

Summary
Recent in vitro studies of human sex chromosome aneuploidy showed that the Xi (“inactive” X) and Y chromosomes broadly modulate autosomal and Xa (“active” X) gene expression. We tested these findings in vivo. Linear modeling of CD4+ T cells and monocytes from individuals with one to three X chromosomes and zero to two Y chromosomes revealed 82 sex-chromosomal and 344 autosomal genes whose expression changed significantly with Xi and/or Y dosage in vivo. Changes in sex-chromosomal expression were remarkably constant in vivo and in vitro; autosomal responses to Xi and/or Y dosage were largely cell-type specific (∼2.6-fold more variation than sex-chromosomal responses). Targets of the sex-chromosomal transcription factors ZFX and ZFY accounted for a significant fraction of these autosomal responses both in vivo and in vitro. We conclude that the human Xi and Y transcriptomes are surprisingly robust and stable, yet they modulate autosomal and Xa genes in a cell-type-specific fashion.
Keywords: sex chromosomes, sex differences, aneuploidy, primary CD4+ T cells, primary monocytes, gene expression, linear modeling, CD4+ T cell activation
Graphical abstract
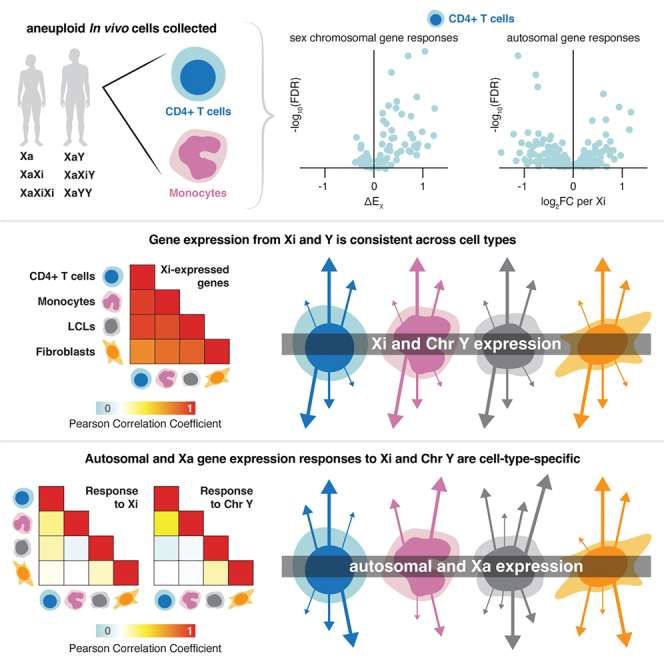
Highlights
-
•
Gene expression in immune cells modeled as a function of Xi and Y chromosome dosage
-
•
Xi and Y transcriptomes are surprisingly consistent across the four cell types examined
-
•
Autosomal responses to Xi and Y copy number are largely cell-type specific
-
•
X- and Y-encoded transcription factors drive these autosomal responses in vivo
Studying primary immune cells with sex chromosome aneuploidy, Blanton et al. gauged impacts of inactive X (Xi) and Y chromosomes on transcription—both sex chromosomal and autosomal—in vivo. Linear modeling reveals that Xi and Y transcriptomes are surprisingly consistent, but Xi and Y modulate autosomal and Xa genes in a cell-type-specific fashion. This suggests that Xi and Y—which distinguish female and male somatic cells—play cell-type-specific roles in modulating gene expression throughout the human body.
Introduction
The somatic cells of human females and males share 45 chromosomes: 22 pairs of autosomes and one X chromosome in the epigenetically “active” state (Xa). The sexes are distinguished by the 46th chromosome, which is either an “inactive” X chromosome (Xi) in somatic cells of most females or a Y chromosome (Chr Y) in males.1,2 Despite the “inactive” label, Xi is transcriptionally active and contributes substantially to female fetal viability.3,4,5 In the reproductive tract, the male-differentiating roles of Chr Y are well documented.6,7,8,9,10 Elsewhere in the human body, we know little about Xi’s and Y’s roles in gene expression and their regulation genome-wide.
We recently assessed how Xi and Y chromosomes impact gene expression across the genome by RNA sequencing of lymphoblastoid cell lines (LCLs) and fibroblasts cultured from individuals with sex chromosome aneuploidy (one to four X chromosomes and zero to four Y chromosomes).11,12 Through linear modeling, we quantified the effects of Xi and Y chromosome dosage on sex-chromosomal11 and autosomal12 gene expression. These studies also identified zinc finger transcription factors encoded by the homologous X-Y gene pair ZFX and ZFY as major drivers of the autosomal gene response.12 In these studies, the highly controlled setting of in vitro culture was both a strength and a limitation: a strength in that the effects of circulating hormones and environmental exposures were minimized and a limitation in that the pertinence of the findings to human cells in vivo remained unknown.
To address this limitation of our previous studies, we assembled parallel collections of primary CD4+ T cells and monocytes, representing the lymphoid and myeloid arms of the immune system, isolated from sex chromosome euploid and aneuploid individuals. We prioritized the study of these peripheral immune cells for two reasons. First, they are among the most accessible primary cells in humans. Second, sex differences within the immune system are pervasive in health and disease. Across hematopoietic lineages, nearly every major immune cell type—from B and T cells in the adaptive immune arm to monocytes and neutrophils in the innate immune arm—displays sex-biased phenotypes.13,14,15,16,17 Immunological sex differences profoundly impact human health, with disparities in the prevalence and presentation of autoimmunity and distinct responses to infections and vaccines.18,19,20 Even in healthy basal conditions, XX and XY individuals show different proportions of circulating immune cells and different immune cell gene expression profiles across the genome.21,22,23,24
To quantitatively assess Xi and Y dosage effects in vivo, we RNA sequenced CD4+ T cells and monocytes from individuals with one, two, or three X chromosomes and zero, one, or two Y chromosomes, constructing linear models of both sex-chromosomal and autosomal gene expression responses to increasing Xi and Y chromosome dosage. As reported here, we found the Xi and Y transcriptomes to be remarkably modular, stable, and robust—not only between the two in vivo cell types but also when compared with the previously studied in vitro LCLs and fibroblasts. In contrast, the effects of Xi and Y dosage on both autosomal and Xa expression were significantly more variable across cell types. These findings indicate that a consistent set of genes expressed on Xi and Y chromosomes drive cell-type-dependent autosomal and Xa responses, with important implications for understanding the molecular mechanisms that underpin sex-biased phenotypes.
Results
Responses of sex-chromosomal genes to sex chromosome dosage in CD4+ T cells and monocytes
To investigate the contribution of sex chromosome constitution to immune cell gene expression, we isolated and performed RNA-seq on CD4+ T cells and monocytes from, respectively, 76 and 72 adults, with one of six different sex chromosome constitutions, comprising one, two, or three X chromosomes together with zero, one, or two Y chromosomes (Figures 1A and 1B; Table S1). The individuals ranged in age from 17 to 70 years, with a median age of 34.5 years. Computational deconvolution of the bulk RNA-seq data confirmed successful enrichment of either CD4+ T cells (estimated purity 80.9% ± 6.7%) or monocytes (81.5% ± 5.8%; Figure S1).25
Figure 1.

Changes in Xi and Chr Y dosage alter expression of X and Y chromosome genes
(A) Experimental design for analysis of primary CD4+ T cells and monocytes from individuals with varying numbers of sex chromosomes.
(B) Chr X and Chr Y dosage series in CD4+ T cells and monocytes.
(C) Schematic of Chr X and Chr Y, indicating locations of pseudoautosomal regions (PAR) in green, non-PAR X (NPX) in orange, NPX genes with NPY homologs in blue, and NPY genes in black.
(D–G) Normalized read counts (x1,000) by sex chromosome dosage for XIST (D), the PAR gene AKAP17A (E), the NPX-NPY homologous pair KDM6A and UTY (F), or the non-Xi- or Chr Y-responsive SLC10A3 (G) in CD4+ T cells. Regression lines with confidence intervals are shown.
(H and I) Volcano plot of ΔEX values for all expressed NPX and PAR genes (H) or ΔEY values of all expressed NPY genes in CD4+ T cells (I). Dotted horizontal lines indicate adjusted p < 0.05. In (H), genes are annotated by class: PAR genes in green, NPX genes with NPY homologs in blue, and NPX genes with no NPY homolog in orange.
We first examined gene expression from Chr X and Chr Y, including genes in the pseudoautosomal region (PAR), the region of sequence identity and meiotic crossing over between Chr X and Chr Y.26 After filtering for genes with at least one transcript per million (TPM) in either euploid karyotype, there were 18 PAR genes expressed in CD4+ T cells and 19 in monocytes, 436 non-PAR X chromosome (NPX) genes expressed in CD4+ T cells and 390 in monocytes, and 14 non-PAR Y chromosome (NPY) genes expressed in CD4+ T cells and 13 in monocytes. As in our prior studies of LCLs and fibroblasts, we used a linear model to assess the modular impact of Chr X or Chr Y dosage on a given gene’s expression, controlling for sequencing batch; donor age had little impact on the model and was excluded from downstream analyses (Figure S2). To quantify the relative expression of a given gene in the presence of additional copies of Xi or Chr Y, we employed the metrics ΔEX or ΔEY, respectively, by dividing the slope of regression (βX or βY, representing the change in expression per copy of Chr X or Y) by the intercept (β0, representing expression in samples with one Chr X or Y). From these ΔEX and ΔEY values, we identified genes whose expression significantly increased with additional copies of Xi or Chr Y (ΔEX or ΔEY > 0), significantly decreased (ΔEX or ΔEY < 0), or remained essentially unchanged (ΔEX or ΔEY ≅ 0).
