

Abstract
INTRODUCTION
Multi‐omics studies in Alzheimer's disease (AD) revealed many potential disease pathways and therapeutic targets. Despite their promise of precision medicine, these studies lacked Black Americans (BA) and Latin Americans (LA), who are disproportionately affected by AD.
METHODS
To bridge this gap, Accelerating Medicines Partnership in Alzheimer's Disease (AMP‐AD) expanded brain multi‐omics profiling to multi‐ethnic donors.
RESULTS
We generated multi‐omics data and curated and harmonized phenotypic data from BA (n = 306), LA (n = 326), or BA and LA (n = 4) brain donors plus non‐Hispanic White (n = 252) and other (n = 20) ethnic groups, to establish a foundational dataset enriched for BA and LA participants. This study describes the data available to the research community, including transcriptome from three brain regions, whole genome sequence, and proteome measures.
DISCUSSION
The inclusion of traditionally underrepresented groups in multi‐omics studies is essential to discovering the full spectrum of precision medicine targets that will be pertinent to all populations affected with AD.
Highlights
Accelerating Medicines Partnership in Alzheimer's Disease Diversity Initiative led brain tissue profiling in multi‐ethnic populations.
Brain multi‐omics data is generated from Black American, Latin American, and non‐Hispanic White donors.
RNA, whole genome sequencing and tandem mass tag proteomicsis completed and shared.
Multiple brain regions including caudate, temporal and dorsolateral prefrontal cortex were profiled.
Keywords: Alzheimer's disease, data descriptor, multi‐omics, precision medicine, proteome, transcriptome, whole genome sequencing
1. BACKGROUND
Alzheimer's disease (AD) is a devastating neurodegenerative disorder that affects millions of people worldwide. 1 While AD is a global health concern, it has been observed that Black Americans (BA) and Latin Americans/Latinos/Hispanics (hereafter referred to as LA), are disproportionately affected by the disease. 2 The prevalence of AD in BA is ≈ twice that of non‐Hispanic Whites (NHW), while LA face a 1.5 times higher risk. Despite these alarming disparities in risk, BA and LA populations remain significantly underrepresented in AD research. 3 , 4 , 5 , 6
This underrepresentation becomes even more apparent in genetic studies, in which large‐scale genome‐wide association studies (GWAS) have yielded valuable insights into AD risk factors and potential therapeutic targets. The largest AD GWAS to date comprises > 1 million individuals 7 and, with other studies, identified 75 genetic risk loci 8 in NHW populations of European ancestry. In contrast, GWAS in BA and LA populations have suffered from limited power, 3 , 4 , 9 , 10 , 11 , 12 , 13 with sample sizes less than one to two orders of magnitude of those for NHW populations. Despite these limitations, genetic studies in BA populations identified novel AD risk loci 13 , 14 , 15 and demonstrated allelic heterogeneity for AD risk genes initially discovered in NHW populations, including TREM2 14 , 16 and ABCA7. 9 , 14 , 17 These findings highlight the knowledge to be gained by studying diverse populations, potentially enabling the development of personalized treatments and interventions for AD using a precision medicine approach, as demonstrated in cancer. 18 , 19
Genetic variant information is necessary but not sufficient to realize the promise of precision medicine. Multi‐omics data from large‐scale, diverse, and deeply phenotyped individuals are required to uncover disease pathways and mechanisms in all affected populations. With a goal to accelerate discovery of candidate drug targets and translate these discoveries to new therapies for AD, the Accelerating Medicines Partnership in Alzheimer's Disease (AMP‐AD) Target Discovery and Preclinical Validation Project was launched in 2014. 20 This effort led to the generation and analysis of RNA sequencing (RNAseq)–based transcriptome, whole genome sequence (WGS), proteome, metabolome, and epigenome data on > 2500 brain samples primarily from NHW donors with AD and non‐AD neuropathologies, and unaffected controls. This vast amount of rich, high‐quality data has been made available to the research community 21 , 22 , 23 , 24 and can be accessed through the AD Knowledge Portal. 20 , 25 This resource has been used to identify or validate potential risk mechanisms in AD and other neurodegenerative diseases (examples can be seen in reference citations 22 , 23 , 24 and 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 ) and led to the nomination of > 600 key driver genes/candidate targets for AD. These target nominations and the associated data, including a set of curated genomic analyses and target druggability information, have been made available via the AMP‐AD open‐source platform Agora (https://agora.ampadportal.org/).
