

Abstract
Plant viral infections cause significant economic losses, totalling $350 billion USD in 2021. With no treatment for virus-infected plants, accurate and efficient diagnosis is crucial to preventing and controlling these diseases. High-throughput sequencing (HTS) enables cost-efficient identification of known and unknown viruses. However, existing diagnostic pipelines face challenges. First, many methods depend on subjectively chosen parameter values, undermining their robustness across various data sources. Second, artifacts (e.g. false peaks) in the mapped sequence data can lead to incorrect diagnostic results. While some methods require manual or subjective verification to address these artifacts, others overlook them entirely, affecting the overall method performance and leading to imprecise or labour-intensive outcomes. To address these challenges, we introduce IIMI, a new automated analysis pipeline using machine learning to diagnose infections from 1583 plant viruses with HTS data. It adopts a data-driven approach for parameter selection, reducing subjectivity, and automatically filters out regions affected by artifacts, thus improving accuracy. Testing with in-house and published data shows IIMI’s superiority over existing methods. Besides a prediction model, IIMI also provides resources on plant virus genomes, including annotations of regions prone to artifacts. The method is available as an R package (iimi) on CRAN and will integrate with the web application www.virtool.ca, enhancing accessibility and user convenience.
Keywords: virus diagnosis, genome mappability, machine learning, read mapping, artifacts in genomic mapping, clean plant program
Introduction
Plant virus infection causes significant economic losses every year. It was reported that in 2014, these viruses resulted in a global crop production loss of around $30 billion [1]. By 2021, the crop production loss globally has increased to more than $220 billion annually [2, 3]. Since no treatments are available, a plant infected by a virus remains infected for the entirety of its life. Perennial crops can have long and productive lifespans if new planting material starts virus-free and methods are employed to detect and remove infected plants before a disease can spread. A reliable and cost-efficient virus diagnostic method is critical for the production and maintenance of virus-free crops in both nurseries and production fields.
In-field visual inspections are commonly utilized to detect a virus infection based on characteristic disease symptoms. Biological indexing is employed in specialized facilities for more in-depth analysis, requiring specific conditions and expert knowledge, and is notably time-consuming [4]. Technological advancements led to serological methods like Enzyme-Linked ImmunoSorbent Assay (ELISA), which is favoured for its cost-effectiveness in testing large sample volumes for a specific virus [5], and PCR method which is known for improved sensitivity and specificity, as well as its limited multiplexing capabilities [4]. However, both of these methods require knowledge of the virus to be detected, and their effectiveness is constrained by plant viruses’ diversity, mutation rates, and quasi-species [6, 7]. Unlike bioassays, ELISA and PCR are unable to detect a novel virus. High-throughput sequencing (HTS) has since emerged as a superior technology, offering the ability to quickly and cost-efficiently identify all known and novel viruses within a sample. HTS stands out for its comprehensive virus detection capabilities, making it an exceptionally effective tool for plant virus screening. This is especially beneficial for crops, where tests for many virus species are required.
Applying HTS in plant virus diagnostics involves three main steps. Since most viruses of interest have RNA genomes, extracting and enriching viral RNA is the first step after collecting plant tissue samples. Total RNA, ribodepleted total RNA, small interfering RNAs, or double-stranded RNA (dsRNA) extraction methods have been used for this purpose [8, 9]. The second step involves sequencing, predominantly using technologies like Illumina and Oxford Nanopore. Specific protocols are employed to convert the RNA into a form that can be sequenced and varies depending on the type of RNA extracted and the sequencing instrument. The third and final step is data analysis, which is critical for interpreting the information gleaned from sequencing. This step requires the building of analysis pipelines and data interpretation schemes.
Various data analysis pipelines have been developed in plant virus diagnostics using HTS technology, utilizing de novo assembly and read mapping strategies. Fastv is a specialized tool developed to identify SARS-CoV-2 [10], which causes COVID-19. It generates unique 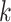 -mers (
-mers (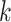 ranging from 3 to 32, with a default of 25) based on the virus’s reference genomes. The tool calculates the edit distance between the sample reads and these
ranging from 3 to 32, with a default of 25) based on the virus’s reference genomes. The tool calculates the edit distance between the sample reads and these 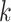 -mers to assess similarities. It is considered a match when the edit distance is less than a defined threshold. If the percentage of reads matched to a particular virus exceeds a predefined threshold, fastv indicates the virus’s presence. While fastv shows high sensitivity and specificity for detecting SARS-CoV-2, its parameters and thresholds are set explicitly for this virus, limiting its effectiveness in identifying plant viruses. VirHunter is a deep learning tool using convolutional neural networks to detect new and known viruses in HTS data [11]. VirHunter excels at identifying anomalies based on
-mers to assess similarities. It is considered a match when the edit distance is less than a defined threshold. If the percentage of reads matched to a particular virus exceeds a predefined threshold, fastv indicates the virus’s presence. While fastv shows high sensitivity and specificity for detecting SARS-CoV-2, its parameters and thresholds are set explicitly for this virus, limiting its effectiveness in identifying plant viruses. VirHunter is a deep learning tool using convolutional neural networks to detect new and known viruses in HTS data [11]. VirHunter excels at identifying anomalies based on 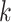 -mer matches to the host genome. However, its precision in identifying specific viruses is somewhat limited, as its published results indicate. PathoScope employs a Bayesian approach to address ambiguous mapping issues and quantify the abundance of sequences mapped to viral genomes [12]. Informed interpretation is required to determine the presence of infection based on the reported abundance by Pathoscope. Although these methods generally provide reasonable accuracy, they all share the common challenge of requiring subjective decision-making and human involvement, which can lead to inconsistent outcomes and a labour-intensive analysis process.
-mer matches to the host genome. However, its precision in identifying specific viruses is somewhat limited, as its published results indicate. PathoScope employs a Bayesian approach to address ambiguous mapping issues and quantify the abundance of sequences mapped to viral genomes [12]. Informed interpretation is required to determine the presence of infection based on the reported abundance by Pathoscope. Although these methods generally provide reasonable accuracy, they all share the common challenge of requiring subjective decision-making and human involvement, which can lead to inconsistent outcomes and a labour-intensive analysis process.
The Canadian Food Inspection Agency (CFIA) has created Virtool, a web application (available at https://www.virtool.ca/) designed to screen for plant viruses as part of a national clean plant program [13]. Currently, Virtool utilizes Bowtie 2 [14] for sequence mapping and Pathoscope [12] for quantifying the abundance of sequences that map to the virus genome. Analysts are tasked with interpreting these abundance reports and, at times, must investigate any unusually high peaks evident in the data visualizations. This multi-step process is not automatable and requires significant human intervention. The research and development described here are part of an ongoing initiative to refine and enhance Virtool. The goal is to simplify the process of plant virus detection used in regulatory and biosecurity contexts, focusing on minimizing the labour required from users and improving the reliability and accuracy of the diagnostic results.
