

Abstract
目的
探索非综合征型唇裂伴或不伴腭裂(non-syndromic cleft lip with or without cleft palate, NSCL/P)全基因组常见遗传变异对NSCL/P风险的影响。
方法
利用全基因组关联研究(genome-wide association study, GWAS)数据,以全基因组单核苷酸多态性(single nucleotide polymorphism, SNP)遗传度和基因组不同分区SNP遗传度评估基因组上常见变异的效应。对GWAS汇总数据进行质量控制,标准包括数据中无缺失值、弱势等位基因频率≥1%、P值在0~1、SNP正负链明确等。利用连锁不平衡得分回归计算NSCL/P的SNP遗传度,采用分层的连锁不平衡得分回归计算基因组编码区、启动子区、内含子区、增强子区和超级增强子区的分区SNP遗传度,并评估不同分区内的富集度,分析工具为LDSC (v1.0.1)软件。
结果
纳入中国人群806个NSCL/P核心家系(2 418人)的GWAS数据,490 593个SNP通过质量控制,被纳入到SNP遗传度的计算中。观测样本中NSCL/P的SNP遗传度为0.55(95%CI: 0.28~0.82), 由于观测样本患病率较高,按中国人群患病率转换为一般人群后SNP遗传度为0.37(95%CI: 0.19~0.55)。SNP遗传度在增强子区的富集度为15.70(P=0.04),在超级增强子区的富集度为3.18(P=0.03)。
结论
基因组常见变异有助于解释一部分中国人群NSCL/P目前未被解释的遗传度,同时中国人群NSCL/P的SNP遗传度在增强子分区和超级增强子分区中显著富集,提示该区域中可能存在未被发现的遗传致病因素。
Keywords: 非综合征型唇裂伴或不伴腭裂, 单核苷酸多态性遗传度, 核心家系
Abstract
Objective
To delve into the intricate relationship between common genetic variations across the entire genome and the risk of non-syndromic cleft lip with or without cleft palate (NSCL/P).
Methods
Utilizing summary statistics data from genome-wide association studies (GWAS), a thorough investigation to evaluate the impact of common variations on the genome were undertook. This involved assessing single nucleotide polymorphism (SNP) heritability across the entire genome, as well as within specific genomic regions. To ensure the robustness of our analysis, stringent quality control measures were applied to the GWAS summary statistics data. Criteria for inclusion encompassed the absence of missing values, a minor allele frequency ≥1%, P-values falling within the range of 0 to 1, and clear SNP strand orientation. SNP meeting these stringent criteria were then meticulously included in our analysis. The SNP heritability of NSCL/P was calculated using linkage disequilibrium score regression. Additionally, hierarchical linkage disequilibrium score regression to partition SNP heritability within coding regions, promoters, introns, enhancers, and super enhancers were employed, and the enrichment levels within different genomic regions using LDSC (v1.0.1) software were further elucidated.
Results
Our study drew upon GWAS summary statistics data obtained from 806 NSCL/P trios, comprising a total of 2 418 individuals from the Chinese population. Following rigorous quality control procedures, 490 593 out of 492 993 SNP were deemed suitable for inclusion in SNP heritability calculations. The observed SNP heritability of NSCL/P was 0.55 (95%CI: 0.28-0.82). Adjusting for the elevated disease pre-valence within our sample, the SNP heritability scaled down to 0.37 (95%CI: 0.19-0.55) based on the prevalence observed in the general Chinese population. Notably, our enrichment analysis unveiled significant enrichment of SNP heritability within enhancer regions (15.70, P=0.04) and super enhancer regions (3.18, P=0.03).
Conclusion
Our study sheds light on the intricate interplay between common genetic variations and the risk of NSCL/P in the Chinese population. By elucidating the SNP heritability landscape across different genomic regions, we contribute valuable insights into the genetic basis of NSCL/P. The significant enrichment of SNP heritability within enhancer and super enhancer regions underscores the potential role of these regulatory elements in shaping the genetic susceptibility to NSCL/P. This paves the way for further research aimed at uncovering novel genetic pathogenic factors underlying NSCL/P pathogenesis.
