

Abstract
Advancements in peptidomics have revealed numerous small open reading frames with coding potential and revealed that some of these micropeptides are closely related to human cancer. However, the systematic analysis and integration from sequence to structure and function remains largely undeveloped. Here, as a solution, we built a workflow for the collection and analysis of proteomic data, transcriptomic data, and clinical outcomes for cancer-associated micropeptides using publicly available datasets from large cohorts. We initially identified 19 586 novel micropeptides by reanalyzing proteomic profile data from 3753 samples across 8 cancer types. Further quantitative analysis of these micropeptides, along with associated clinical data, identified 3065 that were dysregulated in cancer, with 370 of them showing a strong association with prognosis. Moreover, we employed a deep learning framework to construct a micropeptide-protein interaction network for further bioinformatics analysis, revealing that micropeptides are involved in multiple biological processes as bioactive molecules. Taken together, our atlas provides a benchmark for high-throughput prediction and functional exploration of micropeptides, providing new insights into their biological mechanisms in cancer. The HMPA is freely available at http://hmpa.zju.edu.cn.
Keywords: nonclassical peptidome, mass spectrometry, structure prediction, functional annotation, micropeptide database
Introduction
Increasing evidence indicates that canonical untranslated regions and long noncoding ribonucleic acids (lncRNAs) have coding potential [1–4]. The discoveries of novel translation products have gradually clarified their unique functions [5–11], highlighting the importance of identifying coding elements within the genome to fully understand biological regulatory mechanisms in organisms [12]. Beyond cell survival and proliferation [13], the products of these small open reading frames (sORFs) were critical for a variety of physiological and pathological processes, including human cancer progression [14–17], mitochondrial activity and energy metabolism [6], and adiposity [11, 18] and immune responses [19, 20]. These studies have deepened our understanding of the role played by micropeptides in numerous molecular mechanisms, suggesting that the systematic identification and functional characterization of sORFs and novel peptides will contribute to the discovery of novel therapeutic targets for cancer treatment.
With the growing interest in sORFs and advancements in sequencing technology, numerous studies have attempted to systematically identify these molecules [21]. During the early stages, several studies have utilized bioinformatics approaches such as phylogenetic analysis [22], nucleotide and amino acid homology [23], and secondary structure [24] to identify unannotated sORFs in yeast [25], mouse [26] and human beings [2]. However, since thousands of sORFs have been predicted by bioinformatics pipelines, these traditional approaches are considered inefficient for systematic identification. The further development of ribosomal profiling analysis (Ribo-seq) offered a novel strategy for the genome-wide assessment of sORF translation. Ingolia et al. demonstrated extensive ribosomal translation of 4648 genes outside of canonical ORFs [27]. Fritsch et al. revealed that 2994 novel upstream open reading frames (uORFs) including 1406 coding sequence (CDS) overlapping uORFs, present in all human genome [28]. In addition, Van Heesch et al. studied the translation in the human heart tissues by Ribo-seq and identified 1176 lncRNAs with translational potential [29]. These RNA sequencing methods have been applied to the systematic construction of translation profiles across of various cell lines [30]. Additionally, mass spectrometry (MS) proteomics, known for its high sensitivity in detecting sORF translation products [31] has been incorporated into micropeptide recognition strategies [32]. Banfai et al. and Slavoff et al. successfully identified nearly 100 new sORFs in human cell lines through the integration of RNA sequencing (RNA-seq) and MS data [30] or peptidomics strategies [2]. Nevertheless, these methods for sORFs discovery tend to yield scattered information, emphasizing the need for a systematic approach to comprehensively interpret and analyze identified micropeptides, which is crucial for assessing the underlying molecular mechanisms.
Given that the descriptions of identified ncRNA-encoded peptides are scattered across many publications, it is imperative to develop a standardized reference set for micropeptides. Olexiouk et al. established sORF.org, a repository comprising 78 sORFs exhibiting ribosome binding activity identified through Ribo-seq analysis [33], thus laying the groundwork for systematic functional studies in the field. Hao et al. provided the SmProt database, documenting ~2 million peptides [34], primarily derived from systematic screening based on Ribo-seq, along with a limited number of sequences (a few thousand) derived from MS and other experimental approaches. With the increasing quantity of predictive data and published experimental results, some studies have attempted to manually organize and consolidate these contents. For example, Liu et al. established the ncEP database, which predominantly includes low-throughput experimentally validated data, including 80 ncRNA-encoded proteins or peptides from published articles [35]. Similarly, Huang et al. developed the cncRNAdb database, which houses 2600 functional entries from more than 20 species, with experimental support obtained through manual curation [36]. Dragomir et al. further introduced FuncPEP, a database featuring 112 functional peptides, notably incorporating over 100 newly identified micropeptides [32]. With the development of bioinformatics frameworks and molecular biology tools, the characterization of novel peptides has advanced substantially. Disease-associated micropeptides derived from large-scale tissue-level studies were subsequently integrated. Luo et al. introduced SPENCER, a database of approximately 20 000 micropeptides expressed in various clinical tissue samples, which for the first time predicted micropeptides as potential targets for cancer immunotherapy as neoantigens, making this database a valuable resource for studying cancer-associated micropeptides [37]. OpenProt 2.0, published by Leblanc et al., has been instrumental in making deep-learning–based peptide property predictions and adding information about genetic variation [38], thereby promoting the understanding of the micropeptide function. These collaborative efforts have greatly enriched our understanding of sORFs, providing a wealth of resources and tools for further exploration of their critical role in organisms.
However, challenges remain. Most existing micropeptide-related datasets focus on one or more specific omics datasets, making it difficult to effectively integrate and utilize these different databases. This fragmentation poses a significant obstacle to micropeptide retrieval and application. Therefore, integrated identification is the starting point for systematic peptide research. In addition, the currently available clinical information mainly focused on proteomic and genomic data, and the relationship between micropeptide expression and tumor prognosis have not been systematically explored [39]. Publicly available proteomic knowledge bases, such as the Clinical Proteomic Tumor Analysis Consortium (CPTAC), have not been used to identify micropeptides. As a result, there is a critical need to repurpose a vast array of resources to build a comprehensive and continuously updated library of cancer-related micropeptides. In addition, the current lack of high-precision 3D structures limits the analysis of functional mechanisms; therefore, advanced modeling tools, such as Alphafold2 [40], must be integrated for a more detailed analysis of micropeptide structures.