Datasets
Two different datasets are used in this study. The first comprises 261 HTS-sequenced plant samples from a mixture of virus-infected grapevine and fruit trees [15]. The samples contain freeze-dried leaf or fresh leaf tissues stored at −80 C. The extracts are primarily dsRNA and total RNA with a mixture of paired- and single-end RNA-seq reads. The exceptions are 10 small RNA extracts of less than 25 base reads. All sequences were generated using Illumina sequencers in the FASTQ format. Part of this dataset was used for IIMI’s model training, while the rest was used to evaluate IIMI’s performance.
C. The extracts are primarily dsRNA and total RNA with a mixture of paired- and single-end RNA-seq reads. The exceptions are 10 small RNA extracts of less than 25 base reads. All sequences were generated using Illumina sequencers in the FASTQ format. Part of this dataset was used for IIMI’s model training, while the rest was used to evaluate IIMI’s performance.
The second dataset was published by Sukhorukov et al. [11] and accessible on the Recherche Data Gouv website (https://entrepot.recherche.data.gouv.fr/). This data set includes virus infection details for 12 samples across various species, such as grapevines [16], sugar beets [17], and peaches [18]. Two samples are omitted from the analysis due to the lack of publicly available viral genome sequences for the several viruses infecting them.
The virus reference genomes used in this study were sourced from Virtool’s virus database (version 1.4.0). Virtool’s virus database presents a curated collection of genomic data from the NCBI repository by Virtool’s developers. This database version contains 1583 viruses, encompassing 3183 virus segments, including viroids.
The genome of Arabidopsis thaliana (A. thaliana) with the taxonomy ID 3702 was obtained from Ensembl Plants and used in this study as a generic host. It is chosen because it has the most well-characterized plant genome and its concise and low-noise genetic structure. A host genome is instrumental in creating mappability profiles for virus genomes. Where a virus genome region matches a region in the A. thaliana genome, it becomes challenging to determine whether the reads mapped to this region are from the plant or the virus. Consequently, such regions are deemed unreliable for virus diagnostic purposes.
Method—IIMI
IIMI, an innovative and automated analysis pipeline utilizing machine intelligence for plant virus diagnostics, is presented in this section. IIMI comprises four main components, as depicted in Fig. 1. In Component (A), IIMI aligns raw sequencing reads against a database of plant virus genomes, in this case, taken from the Virtool virus database, version 1.4.0, with over 1563 virus genomes, transforming the consolidated HTS data of each sample into approximate coverage profiles for the genomes of all targeted viruses. Component (B) involves IIMI automatically filtering out unreliable regions in the coverage profiles, utilizing a prebuilt mappability profile and the proportions of nucleotides A, T, C, and G in all mapped reads. In Component (C), IIMI extracts features from these coverage profiles, which are crucial for determining whether a specific virus infects a sample. Finally, in Component (D), IIMI builds supervised machine-learning models based on the observed virus infection labels and the features extracted in the previous step. This machine learning model, refined over time, is intended to predict virus infections in future HTS samples. Further details of each component are discussed in the subsequent sections.
Figure 1.
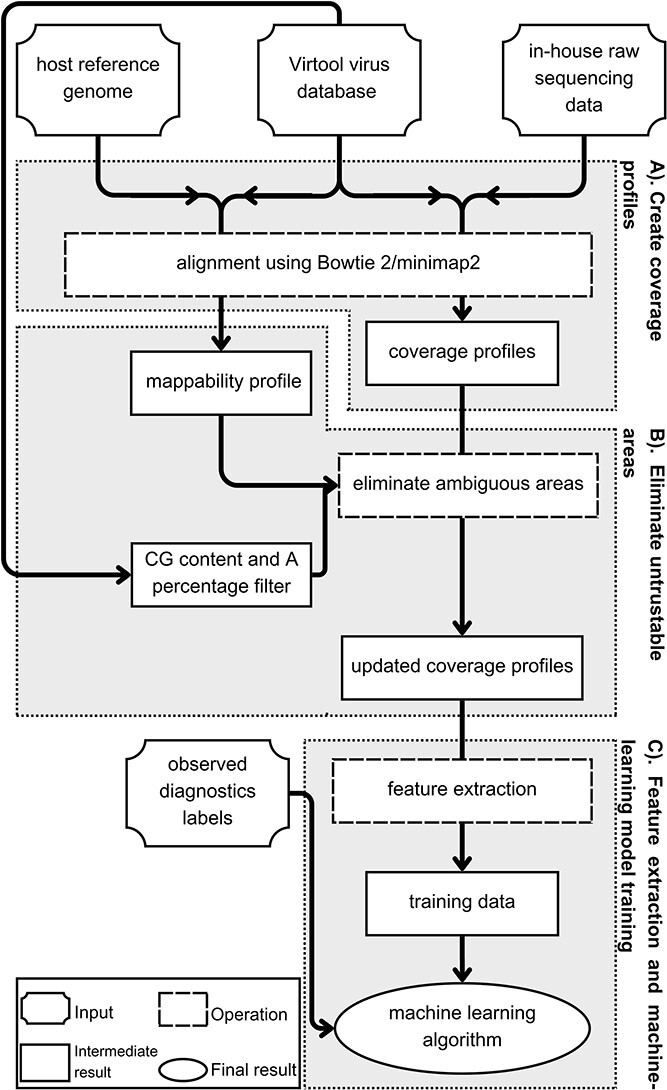
Architecture of IIMI analysis pipeline. The workflow of IIMI is separated into three components: Component (A). Create coverage profiles; Component (B). Eliminate untrustable areas; Component (C). Feature extraction and machine learning model training. Each component is illustrated in a box with a grey background. Inside the box, a diagram represents the workflow for that component.
Create coverage profiles (Component A)
While common approaches rely on identifying  -mer reads in a sample that match a reference virus genome or host genome, our method differs. Reads are mapped to virus reference genomes, generating coverage profiles for diagnosing infection with the corresponding virus.
-mer reads in a sample that match a reference virus genome or host genome, our method differs. Reads are mapped to virus reference genomes, generating coverage profiles for diagnosing infection with the corresponding virus.
A coverage profile is a numeric vector of physical coverage, representing the count of sequencing reads from an HTS sample aligned to each region or base of a virus reference genome. Regions with high sequence coverage, which is the number of unique reads mapped to a position, are characterized by more aligned reads, while those with low sequence coverage have fewer. The likelihood of a virus infecting a sample increases with more reads mapping to its genome. Thus, sequence coverage plays a pivotal role in developing IIMI because it carries information on the possibility of infection by a virus to differentiate between infected and non-infected plant samples, which helps determine what machine-learning features should be extracted.