Keywords: Non-syndromic cleft lip with or without cleft palate, Single nucleotide polymorphism heritability, Case-parent trios
非综合征型唇腭裂(non-syndromic oral clefts, NSOC)是常见的出生缺陷,其中非综合征型唇裂伴或不伴腭裂(non-syndromic cleft lip with or without cleft palate, NSCL/P)是最常见的亚型[1]。中国活产儿唇腭裂患病率约为1.4/1 000[2]。遗传度是评价遗传与环境对表型贡献的指标,它衡量的是遗传因素所能解释表型方差的占比。双生子研究和家系研究均表明遗传因素是NSCL/P重要的危险因素,其遗传度为70%[3-4]。NSCL/P是一种多基因疾病,尽管国内外多个全基因组关联研究(genome-wide association study, GWAS)发现了NSCL/P的阳性位点,但这些潜在致病位点所能解释的遗传度合计仅为20%左右[5], 其遗传病因尚不完全清晰,仍存在未被解释的遗传度,即尚有致病性遗传因素有待挖掘。目前学界认为尚未被解释的遗传度可能是因为许多常见变异因为GWAS效能不足还没有被发现。如果能够得到全基因组所有常见变异所能解释的遗传度,将有助于理解常见变异在疾病遗传病因中的作用。同时,如果发现常见变异在遗传度中的贡献较大,说明还存在许多相关的常见变异位点未被发现,可以通过进一步增大GWAS效能的方式发现更多致病位点[6-7]。因此,探索NSCL/P的GWAS中所有常见变异所能解释的遗传度将为进一步开展遗传病因研究提供基础。
传统上主要采用基于家系[8]或双生子设计[9]对遗传度进行估计。随着生物技术的发展,在人群中开展的GWAS可以获得基因数据。依赖单核苷酸多态性(single nucleotide polymorphism, SNP)位点对遗传度贡献大小的一些前提假设,已经可以利用人群的基因数据计算遗传度,得到全基因组所有常见变异所能解释的遗传度,即SNP遗传度[7]。连锁不平衡得分回归是一种计算SNP遗传度的方法,通过对GWAS汇总数据进行统计建模,其不仅适用于许多以前不能用于SNP遗传度估计的数据,而且大大提高了计算速度[10-11]。此外,目前随着生物学技术的飞速发展,基因组不同分区的功能正在不断被发现[12-15]。由于生物学功能不同,不同分区在疾病发生中发挥的作用大小可能不同。可针对基因组不同分区开展SNP遗传度研究,如果发现某些功能分区中遗传度更为富集,提示该分区中可能存在遗传致病因素,例如Won等[16]在心肌梗死和冠状动脉心脏病的研究中发现调控区域的SNP能解释大部分的SNP遗传度,提示基因表达调控区域可能存在重要的致病因素。量化不同分区中的SNP遗传度大小可以作为一种新型的候选区域策略,为进一步精细定位和机制研究提供新的思路和线索。
全基因组范围内常见变异以及不同功能分区范围内常见变异在NSCL/P遗传病因学中的作用尚不清晰,本研究拟利用806个中国人群核心家系的GWAS数据,计算NSCL/P的SNP遗传度和分区SNP遗传度,探索全基因组常见变异和不同功能分区常见变异所能解释的遗传度。
1. 资料与方法
1.1. GWAS数据
本研究所依据的GWAS包括806个汉族NSCL/P核心家系(包括患儿及其父母双亲,共计2 418人), 其地区和性别分布见表 1。GWAS收集了患者及其父母的生物标本并提取DNA,使用Illumina Human610-Quad v.1_B BeadChip芯片进行基因型检测,检测工作于2009年在美国约翰霍普金斯大学医学院McKusick-Nathans遗传医学研究所遗传病研究中心(Center for Inherited Disease Research, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, USA)完成,详见Beaty等[17]的研究。本研究获得了北京大学生物医学伦理委员会批准(批准号: IRB00001052-15081)。患儿唇腭裂分型由临床遗传学家和医生共同确定,确保纳入患儿均为NSCL/P,纳入家系的父母均签署了书面知情同意书。
表 1.
