

Abstract
The discovery of new pain therapeutics targeting human nociceptive circuitry is an emerging, exciting, and rewarding field. However, current models for evaluating prospective new therapeutics [e.g., animals and two-dimensional (2D) in vitro cultures] fail to fully recapitulate the complexity of human nociceptive neuron and dorsal horn neuron biology, significantly limiting the development of novel pain therapeutics. Here, we report human spinal organoid-on-a-chip devices for modeling the biology and electrophysiology of human nociceptive neurons and dorsal horn interneurons in nociceptive circuitry. Our device can be simply made through the integration of a membrane with a three-dimensional (3D)-printed organoid holder. By combining air–liquid interface culture and spinal organoid protocols, our devices can differentiate human stem cells into human sensori-spinal-cord organoids with dorsal spinal cord interneurons and sensory neurons. By easily transferring from culture well plates to the multiple-electrode array (MEA) system, our device also allows the plug-and-play measurement of organoid activity for testing nociceptive modulators (e.g., mustard oil, capsaicin, velvet ant venom, etc.). Our organoid-on-a-chip devices are cost-efficient, scalable, easy to use, and compatible with conventional well plates, allowing the plug-and-play measurement of spinal organoid electrophysiology. By the integration of human sensory-spinal-cord organoids with our organoid-on-a-chip devices, our method may hold the promising potential to screen and validate novel therapeutics for human pain medicine discovery.
Graphical Abstract
1. INTRODUCTION
Chronic pain is widespread, affecting more than 20% of the population.1 Chronic pain is often associated with a variety of diseases such as diabetes, cancer, arthritis, inflammatory bowel disease (IBD), interstitial cystitis, temporomandibular joint dysfunction (TMJ), etc.,2–4 and there may be no apparent etiology. The clinical treatment of chronic pain is difficult, since commonly used pain therapies often have insufficient efficacy and suffer from many side effects such as liver toxicity, addiction, and person-to-person variability.5 For example, opioids are effective against acute pain but are often a poor choice to treat chronic pain due to their addiction liability and low efficacy.6 In many cases, currently available pain therapies were incidentally discovered during the development of drugs for a different condition, examples include biogenic amine reuptake inhibitors (e.g., antidepressants amitriptyline and duloxetine) or anticonvulsant drugs (e.g., gabapentin).7 There is an emergent and unmet medical need to develop chronic pain treatment with broader efficacy and better tolerability. One current approach by the pharmaceutical industry is to screen hundreds of thousands of compounds to identify a few candidates for lengthy, costly, and complex clinical evaluation before bringing a compound to market. Although the failure rate of new compounds is extremely high (e.g., about a 90% failure rate for these clinical candidates), there is clearly a need to discover new drugs for treating chronic pain. Thus, intensive efforts have been made to better understand pain biology and to explore new mechanisms and discover new drugs for chronic pain treatment. One attractive approach is to target the human nociceptive circuitry that receives input from peripheral neurons and processes these signals in the dorsal horn of the spinal cord. Abnormalities of this processing can lead to chronic pain after tissue damage or during a disease condition.8,9
So far, several approaches have been employed for pain biology research and the development of pain therapeutics. Rodents are the most common model to study pain mechanisms and discover new drug targets. However, rodents differ from humans in terms of genetics, pain etiology, and pain pathology.10,11 For example, chronic pain in temporomandibular disorder (TMD) is linked with human-specific epidermal growth factor receptor (EGFR) gene polymorphisms that are not reflected in mouse genetics.12,13 In addition, numerous potential chronic pain therapies have been developed that are efficacious in rodents but have failed in clinical trials.14,15 To fully capture human pain biology, human volunteers have been tested directly for translational pain medicine. However, human subject pain research is significantly limited due to experimental accessibility, moral concerns, and is not suitable for early-stage drug discovery studies.16,17 Thus, to address the gap between animal models and human volunteers, in vitro two-dimensional (2D) neural cultures have been developed for pain medicine. For example, 2D cultures of induced pluripotent stem cell (iPSC)-derived sensory neurons carrying sodium channel 1.7 (Nav1.7) mutations were generated to model one human chronic pain disorder (erythromelalgia).18 However, the current in vitro sensory neuron-based nociceptive models lack critical dorsal horn neurons in the spinal cord, which are key mediators of peripheral pain signal transmission.19–21 Hence, there is still a tremendous need to develop human cell culture models that can better recapitulate the complexity of human nociceptive circuitry.
Intensive efforts are underway to develop novel protocols to differentiate iPSCs into complex three-dimensional (3D) culture models (e.g., human brain and spinal organoids) that recapitulate the tissue complexity and neuronal electrophysiology of the human CNS.22,23 Pioneering human brain organoid models derived from human pluripotent stem cells (hPSCs) have been developed for mimicking human brain development and modeling neurodevelopmental disorders.24 Additionally, human spinal organoids have been generated from human pluripotent stem cells for mimicking human spinal cord tissues by restricting the direction of differentiation.25 Despite the successful use of these organoids in modeling neurodevelopmental disorders and neurodegenerative diseases, their potential in modeling human pain biology and discovering new pain medicine is largely unexplored due to several reasons: first of all, current human organoid cultures are highly heterogeneous and suffer from issues of standardization and scale-up, limiting their applications in high-throughput screening to better understand pain biology or to develop new therapeutics. Moreover, measuring electrophysiological parameters of human brain organoids is laborious and suffers from various difficulties (e.g., stemming from the lack of organoid–electrode contact, neuron death, or organoid detachment), making it difficult to obtain high-quality electrophysiological recordings from organoids.
To improve the throughput and standardization of human organoids and 3D in vitro cultures, pioneering engineering attempts have been made for the development of organoid-on-a-chip devices and systems.26–32 Droplet microfluidics and microgels have been employed for the massive fabrication of uniform human organoids and 3D cell spheroids such as islet and tumor organoids.33,34 Microfabricated perfusable devices have been reported for the production, growth, and differentiation of human brain organoids,35,36 as well as vascularization and maturation of kidney organoids.37 Moreover, perfusable 3D hydrogel-containing microfabricated devices have been used for the generation of functional human tubular mini-gut cultures.38 Recently, by employing 3D printing technology, our group has developed several organoid scaffolding and perfusion devices for the formation of standardized human organoids.39–44 Additionally, automated microfluidic systems have been developed for high-throughput generation and real-time imaging of large numbers of tumor or gastrointestinal organoids, highlighting their potential for drug screening.45–47 Although organoid-on-a-chip technologies significantly improve current human organoid models, the challenge remains in the culture and electrophysiological measurement of organoids for human pain biology and therapeutic development.
