

Abstract
In recent years, the connection between APOBEC3 cytosine deaminases and cancer mutagenesis has become ever more apparent. This growing awareness and lack of inhibitory drugs has created a distinct need for biochemical tools that can be used to identify and characterize potential inhibitors of this family of enzymes. In response to this challenge, we have developed a Real-time APOBEC3-mediated DNA Deamination (RADD) assay. The RADD assay provides a rapid, real-time fluorescence readout of APOBEC3 DNA deamination and serves as a crucial addition to the existing APOBEC3 biochemical and cellular toolkit. This method improves upon contemporary DNA deamination assays by offering a more rapid and quantifiable readout as well as providing a platform that is readily adaptable to a high-throughput format for inhibitor discovery. In this chapter we provide a detailed guide for the usage of the RADD assay for the characterization of APOBEC3 enzymes and potential inhibitors.
Keywords: APOBEC3, Cancer, Cytidine deaminase, Fluorescence resonance energy transfer (FRET), High-throughput screening (HTS), Real-time assay
1. Introduction
The human APOBEC3 (A3) family is composed of 7 enzymes (A3A, B, C, D, F, G, and H) and plays important roles in the innate immunity against foreign genetic elements such as viruses and transposons. These A3 proteins restrict exogenous nucleic acids by catalyzing hydrolytic deamination of cytosine into uracil (C-to-U) in single-stranded (ss)DNA, thereby inactivating them by hypermutation. Recent evidence has shown that at least two A3 members (A3A and A3B) are capable of targeting cellular genomic DNA in human cancers to elicit C-to-T and C-to-G mutations in preferred trinucleotide contexts (TCA and TCT), resulting in characteristic patterns of mutations referred to as “APOBEC mutational signatures” prevalent in approximately 70% of tumor types. Over the past decade, the expression of A3A/B and/or the presence of APOBEC-mediated mutations has been associated with tumor development, metastasis, drug resistance, and poor clinical outcomes (Burns et al., 2013, Harris, 2015, Petljak et al., 2022, Petljak and Maciejowski, 2020). These associative studies have been supported by work in mice, where the expression of catalytically active human A3A or A3B triggers an accumulation of APOBEC signature mutations and an elevation in tumor numbers (Durfee et al., 2023, Law et al., 2020, Naumann et al., 2023).
These clinical and animal experiments combine to suggest that inhibiting A3A and A3B activities has the potential to improve the effectiveness of conventional anti-cancer therapies by limiting tumor evolution and preventing adverse outcomes such as drug resistance and metastasis. However, despite significant advances made toward understanding the mechanisms by which A3A and A3B drive tumorigenesis and cancer progression, less progress has been made in the development of therapeutics tailored toward inhibiting the mutagenic activity of these enzymes. One of the key factors hampering the identification of small molecule inhibitors of A3A/B has been the lack of real-time assays to rapidly assess APOBEC enzymatic activity.
The Real-time APOBEC3-mediated DNA Deamination (RADD) assay described in this work is positioned to fill the gap left by the current tools used to evaluate A3-mediated DNA deamination. Existing in vitro methods such as the gel-based oligonucleotide cleavage assay involve a deamination step followed by treatment with uracil-DNA glycosylase and harsh basic conditions to cleave fluorescently labeled oligonucleotides (Fig. 1A) (Carpenter et al., 2012, Li et al., 2012, Thielen et al., 2010). Though reliable, this method requires multistep sample processing and gel electrophoresis to separate cleaved and uncleaved DNA for the readout and is therefore incapable of reporting deamination in real-time. Conversely, NMR-based methods are capable of detecting A3-mediated C-to-U conversion in real-time, but these assays require much larger quantities of material and suffer from low throughput and high expense (Barzak et al., 2019, Furukawa et al., 2009, Kvach et al., 2019).
Fig. 1. UDG deamination assay versus the RADD assay.
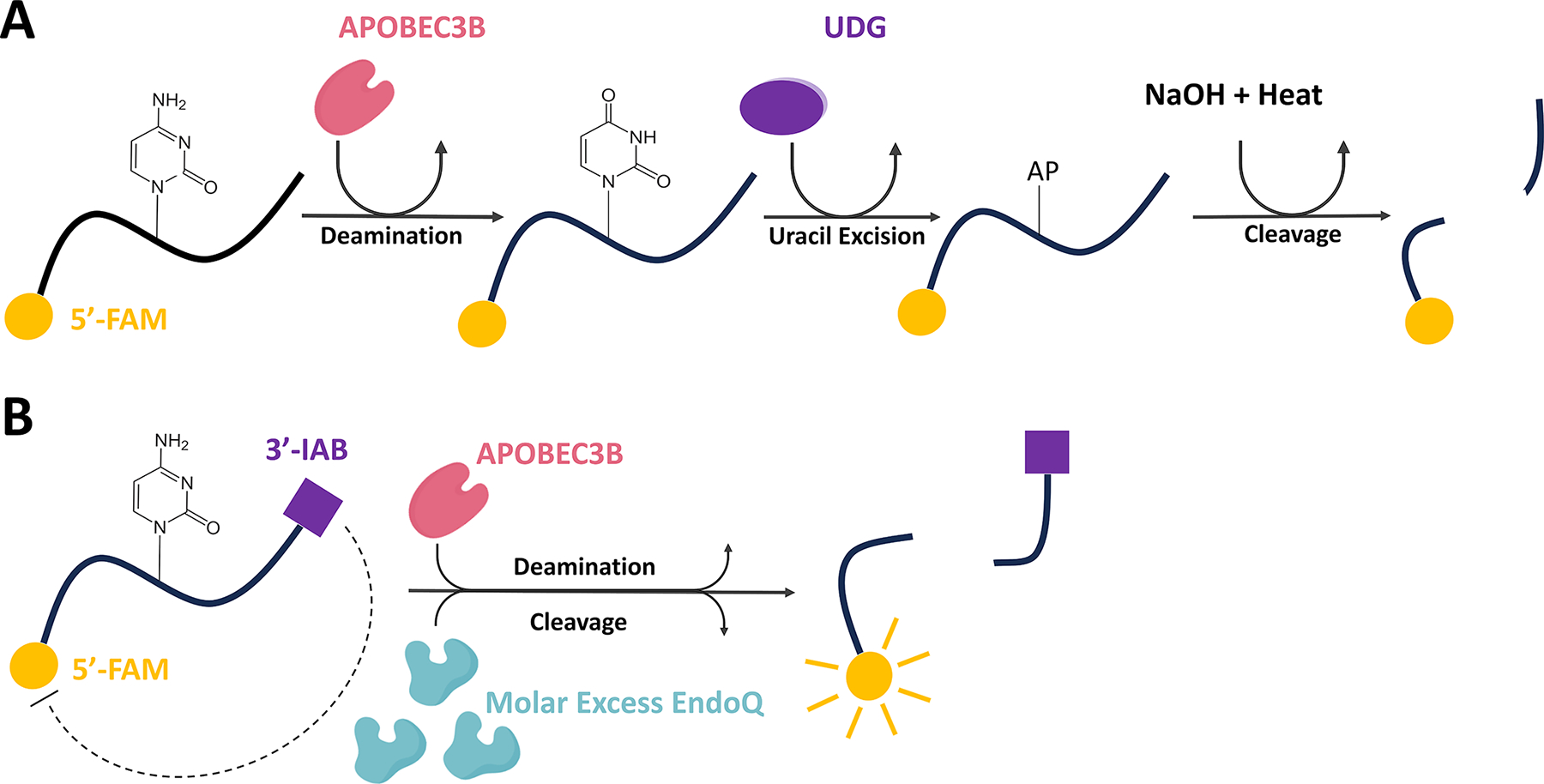
(A) Schematic representing the molecular process involved in traditional UDG-based deamination assays. (B) Schematic of the molecular process which results in real-time fluorescence reporting of APOBEC deamination in a single step through the RADD assay. FAM represents a 5′ 6-carboxyfluorescein fluorophore while IAB represents a 3′ Iowa Black® FQ quencher.
Figure adapted from Belica, C., Carpenter, M. A., Chen, Y., Moeller, N., Harris, R. S., & Aihara, H. (2024). A real-time biochemical assay for quantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry.
As a precursor to the method we describe here, we showed that Pyrococcus furiosus endonuclease Q (EndoQ) can be used to directly cleave DNA adjacent to 2′-deoxyuridine generated by A3B (Shi et al., 2021, Shiraishi et al., 2015). Building upon this discovery, we have developed the RADD assay which incorporates many of the positive aspects of existing methods without notable drawbacks and is capable of rapid quantitative analysis of A3 activities for purified or crude samples. Here we outline the usage of a streamlined in vitro method for monitoring A3-mediated DNA deamination in real-time. This novel method only requires a single A3-containing reaction with a self-quenched fluorescent ssDNA reporter substrate and a large excess of EndoQ, which allows sensitive detection of A3-mediated DNA deamination via a fluorescence plate reader or a gel-based read-out (Fig. 1B).
2. Substrate design
The RADD assay relies on a self-quenched fluorescent single-stranded DNA reporter substrate. The reporter first undergoes deamination and is rapidly cleaved endonucleolytically by an excess of EndoQ at the site of deamination, releasing the fluorophore from the quencher. The original reporter oligo utilized in the development of this assay (TC) is a 15-nucleotide (nt) ssDNA with a single target cytosine in a TCA motif, a 5′ 6-carboxyfluorescein (FAM), and a 3′ Iowa Black® FQ (IAB) quencher (5′-FAM-TAGGTCATTATTGTG-IAB-3′). Position 6 of this ssDNA oligo has the only cytosine base and, therefore, is the only site for A3-catalyzed deamination. The control reporter used to evaluate EndoQ activity (TdU) is nearly identical to the primary reporter apart from a 2′-deoxyuridine at position 6 rather than a 2′-deoxycytidine, allowing for EndoQ cleavage without the need for deamination. In addition to these standard reporters, we have developed a second generation of reporters (TC-ZEN and TdU-ZEN) which contain an internal ZEN™ quencher between nt 9 and 10 in addition to a 3′ IAB quencher (5′-FAM-TAGGTCATT-ZEN-ATTGTG-IAB-3′). These doubly quenched reporters offer improved signals while also reducing background compared to their singly quenched counterparts (Table 1).
Table 1.
List of fluorescent reporter oligonucleotides.
| Reporter oligonucleotides | Sequence (5’ – 3’) |
|---|---|
| TC | FAM-TAGGTCATTATTGTG-IAB |
| TdU | FAM-TAGGTdUATTATTGTG-IAB |
| TC-ZEN | FAM-TAGGTCATT-ZEN-ATTGTG-IAB |
| TdU-ZEN | FAM-TAGGTdUATT-ZEN-ATTGTG-IAB |
The example reactions shown in this article were created using A3B. However, the TC oligo is also compatible with other TC-motif-preferring A3 enzymes such as A3A, and it can also be easily modified to accommodate other A3 family members with different sequence preferences such as A3G. Furthermore, EndoQ is capable of cleaving ssDNA containing either dU or deoxyinosine with no specific sequence preference, making the RADD assay compatible with cytosine and adenine DNA deaminases (Shi et al., 2021, Shiraishi et al., 2015). As a result, it is only necessary to consider the sequence preference of the deaminase you wish to evaluate and modify the reporter according to your specific needs.