806个中国NSCL/P核心家系的地区和性别分布
Regional and gender distribution of 806 NSCL/P trios in China
| Site | Male | Female | Total |
| NSCL/P, non-syndromic cleft lip with or without cleft palate. | |||
| China Taiwan, n | 139 | 94 | 233 |
| Shandong, n | 193 | 81 | 274 |
| Hubei, n | 132 | 55 | 187 |
| Sichuan, n | 75 | 37 | 112 |
| Total, n | 539 | 267 | 806 |
GWAS采用传递不平衡检验(transmission disequilibrium test, TDT)探索基因位点与疾病的关联关系,其结果详见Beaty等[17]的研究。本研究以GWAS汇总统计数据为基础,包含每个SNP位点的等位基因频率、物理位置及其与NSCL/P关联强度。
1.2. 质量控制
对GWAS进行了严格的质量控制,条件包括: (1)缺失率≤10%;(2)弱势等位基因频率≥1%;(3)父母人群符合Hardy-Weinberg平衡(P>0.000 01);(4)孟德尔遗传错误率≤5%。经质量控制后,GWAS数据中共纳入492 993个SNP位点。
本研究在采用LDSC软件进行SNP遗传度计算之前,对GWAS汇总数据进行了二次质量控制,条件包括: (1)无缺失值; (2)弱势等位基因频率≥1%;(3)关联分析P值在0~1;(4)SNP位点正负链明确; (5)数据中SNP能够与参考人群SNP一一对应。
1.3. 统计学分析
1.3.1. SNP遗传度分析
采用基于Python语言开发的LDSC (v1.0.1)软件计算SNP遗传度[10]。以GWAS汇总数据为基础,并以千人基因组计划亚洲人群(481人,1 208 050个SNP位点)的连锁不平衡得分为参考[10],计算GWAS中所有SNP位点所能解释的SNP遗传度。Bulik-Sullivan等[10]经过严格的数学推导证明,可以对连锁不平衡得分进行统计建模:
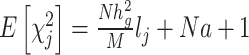 |
1 |
其中,下标j表示第j个SNP。hg2是SNP遗传度,M为染色体固定窗口内的SNP总数目,N为总样本量,a为人群分层及潜在混杂因素等。因变量χj2表示其连锁不平衡得分回归统计量,是第j个SNP与表型关联的卡方统计量。对全基因组所有SNP位点的期望卡方统计量和连锁不平衡得分进行回归分析,可得到SNP遗传度hg2。截距为1(在1附近),表示无混杂,偏离1较远,表示存在混杂。lj为连锁不平衡得分,是一个重要的自变量。利用GWAS汇总数据及单倍型参考数据,对同一条染色体一段区间内(一般为1厘摩尔遗传距离)的两两SNP(第j个SNPj与第k个SNPk)相关系数r进行计算,该区间内合计得到的r2即为某个SNP的连锁不平衡得分lj。lj可表示为:
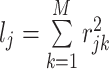 |
2 |
LDSC软件提供了观测尺度和易患性阈值尺度下的SNP遗传度估计结果。由于本研究中NSCL/P患病率较高会相对高估SNP遗传度,所以为了与一般人群实际情况相符,同时便于与其他研究比较,本研究根据一般人群的患病率将其从观测尺度转换到易患性阈值尺度[11]。
1.3.2. 分区SNP遗传度分析
本研究所含分区为基因组编码区、启动子区、增强子区、超级增强子区和内含子区。编码区、启动子区和内含子区的注释来源于UCSC基因组数据库[18],增强子区的注释来源于Hoffman等[19]的研究,超级增强子注释来源于Hnisz等[20]的研究。采用分层的连锁不平衡得分回归计算各分区SNP遗传度[21],并评估各分区SNP遗传度的富集度。SNP遗传度的富集度是指某分区所能解释的SNP遗传度的比例与其所占SNP数目的比例的比。研究使用刀切法评估富集度的统计学意义。
2. 结果
2.1. SNP遗传度分析
经过二次质量控制后,因为基因的正负链不明确排除了2 400个SNP位点,剩余490 593个SNP位点被纳入到SNP遗传度的计算中。在观测尺度下NSCL/P的SNP遗传度为0.55(95%CI: 0.28~0.82), 在易患性阈值尺度下NSCL/P的SNP遗传度为0.37(95%CI: 0.19~0.55), 连锁不平衡得分回归截距为1.004(95%CI: 1.019~0.989)。
2.2. 分区SNP遗传度分析
基于GWAS汇总数据,本研究对基因组编码区、启动子区、内含子区、增强子区和超级增强子区的分区SNP遗传度进行了计算,其结果见表 2。NSCL/P的SNP遗传度在增强子区的富集度为15.70(P=0.04), 表明增强子区内平均每个SNP所能解释的遗传度是其他分区(除增强子区外)的15.70倍,在超级增强子区的富集度为3.18(P=0.03), 表明超级增强子区内平均每个SNP所能解释的遗传度是其他分区(除超级增强子区外)的3.18倍。SNP遗传度在编码区和启动子区虽然也有一定的富集度,但差异无统计学意义。
表 2.
