested itself in the apoLp-III/CT-apoE chimera. While the 99 residues of CT-apoE has sufficient tertiary structure to form ANS binding sites, the 64 amino acid residues of apoA-I are largely unstructured and thus binds ANS poorly. ApoAI and apoE originated from a common ancestral gene, and while they adopted similar α-helical protein architectures, there are important differences that allow these proteins to occupy a unique niche in lipoprotein and lipid metabolism. Their CT domains are a dynamic part of the protein that warrants further study to better understand how these apolipoproteins initiate lipid binding, a process critical for their function in lipid transport processes including reversed cholesterol transport.
Supplementary Material
Funding
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number GM089564 to PMMW, and GM105561 to VN. LMK was supported by a scholarship from the National Institutes of Health under Award Number 5UL1GM118979, 5TL4GM118980, and 5RL5GM118978.
Footnotes
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
The authors declare that they have no conflicts of interest relevant to this study.
Research involving Human Participants and/or Animals: not applicable.
Informed consent: not applicable.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. Additional information will be made available by the corresponding authors on reasonable request.
References
- 1.Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S (2009) Molecular mechanism of apolipoprotein E binding to lipoprotein particles. Biochemistry 48:3025–3032. 10.1021/bi9000694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahley RW (2016) Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med 94:739–746. 10.1007/s00109-016-1427-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Getz GS, Reardon CA (2009) Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res 50:S156–S161. 10.1194/jlr.R800058-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson C, Mau T, Weisgraber KH, Wardell MR, Mahley RW, Agard DA (1994) Salt bridge relay triggers defective LDL receptor binding by a mutant apolipoprotein. Structure 2:713–718 [DOI] [PubMed] [Google Scholar]
- 5.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA (1991) Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 252:1817–1822 [DOI] [PubMed] [Google Scholar]
- 6.Sivashanmugam A, Wang J (2009) A unified scheme for initiation and conformational adaptation of human apolipoprotein E N-terminal domain upon lipoprotein binding and for receptor binding activity. J Biol Chem 284:14657–14666. 10.1074/jbc.M901012200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA (1994) Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem 269:22358–22365 [PubMed] [Google Scholar]
- 8.Raussens V, Fisher CA, Goormaghtigh E, Ryan RO, Ruysschaert JM (1998) The low density lipoprotein receptor active conformation of apolipoprotein E. Helix organization in N-terminal domain-phospholipid disc particles. J Biol Chem 273:25825–25830 [DOI] [PubMed] [Google Scholar]
- 9.Bu G (2009) Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344. 10.1038/nrn2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wetterau JR, Aggerbeck LP, Rall SC, Weisgraber KH (1988) Human apolipoprotein E3 in aqueous solution. I. Evidence for two structural domains. J Biol Chem 263:6240–6248 [PubMed] [Google Scholar]
- 11.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH (2000) Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry 39:11657–11666 . 10.1021/bi000099m [DOI] [PubMed] [Google Scholar]
- 12.Phillips MC (2014) Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 66:616–623. 10.1002/iub.1314 [DOI] [PubMed] [Google Scholar]
- 13.Narayanaswami V, Kiss RS, Weers PMM (2010) The helix bundle: a reversible lipid binding motif. Comp Biochem Physiol, Part A Mol Integr Physiol 155:123–133. 10.1016/j.cbpa.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narayanaswami V, Ryan RO (2000) Molecular basis of exchangeable apolipoprotein function. Biochim Biophys Acta 1483:15–36 [DOI] [PubMed] [Google Scholar]
