

Abstract
Photocurable gelatin‐based hydrogels have established themselves as powerful bioinks in tissue engineering due to their excellent biocompatibility, biodegradability, light responsiveness, thermosensitivity and bioprinting properties. While gelatin methacryloyl (GelMA) has been the gold standard for many years, thiol‐ene hydrogel systems based on norbornene‐functionalized gelatin (GelNB) and a thiolated crosslinker have recently gained increasing importance. In this paper, a highly reproducible water‐based synthesis of GelNB is presented, avoiding the use of dimethyl sulfoxide (DMSO) as organic solvent and covering a broad range of degrees of functionalization (DoF: 20% to 97%). Mixing with thiolated gelatin (GelS) results in the superfast curing photoclick hydrogel GelNB/GelS. Its superior properties over GelMA, such as substantially reduced amounts of photoinitiator (0.03% (w/v)), superfast curing (1–2 s), higher network homogeneity, post‐polymerization functionalization ability, minimal cross‐reactivity with cellular components, and improved biocompatibility of hydrogel precursors and degradation products lead to increased survival of primary cells in 3D bioprinting. Post‐printing viability analysis revealed excellent survival rates of > 84% for GelNB/GelS bioinks of varying crosslinking density, while cell survival for GelMA bioinks is strongly dependent on the DoF. Hence, the semisynthetic and easily accessible GelNB/GelS hydrogel is a highly promising bioink for future medical applications and other light‐based biofabrication techniques.
Keywords: 3D bioprinting, bioinks, cell encapsulation, gelatin, hydrogels, thiol‐ene chemistry, tissue engineering
Development of a highly biocompatible, superfast curing photoclick bioink, comprising norbornene‐ and thiol‐functionalized gelatin (GelNB/GelS). Thiol‐ene photocrosslinking allows a significant reduction in photoinitiator concentration while accelerating hydrogel formation upon irradiation with visible light. Combined with low toxicity of GelNB/GelS precursors and minimal cross‐reactivity, this leads to improved cell survival in extrusion‐based 3D bioprinting compared to the gold standard gelatin methacryloyl.
1. Introduction
The considerable progress of biofabrication techniques including 3D bioprinting in recent years has found increasing utility in life science, particularly in tissue engineering, enabling novel methods to produce three‐dimensional scaffolds that mimic the extracellular matrix (ECM) and promote cell growth in artificial settings.[ 1 , 2 ] The choice of material in 3D bioprinting plays a crucial role hence why great efforts have been done to develop suitable inks.[ 3 , 4 , 5 ] The presence of cells in the bioink greatly increases the demands on ink formulation and printing technology.[ 6 , 7 ] Cell‐containing biomaterials, so‐called bioinks, typically consist of a cellular component, such as single cells, spheroids, organoids, microcarriers, and a biomaterial component that serves as a three‐dimensional scaffold.[ 8 ]
Hydrogels are the most commonly used bioinks due to their high water content, controlled swelling behavior, excellent biocompatibility, advantageous viscoelastic properties and shear‐thinning behavior.[ 9 , 10 ] Even though natural hydrogels, such as gelatin, collagen, hyaluronic acid, chitosan, silk fibroin or Matrigel, exhibit best biocompatibility, their application in 3D bioprinting is challenging since gelation is solely based on physical principles like temperature, electrostatic or hydrophobic interactions.[ 11 ] Hence, polymer modification strategies, especially incorporation of functional groups for photochemical crosslinking, have rendered hydrogel precursors photoresponsive, thus allowing spatiotemporal control over the hydrogel formation process.[ 12 ] Semi‐synthetic, light‐curable bioinks based on methacrylate‐functionalized biopolymers, such as gelatin methacryloyl (GelMA),[ 13 , 14 , 15 , 16 ] collagen methacryloyl (ColMA)[ 17 , 18 , 19 ] and hyaluronic acid methacrylate (HAMA)[ 20 , 21 , 22 , 23 ] have been widely used in tissue engineering. However, Muñoz et al. have shown major drawbacks of photocrosslinking reactions through chain‐growth polymerization mechanism, including side reactions of methacrylate groups with components of the cell surface, the need of high amounts of radicals for initiation, long irradiation times, and the formation of a heterogeneous network structure.[ 24 , 25 ]
Therefore, the development of hydrogels with alternative photocrosslinking strategies, that take place under milder, cell culture suitable and faster curing conditions, have attracted researchers’ interest especially for the 3D bioprinting of large‐scale structures as continuous UV irradiation and increased manufacturing time will lead to higher rates of encapsulated dead cells.[ 26 , 27 , 28 ] Fast crosslinking reactions are indispensable in extrusion‐based bioprinting to reduce UV exposure as well as to ensure homogeneous cell distribution, high structure integrity of the released bioink and fast removal of toxic photoinitiator residues post‐printing.[ 29 , 30 ] A promising approach for rapid sol‐gel transition is based on phototriggered‐imine‐crosslinking (PIC) between photogenerated aldehydes (from o‐nitrobenzene) and amines, having found increased utility in in situ tissue regeneration.[ 31 , 32 ]
Currently, the most commonly used strategy for superfast curing is thiol‐ene chemistry, which is based on the reaction between a thiol and an alkene.[ 33 , 34 ] Cyclic alkenes, especially norbornenes, have gained huge importance in this field.[ 35 ] In contrast to methacrylates, the norbornene group exhibits less toxicity for cells and reacts selectively with thiols through a step‐growth polymerization mechanism. Since homopolymerization on the same gelatin chain is prevented, photocrosslinking results in the formation of a highly defined and homogeneous network.[ 36 ] Such hydrogels would be much more suitable for medical applications as they give rise to well defined and reproducible hydrogel networks in terms of transplantable tissue engineering. Moreover, thiol‐ene reactions are not susceptible to oxygen inhibition and require lower radical concentrations for photochemical initiation.[ 37 ] Various hydrogel systems based on a norbornene‐functionalized biopolymer and a multi‐thiol crosslinker have been developed.[ 38 , 39 , 40 , 41 , 42 , 43 , 44 ] For example, Muñoz et al. have proved increased cell viability and spreading of encapsulated human mesenchymal stem cells (hMSCs) in GelNB/DTT hydrogels compared to the gold standard GelMA.[ 24 ] Furthermore, laser‐based high‐definition bioprinting of GelNB/DTT bioinks has been successfully demonstrated by Dobos et al., who used two‐photon polymerization (2PP) to fabricate 3D structures with sub‐micrometer resolution.[ 45 ] In a study on norbornene‐functionalized gelatin hydrogels by Van Hoorick et al., different thiol crosslinkers were compared in terms of their processability and network properties, highlighting the superior suitability of multifunctional over bifunctional crosslinkers.[ 46 ]
Recently, a crosslinker‐free hydrogel system based on norbornene and thiol‐functionalized gelatin has been published by Sandra Van Vlierberghe and coworkers, demonstrating increased differentiation of adipose‐derived stem cells (ASCs), which were seeded onto extrusion‐based printed scaffolds.[ 47 ] However, limited research on the rheological tunability and toxicity of the uncured GelNB/GelS precursor solution as well as its cell encapsulation capability has been conducted so far, which determines the potential of the bioink for cell‐based printing techniques. Cell encapsulation requires well defined mesh sizes, solvent compatibility, low UV light exposition as well as the absence of cell harming chemistry.[ 48 , 49 ] In addition, high cell survival rates in the precursor solution over a reasonable timescale (minutes to hours) are essential in 3D bioprinting to fabricate complex cell‐laden structures of relevant size.[ 50 , 51 ] When it comes to transplantation and medical application the activation of the immune system has to be minimized or avoided.[ 52 , 53 ] Hence, the goal of the semisynthetic, dual‐functionalized gelatin hydrogel was to be as chemically defined as possible while to be as natural as possible.
In this paper, we present an alternative synthesis route for superfast curing GelNB in water, avoiding the use of dimethyl sulfoxide (DMSO) as organic solvent and resulting in low, medium or high functionalization of gelatin with norbornene groups. Furthermore, we demonstrate the superior performance of the GelNB/GelS hydrogel system over GelMA and GelNB/DTT in 3D cell culture and for the first time its application in extrusion‐based 3D bioprinting. GelNB/GelS hydrogels of adaptable stiffness are profoundly characterized in terms of their degree of functionalization, potential of postsynthetic modification, physicomechanical and rheological properties, biocompatibility, biodegradability, proliferation of encapsulated cells and survival of primary human fibroblasts in 3D bioprinting.
