

Abstract
INTRODUCTION
Frontotemporal dementia (FTD) is characterized by phenotypic and genetic heterogeneities. However, reports on the large Chinese FTD cohort are lacking.
METHODS
Two hundred forty‐eight patients with FTD were enrolled. All patients and 2010 healthy controls underwent next generation sequencing. Plasma samples were analyzed for glial fibrillary acidic protein (GFAP), α‐synuclein (α‐syn), neurofilament light chain (NfL), and phosphorylated tau protein 181 (p‐tau181).
RESULTS
Gene sequencing identified 48 pathogenic or likely pathogenic mutations in a total of 19.4% of patients with FTD (48/248). The most common mutation was the C9orf72 dynamic mutation (5.2%, 13/248). Significantly increased levels of GFAP, α‐syn, NfL, and p‐tau181 were detected in patients compared to controls (all p < 0.05). GFAP and α‐syn presented better performance for diagnosing FTD.
DISCUSSION
We investigated the characteristics of phenotypic and genetic spectrum in a large Chinese FTD cohort, and highlighted the utility of plasma biomarkers for diagnosing FTD.
Highlights
This study used a frontotemporal dementia (FTD) cohort with a large sample size in Asia to update and reveal the clinical and genetic spectrum, and explore the relationship between multiple plasma biomarkers and FTD phenotypes as well as genotypes.
We found for the first time that the C9orf72 dynamic mutation frequency ranks first among all mutations, which broke the previous impression that it was rare in Asian patients.
Notably, it was the first time the C9orf72 G4C2 repeat expansion had been identified via whole‐genome sequencing data, and this was verified using triplet repeat primed polymerase chain reaction (TP‐PCR).
We analyzed the diagnostic accuracy of four plasma biomarkers (glial fibrillary acidic protein [GFAP], α‐synuclein [α‐syn], neurofilament light chain [NfL], and phosphorylated tau protein 181 [p‐tau181]) at the same time, especially for α‐syn being included in the FTD cohort for the first time, and found GFAP and α‐syn had the highest diagnostic accuracy for FTD and its varied subtypes.
Keywords: α‐synuclein, biomarker, Frontotemporal dementia, genetics, glial fibrillary acidic protein, neurofilament light chain, phosphorylated tau protein 181, whole‐genome sequencing
1. BACKGROUND
Frontotemporal dementia (FTD) encompasses a spectrum of dementia syndromes characterized by progressive mental and behavioral abnormalities, executive dysfunction, and language impairment. 1 , 2 , 3 Epidemiological data on FTD in China are lacking, whereas in Western countries, the most prevalent age at onset (AAO) ranges from 45 to 64 years, with an annual incidence of 2.7 to 4.0 per 100,000 individuals, ranking it as the second most common early onset neurodegenerative dementia. 4 , 5 FTD is mainly categorized into three clinical subtypes: behavioral variant FTD (bvFTD), progressive non‐fluent aphasia (PNFA), and semantic dementia (SD). It may coexist with other neurodegenerative diseases, such as progressive supranuclear palsy (PSP), corticobasal syndrome (CBS), chromosome 17‐related FTD and parkinsonism (FTDP‐17), and motor neuron disease (MND), forming distinct clinical subtypes. 2 , 4 Previous studies have indicated a strong genetic link to FTD, with at least 17 pathogenic genes identified, including C9orf72, MAPT, GRN, CHCHD10, TBK1, VCP, CHMP2B, TARDBP, FUS, SIGMAR1, SQSTM1, UBQLN2, OPTN, CCNF, TIA1, CYLD, and TREM2. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Some genes linked to amyotrophic lateral sclerosis (ALS) have also been associated with FTD. 22 , 23 , 24 The most prevalent pathogenic genes are C9orf72, MAPT, and GRN. 25 , 26 , 27 The G4C2 hexanucleotide repeat expansion in the C9orf72 gene is a significant cause of FTD. C9orf72‐related FTD accounts for ≈ 20% to 30% of hereditary FTD and is frequently associated with FTD‐ALS and bvFTD. 25 In 2014, Jiao et al. first reported the dynamic mutation of C9orf72 in a Chinese FTD patient. 3 Subsequent reports on this gene's mutation in Asian populations are relatively scarce. The MAPT gene encodes a microtubule‐binding tau protein, with pathogenic mutations impairing tau–microtubule interactions or disrupting the balance of tau protein subtypes. 28 MAPT accounts for ≈ 5% to 20% of hereditary FTD, with bvFTD being the most common phenotype, followed by FTD‐P. 25 Currently, > 50 pathogenic or likely pathogenic (P/LP) mutations have been identified. The GRN gene encodes progranulin (PGRN), and most P/LP mutations are frameshift, splicing, or nonsense types, 29 leading to PGRN loss of function or haploinsufficiency. 30 GRN accounts for ≈ 5% to 25% of hereditary FTD, with bvFTD and PNFA being common phenotypes. 25 More than 80 P/LP mutations have been discovered in GRN. Besides these three common genes, rare pathogenic genes contribute to < 5% of FTD cases, 5 such as CHCHD10, TBK1, VCP, CHMP2B, TARDBP, FUS, SIGMAR1, SQSTM1, UBQLN2, OPTN, and CCNF 31 et al. Owing to the ethnic genetic heterogeneity within the Chinese population, comprehensive reports with large sample sizes regarding the mutation frequencies of FTD are lacking.
Over the past decade, there has been a significant surge in plasma biomarker research in the field of Alzheimer's disease (AD). Among them, amyloid beta protein 42/40 (Aβ42/40) and phosphorylated tau protein 181 (p‐tau181) are considered potential biomarkers for identifying AD. 32 , 33 In some studies, these biomarkers have been reported in small quantities in FTD. 34 , 35 Several other biomarkers are closely related to FTD. Studies have found that neurofilament light chain (NfL) levels are elevated early in patients with familial FTD and are a precursor to the onset of symptoms, 36 while elevated glial fibrillary acidic protein (GFAP) levels were detected in specific FTD variants compared to those in healthy controls. 37 Moreover, another neurodegenerative disease‐related pathological protein, α‐synuclein (α‐syn), has not been reported in FTD. As a result of the limited sample size and the effect of ethnicity on biomarker levels, there is currently no comprehensive study available on biomarkers in Chinese FTD patient cohorts. Furthermore, the relationship between these biomarker levels and clinical subtypes, as well as genetic mutations, remains unclear.
Current clinical data on FTD in China are limited. In this study, we collected data from patients with FTD at Xiangya Hospital, Central South University, between 2009 and 2023, and used next generation sequencing and plasma biomarker analysis to elucidate clinical features, expand the genetic spectrum, and investigate the association between plasma biomarkers and the disease.
2. METHODS
2.1. Participants
This study included 248 patients clinically diagnosed with FTD, and among them, 24 patients carrying gene mutations through gene‐targeted sequencing in our previous studies, were enrolled for further clinical features and plasma biomarker analysis. 1 , 2 , 3 , 38 , 39 The remaining 224 patients and 2010 healthy controls underwent whole‐genome sequencing (WGS). Biomarker assessments were conducted in 106 patients with FTD and 187 age‐ and sex‐matched control subjects (Table S1 in supporting information). All participants met the established consensus criteria for bvFTD, 40 SD, 41 PNFA, 41 FTD‐P, and FTD‐ALS. All patients were from the Department of Neurology at Xiangya Hospital, Central South University, and healthy controls were from the Xiangya Hospital Health Examination Centre and Community Cohort. Each patient underwent comprehensive neurological evaluations by two experienced neurologists. Informed consent was obtained from all subjects. Detailed information regarding the age of onset, disease progression, family history, and clinical manifestations was collected from both patients and caregivers. The process steps of this study are shown in Figure 1.
FIGURE 1.