Here, to address the above challenges, we report organoid-on-a-chip devices that can standardize the culture and electrophysiological measurement of human dorsal horn spinal organoids for human nociceptive circuitry research. Our human spinal organoid-on-a-chip devices have several unique advantages: (1) the devices can be made through the simple integration of a 3D-printed scaffold with a porous membrane, obviating the need for complicated soft-lithography and polydimethylsiloxane (PDMS) fabrication processes. (2) The devices are easy to use and compatible with multiwell plates, allowing the generation of standardized organoids with enhanced perfusion of nutrients and oxygenation. (3) The devices can also be easily integrated with a multiple-electrode array (MEA) system for plug-and-play and reproducible electrophysiological measurements under multiplexed drug testing, avoiding the adherent culture of organoids onto the electrodes of the MEA. (4) The devices can better model the human pain biology by incorporating human stem cell-derived sensory-spinal-cord organoids with human dorsal spinal cord interneurons and sensory neurons. To test and validate our method, we modeled human responses to pain-evoking substances (e.g., mustard oil, capsaicin, and velvet ant venom) as well as nociceptive modulating drugs (e.g., protoxin II, tetrahydrocannabinol, THC, etc.).
2. EXPERIMENTAL SECTION
2.1. Human Embryonic Stem Cell Culture.
Human embryonic stem cells (WA09) were obtained from WiCell institute following the guidelines of both Indiana University and WiCell institute. WA09 cells were maintained on Matrigel (Corning)-coated six-well plates in mTESR plus medium (Stemcell Technologies) in a humidified CO2 incubator. Medium change was performed every other day. The WA09 cells were passaged every week using ReLeSR (Stemcell Technologies).
2.2. Assembly of Human Spinal Cord Organoids.
Human spinal cord organoids were fabricated by aggregation of ~9000 WA09 cells per EB using a 96-well spheroid microplate (Corning) as we previously described.48,49 Embryonic bodies (EBs) were formed in 100 μL Aggrewell EB formation medium (Stemcell Technologies) supplemented with 10 μM Y-27632 (SelleckChem). After the EB formation (Day 1), the medium was switched to the spinal cord medium I (ScM I) containing 3 μM CHIR-99021 (Stemcell Technologies) and 10 nM retinoic acid (RA) (Sigma-Aldrich). After 4 days of culture in ScM I, the medium was then switched to ScM II containing 5 ng/mL recombinant human BMP4 (Peprotech) and 10 nM retinoic acid to continue culture for 6 days. On day 10, spinal cord organoids were embedded in Matrigel (corning). The medium was switched to ScM III containing 10 μM N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenyl glycine t-butyl ester (DAPT) and kept for an additional of 8 days. During this period, spinal cord organoids were transferred to six-well ultralow attachment plates (Corning) held on an orbital shaker (Benchmark) set at 60 rpm. On Day 18, the medium was finally switched to ScM IV, containing 20 μg/mL ascorbic acid (Sigma-Aldrich) and 1 μM cyclic adenosine monophosphate (cAMP) (Sigma-Aldrich) for continuous culture on the orbital shaker. Unless specified, the medium was refreshed every other day during this process. The detailed medium composition can be found in the Supporting Information, Table S3.
2.3. Design and Fabrication of Organoid-on-a-Chip Devices.
The organoid devices were designed in the AutoCAD software. The dimension of the organoid device was 10 mm in diameter and 3 mm in height. The organoid devices were fabricated via our well-developed 3D printing protocol50 using a stereolithography 3D printer (Form 3B, Formlabs) and FormLabs Clear Resin V4.
2.4. On-Chip Culture of Organoids.
To grow organoid-on-a-chip, the polycarbonate membrane on-chip was first coated with poly-L-lysine (Sigma) overnight, which is followed by laminin (Sigma) (10 μg/mL in dI H2O) coating for 4 h. The organoids were then carefully transferred to the precoated membrane. We then load 500 μL of culture medium to fill the culture chamber. The organoids were then kept in culture on-chip for 3 days until they fully adhered to the membrane before any medium change or electric measurements.
2.5. On-Chip MEA Measurement of Organoids.
The organoids-on-a-chip were placed onto a 24-well Cytoview MEA plate (Axion Biosystems) with 500 μL of medium preloaded into the measuring well. The electric activities were measured by Axion Maestro Edge system in a 37 °C, 5% CO2 chamber. Substances were treated at following concentrations: capsaicin (Sigma-Aldrich) 10 μM, bicuculline (Sigma-Aldrich) 10 μM, MK-801 10 μM, velvet ant venome (1: 500 dilution), kindly provided by Dr. Dan Tracey’s lab at IU Bloomington, AITC 100 μM (Signa Aldrich), recombinant human BDNF (Peprotech) 100 nM, ProTx-II (Tocris Bioscience) 100 nM, Δ9-THC (Cerilliant) 10 μM. The electric signals were recorded at a sampling rate of 12.5 kHz. The spikes from the neurons were then detected using an adaptive threshold crossing algorithm, with 6 times standard deviation (STD) as threshold using the Axion Axis Navigator software. The spike file was then transferred to Axion Neural Metric Tool software for further analysis and raster plot visualization.
2.6. Cryo-Section of Organoids.
To section human spinal organoids, organoids were first washed twice with 1 × PBS (Gibco). They were then treated with 4% paraformaldehyde in PBS (Thermo Scientific) at 4 °C overnight. After fixation, the organoids were then washed twice with 1 × PBS again and cryoprotected overnight in 30% sucrose (w/v) at 4 °C. The organoids were then transferred to a cryomold (Sakura Finetek) and embedded in a Tissue Plus O.C.T compound (Fisher Scientific). The block was then sectioned on a cryostat (Leica) at 30 μm thickness.
2.7. Immunostaining of Organoids.
To characterize human spinal organoid cultures, sectioned samples were first washed twice with 1 × PBS. We then treated the samples with 2N hydrochloric acid (HCl) for 15 min for antigen retrieval. We then washed the samples twice with 1 × PBS and blocked the samples at room temperature for 1 h using homemade blocking buffer (0.3% Triton X-100, 5% normal goat serum in 1 × PBS). The samples were then treated with a primary antibody cocktail in a humidified chamber at 4 °C overnight. The sections were then washed 3 times with 1 × PBS, followed by secondary antibody treatment at room temperature for 1 h. The samples were finally washed and stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and cover-slipped with a gold antifade mounting medium (Invitrogen). Detailed antibody (commercial and made in house-CB1 antibody51) information can be found in the Supporting Information, Table S1.