The evolution of the human X and Y chromosomes from ordinary autosomes has led to three distinct classes of X chromosome genes expressed in immune and other somatic cells. These include (1) PAR genes, (2) NPX genes with diverged homologs on Chr Y, and (3) NPX genes that have lost their homologs on Chr Y (Figure 1C).10,11 Fifteen of the 18 PAR genes expressed in CD4+ T cells and 17 of the 19 PAR genes expressed in monocytes significantly increased expression with Xi dosage, while 10 PAR genes in CD4+ T cells and 15 in monocytes significantly increased expression with Chr Y dosage. Forty-three NPX and 11 NPY genes responded significantly to, respectively, Xi or Chr Y dosage in CD4+ T cells, and 17 NPX and nine NPY genes responded significantly to Xi or Y dosage in monocytes (Tables S2 and S3). Of the 43 NPX genes that responded to Xi dosage in CD4+ T cells, seven possessed an NPY homolog; of the 17 NPX genes that responded to Xi dosage in monocytes, six possessed an NPY homolog. As expected, the XIST lincRNA, which mediates X chromosome inactivation, was expressed in individuals with at least two X chromosomes, and its expression increased with additional Xi chromosomes (Figures 1D and S3A). Genes previously shown to be expressed on both Xa and Xi showed significant positive ΔEX values, including PAR genes and NPX genes with NPY homologs (Figures 1E, 1F, 1H, S3B, and S3C).5,27 In contrast, most expressed NPX genes lacking NPY homologs had ΔEX values near zero, indicating little or no change in expression with increasing Xi dosage (Figures 1G, S3D, S3E, S3G, and S3H). These non-responsive NPX genes included TLR7 and CD40LG, which have roles in immune cell signaling and have been proposed as drivers of female-biased immune phenotypes, but did not change significantly with additional X chromosomes (TLR7 ΔEX = 0.04 ± 0.12 in monocytes; CD40LG ΔEX = −0.003 ± 0.04 in CD4+ T cells; Figure S3D).28,29,30,31,32 Most expressed NPY genes had significant positive ΔEY values of at least 0.5, with essentially no ΔEY values near zero (Figures 1I and S3F). ΔEX values remained consistent when the linear model was restricted to only female or only male samples, indicating that ΔEX values are robust to donor sex (Figure S4). Integrating these results, we find that ∼95% of expressed PAR genes, 80% of expressed NPX genes with an NPY homolog, ∼8.6% of expressed NPX genes with no NPY homolog, and ∼86% of expressed NPY genes demonstrate robust responses to Xi and/or Y dosage in CD4+ T cells and/or monocytes in vivo (Tables S2 and S3).
Responses of X chromosome genes to Xi dosage are highly similar across four cell types
To understand the responses of X chromosome genes to Xi dosage across cell types, we first examined ΔEX and ΔEY values by gene category: PAR, NPX with NPY homologs, and NPX with no NPY homolog. Regardless of cell type, expressed PAR genes displayed strongly positive values for both ΔEX and ΔEY (across four cell types, mean ΔEX = 0.78 ± 0.35 and mean ΔEY = 0.81 ± 0.47; Figures 2A and 2B). While expressed NPX genes with NPY homologs also displayed significant positive responses to Xi dosage in each cell type, their ΔEX values were lower than those of expressed PAR genes (mean = 0.36 ± 0.23), and they showed little or no response to Chr Y dosage. ΔEX values for expressed NPX genes with no NPY homologs were largely near zero, regardless of cell type (mean = 0.03 ± 0.3) (Figure 2B). These findings are consistent with previously published observations: PAR genes are identical on Chrs X and Y, and each additional Xi or Y chromosome adds an increment of PAR gene expression comparable to that of Xa. Expressed NPX genes with NPY homologs are transcribed on both Xa and Xi in somatic cells (contributing to their positive ΔEX values), but their expression on Xi is attenuated compared to that on Xa.11,27 These patterns were evident regardless of whether the cells had been isolated directly from blood (the primary immune cells) or cultured in vitro (the previously collected LCLs and fibroblasts) (Figures 2B and 2C).11
Figure 2.

Responses of X chromosome genes to Xi and Chr Y dosage across gene classes and cell types
(A) Scatterplots of ΔEX versus ΔEY values of X chromosome genes, colored by gene class, in CD4+ T cells and monocytes. Pearson correlation coefficients are shown, colored by corresponding gene class. Dashed line indicates X = Y identity line.
(B) Boxplots showing median (line), interquartile range (IQR; top and bottom of box), and 1.5 × IQR (whiskers) of ΔEX values for X chromosome genes by cell type and gene class.
(C) Boxplots showing median (line), interquartile range (IQR; top and bottom of box), and 1.5 × IQR (whiskers) of ΔEX values for individual PAR and NPX genes with NPY homologs. Asterisks mark two genes with <3 TPM median expression in each cell type.
(D) Proportional bar plot of genes with statistically significant ΔEX values across CD4+ T cells, monocytes, LCLs, and fibroblasts, parsed by the number of cell types in which a given gene’s ΔEX FDR was less than 0.05. Bar plot coloring is as follows: PAR genes in green, NPX genes in orange, NPX genes with NPY homologs in blue.
(E) Scatterplot of ΔEX values for all expressed X chromosome genes in CD4+ T cells and monocytes. Pearson correlation coefficient is shown.
(F and G) Scatterplot of ΔEX values for genes previously annotated as expressed on Xi (“Xi-expressed”) (F) or not expressed from Xi (“Xa-only expressed”) (G) in CD4+ T cells and monocytes. Pearson correlation coefficients are shown.
We next looked across cell types at the genes significantly responsive to Xi dosage. Expressed PAR genes and expressed NPX genes with NPY homologs consistently showed significant responses to Xi dosage across all four cell types. Genes that showed significant responses in only one cell type were in all instances NPX genes with no NPY homolog (Figure 2D). Fewer X chromosome genes reached statistical significance in vivo compared to in vitro, which is likely driven by multiple factors: the in vivo data are subject to uncontrolled environmental variables as the cells are isolated directly from blood, and our in vivo dataset has a more limited range of sex chromosome constitutions and fewer total samples. Sub-sampling analysis revealed that our identification of significantly responsive X chromosome genes was approaching, yet not fully at, saturation, with the majority of significantly responsive genes identified but additional genes likely to be identified with additional samples (Figures S5A and S5B). In all, we see that expressed PAR genes and NPX genes with NPY homologs respond more robustly and reliably to Xi dosage than NPX genes lacking an NPY homolog.
Finally, we compared each X chromosome gene’s ΔEX values across the four cell types to see how those values were affected by somatic cell differentiation. Although fewer X chromosome genes displayed significantly non-zero ΔEX values in the cells collected in vivo than in cultured LCLs and fibroblasts, the ΔEX values in vivo and in vitro were highly correlated (Pearson r range was 0.70–0.86, all p < 2.2e−16, Figures 2E, S6, and S7). Cognizant that ΔEX reflects both gene expression on Xi and modulation of gene expression on Xa, we divided Chr X genes into those previously shown to be expressed on Xi (and on Xa) or silenced on Xi (expressed only on Xa) (as tallied in table S4 in San Roman et al., 2024).5,11,12,27,33,34,35,36 This parsing allowed us to see that the consistency of ΔEX values across cell types was a feature of Xi-expressed genes but not of Xi-silenced genes (“Xa-only expressed”) (Figures 2F, 2G, S6, and S7).
While nearly all genes on Xi are silenced or show attenuated expression, genes on supernumerary Y chromosomes display no such attenuation of their expression.11,12 Examining the impact of additional Y chromosomes in vivo, we found that ΔEY values for NPY genes do not vary as widely as ΔEX values for NPX genes do; rather, median ΔEY values across the NPY genes within each cell type were largely consistent, ranging from 0.65 to 1.07 among broadly expressed NPY genes (Figure S8). These results highlight the distinct effects of adding Xi versus Chr Y to the cell, as supernumerary Y chromosomes show no evidence of dosage compensation through Y inactivation or other mechanisms.
Autosomal responses to sex chromosome dosage in vivo
Having established the direct effects of sex chromosome dosage on sex-chromosomal gene expression, we next investigated genome-wide responses to sex chromosome dosage. Using a linear model to estimate the slope of regression (βX or βY, i.e., the log2 fold change in expression per additional Xi or Y), we modeled log2 normalized read counts, controlling for sequencing batch. In CD4+ T cells, 202 autosomal genes responded significantly to Xi dosage, and 122 autosomal genes responded significantly to Chr Y dosage, with 56 genes responding significantly to both Xi and Chr Y dosage (268 unique Xi- and/or Y-responsive genes in all) (Figures 3A–3D; Table S4). In monocytes, 54 and 46 autosomal genes responded significantly to Xi or Y dosage, respectively, with 11 genes responding significantly to both Xi and Y dosage (89 unique Xi- and/or Y-responsive genes in all) (Figures 3E–3H; Table S4). For both primary cell datasets, saturation analyses revealed that there are likely more autosomal genes that respond to sex chromosome dosage to be found, should the dataset become better powered with samples from more individuals, as is the case for the LCL and fibroblast datasets (Figures S5C–S5F). Specifically, power analyses indicated that the current models detect approximately half of the significantly Xi-responsive autosomal genes in CD4+ T cells and two-thirds of those in monocytes (Figure S9).
Figure 3.

CD4+ T cells and monocytes show significant autosomal gene responses to Xi and Chr Y dosage
(A) Examples of genes positively and negatively responsive to Xi (left) or Chr Y (right) dosage in CD4+ T cells. Regression lines with confidence intervals are shown.
(B and C) Volcano plots of autosomal responses in CD4+ T cells to Xi dosage (84 positively and 118 negatively responsive genes) (B) and Chr Y dosage (52 positively and 70 negatively responsive genes) (C).
(D) Overlap of autosomal genes with significant responses to Xi and Chr Y in CD4+ T cells; p value is from hypergeometric test.
(E) Examples of genes positively and negatively responsive to Xi (left) or Chr Y (right) dosage in monocytes.
(F and G) Volcano plots of autosomal responses in monocytes to Xi dosage (38 positively and 16 negatively responsive genes) (F) and Chr Y dosage (15 positively responsive and 31 negatively responsive genes) (G).
(H) Overlap of autosomal genes with significant responses to Xi and Chr Y dosage in monocytes; p value is from hypergeometric test.