2. Results and Discussion
2.1. Synthesis of GelNB and GelS Hydrogel Precursors
Norbornene‐ (GelNB) and thiol‐functionalized gelatin (GelS) precursors were synthesized separately from gelatin type A by chemical modification of primary amines (Lys, Hyl) to incorporate photocrosslinkable moieties along the gelatin backbone. The percentage of converted amino groups is given by the degree of functionalization (DoF). While a maximum DoF of 97% was reported for GelMA, previous synthetic attempts for GelNB based on carbic anhydride resulted in limited norbornene substitution of only 44 ± 2%.[ 24 , 54 ] In a recently published synthesis approach by Van Hoorick et al., 5‐Norbornene‐2‐carboxylic acid (NBCA) and carbodiimide/N‐hydroxysuccinimde coupling (EDC/NHS) chemistry was used to significantly increase DoF to 90%.[ 39 ] However, the reaction was carried out in DMSO, which had to be completely removed during purification due to its cytotoxic effects.[ 55 , 56 , 57 ]
In this paper, we have developed a new synthesis for GelNB, combining the advantages of water‐based synthesis and high norbornene substitution of the two previous routes (Figure 1A).[ 24 , 39 ] First, NBCA was dissolved in MES buffer at pH 5–6 and converted into a succinimidyl ester by activation with EDC·HCl and NHS. The chosen molar ratio of carboxylic acid, EDC·HCl and NHS was 1:2:1 based on a standard peptide coupling protocol.[ 58 ] After addition of gelatin, pH was raised to 7.5–7.8 to initiate the reaction between the primary amines of gelatin and the succinimidyl ester. The degree of norbornene substitution was controlled by the amount of NBCA (0.3, 2, 10 equiv.) and coupling reagents without changing the molar ratio of 1:2:1. In this context, one equivalent refers to the number of primary amino groups in gelatin (0.266 mmol per gram of gelatin), which was colorimetrically determined by a glycine standard curve (Figure S1, Supporting Information). The DoF was quantified using TNBSA assay (Figure 1B) and verified by 1H NMR spectroscopy (Figure S3, Supporting Information). 0.3 equiv. NBCA resulted in GelNB with low norbornene functionalities (DoF: 20 ± 2%), 2 equiv. to GelNB with medium norbornene modification level (DoF: 53 ± 1%), and 10 equiv. to GelNB with high norbornene substitution (DoF: 97 ± 1%). In the following, hydrogel precursors were defined according to these equivalents as GelNB Low, Medium and High. The herein realized DoF of 97% is – to the best of our knowledge – the highest reported DoF for norbornene‐functionalized gelatin.
Figure 1.
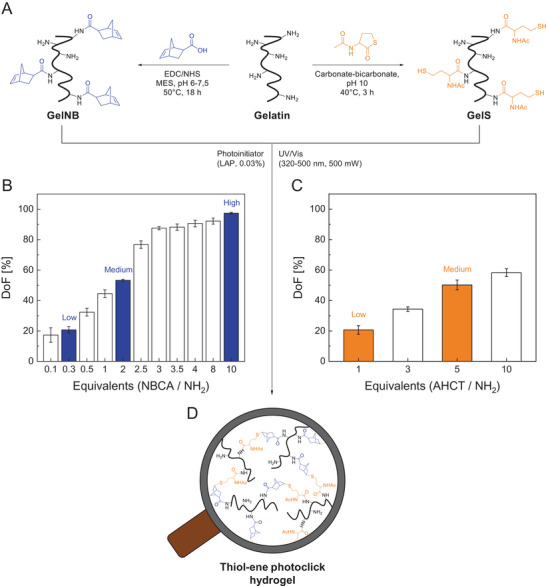
A) Water‐based synthesis of GelNB and GelS from gelatin. Effect of increasing amounts of NBCA and AHCT on the degree of functionalization (DoF) of B) GelNB and C) GelS hydrogel precursors determined by TNBSA assay (mean (n = 8) ± SD). GelNB (Low, Medium, High) and GelS (Low, Medium) precursors used for hydrogel preparation are highlighted in color. D) Photocrosslinked thiol‐ene click hydrogel obtained by exposing the GelNB/GelS hydrogel precursor solution to UV‐visible light.
GelS synthesis was performed using N‐Acetyl‐DL‐homocysteine thiolactone (AHCT) as previously reported by Van Vlierberghe et al. (Figure 1A).[ 59 ] Reaction, purification and storage was done under inert gas atmosphere to prevent disulfide bond formation. The DoF of GelS was controlled by the quantity of the thiolating reagent (1, 5 equiv.) although a considerably lower substitution level was obtained compared to GelNB (Figure 1C). 1 equiv. AHCT resulted in GelS Low (DoF: 20 ± 3%) and 5 equiv. in GelS Medium (DoF: 50 ± 3%). Further increase of the thiolating reagent did not result in any significant increase in DoF due to the growing competition between already incorporated thiols and the natural amines of gelatin. At pH 10, both functional groups act as nucleophiles that can open the thiolactone ring of AHCT. This makes a complete substitution unlikely, or at least requires very high amounts of the thiolating reagent.[ 59 ]
2.2. Thiol‐ene Photoclick Hydrogel GelNB/GelS
It has been shown that thiol and norbornene undergo very fast thiol‐ene click reactions when irradiated with light.[ 43 , 60 ] Thiol‐ene click chemistry was used to selectively crosslink the functionalized gelatin derivatives without the need of an additional crosslinking reagent (Figure 1D). GelNB and GelS were dissolved separately and then mixed equimolarily to ensure equal amounts of photocrosslinkable moieties (norbornene/thiol = 1:1) in the hydrogel precursor solution. After addition of the photoinitiator LAP, photochemical crosslinking reaction was induced by exposure to long‐wave UV light (320–500 nm, 500 mW cm‐2).
As shown above, synthesis of GelNB and GelS hydrogel precursors provided access to DoFs ranging from 20% to 97%. The higher the DoF, the higher was the number of photoreactive moieties available for chemical crosslinking. Hence, the choice of GelNB and GelS precursors allowed control over the degree of crosslinking, which is considered an important tool to fine‐tune the viscoelastic properties of the resulting hydrogels. We aimed to establish and investigate three types of GelNB/GelS hydrogels with low, medium and high crosslinking density as previously described for GelMA in literature.[ 16 , 61 , 62 ] In the following, formulations with equimolar thiol:ene ratio prepared from the precursors 1) GelNB Low + GelS Low, 2) GelNB Medium + GelS Medium, and 3) GelNB High + GelS Medium will be defined as 1) GelNB/GelS Low 2) GelNB/GelS Medium and 3) GelNB/GelS High hydrogels, respectively, after photocrosslinking.
First, swelling behavior and biodegradability of 5% (w/v) GelNB/GelS hydrogels (Low, Medium, High) were characterized, revealing their dependence on the DoF. The crosslinking density greatly affected the equilibrium mass swelling ratio of GelNB/GelS hydrogels (Figure 2A). The swelling capacity of hydrogels decreased with increasing degree of crosslinking due to a tighter and less flexible polymer network structure. An equilibrium swelling ratio of 17.4 and 8.7 was determined for GelNB/GelS Low and GelNB/GelS High, respectively. Enzymatic degradability was assessed by weight loss over a period of 14 days in PBS with 10% FCS (Figure 2B). The presence of protease‐sensitive cleavage sites (MMP sites) along the gelatin molecule ensured degradability of GelNB/GelS hydrogels. It was found that GelNB/GelS Low exhibited an accelerated degradation profile, resulting in complete degradation within 7 days. As the crosslinking density determined diffusion capacity and hence accessibility of enzymes to the inner structure of the hydrogel network, decomposition of lowly crosslinked hydrogels proceeded faster.
Figure 2.
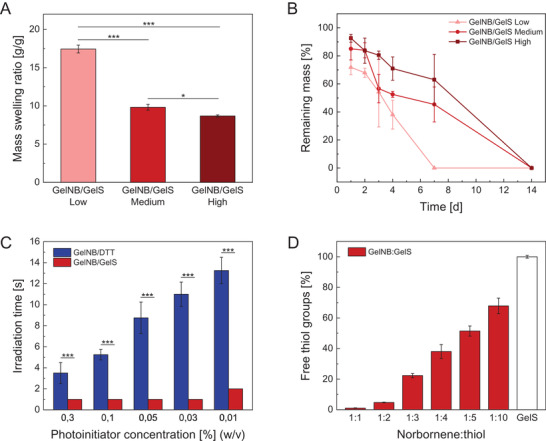
A) Mass swelling ratio of GelNB/GelS hydrogels with increasing crosslinking density (Low, Medium, High) (n = 3). B) Biodegradation of GelNB/GelS hydrogels (Low, Medium, High) in PBS with 10% FCS over a period of 14 days (n = 3). C) Comparison of the two thiol‐ene hydrogel systems GelNB/DTT and GelNB/GelS in terms of curing time and photoinitiator reduction (n = 4). D) Excess thiol functionalities available for chemical modification in GelNB/GelS hydrogels with increasing GelS proportion determined by Ellman's assay (n = 6). Data were presented as mean ± SD and statistically evaluated by C) Student's t‐test and A) one‐way ANOVA. *, **, *** represent p < 0.05, 0.01, and 0.001, respectively.