2.8. Quantitative Polymerase Chain Reaction (qPCR) Analysis of Organoids.
To analyze the gene expression profile of human spinal organoids, we first washed organoids twice with 1 × PBS and extracted RNA using RNeasy Plus Mini Kit (Qiagen). The RNA was immediately reverse transcribed into complementary DNA using qScript cDNA synthesis kit (Quantabio). The cDNA was analyzed by qPCR using SYBR Green real-time PCR master mix (Thermo Fisher). Detailed qPCR primer sequences were described in detail in the Supporting Information, Table S2. The results were analyzed using the ΔΔCt method. As the delta Ct value was calculated by subtracting the target gene Ct value in organoid by the target gene Ct value in day 1 EB. The ΔCt was normalized against the housekeeping gene GAPDH. We repeated each qPCR reaction three times.
2.9. Statistical Analysis.
Statistical analysis was carried out using GraphPad Prism 7. Two sample groups were compared using the Students’ t-test. Three or more sample groups were analyzed using one-way ANOVA followed by Tukey’s post-hoc tests. Statistical significance was denoted as: *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001.
3. RESULTS AND DISCUSSION
3.1. Human Spinal Organoid-on-a-Chip to Model Nociceptive Circuitry.
In humans, the sensation of pain initiates in sensory neurons and is transmitted to the dorsal horn of the spinal cord. There they are integrated by dorsal horn interneurons, with the processed signals transmitted through dorsal horn projection neurons to the thalamus and, eventually, the brain cortex.19 To model the initial steps of the human nociceptive circuitry, we developed organoid-on-a-chip devices with the support of human sensory-spinal-cord-organoids with peripheral sensory neurons and spinal cord interneurons (Figure 1a). The organoid-on-a-chip device consists of a 3D-printed organoid holder and a porous polycarbonate membrane with 8 μm pores for the on-a-chip culture of human spinal organoids. Using our well-established air–liquid interface organoid culture condition,39 our organoid-on-a-chip devices can be used to generate human stem cell-derived sensory-spinal-cord organoids with human dorsal spinal cord interneurons and sensory neurons in well plates. Once neurons were mature in the human spinal organoids, our organoid-on-a-chip devices can be transferred into MEA plates for measuring their electrophysiological responses following treatment with different nociceptive modulators (Figure 1b).
Figure 1.
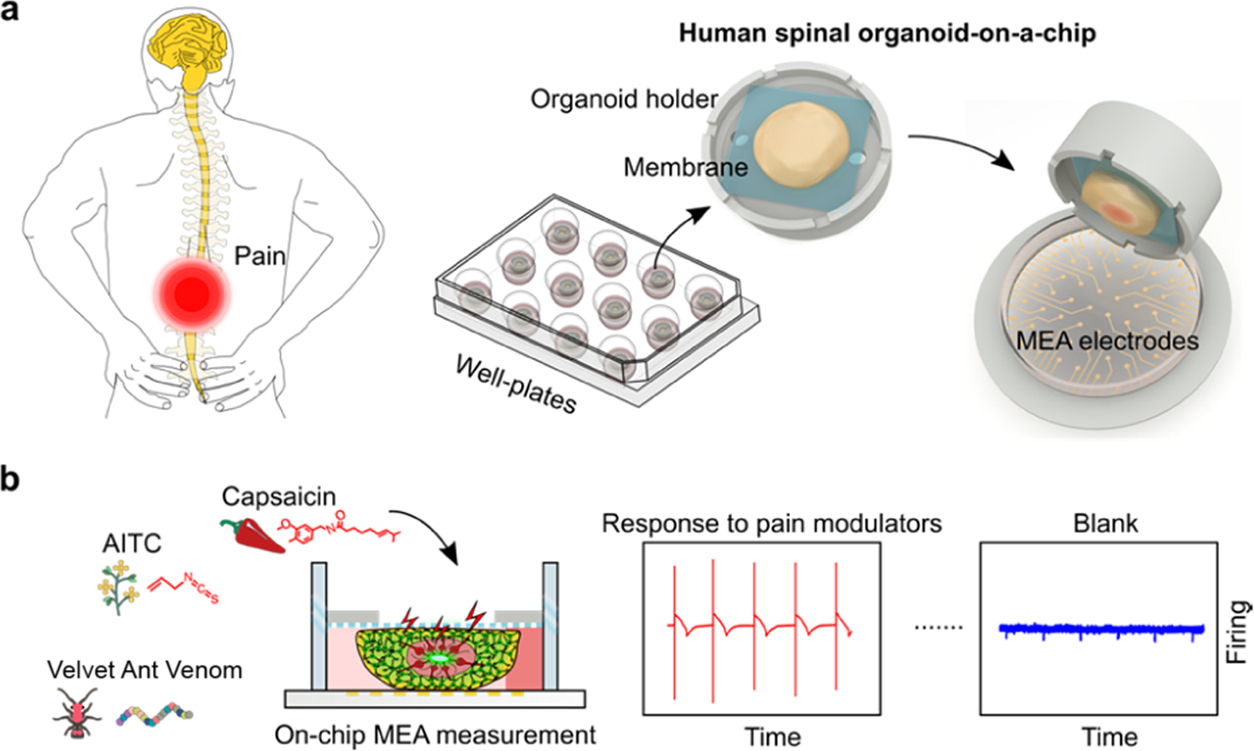
(a) Schematics of human spinal organoid-on-a-chip devices to model human nociceptive circuitry. (b) Illustration of on-chip evaluation of nociceptive stimulating/modulating chemicals using multiple-electrode array (MEA) measurement.
3.2. Development of Human Spinal Organoids.
The human spinal cord develops from the caudal end of the neural tube and specifies along the dorsal-ventral (D-V) axis. The human dorsal spinal cord gives rise to neural crest cells (NCC) that differentiate into sensory neurons and dorsal neural progenitors (dNP) that give rises to interneurons (dI neurons) that modulate sensory inputs and can affect the sensation of pain (Figure 2b). It is known that in vitro Wnt activation combined with retinoic acid (RA) treatment can induce spinal cord development from hPSCs. Treatment with BMP4 further promotes dorsalization of the spinal cord, facilitating dorsal spinal cord interneuron and sensory neuron development.25,52 Adopting these protocols, we developed a human sensory-spinal-cord organoid protocol by treating embryonic bodies (EBs) with GSK-3 inhibitor (CHIR-99021) and RA for 4 days, followed by RA and BMP4-directed dorsalization of the spinal cord for 6 days. From here, human spinal organoids were transferred to the organoid-on-a-chip devices for organoid attachment onto the porous membrane. The attached organoids were cultured under air–liquid interface culture conditions and treated with N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenyl glycine t-butyl ester (DAPT) to promote neuronal differentiation for 8 days. Finally, these organoids were exposed to ascorbic acid and cyclic adenosine monophosphate (cAMP) to further promote neuronal differentiation and maturation (Figure 2a).