(I–K) In CD4+ T cells, correlations of log2 fold-changes per Xi (using full model: all six karyotypes) against Xi or Chr Y dosage in subset models as indicated. Solid black line indicates Deming regression slope, with 95% confidence intervals shaded gray. Pearson’s correlation coefficients are shown; dashed blue lines indicate X = Y identity.
Autosomal responses to sex chromosome dosage are robust to age and hormonal environment
To assess how much donor age impacts sex chromosome responsiveness in autosomal genes, we again generated an alternate linear model that included a term for age in addition to Chr X count, Chr Y count, and sequencing batch. This alternate model revealed 30 age-responsive genes in CD4+ T cells and five age-responsive genes in monocytes, but only two genes in CD4+ T cells (and no genes in monocytes) responded significantly to both age and sex chromosome dosage (Figure S10; Table S5). Including age in the linear model had little impact on the effect sizes of autosomal gene responses to Xi or Chr Y dosage (Figure S10). Accordingly, we modeled autosomal gene expression with no age term in our equations, just as we had done in modeling sex-chromosomal gene expression.
In human embryos, the presence of Chr Y leads to development of testes, which secrete testosterone, while the absence of Chr Y leads to development of ovaries, which secrete estrogen. To examine possible hormonal influences on the sex chromosome response, we re-assessed effects of Xi in samples that had no Chr Y (45,X; 46,XX; and 47,XXX) or that carried one Chr Y (46,XY and 47,XXY), as well as effects of Xi or Chr Y dosage in samples that had one or two Chr Ys (46,XY; 47,XXY; and 47,XYY). These subset models thus focus on samples with comparable hormonal profiles rather than a mix of estrogen- and testosterone-dominant profiles. Despite reduced sample sizes in the subset models, autosomal responses to Xi and Chr Y dosage in the subset and full models were significantly correlated (Figures 3I–3K and S11). The similar responses across the full and subset models indicate that the autosomal response to sex chromosome dosage is not an artifact of the presence or absence of Chr Y; that is, the responses to sex chromosome dosage cannot be explained by estrogen versus testosterone alone.
ZFX and ZFY drive a portion of the autosomal response in vivo
We showed previously that, in cultured cells, the impact of Xi on autosomal gene expression is similar to that of Chr Y.12 This suggested that the autosomal expression patterns were driven by shared factors on Chr X and Chr Y, i.e., PAR genes or NPX-NPY gene pairs. We asked whether these in vitro findings and insights also held in primary CD4+ T cells and monocytes taken directly from the body. Indeed, autosomal gene responses to Xi and Chr Y dosage, in both CD4+ T cells and monocytes, were remarkably correlated for significantly responsive genes and across all expressed genes (Figures 4A, 4B, S12A, and S12B). Thus, PAR genes and NPX-NPY homologous pairs are candidate drivers of the autosomal response both in vitro and in vivo.
Figure 4.

The transcription factor ZFX drives a significant fraction of autosomal response to sex chromosome dosage in immune cells
(A and B) Scatterplot of log2 fold changes per Xi versus per Chr Y of all expressed autosomal genes in CD4+ T cells (A) or limited to genes significantly responsive to Xi dosage (orange), to Chr Y dosage (purple), or both (gray) (B). Solid black line indicates Deming regression slope, with 95% confidence intervals shaded gray. Pearson’s correlation coefficients are shown; dashed blue lines indicate X = Y identity.
(C–F) Bar plots showing percentages of genes with significant responses to Xi and/or Chr Y dosage in CD4+ T cells that were also identified as ZFX direct target genes in MCF7, C4-2B, and HEK-293T cell lines or ZFX responsive in fibroblasts. Genes are parsed by whether they significantly increased or decreased with Xi or Chr Y dosage (“up” or “down”) and were activated or repressed by ZFX (“Act.” or “Rep.”). p values reflect hypergeometric tests to identify significant enrichments of ZFX target genes in Xi- or Chr Y-responsive genes. Each comparison was restricted to genes expressed in both CD4+ T cells and the given in vitro cell line. Asterisks indicate the enrichments’ p values were lower than the Bonferroni-adjusted threshold of 0.001.
Experiments in fibroblasts previously demonstrated a role for the transcription factors ZFX and ZFY, one of the ancestral NPX-NPY pairs, in driving much of the autosomal response in vitro.12 We set out to test whether changing the ZFX and/or ZFY expression level could also explain autosomal gene responses to Xi and Chr Y dosage in vivo. We first compared gene expression profiles from previously generated ZFX and/or ZFY CRISPRi knockdowns in fibroblasts to averaged autosomal responses to Xi and Chr Y dosage in CD4+ T cells and monocytes. Despite the distinct transcriptional profiles of fibroblasts versus CD4+ T cells and monocytes, ∼59% of the autosomal responders to Xi or Chr Y in CD4+ T cells and 56% of those in monocytes were significantly differentially expressed with ZFX and/or ZFY knockdown (p = 1.56e−06 and p = 0.01, respectively) (Figures S12C and S12D).
ZFX is a transcriptional activator that targets thousands of genes across the genome.37,38 We hypothesized that ZFX activates autosomal genes whose expression increases with Xi or Chr Y dosage. Utilizing lists of ZFX target genes (as determined by ZFX knockdown and ZFX binding) in C4-2B, HEK293T, and MCF7 cells, we sorted the targets into those whose expression decreases with ZFX loss (“ZFX activated”) or increases with ZFX loss (“ZFX repressed”).37,38 Autosomal genes whose expression increased with either Xi or Chr Y dosage in CD4+ T cells were significantly enriched for direct activation by ZFX (Figures 4C and 4E). In contrast, genes whose expression decreased with Xi or Chr Y showed no such enrichment (Figures 4C and 4E). When comparing Xi- or Chr Y-responsive genes to the CRISPRi ZFX knockdown in fibroblasts (which, lacking ZFX binding data, should represent a combination of direct and indirect targets of ZFX), we found significant enrichments for both (1) ZFX-activated genes among the positively responsive genes and (2) ZFX-repressed genes among the negatively responsive genes (Figures 4D and 4F). We observed similar trends among genes that responded positively to Xi or Chr Y dosage in monocytes; however, these trends were not statistically significant following multiple hypothesis corrections (Figures S12E–S12H). While these findings are limited by comparing ZFX targets in different cell types, they nonetheless suggest that ZFX and ZFY drive a significant portion of the autosomal response to sex chromosome dosage both in vitro and in vivo—and that additional sex-chromosomal drivers of autosomal gene expression remain to be elucidated.
Autosomal responders to Xi and Chr Y dosage show cell-type-specific functional enrichments
We next examined the annotated functions of the autosomal genes most responsive to Xi and Chr Y dosage in CD4+ T cells and monocytes. One of the monocyte-expressed genes most responsive to Xi dosage was FCGR2B, which encodes a receptor for the Fc region of immunoglobulin gamma complexes and has been implicated in the female-biased autoimmune disease systemic lupus erythematosus (SLE) (Figure 5A).39,40 FCGR2B significantly increased expression with Xi, but not Chr Y, dosage—in contrast to the majority of autosomal genes that display correlated responses to Xi and Chr Y dosage. Consistent with its positive response to Xi (but not Y) dosage, FCGR2B has been previously shown to be expressed more highly in females.41 As an Xi-responsive, female-biased, and disease-associated gene, FCGR2B is a compelling target for mechanistic studies of sex differences in susceptibility to SLE.
Figure 5.

Autosomal responders to Xi and Chr Y dosage are functionally enriched for interferon and TNFα signaling pathways
(A) Normalized read counts (x1,000) by Chr X (top) or Chr Y (bottom) dosage for FCGR2B in monocytes. Regression lines with confidence intervals are shown.
(B) Heatmap of normalized enrichment scores (NES) for Hallmark gene sets in CD4+ T cells and monocytes; significant enrichments are indicated by asterisks (∗p-adj < 0.05; ∗∗p-adj <0.01; ∗∗∗p-adj <0.001). CD4+ T cell pathways and monocyte pathways with significantly concordant Xi and Chr Y responses are in bold.
(C) Heatmap of normalized enrichment scores for Hallmark gene sets in CD4+ T cells in vivo and unstimulated and stimulated CD4+ T cells in vitro; significant enrichments are indicated by asterisks, as above.
(D) Proportions of leading edge genes, in the indicated pathways, that are specific to the Xi response (orange), specific to the Chr Y response (purple), or common to Xi and Chr Y responses (gray). Asterisks indicate significant overlaps of leading edge genes between Xi and Chr Y responses.
(E) Proportions of leading edge genes, in indicated pathways, that are specific to CD4+ T cells (blue), to monocytes (pink), or shared by the two cell types (gray).
To investigate broader functional implications of Xi and Chr Y dosage, we then conducted gene set enrichment analyses. Gene set enrichment analysis of the Hallmark pathways, as defined by the Human Molecular Signatures Database, revealed several significantly enriched gene sets in each primary immune cell type (Figure 5B; Tables S6 and S7).42,43 CD4+ T cells and monocytes had largely distinct pathway enrichments. In CD4+ T cells, interferon alpha (IFNα) and gamma (IFNγ) response genes, complement, and metabolic pathways such as fatty acid metabolism and oxidative phosphorylation all had significantly negative responses to Xi and Chr Y dosage; i.e., expression of these gene sets decreased with increasing numbers of X or Y chromosomes (Figures 5B and S13A–S13C). For the interferon response gene sets, genes at the “leading edge” of the enrichment included factors that augment and mediate interferon signaling, such as CSF2RB, CASP1, CASP4, TNFAIP2, and NMI (Figures S13A and S13B; Table S6). In contrast, monocytes showed significant positive enrichments for pathways such as “TNFα signaling via NF-κB” and “inflammatory response” (Figures 5B, S13D, and S13E). The “inflammatory response” pathway enrichment was led by the interleukin 1 receptor IL1R2 (Figure S13E; Table S7). Notably, significant enrichments for IFNα and IFNγ responses in CD4+ T cells persisted across subset models restricted to karyotypes with or without a Chr Y, indicating that the negative response to sex chromosome dosage is not driven merely by the presence or absence of a Chr Y (or, by proxy, a testosterone-dominant hormonal milieu) (Figure S14; Table S6).