3. Enzyme purification protocols
Here we briefly describe the purification of the A3B constructs used as examples in this work (A3BctdDM and A3BctdWT), P. furiosus EndoQ, and the model A3B inhibitor protein BORF2. A3BctdWT refers to an enzymatically active human A3B C-terminal domain construct while A3BctdDM refers to the same construct with a double mutation (L230K/F308K) for improved solubility. BORF2 is the large subunit of the Epstein-Barr virus (EBV) ribonucleotide reductase which is known to act as an A3B antagonist by tightly binding A3B and preventing DNA deamination activity (Cheng et al., 2019, Shaban et al., 2022).
3.1. Summary of the A3BctdDM and A3BctdWT purification protocol
pET42b(+)GST-6xHis-A3BctdDM and pET42b(+)GST-6xHis-A3BctdWT plasmids (Belica et al., 2024) which contain codon-optimized genes for human A3B residues 187–378 with or without the solubility-enhancing mutations (L230K/F308K) fused to an N-terminal GST-6xHis tag with a HRV-3 C protease cleavage site, were used to transform chemically competent C41(DE3) Escherichia coli The transformed cells were grown on an LB-agarose plate in the presence of 40 μg/mL kanamycin overnight at 37 °C.
All colonies growing on the plate were then resuspended and distributed to four 1 L flasks of LB media containing 40 μg/mL kanamycin. These cultures were grown at 37 °C until an OD600 of 0.8–1.0 was reached. Cultures were then induced with 0.5 mM isopropylβ-D-1-thiogalactopyranoside and 50 μM ZnCl2 at 18 °C for 18 h.
The cultures were then centrifuged at 4000 × g for 30 min at 4 °C. The pelleted cells were resuspended in 66 mL of buffer A (20 mM Tris-HCl pH 7.4, 500 mM NaCl, 5 mM imidazole, 5 mM β-Mercaptoethanol).
Cells were lysed with the addition of 40 mg hen egg white lysozyme followed by sonication. The cell lysate was centrifuged at 64,000 × g, at 4 °C for 1 h, followed by vacuum filtration through a 0.22 μM filter.
The filtered lysate was then passed over a 5 mL Ni-NTA Superflow column (Qiagen). The column was washed extensively with buffer A and subsequently, the bound protein was eluted with a linear gradient of buffer A to B (20 mM Tris-HCl pH 7.4, 500 mM NaCl, 400 mM imidazole, 5 mM β-Mercaptoethanol).
Fractions containing GST-A3Bctd were pooled and treated with the HRV-3 C protease over night at 4 °C to remove the GST-6xHis tag.
The cleaved protein was then concentrated and underwent size exclusion chromatography using a HiLoad Superdex 75 26/600 pg column operating with A3B storage buffer (20 mM Tris-HCl pH 7.4, 200 mM NaCl, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP)). Protein was snap-frozen in liquid nitrogen and stored at −80 °C.
3.2. Summary of the full-length pfuEndoQ purification protocol
A pET24-based expression plasmid for C-terminally 6xHis-tagged full-length P. furiosus EndoQ (Shi et al., 2021) was used to transform chemically competent BL21(DE3) E. coli, which were grown on an LB-agarose plate in the presence of 40 μg/mL kanamycin overnight at 37 °C.
Obtained colonies were resuspended and distributed to four 1 L flasks of LB media containing 40 μg/mL kanamycin. These cultures were grown at 37 °C until an OD600 of 0.8–1.0 was reached. Cultures were then induced with 0.5 mM IPTG and 50 μM ZnCl2 at 18 °C for 18 h.
The cultures were then centrifuged at 4000 × g for 30 min at 4 °C. The cells were resuspended in 66 mL of buffer A (20 mM Tris-HCl pH 7.4, 500 mM NaCl, 5 mM imidazole, 5 mM β-Mercaptoethanol).
Cells were lysed with the addition of 40 mg lysozyme followed by sonication. The cell lysate was centrifuged at 64,000 g, at 4 °C for 1 h, followed by vacuum filtration through a 0.22 μM filter.
The filtered lysate was then passed over a 5 mL Ni-NTA Superflow column (Qiagen). Protein elution was carried out with a gradient mixture of buffer A and buffer B (20 mM Tris-HCl pH 7.4, 500 mM NaCl, 400 mM imidazole, 5 mM β-Mercaptoethanol).
Pooled fractions containing PfuEndoQ were concentrated and underwent size-exclusion chromatography using a HiLoad Superdex 75 26/600 pg column operating with 20 mM Tris-HCl pH 7.4, 200 mM NaCl, and 0.5 mM TCEP. The purified protein was snap-frozen with liquid nitrogen and stored at −80 °C.
3.3. Summary of MBP-BORF2(1–739) purification protocol
Chemically competent BL21(DE3) E. coli was transformed with a pMalX(E)-BORF2(1–739-6xHis) plasmid (Belica et al., 2024) and grown on an LB-agarose plate containing 100 μg/mL ampicillin overnight at 37 °C.
Colonies were then resuspended in water and distributed to 4, 1 L flasks of TB media containing 100 μg/mL ampicillin. These cultures were grown at 37 °C until an OD600 of 0.8–1.0 was reached. Cultures were then induced with 0.5 mM IPTG at 18 °C for 18 h.
The cultures were then centrifuged at 4000 × g for 30 min at 4 °C. The cells were resuspended in 66 mL of a modified buffer A (20 mM Tris-HCl pH 7.4, 500 mM NaCl, 5 mM Imidazole, 5% glycerol, 5 mM β-Mercaptoethanol).
Cells were lysed with the addition of 40 mg lysozyme followed by sonication. The cell lysate was centrifuged at 64,000 × g, at 4 °C for 1 h, followed by vacuum filtration through a 0.22 μM filter.
The filtered lysate was then passed over a 5 mL Ni-NTA Superflow column (Qiagen). Protein elution was carried out with a linear gradient of modified buffer B (20 mM Tris-HCl pH 7.4, 500 mM NaCl, 400 mM imidazole, 5% glycerol, 5 mM β-Mercaptoethanol).
The pooled protein was then concentrated and underwent size-exclusion chromatography using a Superdex 200 increase 10/300 column operating with BORF2 storage buffer (20 mM Tris-HCl pH 7.4, 200 mM NaCl, 5% glycerol, 1 mM TCEP). Protein was snap-frozen with liquid nitrogen and stored at −80 °C.
4. Generalized RADD protocol
This section describes the implementation of the basic RADD protocol which serves as the foundation for all subsequent variations described in later sections. Here we depict an example RADD reaction using 40 μL reaction volumes containing 1 μM A3B C-terminal domain (A3BctdWT), 2 μM pfuEndoQ and 1 μM TC reporter with a follow-up gel-based readout.
4.1. Equipment
Temperature-controlled multimode plate reader (e.g., TECAN Spark 10 M)
Black 96-well half-area plate (e.g., Corning #3993)
PCR Thermocycler (e.g., Bio-Rad T100 Thermal Cycler)
15% TBE-Urea polyacrylamide gel (e.g., Invitrogen Novex gels)
Gel electrophoresis tank (e.g., ThermoFisher XCell SureLock Mini-Cell)
Power supply (e.g., Bio-Rad PowerPac)
Gel imaging instrument (e.g., Bio-Rad Gel Doc EZ, Amersham Typhoon imager)
PCR strip tubes
8-Channel multichannel pipette 2–20 μL (e.g., Gilson PIPETMAN G Multichannel P8×20G)
8-Channel multichannel pipette 20–200 μL (e.g., Gilson PIPETMAN L Multichannel P8×200L)
4.2. Buffers and reagents
10X RT reaction buffer (600 mM Tris-HCl pH 8.0, 0.1% Tween20, 10% DMSO, 10 mM DTT)
Enzyme storage buffer (20 mM Tris-HCl pH 7.4, 200 mM NaCl, 0.5 mM TCEP)
RNase and DNase-free water (Invitrogen)
10 μM A3BctdWT suspended in enzyme storage buffer.
20 μM pfuEndoQ suspended in enzyme storage buffer.
10 μM FAM-TC-IAB reporter suspended in water (Integrated DNA Technologies).
10 μM FAM-TdU-IAB reporter suspended in water (Integrated DNA Technologies).
1X Tris-Borate-EDTA (TBE) running buffer.
DNA loading buffer (95% Formamide, 0.025% Bromophenol blue, 0.025% Xylene cyanol, 5 mM EDTA).
Molecular biology grade formamide (e.g., RPI F11900–0.5)
4.3. Procedure
4.3.1. Plate reader setup
Fluorescence scans should be carried out at 37 °C using an excitation wavelength and bandwidth of 480 and 20 nm respectively. The emission wavelength should be set to 520 nm and the bandwidth set to 20 nm. In this example, instrument gain should be set to 71 and the Z-position of the emitter should be set to 16,156 μM. Total scan time was set for 30 min with 30 s scan intervals. For lower enzyme concentrations, you may choose to increase the total scan time to 60 min. In that scenario, we recommend increasing the scan interval time to 60 s to avoid excessive photobleaching.
Before preparing the reactions, the plate reader should be allowed to warm up to 37 °C with an empty plate inside. This will allow for rapid initiation of the deamination and cleavage reactions upon the addition of the reaction components to the plate.
4.3.2. Enzyme stock preparation
Frozen enzyme stocks should be thawed on ice, followed by centrifugation at 18,000 × g for 10 min at 4 °C to remove insoluble aggregate material then stored on ice until usage. If using high-concentration storage stocks, we suggest checking the concentration of each freshly thawed stock via nanodrop before diluting to the appropriate concentration in enzyme storage buffer.
4.3.3. Reaction mixture preparation
Each 40 μL reaction is comprised of 32 μL reporter master mix and 8 μL enzyme mix. A 1X reporter master mix is composed of 24 μL DNase/RNase-free water, 4 μL 10X RT reaction buffer, and 4 μL 10 μM reporter. After combining the components, the master mixes should be vortexed to ensure homogeneity. In this example, a 3X volume master mix was prepared for both the TC and TdU reporters for a total of four samples (TdU reporter control, TdU + EndoQ, TC reporter control, and TC + A3B + EndoQ) (Table 2).
32 μL of the reporter master mixes should be aliquoted into PCR tubes according to the number of samples for each reporter type. These tubes should then be heated in a thermocycler at 60 °C for 10 min and rapidly cooled on ice for 5 min to ensure all DNA is in a linear, single-stranded form.
After preparing the reporter master mix tubes, a 9 μL enzyme mixture should be prepared for each condition in separate PCR tubes. In this example, the TC control was composed of 4.5 μL enzyme storage buffer + 4.5 μL 20 μM EndoQ. The TdU control was composed of 4.5 μL 10 μM A3BctdWT + 4.5 μL enzyme storage buffer. The enzyme mixture for the TdU + EndoQ mixture was 4.5 μL enzyme storage buffer + 4.5 μL 20 μM EndoQ, and the enzyme mixture for the TC + A3B + EndoQ condition was composed of 4.5 μL 10 μM A3BctdWT and 4.5 μL 20 μM EndoQ (Table 3).
Once the reporter master mix and enzyme mix tubes have been prepared, both sets of tubes should be heated in a thermocycler at 37 °C for 5 min to equilibrate the temperature of both halves of the reaction before mixing.
After the pre-heating step is complete, using a multichannel pipette, combine the 8 μL enzyme mixtures with the corresponding 32 μL reporter master mixtures. Following this step, mix thoroughly by pipetting before transferring each 40 μL to the pre-warmed plate as rapidly as possible and immediately initiate fluorescence scanning (Fig. 2A).
Table 2.