Figure 2.
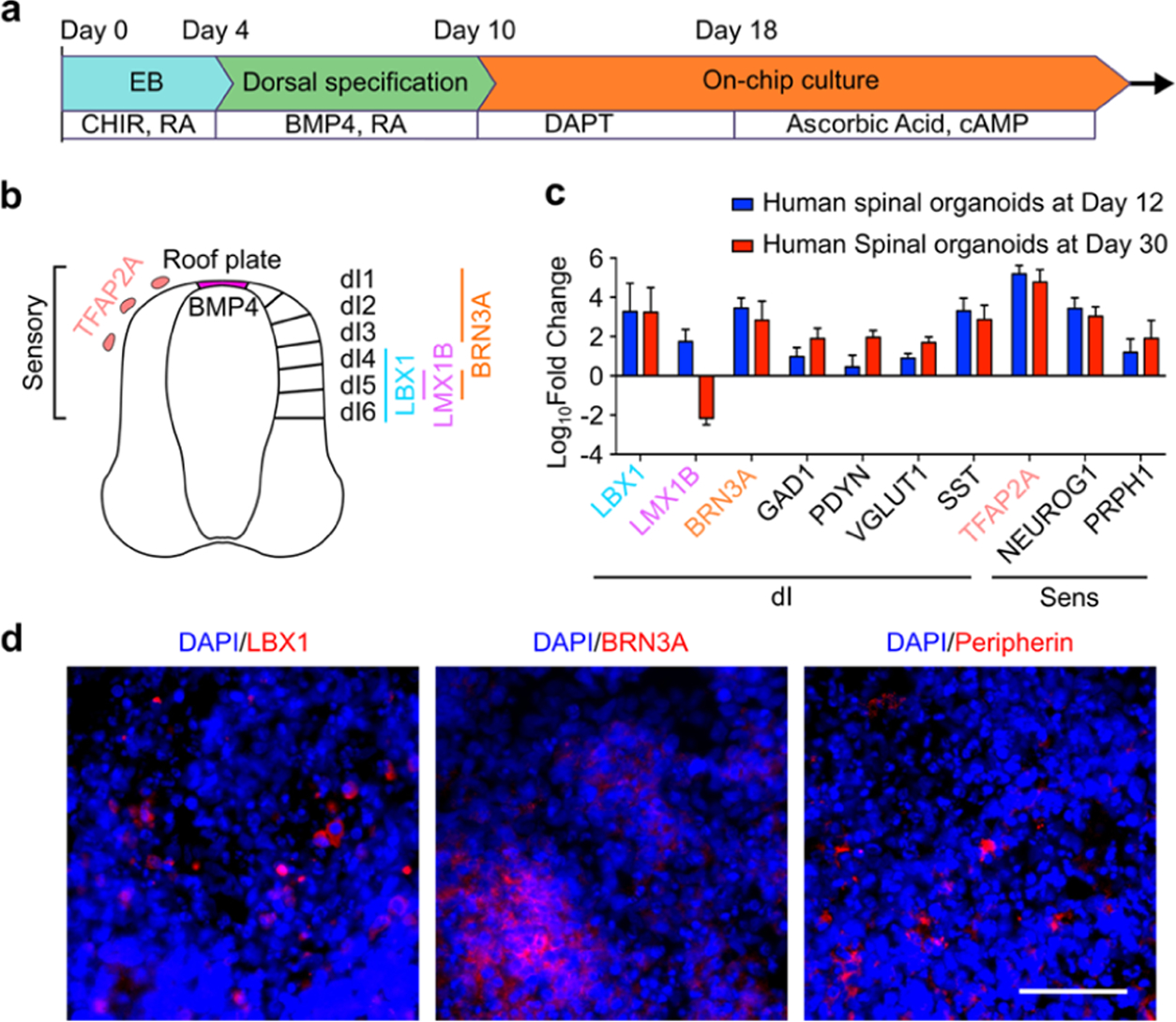
(a) Illustration shows the protocol and timeline of on-a-chip fabrication of human spinal organoids. (b) Illustration of dorsal spinal cord cell types including sensory neurons and interneurons and their corresponding markers. (c) qRT-PCR results of developing dorsal spinal cord sensory neurons and interneurons inside human spinal organoids at day 12 and day 30. (d) Immunofluorescence staining of dorsal spinal cord interneurons (marker: LBX1 and BRN3A) and sensory neurons (marker: PRPH1/peripherin) inside human spinal organoids. Scale bar: 50 μm.
3.3. Characterization of Human Spinal Organoids.
We analyzed the key gene signatures of dI neuron progenitors (dNP) as well as sensory neuron progenitors (NCC) by qRT-PCR. By day 12, key genes were expressed in human spinal organoids showing specification of dNP (LBX1, LMX1B, and BRN3A) as well as NCC (BRN3A, NEUROG1, and TFAP2A) (Figure 2b). After further neuronal differentiation and maturation, we analyzed these organoids again on Day 30. Here, the expression of dNP markers subsided, whereas functional dI neuron markers started to express, such as inhibitory GABAergic interneurons (GAD1) and excitatory glutaminergic neurons (vGlut1). More importantly, markers of subtypes of interneurons involved in modulating nociceptive signals, including somatostatin (SST) and dynorphin (Dyn), were abundantly expressed.53 Additionally, the mature peripheral neuron marker, peripherin (PRPH1), was also detected (Figure 2c). The expression of selected dI neuron and sensory neuron markers was further confirmed by immunofluorescence staining (Figure 2d).
3.4. Plug-and-Play MEA Measurement of Organoids.
To further validate our organoid-on-a-chip method, we also measured electrophysiological responses of on-chip cultured human spinal organoids (e.g., spontaneous firing). Traditionally, the electrophysiology of organoids can be measured by directly loading organoids onto MEA electrodes. However, this requires reducing the medium level to several tens of microliters to prevent organoids from floating, limiting the duration of the analysis. Additionally, the traditional measurements suffer from the limited active MEA electrode number and reproducibility since spheroidal-shaped organoids do not conform well with the flat MEA electrode. An alternative way to measure organoid electrophysiology is to adhere organoids to MEA electrodes for several days before the MEA measurement. However, the adherence success rate is variable and high-quality measurements may be difficult to obtain due to neuronal death when processes adhere and organoid detachment during medium change and drug dosing. Our organoid-on-a-chip devices can address these challenges and enable the plug-and-play and reproducible electrophysiological measurements without any adhesion and prolonged culture of organoids. Using our organoid-on-a-chip devices, on-chip cultured human spinal organoids can be easily transferred from the cell culture well plates to the MEA plates for plug-and-play MEA measurements (Figure 3a). We further compared MEA measurements of off-chip cultured human spinal organoids (or conventional organoids) and on-chip cultured human spinal organoids at days 1 and 7 (Figure 3b,c). We found that on-chip cultured human spinal organoids have more electrical activity and more active electrodes than off-chip cultured human spinal organoids. The better MEA measurements of our organoid-on-a-chip method may stem from the promotion of neuron growth and activity as well as the enhancement of organoid–electrode contact area through the on-chip air–liquid interface culture.