Despite these advances, such multi‐omics studies of AD and related disorders (ADRD) have lacked sampling from BA and LA populations with few exceptions. 54 , 55 To bridge this data and knowledge gap, AMP‐AD investigators launched a diversity initiative to expand molecular profiling of brain tissue to multi‐ethnic donors. We generated WGS, transcriptome, and proteome data; curated and harmonized phenotypic data from BA (n = 306), LA (n = 326), and BA and LA (4) brain donors along with NHWs (n = 252) and other (n = 20) ethnic groups to establish a foundational multi‐omics dataset enriched for BA and LA participants. This study describes this unique dataset made available to the research community. These data will lay the groundwork for bridging the knowledge disparities in AD research and are expected to uncover pathways, molecules, and genetic variants that drive or contribute to AD in these populations. By focusing on these high‐risk populations and leveraging the infrastructure developed by AMP‐AD, this initiative promotes inclusivity in research, is aligned with the broader goal of advancing precision medicine for All of AD in the spirit of the National Institutes of Health All of Us program, 56 and aims to ultimately improve lives of all individuals affected by this devastating disease.
RESEARCH IN CONTEXT
Systematic review: Despite facing a higher risk of Alzheimer's disease (AD), Black and Latino Americans are notably absent from clinical trials and genomic studies, leading to a limited understanding of the disease's impact on these populations. Additionally, there is a lack of diversity in multi‐omics studies and brain tissue profiling efforts, hindering the identification of key mechanisms driving AD in high‐risk groups.
Interpretation: We launched the Accelerating Medicines Partnership in Alzheimer's Disease Diversity Initiative with the objective of performing multi‐omics profiling and analysis of samples from diverse cohorts. We describe transcriptome data from 2224 brain samples, proteome data from 1385 samples, and new whole genome sequencing from 626 samples, primarily from 908 multi‐ethnic donors enriched for Black and Latino American participants. These data are accompanied by harmonized neuropathologic diagnoses of AD (n = 500), control (n = 211), or other (n = 185).
Future directions: Immediate next steps include comprehensive quality control and analysis of these data to identify molecular signatures of AD in diverse populations.
2. METHODS AND RESULTS
2.1. Study populations by biospecimen and data‐contributing institutions
Five AMP‐AD data‐contributing institutions participated in providing brain samples and associated data for the AMP‐AD Diversity Initiative, which is enriched for donors from BA and LA populations. Each of the following institutions (Mayo Clinic, Rush University, Mount Sinai, Columbia University, and Emory University) coordinated the collection of these brain samples from their own networks of affiliated brain banks, cohort studies, and Alzheimer's Disease Research Centers (Table 1, with additional details of racial category and ethnicity per cohort in Table S1 in supporting information). In addition to new donors from the studies and cohorts described below, 307 predominantly NHW (96%) individuals previously characterized in the AMP‐AD 1.0 initiative are described in Table S2 in supporting information. These 307 samples were included in the proteomics to provide more balance to the batches (described in Methods). Of the new collection of samples described below, 62 individuals have newly generated transcriptomic or proteomic data as part of the Diverse Cohorts initiative but only have WGS available from AMP‐AD 1.0.
TABLE 1.
Tissue sample sources by contributing institutions and cohorts.
| Data contributor group | Participating cohorts | N |
|---|---|---|
| Columbia | Columbia ADRC | 61 |
| Biggs Institute Brain Bank | 6 | |
| Estudia Familiar de Influencia Genetica en Alzheimer (EFIGA) | 4 | |
| National Institute on Aging Alzheimer's disease Family Based Study (FBS) | 3 | |
| Washington Heights, Inwood Columbia Aging Project (WHICAP) | 31 | |
| Emory | Emory Goizueta ADRC | 112 |
| Mt Sinai Brain Bank | 22 | |
| UPenn CNDR | 22 | |
| Mayo Clinic | Mayo Clinic Brain Bank | 268 |
| Banner Sun Health Research Institute | 43 | |
| University of Florida (UFL) | 20 | |
| Mt. Sinai | Mt. Sinai Brain Bank | 88 |
| Rush | Clinical Core (CLINCOR) | 67 |
| Latino Core Study (LATC) | 1 | |
| Minority Aging Research Study (MARS) | 32 | |
| Religious Orders Study (ROS) | 56 | |
| Memory and Aging Project (MAP) | 72 | |
| Additional samples sourced from AMP‐AD 1.0 to balance proteomics batches a | ||
| Mt. Sinai | Mt. Sinai Brain Bank | 114 |
| Rush | ROS | 145 |
| MAP | 48 | |
Abbreviation: AD, Alzheimer's disease; ADRC, Alzheimer's Disease Research Center; AMP‐AD, Accelerating Medicines Partnership in Alzheimer's Disease; UPenn CNDR, University of Pennsylvania Center for Neurodegenerative Disease Research; WGS, whole genome sequencing.