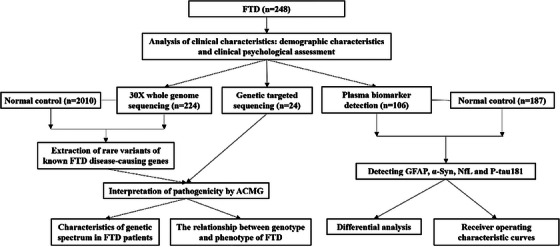
Flow chart of this study. α‐syn, α‐synuclein; ACMG, American College of Medical Genetics and Genomics; FTD, frontotemporal dementia; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; p‐tau181, phosphorylated tau protein 181.
RESEARCH IN CONTEXT
Systematic review: The authors conducted a thorough review of the literature on the current status of the frontotemporal dementia (FTD) clinical and genetic spectrum worldwide. Several pathogenic mutations and elevated levels of various plasma biomarkers have been linked to FTD. Nevertheless, data on the clinical phenotypic features and genetic mutation spectrum of FTD in large Chinese populations are lacking. Additionally, the diagnostic performance of plasma biomarkers for FTD and its subtypes has not been evaluated. These relevant citations are appropriately cited.
Interpretation: Our findings update the clinical and genetic spectrum through an FTD cohort with a large sample size in Asia, and reveal the relationship between multiple plasma biomarkers and FTD phenotypes as well as genotypes. This is an update and supplement to research currently in the public domain.
Future directions: This study demonstrated the clinical and genetic spectrum of Chinese FTD patients. We found for the first time that the C9orf72 dynamic mutation frequency ranks first among all mutations, which broke the previous impression that it was rare in Asian patients. Notably, it was the first time to identify C9orf72 G4C2 repeat expansion via whole‐genome sequencing data, and this was verified using triplet repeat primed polymerase chain reaction. We analyzed the diagnostic accuracy of four plasma biomarkers (glial fibrillary acidic protein [GFAP], α‐synuclein [α‐syn], neurofilament light chain [NfL], and phosphorylated tau protein 181 [p‐tau181]) at the same time, especially for α‐syn being included in the FTD cohort for the first time, and found GFAP and α‐syn had the highest diagnostic accuracy for FTD and its varied subtypes. All biomarker levels were not affected by genetic mutations.
2.2. Gene sequencing
First, purification and quality control of genomic DNA extracted from peripheral blood leukocytes was performed. The genomic DNA was randomly fragmented, end‐repaired, and 3′ adenylated. Subsequently, a single adenine base was added to the 3′ end, followed by DNA adaptor ligation and polymerase chain reaction (PCR) amplification. After quality control of the library, the PCR products were circularized. Sequencing was performed using the BGI platform. We selected genes that highly correlated with FTD for analysis, including C9orf72, MAPT, GRN, CHCHD10, TBK1, VCP, CHMP2B, TARDBP, FUS, SIGMAR1, SQSTM1, UBQLN2, OPTN, CCNF, TIA1, CYLD, TREM2, DCTN1, MATR3, NEK1, C21orf2, HNRNPA1, HNRNPA2B1, KIF5A, GLT8D1, DNAJC7, TUBA4A, ANG, ERBB4, SETX, CHRNA4, and CHRNB4. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 ,
2.3. Bioinformatics processing and variant analysis
We first perform data quality control and filtering on the FASTQ data. Filtered reads were aligned to the human v38 reference genome using BWA 42 and were sorted by samtools. 43 Variant calling was performed using GATK 44 and ANNOVAR 45 was used for variant annotation. Variant frequencies were assessed using public databases, including gnomAD, ExAC, and Kaviar. Twenty‐four predictive software packages, including SIFT, PolyPhen2, LRT, MutationTaster, and PROVEAN, 46 among others, were used to evaluate the effects of the variants. For variants with a minor allele frequency (MAF) between 0 and 0.01 in these databases, classifications followed the American College of Medical Genetics and Genomics (ACMG) guidelines, categorizing variants as pathogenic, likely pathogenic, uncertain significance, likely benign, or benign. 47 Variants interpreted as P/LP were verified in the normal control data.
We used ExpansionHunter software 48 to evaluate C9orf72 repeat expansions in all patients. For patients with C9orf72 repeats exceeding the pathogenic threshold, we performed triplet repeat primed polymerase chain reaction (TP‐PCR) for verification.
2.4. Biomarker testing
Venous blood samples were collected in ethylenediaminetetraacetic acid tubes and centrifuged at 1006.2 (g) for 15 minutes at 4°C within 2 hours post collection. Plasma samples were preserved at −80°C without undergoing freeze–thaw cycles. Quantification of phosphorylated p‐tau181, NfL, α‐syn, and GFAP was conducted using a fully automated single‐molecule detection machine (AST‐Sc‐Lite, AstraBio) following the manufacturer's guidelines (detailed parameters and steps are shown in the supporting information and Table S2). Assay kits R64030, R64040, R64070, and R64060 (Suzhou AstraBio Technology) were used for detecting plasma biomarkers of p‐tau181, NfL, α‐syn, and GFAP, respectively. The individuals who conducted the tests were unaware of the participants' group status.
2.5. Statistical analysis
Continuous variables are presented as mean ± standard deviation or median (interquartile range), and categorical variables as percentages. t tests or analysis of covariance (ANCOVA) were applied to normally distributed data, whereas Mann–Whitney U or Kruskal–Wallis tests were used for non‐normally distributed data. Chi‐square or Fisher exact tests were used to analyze categorical variables. Spearman rank correlation was used for correlation analysis. Plasma biomarker values were log10‐transformed for normalization, followed by ANCOVA and Bonferroni post hoc analysis, with age and sex as covariates for multiple comparisons. Biomarker prediction performance was evaluated using receiver operating characteristic (ROC) curves and area under curve (AUC) values. Multivariate binary logistic regression models predicted disease presence, with AUCs calculated from the predicted probabilities. Statistical analyses were performed using R version 4.3.2. Values of p < 0.05 were deemed statistically significant.
3. RESULTS
3.1. Participant characteristics
A total of 248 patients with FTD were recruited. All participants were of Chinese descent. Patient characteristics are presented in Table 1. Among the participants, 50.8% (126/248) were male and 49.2% (122/248) were female, the median years of education was 9.0 (6.0–12.0) years, the average age at onset (AAO) was 59.4 ± 10.4 years old, and the median disease course was 2.0 (1.0–4.0) years; 29.4% (73/248) of patients had a positive family history. Of the 248 patients, 45.6% (113/248) were diagnosed with bvFTD, 21.0% (52/248) with SD, 15.3% (38/248) with PNFA, 10.1% (25/248) with FTD‐P, and 8.1% (20/248) with FTD‐ALS. The FTD‐P subtype had the earliest AAO (56.6 ± 9.9), followed by PNFA, FTD‐ALS, bvFTD, and SD. We found significant differences in the disease course between different phenotypes (p < 0.05), and after pairwise comparison, it was found that the difference between bvFTD and FTD‐P was still significant (adjusted p < 0.05). There were no significant differences in any of the subtypes with respect to sex, education level, age of onset, or the presence of a positive family history.
TABLE 1.