In this study, we constructed a novel and comprehensive database designed to facilitate the discovery of micropeptides and provide an interface for visualizing their role in cancer progression. This database was constructed using a rigorous discovery framework that combines detailed proteomic data compilation with bioinformatics function prediction. We initially used the Ribotricer search tool on Ensembl-based human reference genome data to construct a reference database comprising ~10 million sORFs translated from known ncRNAs and other noncanonical regions. Using Proteome Discoverer (PD) software, we systematically identified 19 586 micropeptides, including 8945 authentic micropeptides isolated from gastric cancer (GC) tissue samples via an in-house laboratory ultrafiltration concentration system. Moreover, the results of the quantitative analysis revealed differential micropeptide expression in tumor tissues, with 3065 peptides showing dysregulation in tumor tissues compared with adjacent or normal tissues, and 370 exhibiting significant associations with patient overall survival.
Our work is presented in an interactive web interface (hmpa.zju.edu.cn), offering full interpretation and convenient visualization for the high-throughput exploration of annotated micropeptides, providing vital support for precision medicine and postgenomic therapy. In particular, we predicted and presented the structures of all identified micropeptides for the first time via Alphafold2. Subsequently, we constructed a micropeptide–protein network (mPPI) by applying a deep learning algorithm with weighted correlation network analysis to the high-level structural information and integrating these predictions with the peptide–protein network annotations from a protein interaction database. Our work is presented in an interactive web interface (hmpa.zju.edu.cn) that provides full interpretation and convenient visualization for the high-throughput exploration of annotated micropeptides, offering vital support for precision medicine and postgenomic therapy.
Results
The workflow of micropeptide discovery and integration
For systematic dataset mining and analysis, we constructed a workflow (Fig. 1) involving micropeptide discovery and the expansion of functional annotations across different cancers. Multiomics dataset collection, micropeptide discovery, and functional visualization are the three main steps in our workflow. In the initial phase, we amassed a trove of genomic, transcriptomic, and proteomic data, complemented by micropeptide expression profiles and cancer-related clinical phenotypes. These datasets were meticulously preprocessed to ensure compatibility and enhance integrative analysis. In the subsequent phase, we developed in-house scripts to harmonize micropeptide and protein linkages and further enriched our network by annotating it with micropeptides implicated in cancer pathogenesis. In the final phase, we engineered a user-friendly database that enables the exploration and visualization of cancer-related micropeptides and their potential functions, thereby fostering a deeper understanding of micropeptides in oncology.
Figure 1.

Overall design and construction of HMPA. (a) The design framework of HMPA. The process begins by repurposing proteomic datasets encompassing 8 cancer types, 2279 patients, and 3753 samples. Proteomics raw files were subjected to expert curation and analysis using the PD tool to identify of micropeptides. These identified peptides are then integrated with multi-omics expression data from sources such as CPTAC, GDC, and PRIDE, and stored within the HMPA database. Functional insights are derived by exploring the genomic contributions to peptide features. Furthermore, network-based models and STRING analysis are used to predict micropeptide–protein interactions, with metagraphs providing a visual representation of the network. In the realm of computational oncology, various analyses are conducted, including the evaluation of micropeptide and RNA expression, mRNA and peptide expression levels, clinical phenotypes, and genome annotations. (b) Explore of each micropeptide at HMPA. Basic information and function prediction of the HMPA (left) and the tools interface for analysis about expression, clinical data and RNA–protein relation (right).
Rigorous micropeptide discovery procedures
We meticulously curated and re-annotated 3753 proteomic files from eight extensive cancer-focused quantitative proteomic investigations (Table S1 available online at http://bib.oxfordjournals.org/). This process involved the consistent evaluation of data including transcriptomic data, global proteomic data, anonymized clinical data, histologic findings, and treatment outcomes across different cancer types (Fig. 2a). Additionally, we analyzed six pairs of GC tissues and corresponding paracancerous tissues, five normal gastric tissues, and several GC cell lines. We extracted total protein from these tissues or cell lines using both mechanical grinding and ultrasonic disruption methods. High-molecular-weight proteins were subsequently removed via 30/10 kDa ultrafiltration. Our selection criteria specifically targeted studies employing MS1-based protein quantification based on the ionic properties of peptide precursors and utilizing the Thermo Orbitrap MS platform. After reanalyzing all the MS data with PD, we compiled a dataset of 19 586 sORFs, each supported by at least one definitive peptide.
Figure 2.
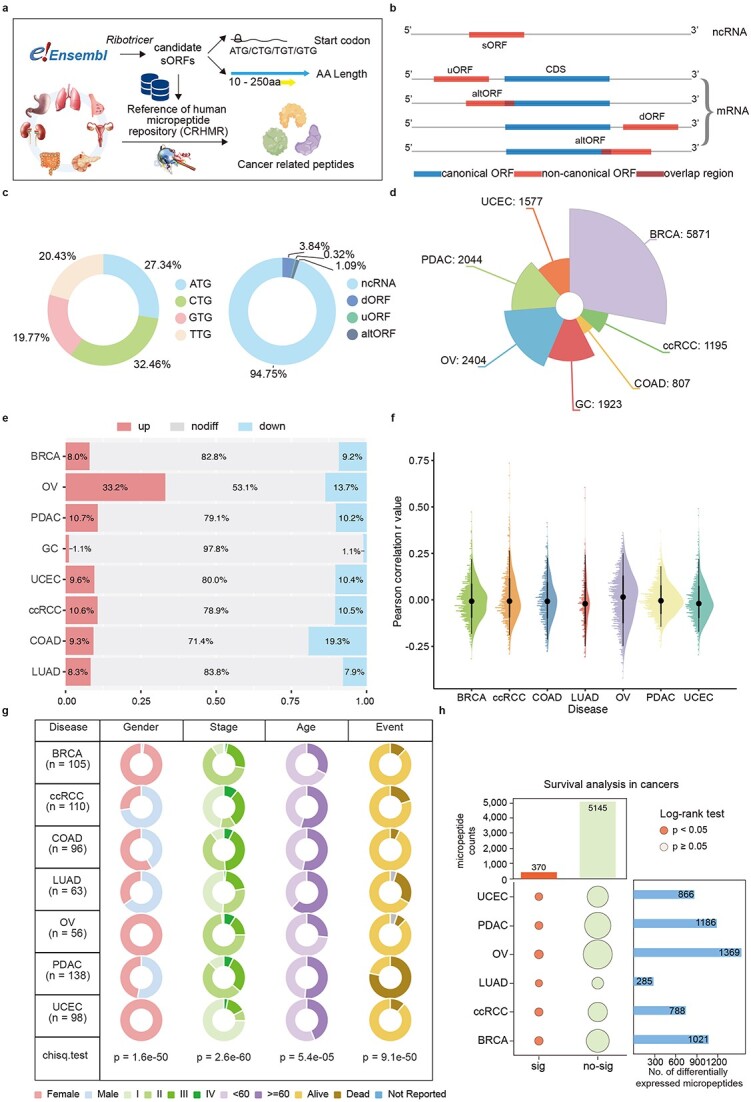
HMPA data analysis pipeline and data overview. (a) Overview of the study design and the data reanalysis pipeline. (b) The construction of ORF classification diagram with CRHMR. (c) The distribution of codon and sORFs main source. sORFs are mainly found on the 3’-UTR and CDS of coding RNAs as well as on non-coding RNA (ncRNAs). (d) A sunburst of micropeptide in different cancer types. The number on each line is the counts of micropeptide in different cancers. (e) The bar chart for deregulation micropeptide in each cancer. The three change levels between tumor and NATs. (f) The raincloud plot for the distribution of expression level of Pearson correlation analysis between RNA and peptide level. (g) Table for clinical information collection about each caner in PDC database. (h) The marginal bubble plot of survival analysis of peptide in different cancer.