These individuals were sourced from AMP‐AD 1.0 tissue repositories and added to the Diverse cohort samples for proteomics processing only, to fully balance batches by race, ethnicity, age, sex, diagnosis (AD), and tissue region. All individuals have accompanying WGS data generated during the AMP‐AD 1.0 initiative; WGS biospecimen data for these individuals can be found in the AD Knowledge Portal (syn53352733).
2.1.1. Mayo Clinic
Brain samples provided by the Mayo Clinic were from three brain banks: Mayo Clinic Florida Brain Bank (n = 268), the Arizona Study of Aging and Neurodegenerative Disorders and the Brain and Body Donation Program at Banner Sun Health (n = 43), and the University of Florida Human Brain and Tissue Bank (n = 20). There were 53 BA, 182 LA, and 96 NHW brain donors. Tissue samples from the superior temporal gyrus (STG), anterior caudate nucleus, and dorsolateral prefrontal cortex (DLPFC) were obtained from the donors. The Mayo Clinic Institutional Review Board (IRB) approved all this work. All donors or their next of kin provided informed consent.
The Mayo Clinic Brain Bank collects brain specimens with neurodegenerative diseases as well as unaffected controls. All donors from the Mayo Clinic Brain Bank underwent neuropathologic evaluation by Dr. Dennis W. Dickson. Neuropathologic AD diagnosis was made according to the NINCDS‐ADRDA criteria, 57 such that all AD donors had Braak neurofibrillary tangle (NFT) stage of ≥ IV and evidence of Thal ≥ 2 amyloid deposits.
The Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program at Banner Sun Health (Banner) has collected brain and whole body donations since 1987. 58 Donors are residents of retirement communities in Phoenix, Arizona, and are typically enrolled when they are cognitively normal, with directed recruitment efforts aimed at individuals with AD, Parkinson's disease, and cancer. Neuropathological diagnosis of AD followed standard National Institute on Aging (NIA) guidelines. 59
University of Florida (UFL) samples were collected through the University of Florida Human Brain and Tissue Bank (UFHBTB). All UFL brains underwent neuropathological diagnosis of AD according to current NIA guidelines, 59 , 60 with any degree of AD neuropathologic change resulting in an AD diagnosis.
2.1.2. Emory University
All samples were collected as part of ongoing studies at Emory's Goizueta Alzheimer's Disease Research Center (ADRC), including participants in the ADRC Clinical Core, the Emory Healthy Brain Study, and the ADRC‐affiliated Emory Cognitive Neurology Clinic. AD cases were consistent with NIA‐Reagan criteria for “high likelihood.” 61 In addition, investigators at Emory reviewed banked tissue samples previously sent to Emory as part of the AMP‐AD 1.0 initiative (but never were submitted for ‐omics generation until now) and included tissues from the University of Pennsylvania Integrated Neurodegenerative Disease Brain Bank 62 and Mount Sinai Brain Bank 23 to maximize the number of BA and LA samples and provide balance in their proteomics batching (as described in Methods). There were 75 BA, 5 LA, and 76 NHW donors with new data generated as part of the Diverse Cohorts initiative. Further, 307 samples with transcriptomics and/or WGS data generated as part of the AMP‐AD 1.0 initiative were added to provide further balance to proteomics batching. Tissue samples were obtained from the anterior caudate nucleus, DLPFC, and the STG. All participants provided informed consent under protocols approved by Emory University's IRB.
2.1.3. Rush University
Multiple longitudinal, epidemiologic cohort studies of aging and the risk of AD are conducted by Rush Alzheimer's Disease Center (RADC) and include Clinical Core (CLINCOR), Latino Core Study (LATC), Minority Aging Research Study (MARS), Religious Orders Study (ROS), and Memory Aging Project (MAP). Most of the participants of these cohorts are older adults aged ≥ 65, encompassing a range of ethnic and demographic backgrounds. They do not have known dementia at enrollment and agree to undergo annual clinical evaluations, with optional brain donation. There were 113 BA, 45 LA, 11 Asian, 49 NHW, 1 American Indian or Alaska Native, 4 American Indian or Alaska Native donors who also identified as Hispanic, and 3 BA donors who also identified as Hispanic. Tissue samples were obtained from the anterior caudate nucleus, DLPFC, and the STG. Informed consent and IRB approvals were obtained under the Rush University IRB. Details for each cohort are as follows.
CLINCOR studies the transition from normal aging to mild cognitive impairment (MCI) to the earliest stages of dementia. Enrollment started in 1992, primarily with individuals diagnosed with dementia. Since 2008, the study has transitioned to consist of primarily BA, most without dementia, who share a common core of risk factors with the other RADC studies. The participants are from the metropolitan Chicago area and outlying suburbs.