NSCL/P的分区SNP遗传度分析
Partitioning SNP heritability results of NSCL/P
| Annotation | Proportion of SNP | Proportion of SNP heritability | Enrichment | P for enrichment |
| NSCL/P, non-syndromic cleft lip with or without cleft palate; SNP, single nucleotide polymorphism. | ||||
| Coding | 0.014 | 0.035 | 2.44 | 0.91 |
| Promoter | 0.046 | 0.144 | 3.12 | 0.70 |
| Intron | 0.388 | 0.289 | 0.74 | 0.63 |
| Enhancer | 0.042 | 0.654 | 15.70 | 0.04 |
| Super-enhancer | 0.167 | 0.532 | 3.18 | 0.03 |
3. 讨论
本研究发现中国人群NSCL/P的SNP遗传度为0.37,同时研究发现SNP遗传度在增强子区和超级增强子区显著富集,富集度分别达到了15.70和3.18。利用GWAS数据,计算SNP遗传度有助于理解常见变异在其病因学中的作用,通过对基因组不同功能分区的SNP遗传度计算有助于进一步的精细定位和生物学机制研究。
常见复杂疾病和表型的SNP遗传度与传统家系研究发现的遗传度仍有一定差距,这部分仍然无法用所有常见变异所解释的遗传度,也被称为“仍然消失的遗传度(still-missing heritability)”[22]。Ludwig等[23]利用Bonn队列1 717名研究对象的GWAS数据,计算出NSCL/P全基因组常见变异所能解释的遗传度为0.32(95%CI: 0.24~0.40),与本研究结果较为接近,而家系研究和双生子研究发现NSCL/P的遗传度约为70%[3-4],参照本研究所得到NSCL/P的SNP遗传度为37%,提示除常见遗传变异外,尚有致病性遗传因素未被发现。“仍然消失的遗传度”存在以下几种可能的解释: (1)存在效应较大的罕见变异,不能通过GWAS识别; (2)存在交互作用,包括基因-基因交互作用、基因-环境交互作用等; (3)通过家系研究所得到的遗传度可能被高估了; (4)表观遗传学等因素会影响疾病表型。未来有必要对这些可能的遗传致病因素进行深入探索。
另外,对于大多数复杂表型来说,GWAS的阳性位点合计解释的遗传度仍低于SNP遗传度。对于NSCL/P,现有阳性结果可解释约20%的遗传度[5],而本研究的结果提示SNP遗传度为37%,即尚有未被发现的潜在致病性常见遗传变异。由于GWAS往往会检测数十万甚至上百万个位点,为了避免出现假阳性结果,设置的统计学显著性阈值非常严苛。既往身高和精神分裂症等众多疾病和表型的SNP遗传度研究提示,GWAS发现的阳性位点合计解释的遗传度低于SNP遗传度,主要是因为某些致病性常见变异由于效应较小而被严格的显著性阈值过滤掉[24-28]。基因组范围内可能还存在更多与NSCL/P有关的常见变异未被发现,可通过增加样本量和减少多重比较等增加研究效能的方法来识别这类常见变异。
本研究通过计算分区SNP遗传度,发现NSCL/P的遗传度在增强子区和超级增强子区较为富集。既往NSCL/P的GWAS阳性结果多位于非编码区域[24-28]。IRF6基因被众多候选基因研究和GWAS证实与NSCL/P有关[29-31], Rahimov等[32]通过多序列比对识别到IRF6基因一个增强子的常见变异(rs642961), 通过关联分析发现其变异与NSCL/P有关,且风险等位基因数量与NSCL/P发生风险存在剂量反应关系,在功能验证中发现该风险等位基因能够破坏转录因子AP-2α的结合位点,且发现其在小鼠的颅面结构中表达,这表明增强子这一调控原件在NSCL/P的遗传病因中可能起到重要作用。2020年,Morris等[33]研究了增强子变异是否与NSCL/P有关,通过对20个增强子进行目标区域测序并开展关联分析,发现位于MSX1基因和SPRY基因附近的增强子与NSCL/P存在显著关联,并进一步通过功能预测发现这些增强子可能会破坏重要的转录因子结合位点。2013年Whyte等[34]在Cell期刊上提出了超级增强子的概念,指出超级增强子是具有转录活性增强子的集合,其可以驱动控制细胞身份基因的表达。Hnisz等[20]对1 675个GWAS中鉴定的5 303个阳性SNP分析发现,93%的阳性SNP位于非编码区域,其中有64%的位点在增强子区域富集,并且富集于超级增强子区域的变异显著多于增强子区域。目前超级增强子的研究主要集中于肿瘤领域[35], 通过开展关联分析推断出与肿瘤相关的超级增强子,从而为进一步确定肿瘤易感基因提供依据。VISTA Enhancer Browser[36]和EnhancerAtlas[37]等大型数据库的开发也为开展增强子和超级增强子研究提供了便利。目前尚未发现有针对超级增强子开展的NSCL/P的病因学研究,本研究发现NSCL/P的SNP遗传度在超级增强子区富集,为进一步开展相关易感基因定位研究提供了依据。
计算SNP遗传度有助于量化常见遗传变异对表型风险的贡献,明确了进一步定位致病性常见变异的必要性,也有助于理解除常见变异外其他遗传因素的贡献程度。SNP遗传度分区较为富集的区域可作为进一步研究的目标区域,可以作为一种新型的候选区域策略。本研究采用连锁不平衡得分回归计算SNP遗传度,其基于GWAS汇总数据,通过每个SNP位点关联研究结果即可计算SNP遗传度,不需要个体的基因型信息,计算效率高。Evans等[38]证明无论GWAS分析时是否很好地控制了环境变量或亲缘关系等因素,连锁不平衡得分回归都能对SNP遗传度进行较为准确的估计。本研究依据的GWAS通过家系设计提高了研究效能[17],得到了每个SNP位点与表型关联的可靠结果,可以作为计算SNP遗传度的数据基础。虽然本研究使用连锁不平衡得分回归计算SNP遗传度发现截距为1.004(在1附近), 表明本研究潜在的混杂偏倚较少,并且研究结果与现仅有的一篇NSCL/P的SNP遗传度结果相近[23],但本研究仍存在以下不足: 本研究采用的连锁不平衡得分来源于千人基因组亚洲人群,尽管中国人群属于亚洲人群,但两者之间仍存在一定的不同,所以用此数据来评估中国人群SNP遗传度时可能会有一定的偏倚; 本研究依据的GWAS基于核心家系设计,尽管在分析时采用了正确的估计方法控制了家系相关等因素,但在使用连锁不平衡得分回归估计SNP遗传度时仍可能有一定的残余混杂; Evans等[38]发现当致病性位点是常见变异时,连锁不平衡得分回归一般会低估约5%~10%的遗传度,当致病位点包含很多罕见变异时则可能低估更多,因此,本研究可能低估了中国人群NSCL/P的SNP遗传度。综上,本研究是对中国人群NSCL/P SNP遗传度的初步探索,研究者可以根据所拥有的数据类型选择相应的模型和算法来对SNP遗传度进行估计。未来也需要应用更大样本量、独立人群、覆盖更多SNP位点的GWAS数据,开发利用不同的统计分析模型对中国人群NSCL/P的SNP遗传度进行更加准确的估计。