Characteristics of clinical features and neuropsychological assessment of the FTD patients in this study (n = 248).
| All (N = 248) | bvFTD (N = 113) | PNFA (N = 38) | SD (N = 52) | FTD‐ALS (N = 20) | FTD‐P (N = 25) | p value | |
|---|---|---|---|---|---|---|---|
| Sex | |||||||
| Male, N (%) | 126 (50.8%) | 59 (52.2%) | 17 (44.7%) | 25 (48.1%) | 15 (75.0%) | 10 (40.0%) | 0.155 a |
| Female, N (%) | 122 (49.2%) | 54 (47.8%) | 21 (55.3%) | 27 (51.9%) | 5 (25.0%) | 15 (60.0%) | |
| Education (years), median (IQR) | 9.0 (6.0–12.0) | 9.0 (6.0–12.0) | 9.00 (6.0–12.0) | 9.0 (6.0–12.0) | 6.0 (6.0–11.3) | 9.0 (9.0–12.0) | 0.540 b |
| AAO (years), mean (std dev) | 59.4 (10.4) | 60.3 (11.6) | 57.0 (10.2) | 61.2 (7.9) | 58.0 (8.7) | 56.6 (9.9) | 0.152 c |
| Disease course (years), median (IQR) | 2.0 (1.0–4.0) | 3.0 (2.0–4.0) | 2.0 (1.0–3.0) | 2.25 (1.8–3.0) | 1.0 (1.0–2.3) | 2.0 (1.1–3.0) | 0.024 b |
| Family history, N (%) | 73 (29.4%) | 30 (26.5%) | 12 (31.6%) | 14 (26.9%) | 8 (40.0%) | 9 (36.0%) | 0.683 a |
| MMSE score, median (IQR) | 14.1 (11.0–16.3) | 14.6 (11.0–19.0) | 11.9 (4.0–15.0) | 14.5 (14.1–14.1) | 13.2 (13.3–14.1) | 14.4 (13.0–15.0) | >0.05 b |
| MoCA score, median (IQR) | 7.2 (1.0–10.0) | 7.7 (1.0–12.0) | 6.1 (0.0–9.0) | 7.7 (3.3–7.2) | 6.0 (2.5–7.8) | 6.8 (1.5–8.5) | >0.05 b |
| ADL sore, median (IQR) | 33.8 (32.8–33.8) | 33.6 (29.0–34.0) | 36.6 (33.8–43.0) | 32.3 (33.8–33.8) | 34.4 (33.8–33.8) | 33.2 (33.8–33.8) | >0.05 b |
| NPI sore, median (IQR) | 22.9 (20.0–22.9) | 22.3 (14.8–22.9) | 25.6 (15.0–31.0) | 22.3 (22.9–22.9) | 24.4 (22.9–23.7) | 22.0 (22.4–22.9) | >0.05 b |
Abbreviations: AAO, age at onset; bvFTD, behavioral variant frontotemporal dementia; FTD, frontotemporal dementia; FTD‐ALS, frontotemporal dementia–amyotrophic lateral sclerosis; FTD‐P, frontotemporal dementia–Parkinson's; IQR, interquartile range; MMSE, Mini‐Mental State Examination; MoCA, Montreal Cognitive Assessment; NPI, Neuropsychiatric Inventory; PNFA, progressive non‐fluent aphasia; SD, semantic dementia; std dev, standard deviation.
Chi‐square test.
Kruskal–Wallis H test.
One‐way analysis of variance.
3.2. Causative gene mutations spectrum
We identified 104 rare mutations linked to FTD‐related genes using WGS. According to the ACMG criteria, 47 24 P/LP mutations across 13 genes were identified in 24 patients. In addition, 56 variants of unknown significance (VUS) were found in 23 genes from 49 patients. Adding the 24 P/LP mutations, which were previously reported by our team using gene‐targeted sequencing, 1 , 2 , 3 , 38 , 39 we identified P/LP and VUS variants in 19.4% (48/248) and 22.6% (56/248) of FTD patients, respectively.
Among the P/LP mutations (Table 2, Figure 2), the three most frequently causative genes were C9orf72 (n = 13; 5.2%), MAPT (n = 12; 4.8%), and CHCHD10 (n = 6; 2.4%), followed by GRN (n = 4, 1.6%), TBK1 (n = 3, 1.2%), VCP (n = 2, 0.8%), ERBB4 (n = 2, 0.8%), FUS (n = 1, 0.4%), CCNF (n = 1, 0.4%), OPTN (n = 1, 0.4%), SQSTM1 (n = 1, 0.4%), SIGMAR1 (n = 1, 0.4%), and PRKAR1B (n = 1, 0.4%). Among the P/LP mutations, we found seven were novel, including one nonsense mutation (GRN: p.W49X), one deletion mutation (CCNF: p.Q297del), one insertion mutation (FUS: p. G230_Y231insG), and four missense mutations (VCP: p.N91K; PRKAR1B: p.R335Q; ERBB4: p.I959T, p.S701R). The information on VUS and their corresponding clinical phenotypes are shown in Table S3 in supporting information. It was noted that 10 cases with C9orf72 repeat expansion were identified using ExpansionHunter software, 48 which were first identified using WGS data, and these findings were further confirmed using TP‐PCR (Figure S1 in supporting information).
TABLE 2.
Clinical characteristics and variant information of patients with pathogenic or likely pathogenic variants.
| Genes | Base change | Protein change | ACMG | Pathogenic level | Novel mutation | Phenotype | Sex | Age at onset (years) | Disease course (years) | Family history |
|---|---|---|---|---|---|---|---|---|---|---|
| C9orf72 | ∖ | ∖ | ∖ | Pathogenic | No | FTD‐ALS | Male | 46 | 1 | Yes |
| No | bvFTD | Male | 65 | 2 | No | |||||
| No | PNFA | Female | 57 | 5 | No | |||||
| No | bvFTD | Female | 49 | 2 | No | |||||
| No | PNFA | Female | 54 | 3 | Yes | |||||
| No | bvFTD | Female | 44 | 7 | Yes | |||||
| No | SD | Female | 44 | 1 | Yes | |||||
| No | bvFTD | Male | 56 | 3 | No | |||||
| No | FTD‐ALS | Female | 44 | 1 | No | |||||
| No | PNFA | Female | 47 | 5 | No | |||||
| No | bvFTD | Female | 72 | 2 | Yes | |||||
| No | bvFTD | Male | 52 | 2 | Yes | |||||
| No | bvFTD | Female | 47 | 2 | No | |||||
| MAPT | c.1537C > G | p.P513A | PM1+PM2+PP1+PP3 | Likely pathogenic | No | bvFTD | Female | 55 | 1 | No |
| No | bvFTD | Male | 77 | 5 | No | |||||
| No | PNFA | Male | 48 | 4 | Yes | |||||
| MAPT | c.14G > A | p.R5H | PS3+PM1+PM2+PM5 | Pathogenic | No | bvFTD | Male | 57 | 5 | No |
| No | bvFTD | Female | 83 | 2 | No | |||||
| MAPT | c.1842T > G | p.N614K | PS3+PM1+PM2+PP3+PP5 | Pathogenic | No | FTD‐P | Female | 40 | 5 | Yes |
| No | FTD‐P | Female | 42 | 5 | No | |||||
| No | FTD‐P | Male | 38 | 1 | Yes | |||||
| MAPT | c.530A > T | p.D177V | PM1+PM2+PP3+PP5 | Likely pathogenic | No | PNFA | Male | 57 | 3 | No |
| No | PNFA | Female | 32 | 4 | Yes | |||||
| MAPT | c.1907C > T | p.P636L | PS3+PM1+PM2+PM5 | Pathogenic | No | SD | Male | 58 | 5 | No |
| No | SD | Male | 54 | 3 | Yes | |||||
| GRN | c.328C > T | p.R110X | PVS1+PM2+PP5 | Pathogenic | No | SD | Female | 61 | 1 | No |
| GRN | c.20G > A | p.W7X | PVS1+PM2+PP3 | Pathogenic | No | bvFTD | Female | 73 | 4 | Yes |
| GRN | c.898C > T | p.Q300X | PVS1+PM2+PP5 | Pathogenic | No | bvFTD | Male | 63 | 4 | No |
| GRN | c.147_159delinsAGC | p.W49X | PVS1+PM2+PP3 | Pathogenic | Yes | SD | Female | 66 | 1 | No |