LATC is a cohort study of cognitive decline aiming to identify risk factors of AD in older Latinos. The participants self‐identified as Latino/Hispanic, and enrollment started in 2015. Recruitment locations include churches, subsidized senior housing facilities, retirement communities, Latino/Hispanic clubs, organizations, and social service centers that cater to seniors in various Chicago neighborhoods and outlying suburbs.
MARS is a cohort study of cognitive decline and risk of AD in older BAs. The recruitment began in 2004, and brain donation in 2010. The participants were recruited from various places, including churches, senior housing facilities, retirement communities, BA clubs, organizations, fraternities and sororities, and social service centers catering to seniors in metropolitan Chicago and outlying suburbs. 63
ROS and MAP are prospective community‐based studies of risk factors for cognitive decline, incident AD dementia, and other health outcomes. ROS began to recruit Catholic nuns, priests, and brothers from across the United States in 1994. MAP started recruiting participants from retirement communities and subsidized senior housing facilities throughout Chicago and northeastern Illinois in 1997. 64 The ROSMAP participants are primarily NHW, with small proportions of BA, Latino, and other racial groups.
2.1.4. Mount Sinai School of Medicine
The Mount Sinai School of Medicine (MSSM) cohort comprises donor brain tissue obtained from the Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB) 23 , 65 . There were 31 BA, 27 LA, and 30 NHW donors. Tissue samples were obtained from the anterior caudate nucleus, DLPFC, and STG. Autopsy protocols were approved by the Mount Sinai and JJ Peters VA Medical Center IRBs, and patient consent for donation was obtained.
2.1.5. Columbia University
Samples were collected from the New York Brain Bank (NYBB) at Columbia University, which was established to collect post mortem human brains to further study neurodegenerative disorders. There were 35 BA donors (one also identified as LA), 68 LA, 1 NHW, and 1 Asian donor. Tissue samples were obtained from the anterior caudate nucleus, dorsolateral/dorsomedial prefrontal cortex, and temporal pole. The appropriate review boards approved this study. The brain tissues contributed by Columbia University come from the following cohorts, brain banks, and studies.
The Columbia Alzheimer's Disease Research Center (Columbia ADRC) cohort consists of clinical participants in the Columbia ADRC who agreed to brain donation. All banked brains have one hemisphere fixed for subsequent diagnostic evaluation, and one hemisphere is banked fresh. For the hemisphere that is banked fresh, we block and freeze regions that are most commonly requested by researchers using liquid nitrogen, and specimens are stored at –80°C. This is performed on all ADRC brain donations, as well as on brains from the additional cohorts described below that also contributed to this study.
National Institute on Aging Alzheimer's Disease Family Based Study (NIA‐AD FBS) has recruited and followed 1756 families with suspected late‐onset AD, including 9682 family members and 1096 unrelated, non‐demented elderly from different racial and ethnic groups. This resource related cooperative agreement has now extended to the recruitment of familial early‐onset AD. The goals of this protocol are to provide rich genetic and biological resources for the scientific community, which includes longitudinal phenotype data, genotyped data, as well as brain tissue, plasma, and peripheral blood mononuclear cells.
Washington Heights, Inwood Columbia Aging Project (WHICAP) includes representative proportions of BA (28%), Caribbean Hispanics (48%), and NHW (24%). Since its inception in 1992, > 6000 participants have enrolled in this program project. Over the length of the project, we have identified environmental, health‐related, and genetic risk factors of disease and predictors of disease progression by collecting longitudinal data on cognitive performance, emotional health, independence in daily activities, blood pressure, anthropometric measures, cardiovascular status, and selected biomarkers in this elderly, multi‐ethnic cohort. WHICAP includes biomarker studies, magnetic resonance imaging (MRI), positron emission tomography scans, and brain tissue.
The Biggs Institute Brain Bank at the University of Texas Health Science Center at San Antonio is a biorepository and research laboratory focused on the pathology of neurodegenerative disorders in the San Antonio metropolitan region and greater South Texas. The Biggs Institute Brain Bank is the central service provider for the South Texas ADRC Neuropathology Core, collecting post mortem brain, spinal cord, cerebrospinal fluid, and dermal tissue from study participants and donors. Brain donation consent was obtained from the donor's legal next of kin prior to the autopsy. Autopsied brain tissue is hemisected, with the left hemibrain (typically) fixed in 10% neutral‐buffered formalin and the right hemibrain (typically) sectioned fresh and preserved at –80°C. After a minimum 4‐week fixation period and post mortem ex vivo MRI, 66 fixed tissue is sectioned and sampled in accordance with NIA‐Alzheimer's Association AD neuropathologic guidelines. For the AMP‐AD Diversity Initiative, frozen tissue (≈ 500 mg) was sampled from the anterior caudate, the middle frontal gyrus (Brodmann Area 9 or DLPFC; at the same level as the anterior caudate), and the STG (at the level of the amygdala) from six brain autopsy cases in the Biggs Institute Brain Bank. All research and tissue‐sharing activities herein were reviewed and approved by the University of Texas Health Science Center at San Antonio IRB and Office of Sponsored Projects.
Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) is a family‐based study initiated in 1998. The study included 683 at‐risk family members from 242 AD‐affected families of Caribbean Hispanic descent, recruited from clinics in the Dominican Republic and the Taub Institute on Alzheimer's Disease and the Aging Brain in New York. An AD case was defined as any individual meeting National Institute of Neurological and Communicative Disorders and Alzheimer's Disease and Related Disorders (NINCDS‐ADRD) criteria 57 for probable or possible late‐onset AD (LOAD).
2.2. Demographic, clinical, and neuropathologic variables collected
Each donor with brain samples included in the AMP‐AD Diversity Initiative was assigned a non‐identifiable individual ID by the contributing institution. For each participant, the same demographic variables were curated: cohort (or initial study group population to which the participant belonged), sex (male or female), self‐reported race (American Indian or Alaska Native, Asian, Black or African American, White, Other), self‐reported ethnicity (a true/false indicator for “is Latin American/Hispanic”), age of death in years (individuals ≥ 90 were designated as “90+” according to Health Insurance Portability and Accountability Act privacy rules), post mortem interval in hours where available, and apolipoprotein E (APOE) genotype.
The results of standard neuropathological assessments previously performed on the donor's brains were also collected from the relevant brain banks and harmonized when possible, following the harmonization protocols established by the Alzheimer's Disease Sequencing Project Phenotype Harmonization Consortium, as noted in their neuropathology data dictionary (https://vmacdata.org/adsp‐phc). Post mortem Thal amyloid stages 67 were available for Mayo Clinic, Emory, and a subset of Rush donors. All other donors were assigned a semi‐quantitative measure of neuritic plaque on a 4‐point scale, the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) score. 68 A semiquantitative measure of the severity of neurofibrillary tangle pathology, Braak stage (values equal to 0, I, II, III, IV, V, or VI) was included for all donors. 69
2.3. Donor characteristics
Donor characteristics varied by the contributing institution (Table 2). The overall median age of all participants was 82 years, with 88.9% of the participants age ≥ 65. A larger proportion of participants were female (59.0%) than male.
TABLE 2.
Donor characteristics by contributing institution.
| Columbia | Emory | Mayo | MSSM | Rush | |
|---|---|---|---|---|---|
| Characteristic | N = 105 | N = 156 | N = 331 | N = 88 | N = 228 |
| Female sex, N (%) | 72 (69%) | 89 (57%) | 166 (50%) | 49 (56%) | 160 (70%) |
| Age at death a in years, median (range) | 84.0 (51–90+) | 73.5 (20–90+) | 80.5 (20–90+) | 82.5 (62–90+) | 86.8 (54–90+) |
| Race b , N (%) | |||||
| Black or African American | 35 (33%) | 75 (48%) | 53 (16%) | 31 (35%) | 116 (51%) |
| Non‐Hispanic White | 1 (1%) | 76 (49%) | 96 (29%) | 30 (34%) | 49 (21%) |
| Other | 68 (65%) | 5 (3%) | 182 (55%) | 27 (31%) | 44 (19%) |
| Asian | 1 (1%) | 0 | 0 | 0 | 11 (5%) |
| American Indian or Alaska Native | 0 | 0 | 0 | 0 | 5 (2%) |
| Missing or unknown | 0 | 0 | 0 | 0 | 3 (1%) |
| Hispanic ethnicity c , N (%) | 69 (66%) | 5 (3%) | 182 (55%) | 27 (31%) | 52 (23%) |
| APOE genotype, N (%) | |||||
| ε2/ε2 | 0 | 1 (1%) | 0 | 1 (1%) | 0 |
| ε2/ε3 | 6 (6%) | 8 (5%) | 22 (7%) | 9 (10%) | 20 (9%) |
| ε2/ε4 | 5 (5%) | 4 (3%) | 7 (2%) | 3 (3%) | 8 (4%) |
| ε3/ε3 | 31 (30%) | 52 (33%) | 184 (56%) | 43 (49%) | 112 (49%) |
| ε3/ε4 | 23 (22%) | 47 (30%) | 98 (30%) | 29 (33%) | 47 (21%) |
| ε4/ε4 | 6 (6%) | 19 (12%) | 20 (6%) | 3 (3%) | 16 (7%) |
| Missing or unknown | 34 (32%) | 25 (16%) | 0 | 0 | 25 (11%) |
| Thal phase, N (%) | |||||
| None | NA | 34 (22%) | 46 (14%) | NA | 22 (10%) |
| Phase 1 | NA | 3 (2%) | 13 (4%) | NA | 28 (12%) |
| Phase 2 | NA | 11 (7%) | 16 (5%) | NA | 10 (4%) |