- 15.Saito H, Dhanasekaran P, Nguyen D, Holvoet P, Lund-Katz S, Phillips MC (2003) Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J Biol Chem 278:23227–23232. 10.1074/jbc.M303365200 [DOI] [PubMed] [Google Scholar]
- 16.Hatters DM, Peters-Libeu CA, Weisgraber KH (2006) Apolipoprotein E structure: insights into function. Trends Biochem Sci 31:445–454. 10.1016/j.tibs.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 17.Choy N, Raussens V, Narayanaswami V (2003) Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J Mol Biol 334:527–539 [DOI] [PubMed] [Google Scholar]
- 18.Aggerbeck LP, Wetterau JR, Weisgraber KH, Wu CS, Lindgren FT (1988) Human apolipoprotein E3 in aqueous solution. II. Properties of the amino- and carboxyl-terminal domains. J Biol Chem 263:6249–6258 [PubMed] [Google Scholar]
- 19.Westerlund JA, Weisgraber KH (1993) Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J Biol Chem 268:15745–15750 [PubMed] [Google Scholar]
- 20.Tanaka M, Vedhachalam C, Sakamoto T, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H (2006) Effect of carboxyl-terminal truncation on structure and lipid interaction of human apolipoprotein E4. Biochemistry 45:4240–4247. 10.1021/bi060023b [DOI] [PubMed] [Google Scholar]
- 21.Sakamoto T, Tanaka M, Vedhachalam C, Nickel M, Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S, Saito H (2008) Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry 47:2968–2977. 10.1021/bi701923h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan D, Li Q, Korando L, Jerome WG, Wang J (2004) A monomeric human apolipoprotein E carboxyl-terminal domain. Biochemistry 43:5055–5064. 10.1021/bi035958w [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Vasudevan S, Sojitrawala R, Zhao W, Cui C, Xu C, Fan D, Newhouse Y, Balestra R, Jerome WG, Weisgraber K, Li Q, Wang J (2007) A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry 46:10722–10732 . 10.1021/bi700672v [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Li Q, Wang J (2011) Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci USA 108:14813–14818. 10.1073/pnas.1106420108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horn JVC, Ellena RA, Tran JJ, Beck WHJ, Narayanaswami V, Weers PMM (2017) Transfer of C-terminal residues of human apolipoprotein A-I to insect apolipophorin III creates a two-domain chimeric protein with enhanced lipid binding activity. Biochim Biophys Acta - Biomembranes 1859:1317–1325. 10.1016/j.bbamem.2017.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makrides SV (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 60: 512–538. https://journals.asm.org/doi/10.1128/mr.60.3.512-538.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta V, Narayanaswami V, Budamagunta MS, Yamamato T, Voss JC, Ryan RO (2006) Lipid-induced extension of apolipoprotein E helix 4 correlates with low density lipoprotein receptor binding ability. J Biol Chem 281:39294–39299. 10.1074/jbc.M608085200 [DOI] [PubMed] [Google Scholar]
- 28.Weers PMM, Van der Horst DJ, Ryan RO (2000) Interaction of locust apolipophorin III with lipoproteins and phospholipid vesicles: effect of glycosylation. Journal of Lipid Research 41:416–423. 10.1016/S0022-2275(20)34480-1 [DOI] [PubMed] [Google Scholar]
- 29.Liu H, Scraba DG, Ryan RO (1993) Prevention of phospholipase-C induced aggregation of low density lipoprotein by amphipathic apolipoproteins. FEBS Lett 316:27–33. 10.1016/0014-5793(93)81730-n [DOI] [PubMed] [Google Scholar]
- 30.Van der Horst DJ, Van Doorn JM, Voshol H, Kanost MR, Ziegler R, Beenakkers AM (1991) Different isoforms of an apoprotein (apolipophorin III) associate with lipoproteins in Locusta migratoria. Eur J Biochem 196:509–517 . 10.1111/j.1432-1033.1991.tb15843.x [DOI] [PubMed] [Google Scholar]
- 31.Narayanaswami V, Wang J, Kay CM, Scraba DG, Ryan RO (1996) Disulfide bond engineering to monitor conformational opening of apolipophorin III during lipid binding. J Biol Chem 271:26855–26862. 10.1074/jbc.271.43.26855 [DOI] [PubMed] [Google Scholar]