Through collaborative analysis, we synthesized a comprehensive dataset detailing the derivations of various sORFs (Fig. 2b). To efficiently access and differentiate the genomic information associated with the micropeptides based on their origins and characteristics, we devised a systematic naming system, ‘pepNo.-Type-Gene’: ‘No.’ represents the sequential numbering from the genomic coordinates of the parent open reading frame; ‘Type’ denotes the ORF classification (u: upstream, alt: alternative, d: downstream, nc: noncoding RNA); and ‘Gene’ specifies the gene or locus of origin.
We evaluated the length of the sORFs, their start codon usage, and the annotation status of the RNAs producing these newly identified micropeptides. The lengths ranged from 10 to 250 amino acids, with 27.3% utilizing the ATG start codon, and RNAs annotated as noncoding accounted for 94.7% of the total (Fig. 2c). Additionally, owing to a significant number of missing proteomic values, we quantified the protein expression in tumor samples and cell lines via standard methods and categorized the micropeptide expression across different samples (Fig. 2d and e). These results highlight the widespread distribution of micropeptides in tissues, underscoring the growing interest in this unique class of biomolecules.
Cancer-associated peptide expression in disease progression and visualization
The HMPA features interactive expression heatmaps that vividly illustrate the peptides expression levels within individual samples. Users can filter the results by disease type and by significant differential expression, which facilitates targeted analysis. Additionally, the tool integrates clinical data with micropeptide expression patterns to predict survival outcomes for cancer patients (Fig. 1b). Users can filter results by cancer type to obtain detailed survival analyses and relevant micropeptide data. The interface also allows users to categorize micropeptides by various clinical indicators, such as stage, age, and sex, aiding in the exploration of how micropeptide expression varies across patient demographics. We explored the potential associations between RNA–peptide correlations and variations in overall survival or progression-free survival across various cancer types (Fig. 2f). A total of 264 RNA-peptide expression pairs were significantly differentially expressed (P value <.05) in the cancer samples. Initially focusing on the TCGA samples with well-documented survival data as a subset of the CPTAC dataset, we pooled all available proteomic TCGA datasets (Fig. 2g). Our analysis revealed a strong correlation between the expression patterns of 303 micropeptide biomarkers and patient survival across all disease-associated micropeptide datasets (Fig. 2h).
Systematic exploration of micropeptide functions
To thoroughly explore micropeptide origins and functions, HMPA systematically collects extensive information across six essential categories (Fig. 1b). These categories include fundamental information on transcripts, subcellular localization, physical attributes, spectral data, advanced structural predictions, and functional annotations. Users can navigate seamlessly through the micropeptide IDs in the results, accessing relevant detailed pages with a simple click. The ‘Summary’ segment furnishes a comprehensive table of gene details, showcasing the gene ID, sORF, description, genomic coordinates, chain, and conformity references, as well as an overview of the coding transcripts. Within the ‘Transcripts’ section, the HMPA presents fundamental insights into transcripts. Additionally, a colorful ‘Subcellular Localization’ module allows the visual exploration of micropeptide characteristics. Recognizing the growing potential of cancer-related micropeptides as novel targets for antitumor interventions, we have forecasted the ‘properties’ of micropeptides, delineating the outcomes in tabular format. Notably, the ‘Mass Spectrometry’ module provides the underlying spectral evidence, enabling users to view the ion peaks and peptide sequence fragments for each annotated mass spectrum. Considering the influence of the RNA secondary structure on translation, we also provide predictions of the micropeptide transmembrane properties, surface accessibility, secondary structure, disorder, and phi/psi dihedral angles, thus providing deeper insights into their potential mechanisms. Furthermore, in the ‘Structure’ section, we provide advanced structural predictions for micropeptides using AlphaFold2; these predicted structures are also available as downloadable resources.
Moreover, we employed sophisticated artificial intelligence tools to model the structure and organization of functional molecules (Fig. 3a). We subsequently performed proteome-wide micropeptide–protein molecular docking to identify potential interacting proteins. This allowed us to examine the characteristics of these interactions and achieve an initial mapping of the mPPI network. Utilizing the functional annotations of these interacting proteins, we performed comprehensive consistency cluster analyses through Gene Ontology (GO) functional and KEGG enrichment analyses for interacting proteins (Fig. 3b and c). Throughout this process, we evaluated the contribution scores of each interaction in the network, weighting, and visualizing their correlations. This analysis lays the groundwork for understanding the biological mechanisms relevant to micropeptide functionality and systematically annotating the multidimensional biological characteristics of micropeptide sequences, structures, localizations, and functions.
Figure 3.