Generalized RADD protocol report master mix.
| 3X TC master mix | 3X TdU master mix | |
|---|---|---|
| 10X RT buffer | 12 μL | 12 μL |
| 10 μM reporter (TC or TdU) | 12 μL | 12 μL |
| DNase/RNase-free H2O | 72 μL | 72 μL |
Table 3.
Generalized RADD protocol enzyme mixes.
| TC control | TdU control | A3B + TC + EndoQ | TdU + EndoQ | |
|---|---|---|---|---|
| 10 μM A3BctdWT | – | 4.5 μL | 4.5 μL | – |
| 20 μM EndoQ | 4.5 μL | – | 4.5 μL | 4.5 μL |
| Storage buffer | 4.5 μL | 4.5 μL | – | 4.5 μL |
Fig. 2. Fluorescence and gel-based readout of the RADD assay.
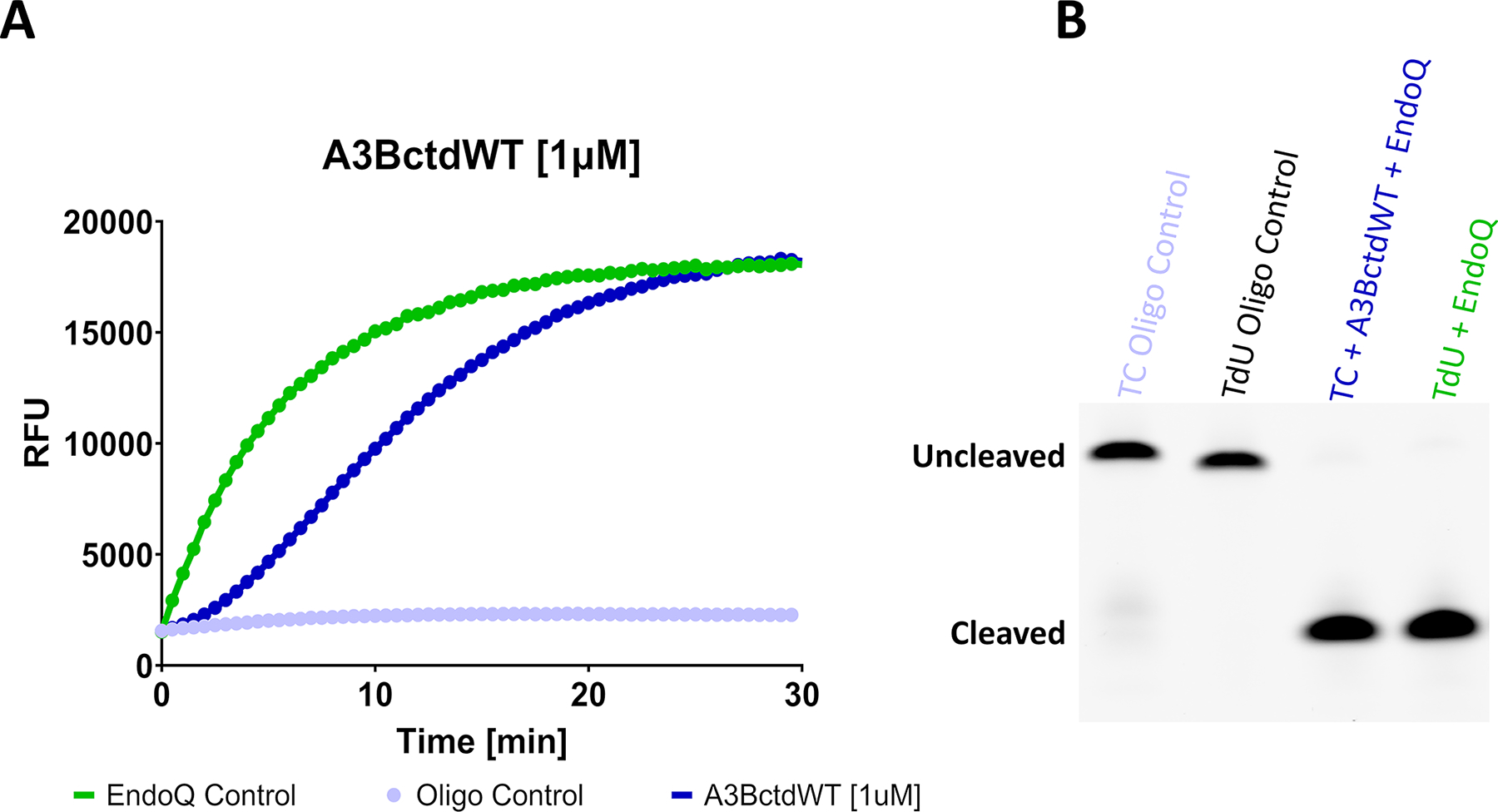
(A) Fluorescence readout of a RADD assay monitoring the deamination activity of 1 μM A3BctdWT. The A3BctdWT reaction (dark blue) contained 1 μM TC reporter and 2 μM EndoQ. The EndoQ control reaction (green) contained A3BctdWT protein storage buffer, 2 μM EndoQ, and 1 μM TdU reporter. The negative control (light blue) contained 2 μM EndoQ, A3BctdWT protein storage buffer, and 1 μM TC reporter. (B) Gel readout of the reactions shown in A. Both the A3BctdWT and EndoQ control reactions show complete substrate processing within 30 min.
Figure adapted from Belica, C., Carpenter, M. A., Chen, Y., Moeller, N., Harris, R. S., & Aihara, H. (2024). A real-time biochemical assay for quantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry.
4.3.4. Gel-based readout
Once fluorescence scanning is complete, eject the plate from the plate reader and transfer 20 μL of each reaction to separate PCR tubes and halt the reaction with the addition of 40 μL formamide. These tubes should then be boiled at 95 °C for 5 min.
Following the boiling step, briefly allow the tubes to cool at room temperature before adding 80 μL of DNA loading buffer and mix by pipetting.
Prepare a 15% TBE-Urea polyacrylamide gel electrophoresis assembly and fill the chamber with 1X TBE. Using a pipette, remove excess urea from the wells by washing with 1X TBE from the chamber.
Slowly add 12 μL of each reaction condition to the wells of the gel, avoiding spillover and run the gel at 300 V for 35 min.
After the gel has been run, it can be imaged using any FAM-compatible fluorescence imaging instrument. Higher molecular weight bands represent uncleaved reporter while lower weight bands represent the cleaved product. In this example, the gel was imaged with an Amersham Typhoon imager using the built-in Cy2 method (Fig. 2B).
4.4. Data processing
When using the standard TC and TdU reporters, some data processing is required to establish a clear picture of the reaction. This is a result of the uncleaved TdU reporter producing more than twice the background fluorescence of the uncleaved TC reporter (Fig. 3). This is easily adjusted for but can also be avoided entirely if using the TC-ZEN and TdU-ZEN reporters.
Calculate the difference in RFU value between the TdU control and the TC control at each time point.
Subtract the value calculated in the previous step from the TdU reporter control and TdU + EndoQ samples. This should result in the total fluorescence of the fully cleaved TC reporter aligning with the total fluorescence of the fully cleaved TdU reporter as seen in figure Fig. 2A.
Fig. 3. Comparison of singly and doubly quenched reporters.
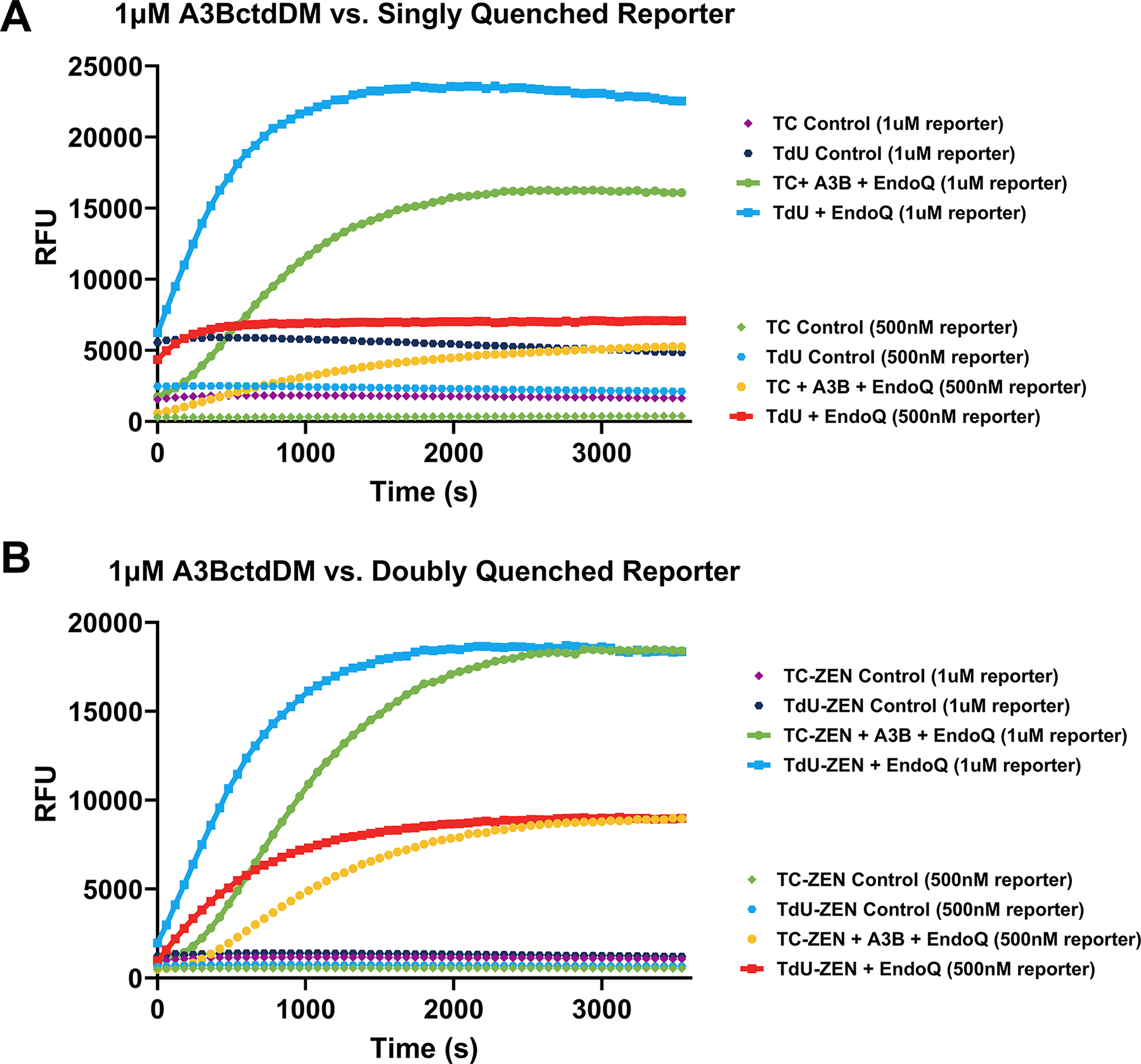
(A) A RADD assay comparing 500 nM and 1 μM standard, singly quenched, TC and TdU reporters without data processing. It is clear that the TdU control signal is significantly higher than that of the TC control. While this is easily corrected for, additional data processing is required after scanning is complete. (B) Depicts the same assay conditions as shown in A, using the doubly quenched TC-ZEN and TdU-ZEN reporters. Even without background correction, the signals from the controls are generally lower than the singly quenched reporters.