Second, we investigated the time scale of the hydrogel formation process, which is an important parameter in additive manufacturing, determining manufacturing time and cellular exposure to UV radiation. GelNB/GelS hydrogels were formed through a step‐growth polymerization mechanism. Numerous studies on reaction kinetics of step‐growth and chain‐growth polymerization mechanisms have been conducted in literature, outlining the superiority of thiol‐ene chemistry.[ 63 , 64 ] Hence, we focused on comparing the two thiol‐ene hydrogel systems GelNB/DTT (High) and GelNB/GelS (High) in terms of curing time and photoinitiator concentration required for photocrosslinking. Since crosslinking reaction of GelMA hydrogels is usually performed at concentrations beyond 0.3% LAP, we took this concentration as our starting point and gradually reduced the amount of photoinitiator to 0.01% while determining curing time for both hydrogel systems (Figure 2C). The decrease of photoinitiator concentration had very little impact on curing time of GelNB/GelS hydrogels. The curing time remained constant over a wide range of LAP concentrations. Only at 0.01%, we observed a slight increase from 1 to 2 s, hence why we defined 0.03% as an appropriate LAP concentration for GelNB/GelS curing. In the crosslinker‐free hydrogel system, one thiol‐ene reaction between a thiol group of GelS and a norbornene group of GelNB was needed to form a covalent bond between gelatin molecules. In contrast, the number of required thiol‐ene reactions was increased to two when using the bifunctional thiol crosslinker DTT. Each thiol group of DTT had to react with a norbornene group of GelNB. Since the norbornene groups could be located either on the same or on adjacent gelatin chains, only a limited number of thiol‐ene reactions led to crosslinks between different polymer chains. Therefore, curing time of GelNB/DTT hydrogels was comparatively longer and increased with reducing amount of LAP.
A further advantage of the GelNB/GelS hydrogel is the possibility of postsynthetic functionalization. While the degree of conversion of methacrylate groups in GelMA hydrogels is either complete or barely controllable, hydrogels comprising two differently functionalized gelatin derivatives do not face such limitation as excess thiol or norbornene groups are not consumed during photocrosslinking.[ 24 , 65 , 66 , 67 ] By varying the norbornene:thiol ratio, excess functional moieties can be used for selective attachment of molecules of interest. We demonstrated a proof of concept using GelNB/GelS (Medium) formulations with increasing GelS proportion from 1:1 to 1:10 (norbornene/thiol). Excess thiol groups in the photocrosslinked hydrogels were detected in a color‐forming reaction by Ellman's reagent, allowing to determine the percentage of unreacted thiol groups (Figure 2D). For the equimolar GelNB/GelS hydrogel formulation (norbornene/thiol = 1:1), a degree of conversion of 99% was assessed, proving high efficiency of the thiol‐ene reaction. Increasing GelS proportion provided a growing number of thiol groups that were available for postsynthetic modification.
2.3. Biocompatibility Study
In a profound biocompatibility study, we aimed to prove the superior properties of the superfast curing GelNB/GelS hydrogel compared to state‐of‐the‐art hydrogel systems, particularly GelNB/DTT and the gold standard GelMA. First, we focused on the biocompatibility of the uncrosslinked gelatin derivatives GelNB and GelS in comparison to GelMA and assessed their toxicity prior to photocrosslinking (Figure 3A). In order to compare only the toxicity of the photoreactive moieties (methacrylate, norbornene, thiol), we used hydrogel precursors that were synthesized from the same gelatin educt, had a comparable DoF (approx. 50%), and were tested at the same polymer concentration (5% (w/v)). Cell viability was determined for different exposure times by a MTT proliferation assay with the human hepatocarcinoma cell line HepG2, which is commonly used as a liver model for in vitro toxicity studies. Since the liver is the main organ involved in metabolism and processing of xenobiotics, HepG2 cells represented an ideal choice for a comprehensive toxicity study on the hydrogel components. It was found that GelNB and GelS precursors exhibited similar biocompatibility over several hours, while cell viability of GelMA decreased substantially after only 2 h. The detrimental effects of GelMA on cell viability was due to side reactions, especially Michael additions, between methacrylate groups and nucleophiles on the cell surface. However, high levels of viability in the uncrosslinked precursor solution over a reasonable period ranging from minutes to hours are indispensable for 3D bioprinting, thus rendering thiol‐ene based bioinks more favorable candidates.
Figure 3.
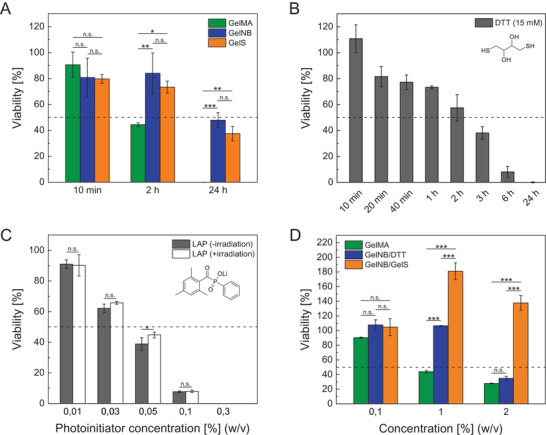
Cytotoxicity assessed by MTT proliferation assays with A, B, D) HepG2 and C) HUVEC of A) hydrogel precursors GelMA, GelNB, and GelS (5% (w/v)), B) crosslinking reagent DTT (15 mm), C) photoinitiator LAP (0.01–0.3%) and D) degradation products of GelMA, GelNB/DTT, and GelNB/GelS hydrogels obtained by collagenase digestion (10 U mL‐1, type 1, from Clostridium histolyticum) (n = 3). Data were presented as mean ± SD and statistically evaluated by C) Student's t‐test and A, D) one‐way ANOVA. *, **, *** represent p < 0.05, 0.01, and 0.001, respectively.
Next, we compared the biocompatibility of the two GelNB‐based hydrogel systems with regard to their thiol component. We were able to demonstrate high cytotoxicity of the crosslinker DTT at a concentration of 15 mm, which is commonly used for crosslinking GelNB hydrogels (Figure 3B).[ 24 ] DTT toxicity was greatly dependent on exposure time, resulting in complete cell death after 24 h. It is known from literature that DTT is unstable and easily undergoes fast air oxidation, which leads to H2O2 production and formation of harmful free radicals and reactive oxygen species, causing oxidative stress for cells.[ 68 , 69 , 70 ] Despite the usage of GelNB/DTT hydrogels in 3D cell culture, their suitability in 3D bioprinting is very limited because bioink preparation and photopolymerization are delayed in time, thus intensifying the adverse effects of the crosslinker DTT. In contrast, GelS was stable in solution for at least two weeks and turned out to be significantly less toxic than DTT at longer exposure times.
As shown above, a major advantage of the crosslinker‐free GelNB/GelS formulation is the reduced amount of harmful photoinitiator from 0.3% to 0.03%. Since LAP remained in the hydrogel after photopolymerization and was only slowly removed by diffusion over time, its toxicity had to be considered. An LD50 of 0.04% LAP after 24 h exposure was determined by a MTT assay with HUVEC (Figure 3C). We also demonstrated that irradiation with UV–vis light for 30 s, which lead to decomposition of the photoinitiator, did not cause any additional toxicity.
Furthermore, we evaluated the toxicity of the degradation products of photocrosslinked GelNB/GelS hydrogels in comparison to GelNB/DTT and GelMA hydrogels by enzymatic decomposition with collagenase (Figure 3D). Although the three hydrogels were gelatin‐based, the effect of the degradation solutions on cell viability greatly differed. We assume that the increased toxicity is due to multiple reasons, including unreacted photoreactive groups, excess DTT crosslinker and especially photoinitiator residues, which were ten times higher in GelMA and GelNB/DTT hydrogels.
2.4. Cell Encapsulation
Gelatin‐based hydrogels have gained great importance as scaffolds for 3D cell culture due to the presence of inherent bioactive sites in the amino acid sequence of gelatin. Arg‐Gly‐Asp (RGD) sequences act as cell‐binding domains and promote cell‐scaffold interaction.[ 43 , 71 , 72 ] In addition, protease‐sensitive sites allow enzymatic degradation and scaffold remodeling, which is necessary for cell motility and spreading within the hydrogel.[ 73 , 74 , 75 , 76 ] Gelatin naturally contains both RGD and matrix metalloproteinase (MMP) sites, thus rendering gelatin‐based hydrogels particularly suitable for tissue reconstruction.[ 16 ]
Normal human dermal fibroblasts (NHDF) were used as a model for cell encapsulation because of their characteristic spindle‐shaped morphology, huge size and their capability to only proliferate when they can attach, elongate and contact other cells in a correct manner. NHDF are also widely used in tissue engineering to prove the material stability as fibroblasts exhibit strong mechanical forces and contract the material when they are properly grown to connective tissue. Moreover, they are usually strongly dependent on a high collagen content in their ECM to attach and stretch correctly. Since gelatin scaffolds closely mimic the natural 3D environment of fibroblasts, the primary cell line was ideally suited to compare different gelatin‐based hydrogel systems. NHDF were embedded in GelNB/GelS hydrogels (Low, Medium, High) and cultured over a period of 14 days to study cell adhesion, spreading, viability, and proliferation behavior. Live/dead staining with calcein‐AM and propidium iodide was performed 1, 7, and 14 days post‐encapsulation, followed by acquisition of z‐stacks (300 µm) through confocal microscopy (Figure 4A). Although high cell viability was observed in GelNB/GelS hydrogels of varying degrees of crosslinking, cell spreading and proliferation proceeded much faster in lowly crosslinked hydrogels. One day post‐encapsulation, the fibroblasts still retained a rounded morphology in GelNB/GelS High, while elongation of NHDF was already initiated in GelNB/GelS Low, resulting in the formation of a dense cell layer after one week. The higher the degree of crosslinking, the more time was needed for NHDF to reach their characteristic morphology. Likewise, biodegradation of the 3D gelatin scaffold was accelerated in low‐degree crosslinked hydrogels, thus reducing the available space for a growing number of cells and contributing to increased cell death in GelNB/GelS Low after 14 days. Furthermore, proliferation of NHDF and HepG2 embedded in GelNB/GelS Low was exemplarily monitored over a period of 21 days by PrestoBlue proliferation assay (Figure 4B). For both primary fibroblast and cancer cell line, cell numbers increased significantly up to 2‐ to 8‐fold.