| CHCHD10 | c.64C > T | p.H22Y | PM1+PM2+PP3+PP5 | Likely pathogenic | No | bvFTD | Male | 54 | 10 | No |
| CHCHD10 | c.67C > T | p.P23S | PM1+PM2+PP3+PP6 | Likely pathogenic | No | bvFTD | Male | 66 | 3 | No |
| CHCHD10 | c.68C > T | p.P23L | PM1+PM2+PP3+PP7 | Likely pathogenic | No | SD | Male | 52 | 4 | No |
| CHCHD10 | c.95C > A | p.A32D | PM1+PM2+PP3+PP8 | Likely pathogenic | No | bvFTD | Male | 76 | 1 | No |
| CHCHD10 | c.170T > A | p.V57E | PM1+PM2+PP3+PP9 | Likely pathogenic | No | bvFTD | Female | 60 | 4 | No |
| CHCHD10 | c.121C > T | p.Q41X | PVS1+PM2+PP3 | Pathogenic | No | bvFTD | Female | 56 | 10 | No |
| TBK1 | c.973dup | p.Y325Lfs*4 | PVS1+PM2+PP3 | Pathogenic | No | PNFA | Male | 61 | 1 | No |
| TBK1 | c.2063_2064delTT | p.L688Rfs*14 | PVS1+PM2+PM4+PP3 | Pathogenic | No | FTD‐ALS | Male | 45 | 1 | Yes |
| TBK1 | c.1959_1960insGT | p.E653fs | PVS1+PM2+PM4+PP3 | Pathogenic | No | SD | Female | 63 | 2 | No |
| VCP | c.273C > G | p.N91K | PM1+PM2+PM5+PP3 | Likely pathogenic | Yes | bvFTD | Male | 59 | 6 | No |
| VCP | c.475C > T | p.R159C | PS3+PM1+PM2+PM5+PP3 | Pathogenic | No | bvFTD | Male | 51 | 7 | No |
| OPTN | c.1402_1407del | p.M468_E469del | PM1+PM2+PM4+PP5 | Likely pathogenic | No | PNFA | Female | 63 | 1 | No |
| SQSTM1 | c.558_559insC | p.V287Rfs*21 | PVS1+PM1+PM4+PP3 | Pathogenic | No | bvFTD | Female | 71 | 1 | No |
| SIGMAR1 | c.26G > A | p.W9X | PVS1+PM1+PM2+PP3 | Pathogenic | No | bvFTD | Female | 74 | 3 | No |
| FUS | c.663_664insGGC | p.G230_Y231insG | PM1+PM2+PM4 | Likely pathogenic | Yes | PNFA | Male | 35 | 1 | No |
| CCNF | c.884_886del | p.Q297del | PM1+PM2+PM4 | Likely pathogenic | Yes | SD | Female | 63 | 3 | Yes |
| ERBB4 | c.2876T > C | p.I959T | PM1+PM2+PP2+PP3 | Likely pathogenic | Yes | PNFA | Male | 70 | 10 | No |
| ERBB4 | c.2101A > C | p.S701R | PM1+PM2+PP2+PP3 | Likely pathogenic | Yes | PNFA | Male | 50 | 1 | No |
| PRKAR1B | c.1004G > A | p.R335Q | PM1+PM2+PM5+PP3 | Likely pathogenic | Yes | bvFTD | Female | 40 | 1 | No |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; bvFTD, behavioral variant frontotemporal dementia; FTD, frontotemporal dementia; FTD‐ALS, frontotemporal dementia–amyotrophic lateral sclerosis; FTD‐P, frontotemporal dementia–Parkinson's; PNFA, progressive non‐fluent aphasia; SD, semantic dementia.
FIGURE 2.
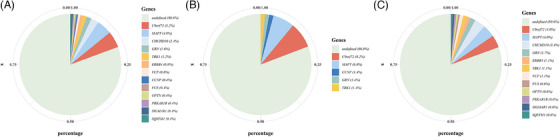
Genetic spectrum of the FTD participants. (A) The frequencies of P/LP mutations in the whole FTD cohort. (B) The frequencies of P/LP mutations in patients with familial FTD. (C) The frequencies of P/LP mutations in patients with sporadic FTD. FTD, frontotemporal dementia; P/LP, pathogenic or likely pathogenic
3.3. Phenotype of FTD‐related gene mutation carriers
The genetic spectrum varied between familial and sporadic cases of FTD. As shown in Figure 2, C9orf72 dynamic mutations (8.2%) emerged as the most common causative gene in familial FTD, followed by MAPT (6.8%). In sporadic FTD, the predominant causative genes were C9orf72 dynamic mutations (4.0%) and MAPT (4.0%), followed by CHCHD10 (3.4%).
Distinct phenotype distributions were observed among different P/LP mutations within the FTD genes (Figure 3A). C9orf72 dynamic mutations were most frequently associated with bvFTD, followed by PNFA and FTD‐ALS, but not with FTD‐P. Mutations in MAPT were more prevalent in bvFTD and were also found in PNFA and SD, but not in FTD‐ALS. Notably, only MAPT mutations were identified in FTD‐P. CHCHD10 mutations were predominantly found in bvFTD. GRN mutations were exclusively observed in bvFTD and SD.
FIGURE 3.
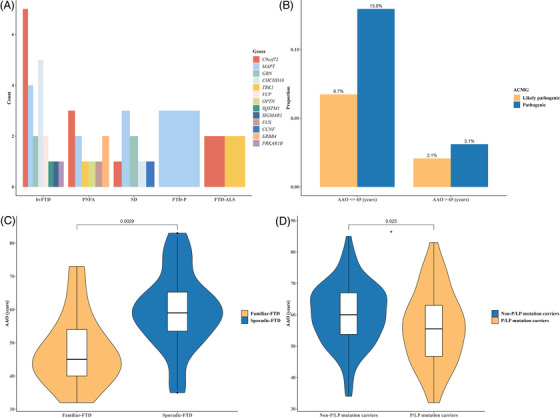
Genotype‐phenotype information of P/LP mutation carriers. (A) Phenotype distribution carrying each P/LP mutation. (B) Distribution of P/LP mutations in different AAO. (C) Comparison of AAO in all familiar and sporadic FTD patients with P/LP mutations. (D) Comparison of AAO between P/LP mutation carriers and non‐P/LP mutation carriers. AAO, age at onset; ACMG, American College of Medical Genetics and Genomics; bvFTD, behavioral variant frontotemporal dementia; FTD, frontotemporal dementia; FTD‐ALS, frontotemporal dementia–amyotrophic lateral sclerosis; FTD‐P, frontotemporal dementia–Parkinson's disease; P/LP, pathogenic or likely pathogenic; PNFA, progressive non‐fluent aphasia; SD, semantic dementia.
Regarding the phenotype, bvFTD exhibited the highest mutation frequency, encompassing C9orf72, MAPT, CHCHD10, GRN, VCP, SIGMAR1, SQSTM1, and PRKAR1B mutations. Patients with PNFA predominantly carried MAPT, C9orf72, and ERBB4 mutations; those with SD carried mutations in MAPT and GRN; those with FTD‐ALS mainly carried mutations in C9orf72 and TBK1; and those with FTD‐P only carried MAPT mutations.
The frequency of P/LP mutations in patients with AAO of ≤ 65 years surpassed that in patients > 65 years (Figure 3B). Additionally, among patients with P/LP mutations, AAO for familial FTD was significantly earlier than that for sporadic FTD (p < 0.01, Figure 3C). We also found that the AAO in P/LP mutation carriers was significantly earlier than in non‐P/LP mutation carriers (p < 0.05, Figure 3D). In contrast, no significant differences were found in sex, disease duration, and family history between P/LP mutation carriers and non‐P/LP mutation carriers.
3.4. Plasma biomarker levels in FTD and its subtypes
The distribution of plasma GFAP, α‐syn, NfL, and p‐tau181 in patients with FTD or its subtypes and controls is presented in Figure 4. Compared to healthy controls, the levels of these four biomarkers were significantly elevated in the FTD group. Concerning its subtypes, the levels of GFAP in bvFTD, SD, and PNFA; NfL in bvFTD, SD, and FTD‐ALS; p‐tau181 in bvFTD and FTD‐P; and α‐syn in bvFTD, SD, PNFA, and FTD‐ALS, were significantly higher than those in the control group.
FIGURE 4.
