

Abstract
We reviewed basal ganglia (BG) dysfunction in Parkinson’s disease (PD) based on recent findings on saccade performance. Hypometria in all saccade paradigms and impaired initiation of internally triggered saccades such as memory guided saccades (MGS) are reported, whereas visually guided saccades (VGS) are relatively spared, although they are also mildly affected. The ability to inhibit unwanted saccades is also impaired. We propose that three major drives converges on SC to determine the saccade abnormalities. The impairment in VGS may be caused by the excessive inhibition of SC due to the increased BG output, whereas for MGS, decreased activity of the frontal cortex-BG circuit may also be involved. The impaired suppression of unwanted saccades may result from the “leaky” inhibition of SC. When PD patients inspect pictures, they end up exploring a smaller area of them with smaller saccades compared to normal subjects. Levodopa slightly prolongs VGS latency and shortens MGS latency, by altering the balance between the direct and indirect pathways of the BG circuit. In contrast, deep brain stimulation of the subthalamic nucleus improves saccade hypometria in both VGS and MGS, presumably by acting relatively directly on the SC-substantia nigra pars reticulata pathway to remove the excessive SC inhibition.
Keywords: Saccade, Basal ganglia, Parkinson’s disease, Superior colliculus, Substantia nigra, Inhibition
1. Introduction
Most of the anatomical connections and physiology of the basal ganglia (BG) within the oculomotor network have been extensively studied in primates (Hikosaka and Wurtz, 1989; Hikosaka et al., 2000), and the existence of a similar oculomotor system organization in humans is supported by means of lesion (see Pierrot-Deseilligny et al., 2004 for review) and neuroimaging studies (O’Driscoll et al., 1995; Sweeney et al., 1996; Brown et al., 2004). These studies provide a firm basis for the clinical application of eye movement examination.
Based on the findings of primate neurophysiology, which were already abundant by the 1980s, clinical studies of eye movement have emerged as a promising non-invasive tool to investigate the underlying brain pathophysiology in various neurological disorders and to help diagnose the patients suffering from such disorders. Saccade recordings have helped to provide a non-invasive measure in the clinical setting to probe the function of the neural system implicated in oculomotor control, especially the BG, and a number of saccade studies have been conducted to clarify characteristic patterns of saccade abnormalities in various neurological disorders (DeJong and Jones, 1971; Teräväinen and Calne, 1980a, b; White et al., 1983; Vidailhet et al., 1994; Briand et al., 1999; Kimmig et al., 2002;Sweeney et al., 1996; Blekher et al., 2009), e.g., saccades of slow velocity in patients with progressive supranuclear palsy (Pierrot-Deseilligny et al., 1989; Vidailhet et al., 1994; Rottach et al., 1996; Rivaud-Péchoux et al., 2000b; Terao et al., 2013). Successful as they were in differentiating neurological disorders through oculomotor measures, many studies served to confirm what had been already found in primate studies rather than being directed at providing a comprehensive insight into the underlying pathophysiology of neurological patients, while other studies address movement disorders such as progressive supranuclear palsy which do not have an appropriate animal model. Even the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model fails to account for all features of PD such as progressive neurodegeneration. Therefore, while miscellaneous findings of saccadic dysfunction in neurological disorders have been reported, both clinical findings of saccade performance and insights gained from basic primate studies should complement each other to arrive at a comprehensive explanation capable of integrating all aspects of saccade abnormalities. For example, although hypometria and prolonged latencies in various oculomotor paradigms are frequently observed both in PD patients and in those with cerebellar ataxia, what difference is there in the underlying pathophysiology between the two categories of neurological disorders? It has long been known that patients with cerebellar ataxia under- or over-shoot the target, then make refixation saccades to the target (Flaherty and Rost, 2007). On the other hand, patients with extra-pyramidal bradykinesia make hypometric saccades, regardless of the task. Obviously, the hypometria in both these disorders cannot share the same origin.
Other types of eye movements are also affected in PD. PD patients even at early stages may show impaired smooth pursuit with reduced gain, both horizontally and vertically, due partly to hypometric catch-up saccades, as well as impaired convergence and combined eye-head tracking (White et al., 1983, 1988; Waterston et al., 1996; Bares et al., 2003; Leigh and Zee, 2006). Interestingly, despite the reduced gain of smooth pursuit, the phase relationship between the eyes and target movement is relatively spared, which implies normal predictive tracking strategies in PD (Bronstein and Kennard, 1985). In this review, we focus on the abnormalities of saccades.
Although the Braak hypothesis (Braak et al., 2003) states that neurodegeneration in PD initially occurs in the brainstem structures including the dorsal motor nucleus of the glossopharyngeal and vagal nerves and anterior olfactory nucleus, within the oculomotor system, the neural degeneration may be considered to be relatively restricted to the striatonigral system at least at its earliest stage (Jellinger, 2001). Thus, PD provides an optimal situation in which we can study how BG dysfunction affects saccade parameters. Recently, however, it is becoming clear that the traditional BG network model for saccade control is too limited to account for many of the behavioral changes observed in this disorder (Watanabe and Munoz, 2011). Herein, we reviewed how saccade studies have arrived at new perspectives on the BG pathophysiology in this disease.
2. Two categories of saccade paradigms used for clinical studies: externally and internally guided saccades
Volitional saccades are categorized into two main classes, those made in response to a visual cue (externally or visually guided saccades in the “broad” sense), which are considered rather “reflexive” in nature, and those made in the absence of such a cue and are therefore considered to be more “voluntary” (internally guided saccades). When we study saccades in the laboratory setting, oculomotor paradigms that correspond to these two types are used: the visually guided (VGS) and memory guided saccade (MGS) tasks (see below) are among the most commonly employed behavioral paradigms.
In the VGS task (in the narrow sense, Fig. 1A), a fixation point is typically turned on in the middle of the dome and subjects are instructed to fixate on it as quickly as possible. It is then turned off after a period, and immediately thereafter, a target point is turned on at a random location at various target eccentricities, typically 5–30 degrees, horizontally to the left or right of the original fixation point. The subjects fixate on the new target as quickly as possible. A variant of VGS is the gap saccade (GS) paradigm, which is identical to the VGS task except that the target spot is turned on 200 ms after the fixation point is extinguished.
Fig. 1.
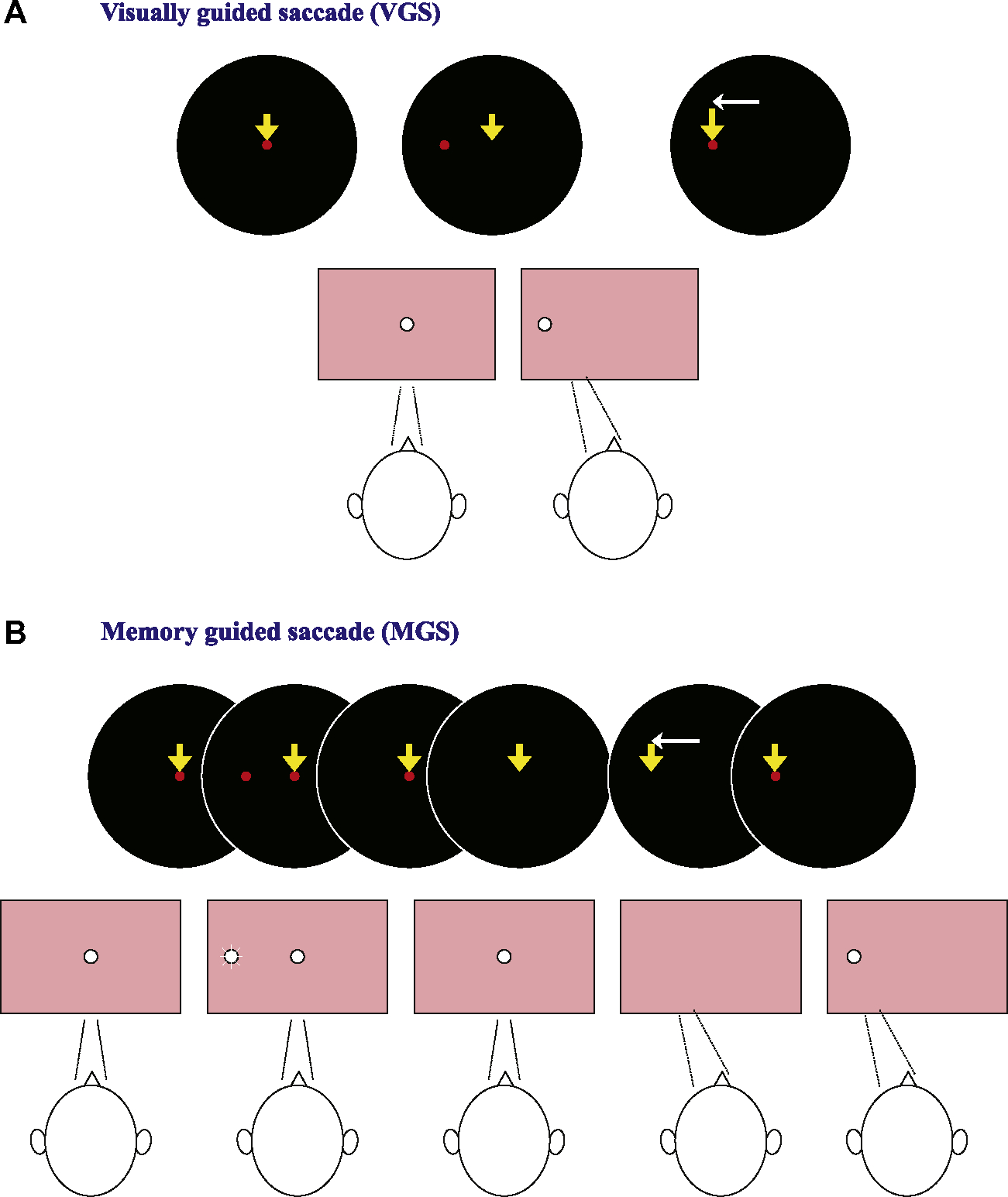
Oculomotor tasks used for clinical saccade studies (A: VGS, B: MGS). In VGS (A), first, a central spot of light which subjects were required to fixate was turned on. After a random period of time (1.2–2.0 s), the fixation spot was extinguished, and a target stimulus simultaneously appeared randomly to the left or right of it. The subjects were to foveate the target as quickly as possible. In the MGS task (B), while the subject fixated the central spot, a peripheral stimulus (“cue”) appeared for a brief period of 50 ms. The subjects were required to maintain visual fixation until the fixation point was turned off (delay period), when the subjects had to make a saccade based on their memory to the spatial location where the cue had appeared.
The MGS task (Fig. 1B), originally developed by Hikosaka and Wurtz (1983), is a task in which the subjects make a saccade according to their memory of the target location (i.e., in the absence of a visual signal). While the subject fixates on the central spot, a peripheral stimulus (“cue”) appears for a brief period (e.g., 50 ms). The subjects are to maintain fixation until the central spot is turned off, at which time they are to make a saccade to the spatial location where the cue appeared. Saccades unintentionally made to the cue during the delay period are called “saccades to cue.” Therefore, this task is suitable not only for assessing the ability to generate voluntary saccades but also for investigating the inhibitory control of saccades; when the latter ability is impaired, there will be an increase in the frequency of saccades to cue.
The antisaccade task (AS) is a task in which the subject, after presentation of a peripheral target, looks away to its mirror position. This requires them to inhibit a reflexive saccade toward the target (termed a prosaccade), and instead generate a voluntary saccade toward a mirror position without a visual stimulus. Thus, this task is considered to fall also into the “voluntary” category. For this task, the frequency of prosaccades is considered to reflect the subject’s ability to suppress unwanted saccades.
3. Superior colliculus as a “bottleneck” of neural structures responsible for the generation of volitional saccades
Generation of saccades, both visually guided and voluntary, are controlled differently by separate parallel descending pathways involving the cerebral cortex (the frontal and parietal areas), BG, and the superior colliculus (SC), and the brainstem saccade generator. Although anatomically, frontal cortical regions such as the frontal eye field (Stanton et al., 1989) and supplementary eye field (Shook et al., 1990) innervate the brainstem saccade generator directly, direct projections from these areas are not considered functionally sufficient to evoke accurate saccades when corresponding sites in the SC are inactivated (Hanes and Wurtz, 2001; Fig. 2, left). Therefore, the SC may constitute the “final common pathway” for purposive saccades, with converging commands arriving through the BG-SC and cortex-SC pathways, including projections both from the frontal and parietal cortices (Hikosaka et al., 2000). The BG process signals from the frontal oculomotor areas which funnel to the SC; outputs from the BG reach the SC via the substantia nigra pars reticulata (SNr). In the clinical setting, since eye movement reflects the output of the BG relatively directly, saccade recordings can provide useful insights into the pathophysiology underlying neurological disorders at the systems level, especially for BG disorders such as PD (Hikosaka et al., 2000; Terao et al., 2011a).
Fig. 2.