Figure 4.
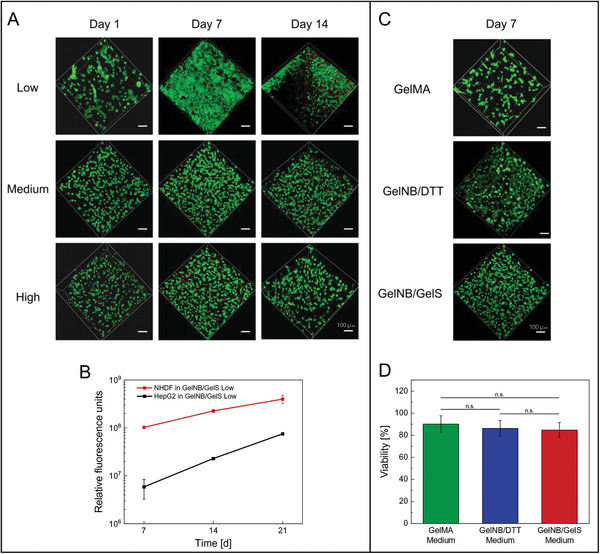
A) Live/dead staining of NHDF embedded in GelNB/GelS hydrogels of varying crosslinking rates (Low, Medium, High) with calcein‐AM (green, live cells) and propidium iodide (red, dead cells) 1, 7, and 14 days post‐encapsulation, followed by confocal microscopy (Leica TCS SPE, scale bar 100 µm). B) Proliferation of encapsulated HepG2 and NHDF in GelNB/GelS (Low) hydrogels over a period of 21 days monitored by PrestoBlue assay (n = 3). C) Live/dead staining of NHDF embedded in GelMA (Medium), GelNB/DTT (Medium) and GelNB/GelS (Medium) hydrogels 7 days post‐encapsulation, followed by confocal microscopy and D) cell viability analysis. Viability was determined by the percentage of live cells over the total cell count (Python software). Data were presented as mean ± SD and statistically evaluated by one‐way ANOVA. *, **, *** represent p < 0.05, 0.01, and 0.001, respectively.
In addition to varying crosslinking rates, we investigated the biological performance of NHDF embedded in GelNB/GelS hydrogels compared to GelNB/DTT and GelMA (Figure 4C,D). After 7 days of cultivation, fibroblasts showed a spindle‐shaped morphology in all three hydrogels without any significant difference in cell viability. Despite the excellent suitability of all three hydrogels for 3D cell culture, it has to be taken into account that photocuring took place immediately after cells had been suspended in the hydrogel precursor solution, thus neglecting any detrimental effects caused by components of the liquid bioink. In 3D bioprinting, however, bioink preparation and photocrosslinking are typically delayed by several minutes based on the size of the layers. Hence, side reaction of methacrylate groups as well as crosslinker and photoinitiator toxicity have to be considered, which strongly affect cell viability as demonstrated in Figure 3.
2.5. Rheological Characterization
Rheological measurements were performed to determine the viscoelastic properties governing 3D printability, particularly shear moduli (storage modulus G’, loss modulus G’’) and shear viscosity of GelNB/GelS hydrogels and their uncrosslinked precursor solutions. We investigated how the DoF of gelatin derivatives affected the shear moduli of photocrosslinked GelNB/GelS hydrogels at 37 °C after swelling (Figure 5A). Rheological data revealed that the elasticity of the hydrogels was related to the number of norbornene and thiol groups involved in photochemical crosslinking. Increasing crosslinking density within the polymer network resulted in an increase of G’ from 126 to 405 to 520 Pa for GelNB/GelS Low, Medium and High, respectively. Corresponding frequency sweep data are provided in the supporting information (Figure S4, Supporting Information). In addition, we determined the impact of amino group functionalization on the temperature‐induced, reversible gelation process by measuring the storage moduli of the three precursor solutions at 14 °C prior to photocrosslinking (Figure 5B). Interestingly, we observed an opposite trend compared to the crosslinked GelNB/GelS hydrogels. Increasing DoF led to a decrease of G’ from 350 to 270 to 190 Pa for GelNB/GelS Low, Medium and High precursor solutions, respectively. We assume that the norbornene group is mainly responsible for this finding. Due to its hydrophobic character and steric hindrance, the norbornene group disturbs physical gelation by hindering gelatin chains from self‐assembling into triple helices at low temperatures.
Figure 5.
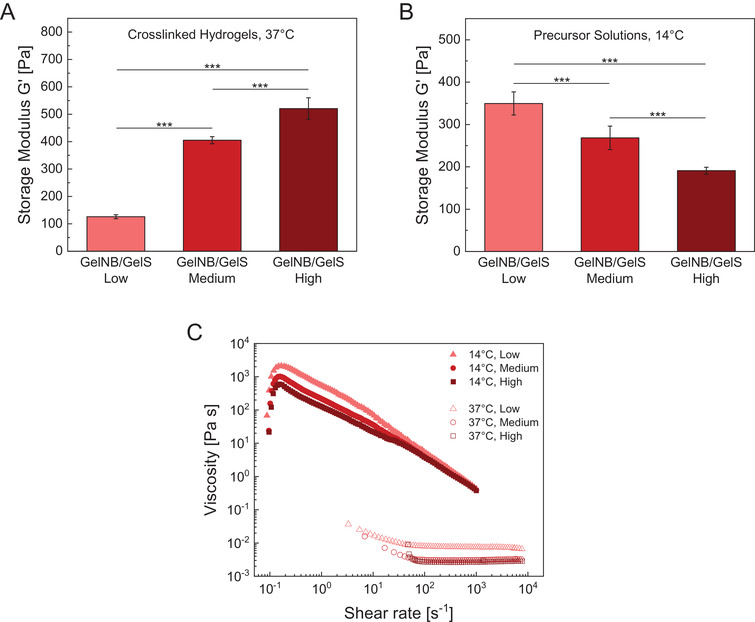
Plateau storage moduli G ′ of A) photocrosslinked GelNB/GelS hydrogels (Low, Medium, High) at 37 °C and B) their uncrosslinked, but physically gelated precursor solutions at 14 °C. Average G ′ were determined from the linear viscoelastic region of frequency sweep measurements. Data were presented as mean ± SD and statistically evaluated by one‐way ANOVA. *, **, *** represent p < 0.05, 0.01, and 0.001, respectively. C) Shear‐viscosity profiles of GelNB/GelS precursor solutions at 37 and 14 °C.
Furthermore, we determined the shear viscosity profiles of uncrosslinked GelNB/GelS solutions to evaluate their suitability for extrusion‐based 3D bioprinting (Figure 5C). Printable inks typically exhibit shear‐thinning behavior, characterized by a decrease in viscosity with increasing shear stress, allowing to extrude viscous solutions through a nozzle and to reduce shear stress on cells. At 37 °C, GelNB/GelS precursor solutions were strongly liquid‐like and displayed almost Newtonian behavior, which means that there was no significant dependence of the viscosity on the shear rate. The measured viscosity of ≈5 mPa s remained nearly constant for shear rates between 102 and 104 s−1. Since such a low viscosity at 37 °C prevents 3D printability, a common approach is to cool the precursor solution near the gelling point of gelatin to exploit the temperature‐dependent viscosity increase. Hence, we also measured the shear‐rate dependent viscosity at 14 °C to evaluate shear‐thinning behavior. All precursor solutions displayed pseudoplastic behavior at 14 °C, as their shear‐viscosity ranged from 2000 Pa s for low shear rates to 0.5 Pa s for high shear rates. This property rendered GelNB/GelS precursor solutions printable and ensured fiber formation during extrusion.