To assess the effects of Xi dosage on cell function, we activated CD4+ T cells from individuals with one or two X chromosomes (45,X; 46XY; 46,XX; and 47,XXY karyotypes) in vitro with beads biotinylated with CD2, CD3, and CD28 antibodies and then performed RNA-seq (Figure S15; Tables S8 and S9). Many transcriptional patterns persisted between the in vivo and unstimulated in vitro CD4+ T cells, including decreased expression of IFNα and IFNγ response genes with increasing Xi copy number (Figure 5C; Table S10). Upon activation, we observed even starker effects of Xi dosage on gene expression, with several cell-cycle-associated gene sets, such as MYC targets, E2F targets, and G2M checkpoint genes, significantly increased with Xi dosage. These results demonstrate functional consequences of Xi dosage in immune cells and suggest that further sex chromosomal effects may be revealed in other states of cell stimulation or perturbation.
We next asked whether Xi dosage and Chr Y dosage similarly impacted the enriched pathways within a given cell type. In pathways with significant responses to both Xi and Chr Y, the genes responsible for these results were largely the same. There were significant overlaps between the sets of “leading edge” genes responsive to Xi and to Chr Y, with generally correlated effect sizes (Figure 5D; Table S11). While these results underscore the broadly similar effects of Xi and Chr Y dosage on autosomal genes within cell types (Figures 4A, 4B, S12A, and S12B), regressions between Xi and Chr Y effects for several pathways were skewed, with unequal responses to Xi versus Chr Y dosage (i.e., pathways with slopes ≠ 1) (Table S11).
In addition to gene pathways that were significantly enriched in only one of the two immune cell types, we observed pathways in which sex chromosome dosage had opposite effects in different cell types. These included the complement and “inflammatory response” gene sets, whose expression increased with sex chromosome dosage in monocytes and decreased in CD4+ T cells (Figure 5B). The “leading edge” genes responsible for the significant enrichments in CD4+ T cells versus monocytes for these gene sets were largely cell-type specific, with no significant overlap between the two cell types (Figure 5E). This indicates that, rather than having opposing effects on the same genes across cell types, sex chromosome dosage impacts different genes within these pathways across cell types. Taken together, these results underscore the cell-type specificity of responses to Xi and Chr Y dosage.
Autosomal responses are more cell-type specific than are sex-chromosomal responses
To investigate differences and similarities between cell types in autosomal responses to Xi and Y dosage, we first examined genes with statistically significant responses. Out of 247 autosomal genes expressed in both CD4+ T cells and monocytes and responsive to Xi dosage in CD4+ T cells and/or monocytes, only nine were significantly responsive in both immune cell types; of 1,857 autosomal genes expressed in all four cell types that responded to Xi dosage in one or more of the four cell types, only two were significantly responsive across CD4+ T cells, monocytes, LCLs, and fibroblasts (Figure 6A). X chromosome responses to Xi dosage were decidedly more consistent: of 124 X chromosome (including PAR and NPX) genes that had a significant ΔEX in at least one of the four cell types, 19 genes responded significantly to Xi dosage in all four cell types. Even more strikingly, five of the seven NPY genes that were expressed in all four cell types had a significant ΔEY in all four cell types, but only one autosomal gene—out of 652—had a significant response to Chr Y dosage in all four cell types (Figures S8 and 6B).
Figure 6.

Autosomal responses to Xi and Chr Y dosage are more cell-type specific than are sex-chromosomal responses
(A and B) UpSet plots showing limited commonalities across cell types of autosomal genes with significant responses to Xi (A) or Chr Y (B). Analyses were restricted to genes expressed in all four cell types.
(C and D) Correlations of Xi (C) and Chr Y (D) log2 fold changes between CD4+ T cells and monocytes. Plotted genes reached statistical significance in either CD4+ T cells or monocytes. Deming regression is indicated by solid black line with 95% confidence intervals shaded gray; dotted blue line indicates X = Y identity. Pearson correlation coefficients are shown.
(E) Heatmaps of Pearson correlation coefficients between ΔEX values of all NPX and PAR genes across four cell types (left); between ΔEX values of all Xi-expressed genes across four cell types (second from left); between ΔEX values of Xa-only expressed genes, with mean absolute ΔEX values of at least 0.1, across four cell types (middle); and between log2 fold changes of all expressed autosomal genes in response to Xi dosage (second from right) or Chr Y dosage (right). Analyses were restricted to genes expressed in all four cell types.
(F) Variation in gene-by-gene responses to Xi or Y dosage, as indicated, across cell types, was calculated by finding coefficients of variation (CV = standard deviation in effect size across cell types/absolute mean effect size across cell types) for genes expressed in all four cell types. Boxplots show median (line), interquartile range (IQR; top and bottom of box), and 1.5 × IQR (whiskers) of CV values. Gene sets are limited to those with absolute mean effect sizes above the indicated values; FDR was calculated from unpaired t tests, and asterisks indicate FDR < 1e−06.
(G) Coefficients of variation for responses to Xi dosage for genes in the indicated classes. Analyses were restricted to genes expressed in all four cell types with an absolute mean effect size >0.1. Boxplots show median (line), interquartile range (IQR; top and bottom of box), and 1.5 × IQR (whiskers) of CV values. FDR was calculated from unpaired t tests.
Although only nine autosomal genes responded significantly to Xi dosage (and only six genes to Chr Y dosage) in both CD4+ T cells and monocytes, the magnitude and direction of response to Xi (or Chr Y) dosage were correlated across all 11,142 expressed autosomal genes—and these correlations became stronger when limited to significant autosomal responders (Figures 6C, 6D, and S16). However, even among significantly Xi-responsive genes, the correlation between CD4+ T cell and monocyte autosomal gene responses was weaker than X chromosome ΔEX correlations between the same cell types. This pattern persisted across all pairwise cell type comparisons, with ΔEX values of X chromosome genes highly correlated between cell types, whereas autosomal gene responses were less concordant (Pearson correlation coefficients ranging from 0.07 to 0.27; Figures 6C–6E and S16). The strong ΔEX correlations between NPX and PAR genes persisted when restricting these analyses to genes expressed on Xi, while genes only expressed on Xa had more cell-type-dependent responses to Xi dosage (Figure 6E). Thus, while X and Y chromosome responses to Xi and Chr Y dosage are preserved across cell types, downstream effects of Xi and Chr Y dosage on autosomal and Xa expression are more cell-type specific. This comports with recent reports on cultured cells, where Xi’s effects in cis appeared more stable than those in trans.11,12,44 Specifically, we find that the responses of autosomal genes to Xi dosage are as much as 2.6 times as variable (between cell types) as those of NPX and PAR genes (false discovery rate [FDR] = 9.07e−09; mean coefficient of variation [CV] for autosomal genes with mean absolute effect sizes >0.25 is 0.90; CV for NPX and PAR genes is 0.35) (Figure 6F). Parsing X chromosome genes by Xi expression, we found that Xi-expressed genes’ variation in response to Xi dosage is significantly lower than that of genes not expressed on Xi (“Xa-only”), while the variability in responses of Xa-only genes was not statistically different from that of autosomal genes (FDR = 0.97) (Figure 6G). Finally, NPY gene responses to Chr Y dosage showed lower variation than autosomal responses to Chr Y dosage (FDR = 0.001) (Figure 6F). In summary, Xi-expressed genes and NPY genes show little variation in this regard across cell types, while Xa-only genes display variation as high as that seen with autosomal genes.
Discussion
We explored the transcriptome-wide impact of Xi and Chr Y dosage in vivo, employing linear modeling of gene expression in primary immune cells of individuals with sex chromosome aneuploidy. We characterized Xi- and Chr Y-responsive genes in vivo and quantified similarities and differences in these responses among four human cell types. For sex-chromosomal gene expression, the metrics ΔEX and ΔEY quantify the impact of Xi and Chr Y with gene-by-gene specificity. The wide range of ΔEX values across X chromosome genes—within a single cell type—underscores the importance of such a metric to gauge Xi’s contributions to X chromosome expression on a gene-by-gene basis. By contrast, the relative consistency of ΔEY values across expressed Y chromosome genes speaks to the absence of special regulatory mechanisms on supernumerary Y chromosomes. Linear modeling also revealed abundant trans effects on autosomal gene expression.
The influence of Xa, Xi, and Y on human immune cell function
Given the variety and extent of sex differences in immune function, immune cells present an especially poignant opportunity to dissect the extent and nature of Xa, Xi, and Y chromosomal influence on human gene expression and cell function. Evidence indicates that both hormones and sex chromosomes drive sex-biased phenotypes, but their roles have not been fully disentangled.45,46,47 Previous studies of sex chromosomal influences on immune cell function have revealed immune dysfunction in sex chromosome aneuploid individuals—including increased risk of autoimmunity in Turner syndrome (45,X) and Klinefelter syndrome (47,XXY)—while other molecular studies have focused on the effects of X-linked immune genes that are expressed from both Xa and Xi.48,49,50,51 For example, the X-linked toll-like receptor 7 gene, TLR7, is biallelically expressed in some human 46,XX plasmacytoid dendritic cells, B cells, and monocytes and in turn has been associated with sex-biased IFNα and IFNγ responses and female-biased autoimmunity.28,50,51,52 Similarly, it has been proposed that heterogeneous expression of other X-linked genes from Xi, including CD40LG, contributes to the female-biased prevalence of lupus.31 In our present study, we did not observe a significant increase in TLR7 transcript levels in monocytes, nor increased CD40LG transcript levels in CD4+ T cells, with additional copies of Xi. These findings run counter to prior expectations that expression from Xi would result in increased total expression with increased Xi dosage. In our bulk RNA-seq analyses, expression of TLR7 and CD40LG from Xi is too modest to have a discernible effect on total expression levels. These findings suggest that other factors likely contribute to female-biased autoimmunity and other sex differences in the immune system.