Figure 3.
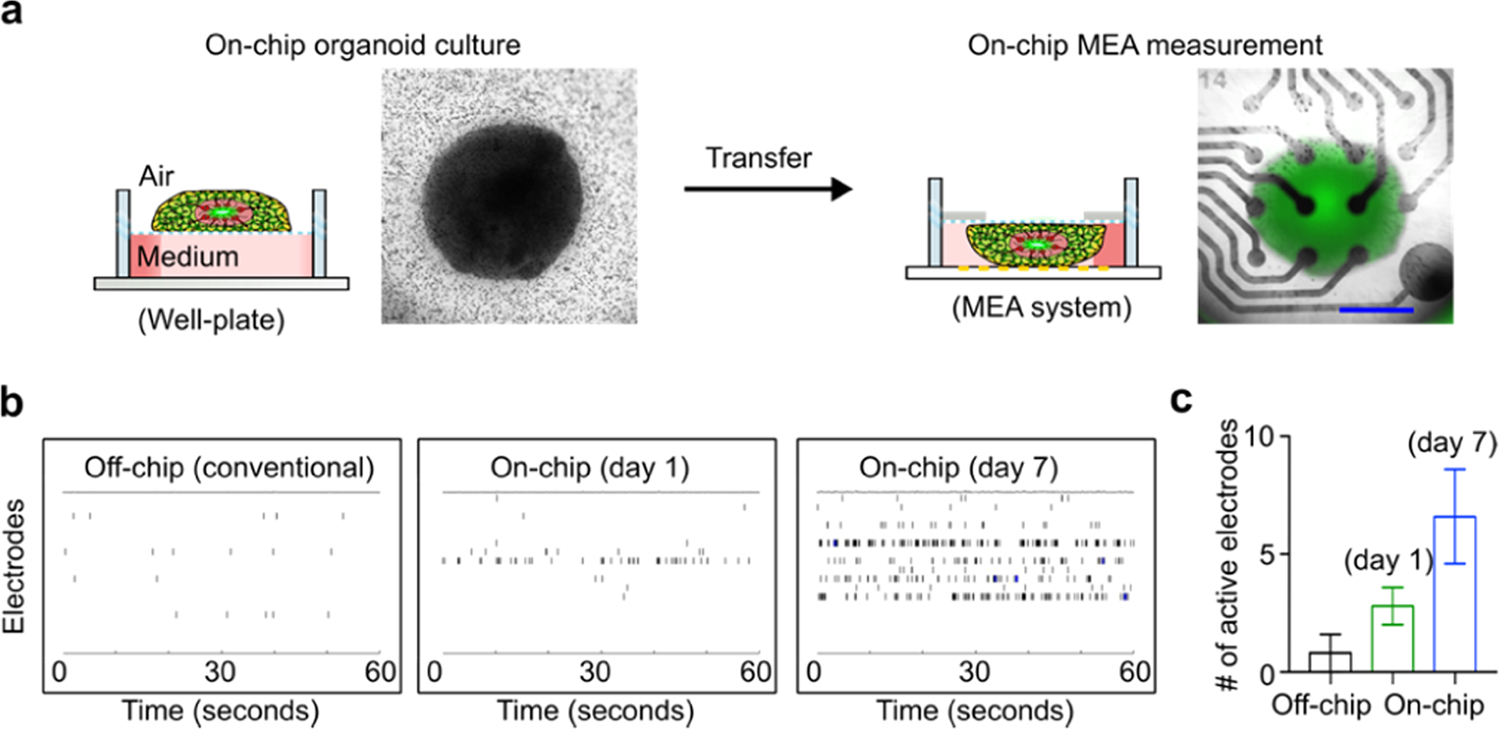
(a) Schematics describe the transferring of an on-chip cultured human spinal organoid for MEA measurement. The bright-field images show the cultured human spinal organoid on the organoid-on-a-chip device and the placement of the human spinal organoid within the device onto MEA electrodes. Representative raster plots (b) and active electrode number (c) of off-chip cultured human spinal organoids (or conventional organoids), on-chip cultured human spinal organoids at day 1, and on-chip cultured human spinal organoids at day 7. Scale bar: 500 μm.
3.5. Modeling Nociception.
To test human nociceptive responses using our method, we dosed human spinal organoids with common pain-evoking reagents. We found that AITC (mustard oil), a natural transient receptor potential ankyrin 1 (TRPA1) agonist, increases the mean firing rate (MFR) of human spinal organoids. Similarly, capsaicin, a transient receptor potential cation channel subfamily V member 1 (TRPV1) agonist, also increases the MFR of human spinal organoids. Additionally, we tested velvet ant venom, a natural venom known to cause extreme pain in humans, on our human spinal organoids and observed a prominent increase in neuronal firing of 156%, indicating the versatility of our model to evaluate pain-stimulating reagents with both well- and lesser-known mechanisms (Figure 4a). Furthermore, our human spinal organoid-on-a-chip responded appropriately to the γ-aminobutyric acid receptor (GABAR) antagonist bicuculline, as well as the N-methyl-D-aspartate receptor (NMDAR) antagonist MK-801, indicating the presence of functional inhibitory and excitatory ionotropic receptors, respectively. Modulation of excitatory or inhibitory ionotropic receptors by bicuculline and MK-801 also increased/decreased the responses of human spinal organoids to capsaicin, respectively (Figure 4b). Finally, thermosensitive ion channels have been implicated as key contributors to the human pain process. To test the response of our model to temperature change, we programmed a temperature increase from 37.0 to 42.0 °C in 30 s inside the MEA incubation chamber and analyzed the firing of our on-chip cultured human spinal organoids in 5 s bins. A temperature-dependent increase in human spinal organoids was observed, indicating its applicability in evaluating thermosensitive nociceptive pathways (Figure 4c).
Figure 4.