5. Determining Michaelis-Menten enzyme kinetics using the RADD assay
This section builds upon the technique described in Section 4 and uses a dilution series of fluorescent TC reporters to derive KM, Vmax, and kcat of A3BctdDM using the Michaelis-Menten model. This example compares 100 nM A3BctdDM versus 100 nM A3BctdWT and a 1/2 FAM-TC-IAB reporter dilution series ranging from 16 μM down to 1 μM. Before undertaking this protocol, it is important to establish the total signal (RFU) produced by reactions containing 1 μM TC reporter after complete reporter cleavage has occurred. To accomplish this, we recommend performing at least three replicates of the assay described in Section 4, ensuring enough enzyme is used to result in complete cleavage of the TC reporter by the end of the scan. You can then determine an average total RFU value of the processed reporter based on the last datapoint. This will allow for a straightforward translation of RFU/s rates to μM/s rates which will become important later in this protocol.
5.1. Equipment
Temperature-controlled multimode plate reader (e.g., TECAN Spark 10 M)
Black 96-well half-area plate (e.g., Corning #3993)
PCR Thermocycler (e.g., Bio-Rad T100 Thermal Cycler)
PCR strip tubes
8-Channel multichannel pipette 2–20 μL (e.g., Gilson PIPETMAN G Multichannel P8×20G)
8-Channel multichannel pipette 20–200 μL (e.g., Gilson PIPETMAN L Multichannel P8×200L)
ICEKAT software (Olp et al., 2020)
5.2. Buffers and reagents
10X RT reaction buffer (600 mM Tris-HCl pH 8.0, 0.1% Tween20, 10% DMSO, 10 mM DTT)
Enzyme storage buffer (20 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.5 mM TCEP)
RNase and DNase-free water (Invitrogen)
1 μM A3BctdDM or 1 μM A3BctdWT suspended in enzyme storage buffer. (See Section 3)
20 μM pfuEndoQ suspended in enzyme storage buffer. (See Section 3)
160 μM FAM-TC-IAB reporter suspended in water (Integrated DNA Technologies).
5.3. Procedure
5.3.1. Plate reader setup enzyme stock preparation
Plate reader setup and enzyme stock preparation step should be performed as described in 4.3.1 Plate reader setup, 4.3.2 Enzyme stock preparation. For this example, you will need at least 32 μL of 1 μM A3BctdDM and 32 μL 20 μM EndoQ.
5.3.2. Reporter dilution series
Due to the high concentration of reporter oligo utilized in this example, it is important to have a negative control for each reporter condition. As a result, all reporter dilutions should be prepared at a 3X volume. In a PCR tube strip, add 24 μL of 160 μM TC reporter oligo to tube 1 followed by the addition of 12 μL DNase/RNase-free water to tubes 2–5.
Remove 12 μL of the TC reporter oligo from tube 1 and combine with the water in tube 2 and ensure the dilution is completely mixed. Repeat this process for the remaining tubes until you have 12 μL of 160, 80, 40, 20, and 10 μM TC reporter oligo (Table 4).
Table 4.
Michaelis-Menten enzyme kinetics reporter dilution series.
| Tube no. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| DNase/RNase-free H2O | – | 12 μL | 12 μL | 12 μL | 12 μL |
| TC reporter | 24 μL (160 μM) | 12 μL (160 μM) | 12 μL (80 μM) | 12 μL (40 μM) | 12 μL (20 μM) |
| Final concentration | 160 μM | 80 μM | 40 μM | 20 μM | 10 μM |
5.3.3. Reaction preparation
In this example, you will have a total of 10 samples: 5 active samples and 5 negative controls. As a result, you should prepare an 11X volume reaction mixture. Combine 44 μL 10X RT reaction buffer and 264 μL of DNase/RNase-free water and ensure the components are thoroughly mixed (Table 5).
In two separate PCR tube-strips (reporter active and reporter control), aliquot 28 μL of the reaction mixture into tubes 1–5 of both active and control tube strips (Table 6).
Using a multichannel pipette, transfer 4 μL of your reporter dilution series into your active and negative control strips so that your highest concentration reporter is in tube #1 and your lowest concentration reporter is in tube 5. Mix well by pipetting up and down. Following this step, all 10 tubes should contain a final volume of 32 μL.
The active and negative control tube-strips should then be transferred to a thermocycler and heated to 60 °C for 10 min, followed by rapid cooling on ice for 5 min to ensure all DNA is single-stranded.
Next, on ice, prepare two additional 5-tube strips labeled enzyme(+) and enzyme(−). In tube #1–5 of the enzyme(+) tube-strip, combine 4.5 μL of 1 μM A3BctdDM and 4.5 μL of 20 μM EndoQ, ensuring the components are thoroughly mixed. In tubes #1–5 of the enzyme(−) tube-strip aliquot 9 μL of enzyme storage buffer (Table 7 & Table 8).
Transfer all four tube-strips reporter active, reporter control, enzyme(+), and enzyme(−) to a thermocycler and pre-heat to 37 °C for 5 min.
After pre-heating, use a multichannel pipette to transfer 8 μL from tubes #1–5 of the enzyme(−) tube-strip to the reporter control tube-strip and mix well, before transferring the full 40 μL samples into the pre-warmed 96-well plate.
Transfer 8 μL from tubes 1–5 of the enzyme(+) tube-strip to the reporter active tube-strip and mix well, before transferring the full 40 μL samples into the pre-warmed 96-well plate before immediately initiating scanning.
Table 5.
RT buffer master mix.
| 11X RT Buffer master mix | |
|---|---|
| 10X RT buffer | 44 μL |
| DNase/RNase-free H2O | 264 μL |
Table 6.
Reporter active and reporter negative tube-strips.
| Tube no. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Report dilution volume (reporter concentration) | 4 μL (160 μM) | 4 μL (80 μM) | 4 μL (40 μM) | 4 μL (20 μM) | 4 μL (10 μM) |
| RT buffer master mix | 28 μL | 28 μL | 28 μL | 28 μL | 28 μL |
Table 7.
Enzyme(+) mix.
| Enzyme (+) tube no. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 μM A3BctdDM | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL |
| 20 μM EndoQ | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL |
| Storage buffer | – | – | – | – | – |
Table 8.
Enzyme(−) mix.
| Enzyme (−) tube no. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 μM A3BctdDM | – | – | – | – | – |
| 20 μM EndoQ | – | – | – | – | – |
| Storage buffer | 9 μL | 9 μL | 9 μL | 9 μL | 9 μL |
5.4. Data processing
After scanning is complete, the reporter control data should be subtracted from the active reactions with respect to the reporter concentration to adjust for background signal.
Using ICEKAT, determine the maximum velocity of the enzyme at each substrate concentration by determining the maximum slope of the data for each condition. These slopes will be in units of relative fluorescence units per second (RFU/s).
To transform these slopes from RFU/s to μM reporter deamination per second (μM/s), you should divide the value of the slopes by the average RFU value of a 40 μL reaction containing 1 μM of fully processed TC substrate (see Section 5 introduction).
These data can then be plotted as μM substrate versus enzyme velocity to determine Km, Vmax, and kcat (Fig. 4)
Fig. 4. Fluorescence and gel-based readout of the RADD assay.
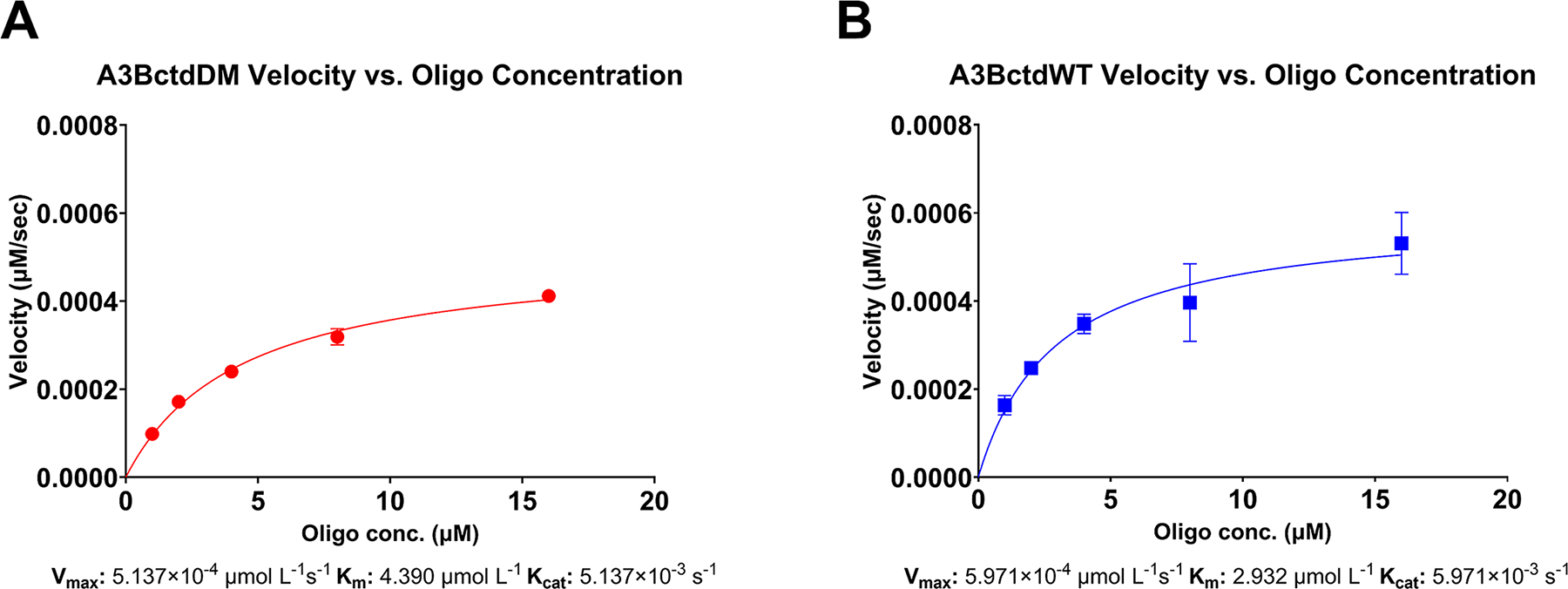
(A) Enzyme velocity of 100 nM A3BctdDM versus a ½ dilution series of 16 μM down to 1 μM reporter oligo (N = 3) fitted to the Michaelis-Menten model (R2 = 0.99). The predicted Vmax was 5.1397 × 10−4 μmol/L/s 95% CI [4.789 × 10−4–5.532 × 10−4], resulting in a Kcat of 5.137 × 10−3 s−1. The Km of A3BctdDM was predicted to be 4.390 μmol/L 95% CI [3.651–5.290]. (B) Enzyme velocity of A3BctdWT versus the same reporter oligo dilution series described above (N = 3), fitted to the Michaelis-Menten model (R2 = 0.87). The predicted Vmax for A3BctdWT was 5.971 × 10−4 μmol/L/s 95 % CI [5.051 × 10−4, 7.204 × 10−4], resulting in a Kcat of 5.971 × 10−3/s. The Km was predicted to be 2.932 μmol/L 95% CI [1.723, 4.943].
Figure adapted from Belica, C., Carpenter, M. A., Chen, Y., Moeller, N., Harris, R. S., & Aihara, H. (2024). A real-time biochemical assay for quantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry.