2.6. 3D Bioprinting
Finally, 3D extrusion‐based bioprinting with NHDF‐laden hydrogel precursor solutions were conducted to evaluate GelNB/GelS bioink performance in comparison to the gold standard GelMA. In a previous study by Tytgat et al., GelNB/GelS scaffolds had been successfully fabricated, however, cells were only seeded onto photocrosslinked hydrogels without resulting in any significant difference in cell viability between GelNB/GelS and GelMA hydrogels.[ 47 ] In our study, we hence focused on the bioprintability of the GelNB/GelS precursor solution. NHDF were suspended in GelNB/GelS (Low, Medium, High) and GelMA (Low, Medium, High) precursor solutions, respectively, and transferred to the cartridge of an extrusion‐based 3D printer. An increase in viscosity of the gelatin‐based bioinks was achieved by cooling the cell‐laden precursor solutions to 22 °C to induce physical gelation, rendering the bioinks printable. A grid structure consisting of four layers served as a model scaffold to ensure uniform nutrient supply (Figure 6A). From the bioink preparation to the completion of the 3D printing process, cells were exposed to a myriad of detrimental effects caused by the components of the precursor solution, the printing process and photochemical crosslinking. Biocompatibility of GelNB/GelS bioinks was evaluated in comparison to the GelMA bioink by post‐printing cell viability analysis, revealing improved cell survival for the thiol‐ene based bioink (Figure 6B,C). While viability considerably decreased with higher crosslinking rates in GelMA Low, Medium and High, respectively (Figure 6B), the GelNB/GelS bioink showed constant cell survival rates of over 84%, regardless of the DoFs of the involved precursors (Figure 6C). The lower viability of GelMA was due to the cross‐reactivity of methacrylate groups which became more significant with increasing DoF. In addition, chain‐growth polymerization mechanism required increased amounts of photoinitiator, radicals and UV irradiation for photocrosslinking, thus reducing cell viability. NHDF in the GelNB/GelS scaffolds displayed high long‐term viability over 80% and underwent considerable morphological changes over a period of 14 days (Figure S5, Supporting Information, Figure 6D). On day 7, spindle‐shaped morphology of NHDF and increasing cell interconnection was observed particularly at the edges of the scaffold. Cell orientation seemed to be determined by the geometry of the structure, since NHDF predominantly aligned along the longitudinal axis of the extruded fiber. Depth coding revealed a uniform cell distribution of encapsulated NHDF within the hydrogel. The presence of RGD domains ensured cell adhesion, resulting in a neglectable number of NHDF that grew on the glass slide. Additionally, the NHDF network stabilized the hydrogel scaffold, hence why the structural integrity of the bioink was easily maintained over 14 days.
Figure 6.
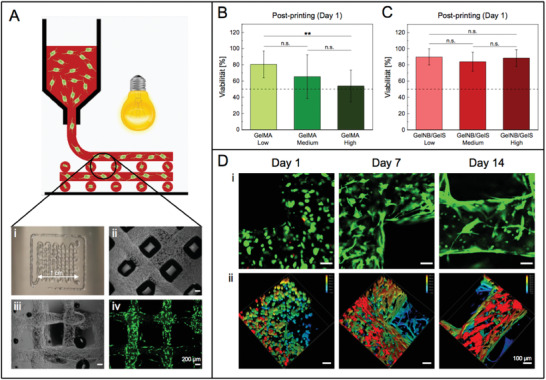
A) 3D bioprinting of a hydrogel grid structure (1 cm × 1 cm) consisting of four layers on a glass slide. Post‐printing cell viability analysis of 3D bioprinted NHDF at day 1 using B) GelMA (Low, Medium, High) and C) GelNB/GelS (Low, Medium, High) bioinks. Viability was determined by the percentage of live cells over the total cell count (Python software). Data were presented as mean ± SD and statistically evaluated by one‐way ANOVA. *, **, *** represent p < 0.05, 0.01, and 0.001, respectively. D) i) Live/dead staining with calcein‐AM (green, live cells) and propdium iodide (red, dead cells) of 3D bioprinted NHDF in the GelNB/GelS (Medium) bioink 1, 7, and 14 days post‐printing, followed by confocal microscopy (Leica TCS SPE, scale bar 100 µm). ii) Distribution of NHDF within the hydrogel along the z‐axis (depth coding, z‐stacks of 300 µm).
In summary, the GelNB/GelS bioink has turned out to be a promising substitute to the gold standard GelMA, particularly due its superior biocompatibility, excellent 3D bioprinting performance as well as highly defined composition for reproducible experiments.
3. Conclusion
In the current study, we developed a novel water‐based synthetic route for superfast curing GelNB/GelS hydrogels for 3D bioprinting. Well defined crosslinking rates in comparison to the gold standard GeMA ensure the reproducible production of GelNB/GelS hydrogels and bioinks for 3D tissue engineering in terms of future industrial process development and medical applications. In addition, the thiol‐ene photoclick hydrogels showed a superior performance and biocompatibility due to the reduction of photoinitiator concentration while at the same time decreasing the curing time in visible light, less side reactions with cell components, an increase in network homogeneity by step‐growth polymerization mechanism, and improved biocompatibility of both hydrogel precursors and degradation products. We were able to remove or at least reduce all detrimental effects on cell viability to a minimum, particularly the toxicity of photopolymers, crosslinker, and photoinitiator, resulting in excellent biocompatibility of all three types of GelNB/GelS precursor solutions for 3D bioprinting. This is a very crucial, but often neglected aspect in biocompatibility evaluation of bioinks, since cells are exposed to the uncrosslinked photopolymers and reagents for a considerable period of time during the 3D bioprinting process. The advantageous properties over state‐of‐the‐art hydrogels and its cost‐effective synthesis hence make the thiol‐ene photoclick hydrogel a promising candidate for further tissue engineering applications and light‐based biofabrication techniques.
4. Experimental Section
Materials
5‐Norbornene‐2‐carboxylic acid and lithium phenyl(2,4,6‐trimethylbenzoyl)phosphinate (LAP) were purchased from TCI Deutschland. 1‐Ethyl‐3‐(3‐dimethylaminopropyl)carbodiimid‐hydrochlorid (EDC·HCl), 2‐(N‐morpholino)ethanesulfonic acid (MES), 1,4‐dithiothreitol (DTT) and ethylene diamine tetraacetic acid (EDTA) were obtained from Carl Roth. DPBS−/−, DMEM, fetal calf serum (FCS), penicillin‐streptomycin (10 000 U mL−1), trypsin‐EDTA (0.05%), phenol red, calcein‐AM and PrestoBlue cell viability reagent were received from Thermo Fisher Scientific. The CellTiter 96 non‐radioactive cell proliferation assay (MTT) was provided by Promega. Dialysis tubes (MWCO 12–14 kDa) were obtained from VWR International. All other chemicals were purchased from Sigma‐Aldrich. Normal human dermal fibroblasts (NHDF), which derived from the same donor and were isolated from the dermis of adult skin, and human umbilical vein endothelial cells (HUVEC) were provided from PromoCell.
Synthesis of Gelatin Methacryloyl
The synthesis and purification of Gelatin Methacryloyl (GelMA) was performed as previously published in literature.[ 13 , 15 ] In brief, 1 g gelatin (type A, gel strength ≈300 g bloom, 0.266 mmol NH2 groups, 1 equiv.) was dissolved in 10 mL DPBS, and the solution was heated to 50 °C. After complete dissolution of gelatin, 39.6 µL methacrylic anhydride (0.266 mmol, 1 equiv.) was added dropwise and the reaction mixture was stirred at 50 °C for 2 h. Then, the solution was diluted with 10 mL dH2O, transferred into dialysis tubes and dialyzed against dH2O at 40 °C for 7 days. The purified solution was frozen at −80 °C and lyophilized (LSL Secfroid, Aclens, Switzerland). The product was obtained as a white solid and stored at −20 °C. GelMA with different degrees of functionalization (Low, Medium, High) was synthesized by varying the amount of methacrylic anhydride (1, 8, 20 equiv. referred to NH2 groups in gelatin).
1H NMR (400 MHz, D2O, δ): 7.60–7.40 (Haromtic, gelatin), 5.88 (1H, Ha, vinyl), 5.64 (1H, Hb, vinyl), 5.24–0.99 (gelatin), 3.20 (bs, 2H, NH2), 2.11 (s, 3H, CH3, Hc).
Water‐Based Synthesis of Norbornene‐Functionalized Gelatin
326 µL 5‐Norborne‐2‐carboxylic acid (368 mg, 2.66 mmol, 10 equiv.) was dissolved in 10–20 mL MES buffer (0.5 m, pH 6) and subsequently activated by addition of 1.02 g EDC·HCl (5.32 mmol, 20 equiv.) and 0.31 g NHS (2.66 mmol, 10 equiv.). The mixture was stirred at 50 °C for 15 min. After adding 1 g gelatin (type A, gel strength ≈300 g bloom, 0.266 mmol NH2 groups, 1 equiv.), pH 7.5–7.8 was adjusted with NaOH (10 m). The reaction mixture was stirred overnight at 50 °C. Afterwards, the solution was centrifuged (2000 rpm, 3 min) to remove undissolved residues. The supernatant was transferred into dialysis tubes and dialyzed against dH2O at 40 °C for 7 days. The purified solution was frozen at −80 °C and lyophilized (LSL Secfroid, Aclens, Switzerland). The product was obtained as a white solid and stored at −20 °C. GelNB with different degrees of functionalization (Low, Medium, High) was synthesized by varying the amounts of 5‐Norborne‐2‐carboxylic acid (0.3, 2, 10 equiv. referred to NH2 groups in gelatin), EDC·HCl (0.6, 4, 20 equiv.) and NHS (0.3, 2, 10 equiv.).
1H NMR (400 MHz, D2O, δ): 7.60–7.40 (Haromatic, gelatin), 6.44 (1H, Ha, endo), 6.39 (1H, Ha’, exo), 6,37 (1H, Hb’, exo), 6.11 (1H, Hb, endo), 5.24–0.99 (gelatin), 3.20 (bs, 2H, NH2).