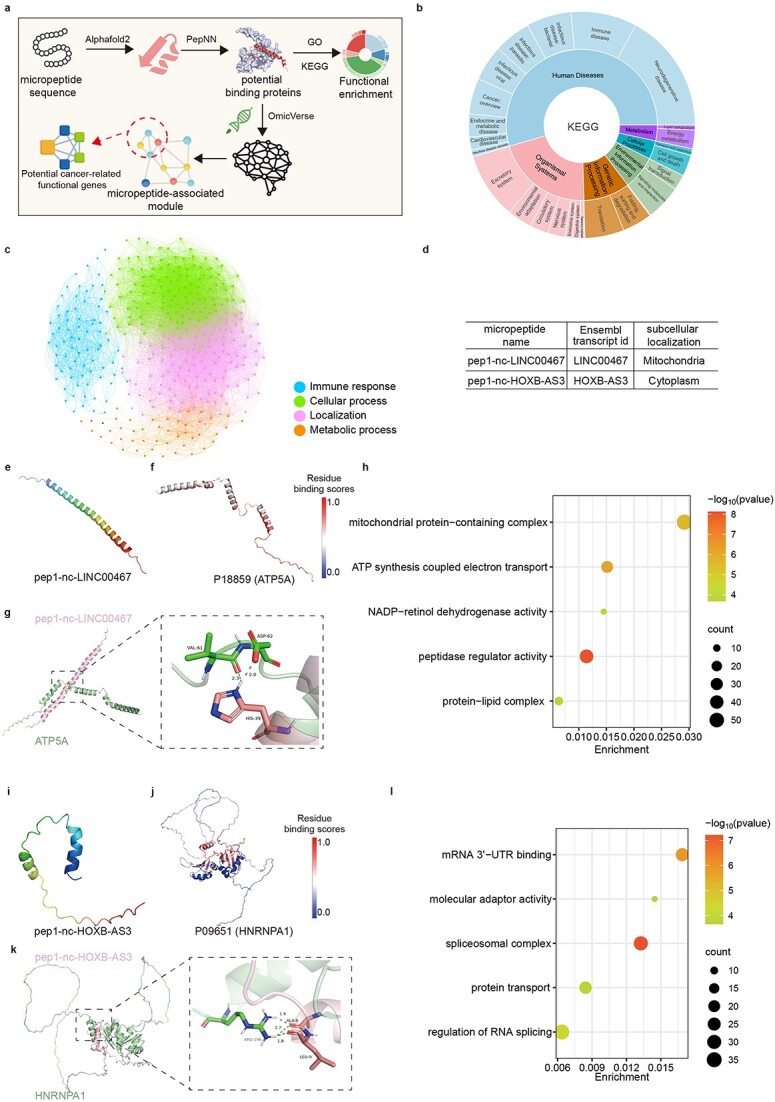
Exploration of micropeptide structures and biological functions. (a) Overview of the micropeptide functional annotation design pipeline. (b) Comprehensive visualization of KEGG pathways by Plotly-sunburst method. (c) GO classification atlas for micropeptides based on potential protein interactions. Candidates are categorized into four groups. (d) Two previous research of micropeptide information in HMPA. (e) The 3D structure of pep1-nc-LINC00467 predicted by AlphaFold2. (f) Visualization ATP5A each residue binding probability colored by B-factor, range for probability is [0,1]. (g) Docking result of pep1-nc-LINC00467 and P18859 by ClusPro. (h) The 3D structure of pep1-nc-HOXB-AS3 predicted by AlphaFold2. (i) Visualization HNRNRA1 each residue binding probability colored by B-factor, range for probability is [0,1]. (j) Docking result of pep1-nc-HOXB-AS3 and P09651 by ClusPro. (k) GO enrichment result for proteins in mPPI network. (l) GO enrichment result for proteins in mPPI interaction network.
Web interface
The HMPA provides a user-friendly web interface, as depicted in the accompanying figure (Fig. 1b). This interface features an advanced browser page, enabling users to navigate the database via a nuanced combination of search criteria. It supports not only the querying of results but also seamless browsing, allowing users to easily access and download all cancer-relevant micropeptides housed within the database. The HMPA homepage features the following interfaces: search, HMPA, tools, genome, downloads, and help. Below, we provide a concise description of each interface.
The HMPA homepage offers both basic and advanced query capabilities to accommodate a wide range of user needs. Users can initiate searches for micropeptides via the ‘Home’ interface, which includes data from the HMPA and other sources. More specialized searches can be performed on the advanced search page by combining terms, such as HMPA No., transcript ID, symbol, genomic position, subcellular localization, and source. This functionality allows users to retrieve precise information tailored to their specific research needs.
The ‘HMPA’ section of the website allows users to explore micropeptides associated with specific cancer types. Detailed information about micropeptides, including their unique ‘HMPA No.’, ‘micropeptide name’, and ‘ORF ID’, is readily accessible. The interface also provides a ‘Change level’ feature, which summarizes the variations in expression between malignant and normal adjacent tissue (NAT) samples according to the data stored in HMPA. Notably, this section includes a database of 1161 micropeptides that CRISPR screening has verified to have tumor-promoting properties. Clicking on the HMPA ID in the expression table directs users to a detailed page for each micropeptide, offering deeper insights into the screening results.
The UCSC Genome Browser is employed to analyze the genomic data, integrating various data sources into a unified view. The tracks displayed in this section include micropeptides, reference genomes, and phyloP conserved data, offering a comprehensive view of genomic intricacies and facilitating advanced genomic research (Fig. 1b). Dedicated to transparency and data sharing, the HMPA includes a download interface segmented into four parts: general information, advanced information, micropeptide expression, micropeptide sequence, micropeptide structure, and micropeptide biological function information. This organization allows users to easily access and download exactly the data they need for further analysis.
Discussion and conclusion
Recent developments in proteomics, transcriptomics, and bioinformatics have elucidated the role of micropeptides in various human cancers [7–9, 41], and investigations of micropeptides in tumors have enhanced our understanding of immunotherapy, diagnostics, and cancer etiology [39]. However, the study of micropeptide biological functions and mechanisms in disease has been constrained by the lack of standardized methods for micropeptide identification and by insufficient data on their functions. In this study, we employed a comprehensive and innovative functional annotation process for micropeptides, utilizing a robust reference database search strategy, and identified 19586 novel sORFs in pancancer proteomic data, including 3065 micropeptides whose dysregulation is associated with cancer. The establishment of this database, which is the most comprehensive to date, not only will help increase the time efficiency of novel peptidomics and data mining but also represents a major step in micropeptide function annotation.
Although micropeptides are increasingly recognized as important regulators of cancer progression [6, 7, 9, 39, 42], how micropeptides can be applied for early therapeutic interventions and to improve cancer survival remains to be further investigated [39]. By reusing stable isotopically labeled TMT and pancancer cohort data, and incorporating clinically relevant variables such as patient age, sex, and survival time, we created a detailed map of cancer-associated micropeptides and identified 370 micropeptides associated with patient OS, suggesting analysis of these peptides may help identify disease-associated features. Our analysis further revealed a significant correlation between patient-level peptide associations and prognosis across multiple cancer types. On the basis of the aforementioned analysis, we comprehensively examined the correlation between micropeptide expression levels and disease progression, which will promote future applications in early cancer diagnosis to facilitate earlier therapeutic interventions and improve survival.
The micropeptide localization and functional features provided in this database are consistent with previous reports. Our methodology for the functional annotation and classification of micropeptides represents a significant advancement in biomarker network analysis (Fig. 3d). For example, the micropeptide named ASAP, encoded by LINC00467 and implicated in mitochondrial activity in colorectal cancer, was named as pep1-nc-LINC00467 in HMPA [6] (Fig. 3e). Consistent with our findings, we observed that pep1-nc-LINC00467 interacted with ATP synthase-coupling factor 6 (UniProt ID: P18859, synonyms: ATP5A, ATP5PF, ATP5J, and ATPM; Fig. 3f and g) and was enriched in functions related to the ‘mitochondrial protein-containing complex’ and ‘oxidative phosphorylation’ pathways (Fig. 3h). Furthermore, pep1-nc-HOXB-AS3 was enriched in the cytoplasm (Fig. 3i) and exhibited potential interaction sites with HNRNPA1 (Uniprot ID: P09651; Fig. 3j and k). Moreover, its enrichment functions included ‘spliceosomal complex’, ‘mRNA 3'-UTR binding’, and ‘regulation of RNA splicing’, consistent with previous findings [43] (Fig. 3l). Overall, our comprehensive analysis advances the understanding of the function of micropeptides and further highlights their potential as targets for early diagnosis and intervention in cancer.