| Phase 3 | NA | 7 (4%) | 20 (6%) | NA | 51 (22%) |
| Phase 4 | NA | 15 (10%) | 23 (7%) | NA | 29 (13%) |
| Phase 5 | NA | 51 (33%) | 131 (40%) | NA | 50 (22%) |
| Missing or unknown | NA | 35 (22%) | 82 (25%) | NA | 38 (17%) |
| CERAD, N (%) | |||||
| None/no AD/C0 | 19 (18%) | 64 (41%) | NA | 18 (20%) | 54 (24%) |
| Sparse/possible/C1 | 15 (14%) | 1 (1%) | NA | 11 (12%) | 20 (9%) |
| Moderate/probable/C2 | 20 (19%) | 6 (4%) | NA | 11 (12%) | 62 (27%) |
| Frequent/definite/C3 | 49 (47%) | 83 (53%) | NA | 47 (53%) | 92 (40%) |
| Missing or unknown | 2 (2%) | 2 (1%) | NA | 1 (1%) | 0 |
| Braak stage, N (%) | |||||
| None | 2 (2%) | 21 (13%) | 15 (5%) | 4 (5%) | 8 (4%) |
| I | 1 (1%) | 27 (17%) | 27 (8%) | 3 (3%) | 14 (6%) |
| II | 4 (4%) | 14 (9%) | 41 (12%) | 11 (12%) | 21 (9%) |
| III | 11 (10%) | 14 (9%) | 58 (18%) | 7 (8%) | 37 (16%) |
| IV | 15 (14%) | 9 (6%) | 23 (7%) | 13 (15%) | 76 (33%) |
| V | 16 (15%) | 20 (13%) | 62 (19%) | 10 (11%) | 56 (25%) |
| VI | 52 (50%) | 49 (31%) | 100 (30%) | 35 (40%) | 16 (7%) |
| Missing or unknown | 4 (4%) | 2 (1%) | 5 (2%) | 5 (6%) | 0 |
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CERAD, Consortium to Establish a Registry for Alzheimer's Disease; MSSM, Mount Sinai School of Medicine; NA, not applicable.
Age at death was reported as 90+ for all individuals > 89 years old.
Self‐reported race. The “Other” category stood for individuals who might have reported themselves to be of Hispanic or Latin ethnicity within a race category (this information is also captured in the Hispanic Ethnicity variable).
Hispanic Ethnicity was captured as a TRUE/FALSE variable. Individuals of any self‐reported race could report Hispanic Ethnicity = TRUE.
2.4. Diagnostic harmonization
AMP‐AD Diversity Initiative has brain biospecimens from archival brain banks (e.g., Mayo Clinic) and from participants who were followed clinically while living before they came to autopsy (e.g., Rush University ROS and MAP cohorts). Donors from archival brain banks may not have a clinical diagnosis, while all donors had neuropathologic variables that enabled neuropathologic diagnosis. Because cohorts had variable clinical and neuropathological diagnostic information regarding AD case status, we chose to determine AD case/control status according to neuropathologic data for purposes of cross‐cohort analysis (Table 3). For all individuals with measures of CERAD and Braak, we calculated a modified NIA Reagan diagnosis of AD, 61 resulting in the following outcomes: no AD, low likelihood of AD, intermediate likelihood of AD, and high likelihood of AD. Mayo Clinic Brain Bank donors, which constituted the largest overall and single brain bank group contributing to the AMP‐AD Diversity Initiative, lacked CERAD scores but had AD diagnoses according to NINCDS‐ADRDA criteria. 57 Mayo Clinic Brain Bank donors were diagnosed as definite AD if they had Braak stage ≥ IV and the presence of amyloid beta (Aβ) plaques as assessed by a single neuropathologist (Dr. Dennis W. Dickson). Mayo Clinic Brain Bank donors were diagnosed as controls if they had Braak stage ≤ III, sparse or no Aβ plaques, and lacked any other neuropathologic diagnosis for neurodegenerative diseases. For all donors, we established the following criteria to achieve a uniform neuropathologic diagnosis of AD and to harmonize AD case/control diagnoses between cohorts as closely as possible: AD diagnosis was assigned to individuals with Braak stage ≥ IV and CERAD measure equal to moderate/probable AD or frequent/definite AD. Control diagnosis was assigned to individuals with Braak stage ≤ III and CERAD measure equal to none/no AD or sparse/possible AD. Any donors who did not fall under these criteria were assigned as “other.” These thresholds, while imperfect, are relatively conservative and also serve to exclude individuals with age‐related tauopathies from having an AD case or control designation.