Creating a coverage profile involves the following four steps: (1) converting reference virus genomes in the Virtool virus database from JSON to FASTA format; (2) mapping HTS reads from the sample to the converted reference genome using Bowtie 2 (version 2.4.4) [14] through employing the -a mode to report all alignments and outputting in SAM format; and (3) using SAMtools [19] to convert SAM output to BAM format, a binary version of SAM. This format includes detailed alignment information, such as each read’s location and mapping score and its alignment to the virus segment [20]. BAM files are also indexed for easy extraction of coverage profiles. Finally, (4) coverage profiles are extracted from the BAM file using the GenomicAlignments R package [21]. The final coverage profile is saved in a Run-Length Encoding (RLE) format, efficiently compressing data sequences without loss. These RLE-formatted coverage profiles are then ready for visualization or as inputs for machine-learning algorithms in diagnosing viral infections.
Figure 2 displays logarithmic-scale coverage profiles for HTS reads from infected and non-infected samples aligned to the genomes of four distinct viruses: Asian prunus virus 2 (APV2), Asian prunus virus 3 (APV3), Broad bean mottle virus (BBMV) RNA 3, and Fusarium poae virus 1 (FpV1) RNA 2. Only the infection with APV2 is confirmed in the plant. The figure illustrates that the coverage profile from an infected sample typically exhibits higher values than those from non-infected samples. Note, to illustrate the idea, coverage profiles visualized in Fig. 2 are selected to have apparent signals. Many other coverage profiles are less clear than those shown above, making diagnosing virus infection a non-trivial work.
Figure 2.

Log-scaled coverage profiles of four viruses from the same infected plant sample. The  -axis is the number of mapped reads in the logarithmic scale, and the
-axis is the number of mapped reads in the logarithmic scale, and the  -axis indicates the position of the virus segment. The red (thicker horizontal) lines at $y$ = 0 in the upper part of the panels indicate the ambiguous regions denoted by the mappability profile. The solid lines at the bottom represent the GC content and the percentage of nucleotides G and C. The dashed lines denote A%, the percentage of A nucleotide. Both GC content and A% are calculated for a series of 75-base
-axis indicates the position of the virus segment. The red (thicker horizontal) lines at $y$ = 0 in the upper part of the panels indicate the ambiguous regions denoted by the mappability profile. The solid lines at the bottom represent the GC content and the percentage of nucleotides G and C. The dashed lines denote A%, the percentage of A nucleotide. Both GC content and A% are calculated for a series of 75-base 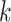 -mers and a step of 1.
-mers and a step of 1.
Automatically eliminate unreliable regions in coverage profiles (Component B)
In Fig. 2, false peaks in the coverage profiles are also evident. These peaks, which arise from factors such as genomic similarities or repetitive sequences, could lead to erroneous infection classifications if not addressed. The coverage profile in Fig. 2(a) corresponds to the known infection by APV2, with extensive mapping across most portions of the virus genome and relatively uniform GC content. Conversely, profiles in Fig. 2(b–d) correspond to viruses not identified in the sample, showing sparse coverage except for sporadic peaks. Specifically, Fig. 2(b) highlights an area in red, pinpointed through mappability profiling as ambiguous due to similarity with other viruses or the host genome. The peak in Fig. 2(c) aligns with an increase in ‘A’ nucleotides, while the peak in Fig. 2(d) aligns with a rise in GC content. These misleading peaks must be removed from the initial coverage profiles to reduce false positives.
Component B is designed to automatically eliminate unreliable regions from the coverage profiles, functioning without manual intervention. Effectively eliminating bad signals prior to constructing a predictive model is a straightforward yet more powerful strategy, offering more advantages than creating intricate machine learning methods to handle such noise. This preliminary step can greatly enhance the accuracy and efficiency of virus diagnostics. Identifying unreliable regions relies on three pieces of precalculated information for each virus reference genome: (1) the proportions of adenine (A), (2) combined cytosine-guanine (CG) contents, and (3) mappability profiles. Next, it is important to explain why these factors can compromise the reliability of coverage profiles in certain genomic regions and describe our approach to automatically identifying them.
Coverage profiles in regions with a high concentration of repetitive adenine (A) sequences or elevated GC content are deemed unreliable, as highlighted by several studies in the field. This unreliability stems from various challenges in sequencing and data analysis. Difficulties in aligning these repetitive sequences can lead to misalignments or coverage gaps [22]. Additionally, interpreting long sequences of a single nucleotide is complicated and often inaccurate [23]. In regions with repetitive A’s or strong GC bonding, PCR amplification, a standard step in sequencing preparation, may be biased due to secondary structure formation, which could inhibit DNA polymerase activity [24] or interfere with sequencing [25]. Specific limitations of sequencing technologies, like determining run lengths in homopolymeric sequences for Illumina or base calling accuracy for Oxford Nanopore, add to these challenges [26]. Mononucleotide repeats are particularly susceptible to sequencing errors, resulting in incorrect insertions or deletions [27]. Furthermore, GC-rich areas will likely encounter sequencing issues, typically manifesting as lower coverage or erroneous reads [28].
A sliding window technique was employed to identify unreliable regions stemming from high percentages of A nucleotide and GC content. A sliding window of 75 bases was employed for each virus reference genome, moving in increments of 1 base across the genome. Windows are labelled as unreliable when the percentage of A or GC content exceeds a set threshold. After completing the scan, all overlapping unreliable windows are consolidated to define the final unreliable regions. This process results in a two-column matrix for every reference virus genome, where each row corresponds to an unreliable region, and the columns indicate the start and end positions of these regions. The threshold value to define a high percentage is 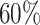 for GC content and
for GC content and 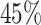 for A percentage, which is empirically selected and will be activated and updated using new samples generated in the near future. These threshold values are similar to the ones already used in the past [29].
for A percentage, which is empirically selected and will be activated and updated using new samples generated in the near future. These threshold values are similar to the ones already used in the past [29].
Ambiguous mapping in genomic sequencing, a challenge stemming from the repetitive nature and similarity between regions of different genomes, significantly impacts the accuracy of coverage profiles. In this scenario, a single read might align to multiple sites across various genomes, leading to uncertainty in pinpointing its true origin. Such uncertainty can result in either overestimating or underestimating coverage in specific genomic regions [30, 31]. Beyond affecting the reliability of coverage profiles, this ambiguity complicates variant identification and introduces potential biases in comparative genomic analyses [22, 32]. Accurately identifying these problematic regions in reference genomes is crucial for mitigating false peaks or valleys in coverage data, enhancing the overall accuracy of genomic studies. Precalculating unmappable regions of each reference genome can save computational resources and enhance the precision of analyses. The success of this approach has been demonstrated in past studies [33, 34]. While these studies address repetitive sequences within a single reference genome, our task extends across a full database of virus genomes, introducing a higher level of complexity.