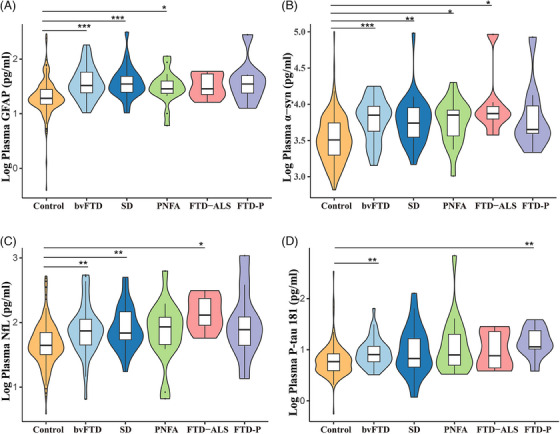
Comparisons of plasma GFAP, α‐syn, NfL, and p‐tau181 across controls and patients with different FTD phenotypes. α‐syn, α‐synuclein; bvFTD, behavioral variant frontotemporal dementia; FTD, frontotemporal dementia; FTD‐ALS, frontotemporal dementia–amyotrophic lateral sclerosis; FTD‐P, frontotemporal dementia–Parkinson's; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; PNFA, progressive non‐fluent aphasia; p‐tau181, phosphorylated tau protein 181; SD, semantic dementia.
We further compared the levels of the four plasma biomarkers among P/LP mutation carriers, VUS carriers, non‐mutation carriers, and normal controls. The levels of GFAP, α‐syn, NfL, and p‐tau181 were not significantly different in P/LP mutation carriers compared to VUS and non‐mutation carriers (Figure 5). Interestingly, NfL and α‐syn levels in P/LP mutation carriers of FTD patients tend to be higher than those in VUS carriers and non‐mutation carriers.
FIGURE 5.
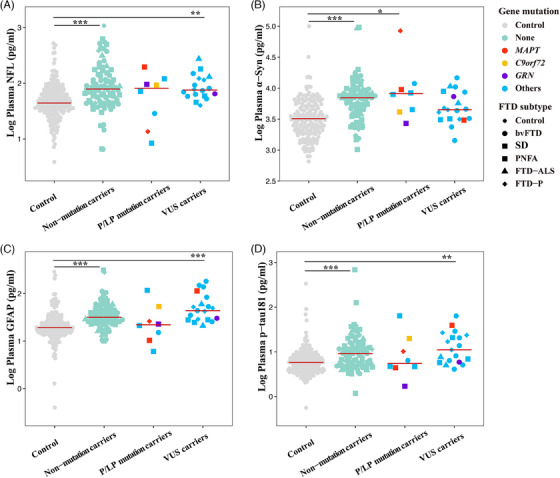
Comparison of plasma NfL, α‐syn, GFAP, and p‐tau181 levels among P/LP mutation carriers, VUS carriers, non‐mutation carriers, and controls. α‐syn, α‐synuclein; bvFTD, behavioral variant frontotemporal dementia; FTD, frontotemporal dementia; FTD‐ALS, frontotemporal dementia–amyotrophic lateral sclerosis; FTD‐P, frontotemporal dementia–Parkinson's disease; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; P/LP, pathogenic or likely pathogenic; PNFA, progressive non‐fluent aphasia; p‐tau181, phosphorylated tau protein 181; SD, semantic dementia; VUS, variants of unknown significance.
3.5. Classification analyses
We further assessed the diagnostic performance of various biomarkers for diagnosing FTD by plotting ROC curves (Figure 6). GFAP and α‐syn levels showed the best performance for diagnosing FTD compared to controls (GFAP: AUC = 0.742, 0.681–0.802; α‐syn: AUC = 0.742, 0.682–0.801), followed by NfL (AUC = 0.707, 0.644–0.801), while p‐tau181 showed the lowest diagnostic power (AUC = 0.68, 0.614–0.745).
FIGURE 6.
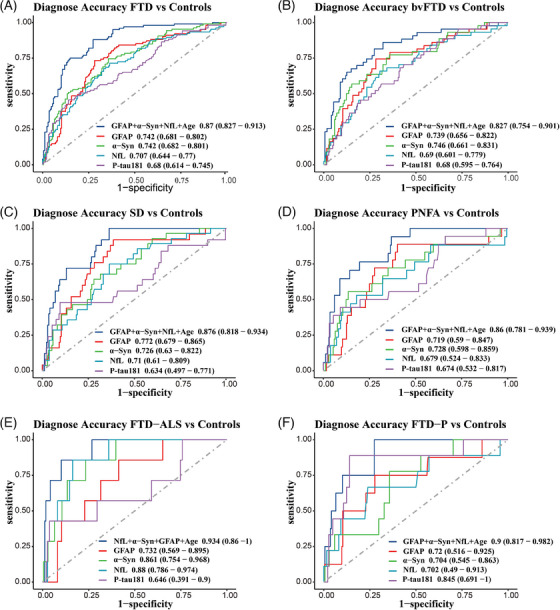
Receiver operating characteristic (ROC) curves of predictive models and individual plasma biomarkers for discriminating FTD and individual clinical subtypes from healthy controls. α‐syn, α‐synuclein; bvFTD, behavioral variant frontotemporal dementia; FTD, frontotemporal dementia; FTD‐ALS, frontotemporal dementia–amyotrophic lateral sclerosis; FTD‐P, frontotemporal dementia–Parkinson's disease; GFAP, glial fibrillary acidic protein; NfL, neurofilament light chain; PNFA, progressive non‐fluent aphasia; p‐tau181, phosphorylated tau protein 181; SD, semantic dementia
For each phenotype of FTD, GFAP and α‐syn levels also performed well, with an AUC > 0.7 while α‐syn reached an AUC of 0.86 when diagnosing FTD‐ALS. In addition, p‐tau181 outperformed GFAP, α‐syn, and NfL in diagnosing the FTD‐P subtype, with an AUC of 0.85. Notably, a combination of GFAP, α‐syn, and NfL levels with age demonstrated the highest diagnostic value (bvFTD: AUC = 0.827, 0.754–0.901; SD: AUC = 0.876, 0.818–0.934; PNFA: AUC = 0.86, 0.781–0.939; FTD‐ALS: AUC = 0.934, 0.86–1; FTD‐P: AUC = 0.9, 0.817–0.982).
4. DISCUSSION
This was a FTD cohort study with a large sample size conducted in Asia, involving phenotypes, genotypes, and diagnostic accuracy evaluation of four plasma biomarkers (GFAP, α‐syn, NfL, and p‐tau181) for FTD.
In terms of phenotypes, numerous studies in White populations have found bvFTD to be the most common phenotype of FTD, followed by PPA, whereas FTD‐ALS and FTD‐P are relatively rare. 27 , 49 , 50 Likewise, we observed that bvFTD accounted for the largest proportion of the Chinese population (45.6%). BvFTD was the most common phenotype among C9orf72 (53.8%), MAPT (33.3%), and CHCHD10 (83.3%) P/LP mutation carriers in our cohort. Additionally, only bvFTD was found in patients with VCP, SQSTM1, SIGMAR1, and PRKA1B P/LP mutations. The PPA phenotype ranks second (PNFA and SD account for 15.3% and 21.0%, respectively). The PNFA phenotype was mainly concentrated in C9orf72 (23.1%), MAPT (16.7%), and TBK1 (33.3%) mutation carriers and was only enriched in ERBB4, FUS, and OPTN mutations. The SD phenotype was more common in GRN (50.0%) P/LP mutation carriers and was the only phenotype in CCNF mutation carriers. All FTD‐P subtypes were concentrated in patients with MAPT mutation. The FTD‐ALS phenotype was concentrated in FTD‐ALS–related gene mutations, including C9orf72, TBK1, OPTN, and SETX, with C9orf72 accounting for the majority, proving the genetic overlap between FTD and ALS.