TABLE 3.
Neuropathologic diagnoses by contributing institution.
| Columbia | Emory | Mayo | MSSM | Rush | |
|---|---|---|---|---|---|
| Outcome | N = 105 | N = 156 | N = 331 | N = 88 | N = 228 |
| NIA Reagan a | |||||
| No AD | 2 (2%) | 21 (13%) | NA | 4 (5%) | 4 (2%) |
| Low likelihood | 30 (29%) | 45 (29%) | NA | 24 (27%) | 78 (34%) |
| Intermediate likelihood | 20 (19%) | 20 (13%) | NA | 16 (18%) | 82 (36%) |
| High likelihood | 48 (46%) | 68 (44%) | NA | 39 (44%) | 64 (28%) |
| Missing or unknown | 5 (5%) | 2 (1%) | NA | 5 (6%) | 0 |
| Derived AD outcome b | |||||
| Control | 16 (15%) | 64 (41%) | 58 (18%) | 22 (25%) | 51 (22%) |
| AD | 66 (63%) | 77 (49%) | 180 (54%) | 52 (59%) | 125 (55%) |
| Other | 18 (17%) | 13 (8%) | 93 (28%) | 9 (10%) | 52 (23%) |
| Missing or unknown | 5 (5%) | 2 (1%) | 0 | 5 (6%) | 0 |
Abbreviations: AD, Alzheimer's disease; CERAD, Consortium to Establish a Registry for Alzheimer's Disease; MSSM, Mount Sinai School of Medicine; NIA, National Institute on Aging.
NIA Reagan score modified in accordance with Bennett et al. 63 : No AD: CERAD = No AD/None and Braak = Stage 0; Low Likelihood: CERAD = No AD/None and Braak ≥ Stage I OR CERAD = Possible/sparse and Braak = any stage OR CERAD = Probable AD/moderate and Braak ≤ Stage II; Intermediate Likelihood: CERAD = Probable/moderate and Braak ≥ Stage III OR CERAD = Definite/frequent and Braak ≥ Stage I and ≤ Stage IV; High Likelihood: CERAD = Definite AD/frequent and Braak ≥ Stage V.
For Mayo patients, this outcome is the reported diagnosis according to Mayo neurologist guidelines, as reported. 57 For all other patients: Control: CERAD = No AD/None or Possible/sparse and Braak ≤ Stage III; AD case: CERAD = Probable/moderate or Definite/frequent and Braak ≥ Stage IV; Other = all other combinations of CERAD and Braak.
NA = not applicable for Mayo patients because Mayo did not report CERAD measures.
2.5. Sampling across brain regions
Different brain regions were sampled to capture differences in molecular profiles, including gene and protein expression across regions occurring at different stages of AD neuropathology (Figure 1). The DLPFC and temporal cortex are regions affected in AD, albeit typically later for DLPFC than the temporal cortex. 69 DLPFC 24 and temporal cortex—especially STG 21 , 23 —were profiled with multi‐omics measurements in AMP‐AD studies of predominantly NHW donors. DLPFC and STG were obtained from all donors in the AMP‐AD Diversity initiative, except those from Columbia, who had temporal pole tissue available instead of STG. The anterior caudate nucleus was selected as a non‐cortical region also affected by AD neuropathology. 70 , 71 The total numbers of samples per tissue per data type and per donor by race and ethnicity are depicted in Figure 2. WGS for 626 donors were generated through the Diverse Cohorts initiative.
FIGURE 1.