Identifying unmappable regions in all reference virus genomes unfolds in the following steps: initially, parameters are set, defining the window size as 75 bases and the step size as 1 base. A sliding window of 75 bases moves along the genome for each reference genome, extracting overlapping 75-mer sequences at each step. These sequences are then mapped to all other genomes, including a self-mapping step, using Bowtie 2. The regions where each 75-mer sequence maps are recorded. After this mapping process, each genome is analyzed to identify ‘unmappable’ regions where a 75-mer sequence maps to multiple locations. Any overlapping 75-mer unmappable regions are merged within each genome. Finally, the algorithm compiles a comprehensive list of these unmappable regions for all reference genomes. This meticulous process results in creating an output file or database, which details the unmappable regions of each genome, tailored for seamless integration into further genomic studies and pipelines. The process is summarized in Algorithm 1.
 |
The described method produces unmappable profiles by comparing virus genomes within the Virtual database. However, it is crucial to incorporate a corresponding host genome into the comparative analysis to diagnose virus infections in various samples. This can be achieved by adapting the process to compare each virus genome exclusively with the host genome, thus generating host-specific unmappable regions. Subsequently, these host-specific unmappable regions need to be integrated with the initial set of unmappable regions derived from the virus-to-virus comparisons. This merged dataset of unmappable regions is essential for the final analysis of virus diagnostics, ensuring accuracy and reliability in detecting virus infections in different host samples.
Please note that executing this algorithm demands considerable computational power, attributed to the sheer volume of genomes and the extensive nature of the mapping procedure. To accommodate this, precalculated mappability profiles using Compute Canada’s high-performance clusters are provided, and these results are integrated into our R package, IIMI. This package offers two distinct versions of unmappable regions: one derived from the Virtool virus database and another that includes the host genome of A. thaliana. These pre-computed profiles in IIMI facilitate efficient and resource-effective analysis for users.
Feature extraction and training machine learning models (Component C)
Coverage profiles, conceptualized as curves or high-dimensional vectors, are not ideal for direct use as inputs in machine learning models, particularly when training data sizes are limited or moderate. Extracting key features from these coverage profiles for machine learning algorithms is more practical for effectively training robust models.
Three types of features are considered for describing coverage profiles (after removing unreliable regions): (1) basic overall summaries, including average and maximum values of coverage profiles and the length of each virus segment. (2) Nucleotide composition percentages, including A, C, T, and GC content in each virus segment. (3) The proportion of the genome with high coverage is vital in minimizing false positives arising from sparse yet high peaks. This is measured as the percentage of the virus genome where the mapped read count exceeds a predetermined threshold, 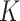 . Since the optimal threshold for ‘high coverage’ is uncertain, features are extracted at various thresholds (i.e.
. Since the optimal threshold for ‘high coverage’ is uncertain, features are extracted at various thresholds (i.e. 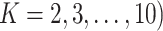 , allowing the machine learning models to select the most effective one automatically. Table 1 outlines these features with a summary of their importance metrics from various classification models, which will be discussed in detail in the Results section.
, allowing the machine learning models to select the most effective one automatically. Table 1 outlines these features with a summary of their importance metrics from various classification models, which will be discussed in detail in the Results section.
Table 1.
Features extracted from mappability-profile processed coverage profiles for the machine-learning process. The median variable importance is summarized from all models trained in experiments described in the Results section. The top 5 features of each method are highlighted in bold font
| Categories | Features | Median Variable Importance | |
|---|---|---|---|
| IIMI-RF | IIMI-X | ||
| Basic overall summaries of the virus genome | average coverage | 0.1003 | 0.0324 |
| maximum coverage | 0.1189 | 0.0294 | |
| length of genome | 0.0917 | 0.0716 | |
| Nucleotide composition percentages | % of A | 0.0482 | 0.0361 |
| % of C | 0.0417 | 0.0203 | |
| % of T | 0.0557 | 0.0285 | |
| GC content | 0.0559 | 0.0359 | |
Percentage of coverage exceeds a certain threshold (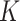 reads) across the whole virus genome reads) across the whole virus genome |
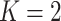
|
0.1894 | 0.6699 |
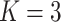
|
0.0967 | 0.0081 | |
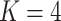
|
0.062 | 0.0012 | |
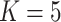
|
0.0484 | 0.0179 | |
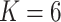
|
0.0367 | 0.0064 | |

|
0.0295 | 0.0009 | |
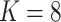
|
0.0363 | 0.0057 | |
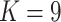
|
0.0475 | 0.0034 | |
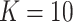
|
0.0657 | 0.0323 | |
Using features extracted from coverage profiles and each profile’s observed virus infection status, we train several widely used supervised machine learning models as part of the IIMI pipeline. Specifically, we consider classification tree [35], random forest [36], XGBoost [37], and elastic net [38]. These four models are separately named IIMI-CT, IIMI-RF, IIMI-X, and IIMI-EN. All models’ predictions are made at the virus segment level. To predict if the plant is affected by a virus, a result is considered positive if at least one virus segment is indicated as positive in the detection.
The rationale for selecting these four machine learning methods is their proven performance in numerous evaluations: random forest, XGBoost, and elastic net are well-known top performers, and the classification tree serves as the building block for the first two methods. We also explored other machine learning methods, including gradient boosting machine [39], naive Bayes [40], logistic regression [38], support vector machine [41], linear discriminant analysis [42], and quadratic discriminant analysis [42]. Complete results for all 10 models are provided in S1 of the Supplementary Material.
Validation of IIMI
To evaluate the performance of IIMI, we conducted computational experiments using real-world data, comparing four variations of IIMI against state-of-the-art methods such as fastv and Pathoscope. Our evaluation consists of two parts: (1) cross-validation with in-house data and (2) independent validation using data from VirHunter. Detailed descriptions of the design and execution of the computational experiments are provided in the rest of the section. The results of these experiments will be discussed in the Results section.
In data analysis using fastv, we adhere to its default setting. Pathoscope only provides coverage profile without binary diagnostic results. We follow the Virtool web application guidelines, which are provided by the CFIA and used for their routine diagnostic works to determine if a virus infects a sample through a two-step process. Initially, the application applies deterministic rules with set thresholds concerning genomic coverage, depth, and weight. A virus is flagged as positive if the genomic coverage exceeds 0.3 and the weight is greater than 0.000006, followed by checking if any virus segment has genomic coverage of at least 0.9 with a median depth over 0.5. Here, genomic coverage represents the virus genome fraction covered by sequencing reads, depth signifies the read count at a particular genome location, and weight is the reads’ proportion mapped to a virus, totalling 1. Subsequently, a manual review is conducted to ensure no cross-contamination, such as confirming the absence of a virus in a plant sample that could be a contaminant from other samples processed in the same or previous sequencing batches, NGS libraries, or RNA extractions. The second step is omitted because it requires manual checks, which are impossible when more than 3000 viruses need to be diagnosed for more than 200 plant samples and replicate such experiments many times.
We use a five-fold cross-validation method to evaluate the methods. Our in-house data is divided into five folds; in each of the five iterations, four folds are used to train IIMI models, while the remaining fold is used as the testing dataset. This ensures that each fold serves as the testing set exactly once.