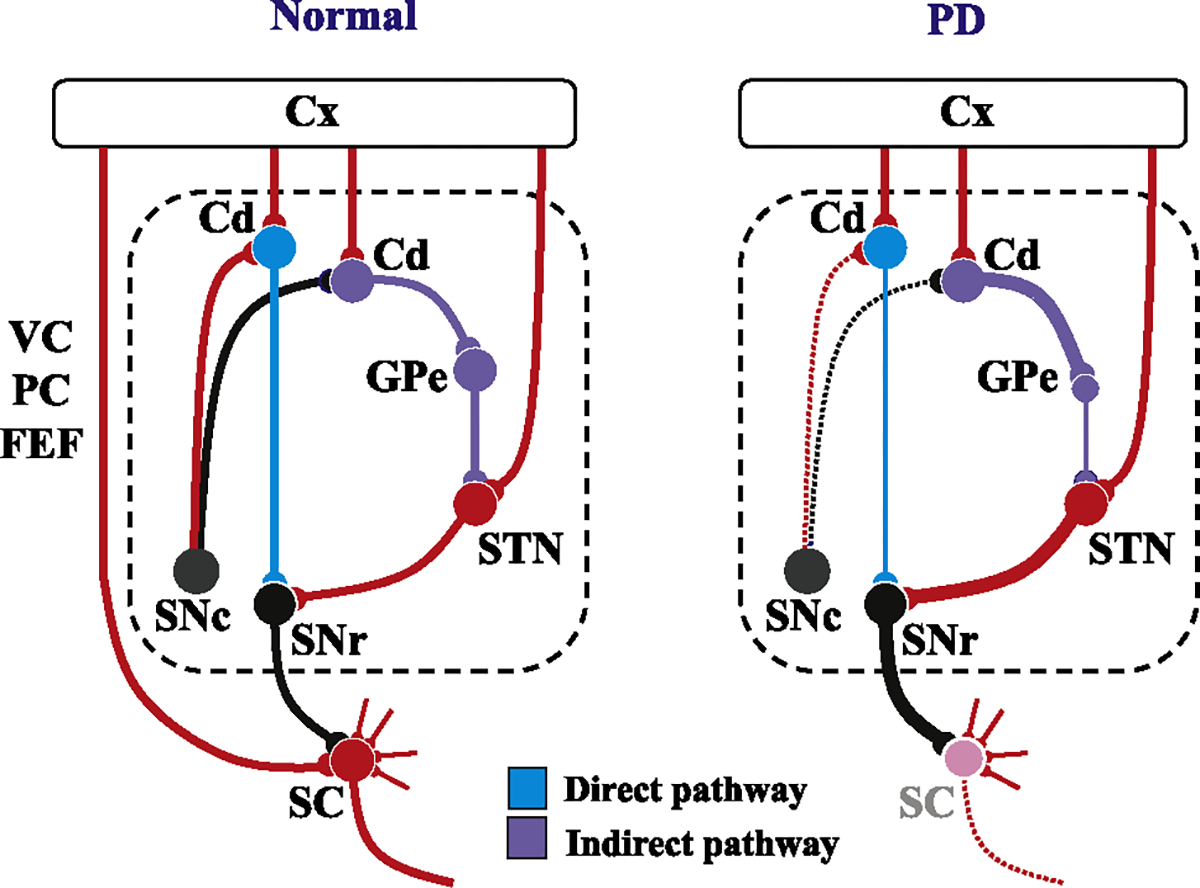
Schematic diagram showing the neural structures depicting the pathophysiology underlying saccade abnormalities in PD. Left figure: Schematic diagram depicting the neural structures involved in the control of saccades in a normal subject. Red lines indicate excitatory connections and black, blue, and purple lines indicate inhibitory connections. The dotted area designates the BG circuit. The blue curves denote the direct pathway and purple curves the indirect pathway (also see panel linked to the bottom of the figure). The pathways involved in the volitional control of saccades all converge onto the SC. Note that most of the inputs converging onto the SC are excitatory, while the only inhibitory input is from the basal ganglia (in the case of saccades, SNr). Some recent studies suggest the existence of direct projections from the prefrontal cortex to the brainstem saccade generator that play an important role in saccade inhibition, but these are not included in this figure. On the other hand, ascending inputs from the SNc to the caudate nucleus change the balance between the activity of the direct and indirect pathways of the BG circuit. Right figure: In PD, as the dopaminergic input from the SNc decreases, the balance between the activity of the direct and indirect pathways shifts in favor of the latter. Note that the inputs from the cortex is omitted in this figure. As a consequence, the SC becomes excessively suppressed compared to the SC in normal subjects (left figure), leading to the suppression of all kinds of saccades mediated by this structure. At the same time, the functioning of the direct pathway of the BG also decreases. These changes may explain why both VGS and MGS are affected in PD, and also why the degree of involvement is much stronger for the latter. Cd: caudate, Cx: the cerebral cortex, SC: superior colliculus, SNc: substantia nigra pars compacta, SNr: substantia nigra pars reticulata, GPe: external segment of the globus pallidus.
The processing involved in MGS, a voluntary saccade, mainly takes place in the frontal lobe, where the motor signal is emitted directly or via the BG to the SC (the direct pathway of the BG circuit). A phasic reduction from the high resting rates of the SNr temporarily releases the saccade cells in the recipient SC, resulting in the generation of voluntary saccades (Hikosaka and Wurtz, 1983, 1985b). Therefore, the performance of MGS reflects the functioning of the direct pathway of the BG circuit implemented through this mechanism of “double inhibition” (Hikosaka et al., 2000).
For VGS and GS, on the other hand, the parietal eye field, including the posterior parietal cortex, mainly integrates visuospatial information to generate a motor signal that is sent to the SC via the parietal lobe-SC pathway (Gaymard et al., 2003b). Thus, the processing involved in VGS is considered largely to bypass the BG circuit.
Note that while most of the inputs reaching the SC are excitatory, the main inhibitory pathway impinging on the SC is from the BG, excluding inhibitory circuits intrinsic in the brainstem and within the SC (see below). Therefore, unlike limb movements, the control of saccades primarily depends on inhibition (or release of inhibition, “disinhibition”). For example, the initiation of MGS is controlled by the tonic inhibition of the SC, which is released only when one wants to make a saccade.
The majority of cortical saccade-related brain areas influence saccade generation by projections to SC neurons both for reflexive and voluntary saccades, by way of the SC’s well-organized motor map representing saccades of different sizes and its well-characterized neuronal elements (Munoz and Fecteau, 2002), but they also play an important role in the inhibitory control of saccades. Above the brainstem level, the BG-SC pathway is believed to be the only inhibitory pathway. Recently, however, the frontal cortical regions are also thought to exert direct inhibition of the brainstem saccade generator (Guitton et al., 1985; Gaymard et al., 2003a; Condy et al., 2004, 2007; Fukushima et al., 1994; Pierrot-Deseilligny, 1991; Pierrot-Deseilligny et al., 2004; Hodgson et al., 2007). The dynamic interactions between the excitatory and inhibitory inputs arriving into the SC determine the target-related activity in the SC, which strongly correlates with the saccade reaction time (Dorris and Munoz, 1998; Munoz and Fecteau, 2002; Fecteau et al., 2004). Therefore, in the following, we looked at these findings from the perspective of the SC.
4. Prominent features of saccade abnormality in Parkinson’s disease
Saccade abnormalities in PD patients have been addressed by a number of studies. Among them, the most consistently noted saccade abnormality in PD patients is that involving markedly hypometric saccades in all types of oculomotor tasks, including VGS and MGS (Fig. 3). Because of these hypometric saccades, PD patients often need to utilize a multi-step sequence to reach the target (DeJong and Jones, 1971; Jones and DeJong, 1971). For voluntary saccades such as MGS, the hypometria is so pronounced that it may be accurately called a fragmentation of gaze shifts (target is reached by several hypometric saccades) (Kimmig et al., 2002). This kind of multiple step pattern is very remarkable in PD and some authors even consider it to be a biomarker in PD (Blekher et al., 2009).
Fig. 3.
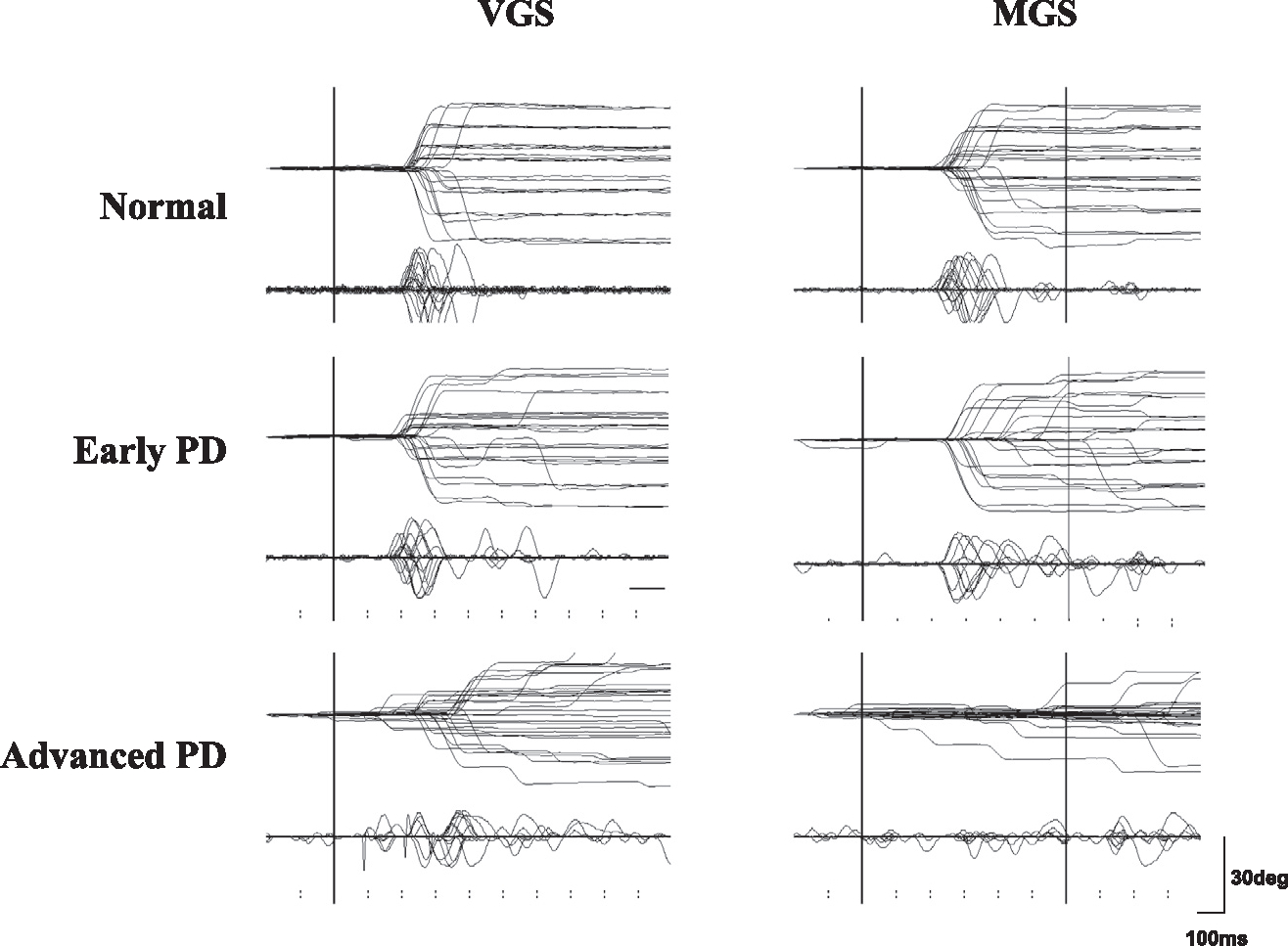
Traces of VGS and MGS. Traces of VGS and MGS in a normal subject (top row) and in a single patient with early and advanced PD (middle and bottom rows). Horizontal electro-oculographic (EOG) recordings were made using two Ag–AgCl gel electrodes of 1.0 cm diameter placed at the bilateral outer canthi with an adhesive margin. Vertical EOG recordings were made using the same electrodes placed above and below the right eye. The signals were fed to a DC amplifier that was low-pass filtered at 20 Hz and then digitized (500 Hz). Twenty to 30 trials of eye movements (upper traces) and velocity curves (lower traces) are superimposed and are time-locked to the presentation of the target (VGS) or to the offset of the central fixation point (MGS). The abscissa is the time axis and the ordinate gives the angle. Note the hypometria both in the VGS and MGS tasks, especially for the latter at advanced disease stages. Initiation of MGS is impaired at advanced stages of PD. Reproduced with permission from Terao et al. (2011a).
Another prominent saccade abnormality in PD patients is that they have difficulty in initiating voluntary saccades such as MGS (Crawford et al., 1989; Nakamura et al., 1994; Vermersch et al., 1994) and AS (Lueck et al., 1990; Kitagawa et al., 1994; Briand et al., 1999; Chan et al., 2005), with decreased accuracy (Bronstein and Kennard, 1985; Ventre et al., 1992; Hikosaka et al., 2000). In contrast, externally triggered saccades to visual targets, whose generation largely bypasses the BG, such as VGS, are relatively spared (White et al., 1983; Rascol et al., 1989; Vidailhet et al., 1994; Briand et al., 2001). The preferential impairment of voluntary saccades such as MGS has been explained by the fact that the BG are more involved in voluntary saccades such as MGS (see previous section; Kori et al., 1995; Fukushima et al., 1994; Briand et al., 1999; Pierrot-Deseilligny et al., 2004). Similarly, for the AS task, another type of voluntary saccade, the latency is delayed, accuracy is reduced, and subjects make increased error saccades toward the target (Kitagawa et al., 1994; Briand et al., 1999).
The abnormality of saccade performance in PD is consistent with the prevailing view on the pathophysiology of PD: that the patients are more impaired in making internally guided movements, which are performed on the basis of an internal representation of the movement pattern, than in making externally guided movements, which are relatively preserved (e.g., VGS; see Berardelli et al., 2001 for review). This apparent dichotomy has also been invoked to explain the treatment effects of dopaminergic drugs and deep brain stimulation (DBS), which were widely believed to improve internally but not externally guided saccades. This simple dichotomy, however, has recently come to be challenged, as we will now discuss.
5. Pathophysiology of PD viewed from the perspective of the superior colliculus
Some saccade abnormalities in PD do not neatly fit the aforementioned dichotomized view of externally versus internally generated saccades. For example, a mild but definite abnormality of reflexive saccades has been demonstrated, including VGS, which was previously thought to be relatively spared (Shibasaki et al., 1979; Rascol et al., 1989; Chambers and Prescott, 2010). Terao et al. (2011a) recorded saccade performance in 66 patients with PD at various stages of the disease, and found that the delay in VGS latency was actually quite a consistent finding. This delay also varied with disease progression (Terao et al., 2011a) and with cognitive impairment (MacAskill et al., 2012).
Therefore, not only voluntary saccades such as MGS but also externally triggered saccades such as VGS are affected in PD, though to a milder degree. These findings are not consistent with the aforementioned externally over internally guided saccade dichotomy. It is thus necessary to revise the pathophysiological view of PD from the perspective of the SC, which comprises the bottleneck region for reflexive and voluntary guided saccades.
Although oversimplified in some respects, the classical model proposed by Alexander and Crutcher (1990) provides relatively clear explanations for many aspects of parkinsonian symptoms. Here dopamine deficiency leads to decreased activity of the direct pathway and increased activity of the inhibitory indirect pathway, especially of the subthalamic nucleus (Bergman et al., 1998), resulting in the increased activity of GABAergic inhibitory projection neurons of the BG output nuclei. The decreased activity of the direct pathway leads to the impaired initiation of voluntary saccades such as MGS, which have been shown to use this pathway for their initiation (see Hikosaka et al., 2000, for review). On the other hand, the internal segment of globus pallidus (GPi) and SNr exhibit overactive tonic firing rates and excessively suppress the thalamus and the SC which is downstream from it (Albin et al., 1995; Fig. 2, right). This may prevent the thalamus from producing quick movements of an appropriate scale, resulting in bradykinesia/akinesia. In terms of oculomotor control, it is thought that SC is excessively inhibited by the overactive SNr and that all types of saccades are thus suppressed. Indeed, electrical stimulation of the SNr, causing the inhibition of the SC, reduces the amplitude of both VGS and MGS in primates (Basso and Liu, 2007). Prolongation of VGS as well as the reduced velocity and amplitude for all types of saccades may thus be explained by the excessive SC inhibition via the BG (Machado and Rafal, 2004). Conversely, deep brain stimulation of the subthalamic nucleus (STN DBS), which is thought to reduce the level of SNr-SC inhibition, improved VGS and MGS in PD patients (see below; Yugeta et al., 2010). In the Yugeta et al. study, hypometria of VGS as well as MGS were improved, supporting the notion that SC serves as the “bottleneck” region for all types of volitional saccades.