FTD typically occurs between 45 and 65 years of age. 27 We report that the average AAO in our cohort was 59.4 ± 10.4 years and FTD‐P had the earliest AAO, followed by PNFA, FTD‐ALS, bvFTD, and SD. A large AAO study found that MAPT (49.5 years) mutation carriers tend to develop the disease earliest, followed by C9orf72 (58.2 years) and GRN (61.3 years). 51 Among P/LP mutation carriers, we found that the average AAO fluctuated in the range of 52.5 to 65.8 years old, with the earliest in patients with C9orf72 (52.5 years) mutations and the latest in patients with GRN (65.6 years) mutations. A positive family history accounted for a high proportion of these patients; 46.2% of patients with C9orf72 mutations had a family history, followed by MAPT and GRN, accounting for 41.7% and 25.0%, respectively. No positive family history was observed in patients with CHCHD10 mutations.
We identified 48 P/LP mutations within FTD genes in 248 patients with FTD (73 familial and 175 familial individuals). Notably, we found that the frequency of C9orf72 dynamic mutation at 5.2% ranked first in our cohort, and reached 8.2% and 4.0% in familial and sporadic patients, respectively. Several studies have reported that C9orf72 dynamic mutation is the most frequent genetic cause of FTD in Europe and North America (3.5%–29.3%), with frequencies ranging from 2.7% to 53.8% and 2.2% to 22.6% in familial and sporadic patients with FTD, respectively. 52 The high frequency of C9orf72 expansion is thought to be related to populations of Caucasian origin. 53 Notably, the C9orf72 mutation is extremely rare in Asia (0%–4.8%). 53 , 54 Studies in China with a large sample size (> 100 participants) have found that ≈ 0% to 1.2% of patients with FTD carry C9orf72 mutations. 1 , 55 , 56 This figure is ≈ 0% to 3.0% in Japan 57 and 0% in Korea. 58 Our results reveal for the first time that the C9orf72 dynamic mutation is the most common in FTD in China. The reason for this contrast may be due to the lack of large cohort studies of FTD in Asian populations. 56 Moreover, screening for G4C2 repeat expansion of C9ofr72 is often ignored in the analysis of WGS data. We used the ExpansionHunter software 48 for the first time to identify C9orf72 dynamic mutation via WGS data, and this was verified using TP‐PCR, which illustrates the potential of WGS data to identify short tandem repeats (STR). The mutation frequencies inferior to C9orf72 were MAPT (4.8%), CHCHD10 (2.4%), and GRN (1.6%). MAPT is common in Chinese patients with FTD, 59 , 60 , 61 accounting for ≈ 5.0% of all hereditary FTD cases. 62 , 63 MAPT mutations ranked second in our cohort. Patients with MAPT mutations mainly present with executive dysfunction, personality and behavioral changes, language impairment, cognitive decline, and movement disorders. 64 Our findings align with these observations, as the most frequent clinical manifestation of MAPT mutations observed in our cohort was bvFTD. The third most frequently mutated gene was CHCHD10, accounting for 2.4%. The estimated frequency of CHCHD10 mutations ranges from 0.3% to 2.6% for FTD in European populations. 26 Previous studies supported that CHCHD10 mutations have a higher frequency in the Chinese population and are most common in sporadic FTD. 64 In addition to the P/LP mutations of the above four genes, we also found 13 other P/LP mutations in 13 patients, distributed in TBK1, VCP, ERBB4, FUS, CCNF, OPTN, SQSTM1, SIGMAR1, and PRKAR1B genes, accounting for 5.2% of all FTD patients. Patients with P/LP mutations in VCP, SQSTM1, SIGMAR1, and PRKAR1B manifested bvFTD, while patients with P/LP mutations in OPTN, FUS, and ERBB4 presented with PNFA, and patients with P/LP mutations in CCNF and TBK1 presented with SD and FTD‐ALS, respectively. These variants prompted us to pay attention to these rare FTD genes in the clinic because patients with FTD carrying these mutations often do not have specific clinical phenotypes or characteristic symptoms. A total of 56 VUS variants were identified, distributed in MAPT, GRN, TBK1, VCP, OPTN, SQSTM1, SIGMAR1, CHMP2B, FUS, CCNF, TIA1, CYLD, DCTN1, ERBB4, TREM2, MATR3, NEK1, DNAJC7, SETX, CHRNA4, CHRNB4, HNRNPA1, and GLT8D1 genes. The pathogenicity of these variants is currently unknown and may be further clarified in the future through validation in other populations, co‐segregation in families, and functional studies.
Biomarkers are pivotal in FTD research. We detected the levels of four plasma biomarkers (GFAP, α‐syn, NfL, and p‐tau181) in our cohort. We found these four biomarkers were significantly elevated among FTD patients compared to controls. At present, the levels of biomarkers in FTD are not fully understood. Recently published articles found that there was no significant difference in GFAP and NfL levels between FTD patients and normal controls, 65 but other studies thought that both were significantly increased in FTD patients. 66 , 67 This may be because GFAP and NfL are possibly involved in the pathogenesis of FTD as non‐specific indicators of immune inflammation and nerve damage. P‐tau181 is considered a potential biomarker for identifying AD, 32 and its increased level in FTD patients indicates that there is a certain overlap between the two in terms of clinical phenotype and pathology. Meanwhile, we analyzed the relationship between FTD and α‐syn for the first time and found that it was significantly increased in FTD patients. α‐syn is thought to be associated with AD and Parkinson's disease (PD). There is a clinical overlap between the FTD‐P phenotype and PD, so our findings may suggest that α‐syn is also linked to FTD. Genetic factors can also affect biomarker levels. For example, a significant increase in NfL is observed in symptomatic GRN mutation carriers, which is much higher than that in patients with sporadic FTD. 66 We compared the biomarker levels among P/LP mutation carriers, VUS carriers, and non‐mutation carriers, and found no significant difference between P/LP mutation carriers and the other two groups. This may be due to the insufficient number of P/LP mutations included. The FTD cohort needs to be further expanded in the future to verify their relationship. We also found that GFAP and α‐syn had the highest diagnostic accuracy for FTD and its subtypes (bvFTD and PPA). GFAP is reportedly involved in FTD, and differences in these markers have been observed in cohorts with different subtypes and genetic mutations. 68 , 69 , 70 , 71 , 72 High plasma GFAP levels predicted future temporal lobe atrophy, decline in cognitive function, and decreased survival in longitudinal analyses. 66 Our results further confirmed the effectiveness of GFAP in diagnosing FTD, indicating that GFAP level showed the best performance in detecting FTD. Currently, there are no relevant reports explaining the exact relationship between α‐syn and FTD. We found that α‐syn performed well in diagnosing each FTD phenotype, perhaps indicating that α‐syn is also involved in the pathogenesis of FTD. As α‐syn accumulation can also occur in glial cells, 73 its role in glial cell activation may also activate FTD‐related neuropathological responses. A recent study revealed that blood NfL is a highly reliable biomarker in FTD, 74 and results from our cohort further support this, especially in diagnosing FTD‐ALS and FTD‐P. Furthermore, we found that p‐tau181 was the best marker for detecting FTD‐P. Sanchez et al. found that p‐tau181 has an increasing trend in patients with FTD and is broadly associated with worse baseline cognitive performance, greater cognitive decline, and/or lower functionality, 65 illustrating that p‐tau181 deserves further study regarding its relationship with FTD, especially in the FTD‐P phenotype.