These five models were then applied to make predictions on two datasets: the in-house test set (assessing performance in similar training and test conditions) and the VirHunter-provided data (evaluating performance across different studies and HTS protocols). The binary infection status predictions were compared with actual infection statuses in both test datasets to construct confusion matrices. From these matrices, four performance metrics were calculated: accuracy (the overall correctness of the model), precision (the correctness of positive predictions), recall (the model’s ability to identify all positive cases), and the 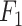 score (a balance between precision and recall). Their definitions and formulas are as follows:
score (a balance between precision and recall). Their definitions and formulas are as follows:
 |
where 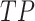 (True Positive) is the count of positive cases correctly predicted,
(True Positive) is the count of positive cases correctly predicted, 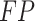 (False Positive) is the count of negatives incorrectly labelled as positive,
(False Positive) is the count of negatives incorrectly labelled as positive, 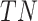 (True Negative) is the count of negatives correctly identified, and
(True Negative) is the count of negatives correctly identified, and 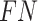 (False Negative) is the count of positives incorrectly labelled as negative. These elements form the confusion matrix, quantifying correct and incorrect predictions. For all four metrics, larger values indicate better classification performance.
(False Negative) is the count of positives incorrectly labelled as negative. These elements form the confusion matrix, quantifying correct and incorrect predictions. For all four metrics, larger values indicate better classification performance.
The experiment is conducted 100 times to determine the reliability of our results, each time using different random divisions of training and test data. This process results in varying sets of training and test data sourced internally, while the data from external studies (VirHunter) remains unchanged. Consequently, 100 values for each evaluation metric across each test data set are obtained. Note that VirHunter’s results are excluded from the visualization below due to its significantly lower performance than other methods, as including it would diminish the visual clarity in distinguishing the remaining methods. Although VirHunter can diagnose known viruses, it is primarily developed to detect novel viruses, partly explaining why its performance on known viruses is not as effective.
Results
Figure 3(a) displays the evaluation results on our internally sourced test data. Each boxplot in the figure is derived from 100 data points gathered across the experiments of 100 different random data splits, with these points also depicted as scatter plots. Among the methods, fastv is noted for identifying the highest number of positives, leading to the highest detection rate in this analysis. However, this comes at the cost of a high false positive rate, resulting in the lowest precision. Consequently, fastv’s 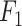 score suffers due to its poor precision, ranking it the lowest. In contrast, Pathoscope adopts a more cautious approach, yielding the lowest recall but the second-highest precision. IIMI-CT, which employs a singular classification tree, balances fastv and Pathoscope regarding precision and recall
score suffers due to its poor precision, ranking it the lowest. In contrast, Pathoscope adopts a more cautious approach, yielding the lowest recall but the second-highest precision. IIMI-CT, which employs a singular classification tree, balances fastv and Pathoscope regarding precision and recall 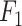 . The ensemble methods IIMI-RF and IIMI-X, which use multiple classification trees through bagging and boosting techniques, respectively, demonstrate superior performance over the single-tree IIMI-CT in all four metrics. According to the in-house data assessment, these two methods perform comparably and emerge as the most effective. The linear model IIMI-EN produces a sparse prediction model, differing largely from other IIMI variants, and its
. The ensemble methods IIMI-RF and IIMI-X, which use multiple classification trees through bagging and boosting techniques, respectively, demonstrate superior performance over the single-tree IIMI-CT in all four metrics. According to the in-house data assessment, these two methods perform comparably and emerge as the most effective. The linear model IIMI-EN produces a sparse prediction model, differing largely from other IIMI variants, and its 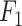 ranks are not impressive. The ranks of methods in their accuracy are very similar to their ranks in
ranks are not impressive. The ranks of methods in their accuracy are very similar to their ranks in 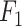 score, suggesting the same winning methods.
score, suggesting the same winning methods.
Figure 3.

Boxplots of evaluation metrics comparing virus detection pipelines based on cross-validation using our in-house HTS data and independent validation using the VirHunter’s HTS data. The 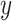 -axis represents the method: fastv, Pathoscope, IIMI-CT, IIMI-EN, IIMI-RF, and IIMI-X. The
-axis represents the method: fastv, Pathoscope, IIMI-CT, IIMI-EN, IIMI-RF, and IIMI-X. The 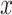 -axis represents the values for the metrics. The smaller lighter dots show the score for each replication. The larger darker dots are outliers of the boxplots. Panel (a) shows the evaluation metrics based on the in-house HTS dataset. Panel (b) shows the evaluation metrics based on the VirHunter HTS dataset.
-axis represents the values for the metrics. The smaller lighter dots show the score for each replication. The larger darker dots are outliers of the boxplots. Panel (a) shows the evaluation metrics based on the in-house HTS dataset. Panel (b) shows the evaluation metrics based on the VirHunter HTS dataset.
Figure 3(b) presents the validation results using external data. The test data provided by VirHunter were created using protocols markedly different from those used for our in-house data. This led to a notable shift in performance rankings. In this external validation, IIMI-X has been identified as the best model. Its consistent good performance from in-house and external data demonstrates it as the best method for virus detection. IIMI-EN ranks as the second-best performing model among the methods, similar to Pathoscope’s performance. IIMI-EN’s performance ranks higher in external validation than it did in the cross-validation of in-house data. This is because the elastic net aims to provide a parsimonious model with few predictors and a simple linear relationship. Simpler models sacrifice prediction accuracy to gain better robustness. Therefore, IIMI-EN is a great alternative method when applied to test data that is quite different from the training data.
To discern which features are more helpful in enhancing virus diagnostic performance, the importance of features is assessed using two distinct metrics: for IIMI-RF, the non-scaled mean decrease in accuracy; for IIMI-X, the percentage contribution of each feature to the model. For both metrics, higher metrics represent more importance.
Table 1 presents the median values of feature importance obtained from models built using 100 random data splits. The top 5 features for each method are highlighted in bold. Complementing this, Fig. 4 offers a detailed visual representation of feature importance. It includes boxplots that capture the range and distribution of feature importance across the models from the 100 random splits, with individual points plotted to denote the importance scores for features within each unique data split. Overall, each feature contributes to the predictive capability of both models to varying extents. The genome-wide coverage percentage of positions with at least 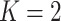 reads and the length of the virus genome have a more pronounced impact on all methods than other features. The coverage percentages of positions with at least
reads and the length of the virus genome have a more pronounced impact on all methods than other features. The coverage percentages of positions with at least 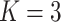 reads are also important for IIMI-RF. The three variables of the basic overall summary of virus genomes rank in the top 5 in their median values for IIMI-RF, whereas two of these variables rank in the top 5 in the median values for IIMI-X. IIMI-X and IIMI-RF are both ensemble methods based on decision trees, but their behaviour in feature selection is quite different. A few variables dominate IIMI-X, while the contributions of the variables in IIMI-RF do not differ dramatically, which can be explained by how these two methods aggregate the trees. The percentage of A nucleotide also emerges as one of the top 5 predictors in IIMI-X. Random Forest averages many deep trees to reduce the model’s variance; hence, deep trees tend to utilize more predictors. In contrast, XGBoost adds up many small trees to sequentially reduce bias; hence, it could involve few predictors (if most trees share common predictors).