6. The pathophysiological basis of hypometric saccades and gaze shift fragmentation in PD
While the excessive suppression of the SC provides a good explanation for the hypometria observed in all types of saccades in PD, it does not sufficiently account for the dissociation in the degree of hypometria or gaze shift fragmentation between externally and internally triggered saccades, with the latter more significantly affected than the former. Kimmig et al. (2002) suggested that such “gaze shift segmentation” occurs because in PD the pre-oculomotor drive through the BG to the SC becomes abnormally weak and/or because the SC is under excessively strong inhibition. We suggest that both of these factors are at work in the hypometria of saccades in PD; the hypometric saccades in PD patients may be caused by two different mechanisms for VGS and MGS. The initiation of sensory driven saccade such as VGS would arise in the posterior parietal cortex that is directly connected to the SC (Pierrot-Deseilligny et al., 2004). Therefore, the abnormality of VGS may be explained mainly by the excessive inhibition of the “bottleneck” SC. In contrast, along with this excessive inhibition, MGS engage two serial inhibitory projections mediated by the frontal cortex (FC) and the BG, finally reaching the SC, as Kimmig et al. (2002) have suggested, and should also reflect the reduced pre-oculomotor drive.
Terao et al. (2011a) looked at how the pre-oculomotor drive and SC inhibition changes with the progression of PD. The VGS amplitude initially became progressively reduced as the disease progressed, although to a much smaller degree than did the MGS amplitude. SC suppression as reflected in VGS may thus comprise only a portion of PD pathophysiology. Although VGS latency and amplitude tended to worsen with the stage of the disease, especially from Hoehn–Yahr (H-Y) stage 1 to 2, after that it tended to stabilize.
At the same time that the VGS latency was slowing and the VGS amplitude was decreasing, the MGS latency progressively increased and MGS amplitude decreased while the MGS success rate declined with the advance of the disease. This suggests that the function of the FC-BG circuit, reflecting the function of the direct pathway of the BG circuit, deteriorated along with the progression of the disease. The success rate of MGS appears to reach a minimum level at H-Y stages 3 to 4 due to an exhausted dopaminergic state. Hypometria in MGS is more pronounced than in VGS, probably because MGS is affected by both the excessive inhibition of the SC and the deteriorated pre-oculomotor drive through the FC-BG circuit.
As an alternative possible explanation for the hypometric saccades in PD, changes in cerebellar function in the presence of BG dysfunction would merit discussion. However, whether this could be addressed directly through saccade recordings is elusive, since saccades in PD are hypometric for almost all types of oculomotor paradigms and hypermetric saccades typical of cerebellar disorders are seldom observed. Brain imaging studies in PD patients using a variety of motor tasks have found hyperactivation in the cerebellum, which is considered to reflect compensation for the BG dysfunction (Rascol et al., 1997; Wu and Hallett, 2005; Yu et al., 2007). On the other hand, resting tremor in PD is considered to arise from a pathological interaction between the BG and the cerebellothalamic circuit, in the presence of dopamine-depleted BG (Helmich et al., 2011). In addition, an interaction is observed in a motor timing task which is considered to involve both the cerebellum and BG (Bares et al., 2011). Husárová et al. (2011) applied a predictive motor timing task to early PD patients and normal controls, which required the subjects to postpone their actions until the right moment (such as in the MGS task) and to adapt their behavior from one trial to the next. PD patients were impaired both in postponing and adapting their motor action. Functional magnetic resonance imaging during the task performance suggested a general “hypoactivation” of both the BG and the cerebellum in early stage PD patients compared to healthy controls. Though differing as regards hypo- and hyperactive cerebellar function, these studies do suggest some interaction between the BG dysfunction and cerebellar function in PD.
Saccade adaptation tasks may be useful to address the interaction of BG dysfunction with the cerebellar function in PD, since saccade accuracy is maintained over time through adaptive ability mediated by the cerebellum. MacAskill and colleagues (2002) studied the performance of saccade adaptation task, both using the VGS and MGS, in PD patients and compared it with that of normal subjects. In this study, adaptation was induced by consistently but imperceptively displacing targets as saccades were made toward them, causing artificial saccadic inaccuracy. They hypothesized that adaptation of MGS would be impaired in PD, since BG dysfunction can disrupt the operation of the prefrontal cortex, while adaptation of VGS would be preserved. Compared to normal subjects, PD patients were less able to modify saccadic size appropriately for MGS (they showed excessive hypometria following the adaptation of MGS), but was normal in adapting the size of VGS. Although saccadic adaptation may involve structures other than the cerebellum, the saccade adaptive ability was generally spared in PD, indicating normal cerebellar function.
7. Saccade disinhibition in PD and its relationship to target eccentricity
Aside from the hypometric saccades in all oculomotor paradigms and the delayed initiation of voluntary saccades, the inhibitory control of saccades is also impaired. PD patients also have difficulty in suppressing unwanted saccades to the novel appearance of visual targets (Joti et al., 2007; van Stockum et al., 2008; van Koningsbruggen et al., 2009), which is also an important function of the BG. For example, PD patients make more saccades to cue in the MGS task, which are saccades unintentionally made to the cue presented during the delay period (Fig. 4). In addition, PD patients experience difficulty in suppressing unwanted saccades of both a reflexive and voluntary nature (e.g., to inhibit saccades during the delay period of the delayed pro- and anti-saccade tasks), indicating a “hyper-reflexivity” of the saccades (Kitagawa et al., 1994; Chan et al., 2005).
Fig. 4.
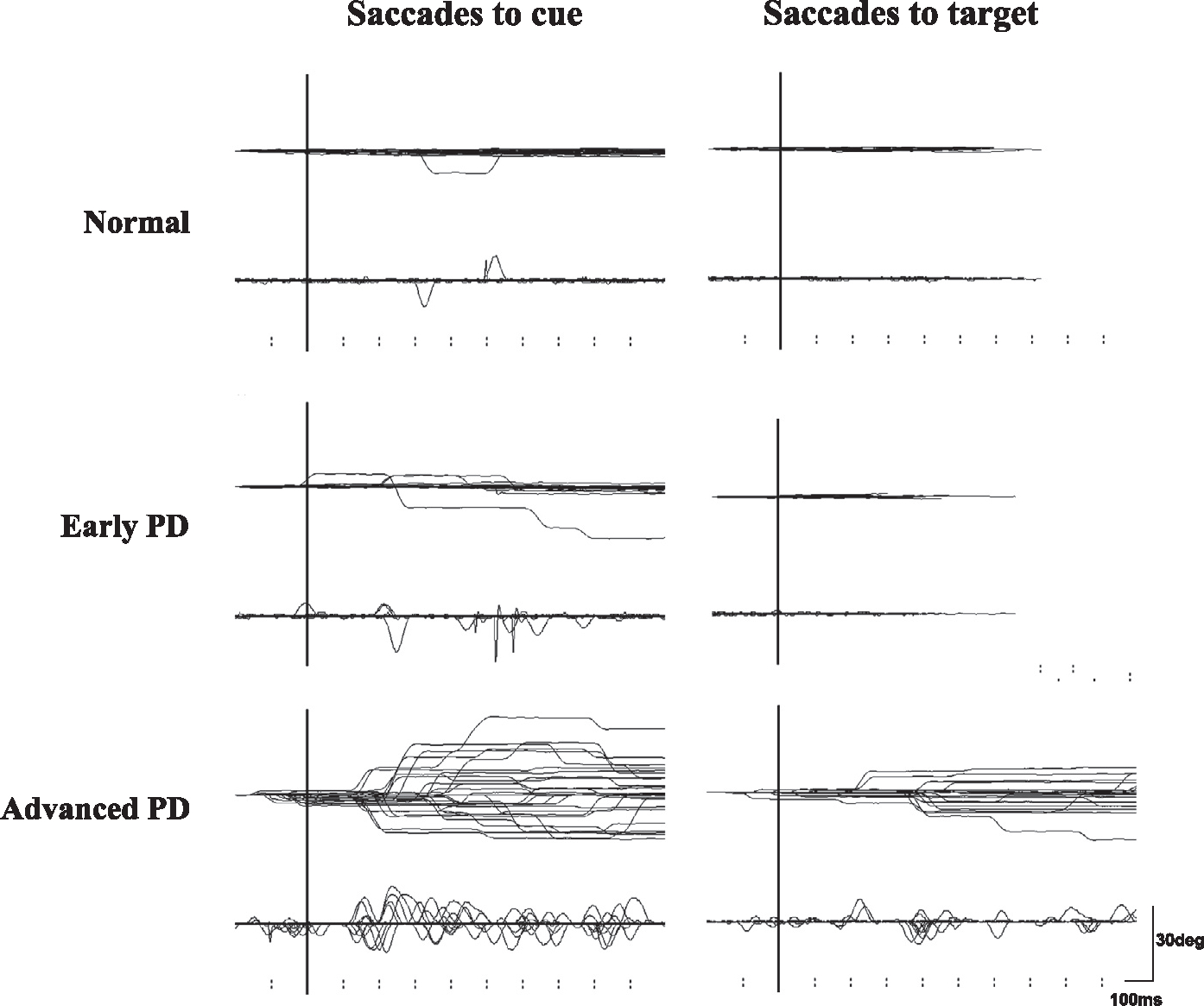
Traces of saccades to cue and saccades to target. Inadvertent eye movements made during the delay period of MGS (saccades to cue) and during the hand reaction time (RT) task (saccades to target) in a normal subject (top row) and in a single patient with early and advanced PD (middle and bottom rows). See text for details of saccades to cue. In the hand RT task, the subjects held a microswitch button which was connected to the microcomputer and allowed them to initiate and terminate a trial by pressing and releasing the button with their thumb. A central spot of light comes on shortly after the subject presses a button, and it stays on throughout each trial. The subjects were instructed to fixate on it continuously. After a random period (2–6.5 s), another spot is illuminated at one of various eccentricities (5–30 degrees) and the subjects release the button as soon as possible while fixating on the central cross point. Inadvertent saccades made to the target are termed “saccades to target.” Note that in PD patients, both the frequencies of saccades to cue and those to target are higher than in normal subjects. The abscissa is the time axis and the ordinate gives the angle. Reproduced with permission from Terao et al. (2013).
The impairment of inhibitory control is difficult to explain within the context of excessive SC suppression, which would naturally result in inhibition of reflexive saccades (Machado and Rafal, 2004). Moreover, it should be noted that the drive to suppress the excitability of the SC and impair saccade initiation and that leading to its “hyper-reflexivity” would counteract one another if they were to converge in the SC.
The impairment of inhibitory control also poses a problem when explaining the changes in VGS latency in PD patients. Excessive inhibition of the SC would tend to prolong the latencies of VGS, as we have seen in the previous section. If “hyper-reflexivity” pre-dominates, however, the latency of VGS would be shortened because of SC disinhibition. In fact, PD patients are known to be able to generate express saccades (very fast reflexive responses), with a latency of 80–120 ms (Biscaldi et al., 1996; Roll et al., 1996), and some studies have found that the generation of externally triggered saccades is usually found to be within the normal range or even faster in early PD patients than in normal subjects (Armstrong et al., 2002; Briand et al., 1999; Chan et al., 2005; Ventre et al., 1992). Using prosaccade (“look towards”), delayed (“wait for cue”) and AS (“look away”) tasks, van Stockum et al. (2008) showed that PD patients made more express saccades in the prosaccade task with a gap (GS), more timing errors in the delayed response task, and more directional errors in the AS task than did the control group. These results indicate an enhancement of externally triggered saccades, in the direction of “hyper-reflexivity”, but in apparent contradiction with the prolonged latency of VGS as discussed in the previous section. Although all these studies point to the facilitation of visually guided “reflexive” saccades, there is no agreement among many of the above studies as to whether the latencies of externally triggered saccades, such as VGS and gap saccades, are shortened or prolonged. Terao et al. (2011a) have shown an increase in the frequency of saccades to cue, but have also demonstrated an increase in the latency of VGS, which at first sight is difficult to reconcile in the context of the “facilitation” and “suppression” of the SC.
These inconsistencies may arise in part from the fact that different studies use different target eccentricities for studying saccades; most of the studies to date used only one or two target eccentricities in the oculomotor paradigms to investigate saccade performance. A recent meta-analytic review of 47 studies (Chambers and Prescott, 2010) suggested that overall, PD patients, especially at early stages, initiate saccades faster than do controls at small target eccentricities, while they respond more slowly for large eccentricities. Using four different target eccentricities for studying saccades, Terao et al. (2011a) also showed that when early PD patients performed VGS, the histograms of latency distribution showed a prominent peak in the early range (110–150 ms) but only for small target eccentricities of 5–10 degrees, whose latency approached that of express saccades. The production of express saccades or short latency saccades at small target eccentricities was considered to be mainly due to an automatic increase in the excitability of SC saccade neurons after the fixation point offset (Biscaldi et al., 1996; Munoz et al., 2000), due to impaired inhibitory control of saccades. These early-latency saccades disappeared at advanced stages and the latency of saccades toward large target eccentricities became more prolonged than for normal subjects. These findings may suggest that saccades toward small eccentricities may especially show a tendency toward disinhibition.
Recent studies show that small and large amplitude saccades are generated by different mechanisms. van Donkelaar et al. (2007) demonstrated that when a peripheral object is foveated by a sequence of multiple saccades, the initial saccade in the sequence is initiated markedly faster than is a single accurate saccade to the same object, and they concluded that multiple saccades are generated in a more automatic manner than are single saccades. To explain this observation, they suggested that large amplitude saccades toward a visual target can become more fully planned when the cortically mediated inhibition is stronger and allows time for the programming to be completed, whereas small amplitude saccades can be more automatically produced (Reddi and Carpenter, 2000; Neggers et al., 2005; Carpenter, 2004).
Thus, it is expected that small amplitude saccades can be initiated with a very short latency unless they are under sufficient inhibition, whereas this may not be the case for large amplitude saccades. In normal subjects, it has been shown that it is more difficult to suppress reflexive saccades (“saccades to cue”) toward targets of 5-degree eccentricity than toward targets of 30-degree eccentricity during the delay period in the MGS task (e.g., Terao et al., 2011a). Moreover, in MGS trials, the delay period (600–1000 ms) allows for enough time to program saccades both of smaller and large amplitudes. Here smaller saccades are even slightly slower to initiate than are larger saccades (Hikosaka et al., 1993, 1995). This implies a stronger inhibition exerted on smaller rather than on larger saccades until they can be finally released. The same is true for voluntary saccades such as AS for which the latency is longer than externally triggered saccades such as VGS and GS (Amador et al., 1998; Terao et al., 2011b).