Synthesis of Thiolated Gelatin
Thiolated Gelatin (GelS) synthesis was performed according to an established protocol.[ 59 ] In brief, 1 g gelatin (type A, gel strength ≈300 g bloom, 0.266 mmol NH2 groups, 1 equiv.) was dissolved in 10–20 mL degassed sodium carbonate‐bicarbonate buffer (0.1 m, pH 10), and the solution was heated to 40 °C under inert argon atmosphere. Then, 0.212 g N‐Acetyl‐DL‐homocysteine thiolactone (1.33 mmol, 5 equiv.) was added. To prevent oxidation of thiol groups, metals were complexed by addition of 1 mm EDTA.[ 77 ] The reaction mixture was stirred at 40 °C for 3 h. Subsequently, the solution was diluted with 10 mL dH2O, transferred into dialysis tubes and dialyzed against dH2O at 40 °C under argon. After 24 h, the purified solution was frozen at −80 °C and lyophilized (LSL Secfroid, Aclens, Switzerland). The product was obtained as a white solid and stored at −80 °C under argon atmosphere. GelS with different degrees of functionalization (Low, Medium) was synthesized by varying the amount of N‐Acetyl‐DL‐homocysteine thiolactone (1, 5 equiv. referred to NH2 groups in gelatin).
1H NMR (400 MHz, D2O, δ): 7.60‐7.40 (Haromatic, gelatin), 5.24‐0.99 (gelatin), 3.20 (bs, 2H, NH2), 2.23 (s, 3H, CH3, Ha).
2,4,6‐Trinitrobenzene Sulfonic Acid Assay
2,4,6‐Trinitrobenzene Sulfonic Acid (TNBSA) assay was performed to determine the percentage of modified free amino groups using protein solutions of 500 µg mL−1 gelatin, GelNB (0.1, 0.3, 0.5, 1, 2, 2.5, 3, 3.5, 4, 8, 10 equiv.) and GelS (1, 3, 5, 10 equiv.). The assay was carried out as previously described.[ 78 ] Blanks were prepared by adding HCl, followed by the addition of TNBSA reagent. The absorption of each sample was measured at 335 nm (SmartSpec 3000, Bio‐Rad Laboratories GmbH, Munich Germany). The degree of functionalization (DoF) was calculated from the ratio of absorption values of functionalized and non‐functionalized gelatin.
| (1) |
Hydrogel Preparation
GelMA and GelNB/DTT hydrogels were fabricated according to published protocols.[ 15 , 24 ] For GelNB/GelS preparation, hydrogel precursors were dissolved separately in DPBS‐/‐. The dissolution process of GelS was accelerated by increasing the temperature to 50–70 °C. Afterwards, GelNB (Low, Medium High) and GelS (Low, Medium) solutions were mixed to ensure equimolar amounts of both functional groups (norbornene/thiol = 1:1) in a 5% (w/v) hydrogel precursor solution (Low, Medium, High). Photoinitiator LAP was added at a concentration of 0.03% (w/v). Hydrogel formation was induced by UV‐visible light exposure (320–500 nm, 500 mW cm‐2) for 1–10 s (Omnicure S2000, igb‐tech, Friedelsheim, Germany).
Mass Swelling Ratio
Mass swelling ratio of 5% (w/v) GelNB/GelS hydrogels (Low, Medium, High) was determined using 200 µL sample volume. Hydrogel preparation was carried out as previously described. Following lyophilization, dry weight was measured, and samples were swollen in DPBS−/− for 48 h at RT. After carefully removing residual water from the surface, hydrogel samples were weighed. The equilibrium mass swelling ratio was calculated from the following equation, where w dry is the weight after lyophilization and w swollen is the weight after complete swelling of the lyophilized samples.
| (2) |
Degradation Assay
In vitro biodegradability of 5% (w/v) GelNB/GelS hydrogels (Low, Medium, High) was evaluated using 200 µL sample volume. Hydrogel preparation was carried out as previously described. Following lyophilization, dry weight was measured, and samples were incubated in DPBS−/− with 10% FCS over a period of 14 days (37 °C, 5% CO2). The degradation solution was exchanged every 2–3 days. After 1, 2, 3, 4, 7, and 14 days, hydrogel samples were lyophilized and weighed. The remaining mass was calculated from the following equation, where wi is the initial dry weight and wt is the dry weight at time t.
| (3) |
Determination of Minimal Photoinitiator Concentration and Curing Time
GelNB/DTT and GelNB/GelS hydrogel precursor solutions with varying LAP concentration (0.3%, 0.1%, 0.05%, 0.03%, 0.01% (w/v)) were prepared. 70 µL of each formulation was irradiated with UV‐visible light as previously described. The curing time was determined by regular checks every second.
Ellman's Assay
Ellman's assay was performed to determine the percentage of free thiol groups in the photocrosslinked GelNB/GelS hydrogels (NB/S = 1:1, 1:2, 1:3, 1:4, 1:5, 1:10). Corresponding GelS hydrogels without GelNB served as controls and were fabricated by cooling the precursor solution at 4 °C for 20 min. The assay was carried out according to an established protocol.[ 79 ] GelNB/GelS hydrogels and their controls were incubated in sodium phosphate buffer (0.1 m, pH 7.4, 1 mm EDTA) in the presence of Ellman's reagent (0.2 mm) at RT for 6 h in the dark. Then, 200 µL of the supernatant was transferred to a 96‐well plate, followed by absorption measurement at 412 nm (SpectraMax ID3, Molecular Devices, San José, USA). The percentage of free thiol groups was calculated according to the following equation.
| (4) |
Rheological Measurements
Dynamic shear oscillatory measurements were performed with a parallel‐plate rheometer (PP25, MCR 501, Anton Paar, Ostfildern‐Scharnhausen, Germany) for crosslinked GelNB/GelS hydrogels (Low, Medium, High) at 37 °C and their uncrosslinked precursor solutions at 14 °C. Frequency sweeps from 0.1 rad s−1 to 100 rad s−1 were measured at a constant amplitude of 1 Pa and a plate‐to‐plate distance of 1 mm. The average G’ of hydrogels and precursor solutions was calculated from the linear plateau. Rotational shear‐viscosity of the precursor solutions was assessed at 37 and 14 °C with shear rates ranging from of 0.1–100 s−1.
MTT Cell Viability Assay
MTT toxicity assays were performed to assess the cytotoxicity of hydrogel precursors (5% (w/v) GelMA, GelNB, GelS), hydrogel degradation products (5% (w/v) GelMA, GelNB/DTT, GelNB/GelS), photoinitiator (0.01–0.3% (w/v) LAP) and crosslinker (15 mm DTT). HepG2 and HUVEC were seeded at the density of 1 × 104 cells per well in a 96‐well plate and cultured overnight (37°C, 5% CO2). The next day, the media was removed and cells were treated with the substances in the indicated concentrations. Different exposure times were tested for hydrogel precursors (10 min, 2 h, 24 h), hydrogel degradation products (72 h), photoinitiator (24 h) and crosslinker (10, 20, 40 min, 1, 2, 3, 6, 24 h). Afterwards, substances were removed, and fresh media was added. From the moment of exposure, cells were incubated for a total period of 72 h. Negative controls for each time point were done by exchanging the media without addition or removal of any substances. After 72 h of incubation, positive controls were treated with 5 µL Triton X‐100 (20% (v/v)). Subsequently, 15 µL MTT solution was added to each well. The reaction was stopped after 3 h by adding 100 µL lysis buffer. Absorption was measured at 595 nm with a microplate spectrophotometer (SpectraMax ID3, Molecular Devices, San José, USA) the next day. A viability of 100% was assigned to the negative control. Cell viability for the tested conditions was calculated in relation to the negative and positive controls.
Cell Encapsulation
HepG2 and NHDF (used between passage 6–8 and a final cell density of 2.5 × 106 cells mL−1 in hydrogels) were suspended in 5% (w/v) GelMA (Medium), GelNB/DTT (Medium) and GelNB/GelS (Low, Medium, High) hydrogel precursor solutions with 0.3% (w/v) or 0.03% (w/v) LAP, respectively. Hydrogel formation was induced by irradiation as previously described. Long‐term cultivation was performed in a 8‐well µ‐slide (ibidi, Gräfelfing, Germany) with 200 µL cell‐laden hydrogels per well. After photopolymerization, hydrogels were covered with 200 µl DMEM (10% FCS, 1% penicillin‐streptomycin) and cultured over a period of 14 days (37 °C, 5% CO2). Cell culture medium was refreshed every 2–3 days.
Live/Dead Staining
Cell viability and spreading of NHDF within the hydrogels were monitored by live/dead staining 1, 7, and 14 days post‐encapsulation. After removal of the supernatant, the cell‐laden hydrogels were stained with calcein‐AM (4 µg mL‐1) and propidium iodide (20 µg mL‐1) for 20 min. Subsequently, the staining solution was removed. Hydrogels were washed several times with DPBS−/− and covered with DMEM. Confocal microscopy (Leica TCS SPE, Leica Microsystems, Wetzlar, Germany) was used to record z‐stacks of 300 µm thickness (step size: 5 µm), which were converted into 3D images with LasX software. 20 images were taken per hydrogel and the number of live and dead cells was counted using Python software. Viability was determined by the percentage of live cells over the total cell count.