The HMPA has an embedded genome ‘browser’ and rich data retrieval, analysis and display tools, allowing users to intuitively search for and analyze multiple types of omics data, such as comparative expression, survival analysis, genome, and conservation data. This integration enables important connections across multidimensional omics big data, providing a comprehensive characterization of each micropeptide from source to function annotation. Increasingly, important micropeptide information is being surfaced, but new data is accompanied by noise, and integrating new data is difficult, and maintenance and updates after the initial database completion are ongoing challenges. Another limitation of the current database is that the number of data samples remains limited, and the data sources are not sufficiently diverse. We will continue to enhance HMPA through ongoing acquisition and analysis of newly published proteomic datasets across various diseases, meticulous refinement of peptide functionality and validation status information, and expansion of our analytical pipeline for genomic mutation data to assess the functional implications of mutations in micropeptides. In conclusion, the HMPA database provides an innovative interactive platform and a pioneering compilation of nonclassical proteins, providing comprehensive insights into the fundamental properties and potential functions of micropeptides. This database serves as a unique public resource for researchers and clinicians seeking to advance the understanding and treatment of cancer.
Materials and methods
Data sources and curation
Using public repositories like PRIDE (https://www.ebi.ac.uk/pride/archive/) and the CPTAC (https://pdc.cancer.gov/pdc/), we combined proteomics data from eight different research. To identify processing factors, intricacies in the experimental design, and sample characteristics, the proteomic data underwent painstaking hand curation. After being painstakingly recorded in sample and data relationship formats, biological metadata was smoothly incorporated into HMPA as distinct tracks for every study. The CPTAC study was specifically chosen because it applied a multiple tandem mass labeling (TMT, iTRAQ) technology to produce proteome insights for various tumor types. Protein quantification based on MS1 is made possible by this novel method, which makes protein quantification dependent on the strength of peptide precursor ions possible. Moreover, the CPTAC research data is enhanced by the additional addition of genetic and clinical information obtained from the same tumor tissues. On the other hand, the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov) provided the transcriptome data. In particular, the GDC RNA-seq experiments CPTAC-2, CPTAC-3, and TCGA-BRCA were utilized. The transcriptome data had been compiled into the GDC framework before our analysis, which guaranteed alignment with the biological metadata present in the proteomic datasets.
Construction of a comprehensive and reliable reference library of human micropeptides
In order to identify micropeptides derived from aggregated MS datasets, we have carefully assembled a comprehensive reference of human micropeptide repository (CRHMR). Assembling the preliminary sORFs database involved utilizing a tool from the Ribotricer software suite (https://github.com/smithlabcode/ribotricer) [41]. The Ensembl database (http://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/) was accessed to obtain a complete copy of the human genome (Hg38, v110) and relevant reference data. This tool facilitated the identification of potential ORFs by processing raw FASTA files alongside GTF annotation files, with key parameters: the ‘—gtf’ parameter was employed to load reference gene annotation files, and the ‘—fasta’ parameter was used to input sequence files. Additionally, the ‘—start_codons’ parameter was set to identify the four typical start codons (ATG/CTG/GTG/TTG), ensuring that the detected ORFs align with the criteria necessary for initiating protein synthesis. Based on the RNA sequence's position relative to the annotated transcript, a comprehensive roster of potential micropeptides was compiled. Peptides overlapping with any annotated proteins were carefully excised, and the remaining peptides were scrutinized using the Basic Local Alignment Search Tool to identify non-replicating human protein sequences. Peptides that closely overlap with proteins, regardless of amino acid sequence similarity, were discarded. Notably, we integrated the ‘strandedness of reads’ parameter for each RNA-encoded micropeptide to guarantee the integrity and authenticity of our micropeptide library. This meticulous step ensures that small peptides that match the transcript exactly are excluded.
Proteomics raw data processing
The raw proteomic files underwent processing utilizing the PD (version 2.5). Raw data and associated sample details were retrieved (Supplementary Table S1 available online at http://bib.oxfordjournals.org/). CPTAC raw data files were procured using research identifiers for various cancer types: colon adenocarcinoma (COAD, PDC000116), breast invasive carcinoma (BRCA, PDC000173), clear cell renal cell carcinoma (ccRCC, PDC000127), pancreatic ductal adenocarcinoma (PDAC, PDC000270), ovarian serous cystadenocarcinoma (OV, PDC000118), lung squamous cell carcinoma (LUAD, PDC000153), and uterine corpus endometrial carcinoma (UCEC, PDC000125). Additionally, the original data file of GC from PRIDE (PXD041392) was obtained. TMT and iTRAQ4 tags from CPTAC, along with label-free raw files from PRIDE, underwent identification and quantification through Sequest HT [44]. Global normalization was applied using recommended search parameters, and quantitative grouping was executed at the peptide level. In comparison to our meticulously constructed reference library, sourced from CRHMR (reviewed entries: 2 969 954), the analysis incorporated complete trypsin specificity with allowance for up to two missed cuts. Fixed modifications included 57.02146 Da for Cys residues and 229.16293 Da for Lys residues. Variable modifications encompassed 15.9949 Da for Met residues, 42.0106 Da for peptide N-terminal, and 229.16293 Da for peptide N-terminal or Ser residues. Mass tolerances for both parent ions and fragments were set at 20 ppm. Protein identification underwent filtration at a 1% false discovery rate, as determined by PD utilizing the embedded decoy sequence.
Micropeptide expression quantification and differential expression analysis
Tumor protein abundance data was utilized for the analysis of differential protein expression. Principal component analysis and unsupervised hierarchical clustering were employed to assess batch effects and sample quality control. An initial filtration step was implemented on the protein abundance data, requiring quantification in at least 20% of the samples for further analysis. Missing values were visualized, and k-nearest neighbor (KNN) interpolation was applied to the data using the R package impute (version 1.76.0) [45]. Previous studies have indicated that KNN interpolation is more effective in TMT proteomic data [46]. Differential protein expression analysis was conducted using the Pandas library (version 0.25.3). The P values between tumors and NATs/normal tissues were adjusted by Benjamini Hochberg method using Wilcoxon rank sum test (unmatched samples) and Wilcoxon signed rank test (matched samples). Proteins that exhibited significant deregulation in tumors compared to NATs were identified using an adjusted threshold of P < .05 and |Log2Fold Change| ≥ 1.