If BG function is compromised and sufficient inhibition is lacking, unwanted smaller amplitude saccades can occur with a very short latency. Indeed, in early PD patients, only saccades made towards small target eccentricities (5–10 degrees) had a shorter latency compared to that of normal subjects until later disease stages, whereas larger saccades showed a latency which was longer than that of normal subjects. Therefore, despite the overall prolongation of VGS latency in PD, smaller saccades of 5–10 degrees tend to be relatively facilitated. A similar trend has also been shown for hereditary progressive dystonia (HPD) in which a mild dopamine deficiency without neuronal degeneration is postulated (Terao et al., 2012). Although visually guided saccades may not be directly triggered by visual inputs, the predominance of smaller saccades may be related to the broader representation within the SC of small amplitude saccades as well as the large input it receives from cortical and subcortical areas (Sparks et al., 2000). Even if the SC were homogenously inhibited by the excessive output from the SNr, smaller saccades would either overcome this inhibition because of their large representation, or the leaky suppression would allow small saccades to be issued, while larger saccades would be greatly suppressed. Consequently, the latency of small amplitude saccades in VGS and GS tasks would be relatively normal or even shortened compared to those of normal subjects, while large amplitude VGS or GS may be slowed in terms of latency.
As previously discussed, hypometric saccades in PD may be largely explained by the excessive SC suppression via the BG. However, considering the effect of target eccentricity referred to above, this could be partially explained by the facilitation of small amplitude saccades, especially when their latency is short, since saccades initially programmed as large amplitude saccades may be prematurely released as small amplitude saccades.
8. Possible mechanisms for the impaired inhibition of saccades
Candidates for inhibitory pathways exerting inhibitory effects on SC include the direct inhibitory projection of the frontal eye field onto the SC, altered BG output from the SNr, actions of omnipause neurons of the midline pontine reticular formation, and the fixation cells of the SC. In the absence of pathological changes in the oculomotor structures in the brainstem and the SC at early stages of PD, the impaired inhibitory control is most likely ascribed to defective suppression of saccade-related collicular cells by the dysfunction of the fronto-nigro-collicular pathway (see Munoz et al., 2000, for review). In primates, injection of bicuculline into the SC has been shown to facilitate the initiation of saccades and an increase of irrepressible saccades made toward the center of the movement field of the SC cells at the injection site (Hikosaka and Wurtz, 1985a, 1989). In humans, patients with HPD also show an increase of saccades to cue in the MGS task (Segawa et al., 1999; Hikosaka et al., 1995; Terao et al., 2011a). Thus, a mild degree of dopamine deficiency may be associated with either an impairment of inhibitory control or “leaky” suppression from the BG (see also the previous section).
An alternative explanation for the inability to suppress unwanted saccades in a context-dependent manner would be dysfunction in the prefrontal cortex, which is presumed to project directly to the collicular saccade cells so as to exert an inhibitory control. The tonic inhibition model of Sereno (1996) suggests that, with respect to orienting, a dysfunction of prefrontal areas would release neural structures involved in reflexive saccades (e.g., the SC) from tonic inhibition, resulting in “hyper-reflexive” responses. Although the pathology of the frontal lobe is not apparent in early PD (Jellinger, 2001), functional impairment of this pathway may gradually set in as the disease progresses and then release the automatic saccade system from a top-down inhibition (Pierrot-Deseilligny et al., 2004; Crevits and de Ridder, 1997). Indeed, for the AS task, the clinical lesion study of Condy et al. (2004) showed that a direct inhibitory control of saccade triggering by the dorsolateral prefrontal cortex (DLPFC) on the SC plays a major role in reflexive saccade suppression required for AS, rather than that by the BG or the thalamus; the number of error saccades in the AS task increased in patients in whom this pathway was impaired, which extends from the prefrontal cortex along the anterior limb, the genu, or the anterior half of the posterior limb of the internal capsule to project onto the SC. This argument is supported by studies of eye movement control in clinical populations with known prefrontal dysfunction, where deficits of response inhibition are also found (Munoz et al., 2003; Nieman et al., 2000; Reuter and Kathmann, 2004; Hodgson et al., 2007). The frequency of saccades to cue increases with the disease stage in PD (Terao et al., 2011a). This finding may be consistent with the increasing dysfunction of the prefrontal cortex in PD patients in advanced stages. It must be acknowledged, however, that uninhibited saccades to cue and short latency visually guided saccades are present in relatively early stages of the disease (Terao et al., 2011a), and the impaired inhibitory control may not be accounted for solely by the prefrontal dysfunction.
Finally, it is also possible that a compensatory increase in activity of the frontal eye field or other oculomotor cortical regions is present secondary to the increased inhibition upon the SC, and this may lead to unsuppressed saccades. It is known that movements driven by external stimuli employ different cortical routes from those driven by an internal trigger. Behaviorally, PD patients in the advanced stage compensate for their motor deficits by using sensory guidance (Brown and Marsden, 1988). Patients are poor at suppressing prepotent behavior in response to sensory stimuli (Praamstra et al., 1998) because information from sensory stimuli relevant to the generation of a response can have rapid access to motor structures so as to facilitate prepotent movements. Structures outside the BG may compensate for the progressive loss of dopamine in PD, through, for example, overactivity of the parietal cortex. Consistent with this view, Shaikh et al. (2011) found abnormally large square wave jerks in PD patients, and ascribed them to a compensatory increase in frontal eye field activity secondary to the increased inhibition upon the SC. SC disinhibition, as indicated by the increased frequency of saccades to cue, may reflect such inputs to the SC from structures outside the BG that become active to compensate for the PD-associated changes in the BG.
In our previous study (Terao et al., 2011a), we considered the possibility that the increased level of saccades to cue in PD patients (i.e., enhancement of the reflexive system) represent balancing mechanisms recruited as a compensation for the impaired voluntary system (Sereno, 1996). If this is the case, restoration of BG function by levodopa (L-dopa) therapy should normalize the frequency of saccades to cue since recruitment of compensatory mechanisms would no longer be necessary if treatment with L-dopa restored the function of the voluntary system. According to this scheme, the frequency of saccades to cue should decrease by L-dopa therapy, but in fact this change was so small that it did not reach significance. However, L-dopa could also normalize the dopaminergic deficiency in PD and also normalize the increase in saccades to cue. We expected this since our study showed that L-dopa reversed the balance between the voluntary system (e.g., MGS) and the reflexive system (e.g., VGS), facilitating the former and suppressing the latter (Yugeta et al., 2008). In any case, L-dopa does not appear to have a significant effect on SC disinhibition as reflected in the frequency of saccades to cue.
Of course, there need not be only a single source of saccade disinhibition. van Stockum et al. (2008) suggested that there are at least two. In the AS task with a delay, timing error rates (i.e., subjects made saccades before the delay period was over), but not directional error rates, were negatively associated with neuropsychological test scores of cognitive function. On the other hand, the higher directional error rates in the AS tasks were associated with higher proportions of express saccades in the prosaccade task. Therefore, they concluded that saccadic disinhibition may not necessarily reflect a general deficit of automatic response inhibition and cognitive impairment in PD, which result in both timing and directional errors as would be caused by prefrontal dysfunction.
The notion that there need not be only a single source of saccade inhibition is supported by the treatment effects on saccade performance. STN DBS decreased the saccades to cue in MGS, but did not affect the frequency of prosaccades in AS (Yugeta et al., 2010). Occurrence of prosaccades in the AS task has been explained by the failure of the inhibitory system to effectively suppress reflexive prosaccades toward the target, especially the prefrontal cortex inhibiting the SC directly or indirectly via the descending pathway (Guitton et al., 1985; Terao et al., 1998; Condy et al., 2004; Fukushima et al., 1994; Pierrot-Deseilligny, 1991; Pierrot-Deseilligny et al., 2004; Hodgson et al., 2007), although some involvement of BG (i.e., caudate nucleus) has also been suggested for AS (Peltsch et al., 2008; Lasker and Zee, 1997; Everling and Fischer, 1998). Therefore, STN DBS may specifically affect the inhibitory mechanism of saccades mediated by the STN-SNr circuit rather than that mediated by the frontal cortex, leaving the frequency of prosaccades unaffected.
To summarize the pathophysiology of PD as discussed so far, we propose that three major drives converging on the SC determine the saccade abnormalities in PD (Terao et al., 2011a). The impairment in visually and memory guided saccades may be caused by the excessive inhibition of the SC due to the increased BG output and the decreased activity of the frontal cortex-BG circuit. The impaired suppression of reflexive saccades may be explained if the excessive inhibition of the SC is “leaky.” Changes in saccade parameters suggest that frontal cortex-BG circuit activity decreases with disease progression, whereas SC inhibition stays relatively mild in comparison throughout the course of the disease.
9. How does the treatment of PD affect saccade performance?
Since saccade parameters have a narrow normal range, saccade performance may also be used to evaluate the effect of treatment in PD (e.g., dopaminergic drugs and deep brain stimulation) on the function of the BG. The simplest prediction would be that the treatment of PD would specifically bring saccade abnormalities back toward the normal range, if not completely. Thus, the prolonged latency and decreased success rate of voluntary saccades such as MGS and AS would be normalized, while the hypometric saccades observed for all types of saccades would increase in amplitude.
Based on the scheme of Alexander and Crutcher (1990), L-dopa and dopamine agonists would be expected to improve the function of the premotor drive by altering the balance between direct and indirect pathways, while the suppression of the SC is alleviated. Similarly, although the mechanism of action of DBS is currently unknown, its effect is likened to the effect of L-dopa and would also function by altering the balance between direct and indirect pathways, leading in turn to the alleviation of the function of the motor and oculomotor loops of the BG-thalamocortical pathway. In the case of saccades, this would lead to the improved initiation of MGS via improved function of the direct pathway on the one hand (e.g., a decrease in latency and an increase in the success rate of MGS), and the increased saccade amplitudes in all oculomotor tasks through the improvement of excessive SC suppression on the other.
Previous reports on the effect of L-dopa, however, are not consistent and sometimes even conflicting. Some show no effect of dopamine treatment, whereas other studies report small effects on latency, gain or amplitude. Surprisingly, early studies showed that L-dopa, while improving motor symptoms, had little effect on saccade performance, including MGS and AS, although it improved their amplitude (Vermersch et al., 1994; Crevits et al., 2000). In contrast, Rascol et al. (1989) reported that L-dopa improves the amplitude of VGS and AS but has no effect on their latency. These inconsistencies may be due to the fact that many of these studies examined only a small number of normal subjects, who were not appropriately age-matched with the patients. This poses a great problem considering the large age-related change in saccade parameters not only during development but also for elderly subjects above 60 years of age, in whom the inter-subject variability increases greatly even in normal population (Munoz et al., 1998).
Studying a sufficient number of subjects with a reliable recording method and a more sophisticated analysis, recent studies have concurred that L-dopa and dopamine agonists shorten the latency of voluntary saccades such as MGS (Michell et al., 2006; Yugeta et al., 2008), while they prolong the latency of both externally guided saccades such as VGS (Michell et al., 2006; Yugeta et al., 2008) or of eye movements tracking the red laser dot in steps as rapidly as possible, which would also be classified as an externally triggered saccade (Hood et al., 2007), although all of these effects are relatively small. Hood et al. report decreased errors in AS, but Yugeta et al. did not observe any effect on AS.
Note that the improvement of voluntary saccades such as MGS was in the expected direction, while the delay in initiating externally triggered saccades was rather unexpected. Although it is difficult to directly explain this observation based on known physiology and anatomy, L-dopa appears to reverse the balance between the voluntary (e.g., MGS) and the reflexive systems (e.g., VGS), facilitating the former and suppressing the latter. This is consistent with the proposal of Brown et al. (1999) that visual inputs to the SC dominate “reflexive” movements by default and allow fast, visually guided saccades to take place toward a suddenly appearing target, but when a planned movement is required instead (e.g., MGS), frontal activity can suppress visually guided saccades and allow time for processing a planned response to a different target. van Stockum et al. (2012) suggested that facilitation of the reflexive saccadic system in PD is not secondary to impairment of the voluntary saccadic system; if so, facilitation of reflexive saccades should co-occur with impaired performance of voluntary saccades in patients with PD. In fact, when they measured performances of reflexive and voluntary saccades in a group of patients prescribed PD (both “on” and “off”) medication, they found strong positive correlations across reflexive and voluntary saccades in the PD group. On this basis, they concluded that the disease process of PD may affect parts of the oculomotor system which are common to reflexive and voluntary saccade generation. However, since both the latencies of reflexive and voluntary saccades may include processes which are unrelated to the BG function, the “common process” may not directly reflect the function of the BG. Although the disease process of PD may affect parts of the oculomotor system subserving both voluntary and reflexive saccade generation (e.g., SC), the effect of L-dopa may be to tip the balance in favor of the voluntary system.
The effects of L-dopa on saccade amplitude are not consistent. L-dopa improves the amplitudes of VGS and AS but it has no effect on their latencies (Rascol et al., 1989). On the other hand, Yugeta et al. (2008) demonstrated no change in the amplitude of saccades, both of VGS and MGS as well as AS, which is similar to the findings of Vermersch et al. (1994) and Crevits et al. (2000). Although there do appear to be small discrepancies among studies, the overall consensus appears to be that no great change in amplitude is induced by L-dopa. If this is true, it may indicate that L-dopa does not significantly affect the activity of the SC, which is the final common pathway for saccade generation.
Nowadays, DBS, especially of the STN, has become the mainstay of the stereotactic operation in PD. As mentioned above, the effect of DBS has been likened to the effect of dopaminergic drugs, since the benefits of DBS are usually not expected in patients who are not currently taking dopaminergic drugs. Consistently, previous studies have shown that DBS improves voluntary saccades more than it does reflexives saccades. For example, Rivaud-Péchoux et al. (2000a) showed that the gain of the first saccade and the final eye position were improved in MGS in eight bilateral STN DBS cases, while VGS was unaffected. However, Yugeta et al. (2010) studied 32 PD patients and showed changes in four different oculomotor tasks: VGS, GS (externally triggered movements), and MGS tasks, as well as AS tasks (internally triggered movements) (Fig. 5). DBS improved patients’ performance of saccade initiation in all tasks, with shorter latencies, and increased amplitudes and velocities, except for MGS latency. The improved amplitude of MGS as well as of AS was also shown by Fawcett et al. (2010). Furthermore, Kumru et al. (2004) and Sauleau et al. (2008) also showed that STN DBS enhances amplitude and shortens latency of eye (head restrained), and of eye and head (head unrestrained) in the absence of dopaminergic medication. These observations challenge the prevailing notion that DBS preferentially improves internally guided saccades but not externally guided saccades; they are both improved.
Fig. 5.