6. Rapid first-order rate constant determination using the RADD assay
This section describes an alternative method for estimating the deamination rate of A3B. While the Michaelis-Menten model can be used to identify a wider range of kinetic variables, the consecutive biochemical reactions model offers a more rapid determination of the first-order rate constant and may be a marginally more accurate estimation as it takes the consecutive nature of the RADD reaction into account. To accomplish this, the RADD assay can be performed in a single-turnover condition, using an A3B concentration well above the and in excess over the reporter substrate. Such conditions ensure that the majority of the substrate is bound to the enzyme at any given time, allowing to be approximately equivalent to . Equation 1 describes the consecutive biochemical reaction model. The variable represents the concentration of processed substrate at a given time (), while [] describes the concentration of unprocessed substrate at the beginning of the reaction. The variables and represent the first-order rate constants of A3B and EndoQ respectively. In this example, we performed the RADD assay using 5 μM A3BctdDM, 2 μM EndoQ, 1 μM FAM-TC-IAB and 1 μM FAM-TC-IAB.
| (1) |
6.1. Equipment
Temperature-controlled multimode plate reader (e.g., TECAN Spark 10 M)
Black 96-well half-area plate (e.g., Corning #3993)
PCR Thermocycler (e.g., Bio-Rad T100 Thermal Cycler)
PCR strip tubes
8-Channel multichannel pipette 2–20 μL (e.g., Gilson PIPETMAN G Multichannel P8×20G)
8-Channel multichannel pipette 20–200 μL (e.g., Gilson PIPETMAN L Multichannel P8×200L)
GraphPad Prism or comparable software
6.2. Buffers and reagents
10X RT reaction buffer (600 mM Tris-HCl pH 8.0, 0.1% Tween-20, 10% DMSO, 10 mM DTT)
RNase and DNase-free water (Invitrogen)
50 μM A3BctdDM suspended in enzyme storage buffer. (See Section 3)
20 μM EndoQ suspended in enzyme storage buffer. (See Section 3)
10 μM FAM-TC-IAB reporter suspended in water (Integrated DNA Technologies).
10 μM FAM-TdU-IAB reporter suspended in water (Integrated DNA Technologies).
6.3. Procedure
6.3.1. Plate reader setup and enzyme stock preparation
Plate reader setup and enzyme stock preparation step should be performed as described in 4.3.1 Plate reader setup, 4.3.2 Enzyme stock preparation.
6.3.2. Reaction preparation
Each 40 μL reaction will be comprised of 32 μL reporter master mix and 8 μL enzyme mix. In this example, there are a total of four reaction conditions (TdU reporter control, TC reporter control, TdU + EndoQ, and TC + A3B + EndoQ). All conditions in this assay should be performed in triplicate. As a result, a 7X volume reporter master mix is composed of 168 μL DNase/RNase free water, 28 μL 10X RT reaction buffer, and 28 μL 10 μM reporter for both TdU and TC reporter (Table 9). After combining the components, the master mixes should be vortexed to ensure homogeneity.
96 μL of the reporter master mixes should be aliquoted into four PCR tubes, two containing TdU master mix, and two containing TC master mix. These tubes should then be heated in a thermocycler at 60 °C for 10 min and rapidly cooled on ice for 5 min to ensure all DNA is in a linear, single-stranded form.
After preparing the reporter master mix tubes, a 24 μL enzyme mixture should be prepared for each condition in separate PCR tubes. In this example, the enzyme mixtures for the TC and TdU reporter controls will contain 24 μL of enzyme storage buffer. The enzyme mixture for the TdU + EndoQ mixture is composed of 12 μL enzyme storage buffer + 12 μL 20 μM EndoQ, and the enzyme mixture for the TC + A3B + EndoQ condition was composed of 12 μL 50 μM A3BctdDM and 12 μL 20 μM EndoQ.
Once the reporter master mix and enzyme mix tubes have been prepared, both sets of tubes should be heated in a thermocycler at 37 °C for 5 min to equilibrate the temperature of both halves of the reaction before mixing.
After the pre-heating step is complete, using a multichannel pipette, combine the 24 μL enzyme mixtures with the corresponding 96 μL reporter master mixtures. Following this step, mix thoroughly by pipetting before transferring 40 μL of each reaction condition to the pre-warmed plate as rapidly as possible. Repeat this for the remaining replicates, changing pipette tips after each transfer. Immediately initiate fluorescence scanning.
Table 9.
TC and TdU reporter master mixes.
| 7X TC reporter master mix | 7X TdU reporter master mix | |
|---|---|---|
| 10X RT buffer | 28 μL | 28 μL |
| 10 μM reporter oligo | 28 μL (TC) | 28 μL (TdU) |
| DNase/RNase-free H2O | 168 μL | 168 μL |
6.4. Data processing
The initial data processing in this section can be performed in Microsoft Excel or any comparable software. For the kinetic analysis portion of this section, we highly recommend GraphPad Prism as it will allow for the most streamlined determination of first-order rate constants.
Begin by adjusting for the additional background signal of the TdU reporter. To do this, subtract the difference between the TdU oligo control and the TC oligo control from the TdU + EndoQ data for each replicate (This step is not necessary if using the TdU-ZEN and TC-ZEN reporter oligos).
To remove all background signals from the data, subtract the values of the TC oligo controls from both the TdU + EndoQ and the TC + A3B + EndoQ data for each replicate. This will reduce the starting signal of the TC + A3B + EndoQ data to ~0. It should be noted that the TdU + EndoQ data starting signal will never reach 0. This observation is due to EndoQ immediately cleaving TdU substrate upon addition to the reaction and before plate reading can begin.
Next, the fluorescence data must be converted from RFU to μM processed substrate. To do this, divide the TdU + EndoQ reaction by the maximum RFU value from the TdU + EndoQ dataset for each replicate. Repeat this process for the TC + A3B + EndoQ reactions, dividing by the maximum RFU value from the TC + A3B + EndoQ from each dataset.
The first order rate constant for EndoQ will serve as the variable in the model equation. To calculate this, the values calculated in the previous step should be transferred into GraphPad Prism. The X values should be the time in seconds and the Y values should be μM processed TdU (in triplicate).
In GraphPad Prism 10, select Analyze -> Regression and Curves -> Non-linear Regression (Curve Fit) -> Model -> Exponential -> One-phase association. Assuming a good fit is achieved, the resulting K value will be the kcat for EndoQ (Fig. 5A).
To calculate (kcat of A3BctdDM), begin by transferring the TC + A3B + EndoQ data into GraphPad Prism. The X values should be the time in seconds and the Y values should be μM processed TC (in triplicate).
In GraphPad Prism, select Analyze -> Regression and Curves -> Non-linear Regression (Curve Fit) -> Model -> New.
In the menu at the top of this box, select “Explicit equation: Y = a function of X and parameters”. Enter Equation 1 as seen above into the text box. The rules for the A and variables can be input as 1 and 0 respectively, with the setting “Initial value, to be fit” for both. The A variable should be constrained to 1.0 as 1 μM is the initial concentration of the reporter oligo.
Finally, for the TC + A3B + EndoQ data, select Analyze -> Regression and Curves -> Non-linear Regression (Curve Fit) -> Model -> User-defined functions -> Your function. The value will represent the kcat of A3B (Fig. 5B).
Fig. 5. Estimating first-order rate constants using RADD and sequential enzymatic reaction modeling.
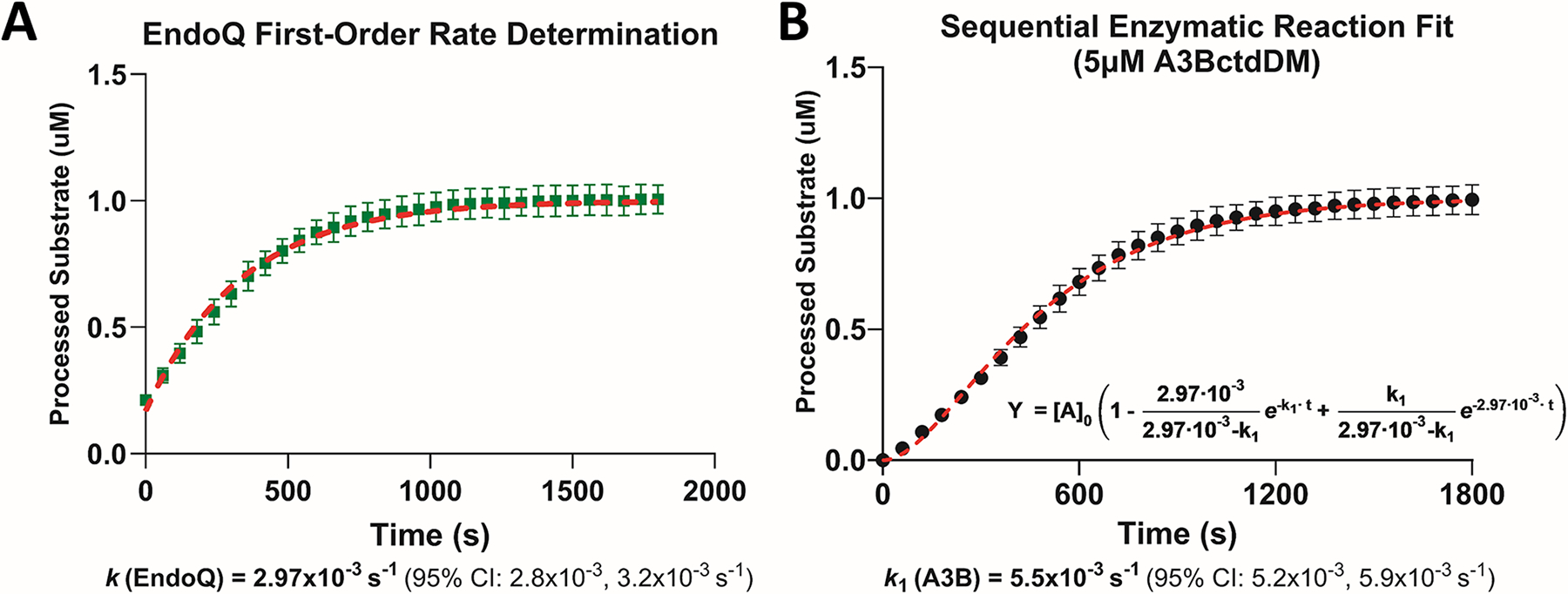
(A) Estimating the kcat of EndoQ by measuring the kobs of EndoQ in a single-turnover condition (N = 3). The kcat of EndoQ was determined to be 2.97 × 10−3/s 95% CI [2.8 × 10−3–3.2 × 10−3] (R2 = 0.97). (B) Estimating the kcat of A3BctdDM by measuring the deamination kobs at 5 μM A3BctdDM (N = 3). Using the kcat of EndoQ as k2 in the sequential enzyme reaction equation, the model showed an excellent fit to the A3BctdDM data (R2 = 0.98) and predicted a kcat of 5.5 × 10−3/s 95% CI [5.2 × 10−3–5.9 × 10−3/s].
Figure adapted from Belica, C., Carpenter, M. A., Chen, Y., Moeller, N., Harris, R. S., & Aihara, H. (2024). A real-time biochemical assay for quantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry.
7. Characterizing A3B inhibitors with the RADD assay
In this section, we will outline the steps required to evaluate potential inhibitors of A3B. In this example, we utilize the EBV large ribonucleotide reductase subunit BORF2 as a model inhibitor. In this example reaction, the activity of A3BctdDM was measured against a dilution series of BORF2 to determine how effective our assay was at detecting inhibition. This setup utilized 500 nM A3BctdDM, and was incubated in the presence of 78, 155, 310, 450, and 620 nM BORF2 (Fig. 6A).
Fig. 6. Measuring deaminase inhibition using the RADD assay.