PrestoBlue Proliferation Assay
Cell proliferation of HepG2 and NHDF in GelNB/GelS hydrogels was monitored by PrestoBlue assay in multiwell plates by measuring metabolic activity 7, 14, and 21 days post‐encapsulation. Cell‐free hydrogels served as corresponding blanks. After removal of the supernatant, hydrogels were washed with DPBS−/−, followed by the addition of PrestoBlue solution (DMEM/PrestoBlue = 9:1) and incubation for 2 h (37 °C, 5% CO2). Afterwards, fluorescence was measured (Ex/Em: 560 nm / 600 nm, integration time: 400 ms) with a microplate spectrophotometer (SpectraMax ID3, Molecular Devices, San José, USA).
Extrusion‐Based 3D Bioprinting
3D bioprinting was performed with an extrusion‐based 3D printer (3D Discovery Gen 1, regenHU, Villaz‐St‐Pierre, Switzerland) equipped with a cartridge system (Nordson EFD, Erkrath, Germany), a dispensing nozzle (cone, diameter: 0.15 mm, Vieweg GmbH, Kranzberg, Germany) and a unichiller (Pilote ONE, Peter Huber Kältemaschinenbau AG, Offenburg, Germany). For 3D bioprinting, 5% (w/v) GelMA (Low, Medium, High) and GelNB/GelS (Low, Medium, High) bioinks with NHDF (2.5 × 106 cells mL−1) were used. After cells had been suspended in the precursor solution, the bioink was transferred to the cartridge, which was sealed and stored upside down to avoid bubble formation. The cartridge was cooled to 22 °C for 30 min to induce an increase in viscosity due to physical gelation. Extrusion‐based printing was carried out at 22 °C, 0.075 MPa and a printing speed of 20 mm min−1, resulting in a cell‐laden 3D scaffold. Each layer of the grid structure (1 cm × 1 cm, four layers) was photocrosslinked for 20 s (315–400 nm, 13.6 W, OSRAM Ultra Vitalux 300W E27, Osram‐Licht AG, Munich, Germany). The printed structure on the glass slide was washed with DPBS−/−, transferred to a petri dish and further cultivated in DMEM (10% FCS, 1% penicillin‐streptomycin, 37 °C, 5% CO2). Live/dead staining was performed 1, 7, and 14 days post‐encapsulation, followed by confocal microscopy and cell viability analysis.
Data Analysis and Statistics
Data analysis and statistics were carried out using OriginPro 2020. All data are presented as mean ± standard deviation (SD). Unless stated otherwise, the value of n is defined as the number of repeat attempts performed. A two‐sample independent Student's t‐test was conducted when two average values were compared. If more average values needed to be compared, one‐way analysis of variance (ANOVA) with Bonferroni correction was carried out across groups. In all cases, significance was defined as p < 0.05 (*p < 0.05, **p < 0.01, and ***p < 0.001).
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
T.G., S.H., and X.K. contributed equally to this work. This research has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany´s Excellence Strategy via the Excellence Cluster 3D Matter Made to Order (EXC‐2082/1 – 390761711) and by the Bundesministerium für Bildung und Forschung (BMBF) by the KMU‐NetC 3D‐Bio‐Net, (FKZ 03VNE1034D). The authors acknowledge financial support by the Helmholtz program “BioInterfaces in Technology and Medicine” (BIFTM). T.G, S.H., R.P., and T.M. would like to thank for financial support. T.G. is funded by a Kekulé Fellowship of the Chemical Industry Fund (FCI). S.H. kindly acknowledges funding received by the Landesgraduiertenförderung. R.P. thanks the Jürgen Manchot Foundation for financial support. T.M. acknowledges funding from the Carl Zeiss Foundation.
Open access funding enabled and organized by Projekt DEAL.
Göckler T., Haase S., Kempter X., Pfister R., Maciel B. R., Grimm A., Molitor T., Willenbacher N., Schepers U., Tuning Superfast Curing Thiol‐Norbornene‐Functionalized Gelatin Hydrogels for 3D Bioprinting. Adv. Healthcare Mater. 2021, 10, 2100206. 10.1002/adhm.202100206
Data Availability Statement
Research data are not shared.
References
- 1. Groll J., Boland T., Blunk T., Burdick J. A., Cho D. W., Dalton P. D., Derby B., Forgacs G., Li Q., Mironov V. A., Moroni L., Nakamura M., Shu W. M., Takeuchi S., Vozzi G., Woodfield T. B. F., Xu T., Yoo J. J., Malda J., Biofabrication 2016, 8, 013001 [DOI] [PubMed] [Google Scholar]
- 2. Bedell M. L., Navara A. M., Du Y. Y., Zhang S. M., Mikos A. G., Chem. Rev. 2020, 120, 10744. [DOI] [PubMed] [Google Scholar]
- 3. Malda J., Visser J., Melchels F. P., Jungst T., Hennink W. E., Dhert W. J., Groll J., Hutmacher D. W., Adv. Mater. 2013, 25, 5011. [DOI] [PubMed] [Google Scholar]
- 4. Arslan‐Yildiz A., El Assal R., Chen P., Guven S., Inci F., Demirci U., Biofabrication 2016, 8, 014103. [DOI] [PubMed] [Google Scholar]
- 5. Panwar A., Tan L. P., Molecules 2016, 21, 685.27231892 [Google Scholar]
- 6. Donderwinkel I., van Hest J. C. M., Cameron N. R., Polym. Chem. 2017, 8, 4451. [Google Scholar]
- 7. Holzl K., Lin S. M., Tytgat L., Van Vlierberghe S., Gu L. X., Ovsianikov A., Biofabrication 2016, 8, 032002 [DOI] [PubMed] [Google Scholar]
- 8. Groll J., Burdick J. A., Cho D. W., Derby B., Gelinsky M., Heilshorn S. C., Jungst T., Malda J., Mironov V. A., Nakayama K., Ovsianikov A., Sun W., Takeuchi S., Yoo J. J., Woodfield T. B. F., Biofabrication 2018, 11, 013001. [DOI] [PubMed] [Google Scholar]
- 9. Zhu J. M., Marchant R. E., Expert Rev. Med. Devices 2011, 8, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guvendiren M., Lu H. D., Burdick J. A., Soft Matter 2012, 8, 260. [Google Scholar]
- 11. Hospodiuk M., Dey M., Sosnoski D., Ozbolat I. T., Biotechnol. Adv. 2017, 35, 217. [DOI] [PubMed] [Google Scholar]
- 12. Qin X.‐H., Ovsianikov A., Stampfl J., Liska R., BioNanomaterials 2014, 15, 49. [Google Scholar]
- 13. Van Den Bulcke A. I., Bogdanov B., De Rooze N., Schacht E. H., Cornelissen M., Berghmans H., Biomacromolecules 2000, 1, 31. [DOI] [PubMed] [Google Scholar]
- 14. Benton J. A., DeForest C. A., Vivekanandan V., Anseth K. S., Tissue Eng. Part A 2009, 15, 3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yue K., Trujillo‐de Santiago G., Alvarez M. M., Tamayol A., Annabi N., Khademhosseini A., Biomaterials 2015, 73, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nichol J. W., Koshy S. T., Bae H., Hwang C. M., Yamanlar S., Khademhosseini A., Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brinkman W. T., Nagapudi K., Thomas B. S., Chaikof E. L., Biomacromolecules 2003, 4, 890. [DOI] [PubMed] [Google Scholar]
- 18. Ravichandran R., Islam M. M., Alarcon E. I., Samanta A., Wang S., Lundstrom P., Hilborn J., Griffith M., Phopase J., J. Mater. Chem. B 2016, 4, 318. [DOI] [PubMed] [Google Scholar]
- 19. Gaudet I. D., Shreiber D. I., Biointerphases 2012, 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smeds K. A., Pfister‐Serres A., Miki D., Dastgheib K., Inoue M., Hatchell D. L., Grinstaff M. W., J. Biomed. Mater. Res. 2001, 54, 115. [DOI] [PubMed] [Google Scholar]
- 21. Baier Leach J., Bivens K. A., Patrick C. W. Jr., Schmidt C. E., Biotechnol. Bioeng. 2003, 82, 578. [DOI] [PubMed] [Google Scholar]
- 22. Burdick J. A., Chung C., Jia X., Randolph M. A., Langer R., Biomacromolecules 2005, 6, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Masters K. S., Shah D. N., Leinwand L. A., Anseth K. S., Biomaterials 2005, 26, 2517. [DOI] [PubMed] [Google Scholar]
- 24. Munoz Z., Shih H., Lin C. C., Biomater. Sci. 2014, 2, 1063. [DOI] [PubMed] [Google Scholar]