In addition to studying the transcriptional profiles of CD4+ T cells in vivo, we measured the effects of Xi dosage on transcription upon CD4+ T cell activation in vitro. We discovered effects on gene expression that persisted in vivo and in vitro, including the IFNα and IFNγ response pathways, as well as context-specific gene expression patterns, such as TNFα and IL6 JAK/STAT3 signaling. Activation of CD4+ T cells in vitro revealed a host of additional functions impacted by Xi dosage. Upon stimulation, CD4+ T cells that possessed an Xi had higher expression of proliferative and metabolic genes than CD4+ T cells lacking an Xi. These results highlight specific pathways through which Xi may influence CD4+ T cell responses to activation and, in turn, contribute to sex-biased immune phenotypes.
Xi, Xa, and Y: An emerging understanding of gene regulation in cis and trans
Having systematically analyzed the expression of protein-coding Xa, Xi, Y, and autosomal genes across four cell types, we can identify some general patterns regarding the sex chromosomes’ wide-ranging roles in regulating the genome. Four major findings appear to hold in vivo and in vitro, and they generalize across cell types: (1) Xi-expressed genes display stable ΔEX values, (2) Y chromosome genes display stable ΔEY values, (3) Xa and autosomal responses to Xi and Y display cell-type specificity, and (4) Xa and autosomal responses are driven by a small number of widely expressed NPX-NPY gene pairs.
First, we observe a striking consistency in the responses of Xi-expressed genes to Xi dosage, such that, gene by gene, ΔEX values are remarkably stable across cell types, both in vivo and in vitro. The simplest interpretation is that, for each Xi-expressed gene, the ratio of Xi to Xa transcription is conserved among somatic cell types. The molecular determinants of these ratios and their stability are presently unknown.
Second, we see a comparable or even greater consistency in the responses of Y chromosome genes to Chr Y dosage, with ΔEY values for each gene being strikingly similar across cell types, in vivo and in vitro. With median ΔEY values of broadly expressed NPY genes ranging from 0.65 to 1.07, our findings are consistent with the absence of Y inactivation or other dosage compensation mechanisms in vivo and in vitro.
Third, while transcription from Xi and Chr Y is stable and robust across cell types, the transcriptional responses of autosomal and Xa-only genes are cell-type specific. In this critical epigenetic respect, Xa behaves like an autosome and not like its genetically identical twin, Xi. Remarkably, the Xi and Y chromosomes can elicit cell-type-specific responses on autosomes and Xa even when the Xi and Y transcriptomes do not vary substantially among cell types.
Fourth, evidence is growing that a small, fixed set of Xi- and Y-expressed genes drive these cell-type-specific autosomal and Xa responses to Xi and Chr Y dosage. In each of the four studied cell types, Xi and Chr Y have comparable effects on genome-wide expression, indicating that regardless of cellular identity, factors expressed by both Xi and Chr Y drive most of the genome-wide response. The observation that Xi vs. Chr Y effects are similar but not identical (Figures 4A, 4B, S12A, and S12B) aligns well with evidence implicating the diverged NPX-NPY genes, such as ZFX and ZFY, rather than PAR genes, which are identical on Chrs X and Y.12 We hypothesize that the cell-type specificity of autosomal and Xa responses to conserved Xi- and Y-expressed drivers is the result of differences between cell types in chromatin accessibility and autosomal cofactors. This hypothesis should now be explored experimentally.
Taken together, these findings provide a counterpoint and complement to the well-established concept of “variable escape” from X chromosome inactivation, which posits that the set of X chromosome genes expressed from Xi varies substantially across cell types.5,33,36,53,54 While our study of four cell types does not exclude the occurrence of biologically critical variation in the Xi transcriptome among these and other cell types, our findings do not provide direct evidence of such variation. Instead, our findings show that the response of autosomal and Xa-only genes is the more cell-type-specific phenomenon—at least among the four cell types that we have analyzed. Studies of additional somatic cell types will be required to resolve this important question.
Conclusion
Through linear modeling of a diverse array of naturally occurring sex chromosome constitutions, we have characterized quantitatively the in vivo impact of Xi and Chr Y on human gene expression in two immune cell types. We find consistent, conserved regulation of Xi and Chr Y gene expression across distinct cell types but divergent effects of Xi and Chr Y on autosomal and Xa gene expression. Xi and Chr Y have comparable regulatory effects on transcription genome-wide, both in vitro and in vivo, driven in part by the X- and Y-encoded homologous pair of transcription factors ZFX and ZFY. The studies reported here establish a foundation for fully dissecting how Xi and Chr Y affect transcription and cell function throughout the body.
Limitations of the study
We found sex-chromosomal and autosomal expression responses to sex chromosome dosage in primary immune cells. These transcriptomic results are at the resolution of bulk cell populations and do not capture expression heterogeneity within cell types. The primary cell types included in the current study are limited to the hematopoietic lineage and may not fully represent gene expression responses in cell types of other developmental lineages and other tissues. We assessed the effects of sex steroids on gene expression indirectly, using the presence or absence of Chr Y to subset samples as “testosterone dominant” or “estrogen dominant”; further studies will be needed to more directly disentangle the effects of sex steroid concentrations and sex chromosome constitution. As described in our saturation and power analyses, we have not identified all genes that respond to sex chromosome dosage, and additional sex-chromosome-responsive genes would likely be revealed with additional samples from sex chromosome aneuploid individuals. Our analysis of ZFX target genes utilized gene expression data from in vitro cell lines, which may not fully reflect ZFX and ZFY targets in primary CD4+ T cells and monocytes. Finally, while our gene set enrichment analysis shows that sex chromosome dosage impacts gene expression in a number of functional pathways, additional in vitro assays will be needed to characterize functional consequences.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human CD4+ T cell Isolation Kit | Miltenyi Biotec | Cat#130-096-533; RRID: AB_2916089 |
| Human Monocyte Isolation Kit | Miltenyi Biotec | Cat#130-091-153 |
| T cell Activation/Expansion kit, human | Miltenyi Biotect | Cat#130-091-441 |
| Biological Samples | ||
| Primary CD4+ T cells | This paper | N/A |
| Primary Monocytes | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium heparin tubes | BD Vacutainer | GTIN:00382903678747 |
| FBS | Hyclone | Cat#SH30071 |
| DMSO | Sigma-Aldrich | Cat#D8418 |
| Trizol Reagent | Life Technologies | Cat#15596026 |
| Chloroform | Millipore Sigma | Cat#CX1055-6 |
| Acid Phenol:Chloroform | Life Technologies | Cat#AM9722 |
| Glycoblue | Life Technologies | Cat#AM9515 |
| Sodium acetate | Invitrogen | Cat#AM9740 |
| RNase-free ethanol | PharmCo | Cat#111000200 |
| RNase-free water | Ambion | Cat#AM9937 |
| RPMI Medium 1640 | Gibco Life Tech | Cat#31800-089 |
| AB Serum | Valley Biomedical | Cat#HP1022HI |
| IL-2 | Corning | Cat#356043 |
| Critical Commercial Assays | ||
| Qubit RNA HS Assay Kit | ThermoFisher | Cat#Q32855 |
| Fragment Analyzer RNA Kit | Agilent | Cat#DNF-471 |
| KAPA mRNA HyperPrep Kit | Roche | Cat#KK8581 |
| PippinHT 2% Agarose Gel Cassettes | Sage Science | Cat#HTC2010 |
| Quick RNA Microprep kit | Zymo | Cat#R1050 |
| xGen RNA Library Preparation kit | IDT | Cat#10009814 |
| Deposited Data | ||
| Raw, de-identified RNA-Seq data | This paper | dbGaP: phs002481 |
| Processed data | This paper | https://doi.org/10.5281/zenodo.11889919 |
| Custom GENCODE v24 transcriptome annotation | Godfrey et al.55 | https://doi.org/10.5281/zenodo.3627233 |
| RNA-Seq LCLs | San Roman et al.11; San Roman et al.12 | dbGaP: phs002481 |
| RNA-Seq Fibroblasts | San Roman et al.11; San Roman et al.12 | dbGaP: phs002481 |
| Molecular Signatures Database (MSigDB) v2022.1.Hs | Subramanian et al.42; Liberzon et al.56 | https://www.gseamsigdb.org/gsea/msigdb/ |
| ZFX ChIP-seq HCT116 | ENCODE | ENCFF858YWR, ENCFF102UZB |
| ZFX ChIP-seq C4-2B | ENCODE | ENCFF160AXQ, ENCFF264NUJ |
| ZFX- ChIP-seq MCF-7 | ENCODE | ENCFF861DOL, ENCFF429AXS |
| ZFX ChIP-Seq HEK-293T | ENCODE | ENCFF292HYE, ENCFF625XHP |
| RNA-seq HCT-116 | ENCODE | ENCFF435PHM |
| RNA-Seq C4-2B | Rhie et al.37 | GSE102616 |
| RNA-Seq MCF-7 | ENCODE | ENCFF721BRA |
| RNA-Seq MCF-7 | Rhie et al.37 | GSE102616 |
| RNA-Seq HEK-293T | Ni et al.38 | GSE145160 |
| Allelic expression data in fibroblasts and hybrid cell lines | Carrel et al.33 | N/A |
| Allelic expression data in LCLs and fibroblasts from paired genomic and cDNA SNP-chips | Cotton et al.34 | N/A |
| Allelic expression data from bulk and single LCLs in GTEx | Tukiainen et al.27 | N/A |
| Allelic expression data in single fibroblasts | Garieri et al.35 | N/A |
| Allelic expression data in bulk LCLs | Sauteraud et al.36 | N/A |
| Software and Algorithms | ||
| R v3.6.3 | The R Foundation | https://www.rproject.org |
| kallisto v0.42.5 | Bray et al.57 | https://pachterlab.github.io/kallisto/ |
| tximport v1.30.0 | Soneson et al.58 | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| DESeq2 v1.26.0 | Love et al.59 | https://bioconductor.org/packages/release/bi oc/html/DESeq2.html |
| BEDTools v2.26.0 | Quinlan et al.60 | https://bedtools.readthedocs.io/ |
| fgsea v1.28.0 | Korotkevich et al.61 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Other | ||
| 5Prime Gel Heavy PhaseLock tubes | Quantabio | Cat#2302830 |
| Thermomixer | Eppendorf | N/A |
| Qubit 4 Fluorometer | ThermoFisher | N/A |
| 5200 Fragment Analyzer System | Agilent | N/A |
| PippinHT System | Sage Sciences | N/A |
| HiSeq 2500 | Illumina | N/A |
| Cell Countess II | ThermoFisher | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David C. Page (dcpage@wi.mit.edu).