reads are also important for IIMI-RF. The three variables of the basic overall summary of virus genomes rank in the top 5 in their median values for IIMI-RF, whereas two of these variables rank in the top 5 in the median values for IIMI-X. IIMI-X and IIMI-RF are both ensemble methods based on decision trees, but their behaviour in feature selection is quite different. A few variables dominate IIMI-X, while the contributions of the variables in IIMI-RF do not differ dramatically, which can be explained by how these two methods aggregate the trees. The percentage of A nucleotide also emerges as one of the top 5 predictors in IIMI-X. Random Forest averages many deep trees to reduce the model’s variance; hence, deep trees tend to utilize more predictors. In contrast, XGBoost adds up many small trees to sequentially reduce bias; hence, it could involve few predictors (if most trees share common predictors).
Figure 4.
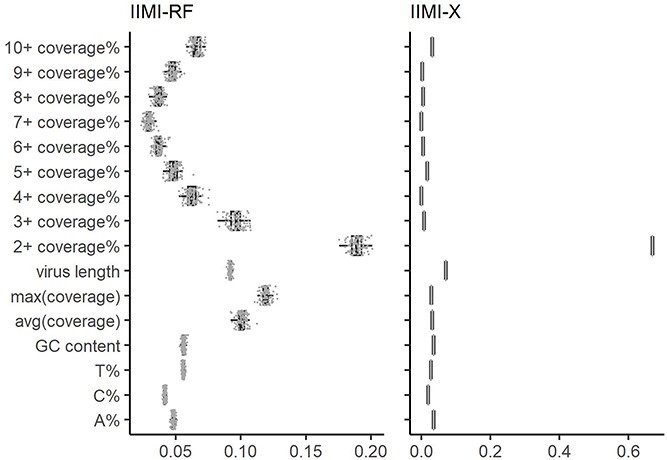
Boxplots of variable importance using IIMI-RF and IIMI-X based on the in-house HTS dataset. Three models use different calculation metrics. IIMI-RF uses mean decrease in accuracy; IIMI-X uses the proportion of contribution to the model. The grey points are each replication of the feature’s importance.
The results above demonstrate IIMI’s outstanding performance in accurately diagnosing viral infections. To highlight IIMI’s ability to process a large number of samples within a reasonable time frame in a real-world setting, we summarize the computing time of IIMI in Table 2. Note that runtime does not include time used in the alignment step since that depends on the standard aligners such as Bowtie and minimap. The second column shows the time needed to train IIMI methods using 261 in-house samples. The two tree-based approaches (IIMI-RF and IIMI-X) take less than 4 seconds, and IIMI-EN takes 82 seconds using a standard personal computer. The third column of Table 2 shows the average testing speed on VirHunter samples. It takes 0.0146 (IIMI-EN) to 0.0627 seconds (IIMI-RF) to diagnose the infection status of 1538 viruses in one sample. That is, within 1 second, IIMI can diagnose 16 (IIMI-EN) to 68 (IIMI-RF) samples. In addition to computing time, we also investigate the maximum memory requirement for running IIMI. Because most aligners (such as Bowtie 2) can process the sequences one by one without loading all of them in the memory, IIMI’s memory requirements are primarily decided by the need to fit machine learning models with the given samples. We investigated the memory usage of running one sample in the in-house data, which takes up to 100 MB for each sample. Processing each sample of our in-house data from the BAM file (output of Bowtie) into RLE format requires up to 500 MB. The feature engineering step for converting RLE data into 16 features requires 2 MB of memory. Hence, the maximum memory requirement is determined by sample sizes and the machine learning model (e.g., IIMI-RF or IIMI-X). When training our model with 261 samples, the maximum memory usage for IIMI-RF is around 400 MB, for IIMI-X, it is around 30 MB, and for IIMI-EN, it is 600 MB. Thus, we recommend having more memory available than the maximum memory usage. Once the model is trained, applying it to classify new samples requires very little memory. If a huge number of new samples need to be classified, we can batch this job to reduce memory requirements (when needed). These results highlight IIMI’s potential for rapid and efficient virus detection in a large number of plant samples, even with limited computational resources.
Table 2.
Runtime of IIMI (excluding runtime of alignment step) on a personal computer. We trained the in-house plant data with IIMI-RF, IIMI-X, and IIMI-EN and tested on the external data one sample at a time with all three methods
| Method | Training time (sec) | Mean testing time (sec) |
|---|---|---|
| IIMI-RF | 3.5744 | 0.0249 |
| IIMI-X | 2.6796 | 0.0627 |
| IIMI-EN | 81.6809 | 0.0146 |
Discussion and conclusion
In the computer experiments, accuracy as an evaluation metric was included in the analysis; however, it may not always be the most insightful metric, particularly in the context of this work. This is because the dataset exhibits a significant imbalance, with samples infected with only a small subset of the 1583 viruses in the database that was used. Therefore, a method could achieve high accuracy with reasonable false positive rates but not fully capture the method’s effectiveness. Metrics like precision, recall, and  score, which directly evaluate the positive detections of methods, are more suited to our situation.
score, which directly evaluate the positive detections of methods, are more suited to our situation.
The results indicate that the IIMI models have a significant edge over other methods in terms of performance. When analyzing test samples with sequencing data generated using protocols similar to the training samples, such as in our internal data cross-validation, it is recommended to use IIMI-RF or IIMI-X. For test data derived from different protocols, results from our external data validation indicate that IIMI-X is the best choice. Alternatively, IIMI-EN is also a good option when test data is quite different from training data due to its robustness. In addition to their superior precision in diagnosis, the IIMI models offer other benefits over alternative methods. They use coverage profiles rather than 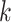 -mer sequences and leverage pre-trained models, enabling the rapid and simultaneous diagnosis of many samples against a large virus reference database. This starkly contrasts with methods like VirHunter, which are limited to processing one sample at a time. Furthermore, IIMI models proactively identify and precalculate unreliable regions in virus reference genomes. This foresight eliminates the need for manual intervention to judge false peaks or valleys, a requirement in methods like Pathoscope, thus providing more consistent results and reducing the labour intensity of the diagnostic process.
-mer sequences and leverage pre-trained models, enabling the rapid and simultaneous diagnosis of many samples against a large virus reference database. This starkly contrasts with methods like VirHunter, which are limited to processing one sample at a time. Furthermore, IIMI models proactively identify and precalculate unreliable regions in virus reference genomes. This foresight eliminates the need for manual intervention to judge false peaks or valleys, a requirement in methods like Pathoscope, thus providing more consistent results and reducing the labour intensity of the diagnostic process.