Transcriptomics raw data processing and quantification
In this study, we further validated our findings using data from GDC cohort. We downloaded and unified raw RNA sequencing data from 2279 cases across 7 cancer types, including COAD (N = 105), BRCA (N = 1095), ccRCC (N = 261), PDAC (N = 170), OV (N = 71), LUAD (N = 337), and UCEC (N = 240). A standardized pipeline was previously used to process transcriptomics data [47]. In a nutshell, STAR (v 2.7.10b) was used to perform genomics alignment, and the TPM (transcript per million) values were to determine normalized gene abundance. Lastly, the average TPM abundance in instances when the repeated observations across datasets.
Messenger ribonucleic acid-peptide correlation analysis
We concentrated on RNA-seq data encoding micropeptide genes and proteomic expression levels of corresponding micropeptides in order to compare mRNA expression and protein abundance of various samples. To compute the correlation at the gene level, all protein isomers are considered as a single gene. The peptide id is linked to the gene name, and only genes or peptide with <85% missing values (NAs) are taken into consideration for study. The Pearson correlation coefficient (r) is the primary measure of correlation discussed in this study. According to the likelihood of creating a linear relationship between the variables established by the normalization approach and reducing any bias resulting from the normalization techniques used while processing proteomic and RNA sequencing data, we opted for Pearson correlation.
Identifying clinical features
Clinical data were procured from the GDC and harmonized with the CPTAC, including sex, age at diagnosis, tumor stage, demographics, and clinical details for each tumor. This encompassed demographic and histopathological details alongside comprehensive treatment and patient outcome records, incorporating genotype and clinical parameters. The prognostic outcomes are elegantly showcased in a graphical rendition, delineating proclivities toward disorder and corresponding functional annotations.
Network integration and scoring
Leveraging artificial intelligence tools and algorithms, we predicted and simulated the structure of the designated micropeptides. Subsequently, we analyzed the entire interactome based on the simulation outcomes. This approach enabled the construction of an mPPI functional network rooted in the interplay among biological molecules. We utilized the PepNN model to assess the quantification score and weight (weighting factor for PepNN-Struct: 0.955) indicating the probability of peptide-binding modules [48]. A more stringent peptide recognition module (prm) score was subsequently employed to sift through proteins for potential interactions.
Next, utilizing a novel algorithmic strategy, we delineated the complex web of interactions from a corpus of text data, employing a threshold-based filtering mechanism to enhance the relevance of the extracted interactions. We harnessed the capabilities of the OmicVerse [49] library and correlated the String-DB data [50] to predict and augment the interaction data with high fidelity, thereby generating a comprehensive map of mPPI that was subsequently transformed into a directed graph for topological analysis.
To quantitatively assess the reliability of the mPPI, we developed a scoring system that integrates the path analysis within the network. By calculating the average path score for each node, we effectively captured the overall connectivity and influence of the associated proteins within the network. The formula for path score calculation in a graph 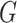 is as follows:
is as follows:
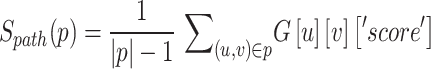 |
Where 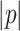 is the number of edges in path
is the number of edges in path 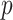 , and
, and 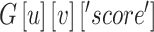 is the score of edge
is the score of edge 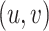 in the graph.
in the graph.
This metric was derived from the summation of edge scores along all simple paths emanating from a node, normalized by the total number of paths. The average path score Sn for micropeptide node n is
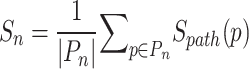 |
Where 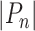 is the total number of simple paths from node
is the total number of simple paths from node 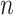 to others. We further refined our analysis by applying a threshold to the computed scores, thereby filtering the mPPI and enriching the dataset with a subset of interactions that demonstrated a higher level of network support. Subsequently, based on the degree of association between micropeptides and proteins, we iterated through each potentially interacting protein and organized proteins with associations, utilizing an interaction-based protein grouping approach. Furthermore, we functionally annotated the micropeptides based on the GO functional clustering results of the interacting proteins.
to others. We further refined our analysis by applying a threshold to the computed scores, thereby filtering the mPPI and enriching the dataset with a subset of interactions that demonstrated a higher level of network support. Subsequently, based on the degree of association between micropeptides and proteins, we iterated through each potentially interacting protein and organized proteins with associations, utilizing an interaction-based protein grouping approach. Furthermore, we functionally annotated the micropeptides based on the GO functional clustering results of the interacting proteins.
Functional annotation of micropeptides
The conservation score was computed using the default parameters of phastCons, utilizing an index of 100 mammalian datasets retrieved from the UCSC database (https://hgdownload.cse.ucsc.edu/goldenpath/hg38/phyloP100way/) [22]. TMHMM-2.0 is an intricately crafted neural network tool tailored precisely to forecast the membrane topology and inner disorder region of transmembrane proteins through deep learning analysis of protein sequences [51]. Deeploc2 represents a neural deep learning approach employed for forecasting the subcellular localization of proteins [52]. This model undergoes training on datasets encompassing human and eukaryotic proteins, subsequently validated through experiments annotating nine classification signals. The micropeptide sequences from the HMPA database are predicted using Deeploc2, and the outcomes are seamlessly integrated into the website. We employed the NetSurfP3 server to anticipate the surface accessibility, secondary structure, disorder, and phi/psi dihedral angles of amino acids within the amino acid sequence [53]. The spatial configuration of all micropeptides was forecasted utilizing AlphaFold2.
Database and web interface implementation
MySQL tables are used by HMPA to store and manage all of its metadata. Java programming serves as the foundation for the server back-end, while HTML, CSS, and JavaScript are used to create the web front-end experience. ECharts (https://echarts.apache.org/) creates several statistical charts for the website in order to visualize all of the analysis findings.
Key Points
The HMPA provides a novel framework leveraging proteomic, transcriptomic, and clinical data from public cohorts to comprehensively analyze cancer-associated micropeptides.
For the first time, survival information was integrated with micropeptide analysis, revealing associations between micropeptides and cancer prognosis and potential for early diagnosis.
Artificial intelligence and AlphaFold2-predicted structures are used to map micropeptide interactions and predict micropeptide functions within networks.
The HMPA database provides a state-of-the-art interactive platform and a pioneering compilation of nonclassical proteins, providing comprehensive insights into the fundamental properties and potential functions of micropeptides.
Supplementary Material
Acknowledgements
The authors would like to thank Prof. Yuanchao Xue from Chinese Academy of Sciences for valuable comments and support throughout this study.
Conflict of interest: The authors declare that they have no competing interests.