Materials availability
There are restrictions on the availability of the CD4+ T cell and monocyte cells in this study, as the studied cells are primary cells that have not been stably transformed into cell lines.
Data and code availability
-
•
Raw RNA-Seq data has been deposited to dpGAP and processed data has been deposited at github. Both are publicly available as of the date of publication. Accession numbers and DOIs are listed in the key resources table.
-
•
This paper analyzes existing, publicly available data. Accession numbers for these datasets are listed in the key resources table.
-
•
Original code has been deposited at github (https://github.com/lvblanton/sca-immune) and is publicly available as of the date of publication (https://doi.org/10.5281/zenodo.11889920).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Human subjects
Recruitment of euploid and sex chromosome aneuploid individuals has been previously described, but is restated briefly here: Seventy-two adults (18+) were recruited through a joint IRB-approved study between the NIH Clinical Center (12-HG-0181) and Whitehead Institute; 3 adults were recruited through Massachusetts General Hospital and 4 adults were recruited through Boston Children’s Hospital (Protocol #1706013503). Informed consent was obtained from all participants. Extensive karyotyping was performed on primary blood cells derived from sex chromosome aneuploid individuals recruited through the NIH clinical center; of 1000 karyotyped cells, the dominant karyotype for the diagnosed aneuploidy was, on average, 94% ± 8.7% of cells.
Isolation of peripheral blood mononuclear cells
Whole blood was collected in BD Vacutainer Sodium Heparin tubes (GTIN: 00382903678747) and enriched for CD4+ T cells and monocytes on the same day as collection. First, peripheral blood mononuclear cells were isolated by transferring whole blood to a 50 mL conical tube and adding 1X PBS without calcium and magnesium to a total volume of 40 mL. After capping the tube and inverting the blood mixture 5–6 times, 10.5 mL of lymphocyte separation media (LSM; MP Biomedicals cat. #50494) was added to the to the tube by placing the pipette directly into the blood mixture and slowly releasing the LSM by gravity. The blood mixture was spun at 1500 rpm for 30 min, at room temperature, with no brake. Without disturbing the separated layers, the cloudy buffy coat of PBMCs were transferred to a new tube and washed with 1X PBS for 10 min at 1500 rpm at room temperature.
Enrichment of CD4+ T cells and monocytes from PBMCs
PBMC cells were split in equal volumes and enriched for CD4+ T cells using the Miltenyi Biotec Human CD4+ T cell Isolation Kit (cat# 130-096-533) and for monocytes using the Miltenyi Biotec Human Monocyte Isolation Kit (cat# 130-091-153), following the manufacturer’s protocol. Following enrichment, each cell type was resuspended in freezing media (FBS +10% DMSO) and stored at −80C° until RNA extraction.
Method details
RNA extraction
For RNA extraction, 1 million cells per sample were spun down for 5 min at 13,000rpm and resuspended in 0.5 mL TRIzol Reagent (Life Technologies #15596026). Following a 5 min incubation at room temperature, each sample was transferred to a 2 mL 5Prime Gel Heavy PhaseLock tube (Quantabio #2302830) and given 0.1 mL of chloroform. Samples were then incubated for 5 min at room temperature on a Thermomixer (Eppendorf) at 1100rpm, then spun for 5 min at 13,000rpm, room temperature. The aqueous phase, containing RNA, was transferred to a new PhaseLock tube and given 0.2 mL acid phenol:chloroform (Life Technologies AM 9722), followed by a second 5 min Thermomixer incubation. Samples were again spun to separate phases, and the aqueous phase was again transferred to a new Eppendorf tube. RNA was precipitated overnight at −20C with 0.5 μL Glycoblue (Life Technologies AM9515), 20 μL 3M sodium acetate, pH 5.5, and 0.5 mL RNase-free ethanol, and resuspended in 15 μL RNase-free water. RNA concentrations were quantified using the RNA HS Qubit kit (ThermoFisher).
RNA sequencing and analysis
RNA sequencing libraries were prepared at the Whitehead Genome Technology Center, using the KAPA HyperPrep kit, followed by size selection for 300-600bp fragments with the PippinHT system (Sage Science) and 2% agarose gel. Paired 100x100bp reads were sequenced on the Illumina HiSeq 2500 or NovaSeq 6000. Following RNA sequencing, data was pseudoaligned using kallisto (v0.42.5), using the --bias flag, to a custom human genome based on the GENCODE v24 Basic annotation, as detailed previously.11,12 kallisto results were imported into R for downstream analysis using the tximport package. We restricted our analysis to protein-coding genes and lincRNAs, with the exception of also including the Y chromosome annotated pseudogenes PRKY and TXLNGY. We filtered for expression by a median TPM >1 in either XX or XY samples within a given cell type.
CIBERSORTx analysis of cell type populations
To validate successful CD4+ T cell and monocyte enrichment, CIBERSORTx was used to impute cell fractions from the bulk RNA-Seq samples.25 Following kallisto pseudoalignment, TPMs were extracted using the tximport package and uploaded to CIBERSORTx (https://cibersortx.stanford.edu/). Cell fractions were imputed using the LM22 signature matrix file as reference for 22 immune cell types, with batch correction (B-mode) enabled.
Linear modeling of sex-chromosomal genes
Linear models for each expressed X chromosome and Y chromosome gene were generated using the lm() function in R, as previously described in San Roman et al., 2023 and briefly restated here. For NPX and PAR genes, normalized read counts from all samples were inputted to the model, with terms for the number of Xi, the number of Chr Y, and sequencing batch. For NPY genes, karyotypes with at least 1 Chr Y were inputted to model the number of Xi, the number of Chr Y minus one, and sequencing batch.
ΔEX and ΔEY values were calculated by dividing the βX or βY, respectively, by the corresponding β0 (i.e., the intercept, representing expression from 45,X samples for NPX and PAR linear models and from 46,XY and 47,XXY samples for NPY linear models). To calculate ΔEX and ΔEY for XIST (which is only expressed in karyotypes with at least two Chr X), βX or βY were divided by β0 + βX. p values were adjusted for multiple hypothesis correction using the Benjamini-Hochberg method; genes with p-adjusted <0.05 were judged to be significant.
For gene classes, we defined PAR genes as those from PAR1. “NPX genes with NPY homolog” were defined as non-PAR X chromosome genes with an expressed NPY homolog (including NPY homologs currently annotated as pseudogenes). Note that for plotting purposes, the ΔEX values for four X chromosome genes (ZFX-AS1, PRKX-AS1, and EIF1AX-AS1 in CD4+ T cells and BEX1 in fibroblasts) with outlying ΔEX values greater than 2 – often inflated from low expression that led to small β0 denominators –were excluded from figures.
To examine the impact of age on the response to Xi or Chr Y, identical linear models were generated with the exception that age, in years, of the donor at time of sample collection was included as an additional term in the model.
Linear modeling of autosomal genes
For autosomal genes, log2 normalized read counts were used to model for Chr X number, Chr Y number, and sequencing batch via DESeq2; the “results” function within DESeq2 was then used to identify specific responses to Xi or Chr Y. Genes with adjusted p-values <0.05 (as output by DESeq2) were called as significantly responsive to Xi or Chr Y. To examine the impact of age on the response to Xi or Chr Y, identical DESeq2 linear models were generated with the exception that age, in years, of the donor at time of sample collection was included as an additional term.
Saturation analysis
Separate saturation analyses were performed for X chromosome genes (NPX and PAR) and autosomal genes, consistent with the linear modeling approaches used to generate the sex-chromosomal versus autosomal models as described above. Size-n subsets were randomly generated, without replacement, 100 times for each sample size, n, and model matrixes were assessed to be full rank. Genes with adjusted p-value < 0.05 were called as significant.
Power analysis
To estimate the power of the linear models to identify all significantly responsive genes, read counts for 100 genes from the sex chromosome aneuploidy cohort were simulated to generate effect sizes (log2 fold-changes) ranging from 0.05 to 1. To simulate the data, karyotype labels were first shuffled to remove expression dependencies on Chrs X or Y count, and then new, artificial read counts were generated such that for a given set of 100 random genes, read counts were altered to reflect the desired effect size. DESeq2 models on the simulated data were iteratively run 20 times for each effect size and compared with the actual DESeq2 results to find the fraction of true positive results (TPRs). After calculating median TPRs for each effect size in response to either Xi or Chr Y, in each cell type (CD4+ T cells and monocytes), power for each effect size was estimated by fitting an exponential curve. Using the curved fit, we then estimated the number of unobserved genes by applying the power for each effect size to the number of observed significantly responsive genes, by cell type and sex chromosome dosage.
Annotation of Xi-expressed and Xa-only expressed genes
A meta-analysis of previously published annotations of X-inactivation status was described in detail in San Roman et al., 2023 (summarized in table S6 of that study) and further extended in San Roman et al., 2024.11,12 We used the expanded analysis in table S4 from San Roman et al., 2024 to annotate genes in the present study. Briefly, allelic ratio (AR) data was summarized across five studies utilizing various AR methodologies, including data from cDNA SNP-chips in skewed LCLs and fibroblasts, single-cell RNA-Seq in LCLs, single-cell RNA-Seq in fibroblasts, and allele-specific bulk RNA-Seq in LCLs and fibroblasts with skewed X-inactivation.5,11,27,33,34,35,36 Genes with evidence of expression from Xi in more than half of the studies, as well as those with AR ≥ 0.1 and with evidence of expression from Xi in at least one study, were designated as “Xi-expressed”. Genes that showed no expression from Xi across all studies, as well as those showing no Xi expression in at least half of studies, plus ARs <0.1, were called as “Xa-only expressed”. In all, 107 genes are annotated as “Xi-expressed” and 460 genes as “Xa-only expressed”; 398 genes were not captured in the analyzed studies (“no call”).