However, it is important to note a limitation of IIMI. The method is not well-suited for detecting viroids due to their extremely short length and circular structure. We evaluate IIMI on only viroids, which is included in S2 in the Supplementary Materials, revealed that while IIMI performs well in detecting viroids in our in-house data, we cannot confidently claim that IIMI works well on viroids detection when it comes to external data. We need more data in in-house data to train IIMI on viroids to guarantee good performance. Thus, IIMI is currently optimized for virus detection rather than viroid detection. In future work, if we have more plant data infected with viroids, we can expand IIMI by building another model specifically targeting viroids. Since the current version of IIMI is based on a supervised machine learning model, it cannot detect infections of uncharacterized viruses. We have several ideas to address this problem, which is part of our ongoing work. It is always challenging to detect viral sequences that have many variations. IIMI has made efforts to address this issue by focusing on the detection model at the segment level. We report infection of a virus if any segment on its genome is detected as positive. Thus, IIMI is robust to variations at the segment level (i.e. when the virus genome has three segments, and two of them show up in the infected sample). However, if substantial variation occurs within a segment, the performance of IIMI can be compromised.
After extensive in-house testing, the three trained models—IIMI-EN, IIMI-RF, IIMI-X—will be integrated into the Virtool web analysis platform. Users will be able to upload sequencing data for diagnosis, benefiting from fastv, batch processing capabilities due to the pre-trained nature of the models. The R package IIMI will further refine these models with a broader range of samples and host-specific information, enhancing Virtool’s diagnostic accuracy. Both the Virtool web platform and the IIMI package are publicly accessible.
This research utilized the A. thaliana genome as a generic host. Employing specific host genomes, where available, could refine the annotation of unreliable regions on virus reference genomes, thereby improving diagnostic accuracy. Two versions of these regions are offered in IIMI: one derived solely from virus genomes and another combining A. thaliana and virus genomes.
In conclusion, this work yields three significant outcomes: (1) pre-trained models with excellent classification performance for immediate virus diagnosis; (2) an analysis pipeline and R package for updating these models with more diverse samples and host information; and (3) a database of 1583 virus reference genomes with annotated unreliable regions, a valuable standalone resource for other research. Our dedication to open research and global resource sharing is fundamental to our approach. To facilitate this, our analytical tools and database are publicly accessible, with detailed access information provided in the availability section at the end of this paper.
This research is part of the CLEan plAnt extractioN SEquencing Diagnostics project, aimed at safeguarding Canada’s wine and grape industry [43]. The findings will be employed for grapevine virus screening of domestic, imported, and exported grapevines. IIMI will be integrated into Virtool, a web-based application with an established user base, for plant virus detection in multiple crops, including grapevine.
Key Points
We proposed IIMI, a system that leverages machine learning algorithms to streamline the process of diagnosing viral infections using genome sequencing data.
IIMI is designed to automatically correct artifacts in genomic data, thereby enhancing diagnostic accuracy, and ensuring more reliable, consistent decision-making while also reducing the demand for manual labor.
The IIMI package has a built-in extensive, easily accessible database of curated virus genomes, streamlining the diagnostic process for plant samples and accommodating the batch processing of multiple samples.
IIMI offers a distinct mappability profile for each virus genome, generated by our algorithm. This resource is crucial for other researchers, as it highlights areas where coverage profiles may be unreliable.
Supplementary Material
Contributor Information
Haochen Ning, Department of Mathematics and Statistics, University of Victoria, 3800 Finnerty Road (Ring Road), BC V8P 5C2, Canada.
Ian Boyes, Canadian Food Inspection Agency, Centre for Plant Health, 8801 Saanich Road E., North Saanich, BC V8L 1H3, Canada.
Ibrahim Numanagić, Department of Computer Science, University of Victoria, 3800 Finnerty Road (Ring Road), BC V8P 5C2, Canada.
Michael Rott, Canadian Food Inspection Agency, Centre for Plant Health, 8801 Saanich Road E., North Saanich, BC V8L 1H3, Canada.
Li Xing, Department of Mathematics and Statistics, University of Saskatchewan, 106 Wiggins Road, Saskatoon, SK S7N 5E6, Canada.
Xuekui Zhang, Department of Mathematics and Statistics, University of Victoria, 3800 Finnerty Road (Ring Road), BC V8P 5C2, Canada.
Conflict of interest
None declared.
Availability
R package: iimi is available on the Comprehensive R Archive Network (https://cran.r-project.org/web/packages/iimi/index.html). The package includes functions to implement our algorithm and information about unreliable regions.
Database: Virus genome sequences. It is available on the GitHub website: https://github.com/virtool/ref-plant-viruses.
Funding
This work is supported by NSERC Discovery # RGPIN-04973 (to I.N.), NSERC DG # RGPIN-2021-03530 (to L.X.), the Canada Research Chair #CRC-2021-00232 (to X.Z.), Michael Smith Health Research BC Scholar: # SCH-2022-2553(to X.Z.), and a Genomic Applications Partnership Program award, 189GRP, from Genome British Columbia and Genome Canada (to X.Z., M.R.). This research was enabled in part by computational resource support provided by Westgrid (https://www.westgrid.ca) and the Digital Research Alliance of Canada (https://alliancecan.ca).
Author contributions
HN contributed to preparing the initial draft and conducting the computer experiments. XZ, MR, and LX were instrumental in conceptualizing the study, designing the research methodology, and supervising students involved in the project. XZ and MR contribute to secure funding for the research. IB prepared the Virtool virus database and guided using the Virtool platform. IN guided sequence alignment work. HN, IB, LX, and XZ contribute to creating the R package iimi. All authors participated in revising the manuscript and have given their approval for the final version.