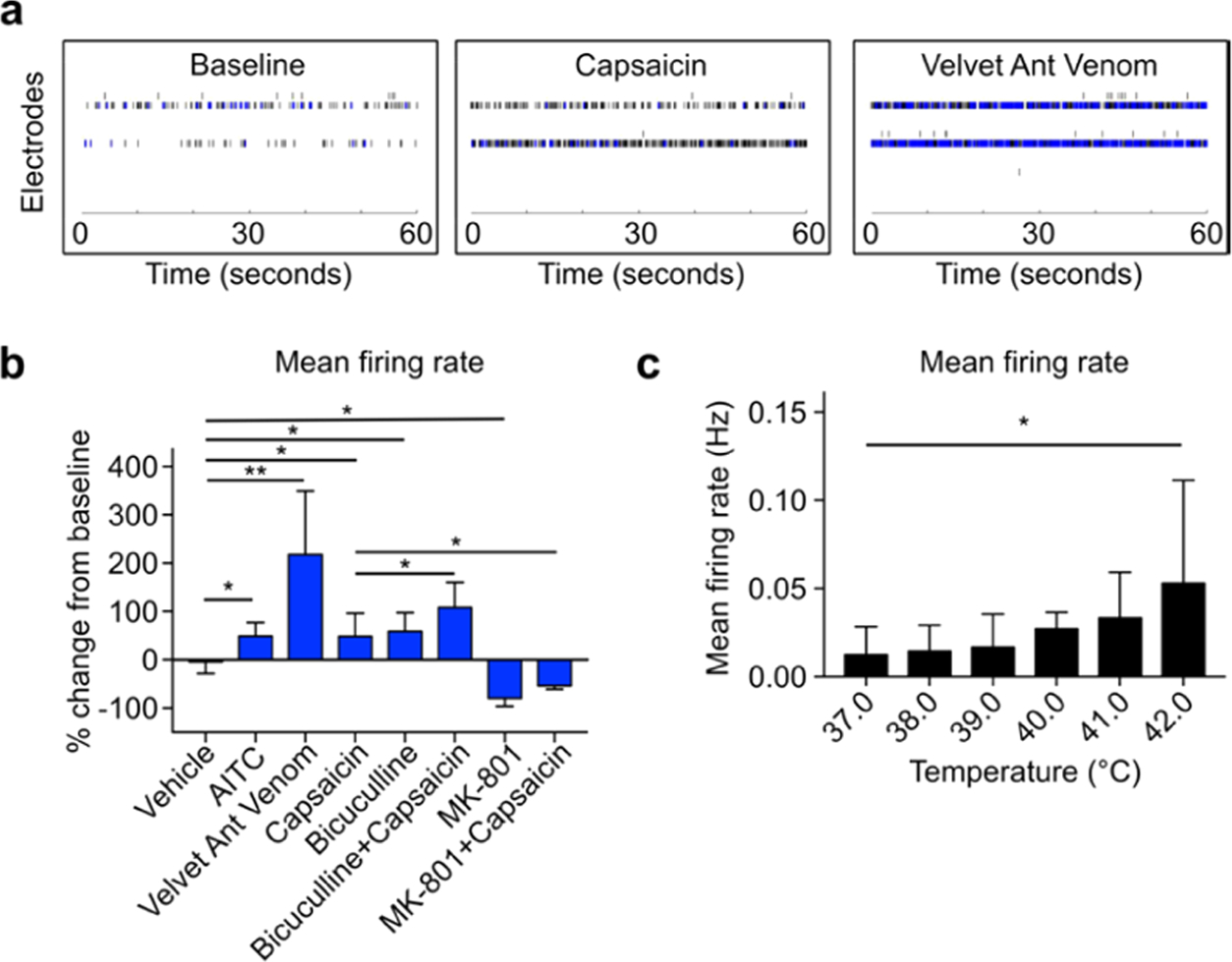
(a) Representative raster plots of on-chip cultured human spinal organoids at baseline and treated with capsaicin or velvet ant venom. (b) Mean firing rate change from baseline (percentage) by various pain stimulating and interneuron modulating reagents. (c) Mean firing rate change of on-chip cultured human spinal organoids as the bath temperature increased.
3.6. Testing Nociceptive Modulators.
To explore our model for investigating potential pain treatments, we tested various nociceptive modulators using our human spinal organoid-on-a-chip devices. Brain-derived neurotrophic factor (BDNF) has been shown to play central roles in both inflammatory and neuropathic pain.54 After treatment with BDNF for 30 min, on-chip human spinal organoids had a dramatic increase in mean firing rate (MFR, 380.8% increase). BDNF also augmented the responses of human spinal organoids to capsaicin (1071% increase in MFR). This indicated that our human spinal organoid-on-a-chip model could recapitulate in vivo features of nociceptive circuitry as observed in animals as well as human ex vivo tissue cultures. Additionally, pro-toxin II (ProTx-II), a selective sodium channel v1.7 (Nav1.7) blocker, was shown to be a promising alternative to opioids for pain relief. We tested whether dosing with ProTx-II could reduce neuron firing in human spinal organoids. Indeed, after ProTx-II dosing, the responses of human spinal organoids to capsaicin were completely abolished (−17.46% change in MFR in the ProTx-II + Capsaicin group versus +90.71% in the Capsaicin alone group). Finally, cannabinoids such as tetrahydrocannabinol (THC) have been indicated as promising candidates for alternative pain-relief drugs.55 To test our model with cannabinoid treatment, we first confirmed that neuron populations within our human spinal organoids expressed the CB1 receptor. Immunofluorescence staining revealed that human spinal organoid neurons widely express CB1 in a variety of neurons, including sensory (CGRP), excitatory (vGlut1), and inhibitory (GAD67) neurons, recapitulating in vivo results as seen in animal studies (Figure 5a).56–58 We then examined whether the CB1 agonist THC could be applied to modulate organoid responses to nociception. After dosing with THC, the MFR of human spinal organoids significantly decreased (−52.35%). Additionally, cotreatment of THC and capsaicin abolished capsaicin-elicited MFR increase (−22.31% in THC + Capsaicin group versus + 90.71% in capsaicin alone) (Figure 5b).
Figure 5.

(a) Immunofluorescence staining of CB1 expression in sensory neurons (CGRP+), inhibitory neurons (GAD67+), and excitatory (vGlut1+) neurons inside human spinal organoids. (b) Electrical responses of on-chip cultured human spinal organoids to nociceptive modulation reagents including BDNF, ProTx-II, and THC. Scale bar: 50 μm.