Contributor Information
Xinwan Su, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Chengyu Shi, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Fangzhou Liu, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Manman Tan, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Ying Wang, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Linyu Zhu, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Yu Chen, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Meng Yu, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Xinyi Wang, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China.
Jian Liu, Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, 718 East Haizhou Rd., Haining, Zhejiang 314400, China.
Yang Liu, Institute of Immunology, Zhejiang University School of Medicine, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310009, China.
Weiqiang Lin, International School of Medicine, International Institutes of Medicine, The 4th Affiliated Hospital of Zhejiang University School of Medicine, No. N1, Shangcheng Avenue, Yiwu, Zhejiang 322000, China.
Zhaoyuan Fang, Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, 718 East Haizhou Rd., Haining, Zhejiang 314400, China; The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Shangcheng District, Hangzhou, Zhejiang 310000, China.
Qiang Sun, International School of Medicine, International Institutes of Medicine, The 4th Affiliated Hospital of Zhejiang University School of Medicine, No. N1, Shangcheng Avenue, Yiwu, Zhejiang 322000, China.
Tianhua Zhou, Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Department of Cell Biology and Program in Molecular Cell Biology, Zhejiang University School of Medicine, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON M5S 1A8, Canada.
Aifu Lin, MOE Laboratory of Biosystem Homeostasis and Protection, College of Life Sciences, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Cancer Center, Zhejiang University, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China; Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310000, China; International School of Medicine, International Institutes of Medicine, The 4th Affiliated Hospital of Zhejiang University School of Medicine, No. N1, Shangcheng Avenue, Yiwu, Zhejiang 322000, China; Future Health Laboratory, Innovation Center of Yangtze River Delta, Zhejiang University, 828 Zhongxing Road, Xitang District, Jiashan, Zhejiang, 314100, China; Key Laboratory for Cell and Gene Engineering of Zhejiang Province, 866 Yuhangtang Road, West Lake District, Hangzhou, Zhejiang 310058, China.
Funding
This study was funded by the Scientific and Technological Innovation 2030—Major Projects (2023ZD0507500), National Science Fund for Distinguished Young Scholars (32225014), 'Lingyan' R&D Research and Development Project (2023C03023), National Key R&D Program of China (2021YFC2700903), National Natural Science Foundation of China (81672791 and 81872300), Zhejiang Provincial Natural Science Fund for Distinguished Young Scholars of China (LR18C060002), the Fundamental Research Funds for the Central Universities (K20220228) and the Young Scientists Fund of the National Natural Science Foundation of China (8240112554).
Data availability
HMPA is available freely for public from hmpa.zju.edu.cn.
Code availability
All scripts associated with analysis for micropeptide discovery and annotation can be found at: https://github.com/suxww/HMPA.git.
Author contributions
A.L., T.Z., and Q.S. conceived and supervised the project. X.S. analyzed the data, gathered the results, and drafted the manuscript. C.S. and F.L. participated project discussion and data analysis, constructed the database, and edited the manuscript. C.S. and X.S. performed mass spectrum analysis. M.T., Y.W., Y.C., L.Z., M.Y., and X.W. participated database construction. A.L., T.Z., Q.S., Z.F., W.L., Y.L., and J.L. participated discussion and data analysis. All authors read and approved the final manuscript.
References
- 1. Hon C-C, Ramilowski JA, Harshbarger J. et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017;543:199–204. 10.1038/nature21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slavoff SA, Mitchell AJ, Schwaid AG. et al. Peptidomic discovery of short open reading frame–encoded peptides in human cells. Nat Chem Biol 2013;9:59–64. 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong X, Zhang K, Xun C. et al. Small open reading frame-encoded micro-peptides: an emerging protein world. IJMS 2023;24:10562. 10.3390/ijms241310562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuanyuan J, Xinqiang Y. Micropeptides identified from human genomes. J Proteome Res 2022;21:865–73. 10.1021/acs.jproteome.1c00889. [DOI] [PubMed] [Google Scholar]
- 5. Bhati KK, Kruusvee V, Straub D. et al. Global analysis of cereal microProteins suggests diverse roles in crop development and environmental adaptation. G3 (Bethesda) 2020;10:3709–17. 10.1534/g3.120.400794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ge Q, Jia D, Cen D. et al. Micropeptide ASAP encoded by LINC00467 promotes colorectal cancer progression by directly modulating ATP synthase activity. J Clin Investig 2021;131:e152911. 10.1172/JCI152911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li M, Li X, Zhang Y. et al. Micropeptide MIAC inhibits HNSCC progression by interacting with aquaporin 2. J Am Chem Soc 2020;142:6708–16. 10.1021/jacs.0c00706. [DOI] [PubMed] [Google Scholar]
- 8. Zhang C, Zhou B, Gu F. et al. Micropeptide PACMP inhibition elicits synthetic lethal effects by decreasing CtIP and poly(ADP-ribosyl)ation. Mol Cell 2022;82:1297–1312.e8. 10.1016/j.molcel.2022.01.020. [DOI] [PubMed] [Google Scholar]
- 9. Pang Y, Liu Z, Han H. et al. Peptide SMIM30 promotes HCC development by inducing SRC/YES1 membrane anchoring and MAPK pathway activation. J Hepatol 2020;73:1155–69. 10.1016/j.jhep.2020.05.028. [DOI] [PubMed] [Google Scholar]
- 10. Papaioannou D, Petri A, Dovey OM. et al. Publisher correction: the long non-coding RNA HOXB-AS3 regulates ribosomal RNA transcription in NPM1-mutated acute myeloid leukemia. Nat Commun 2020;11:204. 10.1038/s41467-019-13969-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee C, Zeng J, Drew BG. et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab 2015;21:443–54. 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Birney E, Stamatoyannopoulos JA, Dutta A. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007;447:799–816. 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prensner JR, Enache OM, Luria V. et al. Noncanonical open reading frames encode functional proteins essential for cancer cell survival. Nat Biotechnol 2021;39:697–704. 10.1038/s41587-020-00806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer 2018;18:5–18. 10.1038/nrc.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du Z, Fei T, Verhaak RGW. et al. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol 2013;20:908–13. 10.1038/nsmb.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Zeng S, Wu M. Novel insights into noncanonical open reading frames in cancer. Biochim Biophys Acta Rev Cancer 2022;1877:188755. 10.1016/j.bbcan.2022.188755. [DOI] [PubMed] [Google Scholar]
- 17. Chen Y, Ho L, Tergaonkar V. sORF-encoded MicroPeptides: new players in inflammation, metabolism, and precision medicine. Cancer Lett 2021;500:263–70. 10.1016/j.canlet.2020.10.038. [DOI] [PubMed] [Google Scholar]