ZFX direct target enrichment analysis
To generate lists of ZFX direct target genes, we utilized ZFX ChIP-Seq data from four cell lines available via ENCODE4 v1.6.1 (HCT 116, C4-2B, MCF-7, and HEK-293T), as well as RNA-Seq of ZFX knockdown experiments in the same cell types from ENCODE or GEO and RNA-Seq data from ZFX knockdown in fibroblasts.12,37,38 When available, we downloaded pre-processed RNA-Seq data, but otherwise processed fastq files using kallisto. To identify ZFX binding at promoters, we used the BEDTools closest function to map ZFX peaks to the nearest annotated transcription start site in the GENCODE v24 comprehensive annotation. Genes that 1) had ZFX bound to the promoter and 2) were significantly differentially expressed upon ZFX knockdown were classified as “direct target genes”. After direct ZFX target genes were identified for each cell type, other genes that decreased expression with ZFX loss were classified as “ZFX activated”, while genes that increased expression with ZFX loss were classified as “ZFX repressed”. We then found the intersection of “ZFX activated” and “ZFX repressed” genes with genes that either increase or decrease with Xi or Chr Y dosage, and performed hypergeometric tests using the phyper function in R to establish whether there was a significant overlap between gene lists.
Gene set enrichment analysis
Gene set enrichment analysis was conducted in R with the fgsea package, using the 50 Hallmark pathways (v7.1) downloaded from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb).42,43 Analysis was restricted to autosomal protein-coding and lincRNA genes, which were ranked by each gene’s t-statistic from the DESeq2 models for Chr X or Y count.
In vitro activation of CD4+ T cells
Frozen CD4+ T cell stocks from individuals with 45,X, 46,XX, 46,XY, or 47,XXY karyotypes were thawed for 2–3 min in a 37°C water bath, followed by gentle addition of media (RPMI +10% AB serum) to dilute the DMSO in the freezing media. Cell viability and concentrations were measured on a Countess Cell Counter. Cell concentrations were then normalized across samples to 2.77 x 106 cells/mL, such that final culture concentrations (following the addition of activation beads or media) would be 2.5 x 106 cells/mL. Activation beads were prepared following manufacturer’s instructions (Miltenyi T cell Activation/Expansion kit, human, cat.# 130-091-441). Cells were plated in a flat-bottom 96-well culture plate (∼300 μL/well) and either cultured in RPMI +10% AB serum + IL-2 alone, or RPMI +10% AB serum + IL-2 + biotinylated activation beads, following manufacturer’s instructions. After plating, cells were incubated at 37C° +5% CO2 for 24 h prior to collection for RNA-Seq. RNA was extracted from each sample using the Zymo Quick RNA Microprep kit (cat. R1050), and RNA-Seq libraries were made via the IDT xGen RNA Library Preparation kit (cat. 10009814).
Quantification and statistical analysis
We used various statistical tests to calculate p-values as indicated in method details, figure legends, or text, where appropriate. To calculate all statistics and generate plots, we used R software, version 3.6.3.31 We considered results statistically significant when p < 0.05 or, when using multiple hypothesis correction, adjusted-p <0.05 or FDR<0.05. We used Deming regressions to compare variables that were measured with error (e.g., log2 fold-change per Xi vs. log2 fold-change per Chr Y). We calculated Deming regressions using the R package “deming” v1.4. For weighted Deming regressions, we included error values using the “xstd” and “ystd” arguments.
Acknowledgments
We thank members of the Page Laboratory, especially Daniel W. Bellott, for critical reading and comments on the manuscript, and Jorge Adarme and Susan Tocio for laboratory support. We thank the Whitehead Genome Technology Core facility for library preparation and sequencing.
Funding is as follows: Lallage Feazel Wall Damon Runyon Cancer Research Foundation Fellowship (L.V.B.), National Institutes of Health grant F32HD091966 (A.K.S.R.), Simons Foundation Autism Research Initiative – Collaboration Award (D.C.P. and J.F.H.), National Institutes of Health grant U01HG0007587 (D.C.P. and M.M.), Howard Hughes Medical Institute (D.C.P.), and National Human Genome Research Institute Intramural Research Program (M.M. and D.K.). Contents are the authors’ sole responsibility and do not necessarily represent official NIH views. Philanthropic support was from The Brit Jepson d'Arbeloff Center on Women’s Health, Arthur W. and Carol Tobin Brill, Matthew Brill, Charles Ellis, The Brett Barakett Foundation, Howard P. Colhoun Family Foundation, Seedlings Foundation, and Knobloch Family Foundation.
Author contributions
Conceptualization: L.V.B. and D.C.P. Data curation: L.V.B. and A.K.S.R. Formal analysis: L.V.B., A.K.S.R., and H.S. Funding acquisition: L.V.B., A.K.S.R., J.F.H., M.M., and D.C.P. Investigation: L.V.B., G.W., and T.T.P. Methodology: L.V.B., G.W., A.K.S.R., and H.S. Project administration: L.V.B., G.W., J.F.H., L.B., A.K.S.R., A.B., P.K., and N.B. Resources: A.B., P.K., N.B., A.E.L., and M.M. Software: L.V.B., A.K.S.R., A.K.G., and H.S. Supervision: L.V.B., A.K.S.R., J.F.H., L.B., A.E.L., D.K., M.M., and D.C.P. Validation: L.V.B., A.K.S.R., and H.S. Visualization: L.V.B. Writing – original draft preparation: L.V.B. and D.C.P. Writing – review and editing: L.V.B., A.K.S.R., J.F.H., and D.C.P.
Declaration of interests
The authors declare no competing interests.
Published: August 6, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100628.
Supplemental information
References
- 1.Ohno S. Monographs on Endocrinology. Springer Berlin; 1967. Sex Chromosomes and Sex-Linked Genes. [DOI] [Google Scholar]
- 2.Lyon M.F. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet. 1962;14:135–148. [PMC free article] [PubMed] [Google Scholar]
- 3.Hook E.B., Warburton D. The distribution of chromosomal genotypes associated with Turner’s syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum. Genet. 1983;64:24–27. doi: 10.1007/bf00289473. [DOI] [PubMed] [Google Scholar]
- 4.Hook E.B., Warburton D. Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 2014;133:417–424. doi: 10.1007/s00439-014-1420-x. [DOI] [PubMed] [Google Scholar]
- 5.Balaton B.P., Cotton A.M., Brown C.J. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 2015;6:35. doi: 10.1186/s13293-015-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reijo R., Lee T.-Y., Salo P., Alagappan R., Brown L.G., Rosenberg M., Rozen S., Jaffe T., Straus D., Hovatta O., et al. Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA–binding protein gene. Nat. Genet. 1995;10:383–393. doi: 10.1038/ng0895-383. [DOI] [PubMed] [Google Scholar]
- 7.Lahn B.T., Page D.C. Functional coherence of the human Y chromosome. Science. 1997;278:675–680. doi: 10.1126/science.278.5338.675. [DOI] [PubMed] [Google Scholar]
- 8.Blanco P., Shlumukova M., Sargent C.A., Jobling M.A., Affara N., Hurles M.E. Divergent outcomes of intrachromosomal recombination on the human Y chromosome: male infertility and recurrent polymorphism. J. Med. Genet. 2000;37:752–758. doi: 10.1136/jmg.37.10.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skaletsky H., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 10.Bellott D.W., Hughes J.F., Skaletsky H., Brown L.G., Pyntikova T., Cho T.-J., Koutseva N., Zaghlul S., Graves T., Rock S., et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature. 2014;508:494–499. doi: 10.1038/nature13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.San Roman A.K., Godfrey A.K., Skaletsky H., Bellott D.W., Groff A.F., Harris H.L., Blanton L.V., Hughes J.F., Brown L., Phou S., et al. The human inactive X chromosome modulates expression of the active X chromosome. Cell Genom. 2023;3 doi: 10.1016/j.xgen.2023.100259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.San Roman A.K., Skaletsky H., Godfrey A.K., Bokil N.V., Teitz L., Singh I., Blanton L.V., Bellott D.W., Pyntikova T., Lange J., et al. The human Y and inactive X chromosomes similarly modulate autosomal gene expression. Cell Genom. 2024;4 doi: 10.1016/j.xgen.2023.100462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Márquez E.J., Chung C.-H., Marches R., Rossi R.J., Nehar-Belaid D., Eroglu A., Mellert D.J., Kuchel G.A., Banchereau J., Ucar D. Sexual-dimorphism in human immune system aging. Nat. Commun. 2020;11:751–817. doi: 10.1038/s41467-020-14396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang M.A., Rego D., Moshkova M., Kebir H., Chruscinski A., Nguyen H., Akkermann R., Stanczyk F.Z., Prat A., Steinman L., Dunn S.E. Peroxisome proliferator-activated receptor (PPAR)α and -γ regulate IFNγ and IL-17A production by human T cells in a sex-specific way. Proc National Acad Sci USA. 2012;109:9505–9510. doi: 10.1073/pnas.1118458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beenakker K.G.M., Westendorp R.G.J., Craen A.J.M. de, Chen S., Raz Y., Ballieux B.E.P.B., Nelissen R.G.H.H., Later A.F.L., Huizinga T.W., Slagboom P.E., et al. Men Have a Stronger Monocyte-Derived Cytokine Production Response upon Stimulation with the Gram-Negative Stimulus Lipopolysaccharide than Women: A Pooled Analysis Including 15 Study Populations. J. Innate Immun. 2019;12:1–12. doi: 10.1159/000499840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campesi I., Montella A., Franconi F. Human monocytes respond to lipopolysaccharide (LPS) stimulation in a sex-dependent manner. J. Cell. Physiol. 2022;237:580–588. doi: 10.1002/jcp.30503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta S., Nakabo S., Blanco L.P., O’Neil L.J., Wigerblad G., Goel R.R., Mistry P., Jiang K., Carmona-Rivera C., Chan D.W., et al. Sex differences in neutrophil biology modulate response to type I interferons and immunometabolism. Proc. Natl. Acad. Sci. USA. 2020;117:16481–16491. doi: 10.1073/pnas.2003603117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitacre C.C. Sex differences in autoimmune disease. Nat. Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 19.Eaton W.W., Rose N.R., Kalaydjian A., Pedersen M.G., Mortensen P.B. Epidemiology of autoimmune diseases in Denmark. J. Autoimmun. 2007;29:1–9. doi: 10.1016/j.jaut.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein S.L., Marriott I., Fish E.N. Sex-based differences in immune function and responses to vaccination. Trans. R. Soc. Trop. Med. Hyg. 2015;109:9–15. doi: 10.1093/trstmh/tru167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee B., Yap H., Chew F., Quah T., Prabhakaran K., Chan G.S.H., Wong S., Seah C. Age- and sex-related changes in lymphocyte subpopulations of healthy Asian subjects: From birth to adulthood. Cytometry. 1996;26:8–15. doi: 10.1002/(sici)1097-0320(19960315)26:1<8::aid-cyto2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 22.Uppal S.S., Verma S., Dhot P.S. Normal values of CD4 and CD8 lymphocyte subsets in healthy indian adults and the effects of sex, age, ethnicity, and smoking. Cytometry B Clin. Cytom. 2003;52:32–36. doi: 10.1002/cyto.b.10011. [DOI] [PubMed] [Google Scholar]