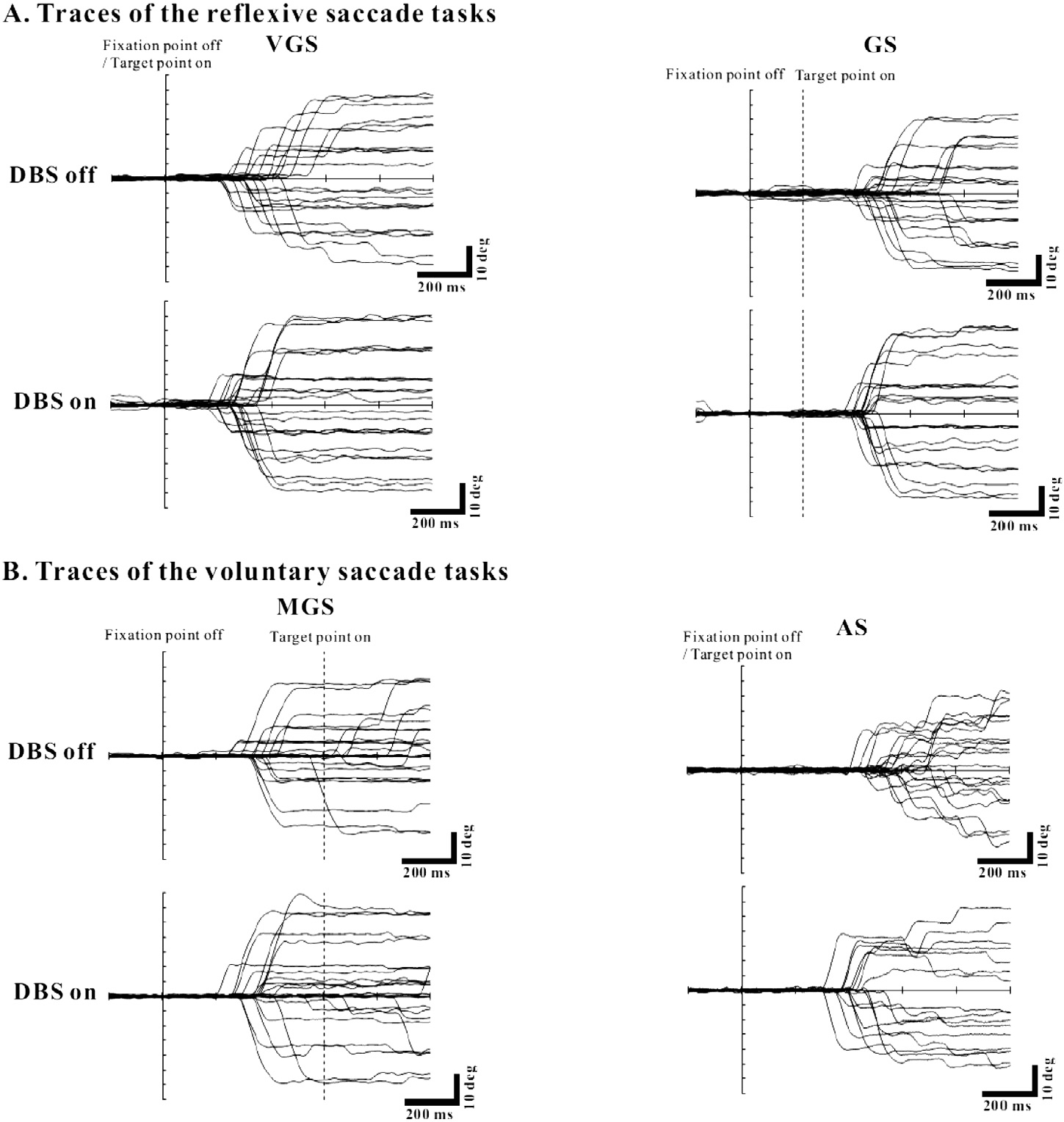
The effect of STN DBS on saccades. PD patients performed four oculomotor tasks: VGS, GS (A), MGS, and AS (B). The traces show saccade recordings in a single PD patient when DBS was off (upper traces) and on (lower traces). Conventions as in Fig. 3. See the scale linked to each figure. For all tasks, the amplitude of saccades was improved when DBS was on relative to when it was off. The PD patients continued to take their regular doses of PD medication. Reproduced with permission from Yugeta et al. (2010).
The most widely-held interpretation for the effect of DBS has been that electrical stimulation of the STN, which is known to have a powerful influence on SNr, enhances both the descending facilitation passing from the cortex to the SC via the BG and also the tonic background inhibition that normally suppresses unwanted early responses (see Temel et al., 2008 for review; Antoniades et al., 2012). To explain the observation that all types of saccades, regardless of whether they are internally or externally guided, were improved, Yugeta et al. (2010) considered these changes to be mediated by the alteration of the information processed by the SC, a pivotal visuomotor structure involved in both voluntary and reflexive saccades. Namely, STN DBS improves the amplitudes of all types of saccades by reversing the excessive inhibition of the SC toward its normal level by targeting the overactive STN-SNr circuit; STN DBS may act on the STN-SNr pathway relatively directly and normalize the function of the “bottleneck” SC, whereas it may not act primarily on the premotor drive through the direct pathway of the BG circuit. In other words, STN DBS has a more direct access to the SC than does L-dopa.
The globus pallidus (GP) is another target often selected for stereotactic surgery in PD. Until recently, few studies had been conducted that combined pallidotomy and pallidal DBS with recordings of saccade performance. Pallidotomy improves motor functions, but appears to show only limited effects on saccadic responses. Blekher et al. (2000) showed that pallidotomy decreases the peak saccadic velocity of internally mediated saccades. Similarly, O’Sullivan et al. (2003) showed that pallidotomy disrupts ocular fixation, although there was no difference in saccadic latency or accuracy, the number of saccadic anticipations, or the ability to generate predictive saccades.
Meanwhile, pallidal DBS improved latency of reflexive saccades and delayed latency of voluntary saccades in Huntington’s disease, and also reduced patient’s ability to suppress unwanted saccades decreased with stimulation, disrupting fixation (Fawcett et al., 2005). At the moment, little is known about the role played by the GP in oculomotor control, blocked function of GPi that receive oculomotor information from the caudate nucleus was considered cause the increase in incorrect saccades, because an inability to suppress unwanted saccades is considered a failure of the volitional saccade system (Briand et al., 1999). Alternatively, because reflexive saccades are mediated by direct cortical action of the parietal and also the frontal eye field on SC (see introduction), the improved performance of VGS with DBS may be due to the restoration of visual and/or oculomotor cortical function (Fawcett et al., 2005). Because GPi sends inhibitory projections to the brain-stem, it is also possible that DBS-mediated improvements are due to disinhibition of brainstem oculomotor neurons. Notably, a similar fixation instability was also demonstrated in PD patients with STN DBS (Wark et al., 2008), but Yugeta et al. (2010) showed that STN DBS decreases saccades to cue in the MGS task. Therefore, the effect of DBS on saccade performance may vary for different targets of stereotactic surgery, but further investigation is needed in order to clarify this issue.
Although the precise mechanisms of action of various treatment modalities are unknown, saccade analysis based on the LATER model (Linear Approach to Threshold with Ergodic Rate, or saccadometry) might provide some insight into what is taking place at the neuronal level. The neuronal discharge of some saccade neurons in the frontal eye field or the SC increases gradually and triggers saccades when their activity exceeds a fixed threshold, although the threshold can vary according to the context in which saccades are made (see, for example, Hanes and Schall, 1996; Paré and Hanes, 2003; Dorris et al., 1997). Similarly in the LATER model, the latency of a single saccade in response to the target appearance can be understood as a decision process, in which saccades are triggered when the decision signal reaches a threshold. The variations in reaction time can be explained either by (i) variations in the rate of (visual) information processing or (ii) the threshold for the decision (see Carpenter, 2004; Temel et al., 2008, for review). Based on the LATER model, the effect of L-dopa on neural decision making was to increase the criterion level of evidence required before the decision to move is made (Michell et al., 2006). On the other hand, the effects of STN DBS on saccade latency can be explained by an acceleration of the processing rate of the visual information, rather than by a changing of the starting and threshold levels for decision making (Temel et al., 2008). In explaining the acceleration of the processing of the decision signal, Temel et al. suggested that electrical stimulation of the STN, which is known to have a powerful influence on SNr, enhances the descending facilitation that passes from the cortex to the SC via the BG, thus increasing the mean rate of rise of the decision signal while also decreasing the tonic background inhibition that normally suppresses unwanted early responses. Therefore, according to their theory, the effect of STN DBS is two-fold, facilitating the pre-oculomotor drive from the frontal cortex to SC and also removing the excessive inhibition of the SC. This is somewhat at variance with the findings of Yugeta et al. (2010) who suggested that the main action of STN DBS is to remove the excessive inhibition on the SC by acting on the STN-SNr pathway.
In summary, although L-dopa and other dopaminergic drugs as well as STN DBS are both effective in improving the motor symptoms of PD, their actions on the BG as well as on the relevant cortical areas differ. L-dopa changes the balance between the function of the BG in favor of the direct pathway, leading to the shortened latency and increased success rate of MGS. It does not, however, have a significant effect on saccade amplitude. In contrast, DBS, at least STN DBS, was considered to act relatively directly on the STN-SNr pathway. Thus, although the effect of DBS has generally been likened to that of DBS, its demonstrated effects were clearly different from those of L-dopa. These effects may be specific to STN DBS, and the effects of DBS on other targets may be different. These are certainly issues that warrants investigation in the future.
Again, these findings provide evidence against the simple dichotomy of BG function, between externally triggered and internally triggered movements, with the idea that the latter is served more by the BG, at least regarding the oculomotor pathways. Further understanding of somatomotor and oculomotor control mechanisms by the BG would be expected to enable better treatment strategies which combine electrophysiological and pharmacological approaches.
10. Behavioral correlates of the abnormal saccades in a more natural setting
Few studies to date have investigated the behavioral correlates of abnormal saccades in BG disorders in a more normal setting that resemble daily life. In monkeys, Kato et al. (1995) and Miyashita et al. (1995) reported deficits in spontaneous saccades with local dopamine depletion in the unilateral caudate nucleus by infusion of MPTP, which could serve as a model for PD. In these monkeys, spontaneous saccades became less frequent toward the visual field contralateral to the injection, and the area scanned by the saccades became narrower and shifted to the hemifield ipsilateral to the infusion site. Although the role of the BG in spontaneous saccades, such as the ones required for visual scanning in this study, is probably nonspecific, the results were taken to suggest that local deprivation of the dopaminergic innervation in the caudate nucleus facilitates neuronal activity of the SNr leading to suppression of saccadic eye movements, which would have gated the saccades whose commands may probably originate in the cerebral cortical areas. In an accompanying paper, Kori et al. (1995) showed in the same monkeys that the latency was consistently prolonged for the contralateral MGS. The amplitude and velocity of saccades also decreased in a contralateral direction, both for MGS and VGS.
In humans, Matsumoto et al. (2011) reported the visual scanning pattern of the gaze when subjects inspected pictures of varying complexity for later recall (Fig. 6). The heads of the subjects were fixed, and saccades were needed to explore the picture. Regardless of the complexity of the picture, patients scanned the picture with smaller amplitude saccades compared to normal subjects, and they subsequently ended up exploring a smaller area of the picture. Interestingly, the smaller area scanned was most prominent with simpler pictures and the patients approached normal behavior as the pictures viewed became more complex (see also Terao et al., 2002). Nevertheless, the area covered by PD patients’ gaze was still smaller than that for normal subjects. Patients also made fewer saccades and had longer fixation times, just as in the MPTP-injected monkeys of the Kato et al. (1995) and Miyashita et al. (1995) study, but the smaller area scanned was mainly ascribed to the small amplitude of saccades during the visual exploration. Therefore, similarly to the monkeys above, the potential saccadic signals impinging on the SC would be gated in PD patients in whom the SC is excessively inhibited by the increased BG output, thus leading to the hypometria of saccades and narrower areas scanned.
Fig. 6.
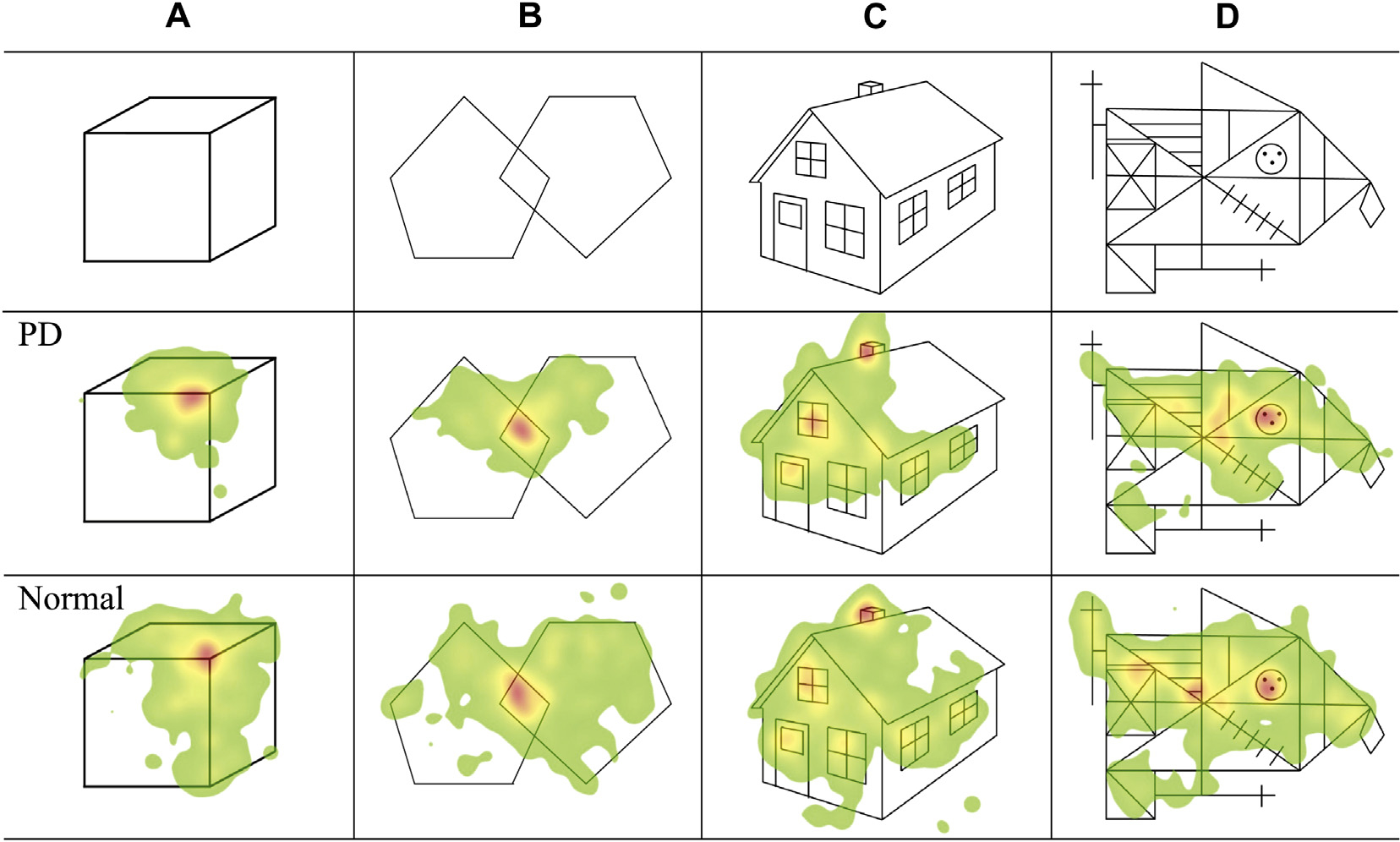
Ocular bradykinesia impairs visual exploration in Parkinson’s disease. The figures show color-coded maps (heat maps) of the distribution of fixation when normal subjects and PD patients view pictures of increasing complexity (from left to right). A: image 1 (a cube), B: image 2 (two pentagons combined in different orientations), C: image 3 (a house), D: image 4 (the Rey–Osterrieth complex figure). Heat maps, or graphical color-coded maps showing the distribution of eye-fixation positions, were created for each image. Heat maps color-coded in red, yellow, and green according to the duration of fixation were overlaid onto each of the images. The upper panels show the images presented to the subjects. The middle panels show the heat maps in PD patients and the lower panels those in normal subjects. In all images, the distribution of eye-fixation position (green-colored area) in PD patients was narrower than that in normal subjects. Reproduced with permission from Matsumoto et al. (2011).