References
- 1. Sastry K, Zitter T. Management of Virus and Viroid Diseases of crops in the tropics. In: Plant Virus and Viroid Diseases in the Tropics, Epidemiology and Management. vol. 2. 1st ed.. Dordrecht: Springer Dordrecht; 2014, 149–480. Available from: 10.1007/978-94-007-7820-7_2. [DOI] [Google Scholar]
- 2. Ogolla E. Invasive Pest Spread another Fallout from Climate Change, UN-Backed Study Finds [Internet]. [place unknow]: United Nations, 2021. [cited 2024 July 4]. Available from: https://news.un.org/en/story/2021/06/1093202. [Google Scholar]
- 3. International Plant Protection Convention Secretariat . Scientific Review of the Impact of Climate Change on Plant Pests. Rome: Food and Agriculture Organization of the United Nations on behalf of the International Plant Protection Convention Secretariat, 2020. Available from: https://openknowledge.fao.org/items/8a6aa7fd-8e86-4c95-886a-ce52f1f6d01d. [Google Scholar]
- 4. Baranwal V, Kapoor R, Kumar S. et al.. Recent advances of virus diagnostics in horticultural crops. In: Awasthi A (ed.), Applied Plant Virology: Advances, Detection, and Antiviral Strategies. Cambridge (MA): Academic Press; 2020. 27–37. Available from: https://www.sciencedirect.com/science/article/pii/B9780128186541000025, 10.1016/B978-0-12-818654-1.00002-5. [DOI] [Google Scholar]
- 5. Clark M, Adams A. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J Gen Virol 1977; 34:475–83. 10.1099/0022-1317-34-3-475. [DOI] [PubMed] [Google Scholar]
- 6. Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci 2016; 73:4433–48. 10.1007/s00018-016-2299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Domingo E, Perales C. Viral quasispecies. PLoS Genet 2019; 15:e1008271. 10.1371/journal.pgen.1008271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaafar Y, Ziebell H. Comparative study on three viral enrichment approaches based on RNA extraction for plant virus/viroid detection using high-throughput sequencing. PloS One 2020; 15:e0237951. 10.1371/journal.pone.0237951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lebas B, Adams I, Al Rwahnih M. et al.. Facilitating the adoption of high-throughput sequencing technologies as a plant pest diagnostic test in laboratories: a step-by-step description. Bull OEPP 2022; 52:394–418. 10.1111/epp.12863. [DOI] [Google Scholar]
- 10. Chen S, He C, Li Y. et al.. A computational toolset for rapid identification of SARS-CoV-2, other viruses, and microorganisms from sequencing data. Brief Bioinform 2021; 22:924. 10.1093/bib/bbaa231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sukhorukov G, Khalili M, Gascuel O. et al.. VirHunter: a deep learning-based method for detection of novel RNA viruses in plant sequencing data. Front Bioinform 2022; 2:1–12. 10.3389/fbinf.2022.867111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hong C, Manimaran S, Shen Y. et al.. PathoScope 2.0: a complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014; 2:33. 10.1186/2049-2618-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boyes I, Hoffmann R, Rott M. Virtool: Viral Infection Diagnostics Using Next-Generation Sequencing [Internet]. [place unknown: publisher unknown]; 2020[cited 2023 Nov 29]. Available from: https://www.virtool.ca/.
- 14. Langmead B, Salzberg S. Fast gapped-read alignment with bowtie 2. Nat Methods 2012; 9:357–9. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. National Library of Medicine . NCBI Virus [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information [cited 2024 Apr 1]. Available from: https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=10239.
- 16. Khalili M, Marais-Colombel A, Lefebvre M. et al.. Peach Sequencing Data (RNAseq) [Internet]. [place unknown]: Recherche Data Gouv; 2022. Available from: 10.15454/GWDPIN. [DOI] [Google Scholar]
- 17. Candresse T, Marais-Colombel A, Brault V. Sugar Beet Sequencing Data (RNAseq) [Internet]. [place unknown]: Recherche Data Gouv; 2022. Available from: 10.15454/MK1JIW. [DOI] [Google Scholar]
- 18. Candresse T, Marais-Colombel A, Faure C. et al.. Grapevine Sequencing Data (RNAseq) [Internet]. [place unknown]: Recherche Data Gouv; 2022. Available from: 10.15454/KUYAT9. [DOI] [Google Scholar]
- 19. Danecek P, Bonfield J, Liddle J. et al.. Twelve years of SAMtools and BCFtools. GigaScience 2021; 10:1–4. 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Handsaker B, Wysoker A. et al.. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078–9. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lawrence M, Huber W, Pagès H. et al.. Software for computing and annotating genomic ranges. PLoS Comput Biol 2013; 9:e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Treangen T, Salzberg S. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 2011; 13:36–46. 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ross M, Russ C, Costello M. et al.. Characterizing and measuring bias in sequence data. Genome Biol 2013; 14:R51. 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aird D, Ross M, Chen W. et al.. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 2011; 12:R18. 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Basile W, Sachenkova O, Light S. et al.. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput Biol 2017; 13:e1005375. 10.1371/journal.pcbi.1005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodwin S, McPherson J, McCombie W. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016; 17:333–51. 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Metzker M. Sequencing technologies - the next generation. Nat Rev Genet 2010; 11:31–46. 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 28. Benjamini Y, Speed T. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 2012; 40:e72. 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261–82. 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 30. Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008; 18:1851–8. 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C, Williams B, Pertea G. et al.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511–5. 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DePristo M, Banks E, Poplin R. et al.. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43:491–8. 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang X, Robertson G, Krzywinski M. et al.. PICS: probabilistic inference for ChIP-seq. Biometrics 2011; 67:151–63. 10.1111/j.1541-0420.2010.01441.x. [DOI] [PubMed] [Google Scholar]
- 34. Zhang X, Robertson G, Woo S. et al.. Probabilistic inference for nucleosome positioning with MNase-based or sonicated short-read data. PloS One 2012; 7:e32095. 10.1371/journal.pone.0032095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ripley B. Tree: Classification and Regression Trees [internet]. Vienna, Austria: R Foundation for Statistical Computing; 2023. R Package Version 1.0-43. Available from: https://CRAN.R-project.org/package=tree.
- 36. Liaw A, Wiener M. Classification and regression by randomForest. R News 2002; 2:18–22. Available from: https://CRAN.R-project.org/doc/Rnews/. [Google Scholar]
- 37. Chen T, He T, Benesty M. et al.. XGBoost: eXtreme Gradient Boosting [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2023. R package version 1.7.5.1. Available from: https://CRAN.R-project.org/package=xgboost.
- 38. Kuhn M. Building predictive models in R using the caret package. J Stat Softw 2008; 28:1–26. 10.18637/jss.v028.i05.27774042 [DOI] [Google Scholar]
- 39. Ridgeway F. GBM developers. GBM: generalized boosted regression models. [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2024. R package version 2.2.2. Available from: https://CRAN.R-project.org/package=gbm.
- 40. Majka M. Naivebayes: high performance implementation of the naive Bayes algorithm in R. [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2024. R package version 1.0.0. Available from: https://CRAN.R-project.org/package=naivebayes.
- 41. Meyer D, Dimitriadou E, Hornik K. et al.. e1071: misc functions of the Department of Statistics, Probability Theory Group (formerly: E1071), TU Wien [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2023. R package version 1.7-14. Available from: https://CRAN.R-project.org/package=e1071.
- 42. Venables W, Ripley B. In: Chambers J, Eddy W, Härdle W, Sheather S, Tierney L, editors. Modern Applied Statistics with S. 4th ed. New York: Springer; 2002. ISBN 0-387-95457-0. Available from: https://www.stats.ox.ac.uk/pub/MASS4/, 10.1007/978-0-387-21706-2. [DOI] [Google Scholar]
- 43. Canada Food Inspection Agency . Collaborating with Genome Specialists to Protect Canadaâ&Euro;Plant Health [Internet]; 2021[cited 2023 Dec 11]. Available from:https://inspection.canada.ca/inspect-and-protect/plant-health/collaborating-with-genome-specialists-to-protect-c/eng/1615827016552/1615827158496.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.