In conclusion, based on a large cohort of Chinese patients with FTD, this study updated the characteristics of the FTD phenotypic and genetic spectrum, and found that C9orf72 dynamic mutation is the most common mutation type of FTD in Chinese patients with FTD. It also underscored the diagnostic utility of these biomarkers in identifying FTD.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent. Written informed consent was obtained from each participant or their legal representatives. The study was approved by the ethics committee of Xiangya Hospital of the Central South University in China (equivalent to an institutional review board) with the ethics number 2022020483.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
Lu Shen and Bin Jiao had full access to all of the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis. Bin Jiao, Lu Shen, Beisha Tang, Shilin Luo, and Junling Wang designed the study. Participant recruitment and data collection: Tianyan Xu, Ling Weng, Cong Zhang, Xuewen Xiao, Qijie Yang, Yuan Zhu, Yafang Zhou, Xinxin Liao. The plasma biomarker detection was performed by Tianyan Xu and Cong Zhang. The data was analyzed by Tianyan Xu, Ling Weng, and Cong Zhang. Tianyan Xu drafted the manuscript. Critical revision of the manuscript was made by Tianyan Xu, Shilin Luo, Junling Wang, Beisha Tang, Bin Jiao, and Lu Shen. Lu Shen and Bin Jiao supervised the completion of this study. This study was supported by the STI2030‐Major Projects (No. 2021ZD0201803), the National Natural Science Foundation of China (No. U22A20300, 82071216, 82371434), the Science and Technology Major Project of Hunan Province (2021SK1020), Outstanding Youth Fund of Hunan Provincial Natural Science Foundation (2024JJ2097), Hunan Health Commission (20232460), Hunan Innovative Province Construction Project (2021SK1010), Hunan Provincial Natural Science Foundation of China (2023JJ40792), and the Grant of National Clinical Research Center for Geriatric Disorders, Xiangya Hospital (2022LNJJ16).
Xu T, Weng L, Zhang C, et al. Genetic spectrum features and diagnostic accuracy of four plasma biomarkers in 248 Chinese patients with frontotemporal dementia. Alzheimer's Dement. 2024;20:7281–7295. 10.1002/alz.14215
Contributor Information
Bin Jiao, Email: jiaobin@csu.edu.cn.
Lu Shen, Email: shenlu@csu.edu.cn.
REFERENCES
- 1. Jiao B, Liu H, Guo L, et al. The role of genetics in neurodegenerative dementia: a large cohort study in South China. NPJ Genom Med. 2021;6(1):69. doi: 10.1038/s41525-021-00235-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jiao B, Sun Q, Yuan Z, et al. Rare TBK1 variants in patients with frontotemporal dementia and amyotrophic lateral sclerosis in a Chinese cohort. Transl Neurodegener. 2018;7(1):31. doi: 10.1186/s40035-018-0136-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiao B, Tang B, Liu X, et al. Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiol Aging. 2014;35(4):936.e19‐936.e22. doi: 10.1016/j.neurobiolaging.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 4. Gaweda‐Walerych K, Sitek EJ, Borczyk M, et al. A patient with corticobasal syndrome and progressive non‐fluent aphasia (CBS‐PNFA), with variants in ATP7B, SETX, SORL1, and FOXP1 Genes. Genes. 2022;13(12):2361. doi: 10.3390/genes13122361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 Causes Chromosome 9p‐Linked FTD and ALS. Neuron. 2011;72(2):245‐256. doi: 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815‐825. doi: 10.1002/ana.410430617 [DOI] [PubMed] [Google Scholar]
- 7. Baker M, Mackenzie IR, Pickering‐Brown SM, et al. Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916‐919. doi: 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- 8. Bannwarth S, Ait‐El‐Mkadem S, Chaussenot A, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137(8):2329‐2345. doi: 10.1093/brain/awu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci. 2015;18(5):631‐636. doi: 10.1038/nn.4000 [DOI] [PubMed] [Google Scholar]
- 10. Watts GDJ, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet. 2004;36(4):377‐381. doi: 10.1038/ng1332 [DOI] [PubMed] [Google Scholar]
- 11. Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806‐808. doi: 10.1038/ng1609 [DOI] [PubMed] [Google Scholar]
- 12. Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409‐416. doi: 10.1016/S1474-4422(08)70071-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(11):2922‐2931. doi: 10.1093/brain/awp214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luty AA, Kwok JBJ, Dobson‐Stone C, et al. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration–motor neuron disease. Ann Neurol. 2010;68(5):639‐649. doi: 10.1002/ana.22274 [DOI] [PubMed] [Google Scholar]
- 15. Appel SH, Rowland LP. Amyotrophic lateral sclerosis, frontotemporal lobar dementia, and p62: a functional convergence? Neurology. 2012;79(15):1526‐1527. doi: 10.1212/WNL.0b013e31826e26ec [DOI] [PubMed] [Google Scholar]
- 16. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature. 2011;477(7363):211‐215. doi: 10.1038/nature10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pottier C, Bieniek KF, Finch N, et al. Whole‐genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130(1):77‐92. doi: 10.1007/s00401-015-1436-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams KL, Topp S, Yang S, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. 2016;7(1):11253. doi: 10.1038/ncomms11253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mackenzie IR, Nicholson AM, Sarkar M, et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron. 2017;95(4):808‐816.e9. doi: 10.1016/j.neuron.2017.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dobson‐Stone C, Hallupp M, Shahheydari H, et al. CYLD is a causative gene for frontotemporal dementia – amyotrophic lateral sclerosis. Brain. 2020;143(3):783‐799. doi: 10.1093/brain/awaa039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogonowski N, Santamaria‐Garcia H, Baez S, et al. Frontotemporal dementia presentation in patients with heterozygous p.H157Y variant of TREM2 . J Med Genet. 2023;60(9):894‐904. doi: 10.1136/jmg-2022-108627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brenner D, Weishaupt JH. Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol. 2019;32(5):735‐739. doi: 10.1097/WCO.0000000000000737 [DOI] [PubMed] [Google Scholar]
- 23. Jankovska N, Matej R. Molecular pathology of ALS: what we currently know and what important information is still missing. Diagnostics. 2021;11(8):1365. doi: 10.3390/diagnostics11081365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirola L, Mukherjee A, Mutsuddi M. Recent updates on the genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Mol Neurobiol. 2022;59(9):5673‐5694. doi: 10.1007/s12035-022-02934-z [DOI] [PubMed] [Google Scholar]
- 25. Grossman M, Seeley WW, Boxer AL, et al. Frontotemporal lobar degeneration. Nat Rev Dis Primers. 2023;9(1):40. doi: 10.1038/s41572-023-00447-0 [DOI] [PubMed] [Google Scholar]
- 26. Younes K, Miller BL. Frontotemporal Dementia. Psychiatr Clin North Am. 2020;43(2):331‐344. doi: 10.1016/j.psc.2020.02.006 [DOI] [PubMed] [Google Scholar]
- 27. Erkkinen MG, Kim MO, Geschwind MD. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2018;10(4):a033118. doi: 10.1101/cshperspect.a033118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Creekmore BC, Watanabe R, Lee EB. Neurodegenerative disease tauopathies. Annu Rev Pathol Mech Dis. 2024;19(1):345‐370. doi: 10.1146/annurev-pathmechdis-051222-120750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buccellato FR, D'Anca M, Tartaglia GM, Del Fabbro M, Galimberti D. Frontotemporal dementia: from genetics to therapeutic approaches. Expert Opinion on Investigational Drugs. 2024;33(6):561‐573. doi: 10.1080/13543784.2024.2349286 [DOI] [PubMed] [Google Scholar]
- 30. Huang M, Modeste E, Dammer E, et al. Network analysis of the progranulin‐deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol Commun. 2020;8(1):163. doi: 10.1186/s40478-020-01037-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deleon J, Miller BL. Frontotemporal dementia. Handbook of Clinical Neurology. Elsevier; 2018:409‐430. doi: 10.1016/B978-0-444-64076-5.00027-2 [DOI] [PubMed] [Google Scholar]