Importantly, the scanned area significantly correlated with the saccade amplitude in most images. Matsumoto et al. (2012) went on to correlate the parameters of saccade visual scanning in this task with those obtained in more conventional oculomotor tasks (VGS and MGS), which were reduced in both cases. Only the amplitude of MGS, but not that of VGS, correlated significantly with the saccade amplitude during the visual exploration of pictures in the same subjects. These results may indicate that BG dysfunction could reduce the amplitude of saccades made during visual scanning, which are generated with a mechanism similar to that of voluntary saccades. Although the exact neural mechanism for generating scanning saccades is unknown, primate studies demonstrated that some cells related to MGS in the caudate nucleus, located in the input portion of the BG that is related to eye movement, were also found to discharge while the monkey was searching for a visual target (Hikosaka et al., 1989). Hallett (2011) argued that what Matsumoto observed in PD patients may represent a counterpart of bradykinesia and/or akinesia for gaze movements.
Of course, the above is but one example of behavioral correlates of the abnormal saccades in PD. Since the experiments of Matsumoto et al. (2011) were all conducted in the “on” state, more studies are warranted to determine what would happen under treatment, such as L-dopa therapy or DBS. Saccade measurements in PD would serve to clarify the treatment effects very clearly under similar ecological settings.
Another behavioral correlate of interest would be whether the saccade abnormality could affect patient’s stance or postural instability. The inability to generate saccades can compromise safe ambulation, since anticipatory saccades should occur normally in situations that involve changing the direction of walking (Hollands et al., 2002) or prior to obstacle avoidance (Di Fabio et al., 2003). When saccades are not generated infrequently, there is an increase in risk for falling (Di Fabio et al., 2005). There are also reports on the improvement of gaze control after balance and eye movement training in patients with progressive supranuclear palsy (Zampieri and Di Fabio, 2009). Although these findings may hold to some extent to PD, as yet there are only a limited number of studies presumably because ocular motility is relatively mild in the early stages of this disorder. Anastasopoulos et al. (2011) found an altered eye-to-foot coordination in standing parkinsonian patients during large gaze and whole-body reorientations. When standing early PD patients could reorient eyes and body to illuminated targets of eccentricities up to ±180 degrees, they were able to assemble multisegmental movements of the eyes, head, trunk, although trunk dyskinesia prolonged the target acquisition time. Preserved eye motion was used to compensate for the trunk slowness. However, since the deficit in ocular motility becomes a problem at relatively advanced stages of PD, postural instability and falls may aggravate when this compensation breaks down as the disease progresses.
11. Basal ganglia dysfunction in Parkinson’s disease as viewed from saccade performance
We have discussed above how the SC serves as a “bottleneck” for all kinds of volitional saccades. The excessive inhibition of the SC would also affect the performance of not only voluntary but also of reflexive saccades, and prolong the latency of all types of saccades mediated by SC. Some researchers point out that this classical model is too limited to account for recent evidence emerging from primate studies (Watanabe and Munoz, 2011), although as we have seen, at least in the clinical setting, the classical model of the BG circuit is quite good at explaining most of the saccade abnormalities in PD. Still, we need to revise this classical model to account for some aspects of recent findings, especially the impairment of inhibitory control of saccades in PD.
We would like to point out that there are at least two major limitations to the classical model that need to be overcome in order to accommodate all the findings of saccade abnormalities. The first is the limitation of the rate or the tonic inhibition model of the BG. The model posits that in PD, the high frequency activity of the SNr excessively suppresses the SC. This leads to the prediction that the saccades to cue would decrease. Indeed, what we observe in PD is quite the opposite of this (see above). Furthermore, our previous study showed that STN DBS decreases the frequency of saccades to cue (Yugeta et al., 2010), meaning that it restores the function of the BG in inhibitory saccade control. Again, if, as discussed above, STN DBS acts directly on the STN-SNr pathway and restores the excitability of the SC from excessive inhibition, we would rather expect that STN DBS should increase the frequency of saccades. To overcome this apparent contradiction, we have interpreted this finding as consistent with a view that STN DBS may set the functional level of the SC appropriate both for saccade initiation and inhibition through this pathway by normalizing the excitability of the SC (Yugeta et al., 2010), thereby decreasing the occurrence of saccades to cue. However, the exact mechanism by which inhibitory control of movement is implemented in the normal brain and what makes it abnormal under dopamine deficient states cannot be explained clearly under the rate model.
Departing from the classical rate model, we should take into consideration not only the “rate” of BG activity but also its pattern (Brown, 2003). A pathological task-related modulation of oscillation within the BG is one possible explanation for the impairment of the inhibitory control of saccades. In normal monkeys performing a saccade task toward a visual target, local field potential synchronization in the β range also showed a task-related decrease, but only for recording sites that were directly engaged in the task (Courtemanche et al., 2003). This suggested that local field oscillations serve the function of filtering striatal input–output transmission (Hutchison et al., 2004). This is also the case with voluntary movements of the hand; β band oscillations are reduced prior to and during self- and externally paced voluntary movements (Cassidy et al., 2002; Williams et al., 2003; Kühn et al., 2004; Doyle et al., 2005; Kempf et al., 2007). Conversely, increased β activity is involved in movement inhibition; in a go/no-go task, the β suppression following the go/no-go signal was prematurely terminated and reverted to an early b power increase (Kühn et al., 2004). There is growing evidence that PD is associated with pathological synchronous oscillatory activity in the BG, primarily in the β range (11–30 Hz) (Bergman et al., 1998; Wichmann et al., 1999a,b; Nini et al., 1995; Raz et al., 1996; Brown, 2003). Recently, Sarma et al. (2012) showed that cues suppress pathological BG activity (β-oscillations) in STN through activation of the corticostriatal pathway. In a two-step motor task, subjects were told to perform a joy-stick movement either in response to a cue presented at a predicted and unpredicted time, or when no cue was presented at a predicted time and subjects had to make a self-initiated movement instead. The decrease in β-oscillations and increase in directional tuning (i.e., the neurons are more likely to fire in one direction more than at least one other direction) was observed only when a cue was presented at a predicted time and when subjects had to initiate a self-triggered movement when no cue was presented at the predicted time. In contrast, no modulation of oscillation and directional tuning was found during trials in which cues were presented at an unpredicted time. This pattern of results suggests that during motor planning either a predictably timed external cue or an internally generated cue (i.e., generated by the absence of a cue) trigger the execution of a motor planning in the premotor cortex, whose increased activation then suppresses pathological activity in STN through direct pathways, leading to motor facilitation in PD. If the modulation of activity is compromised, this would result in an inability to suppress unwanted saccades to the visual signal.
It should be noted, however, that neither tonic nor phasic disinhibition related to oscillatory activity in the BG can explain why the timing of periods of low inhibition just happen to coincide with the time when saccades to cue might take place involuntarily in the MGS task; a phasic disinhibition can occur any time after the presentation of the cue if this is caused by the ongoing oscillation. Therefore, introducing the abnormal discharge pattern in the BG and other neural structures still does not totally explain the defects in saccade inhibition. The precise mechanism warrants future investigation.
Another important limitation of the model is the role of BG in modulating saccades in a context dependent manner (Vokoun et al., 2011). Basso and Liu (2007) demonstrated that SNr stimulation results in only subtle effects on visually guided saccades, whereas it has more profound effects on MGS (i.e., saccades made in the absence of visual cue), also reducing the likelihood of its successful performance by around 10%. Furthermore, electrical stimulation of the SNr causes reliable reduction of the activity of SC build-up neurons, although it also leads to a decrease in saccade latency with reduced variability (Liu and Basso, 2008). During some tasks, SNr and SC neuronal activity are not mirror images of each other (Basso and Wurtz, 2002). To explain the apparent discrepancy in effects, Basso et al. suggested that the SNr initially inhibits saccade-related activity in build-up and burst neurons of the SC through its direct inputs and then disinhibits the SC via inhibitory interneurons.
Accumulating evidence suggests that, rather than simply serving to generate a saccade, both the SNr and the SC may play a role closer to saccade target selection. For example, Basso and Wurtz (1997, 1998, 2002) found that some neurons in the SC reflect changes in the probability that a particular target will be identified for saccade generation; the higher the probability that a particular location will be selected as a saccade target, the larger and longer will be the pause in SNr neuronal activity preceding saccade generation. Thus, the SNr may play a role in target selection because the pause predicted saccade choices independent of the cue and sometimes occurred well before saccade initiation (see Schall and Thompson, 1999 for review). Invoking the formulation of Jeannerod (2006), this implies that the goal of the saccades which are represented in the SC may not correspond to the actual state of the world, but rather corresponds to a possible state of saccade generation which will arise if the saccade were effective. Rather than directly issuing motor output for saccades, the multiple inputs converging onto the SC could be conceived of as an “interface” to ensure versatility of action depending on the context (i.e., to select and implement the possible candidate of motor outputs in favor of the most appropriate for the situation in which the motor action is to be executed, and then switching off the other alternatives). In the case of saccades, this allows one to turn their gaze immediately in the direction of the visual signal, delay the execution of the saccade instead of responding to it immediately, look in the direction where there is no visual input, or even select from among more than one potential target. While some types of saccades, such as MGS, are directly driven by the “double inhibition” pathway through the frontal cortex-caudate-SNr-SC, it is important to remember that the main function of the BG is not to directly implement concrete motor outputs but to modulate functional processes occurring outside the BG. In order to see how these new findings in basic neuroscience can be reconciled with the clinical findings, the role of the STN-SNr pathway in generating volitional saccades may need to be reconsidered in light of the new perspectives as discussed in the present review.
In summary, we have shown that saccade performance could be used to probe the dysfunction of BG in PD. Instead of the widely-held dichotomy of impaired voluntary versus relatively spared reflexive saccades in PD, most of the saccade abnormalities in PD could be explained by postulating various drives converging on the SC, including those from the BG. The differential treatment effects of L-dopa and DBS suggested that the various treatment modalities target different parts of the BG circuit the oculomotor system. On the other hand, some saccade abnormalities such as the impairment in inhibitory control of saccades still remain to be explained. Further elucidation the pathophysiological basis of PD and the mechanism of action of various therapies will be useful in establishing more effective treatment strategies in this disorder, and enable us to use eye movement recording as an indicator of treatment effects.
HIGHLIGHTS.
We review the pathophysiology of basal ganglia (BG) dysfunction in Parkinson’s disease (PD) based on saccade performance.
Saccade abnormalities in PD may be caused by the excessive inhibition of the superior colliculus (SC) due to the increased BG output and decreased activity of the frontal cortex-BG circuit, as well as impaired suppression of reflexive saccades that may be explained by the “leaky” suppression of the SC.
Treatment of PD, such as L-dopa therapy and deep brain stimulation, works by normalizing these abnormal BG functions, but in different ways.
Acknowledgements
This work was partly supported by grants from a Research Project Grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and research funds from the Magnetic Health Science Foundation and the Kato Memorial Trust for Nambyo Research, Japan. This work was partly supported by the intramural research program at the National Eye Institute (Dr. Okihide Hikosaka).