- 25. Tibbitt M. W., Kloxin A. M., Sawicki L., Anseth K. S., Macromolecules 2013, 46, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin R. Z., Chen Y. C., Moreno‐Luna R., Khademhosseini A., Melero‐Martin J. M., Biomaterials 2013, 34, 6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bertassoni L. E., Cardoso J. C., Manoharan V., Cristino A. L., Bhise N. S., Araujo W. A., Zorlutuna P., Vrana N. E., Ghaemmaghami A. M., Dokmeci M. R., Khademhosseini A., Biofabrication 2014, 6, 024105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Masuma R., Kashima S., Kurasaki M., Okuno T., J. Photoch. Photobio. B 2013, 125, 202. [DOI] [PubMed] [Google Scholar]
- 29. Lim K. S., Galarraga J. H., Cui X., Lindberg G. C. J., Burdick J. A., Woodfield T. B. F., Chem. Rev. 2020, 120, 10662. [DOI] [PubMed] [Google Scholar]
- 30. Knowlton S., Yenilmez B., Anand S., Tasoglu S., Bioprinting 2017, 5, 10. [Google Scholar]
- 31. Hong Y., Zhou F., Hua Y., Zhang X., Ni C., Pan D., Zhang Y., Jiang D., Yang L., Lin Q., Zou Y., Yu D., Arnot D. E., Zou X., Zhu L., Zhang S., Ouyang H., Nat. Commun. 2019, 10, 2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang Y., Zhang J., Liu Z., Lin Q., Liu X., Bao C., Wang Y., Zhu L., Adv. Mater. 2016, 28, 2724. [DOI] [PubMed] [Google Scholar]
- 33. McCall J. D., Anseth K. S., Biomacromolecules 2012, 13, 2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cramer N. B., Bowman C. N., J. Polym. Sci. Pol. Chem. 2001, 39, 3311. [Google Scholar]
- 35. Lin C. C., Ki C. S., Shih H., J. Appl. Polym. Sci. 2015, 132, 41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pereira R. F., Bartolo P. J., Engineering 2015, 1, 090. [Google Scholar]
- 37. Hoyle C. E., Bowman C. N., Angew. Chem. Int. Ed. 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]
- 38. Greene T., Lin C. C., ACS Biomater. Sci. Eng. 2015, 1, 1314. [DOI] [PubMed] [Google Scholar]
- 39. Van Hoorick J., Gruber P., Markovic M., Rollot M., Graulus G. J., Vagenende M., Tromayer M., Van Erps J., Thienpont H., Martins J. C., Baudis S., Ovsianikov A., Dubruel P., Van Vlierberghe S., Macromol. Rapid Commun. 2018, 39, 1800181. [DOI] [PubMed] [Google Scholar]
- 40. Gramlich W. M., Kim I. L., Burdick J. A., Biomaterials 2013, 34, 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Acosta‐Velez G. F., Linsley C. S., Craig M. C., Wu B. M., Bioengineering (Basel) 2017, 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aimetti A. A., Machen A. J., Anseth K. S., Biomaterials 2009, 30, 6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fairbanks B. D., Schwartz M. P., Halevi A. E., Nuttelman C. R., Bowman C. N., Anseth K. S., Adv. Mater. 2009, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shih H., Lin C. C., Biomacromolecules 2012, 13, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dobos A., Van Hoorick J., Steiger W., Gruber P., Markovic M., Andriotis O. G., Rohatschek A., Dubruel P., Thurner P. J., Van Vlierberghe S., Baudis S., Ovsianikov A., Adv. Healthcare Mater. 2020, 9, 1900752. [DOI] [PubMed] [Google Scholar]
- 46. Van Hoorick J., Dobos A., Markovic M., Gheysens T., Van Damme L., Gruber P., Tytgat L., Van Erps J., Thienpont H., Dubruel P., Ovsianikov A., Van Vlierberghe S., Biofabrication 2021, 13, 1. [DOI] [PubMed] [Google Scholar]
- 47. Tytgat L., Van Damme L., Van Hoorick J., Declercq H., Thienpont H., Ottevaere H., Blondeel P., Dubruel P., Van Vlierberghe S., Acta Biomater. 2019, 94, 340. [DOI] [PubMed] [Google Scholar]
- 48. Seliktar D., Science 2012, 336, 1124. [DOI] [PubMed] [Google Scholar]
- 49. Nicodemus G. D., Bryant S. J., Tissue Eng. Part B 2008, 14, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun W., Starly B., Daly A. C., Burdick J. A., Groll J., Skeldon G., Shu W. M., Sakai Y., Shinohara M., Nishikawa M., Jang J., Cho D. W., Nie M. H., Takeuchi S., Ostrovidov S., Khademhosseini A., Kamm R. D., Mironov V., Moroni L., Ozbolat I. T., Biofabrication 2020, 12, 022002. [DOI] [PubMed] [Google Scholar]
- 51. Gungor‐Ozkerim P. S., Inci I., Zhang Y. S., Khademhosseini A., Dokmeci M. R., Biomater. Sci. 2018, 6, 915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Catoira M. C., Fusaro L., Di Francesco D., Ramella M., Boccafoschi F., J. Mater. Sci. Mater. Med. 2019, 30, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alvarado‐Velez M., Pai S. B., Bellamkonda R. V., IEEE Trans. BioMed. Eng. 2014, 61, 1474. [DOI] [PubMed] [Google Scholar]
- 54. Shirahama H., Lee B. H., Tan L. P., Cho N. J., Sci. Rep. 2016, 6, 31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galvao J., Davis B., Tilley M., Normando E., Duchen M. R., Cordeiro M. F., FASEB J. 2014, 28, 1317. [DOI] [PubMed] [Google Scholar]
- 56. Yi X., Liu M., Luo Q., Zhuo H., Cao H., Wang J., Han Y., FEBS Open Bio 2017, 7, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. de Abreu Costa L., Henrique Fernandes Ottoni M., Dos Santos M. G., Meireles A. B., Gomes de Almeida V., de Fatima Pereira W., Alves de Avelar‐Freitas B., Eustaquio Alvim Brito‐Melo G., Molecules 2017, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. ThermoScientific , Crosslinking Technical Handbook, 2012. http://tools.thermofisher.com/content/sfs/brochures/1602163-Crosslinking-Reagents-Handbook.pdf [Google Scholar]
- 59. Van Vlierberghe S., Schacht E., Dubruel P., Eur. Polym. J. 2011, 47, 1039. [Google Scholar]
- 60. Dorsey T. B., Grath A., Wang A., Xu C., Hong Y., Dai G., Bioact. Mater. 2018, 3, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pepelanova I., Kruppa K., Scheper T., Lavrentieva A., Bioengineering (Basel) 2018, 5, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li X., Chen S., Li J., Wang X., Zhang J., Kawazoe N., Chen G., Polymers (Basel) 2016, 8, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cramer N. B., Davies T., O'Brien A. K., Bowman C. N., Macromolecules 2003, 36, 4631. [Google Scholar]
- 64. Northrop B. H., Coffey R. N., J. Am. Chem. Soc. 2012, 134, 13804. [DOI] [PubMed] [Google Scholar]
- 65. Madl C. M., Heilshorn S. C., Adv. Funct. Mater. 2018, 28, 1706046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gauthier M. A., Gibson M. I., Klok H. A., Angew. Chem. Int. Ed. Engl. 2009, 48, 48. [DOI] [PubMed] [Google Scholar]
- 67. Goldmann A. S., Glassner M., Inglis A. J., Barner‐Kowollik C., Macromol. Rapid Commun. 2013, 34, 810. [DOI] [PubMed] [Google Scholar]
- 68. Stevens R., Stevens L., Price N. C., Biochem. Educ. 1983, 11, 70. [Google Scholar]
- 69. Munday R., Free Radic. Biol. Med. 1989, 7, 659. [DOI] [PubMed] [Google Scholar]
- 70. Held K. D., Sylvester F. C., Hopcia K. L., Biaglow J. E., Radiat. Res. 1996, 145, 542. [PubMed] [Google Scholar]
- 71. Ruoslahti E., Annu. Rev. Cell Dev. Biol. 1996, 12, 697. [DOI] [PubMed] [Google Scholar]
- 72. Yang F., Williams C. G., Wang D. A., Lee H., Manson P. N., Elisseeff J., Biomaterials 2005, 26, 5991. [DOI] [PubMed] [Google Scholar]
- 73. Lutolf M. P., Lauer‐Fields J. L., Schmoekel H. G., Metters A. T., Weber F. E., Fields G. B., Hubbell J. A., Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lutolf M. P., Raeber G. P., Zisch A. H., Tirelli N., Hubbell J. A., Adv. Mater. 2003, 15, 888. [Google Scholar]
- 75. Benton J. A., Fairbanks B. D., Anseth K. S., Biomaterials 2009, 30, 6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Larsen M., Artym V. V., Green J. A., Yamada K. M., Curr. Opin. Cell Biol. 2006, 18, 463. [DOI] [PubMed] [Google Scholar]
- 77. Kelly S. T., Zydney A. L., Biotechnol. Bioeng. 1994, 44, 972. [DOI] [PubMed] [Google Scholar]
- 78. Kale R., Bajaj A., J. Young Pharm. 2010, 2, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. ThermoScientific , User Guide: Ellman's Reagent, 2011. https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFSAssets%2FLSG%2Fmanuals%2FMAN0011216_Ellmans_Reag_UG.pdf&title=VXNlciBHdWlkZTogIEVsbG1hbiZyc3F1bztzIFJlYWdlbnQ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.