- 32.Weers PMM, Kay CM, Oikawa K, Wientzek M, Van der Horst DJ, Ryan RO (1994) Factors affecting the stability and conformation of Locusta migratoria apolipophorin III. Biochemistry 33:3617–3624. 10.1021/bi00178a019 [DOI] [PubMed] [Google Scholar]
- 33.Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM (1994) The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem 45:303–369. 10.1016/s0065-3233(08)60643-9 [DOI] [PubMed] [Google Scholar]
- 34.Lek MT, Cruz S, Ibe NU, Beck WHJ, Bielicki JK, Weers PMM, Narayanaswami V (2017) Swapping the N- and C-terminal domains of human apolipoprotein E3 and AI reveals insights into their structure/activity relationship. PLoS One 12:e0178346. 10.1371/journal.pone.0178346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan RO, Oikawa K, Kay CM (1993) Conformational, thermodynamic, and stability properties of Manduca sexta apolipophorin III. J Biol Chem 268:1525–1530 [PubMed] [Google Scholar]
- 36.Weers PMM, Abdullahi WE, Cabrera JM, Hsu T-C (2005) Role of buried polar residues in helix bundle stability and lipid binding of apolipophorin III: destabilization by threonine 31. Biochemistry 44:8810–8816. 10.1021/bi050502v [DOI] [PubMed] [Google Scholar]
- 37.Dwivedi P, Rodriguez J, Ibe NU, Weers PMM (2016) Deletion of the N- or C-terminal helix of apolipophorin III to create a four-helix bundle protein. Biochemistry 55:3607–3615. 10.1021/acs.biochem.6b00381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weers PMM, Narayanaswami V, Choy N, Luty R, Hicks L, Kay CM, Ryan RO (2003) Lipid binding ability of human apolipoprotein E N-terminal domain isoforms: correlation with protein stability? Biophys Chem 100:481–492 [DOI] [PubMed] [Google Scholar]
- 39.Weers PMM, Kay CM, Ryan RO (2001) Conformational changes of an exchangeable apolipoprotein, apolipophorin III from Locusta migratoria, at low pH: correlation with lipid binding. Biochemistry 40:7754–7760. 10.1021/bi010410f [DOI] [PubMed] [Google Scholar]
- 40.Soulages JL, Bendavid OJ (1998) The lipid binding activity of the exchangeable apolipoprotein apolipophorin-III correlates with the formation of a partially folded conformation. Biochemistry 37:10203–10210. 10.1021/bi980622l [DOI] [PubMed] [Google Scholar]
- 41.Weers PMM, Narayanaswami V, Ryan RO (2001) Modulation of the lipid binding properties of the N-terminal domain of human apolipoprotein E3: apoE-NT lipid binding interactions. Eur J Biochem 268:3728–3735. 10.1046/j.1432-1327.2001.02282.x [DOI] [PubMed] [Google Scholar]
- 42.Fabilane CS, Nguyen PN, Hernandez RV, Nirudodhi S, Duong M, Maier CS, Narayanaswami V (2016) Mechanism of lipid binding of human apolipoprotein E3 by hydrogen/deuterium exchange/mass spectrometry and fluorescence polarization. Protein Pept Lett 23:404–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, Hernandez RV, Tran TN, Nirudodhi S, Beck WHJ, Maier CS, Narayanaswami V (2018) Ordered opening of LDL receptor binding domain of human apolipoprotein E3 revealed by hydrogen/deuterium exchange mass spectrometry and fluorescence spectroscopy. Biochim Biophys Acta - Proteins and Proteomics 1866:1165–1173. 10.1016/j.bbapap.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saito H, Lund-Katz S, Phillips MC (2004) Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Progress in Lipid Research 43:350–380. 10.1016/j.plipres.2004.05.002 [DOI] [PubMed] [Google Scholar]
- 45.Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Lund-Katz S, Phillips MC (2001) Lipid binding-induced conformational change in human apolipoprotein E. Evidence for two lipid-bound states on spherical particles. J Biol Chem 276:40949–40954. 10.1074/jbc.M106337200 [DOI] [PubMed] [Google Scholar]
- 46.Raussens V, Drury J, Forte TM, Choy N, Goormaghtigh E, Ruysschaert J-M, Narayanaswami V (2005) Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem J 387:747–754. 10.1042/BJ20041536 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. Additional information will be made available by the corresponding authors on reasonable request.