- 18. Martinez TF, Lyons-Abbott S, Bookout AL. et al. Profiling mouse brown and white adipocytes to identify metabolically relevant small ORFs and functional microproteins. Cell Metab 2023;35:166–183.e11. 10.1016/j.cmet.2022.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jackson R, Kroehling L, Khitun A. et al. The translation of non-canonical open reading frames controls mucosal immunity. Nature 2018;564:434–8. 10.1038/s41586-018-0794-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ouspenskaia T, Law T, Clauser KR. et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol 2022;40:209–17. 10.1038/s41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brunet MA, Brunelle M, Lucier J-F. et al. OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes. Nucleic Acids Res 2019;47:403–410. 10.1093/nar/gky936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siepel A, Bejerano G, Pedersen JS. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005;15:1034–50. 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patraquim P, Mumtaz MAS, Pueyo JI. et al. Developmental regulation of canonical and small ORF translation from mRNAs. Genome Biol 2020;21:128. 10.1186/s13059-020-02011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gligorijević V, Renfrew PD, Kosciolek T. et al. Structure-based protein function prediction using graph convolutional networks. Nat Commun 2021;12:3168. 10.1038/s41467-021-23303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kastenmayer JP, Ni L, Chu A. et al. Functional genomics of genes with small open reading frames (sORFs) in S. Cerevisiae. Genome Res 2006;16:365–73. 10.1101/gr.4355406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crappé J, Van Criekinge W, Trooskens G. et al. Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC Genomics 2013;14:648. 10.1186/1471-2164-14-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingolia NT, Ghaemmaghami S, Newman JRS. et al. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009;324:218–23. 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fritsch C, Herrmann A, Nothnagel M. et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res 2012;22:2208–18. 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Heesch S, Witte F, Schneider-Lunitz V. et al. The translational landscape of the human heart. Cell 2019;178:242–260.e29. 10.1016/j.cell.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 30. Bánfai B, Jia H, Khatun J. et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res 2012;22:1646–57. 10.1101/gr.134767.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cassidy L, Kaulich PT, Maaß S. et al. Bottom-up and top-down proteomic approaches for the identification, characterization, and quantification of the low molecular weight proteome with focus on short open reading frame-encoded peptides. Proteomics 2021;21:e2100008. 10.1002/pmic.202100008. [DOI] [PubMed] [Google Scholar]
- 32. Dragomir MP, Manyam GC, Ott LF. et al. FuncPEP: a database of functional peptides encoded by non-coding RNAs. Noncoding RNA 2020;6:41. 10.3390/ncrna6040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olexiouk V, Van Criekinge W, Menschaert G. An update on sORFs.org: a repository of small ORFs identified by ribosome profiling. Nucleic Acids Res 2018;46:D497–502. 10.1093/nar/gkx1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hao Y, Zhang L, Niu Y. et al. SmProt: a database of small proteins encoded by annotated coding and non-coding RNA loci. Brief Bioinform 2018;19:636–643. 10.1093/bib/bbx005. [DOI] [PubMed] [Google Scholar]
- 35. Liu H, Zhou X, Yuan M. et al. ncEP: a manually curated database for experimentally validated ncRNA-encoded proteins or peptides. J Mol Biol 2020;432:3364–8. 10.1016/j.jmb.2020.02.022. [DOI] [PubMed] [Google Scholar]
- 36. Huang Y, Wang J, Zhao Y. et al. cncRNAdb: a manually curated resource of experimentally supported RNAs with both protein-coding and noncoding function. Nucleic Acids Res 2021;49:D65–70. 10.1093/nar/gkaa791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luo X, Huang Y, Li H. et al. SPENCER: a comprehensive database for small peptides encoded by noncoding RNAs in cancer patients. Nucleic Acids Res 2022;50:D1373–81. 10.1093/nar/gkab822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leblanc S, Yala F, Provencher N. et al. OpenProt 2.0 builds a path to the functional characterization of alternative proteins. Nucleic Acids Res 2024;52:D522–8. 10.1093/nar/gkad1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Setrerrahmane S, Li M, Zoghbi A. et al. Cancer-related micropeptides encoded by ncRNAs: promising drug targets and prognostic biomarkers. Cancer Lett 2022;547:215723. 10.1016/j.canlet.2022.215723. [DOI] [PubMed] [Google Scholar]
- 40. Jumper J, Evans R, Pritzel A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choudhary S, Li W, Smith D. et al. Accurate detection of short and long active ORFs using Ribo-seq data. Bioinformatics 2020;36:2053–9. 10.1093/bioinformatics/btz878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu S, Wang J-Z, Chen D. et al. An oncopeptide regulates m6A recognition by the m6A reader IGF2BP1 and tumorigenesis. Nat Commun 2020;11:1685. 10.1038/s41467-020-15403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang J-Z, Chen M, Chen D. et al. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol Cell 2017;68:171–184.e6. 10.1016/j.molcel.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 44. Diament BJ, Noble WS. Faster SEQUEST searching for peptide identification from tandem mass spectra. J Proteome Res 2011;10:3871–9. 10.1021/pr101196n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hastie T, Tibshirani R, Narasimhan B. et al. Impute: Imputation for microarray data. 2023. 10.18129/B9.bioc.impute. [DOI]
- 46. Wang J, Yu W, D’Anna R. et al. Pan-cancer proteomics analysis to identify tumor-enriched and highly expressed cell surface antigens as potential targets for cancer therapeutics. Mol Cell Proteomics 2023;22:100626. 10.1016/j.mcpro.2023.100626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu F, Tian T, Zhang Z. et al. Long non-coding RNA SNHG6 couples cholesterol sensing with mTORC1 activation in hepatocellular carcinoma. Nat Metab 2022;4:1022–40. 10.1038/s42255-022-00616-7. [DOI] [PubMed] [Google Scholar]
- 48. PepNN: a deep attention model for the identification of peptide binding sites. Commun Biol 2022;5:503. https://www.nature.com/articles/s42003-022-03445-2. [DOI] [PMC free article] [PubMed]
- 49. Zeng Z, Ma Y, Hu L. et al. OmicVerse: a single pipeline for exploring the entire transcriptome universe. Nat Commun 2024;15:59832023. 10.1101/2023.06.06.543913. [DOI]
- 50. Szklarczyk D, Kirsch R, Koutrouli M. et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023;51:D638–46. 10.1093/nar/gkac1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Möller S, Croning MDR, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 2001;17:646–53. 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- 52. Thumuluri V, Almagro Armenteros JJ, Johansen AR. et al. DeepLoc 2.0: multi-label subcellular localization prediction using protein language models. Nucleic Acids Res 2022;50:W228–34. 10.1093/nar/gkac278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Høie MH, Kiehl EN, Petersen B. et al. NetSurfP-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res 2022;50:W510–5. 10.1093/nar/gkac439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
HMPA is available freely for public from hmpa.zju.edu.cn.