4. CONCLUSIONS
In summary, we have developed a novel human spinal organoid-on-a-chip method for modeling human nociceptive circuitry. Our 3D-printed organoid-on-a-chip devices are easy to fabricate, cost-efficient, and scalable, which facilitates oxygen and nutrient perfusion, and they can be readily integrated with a multiple-electrode array (MEA) system for plug-and-play and reproducible electrophysiological measurements. Additionally, we incorporated human stem cell-derived sensory-spinal cord organoids with human dorsal spinal cord interneurons and sensory neurons to better recapitulate the complexity of human nociceptive circuitry. We demonstrated the validity of our model by measuring their electrical responses to well-characterized pain-evoking drugs such as capsaicin and AITC, as well as a less-explored, naturally occurring pain-stimulating chemical: velvet ant venom. Additionally, we demonstrated the utility of our model to test novel, nonopioid pain-modulating drugs by measuring their responses to ProTx-II and THC. To conclude, our human spinal organoid chip serves as an easy, scalable, and cost-efficient model of human nociceptive circuitry, holding the promising potential to screen and validate novel drugs for treating pain.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the departmental startup funds of Indiana University Bloomington and the National Institute of Health Awards (R03EB030331 and DP2AI160242). The authors also thank the Indiana University Imaging Center for using their microscopes (NIH1S10OD024988).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c04641.
Additional tables and figures for characterization of viability and baseline firing rate of organoids-on-a-chip; antibody used in immunofluorescence staining; primer sequences for qPCR analysis; and human spinal organoid protocol medium compositions (PDF)
Contributor Information
Zheng Ao, Department of Intelligent Systems Engineering, Indiana University, Bloomington, Indiana 47405, United States.
Hongwei Cai, Department of Intelligent Systems Engineering, Indiana University, Bloomington, Indiana 47405, United States.
Zhuhao Wu, Department of Intelligent Systems Engineering, Indiana University, Bloomington, Indiana 47405, United States.
Jonathan Krzesniak, Department of Intelligent Systems Engineering, Indiana University, Bloomington, Indiana 47405, United States.
Chunhui Tian, Department of Intelligent Systems Engineering, Indiana University, Bloomington, Indiana 47405, United States.
Yvonne Y. Lai, Gill Center for Biomolecular Science, and Department of Psychological and Brain Sciences, Indiana University, Bloomington, Indiana 47405, United States
Ken Mackie, Gill Center for Biomolecular Science, and Department of Psychological and Brain Sciences, Indiana University, Bloomington, Indiana 47405, United States.
Feng Guo, Department of Intelligent Systems Engineering, Indiana University, Bloomington, Indiana 47405, United States.
REFERENCES
- (1).Bouhassira D; Lantéri-Minet M; Attal N; Laurent B; Touboul C Pain 2008, 136, 380–387. [DOI] [PubMed] [Google Scholar]
- (2).Bielefeldt K; Davis B; Binion DG Inflammatory Bowel Dis. 2009, 15, 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Feldman EL; Callaghan BC; Pop-Busui R; Zochodne DW; Wright DE; Bennett DL; Bril V; Russell JW; Viswanathan V Nat. Rev. Dis. Primers 2019, 5, No. 41. [DOI] [PubMed] [Google Scholar]
- (4).Kalso E; Edwards JE; Moore RA; McQuay HJ Pain 2004, 112, 372–380. [DOI] [PubMed] [Google Scholar]
- (5).Bosilkovska M; Walder B; Besson M; Daali Y; Desmeules J Drugs 2012, 72, 1645–1669. [DOI] [PubMed] [Google Scholar]
- (6).Manchikanti L; Helm S 2nd; Fellows B; Janata JW; Pampati V; Grider JS; Boswell MV Pain Physician 2012, 15, ES9–E38. [PubMed] [Google Scholar]
- (7).Moore RA; Wiffen PJ; Derry S; Rice AS Cochrane Database Syst. Rev. 2014, CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dubin AE; Patapoutian AJ Clin. Invest. 2010, 120, 3760–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Baccei ML Children 2016, 3, No. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gregory NS; Harris AL; Robinson CR; Dougherty PM; Fuchs PN; Sluka KA J. Pain 2013, 14, 1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Abboud C; Duveau A; Bouali-Benazzouz R; Massé K; Mattar J; Brochoire L; Fossat P; Boué-Grabot E; Hleihel W; Landry MJ Neurosci. Methods 2020, No. 108997. [DOI] [PubMed] [Google Scholar]
- (12).Martin LJ; Smith SB; Khoutorsky A; Magnussen CA; Samoshkin A; Sorge RE; Cho C; Yosefpour N; Sivaselvachandran S; Tohyama S; Cole T; Khuong TM; Mir E; Gibson DG; Wieskopf JS; Sotocinal SG; Austin JS; Meloto CB; Gitt JH; Gkogkas C; Sonenberg N; Greenspan JD; Fillingim RB; Ohrbach R; Slade GD; Knott C; Dubner R; Nackley AG; Ribeiro-da-Silva A; Neely GG; Maixner W; Zaykin DV; Mogil JS; Diatchenko LJ Clin. Invest. 2017, 127, 3353–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Khoutorsky A; Price TJ Trends Neurosci. 2018, 41, 100–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yekkirala AS; Roberson DP; Bean BP; Woolf CJ Nat. Rev. Drug Discovery 2017, 16, 545–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Middleton SJ; Barry AM; Comini M; Li Y; Ray PR; Shiers S; Themistocleous AC; Uhelski ML; Yang X; Dougherty PM; et al. Brain 2021, 144, 1312–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mouraux A; Bannister K; Becker S; Finn DP; Pickering G; Pogatzki-Zahn E; Graven-Nielsen T Eur. J. Pain 2021, 25, 731–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Katz N; Dworkin RH; North R; Thomson S; Eldabe S; Hayek SM; Kopell BH; Markman J; Rezai A; Taylor RS Pain 2021, 162, 1935–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mis MA; Yang Y; Tanaka BS; Gomis-Perez C; Liu S; Dib-Hajj F; Adi T; Garcia-Milian R; Schulman BR; Dib-Hajj SD; Waxman SG J. Neurosci. 