通过评估中国人群的SNP遗传度和分区SNP遗传度,本研究提示尚有致病性常见变异未被发现,并且SNP遗传度主要富集在增强子区和超级增强子区。未来可以采用更大样本量等增加研究效能的方法来识别未被发现的常见变异,也可以针对遗传度较为富集的分区进行大样本的靶向捕获测序,探索该目标分区内的遗传变异与NSCL/P的关联。
Funding Statement
国家自然科学基金(81102178)
Supported by the National Natural Science Foundation of China (81102178)
Footnotes
利益冲突 所有作者均声明不存在利益冲突。
作者贡献声明 薛恩慈: 提出研究思路,设计研究方案,撰写论文; 陈曦、王雪珩、王斯悦、王梦莹: 设计研究方案; 李劲、秦雪英、武轶群、李楠、李静、周治波: 收集、分析、整理数据; 朱洪平: 收集、分析、整理数据,总体把关和审定论文; 吴涛: 提出研究思路,撰写论文,总体把关和审定论文; 陈大方、胡永华: 总体把关和审定论文。
References
- 1.Mossey PA, Little J, Munger RG, et al. Cleft lip and palate. Lancet. 2009;374(9703):1773–1785. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- 2.Wang M, Yuan Y, Wang Z, et al. Prevalence of orofacial clefts among live births in China: A systematic review and meta-analysis. Birth Defects Res. 2017;109(13):1011–1019. doi: 10.1002/bdr2.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sivertsen A, Wilcox AJ, Skjaerven R, et al. Familial risk of oral clefts by morphological type and severity: population based cohort study of first degree relatives. BMJ. 2008;336(7641):432–434. doi: 10.1136/bmj.39458.563611.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosen D, Bille C, Petersen I, et al. Risk of oral clefts in twins. Epidemiology. 2011;22(3):313–319. doi: 10.1097/EDE.0b013e3182125f9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leslie EJ, Carlson JC, Shaffer JR, et al. Association studies of low-frequency coding variants in nonsyndromic cleft lip with or without cleft palate. Am J Med Genet A. 2017;173(6):1531–1538. doi: 10.1002/ajmg.a.38210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11(6):446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang J, Zeng J, Goddard ME, et al. Concepts, estimation and interpretation of SNP-based heritability. Nat Genet. 2017;49(9):1304–1310. doi: 10.1038/ng.3941. [DOI] [PubMed] [Google Scholar]
- 8.Eaves LJ, Last KA, Young PA, et al. Model-fitting approaches to the analysis of human behaviour. Heredity (Edinb) 1978;41(3):249–320. doi: 10.1038/hdy.1978.101. [DOI] [PubMed] [Google Scholar]
- 9.Keller MC, Coventry WL. Quantifying and addressing parameter indeterminacy in the classical twin design. Twin Res Hum Genet. 2005;8(3):201–213. doi: 10.1375/twin.8.3.201. [DOI] [PubMed] [Google Scholar]
- 10.Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu H, Zhou X. Statistical methods for SNP heritability estimation and partition: A review. Comput Struct Biotechnol J. 2020;18:1557–1568. doi: 10.1016/j.csbj.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Consortium GP. An integrated map of genetic variation from 1 092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.GTEx Consortium Human genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348(6235):648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roadmap Epigenomics Consortium Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Won HH, Natarajan P, Dobbyn A, et al. Disproportionate contributions of select genomic compartments and cell types to genetic risk for coronary artery disease. PLoS Genet. 