References
- Albin RL, Young AB, Penney JB. The functional anatomy of disorders of the basal ganglia. Trends Neurosci 1995;18:63–4. [PubMed] [Google Scholar]
- Armstrong IT, Chan F, Riopelle RJ, Munoz DP. Control of saccades in Parkinson’s disease. Brain Cogn 2002;49:198–201. [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 1990;13:266–71. [DOI] [PubMed] [Google Scholar]
- Amador N, Schlag-Rey M, Schlag J. Primate antisaccades. I. Behavioral characteristics. J Neurophysiol 1998;80:1775–86. [DOI] [PubMed] [Google Scholar]
- Anastasopoulos D, Ziavra N, Savvidou E, Bain P, Bronstein AM. Altered eye-to-foot coordination in standing parkinsonian patients during large gaze and whole-body reorientations. Mov Disord 2011;26:2201–11. [DOI] [PubMed] [Google Scholar]
- Antoniades CA, Carpenter RHS, Yasin Temel Y. Deep brain stimulation of the subthalamic nucleus in Parkinson’s disease: similar improvements in saccadic and manual responses. Neuroreport 2012;23:179–83. [DOI] [PubMed] [Google Scholar]
- Bares M, Brázdil M, Kanovský P, Jurák P, Daniel P, Kukleta M, Rektor I. The effect of apomorphine administration on smooth pursuit ocular movements in early Parkinsonian patients. Parkinsonism Relat Disord 2003;9:139–44. [DOI] [PubMed] [Google Scholar]
- Bares M, Lungu OV, Liu T, Waechter T, Gomez CM, Ashe J. The neural substrate of predictive motor timing in spinocerebellar ataxia. Cerebellum 2011;10:233–44. [DOI] [PubMed] [Google Scholar]
- Basso MA, Liu P. Context-dependent effects of substantia nigra stimulation on eye movements. J Neurophysiol 2007;97:4129–42. [DOI] [PubMed] [Google Scholar]
- Basso MA, Wurtz RH. Modulation of neuronal activity by target uncertainty. Nature 1997;389:66–9. [DOI] [PubMed] [Google Scholar]
- Basso MA, Wurtz RH. Modulation of neuronal activity in superior colliculus by changes in target probability. J Neurosci 1998;18:7519–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso MA, Wurtz RH. Neuronal activity in substantia nigra pars reticulata during target selection. Neuroscience 2002;22:1883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardelli A, Rothwell JC, Thompson PD, Hallett M. Pathophysiology of bradykinesia in Parkinson’s disease. Brain 2001;124:2131–46. [DOI] [PubMed] [Google Scholar]
- Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, et al. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci 1998;21:32–8. [DOI] [PubMed] [Google Scholar]
- Biscaldi M, Fischer B, Stuhr V. Human express saccade makers are impaired at suppressing visually evoked saccades. J Neurophysiol 1996;76:199–214. [DOI] [PubMed] [Google Scholar]
- Blekher T, Siemers E, Abel LA, Yee RD. Eye movements in Parkinson’s disease: before and after pallidotomy. Invest Ophthalmol Vis Sci 2000;41:2177–83. [PubMed] [Google Scholar]
- Blekher T, Weaver M, Rupp J, Nichols WC, Hui SL, Gray J, et al. Multiple step pattern as a biomarker in Parkinson disease. Parkinsonism Relat Disord 2009;15:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- Briand KA, Strallow D, Hening W, Poizner H, Sereno AB. Control of voluntary and reflexive saccades in Parkinson’s disease. Exp Brain Res 1999;129:38–48. [DOI] [PubMed] [Google Scholar]
- Briand KA, Hening W, Poizner H, Sereno AB. Automatic orienting of visuospatial attention in Parkinson’s disease. Neuropsychologia 2001;39:1240–9. [DOI] [PubMed] [Google Scholar]
- Bronstein AM, Kennard C. Predictive ocular motor control in Parkinson’s disease. Brain 1985;108:925–40. [DOI] [PubMed] [Google Scholar]
- Brown J, Bullock D, Grossberg S. How the basal ganglia use parallel excitatory and inhibitory learning pathways to selectively respond to unexpected rewarding cues. J Neurosci 1999;19:10502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson’s disease. Mov Disord 2003;18:357–63. [DOI] [PubMed] [Google Scholar]
- Brown RG, Marsden CD. Internal versus external cues and the control of attention in Parkinson’s disease. Brain 1988;111:323–45. [DOI] [PubMed] [Google Scholar]
- Brown MR, DeSouza JF, Goltz HC, Ford K, Menon RS, Goodale MA, et al. Comparison of memory and visually guided saccades using event-related fMRI. J Neurophysiol 2004;91:873–89. [DOI] [PubMed] [Google Scholar]
- Carpenter RHS. Contrast, probability, and saccadic latency: evidence for independence of detection and decision. Curr Biol 2004;14:1576–80. [DOI] [PubMed] [Google Scholar]
- Cassidy M, Mazzone P, Oliviero A, Insola A, Tonali P, Di Lazzaro, et al. Movement-related changes in synchronization in the human basal ganglia. Brain 2002;125:1235–46. [DOI] [PubMed] [Google Scholar]
- Chambers JM, Prescott TJ. Response times for visually guided saccades in persons with Parkinson’s disease: a meta-analytic review. Neuropsychologia 2010;48:887–99. [DOI] [PubMed] [Google Scholar]
- Chan F, Armstrong IT, Pari G, Riopelle RJ, Munoz DP. Deficits in saccadic eye-movement control in Parkinson’s disease. Neuropsychologia 2005;43:784–96. [DOI] [PubMed] [Google Scholar]
- Condy C, Rivaud-Péchoux S, Ostendorf F, Ploner CJ, Gaymard B. Neural substrate of antisaccades: role of subcortical structures. Neurology 2004;63:1571–8. [DOI] [PubMed] [Google Scholar]
- Condy C, Wattiez N, Rivaud-Péchoux S, Tremblay L, Gaymard B. Antisaccade deficit after inactivation of the principal sulcus in monkeys. Cereb Cortex 2007;17:221–9. [DOI] [PubMed] [Google Scholar]
- Courtemanche R, Fujii N, Graybiel AM. Synchronous, focally modulated beta-band oscillations characterize local field potential activity in the striatum of awake behaving monkeys. J Neurosci 2003;23:11741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T, Goodrich S, Henderson L, Kennard C. Predictive responses in Parkinson’s disease: manual keypresses and saccadic eye movements to regular stimulus events. J Neurol Neurosurg Psychiatry 1989;52:1033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevits L, De Ridder K. Disturbed striatoprefrontal mediated visual behaviour in moderate to severe parkinsonian patients. J Neurol Neurosurg Psychiatry 1997;63:296–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevits L, Versijpt J, Hanse M, De Ridder K. Antisaccadic effects of a dopamine agonist as add-on therapy in advanced Parkinson’s patients. Neuropsychobiology 2000;42:202–6. [DOI] [PubMed] [Google Scholar]
- DeJong JD, Jones GM. Akinesia, hypokinesia, and bradykinesia in the oculomotor system of patients with Parkinson’s disease. Exp Neurol 1971;32:58–68. [DOI] [PubMed] [Google Scholar]
- Di Fabio RP, Greany J, Zampieri C. Saccade-stepping interactions revise the motor plan for obstacle avoidance. J Mot Behav 2003;35:383–97. [DOI] [PubMed] [Google Scholar]
- Di Fabio RP, Zampieri C, Henke J, Olsen K, Rickheim D, Russell M. Influence of elderly executive cognitive function on attention in the lower visual field during step initiation. Gerontology 2005;51:94–107. [DOI] [PubMed] [Google Scholar]
- Dorris MC, Paré M, Munoz DP. Neuronal activity in monkey superior colliculus related to the initiation of saccadic eye movements. J Neurosci 1997;17:8566–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorris MC, Munoz DP. Saccadic probability influences motor preparation signals and time to saccadic initiation. J Neurosci 1998;18:7015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LM, Kuhn AA, Hariz M, Kupsch A, Schneider GH, Brown P. Levodopa-induced modulation of subthalamic beta oscillations during self-paced movements in patients with Parkinson’s disease. Eur J Neurosci 2005;21:1403–12. [DOI] [PubMed] [Google Scholar]
- Everling S, Fischer B. The antisaccade: a review of basic research and clinical studies. Neuropsychologia 1998;36:885–99. [DOI] [PubMed] [Google Scholar]
- Fawcett AP, González EG, Moro E, Steinbach MJ, Lozano AM, Hutchison WD. Subthalamic nucleus deep brain stimulation improves saccades in Parkinson’s disease. Neuromodulation 2010;13:17–25. [DOI] [PubMed] [Google Scholar]
- Fawcett AP, Moro E, Lang AE, Lozano AM, Hutchison WD. Pallidal deep brain stimulation influences both reflexive and voluntary saccades in Huntington’s disease. Mov Disord 2005;20:371–86. [DOI] [PubMed] [Google Scholar]
- Fecteau JH, Bell AH, Munoz DP. Neural correlates of the automatic and goal-driven biases in orienting spatial attention. J Neurophysiol 2004;92:1728–37. [DOI] [PubMed] [Google Scholar]
- Flaherty AW, Rost NS. The Massachusetts General Hospital Handbook of Neurology. 2nd ed. New York: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- Fukushima J, Fukushima K, Miyasaka K, Yamashita I. Voluntary control of saccadic eye movements in patients with frontal cortical lesions and parkinsonian patients in comparison with that in schizophrenics. Biol Psychiatry 1994;36:21–30. [DOI] [PubMed] [Google Scholar]
- Gaymard B, François C, Ploner CJ, Condy C, Rivaud-Péchoux S. A direct prefrontotectal tract against distractibility in the human brain. Ann Neurol 2003a;53:542–5. [DOI] [PubMed] [Google Scholar]
- Gaymard B, Lynch J, Ploner CJ, Condy C, Rivaud-Péchoux S. The parieto-collicular pathway: anatomical location and contribution to saccade generation. Eur J Neurosci 2003b;17:1518–26. [DOI] [PubMed] [Google Scholar]
- Guitton D, Buchtel HA, Douglas RM. Frontal lobe lesions in man cause difficulties in suppressing reflexive glances and in generating goal-directed saccades. Exp Brain Res 1985;58:455–72. [DOI] [PubMed] [Google Scholar]
- Hallett M Bradykinesia: why do Parkinson’s patients have it and what trouble does it cause? Mov Disord 2011;26:1579–81. [DOI] [PubMed] [Google Scholar]
- Hanes DP, Schall JD. Neural control of voluntary movement initiation. Science 1996;274:427–30. [DOI] [PubMed] [Google Scholar]
- Hanes DP, Wurtz RH. Interaction of the frontal eye field and superior colliculus for saccade generation. J Neurophysiol 2001;85:804–15. [DOI] [PubMed] [Google Scholar]
- Helmich RC, Janssen MJ, Oyen WJ, Bloem BR, Toni I. Pallidal dysfunction drives a cerebellothalamic circuit into Parkinson tremor. Ann Neurol 2011;69:269–81. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Fukuda H, Segawa M, Nomura Y. Voluntary saccades in normal and in dopamine-deficient subjects. In: Segawa M, Nomura Y, editors. Age-related dompamine-dependent disorders. Monographs in neural sciences, vol. 14. Basel: Karger; 1995. p. 59–68. [Google Scholar]
- Hikosaka O, Fukuda H, Kato M, Uetake K, Nomura Y, Segawa M. Deficits in saccadic eye movements in hereditary progressive dystonia with marked diurnal fluctuation. In: Segawa M, editor. Hereditary progressive dystonia with marked diurnal fluctuation. Nashville: Parthenon Publishing Group; 1993. p. 159–77. [Google Scholar]
- Hikosaka O, Sakamoto M, Usui S. Functional properties of monkey caudate neurons. I. Activities related to saccadic eye movements. J Neurophysiol 1989;61:780–98. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Takikawa Y, Kawagoe R. Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol Rev 2000;80:953–78. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Wurtz RH. Visual and oculomotor functions of monkey substantia nigra pars reticulata. III. Memory-contingent visual and saccade responses. J Neurophysiol 1983;49:1268–84. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Wurtz RH. Modification of saccadic eye movements by GABA-related substances. I. Effect of muscimol and bicuculline in monkey superior colliculus. J Neurophysiol 1985a;53:266–91. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Wurtz RH. Modification of saccadic eye movements by GABA-related substances. II. Effects of muscimol in monkey substantia nigra pars reticulata. J Neurophysiol 1985b;53:292–308. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Wurtz RH. The basal ganglia. Rev Oculomot Res 1989;3:257–81. [PubMed] [Google Scholar]
- Hodgson T, Chamberlain M, Parris B, James M, Gutowski N, Husain M, et al. The role of the ventrolateral frontal cortex in inhibitory oculomotor control. Brain 2007;130:1525–37. [DOI] [PubMed] [Google Scholar]
- Hollands MA, Patla AE, Vickers JN. “Look where you’re going!”: gaze behaviour associated with maintaining and changing the direction of locomotion. Exp Brain Res 2002;143:221–30. [DOI] [PubMed] [Google Scholar]
- Hood AJ, Amador SC, Cain AE, Briand KA, Al-Refai AH, Schiess MC, et al. Levodopa slows prosaccades and improves antisaccades: an eye movement study in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2007;78:565–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husárová I, Lungu OV, Mareček R, Mikl M, Gescheidt T, Krupa P, Bareš M. Functional imaging of the cerebellum and basal ganglia during predictive motor timing in early Parkinson’s disease. J Neuroimaging 2011. 10.1111/j.1552-6569.2011.00663.x. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Dostrovsky JO, Walters JR, Courtemanche R, Boraud T, Goldberg J, et al. Neuronal oscillations in the basal ganglia and movement disorders: evidence from whole animal and human recordings. J Neurosci 2004;24:9240–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeannerod M Representations for actions. In: Jeannerod M, editor. Motor cognition. Oxford: Oxford University Press; 2006. p. 1–21. [Google Scholar]
- Jellinger KA. The pathology of Parkinson’s disease. Adv Neurol 2001;86:55–72. [PubMed] [Google Scholar]
- Jones GM, DeJong JD. Dynamic characteristics of saccadic eye movements in Parkinson’s disease. Exp Neurol 1971;31:17–31. [DOI] [PubMed] [Google Scholar]
- Joti P, Kulashekhar S, Behari M, Murthy A. Impaired inhibitory oculomotor control in patients with Parkinson’s disease. Exp Brain Res 2007;177:447–57. [DOI] [PubMed] [Google Scholar]
- Kato M, Miyashita N, Hikosaka O, Matsumura M, Usui S, Kori A. Eye movements in monkeys with local dopamine depletion in the caudate nucleus. I. Deficits in spontaneous saccades. J Neurosci 1995;15:912–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf F, Kühn AA, Kupsch A, Brucke C, Weise L, Schneider GH, et al. Premovement activities in the subthalamic area of patients with Parkinson’s disease and their dependence on task. Eur J Neurosci 2007;25:3137–45. [DOI] [PubMed] [Google Scholar]
- Kimmig H, Haussmann K, Mergner T, Lucking CH. What is pathological with gaze shift fragmentation in Parkinson’s disease? J Neurol 2002;249:683–92. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Fukushima J, Tashiro K. Relationship between antisaccades and the clinical symptoms in Parkinson’s disease. Neurology 1994;44:2285–9. [DOI] [PubMed] [Google Scholar]
- Kori A, Miyashita N, Kato M, Hikosaka O, Usui S, Matsumura M. Eye movements in monkeys with local dopamine depletion in the caudate nucleus. II. Deficits in voluntary saccades. J Neurosci 1995;15:928–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn A, Williams D, Kupsch A, Limousin P, Hariz M, Schneider GH, et al. Event related beta desynchronization in human subthalamic nucleus correlates with motor performance. Brain 2004;127:735–46. [DOI] [PubMed] [Google Scholar]
- Kumru H, Summerfield C, Valldeoriola F, Valls-Solé J. Effects of subthalamic nucleus stimulation on characteristics of EMG activity underlying reaction time in Parkinson’s disease. Mov Disord 2004;19:94–100. [DOI] [PubMed] [Google Scholar]