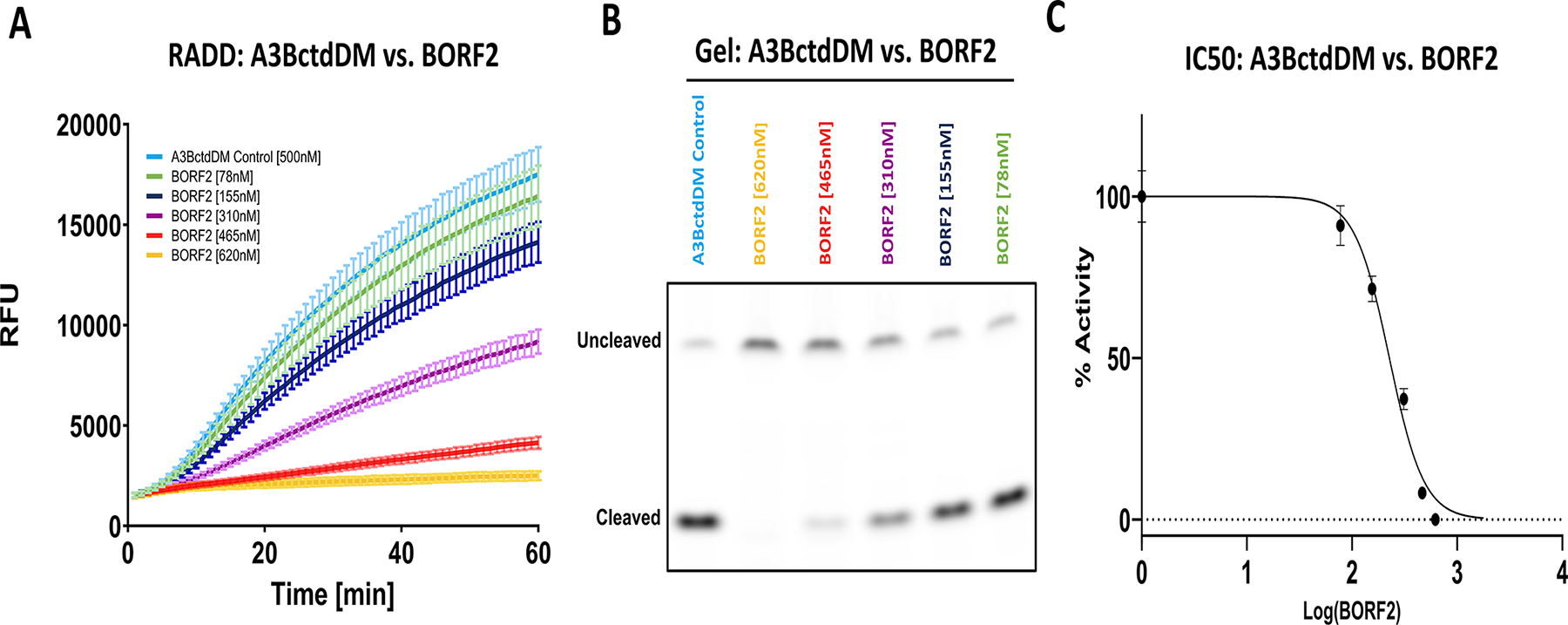
(A) The fluorescence readout of 500 nM A3BctdDM versus a dilution series of EBV BORF2 between 620 and 78 nM (N = 3). (B) The gel readout of the BORF2 inhibition assay confirms decreased substrate processing proportional to BORF2 concentration. (C) IC50 graph displaying the percent-activity of 500 nM A3BctdDM versus the concentration of BORF2 (R2 = 0.98). The top and bottom of the curve were constrained to 100% and 0%, respectively. The IC50 of BORF2 was predicted to be 226.4 nM, 95% CI [205.1–249.1].
Figure adapted from Belica, C., Carpenter, M. A., Chen, Y., Moeller, N., Harris, R. S., & Aihara, H. (2024). A real-time biochemical assay for quantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry.
7.1. Equipment
Temperature-controlled multimode plate reader (e.g., TECAN Spark 10 M)
Black 96-well half-area plate (e.g., Corning #3993)
15% TBE-Urea polyacrylamide gel (e.g., Invitrogen Novex gels)
Gel electrophoresis tank (e.g., ThermoFisher XCell SureLock Mini-Cell)
Power supply (e.g., Bio-Rad PowerPac)
Gel imaging instrument (e.g., Bio-Rad Gel Doc EZ, Amersham Typhoon imager)
PCR Thermocycler (e.g., Bio-Rad T100 Thermal Cycler)
PCR strip tubes
8-Channel multichannel pipette 2–20 μL (e.g., Gilson PIPETMAN G Multichannel P8×20G)
8-Channel multichannel pipette 20–200 μL (e.g., Gilson PIPETMAN L Multichannel P8×200L)
GraphPad Prism or comparable software
7.2. Buffers and reagents
10X RT reaction buffer (600 mM Tris-HCl pH 8.0, 0.1% Tween20, 10% DMSO, 10 mM DTT)
RNase and DNase-free water (Invitrogen)
Inhibitor storage buffer or solvent (variable)
5 μM A3BctdDM suspended in enzyme storage buffer. (See Section 3)
20 μM EndoQ suspended in enzyme storage buffer. (See Section 3)
6.2 μM BORF2 (See Section 3)
10 μM FAM-TC-IAB reporter suspended in water (Integrated DNA Technologies).
1X Tris-Borate-EDTA (TBE) running buffer.
Enzyme storage buffer (20 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.5 mM TCEP)
DNA loading buffer (95% Formamide, 0.025% Bromophenol blue, 0.025% Xylene cyanol, 5 mM EDTA).
Molecular biology grade formamide (e.g., RPI F11900–0.5)
7.3. Procedure
7.3.1. Plate reader setup and enzyme stock preparation
Plate reader setup and enzyme stock preparation step should be performed as described in 4.3.1 Plate reader setup, 4.3.2 Enzyme stock preparation, 4.3.3 Reaction mixture preparation.
7.3.2. Inhibitor dilution series preparation
High concentration inhibitors can result in higher background fluorescence, so it is important to have a negative control for each inhibitor condition. As a result, all inhibitor dilutions should be prepared at a 3X volume. In a PCR tube strip, create a dilution series ranging from highest in tube 1 to lowest in tube 5, and a no-inhibitor control (inhibitor storage buffer) in tube 6. Each tube should contain at least 12 μL of inhibitor or control buffer (Table 10).
Table 10.
Example BORF2 dilution series.
| Tube no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Storage buffer/solvent | – | 3 μL | 6 μL | 9 μL | 10.5 μL | 12 μL |
| BORF2 stock (6.20 μM) | 12 μL | 9 μL | 6 μL | 3 μL | 1.5 μL | – |
| Final concentration | 6.20 μM | 4.65 μM | 3.10 μM | 1.55 μM | 0.78 μM | 0 μM |
7.3.3. Reaction preparation
Each 40 μL reaction will be comprised of 28 μL reporter master mix and 12 μL enzyme mix. In this example, there are a total of 12 reactions, 6 containing TC reporter + A3B + EndoQ + BORF2 and 6 controls containing TC + BORF2. As a result, a 13X reporter master mix needed. This master mix is composed of 260 μL DNase/RNase-free water, 52 μL 10X RT reaction buffer, and 52 μL 10 μM TC reporter (Table 11). After combining the components, the master mix should be vortexed to ensure homogeneity.
The reporter master mix should be aliquoted into two separate PCR tube strips (active and control). Aliquot 28 μL of the reporter master mix into tubes 1–6 of the active tube strip. Repeat this process for the control tube strip.
These reporter tube strips should then be heated in a thermocycler at 60 °C for 10 min and rapidly cooled on ice for 5 min to ensure all DNA is in a linear, single-stranded form.
After preparing the reporter master mix tubes, a 13.5 μL enzyme mixture should be prepared for each condition. In this example we created two separate tube strips, one for the conditions containing A3B + EndoQ + BORF2 (enzyme(+)) and another for the controls containing only BORF2 (enzyme(−)). In tubes 1–6 of the enzyme(+) tube strip, combine 4.5 μL of 5 μM A3BctdDM, and 4.5 μL 20 μM EndoQ. In tubes 1–6 of the enzyme(−) tube strip add 9 μL of enzyme storage buffer.
Using a multichannel pipette, remove 4.5 μL from tubes 1–6 of the BORF2 dilution series and combine with tube 1–6 of the enzyme(+) tube strip (Table 12), ensuring the contents are mixed well. Repeat this process, this time adding BORF2 to tubes 1–6 of the enzyme(−) tube (Table 13).
Transfer the active and negative reporter mix tube strips as well as the enzyme(+) and enzyme(−) tube strips to a thermocycler. Heat at 37 °C for 5 min. This will equilibrate the temperature of the reaction components as well as provide an opportunity for the inhibitor to bind the A3B.
Using a multichannel pipette, transfer 12 μL from tubes 1–6 of the enzyme(−) strip to tubes 1–6 of the negative reporter mix strip. Mix the components thoroughly by pipetting then transfer to the pre-warmed 96-well plate.
Repeat the process from the previous step, this time combining 12 μL from tubes 1–6 of the enzyme(+) strip to tubes 1–6 of the active reporter mix strip. Mix the components thoroughly by pipetting then transfer to the pre-warmed 96-well plate.
Return the plate to the plate reader and immediately initiate scanning.
Table 11.
TC reporter master mix.
| Reporter master mix | 13X TC reporter master mix |
|---|---|
| 10X RT buffer | 52 μL |
| 10 μM TC reporter oligo | 52 μL |
| DNase/RNase-free H2O | 260 μL |
Table 12.
Enzyme(+) mix.
| Enzyme (+) tube no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 5 μM A3BctdDM | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL |
| 20 μM EndoQ | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL |
| BORF2 dilution | 4.5 μL (6.20 μM) | 4.5 μL (4.65 μM) | 4.5 μL (3.10 μM) | 4.5 μL (1.55 μM) | 4.5 μL (0.78 μM) | 4.5 μL (0 μM) |
| Enzyme storage buffer | – | – | – | – | – | – |
Table 13.
Enzyme(−) mix.
| Enzyme (−) tube no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 5 μM A3BctdDM | – | – | – | – | – | – |
| 20 μM EndoQ | – | – | – | – | – | – |
| BORF2 dilution | 4.5 μL (6.20 μM) | 4.5 μL (4.65 μM) | 4.5 μL (3.10 μM) | 4.5 μL (1.55 μM) | 4.5 μL (0.78 μM) | 4.5 μL (0 μM) |
| Enzyme storage buffer | 9 μL | 9 μL | 9 μL | 9 μL | 9 μL | 9 μL |
7.3.4. Gel-based readout
Once fluorescence scanning is complete, eject the plate from the plate reader and transfer 20 μL of each A3B-containing reaction to separate PCR tubes and halt the reaction with the addition of 40 μL formamide. These tubes should then be boiled at 95 °C for 5 min.
Following the boiling step, briefly allow the tubes to cool at room temperature before adding 80 μL of DNA loading buffer and mix by pipetting.
Prepare a 15% TBE-Urea polyacrylamide gel electrophoresis assembly and fill the chamber with 1X TBE. Using a pipette, remove excess urea from the wells by washing with 1X TBE from the chamber.
Slowly add 12 μL of each reaction condition to the wells of the gel, avoiding spillover and run the gel at 300 V for 35 min.