- 32. Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer's disease. Biomarkers Med. 2012;6(4):455‐476. doi: 10.2217/bmm.12.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewczuk P, Kornhuber J. Neurochemical dementia diagnostics in Alzheimer's disease: where are we now and where are we going? Expert Rev Proteomics. 2011;8(4):447‐458. doi: 10.1586/epr.11.37 [DOI] [PubMed] [Google Scholar]
- 34. Lehmann S, Schraen‐Maschke S, Vidal JS, et al. Plasma phosphorylated tau 181 predicts amyloid status and conversion to dementia stage dependent on renal function. J Neurol Neurosurg Psychiatry. 2023;94(6):411‐419. doi: 10.1136/jnnp-2022-330540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foiani MS, Cicognola C, Ermann N, et al. Searching for novel cerebrospinal fluid biomarkers of tau pathology in frontotemporal dementia: an elusive quest. J Neurol Neurosurg Psychiatry. 2019;90(7):740‐746. doi: 10.1136/jnnp-2018-319266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Staffaroni AM, Quintana M, Wendelberger B, et al. Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat Med. 2022;28(10):2194‐2206. doi: 10.1038/s41591-022-01942-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci. 2018;19(11):687‐700. doi: 10.1038/s41583-018-0067-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiao B, Xiao T, Hou L, et al. High prevalence of CHCHD10 mutation in patients with frontotemporal dementia from China. Brain. 2016;139(4):e21‐e21. doi: 10.1093/brain/awv367 [DOI] [PubMed] [Google Scholar]
- 39. Tang M, Gu X, Wei J, et al. Analyses MAPT, GRN, and C9orf72 mutations in Chinese patients with frontotemporal dementia. Neurobiol Aging. 2016;46:235.e11‐235.e15. doi: 10.1016/j.neurobiolaging.2016.05.013 [DOI] [PubMed] [Google Scholar]
- 40. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456‐2477. doi: 10.1093/brain/awr179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006‐1014. doi: 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589‐595. doi: 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078‐2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164‐e164. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J, Shi L, Zhang K, et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018;46(D1):D1039‐D1048. doi: 10.1093/nar/gkx1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dolzhenko E, Deshpande V, Schlesinger F. ExpansionHunter: a sequence‐graph‐based tool to analyze variation in short tandem repeat regions. Bioinformatics. 2019;35(22):4754‐4756. doi: 10.1093/bioinformatics/btz431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burrell JR, Halliday GM, Kril JJ, et al. The frontotemporal dementia‐motor neuron disease continuum. The Lancet. 2016;388(10047):919‐931. doi: 10.1016/S0140-6736(16)00737-6 [DOI] [PubMed] [Google Scholar]
- 50. Leveille E, Ross OA, Gan‐Or Z. Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Relat Disord. 2021;90:142‐154. doi: 10.1016/j.parkreldis.2021.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19(2):145‐156. doi: 10.1016/S1474-4422(19)30394-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cruts M, Gijselinck I, Van Langenhove T, Van Der Zee J, Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36(8):450‐459. doi: 10.1016/j.tins.2013.04.010 [DOI] [PubMed] [Google Scholar]
- 53. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol. 2012;11(4):323‐330. doi: 10.1016/S1474-4422(12)70043-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Woollacott IOC, Mead S. The C9ORF72 expansion mutation: gene structure, phenotypic and diagnostic issues. Acta Neuropathol. 2014;127(3):319‐332. doi: 10.1007/s00401-014-1253-7 [DOI] [PubMed] [Google Scholar]
- 55. Che XQ, Lin GZ, Liu XH, Wang G, Zhao QH, Ren RJ. Genetic and neuroimaging analysis of SIGMAR1 for frontotemporal dementia. JAD. 2023;95(2):469‐475. doi: 10.3233/JAD-221195 [DOI] [PubMed] [Google Scholar]
- 56. Dong L, Wang J, Liu C, et al. Genetic spectrum and clinical heterogeneity of Chinese frontotemporal dementia patients: data from PUMCH Dementia Cohort. JAD. 2022;89(3):893‐901. doi: 10.3233/JAD-220594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Konno T, Shiga A, Tsujino A, et al. Japanese amyotrophic lateral sclerosis patients with GGGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry. 2013;84(4):398‐401. doi: 10.1136/jnnp-2012-302272 [DOI] [PubMed] [Google Scholar]
- 58. Kim EJ, Kim YE, Jang JH, et al. Analysis of frontotemporal dementia, amyotrophic lateral sclerosis, and other dementia‐related genes in 107 Korean patients with frontotemporal dementia. Neurobiol Aging. 2018;72:186.e1‐186.e7. doi: 10.1016/j.neurobiolaging.2018.06.031 [DOI] [PubMed] [Google Scholar]
- 59. Mann DMA, Snowden JS. Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype. Brain Pathology. 2017;27(6):723‐736. doi: 10.1111/bpa.12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Strang KH, Golde TE, Giasson BI. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. 2019;99(7):912‐928. doi: 10.1038/s41374-019-0197-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rademakers R, Cruts M, Van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24(4):277‐295. doi: 10.1002/humu.20086 [DOI] [PubMed] [Google Scholar]
- 62. Neumann M, Mackenzie IRA. Review: neuropathology of non‐tau frontotemporal lobar degeneration. Neuropathology Appl Neurobio. 2019;45(1):19‐40. doi: 10.1111/nan.12526 [DOI] [PubMed] [Google Scholar]
- 63. Wang J, Wang B, Zhou T. The advance on frontotemporal dementia (FTD)’s neuropathology and molecular genetics. Mediators Inflamm. 2022;2022:1‐6. doi: 10.1155/2022/5003902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiang Y, Jiao B, Xiao X, Shen L. Genetics of frontotemporal dementia in China. Amyotrophic Lateral Scler Frontotemporal Degeneration. 2021;22(5‐6):321‐335. doi: 10.1080/21678421.2021.1880596 [DOI] [PubMed] [Google Scholar]
- 65. Sanchez E, Wilkinson T, Coughlan G, et al. Association of plasma biomarkers with cognition, cognitive decline, and daily function across and within neurodegenerative diseases: results from the Ontario neurodegenerative disease research initiative. Alzheimer's & Dementia. 2024;20(3):1753‐1770. doi: 10.1002/alz.13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hsiao‐Nakamoto J, Chiu CL, VandeVrede L, et al. Alterations in Lysosomal, Glial and Neurodegenerative Biomarkers in Patients with Sporadic and Genetic Forms of Frontotemporal Dementia. Published online February 12, 2024. doi: 10.1101/2024.02.09.579529 [DOI]
- 67. Benedet AL, Milà‐Alomà M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471. doi: 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Boeve BF, Boxer AL, Kumfor F, Pijnenburg Y, Rohrer JD. Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022;21(3):258‐272. doi: 10.1016/S1474-4422(21)00341-0 [DOI] [PubMed] [Google Scholar]
- 69. Álvarez‐Sánchez L, Peña‐Bautista C, Ferré‐González L, et al. Assessment of plasma and cerebrospinal fluid biomarkers in different stages of Alzheimer's disease and frontotemporal dementia. IJMS. 2023;24(2):1226. doi: 10.3390/ijms24021226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chouliaras L, Thomas A, Malpetti M, et al. Differential levels of plasma biomarkers of neurodegeneration in Lewy body dementia, Alzheimer's disease, frontotemporal dementia and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2022;93(6):651‐658. doi: 10.1136/jnnp-2021-327788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cousins KAQ, Shaw LM, Chen‐Plotkin A, et al. Distinguishing frontotemporal lobar degeneration tau from TDP‐43 using plasma biomarkers. JAMA Neurol. 2022;79(11):1155. doi: 10.1001/jamaneurol.2022.3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thijssen EH, Verberk IMW, Kindermans J, et al. Differential diagnostic performance of a panel of plasma biomarkers for different types of dementia. Alz & Dem Diag Ass & Dis Mo. 2022;14(1):e12285. doi: 10.1002/dad2.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kwon S, Iba M, Kim C, Masliah E. Immunotherapies for aging‐related neurodegenerative diseases—emerging perspectives and new targets. Neurotherapeutics. 2020;17(3):935‐954. doi: 10.1007/s13311-020-00853-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Linnemann C, Wilke C, Mengel D, et al. NfL reliability across laboratories, stage‐dependent diagnostic performance and matrix comparability in genetic FTD: a large GENFI study. J Neurol Neurosurg Psychiatry. 2024:jnnp‐2023‐332464. doi: 10.1136/jnnp-2023-332464 Published online January 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information