- 23.Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G., et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 2018;175:1701–1715.e16. doi: 10.1016/j.cell.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ecker S., Chen L., Pancaldi V., Bagger F.O., Fernández J.M., Pau E.C. de S., Juan D., Mann A.L., Watt S., Casale F.P., et al. Genome-wide analysis of differential transcriptional and epigenetic variability across human immune cell types. Genome Biol. 2017;18 doi: 10.1186/s13059-017-1156-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell-type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooke H.J., Brown W.R.A., Rappold G.A. Hypervariable telomeric sequences from the human sex chromosomes are pseudoautosomal. Nature. 1985;317:687–692. doi: 10.1038/317687a0. [DOI] [PubMed] [Google Scholar]
- 27.Tukiainen T., Villani A.-C., Yen A., Rivas M.A., Marshall J.L., Satija R., Aguirre M., Gauthier L., Fleharty M., Kirby A., et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550:244–248. doi: 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berghöfer B., Frommer T., Haley G., Fink L., Bein G., Hackstein H. TLR7 ligands induce higher IFN-alpha production in females. J. Immunol. 2006;177:2088–2096. doi: 10.4049/jimmunol.177.4.2088. [DOI] [PubMed] [Google Scholar]
- 29.Deane J.A., Pisitkun P., Barrett R.S., Feigenbaum L., Town T., Ward J.M., Flavell R.A., Bolland S. Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Souyris M., Mejía J.E., Chaumeil J., Guéry J.-C. Female predisposition to TLR7-driven autoimmunity: gene dosage and the escape from X chromosome inactivation. Semin. Immunopathol. 2019;41:153–164. doi: 10.1007/s00281-018-0712-y. [DOI] [PubMed] [Google Scholar]
- 31.Lu Q., Wu A., Tesmer L., Ray D., Yousif N., Richardson B. Demethylation of CD40LG on the Inactive X in T Cells from Women with Lupus. J. Immunol. 2007;179:6352–6358. doi: 10.4049/jimmunol.179.9.6352. [DOI] [PubMed] [Google Scholar]
- 32.Wang J., Syrett C.M., Kramer M.C., Basu A., Atchison M.L., Anguera M.C. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc. Natl. Acad. Sci. USA. 2016;113:E2029–E2038. doi: 10.1073/pnas.1520113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 34.Cotton A.M., Price E.M., Jones M.J., Balaton B.P., Kobor M.S., Brown C.J. Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 2015;24:1528–1539. doi: 10.1093/hmg/ddu564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garieri M., Stamoulis G., Blanc X., Falconnet E., Ribaux P., Borel C., Santoni F., Antonarakis S.E. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc. Natl. Acad. Sci. USA. 2018;115:13015–13020. doi: 10.1073/pnas.1806811115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauteraud R., Stahl J.M., James J., Englebright M., Chen F., Zhan X., Carrel L., Liu D.J. Inferring genes that escape X-Chromosome inactivation reveals important contribution of variable escape genes to sex-biased diseases. Genome Res. 2021;31:1629–1637. doi: 10.1101/gr.275677.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhie S.K., Yao L., Luo Z., Witt H., Schreiner S., Guo Y., Perez A.A., Farnham P.J. ZFX acts as a transcriptional activator in multiple types of human tumors by binding downstream of transcription start sites at the majority of CpG island promoters. Genome Res. 2018;28:310–320. doi: 10.1101/gr.228809.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni W., Perez A.A., Schreiner S., Nicolet C.M., Farnham P.J. Characterization of the ZFX family of transcription factors that bind downstream of the start site of CpG island promoters. Nucleic Acids Res. 2020;48:5986–6000. doi: 10.1093/nar/gkaa384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su K., Yang H., Li X., Li X., Gibson A.W., Cafardi J.M., Zhou T., Edberg J.C., Kimberly R.P. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J. Immunol. 2007;178:3272–3280. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin X., Kim K., Suetsugu H., Bang S.-Y., Wen L., Koido M., Ha E., Liu L., Sakamoto Y., Jo S., et al. Meta-analysis of 208370 East Asians identifies 113 susceptibility loci for systemic lupus erythematosus. Ann. Rheum. Dis. 2021;80:632–640. doi: 10.1136/annrheumdis-2020-219209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gal-Oz S.T., Maier B., Yoshida H., Seddu K., Elbaz N., Czysz C., Zuk O., Stranger B.E., Ner-Gaon H., Shay T. ImmGen report: sexual dimorphism in the immune system transcriptome. Nat. Commun. 2019;10:4295–4314. doi: 10.1038/s41467-019-12348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu S., Akula N., Reardon P.K., Russ J., Torres E., Clasen L.S., Blumenthal J., Lalonde F., McMahon F.J., Szele F., et al. Aneuploidy effects on human gene expression across three cell types. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2218478120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammad I., Starskaia I., Nagy T., Guo J., Yatkin E., Väänänen K., Watford W.T., Chen Z. Estrogen receptor α contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 2018;11 doi: 10.1126/scisignal.aap9415. [DOI] [PubMed] [Google Scholar]
- 46.Sellau J., Groneberg M., Fehling H., Thye T., Hoenow S., Marggraff C., Weskamm M., Hansen C., Stanelle-Bertram S., Kuehl S., et al. Androgens predispose males to monocyte-mediated immunopathology by inducing the expression of leukocyte recruitment factor CXCL1. Nat. Commun. 2020;11:3459–3516. doi: 10.1038/s41467-020-17260-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walecki M., Eisel F., Klug J., Baal N., Paradowska-Dogan A., Wahle E., Hackstein H., Meinhardt A., Fijak M. Androgen receptor modulates Foxp3 expression in CD4+CD25+Foxp3+ regulatory T-cells. Mol. Biol. Cell. 2015;26:2845–2857. doi: 10.1091/mbc.e14-08-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jørgensen K.T., Rostgaard K., Bache I., Biggar R.J., Nielsen N.M., Tommerup N., Frisch M. Autoimmune diseases in women with Turner’s Syndrome. Arthritis Rheum. 2010;62:658–666. doi: 10.1002/art.27270. [DOI] [PubMed] [Google Scholar]
- 49.Panimolle F., Tiberti C., Spaziani M., Riitano G., Lucania G., Anzuini A., Lenzi A., Gianfrilli D., Sorice M., Radicioni A.F. Non-organ-specific autoimmunity in adult 47,XXY Klinefelter patients and higher-grade X-chromosome aneuploidies. Clin. Exp. Immunol. 2021;205:316–325. doi: 10.1111/cei.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagen S.H., Henseling F., Hennesen J., Savel H., Delahaye S., Richert L., Ziegler S.M., Altfeld M. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human pDC Level. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Souyris M., Cénac C., Azar P., Daviaud D., Canivet A., Grunenwald S., Pienkowski C., Chaumeil J., Mejía J.E., Guéry J.-C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018;3 doi: 10.1126/sciimmunol.aap8855. [DOI] [PubMed] [Google Scholar]
- 52.Brown G.J., Cañete P.F., Wang H., Medhavy A., Bones J., Roco J.A., He Y., Qin Y., Cappello J., Ellyard J.I., et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605:349–356. doi: 10.1038/s41586-022-04642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peeters S.B., Cotton A.M., Brown C.J. Variable escape from X-chromosome inactivation: Identifying factors that tip the scales towards expression. Bioessays. 2014;36:746–756. doi: 10.1002/bies.201400032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zito A., Roberts A.L., Visconti A., Rossi N., Andres-Ejarque R., Nardone S., Moustafa J.S.E.-S., Falchi M., Small K.S. Escape from X-inactivation in twins exhibits intra- and inter-individual variability across tissues and is heritable. PLoS Genet. 2023;19 doi: 10.1371/journal.pgen.1010556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Godfrey A.K., Naqvi S., Chmátal L., Chick J.M., Mitchell R.N., Gygi S.P., Skaletsky H., Page D.C. Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 2020 doi: 10.1101/gr.261248.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 58.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2016;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. 2021. Fast gene set enrichment analysis. 060012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw RNA-Seq data has been deposited to dpGAP and processed data has been deposited at github. Both are publicly available as of the date of publication. Accession numbers and DOIs are listed in the key resources table.
-
•
This paper analyzes existing, publicly available data. Accession numbers for these datasets are listed in the key resources table.
-
•
Original code has been deposited at github (https://github.com/lvblanton/sca-immune) and is publicly available as of the date of publication (https://doi.org/10.5281/zenodo.11889920).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.