2015;11(10):e1005622. doi: 10.1371/journal.pgen.1005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaty TH, Murray JC, Marazita ML, et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42(6):525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haeussler M, Zweig AS, Tyner C, et al. The UCSC genome browser database: 2019 update. Nucleic Acids Res. 2019;47(D1):D853–D858. doi: 10.1093/nar/gky1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman MM, Ernst J, Wilder SP, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41(2):827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finucane HK, Bulik-Sullivan B, Gusev A, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47(11):1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witte JS, Visscher PM, Wray NR. The contribution of genetic variants to disease depends on the ruler. Nat Rev Genet. 2014;15(11):765–776. doi: 10.1038/nrg3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludwig KU, Böhmer AC, Bowes J, et al. Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip ± cleft palate and cleft palate only. Hum Mol Genet. 2017;26(4):829–842. doi: 10.1093/hmg/ddx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46(11):1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pantelis C, Papadimitriou GN, Papiol S, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Y, Zuo X, He M, et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat Commun. 2017;8:14364. doi: 10.1038/ncomms14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suazo J, Recabarren AS, Marín NR, et al. Association between IRF6 variants and nonsyndromic cleft lip with or without cleft palate in Chile. Reprod Sci. 2020;27(10):1857–1862. doi: 10.1007/s43032-020-00203-9. [DOI] [PubMed] [Google Scholar]
- 31.Zucchero TM, Cooper ME, Maher BS, et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351(8):769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]
- 32.Rahimov F, Marazita ML, Visel A, et al. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet. 2008;40(11):1341–1347. doi: 10.1038/ng.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris VE, Hashmi SS, Zhu L, et al. Evidence for craniofacial enhancer variation underlying nonsyndromic cleft lip and palate. Hum Genet. 2020;139(10):1261–1272. doi: 10.1007/s00439-020-02169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gröschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 36.Visel A, Minovitsky S, Dubchak I, et al. VISTA enhancer browser: A database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35(Database issue):D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao T, Qian J. EnhancerAtlas 2.0: An updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2020;48(D1):D58–D64. doi: 10.1093/nar/gkz980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans LM, Tahmasbi R, Vrieze SI, et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat Genet. 2018;50(5):737–745. doi: 10.1038/s41588-018-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]