2019, 39, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Peirs C; Seal RP Science 2016, 354, 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Peirs C; Williams S-PG; Zhao X; Walsh CE; Gedeon JY; Cagle NE; Goldring AC; Hioki H; Liu Z; Marell PS; et al. Neuron 2015, 87, 797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Petitjean H; Pawlowski SA; Fraine SL; Sharif B; Hamad D; Fatima T; Berg J; Brown CM; Jan L-Y; Ribeiro-da-Silva A; et al. Cell Rep. 2015, 13, 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lancaster MA; Knoblich JA Nat. Protoc. 2014, 9, 2329–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Trujillo CA; Gao R; Negraes PD; Gu J; Buchanan J; Preissl S; Wang A; Wu W; Haddad GG; Chaim IA; et al. Cell Stem Cell 2019, 25, 558–569.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chiaradia I; Lancaster MA Nat. Neurosci. 2020, 23, 1496–1508. [DOI] [PubMed] [Google Scholar]
- (25).Ogura T; Sakaguchi H; Miyamoto S; Takahashi J Development 2018, 145, No. dev162214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Park SE; Georgescu A; Huh D Science 2019, 364, 960–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Takebe T; Wells JM Science 2019, 364, 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sontheimer-Phelps A; Hassell BA; Ingber DE Nat. Rev. Cancer 2019, 19, 65–81. [DOI] [PubMed] [Google Scholar]
- (29).Yu F; Hunziker W; Choudhury D Micromachines 2019, 10, No. 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Velasco V; Shariati SA; Esfandyarpour R Microsyst. Nanoeng. 2020, 6, No. 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhao Y; Demirci U; Chen Y; Chen P Lab Chip 2020, 20, 1531–1543. [DOI] [PubMed] [Google Scholar]
- (32).Fan H; Demirci U; Chen PJ Hematol. Oncol. 2019, 12, No. 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liu H; Wang Y; Wang H; Zhao M; Tao T; Zhang X; Qin J Adv. Sci. 2020, 7, No. 1903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang H; Liu HT; Zhang X; Wang YQ; Zhao MQ; Chen WW; Qin JH ACS Appl. Mater. Interfaces 2021, 13, 3199–3208. [DOI] [PubMed] [Google Scholar]
- (35).Wang Y; Wang L; Zhu Y; Qin J Lab Chip 2018, 18, 851–860. [DOI] [PubMed] [Google Scholar]
- (36).Zhang M; Wang P; Luo R; Wang Y; Li Z; Guo Y; Yao Y; Li M; Tao T; Chen W Adv. Sci. 2021, 8, No. 2002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Homan KA; Gupta N; Kroll KT; Kolesky DB; Skylar-Scott M; Miyoshi T; Mau D; Valerius MT; Ferrante T; Bonventre JV; Lewis JA; Morizane R Nat. Methods 2019, 16, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nikolaev M; Mitrofanova O; Broguiere N; Geraldo S; Dutta D; Tabata Y; Elci B; Brandenberg N; Kolotuev I; Gjorevski N; Clevers H; Lutolf MP Nature 2020, 585, 574–578. [DOI] [PubMed] [Google Scholar]
- (39).Ao Z; Cai H; Havert DJ; Wu Z; Gong Z; Beggs JM; Mackie K; Guo F Anal. Chem. 2020, 92, 4630–4638. [DOI] [PubMed] [Google Scholar]
- (40).Cai H; Ao Z; Hu L; Moon Y; Wu Z; Lu H-C; Kim J; Guo F Analyst 2020, 145, 6243–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cai H; Ao Z; Wu Z; Song S; Mackie K; Guo F Lab Chip 2021, 21, 2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wu Z; Cai H; Ao Z; Xu J; Heaps S; Guo F Adv. Mater. Technol. 2021, 6, No. 2000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wu Z; Gong Z; Ao Z; Xu J; Cai H; Muhsen M; Heaps S; Bondesson M; Guo S; Guo F ACS Appl. Bio Mater. 2020, 3, 6273–6283. [DOI] [PubMed] [Google Scholar]
- (44).Chen B; Wu Y; Ao Z; Cai H; Nunez A; Liu Y; Foley J; Nephew K; Lu X; Guo F Lab Chip 2019, 19, 1755–1763. [DOI] [PubMed] [Google Scholar]
- (45).Schuster B; Junkin M; Kashaf SS; Romero-Calvo I; Kirby K; Matthews J; Weber CR; Rzhetsky A; White KP; Tay S Nat. Commun. 2020, 11, No. 5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Brassard JA; Nikolaev M; Hübscher T; Hofer M; Lutolf MP Nat. Mater. 2021, 20, 22–29. [DOI] [PubMed] [Google Scholar]
- (47).Brandenberg N; Hoehnel S; Kuttler F; Homicsko K; Ceroni C; Ringel T; Gjorevski N; Schwank G; Coukos G; Turcatti G; Lutolf MP Nat. Biomed. Eng. 2020, 4, 863–874. [DOI] [PubMed] [Google Scholar]
- (48).Cai H; Wu Z; Ao Z; Nunez A; Chen B; Jiang L; Bondesson M; Guo F Biofabrication 2020, 12, No. 035025. [DOI] [PubMed] [Google Scholar]
- (49).Ao Z; Cai H; Wu Z; Ott J; Wang H; Mackie K; Guo F Lab Chip 2021, 21, 688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wu Z; Gong Z; Ao Z; Xu J; Cai H; Muhsen M; Heaps S; Bondesson M; Guo S-S; Guo F ACS Appl. Bio Mater. 2020, 3, 6273–6283. [DOI] [PubMed] [Google Scholar]
- (51).Bodor ÁL; Katona I; Nyíri G; Mackie K; Ledent C; Hájos N; Freund TF J. Neurosci. 2005, 25, 6845–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Duval N; Vaslin C; Barata TC; Frarma Y; Contremoulins V; Baudin X; Nedelec S; Ribes VC Development 2019, 146, No. dev175430. [DOI] [PubMed] [Google Scholar]
- (53).Duan B; Cheng L; Bourane S; Britz O; Padilla C; Garcia-Campmany L; Krashes M; Knowlton W; Velasquez T; Ren X; Ross; Sarah E; Lowell; Bradford B; Wang Y; Goulding M; Ma Q Cell 2014, 159, 1417–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Obata K; Noguchi K Neurosci. Res. 2006, 55, 1–10. [DOI] [PubMed] [Google Scholar]
- (55).Johnson JR; Burnell-Nugent M; Lossignol D; Ganae-Motan ED; Potts R; Fallon MT J. Pain Symptom Manage. 2010, 39, 167–179. [DOI] [PubMed] [Google Scholar]
- (56).Mitrirattanakul S; Ramakul N; Guerrero AV; Matsuka Y; Ono T; Iwase H; Mackie K; Faull KF; Spigelman I Pain 2006, 126, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Yang F; Xu Q; Shu B; Tiwari V; He SQ; Vera-Portocarrero LP; Dong X; Linderoth B; Raja SN; Wang Y; Guan Y Pain 2016, 157, 2582–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sanudo-Pena M; Strangman N; Mackie K; Walker J; Tsou K Acta Pharmacol. Sin. 1999, 20, 1115–1120. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.