After the gel has been run, it can be imaged using any FAM-compatible fluorescence imaging instrument. Higher molecular weight bands represent uncleaved reporter while lower weight bands represent the cleaved product. In this example, the gel was imaged with an Amersham Typhoon imager using the built-in Cy2 method (Fig. 6B).
7.4. Data processing
Begin by subtracting the RFU values of the reporter controls from the corresponding A3B reactions containing the same concentration of BORF2. This will adjust for any unwanted background signal created by the inhibitor.
Using ICEKAT, determine the maximum slope of the curves for each inhibitor condition.
Create a data set where the first column is the concentrations of BORF ranging from 0, to the highest concentration and the second column is the slope of the data for that inhibitor condition.
Divide each slope by the value of the slope corresponding to the 0-inhibitor condition and multiply by 100 to convert your data into % activity.
Convert the inhibitor concentration values to log scale. For the 0-inhibitor concentration, take the log of 0.01.
Graph the data as log([inhibitor]) versus % activity. If using GraphPad Prism, the IC50 can be rapidly determined using the log (inhibitor) versus normalized response non-linear fit model (Fig. 6C).
8. Using the RADD assay to detect A3B activity in cell lysate
In this section, we describe the usage of the RADD assay to detect A3B activity in HEK293T lysate. As these cells do not endogenously express A3B at detectable levels (Jalili et al., 2020), the HEK293T were transfected with an HA-tagged full-length A3B expression vector A3B-HA_pcDNA3.1 (Hultquist et al., 2011). While the previous protocols described in this work allow for the usage of either the singly or doubly quenched TC and TdU reporter oligos, we have found that the doubly quenched oligos (TC-ZEN and TdU-ZEN) are better able to resist non-specific degradation and thus are crucial for this method. It should also be noted that the cell lysis buffer described here contains EDTA, which may have a negative effect on both A3B and EndoQ activity as they are both metal-dependent enzymes.
8.1. Equipment
Temperature-controlled multimode plate reader (e.g., TECAN Spark 10 M)
Black 96-well half-area plate (Corning #3993)
PCR Thermocycler (e.g., Bio-Rad T100 Thermal Cycler)
PCR strip tubes (e.g., Fisher Scientific 14–230-205)
8-Channel multichannel pipette 2–20 μL (e.g., Gilson PIPETMAN G Multichannel P8×20G)
8-Channel multichannel pipette 20–200 μL (e.g., Gilson PIPETMAN L Multichannel P8×200L)
Sonicator water bath (e.g., Brandson Ultrasonics CPX952116R).
Refrigerated table-top centrifuge (e.g., Eppendorf 5427R)
Tabletop nutating mixer (e.g., VWR nutating mixer)
100 mm cell culture dishes (e.g., Falcon 353003)
Cell culture incubator at 37 °C, 5% CO2 (e.g., Thermo Fisher Forma II Water Jacket)
8.2. Buffers and reagents
10X RT reaction buffer (600 mM Tris-HCl pH 8.0, 0.1% Tween20, 10% DMSO, 10 mM DTT)
RNase and DNase-free water (e.g., Invitrogen)
10 μM FAM-TC-ZEN-IAB reporter suspended in water (Integrated DNA Technologies).
10 μM FAM-TdU-ZEN-IAB reporter suspended in water (Integrated DNA Technologies).
A3B-HA_pcDNA3.1 vector
Empty pcDNA3.1 vector
1X Tris-Borate-EDTA (TBE) running buffer.
Enzyme storage buffer (20 mM Tris-HCl pH 7.5, 200 mM NaCl, 0.5 mM TCEP)
DNA loading buffer (95% Formamide, 0.025% Bromophenol blue, 0.025% Xylene cyanol, 5 mM EDTA)
Molecular biology grade formamide (e.g., RPI F11900–0.5)
HEK293T cells (ATCC)
TransIT-LT1 (MirusBio MIR2306)
Complete growth media (e.g., Gibco DMEM)
Serum-reduced media (e.g., Gibco Opti-MEM)
1X Phosphate-buffered saline with 0.1 mM EDTA
1X Phosphate-buffered saline without EDTA
Trypsin-EDTA (0.05%) (e.g., Gibco 25300054)
EDTA-free protease inhibitor tablets (e.g., Sigma-Aldrich 1187358000)
RNase A (e.g., Thermo Fisher 12091021)
HED Lysis buffer (25 mM HEPES pH 7.4, 15 mM EDTA, 10% glycerol)
8.3. Procedure
8.3.1. Cell culture and transfection
Add ~1.2 million cells in 15.5 mL complete growth media into two 10 cm cell culture dishes (A3B and vector control) and incubate overnight at 37 °C.
Combine 15 μg A3B-HA_pcDNA3.1 vector DNA with 1.5 mL serum-reduced media.
Combine 15 μg empty pcDNA3.1 vector DNA with 1.5 mL serum-reduced media.
Mix tubes gently by pipetting up and down.
Add 45 μL room-temperature TransIT-LT1 to each tube and mix via pipetting.
Incubate both tubes at room temperature for 30 min.
Add the TransIT-293 Reagent:DNA complexes to the corresponding plates drop-wise, evenly distributing the volume across the plates.
Incubate plates at 37 °C for 48 h.
Wash both plates with warm 1X PBS/EDTA to remove media.
Release cells with 1 mL pre-warmed Trypsin-EDTA.
Count cells and centrifuge at 1000 × g for 3 min.
Remove supernatant and wash cell pellets with 1 mL 1X PBS without EDTA.
Centrifuge at 1000 × g for 3 min and remove supernatant.
Store cells pellets at −20 °C until use. Freeze-thaw will also assist in the lysis process.
8.3.2. Cell lysis
Thaw cell pellets on ice.
Add protease inhibitor to the HED lysis buffer and invert until completely dissolved.
Dilute cells to ~70,000 cells/μL in chilled HED lysis buffer.
Lyse cells in sonicating water bath for 20 min.
Add RNase A to a final concentration of 100 μg/mL.
Incubate on a nutating mixer for 1 h at room temperature.
Clarify the lysate by centrifuging at 18,000 × g for 10 min at 4 °C
Remove and store the supernatant, taking care not to disturb the insoluble pellet.
Keep the supernatant on ice until use.
8.3.3. Plate reader setup and enzyme stock preparation
8.3.4. Lysate dilution preparation
In a PCR strip tube, add 9 μL A3B lysate to tube 1 and 4.5 μL HED lysis buffer to tubes 2–3. Serially dilute 4 μL A3B lysate from tube 1 into tubes 2 and 3 (Table 14).
In a new PCR tube-strip, repeat the process described in step 1, this time using the vector control lysate (Table 15).
Store both tube strips on ice.
Table 14.
A3B-expressing cell dilution series.
| A3B cell dilution tube no. | 1 | 2 | 3 |
|---|---|---|---|
| HED lysis buffer | – | 4.5 μL | 4.5 μL |
| A3B cell lysate | 9 μL | 4.5 μL (tube 1) | 4.5 μL (tube 2) |
Table 15.
Control cell dilution series.
| Control cell dilution tube No. | 1 | 2 | 3 |
|---|---|---|---|
| HED lysis buffer | – | 4.5 μL | 4.5 μL |
| Control cell lysate | 9 μL | 4.5 μL (tube 1) | 4.5 μL (tube 2) |
8.3.5. Reaction preparation
Prepare a 2X volume TdU-ZEN reporter master mix by combining 8 μL of the 10X RT reaction buffer with 8 μL of 10 μM TdU-ZEN reporter, and 48 μL of DNase/RNase free water (Table 16).
Prepare an 8X TC-ZEN reporter master mix by combining 32 μL of the 10X RT reaction buffer with 32 μL 10 μM TC-ZEN reporter, and 192 μL DNase/RNase free water (Table 16).
In a PCR tube strip, add 32 μL TC-ZEN master mix to tubes 1, and 3–8. Following this, add 32 μL TdU-ZEN master mix to tube 2. Tube 1 will be your TC-ZEN background control and tube 2 will be your EndoQ control. Tubes 3–8 will be your A3B / vector control dilution series.
Transfer tube strip to a thermocycler and heat to 60 °C for 10 min, followed by rapid cooling on ice.
To create the enzyme mixes, begin by adding 9 μL HED lysis buffer to tube 1, and 4.5 μL HED lysis buffer into tube 2. Next, add 4.5 μL 20 μM EndoQ to tubes 2–8.
Using a multichannel pipette transfer 4.5 μL of the A3B lysate dilution series to tubes 3–5 of the enzyme mix tube strip.
Repeat this process, this time transferring 4.5 μL of the vector control lysate dilution series to tubes 6–8 of the enzyme mix tube-strip. Following this step, all tubes in the enzyme mix strip should contain a total volume of 9 μL (Table 17).
Transfer the reporter master mix tube strip and enzyme mix strip to a thermocycler and heat at 37 °C for 5 min.
Using a multichannel pipette, transfer 8 μL from each tube of the enzyme mix strip to the corresponding tubes in the reporter master mix step. Ensure all tubes are thoroughly mixed.
Eject the pre-warmed 96-well plate from the plate reader and transfer the full reaction volumes to the plate.
Return the plate to the plate reader and immediately initiate scanning (Fig. 7A).
Table 16.
TC-ZEN and TdU-ZEN reporter master mixes.
| 2X TdU-ZEN reporter master mix | 8X TC-ZEN reporter master mix | |
|---|---|---|
| 10X RT buffer | 8 μL | 32 μL |
| 10 μM reporter oligo | 8 μL (TdU-ZEN) | 32 μL (TC-ZEN) |
| DNase/RNase-free H2O | 48 μL | 192 μL |
Table 17.
Cell lysate enzyme mixes.
| Tube no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| HED lysis buffer | 9 μL | 4.5 μL | – | – | – | – | – | – |
| 20 μM EndoQ | – | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL | 4.5 μL |
| Cell lysate | – | – | 4.5 μL (A3B) | 4.5 μL (A3B 1:2) | 4.5 μL (A3B 1:4) | 4.5 μL (Control) | 4.5 μL (Control 1:2) | 4.5 μL (Control 1:4) |
Fig. 7. Measuring A3B activity in HEK293T cell lysate.