- Lasker AG, Zee DS. Ocular motor abnormalities in Huntington’s disease. Vision Res 1997;37:3639–45. [DOI] [PubMed] [Google Scholar]
- Leigh RJ, Zee DS. The neurology of eye movements. 4th ed. Oxford: Oxford University Press; 2006. [Google Scholar]
- Liu P, Basso MA. Substantia nigra stimulation influences monkey superior colliculus neuronal activity bilaterally. J Neurophysiol 2008;100:1098–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueck CJ, Tanyeri S, Crawford TJ, Henderson L, Kennard C. Antisaccades and remembered saccades in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1990;53:284–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill MR, Graham CF, Pitcher TL, Myall DJ, Livingston L, van Stockum S, Dalrymple-Alford JC, Anderson TJ. The influence of motor and cognitive impairment upon visually-guided saccades in Parkinson’s disease. Neuropsychologia 2012;50:3338–47. [DOI] [PubMed] [Google Scholar]
- MacAskill MR, Anderson TJ, Jones RD. Adaptive modification of saccade amplitude in Parkinson’s disease. Brain 2002;125:1570–82. [DOI] [PubMed] [Google Scholar]
- Machado L, Rafal RD. Control of fixation and saccades in humans with chronic lesions of oculomotor cortex. Neuropsychology 2004;18:115–23. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Terao Y, Furubayashi T, Yugeta A, Fukuda H, Emoto M, et al. Basal ganglia dysfunction reduces saccade amplitude during visual scanning in Parkinson’s disease. Basal Ganglia 2012;2:73–8. [Google Scholar]
- Matsumoto H, Terao Y, Furubayashi T, Yugeta A, Fukuda H, Emoto M, et al. Small saccades restrict visual scanning area in Parkinson’s disease. Mov Disord 2011;26:1619–26. [DOI] [PubMed] [Google Scholar]
- Michell AW, Xu Z, Fritz D, Lewis SJ, Foltynie T, Williams-Gray CH, et al. Saccadic latency distributions in Parkinson’s disease and the effects of L-dopa. Exp Brain Res 2006;174:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita N, Hikosaka O, Kato M. Visual hemineglect induced by unilateral striatal dopamine deficiency in monkeys. Neuroreport 1995;6:1257–60. [DOI] [PubMed] [Google Scholar]
- Munoz DP, Armstrong IT, Hampton KA, Moore KD. Altered control of visual fixation and saccadic eye movements in attention-deficit hyperactivity disorder. J Neurophysiol 2003;90:503–14. [DOI] [PubMed] [Google Scholar]
- Munoz DP, Broughton JR, Goldring JE, Armstrong IT. Age-related performance of human subjects on saccadic eye movement tasks. Exp Brain Res 1998;121:391–400. [DOI] [PubMed] [Google Scholar]
- Munoz DP, Dorris MC, Paré M, Everling S. On your mark, get set: brainstem circuitry underlying saccadic initiation. Can J Physiol Pharmacol 2000;78:934–44. [PubMed] [Google Scholar]
- Munoz DP, Fecteau JH. Vying for dominance: dynamic interactions control visual fixation and saccadic initiation in the superior colliculus. Prog Brain Res 2002;140:3–19. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Bronstein AM, Lueck C, Marsden CD, Rudge P. Vestibular, cervical and visual remembered saccades in Parkinson’s disease. Brain 1994;117:1423–32. [DOI] [PubMed] [Google Scholar]
- Neggers SF, Raemaekers MA, Lampmann EE, Postma A, Ramsey NF. Cortical and subcortical contributions to saccade latency in the human brain. Eur J Neurosci 2005;21:2853–63. [DOI] [PubMed] [Google Scholar]
- Nieman DH, Bour LJ, Linszen DH, Goede J, Koelman JH, Gersons BP, et al. Neuropsychological and clinical correlates of antisaccade task performance in schizophrenia. Neurology 2000;54:866–71. [DOI] [PubMed] [Google Scholar]
- Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of Parkinson. J Neurophysiol 1995;74:1800–5. [DOI] [PubMed] [Google Scholar]
- O’Sullivan JD, Maruff P, Tyler P, Peppard RF, McNeill P, Currie J. Unilateral pallidotomy for Parkinson’s disease disrupts ocular fixation. J Clin Neurosci 2003;10:181–5. [DOI] [PubMed] [Google Scholar]
- O’Driscoll GA, Alpert NM, Matthysse SW, Levy DL, Rauch SL, Holzman PS. Functional neuroanatomy of antisaccade eye movements investigated with positron emission tomography. Proc Natl Acad Sci USA 1995;92:925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré M, Hanes DP. Controlled movement processing: superior colliculus activity associated with countermanded saccades. J Neurosci 2003;23:6480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltsch A, Hoffman A, Armstrong I, Pari G, Munoz DP. Saccadic impairments in Huntington’s disease. Exp Brain Res 2008;186:457–69. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny C, Rivaud S, Pillon B, Fournier E, Agid Y. Lateral visually-guided saccades in progressive supranuclear palsy. Brain 1989;112:471–87. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny C Cortical control of saccades in man. Acta Neurol Belg 1991;91:63–79. [DOI] [PubMed] [Google Scholar]
- Pierrot-Deseilligny C, Milea D, Müri R. Eye movement control by the cerebral cortex. Curr Opin Neurol 2004;17:17–25. [DOI] [PubMed] [Google Scholar]
- Praamstra P, Stegeman DF, Cools AR, Horstink MW. Reliance on external cues for movement initiation in Parkinson’s disease. Evidence from movement-related potentials. Brain 1998;121:167–77. [DOI] [PubMed] [Google Scholar]
- Rascol O, Clanet M, Montastruc JL, Simonetta M, Soulier-Esteve MJ, Doyon B, et al. Abnormal ocular movements in Parkinson’s disease: evidence for involvement of dopaminergic systems. Brain 1989;112:1193–214. [DOI] [PubMed] [Google Scholar]
- Rascol O, Sabatini U, Fabre N, Brefel C, Loubinoux I, Celsis P, et al. The ipsilateral cerebellar hemisphere is overactive during hand movements in akinetic parkinsonian patients. Brain 1997;120:103–10. [DOI] [PubMed] [Google Scholar]
- Raz A, Feingold A, Zelanskaya V, Vaadia E, Bergman H. Neuronal synchronization of tonically active neurons in the striatum of normal and parkinsonian primates. J Neurophysiol 1996;76:2083–8. [DOI] [PubMed] [Google Scholar]
- Reddi BA, Carpenter RH. The influence of urgency on decision time. Nat Neurosci 2000;3:827–30. [DOI] [PubMed] [Google Scholar]
- Rivaud-Péchoux S, Vermersch AI, Gaymard B, Ploner CJ, Bejjani BP, Damier P, et al. Improvement of memory guided saccades in parkinsonian patients by high frequency subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry 2000a;68:381–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivaud-Péchoux S, Vidailhet M, Gallouedec G, Litvan I, Gaymard B, Pierrot-Deseilligny C. Longitudinal ocular motor study in corticobasal degeneration and progressive supranuclear palsy. Neurology 2000b;54:1029–32. [DOI] [PubMed] [Google Scholar]
- Reuter B, Kathmann N. Using saccade tasks as a tool to analyze executive dysfunctions in schizophrenia. Acta Psychol (Amst) 2004;115:255–69. [DOI] [PubMed] [Google Scholar]
- Roll A, Wierzbicka MM, Wolf W. The “gap paradigm” leads to express-like saccadic reaction times in Parkinson’s disease. Exp Brain Res 1996;111:131–8. [DOI] [PubMed] [Google Scholar]
- Rottach KG, Riley DE, DiScenna AO, Zivotofsky AZ, Leigh RJ. Dynamic properties of horizontal and vertical eye movements in parkinsonian syndromes. Ann Neurol 1996;39:368–77. [DOI] [PubMed] [Google Scholar]
- Sarma SV, Cheng ML, Eden U, Williams Z, Brown EN, Eskandar E. The effects of cues on neurons in the basal ganglia in Parkinson’s disease. Front Integr Neurosci 2012;6:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauleau P, Pollak P, Krack P, Courjon JH, Vighetto A, Benabid AL, et al. Subthalamic stimulation improves orienting gaze movements in Parkinson’s disease. Clin Neurophysiol 2008;119:1857–63. [DOI] [PubMed] [Google Scholar]
- Schall JD, Thompson KG. Neural selection and control of visually guided eye movements. Ann Rev Neurosci 1999;22:241–59. [DOI] [PubMed] [Google Scholar]
- Segawa M, Nishiyama N, Nomura Y. Dopa-responsive dystonic parkinsonism – pathophysiologic considerations. Adv Neurol 1999;80:389–400. [PubMed] [Google Scholar]
- Sereno AB. Parsing cognitive processes: psychological and neurophysiological constraints. In: Matthysse S, Levy DL, Kagan J, Benes FM, editors. Psychopathology: the evolving science of mental disorder. New York: Cambridge University Press; 1996. p. 407–32. [Google Scholar]
- Shibasaki H, Tsuji S, Kuroiwa Y. Oculomotor abnormalities in Parkinson’s disease. Arch Neurol 1979;36:360–4. [DOI] [PubMed] [Google Scholar]
- Shook BL, Schlag-Rey M, Schlag J. Primate supplementary eye field: I. Comparative aspects of mesencephalic and pontine connections. Comp Neurol 1990;301:618–42. [DOI] [PubMed] [Google Scholar]
- Shaikh AG, Xu-Wilson M, Grill S, Zee DS. ‘Staircase’ square-wave jerks in early Parkinson’s disease. Br J Ophthalmol 2011;95:705–9. [DOI] [PubMed] [Google Scholar]
- Sparks D, Rohrer WH, Zhang Y. The role of the superior colliculus in saccade initiation: a study of express saccades and the gap effect. Vision Res 2000;40:2763–77. [DOI] [PubMed] [Google Scholar]
- Stanton GB, Deng SY, Goldberg ME, McMullen NT. Cytoarchitectural characteristic of the frontal eye fields in macaque monkeys. J Comp Neurol 1989;282:415–27. [DOI] [PubMed] [Google Scholar]
- Sweeney JA, Mintun MA, Kwee S, Wiseman MB, Brown DL, Rosenberg DR, et al. Positron emission tomography study of voluntary saccadic eye movements and spatial working memory. J Neurophysiol 1996;75:454–68. [DOI] [PubMed] [Google Scholar]
- Temel Y, Visser-Vandewalle V, Carpenter RHS. Saccadic latency during electrical stimulation of the human subthalamic nucleus. Curr Biol 2008;16:R412–4. [DOI] [PubMed] [Google Scholar]
- Terao Y, Andersson NE, Flanagan JR, Johansson RS. Engagement of gaze in capturing targets for future sequential manual actions. J Neurophysiol 2002;88:1716–25. [DOI] [PubMed] [Google Scholar]
- Terao Y, Fukuda H, Shirota Y, Yugeta A, Yoshioka M, Suzuki M, et al. Deterioration of horizontal saccades in progressive supranuclear palsy. Clin Neurophysiol 2013;124:354–63. [DOI] [PubMed] [Google Scholar]
- Terao Y, Fukuda H, Ugawa Y, Hikosaka O, Furubayashi T, Hanajima R, et al. Visualization of the information through human oculomotor cortical regions by transcranial magnetic stimulation. J Neurophysiol 1998;80:936–46. [DOI] [PubMed] [Google Scholar]
- Terao Y, Fukuda H, Yugeta A, Hikosaka O, Hanajima R, Ugawa Y, et al. Saccade abnormalities in Segawa disease – pathophysiological difference between postural and action dystonia. Mov Disord 2012;27(Suppl. 1):S70. [Google Scholar]
- Terao Y, Fukuda H, Yugeta A, Hikosaka O, Nomura Y, Segawa M, et al. Initiation and inhibitory control of saccades with the progression of Parkinson’s disease – changes in three major drives converging on the superior colliculus. Neuropsychologia 2011a;49:1794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao Y, Furubayashi T, Fukuda H, Hanajima R, Yugeta A, Hamada M, et al. Frontal cortical regions controlling small and large amplitude saccades: a TMS study. Basal Ganglia 2011b;1:221–9. [Google Scholar]
- Teräväinen H, Calne DB. Studies of parkinsonian movement: 1. Programming and execution of eye movements. Acta Neurol Scand 1980a;62:137–48. [DOI] [PubMed] [Google Scholar]
- Teräväinen H, Calne DB. Studies of parkinsonian movement: 2. Initiation of fast voluntary eye movement during postural disturbance. Acta Neurol Scand 1980b;62:149–57. [DOI] [PubMed] [Google Scholar]
- van Donkelaar P, Saavedra S, Woollacott M. Multiple saccades are more automatic than single saccades. J Neurophysiol 2007;97:3148–51. [DOI] [PubMed] [Google Scholar]
- van Koningsbruggen MG, Pender T, Machado L, Rafal RD. Impaired control of the oculomotor reflexes in Parkinson’s disease. Neuropsychologia 2009;47:2909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Stockum S, MacAskill M, Anderson T, Dalrymple-Alford J. Don’t look now or look away: two sources of saccadic disinhibition in Parkinson’s disease? Neuropsychologia 2008;46:3108–15. [DOI] [PubMed] [Google Scholar]
- van Stockum S, MacAskill MR, Anderson TJ. Impairment of voluntary saccades and facilitation of reflexive saccades do not co-occur in Parkinson’s disease. J Clin Neurosci 2012;19:1119–24. [DOI] [PubMed] [Google Scholar]
- Ventre J, Zee DS, Papageorgiou H, Reich S. Abnormalities of predictive saccades in hemi-Parkinson’s disease. Brain 1992;115:1147–65. [DOI] [PubMed] [Google Scholar]
- Vidailhet M, Rivaud S, Gouider-Khouja N, Pillon B, Bonnet AM, Gaymard B, et al. Eye movements in parkinsonian syndromes. Ann Neurol 1994;35:420–6. [DOI] [PubMed] [Google Scholar]
- Vermersch AI, Rivaud S, Vidailhet M, Bonnet AM, Gaymard B, Agid Y, et al. Sequences of memory guided saccades in Parkinson’s disease. Ann Neurol 1994;35:487–90. [DOI] [PubMed] [Google Scholar]
- Vokoun CR, Mahamed S, Basso MA. Saccadic eye movements and the basal ganglia. In: Liversedge S, Gilchrist I, Everling S, editors. Handbook of eye movement. Oxford: Oxford University Press; 2011. p. 215–33. [Google Scholar]
- Wark HA, Garell PC, Walker AL, Basso MA. A case report on fixation instability in Parkinson’s disease with bilateral deep brain stimulation implants. J Neurol Neurosurg Psychiatry 2008;79:443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Munoz DP. Probing basal ganglia functions by saccade eye movements. Eur J Neurosci 2011;33:2070–90. [DOI] [PubMed] [Google Scholar]
- Waterston JA, Barnes GR, Grealy MA, Collins S. Abnormalities of smooth eye and head movement control in Parkinson’s disease. Ann Neurol 1996;39:749–60. [DOI] [PubMed] [Google Scholar]
- White OB, Saint-Cyr JA, Tomlinson RD, Sharpe JA. Ocular motor deficits in Parkinson’s disease. II. Control of the saccadic and smooth pursuit systems. Brain 1983;106:571–87. [DOI] [PubMed] [Google Scholar]
- White OB, Saint-Cyr JA, Tomlinson RD, Sharpe JA. Ocular motor deficits in Parkinson’s disease. III. Coordination of eye and head movements. Brain 1988;111:115–29. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. I. Functional properties in intact animals. J Neurophysiol 1999a;72:494–506. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp Brain Res 1999b;125:397–409. [DOI] [PubMed] [Google Scholar]
- Williams D, Kuhn A, Kupsch A, Tijssen M, van Bruggen G, Speelman H, et al. Behavioural cues are associated with modulations of synchronous oscillations in the human subthalamic nucleus. Brain 2003;126:1975–85. [DOI] [PubMed] [Google Scholar]
- Wu T, Hallett M. A functional MRI study of automatic movements in patients with Parkinson’s disease. Brain 2005;15:15. [DOI] [PubMed] [Google Scholar]
- Yugeta A, Terao Y, Fukuda H, Ugawa Y. Effects of levodopa on saccade performance in Parkinson’s disease. Mov Disord 2008;23(Suppl. 1):S296. [Google Scholar]
- Yugeta A, Terao Y, Fukuda H, Hikosaka O, Yokochi F, Okiyama R, et al. Effects of STN stimulation on the initiation and inhibition of saccade in Parkinson disease. Neurology 2010;74:743–8. [DOI] [PubMed] [Google Scholar]
- Yu H, Sternad D, Corcos DM, Vaillancourt DE. Role of hyperactive cerebellum and motor cortex in Parkinson’s disease. Neuroimage 2007;35:222–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri C, Di Fabio RP. Improvement of gaze control after balance and eye movement training in patients with progressive supranuclear palsy: a quasi-randomized controlled trial. Arch Phys Med Rehabil 2009;90:263–70. [DOI] [PubMed] [Google Scholar]