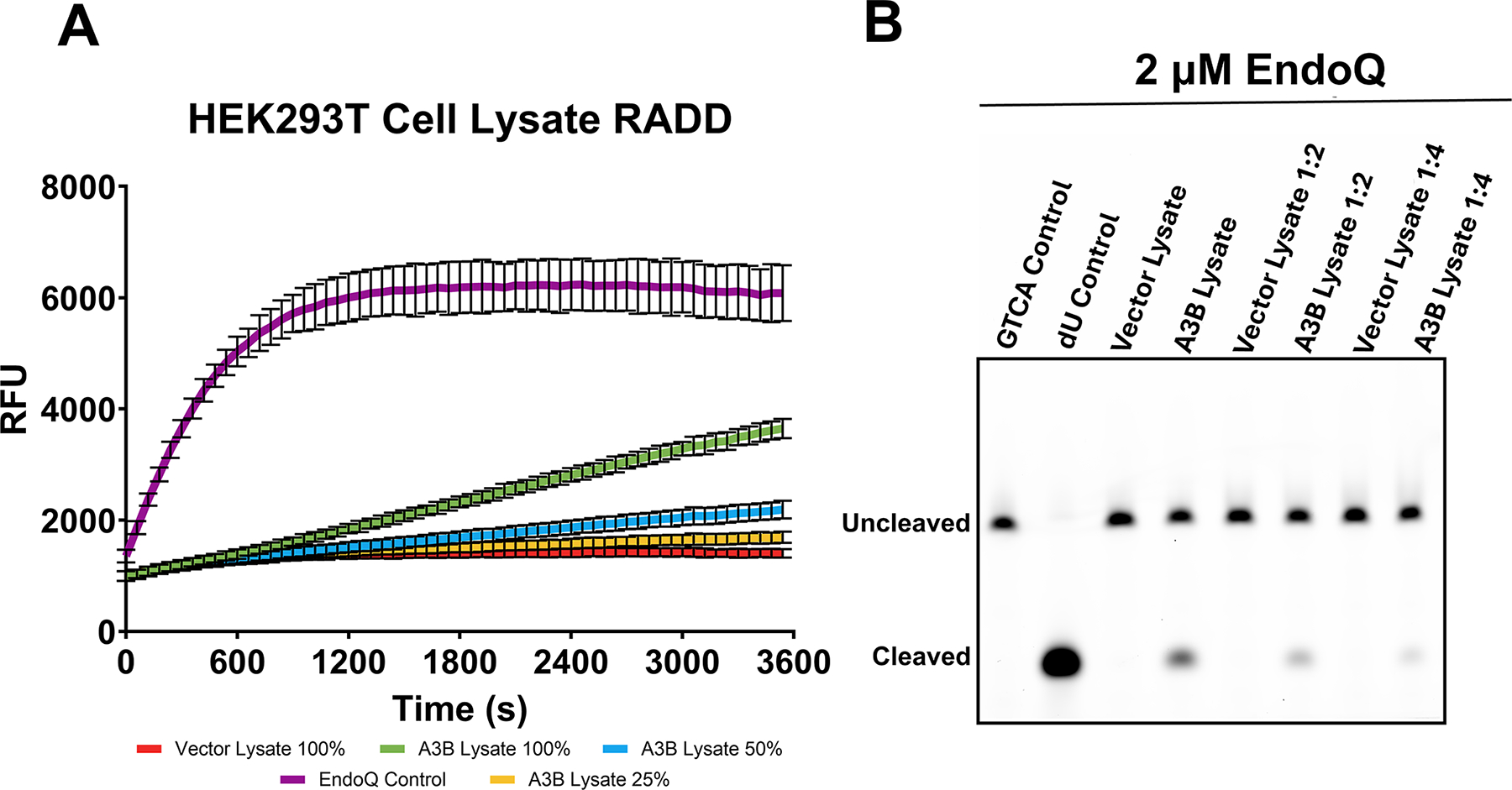
(A) Real-time deamination readout of reactions containing various dilutions of A3B-expressing human cell lysate and controls transfected with empty vector (N = 3 for each condition). The trace in purple represents the EndoQ control reaction containing 1 μM TdU-ZEN, 2 μM EndoQ, and lysis buffer. The green, blue, and yellow traces represent 1 μM TC-ZEN reporter in the presence of undiluted, 1:2, or 1:4-diluted A3B-expressing cell lysate, respectively. The red trace represents the negative control, containing undiluted control cell lysate. All reactions contained 2 μM EndoQ. (B) After fluorescence scanning, the reactions were run on a 15% TBE-Urea gel. None of the control cell lanes showed any substrate processing (lower band) while the A3B-containing samples showed decreased substrate processing in-line with increased dilution.
Figure adapted from Belica, C., Carpenter, M. A., Chen, Y., Moeller, N., Harris, R. S., & Aihara, H. (2024). A real-time biochemical assay forquantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry.
8.3.6. Gel-based readout
Once fluorescence scanning is complete, eject the plate from the plate reader and transfer 20 μL of each reaction to separate PCR tubes and halt the reaction with the addition of 40 μL formamide. These tubes should then be boiled at 95 °C for 5 min.
Following the boiling step, briefly allow the tubes to cool at room temperature before adding 80 μL of DNA loading buffer and mix by pipetting.
Prepare a 15% TBE-Urea polyacrylamide gel electrophoresis assembly and fill the chamber with 1X TBE. Using a pipette, remove excess urea from the wells by washing with 1X TBE from the chamber.
Slowly add 12 μL of each reaction condition to the wells of the gel, avoiding spillover and run the gel at 300 V for 35 min.
After the gel has been run, it can be imaged using any FAM-compatible fluorescence imaging instrument. Higher molecular weight bands represent uncleaved reporter while lower weight bands represent the cleaved product. In this example, the gel was imaged with an Amersham Typhoon imager using the built-in Cy2 method (Fig. 7B).
8.3.7. Data processing
In this example, very little data processing is required. If desired, background fluctuations can be removed by subtracting the TC-ZEN control data from all samples.
If desired, background signal produced by the vector lysate can also be removed by subtracting the vector control dilution values from the corresponding A3B conditions. In this example the data was left unadjusted to highlight the differences between the A3B containing reaction and the vector control reactions.
9. Conclusions
Here, using A3B as an example, we show that the RADD assay is a powerful tool for measuring DNA deaminase kinetics and inhibition in real-time. This assay benefits from single-step simplicity while still providing sensitive real-time data much more quickly than alternative biochemical or cell-based methods. Using the RADD assay, we have been able to detect A3B deamination activity in real-time, determine the kinetic parameters of A3B mutants, and quantify inhibition. The assay format is highly flexible, allowing for a wide variety of A3B-related use-cases. With its plate-based nature and its high Z′-factor (Belica et al., 2024, Zhang et al., 1999) the RADD assay also offers potential for usage in high-throughput screening. Based on the results shown here and in our previous work describing this assay, the RADD assay is likely to become a useful addition to the A3 biochemical toolbox (Grillo et al., 2022). Furthermore, sequences of the ssDNA reporters utilized in this assay can be modified, potentially offering uses for deaminases outside the APOBEC family, including the deluge of bacterial deaminase toxins with demonstrated utilities in a variety of applications (de Moraes et al., 2021, Huang et al., 2023, Vaisvila et al., 2024) and the evolved DNA adenine or cytosine deaminases developed for precision genome editing (Gaudelli et al., 2017, Neugebauer et al., 2023).
Acknowledgments
This work was supported by grants from the US National Institutes of Health, [P01CA234228] to R.S.H. and H.A and [R35GM118047] to H.A, and a Recruitment of Established Investigators Award from the Cancer Prevention and Research Institute of Texas CPRIT [RR220053] to R.S.H. C.B. was supported by the UMN Institute of Molecular Virology and the US National Institutes of Health T32 training program [T32AI083196]. R.S.H. is the Ewing Halsell President’s Council Distinguished Chair at University of Texas San Antonio and an Investigator of the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Barzak FM, Harjes S, Kvach MV, Kurup HM, Jameson GB, Filichev VV, & Harjes E (2019). Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2′-deoxyzebularine. Organic & Biomolecular Chemistry, 17, 9435–9441. [DOI] [PubMed] [Google Scholar]
- Belica C, Carpenter MA, Chen Y, Moeller N, Harris RS, & Aihara H (2024). A real-time biochemical assay for quantitative analyses of APOBEC-catalyzed DNA deamination. Journal of Biological Chemistry, 300, 107410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, & Harris RS (2013). Evidence for APOBEC3B mutagenesis in multiple human cancers. Nature Genetics, 45, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, ... Harris RS (2012). Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A*. Journal of Biological Chemistry, 287, 34801–34808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, ... Harris RS (2019). Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nature microbiology, 4, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moraes MH, Hsu F, Huang D, Bosch DE, Zeng J, Radey MC, ... Mougous JD (2021). An interbacterial DNA deaminase toxin directly mutagenizes surviving target populations. eLife, 10, e62967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee C, Temiz NA, Levin-Klein R, Argyris PP, Alsøe L, Carracedo S, ... Harris RS (2023). Human APOBEC3B promotes tumor development in vivo including signature mutations and metastases. Cell Reports Medicine, 4, 101211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A, Nagata T, Matsugami A, Habu Y, Sugiyama R, Hayashi F, ... Katahira M (2009). Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. The EMBO Journal, 28, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, & Liu DR (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature, 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo MJ, Jones KFM, Carpenter MA, Harris RS, & Harki DA (2022). The current toolbox for APOBEC drug discovery. Trends in Pharmacological Sciences, 43, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS (2015). Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Research, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Lin Q, Fei H, He Z, Xu H, Li Y, ... Gao, C. (2023). Discovery of deaminase functions by structure-based protein clustering. Cell, 186, 3182–3195.e14. [DOI] [PubMed] [Google Scholar]
- Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, & Harris RS (2011). Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1∇. Journal of Virology, 85, 11220–11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili P, Bowen D, Langenbucher A, Park S, Aguirre K, Corcoran RB, ... Buisson R (2020). Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nature Communications, 11, 2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvach MV, Barzak FM, Harjes S, Schares HAM, Jameson GB, Ayoub AM, ... Harjes E (2019). Inhibiting APOBEC3 activity with single-stranded DNA containing 2′-Deoxyzebularine analogues. Biochemistry, 58, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law EK, Levin-Klein R, Jarvis MC, Kim H, Argyris PP, Carpenter MA, ... Harris RS (2020). APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. The Journal of Experimental Medicine, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Shandilya SMD, Carpenter MA, Rathore A, Brown WL, Perkins AL, ... Harris RS (2012). First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chemical Biology, 7, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann JA, Argyris PP, Carpenter MA, Gupta HB, Chen Y, Temiz NA, ...Harris RS (2023). DNA deamination is required for human APOBEC3A-driven hepatocellular carcinoma in vivo. International Journal of Molecular Sciences, 24, 9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer ME, Hsu A, Arbab M, Krasnow NA, McElroy AN, Pandey S, ... Liu DR (2023). Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nature Biotechnology, 41, 673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olp MD, Kalous KS, & Smith BC (2020). ICEKAT: An interactive online tool for calculating initial rates from continuous enzyme kinetic traces. BMC Bioinformatics, 21, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M, Dananberg A, Chu K, Bergstrom EN, Striepen J, von Morgen P, ... Maciejowski J (2022). Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature, 607, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M, & Maciejowski J (2020). Molecular origins of APOBEC-associated mutations in cancer. DNA Repair, 94, 102905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaban NM, Yan R, Shi K, Moraes SN, Cheng AZ, Carpenter MA, ... Harris RS (2022). Cryo-EM structure of the EBV ribonucleotide reductase BORF2 and mechanism of APOBEC3B inhibition. Science Advances, 8, 2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K, Moeller NH, Banerjee S, McCann JL, Carpenter MA, Yin L, ... Aihara H (2021). Structural basis for recognition of distinct deaminated DNA lesions by endonuclease Q. Proceedings of the National Academy of Sciences of the United States of America, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi M, Ishino S, Yamagami T, Egashira Y, Kiyonari S, & Ishino Y (2015). A novel endonuclease that may be responsible for damaged DNA base repair in Pyrococcus furiosus. Nucleic Acids Research, 43, 2853–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielen BK, McNevin JP, McElrath MJ, Hunt BVS, Klein KC, & Lingappa JR (2010). Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. The Journal of Biological Chemistry, 285, 27753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisvila R, Johnson SR, Yan B, Dai N, Bourkia BM, Chen M, ... Sun Z (2024). Discovery of cytosine deaminases enables base-resolution methylome mapping using a single enzyme. Molecular Cell, 84, 854–866.e7. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TDY, & Oldenburg KR (1999). A simple statistical parameter for use in evaluation and validation of high throughput screening assays. SLAS Discovery, 4, 67–73. [DOI] [PubMed] [Google Scholar]