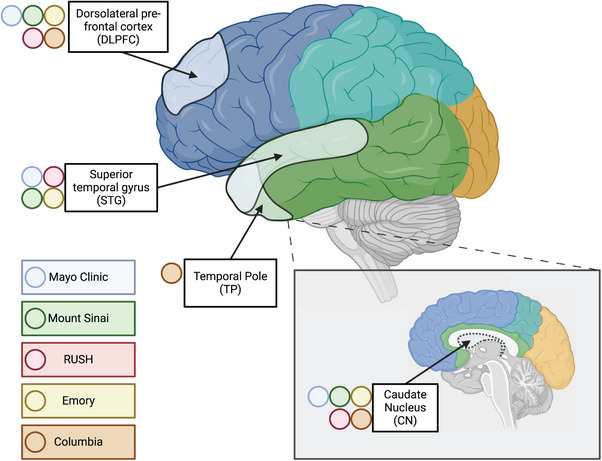
Profiled brain regions. Approximate location of tissue in brain regions sampled for molecular profiling, including RNAseq, WGS, and proteomics. Tissue from the dorsolateral prefrontal cortex (Brodmann areas 8, 9, and/or 46) and caudate nucleus were contributed by all sites, including Mayo Clinic, Mt. Sinai, Columbia, Rush, and Emory. In contrast, tissue from the superior temporal gyrus (Brodmann 22) was provided by all sites except Columbia, which had only the temporal pole available for this lobe. RNAseq, RNA sequencing; WGS, whole genome sequencing. Lobes are demarcated by their different colors shown on the brain schematic. The sites that provided samples from the specific brain regions are shown with the colored circles.
FIGURE 2.
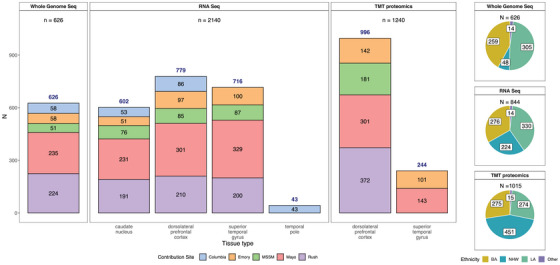
Data types by tissue, site, and individual race and ethnicity. Bar graph depicting the number of samples profiled by each assay (whole genome sequencing, RNAseq, or TMT proteomics). Whole genome sequencing data were generated for 626 donors from various contributing sites (an additional 411 donors had WGS from AMP‐AD 1.0 efforts, not shown here). Similarly, 2140 unique transcriptomics profiles from RNAseq of the caudate nucleus (n = 602), dorsolateral prefrontal cortex (n = 779), superior temporal gyrus (716), and temporal pole (n = 43) from 844 donors were generated. Samples sent to other sites for the swap study are not included. A lone superior temporal gyrus RNAseq sample from Columbia was also not included in this summary. One thousand two hundred forty unique TMT proteomes from dorsolateral prefrontal cortex (n = 996) and superior temporal gyrus (n = 244) were generated from 1015 donors. These include the 307 samples from the AMP‐AD 1.0 efforts to balance batches, as described in the Methods section. Pie charts on the right show the number of donors profiled by ethnoracial categories (BA = Black American, NHW = non‐Hispanic White, LA = Latin American, and Other). These categories were defined as follows: donors whose race was encoded as “Black or African American” and ethnicity as "isHispanic = FALSE" in the individual metadata were treated as "BA." Those with race encoded as White and ethnicity as "isHispanic = FALSE" were categorized as "NHW." The remaining donors, for whom ethnicity was encoded as "isHispanic = TRUE" were treated as "LA." All remaining donors from various other races were encoded as "Other." AMP‐AD, Accelerating Medicines Partnership in Alzheimer's Disease; MSSM, Mount Sinai School of Medicine; RNAseq, RNA sequencing; TMT, isobaric tandem mass tag; WGS, whole genome sequencing.
It should be noted that WGS for an additional 408 donors for whom ‐omics measures were generated in this study was readily available from the AMP‐AD 1.0 initiative. Also, as mentioned earlier, Emory included samples from an additional 307 predominantly NHW donors from AMP‐AD 1.0 to balance proteomics batches. The overlap between data contributor sites was generally highest for DLPFC and STG.
2.6. DNA extraction
All DNA extractions were done from the DLPFC for subsequent WGS. Mayo Clinic extracted DNA for all samples from the Mayo Clinic, Banner Sun Health, UFL, and Emory University Brain Banks. DNA was manually extracted from frozen brain tissue and was isolated using the AutoGen245T Reagent Kit (Part #agkt245td) according to the manufacturer's protocol, including an Rnase step (Qiagen, Cat# 19101) after tissue digestion. DNA was quantified for amount and purity using the Nanodrop Spectrophotometer (ThermoFisher) and Qubit 2.0 Fluorometer (ThermoFisher); 1875 ng per donor were transferred on dry ice to the New York Genome Center (NYGC) for whole genome library preparation and sequencing (WGS). For all other samples, DNA extraction was performed at the NYGC. In brief, for Rush and Mount Sinai samples, 25 mg of tissue was homogenized using a Qiagen Buffer ATL/Proteinase K with overnight incubation at 56 degrees Celsius. DNA was extracted using the Qiagen QIAamp DNA Mini Kit (Qiagen, 51304), and a Qiagen QIAamp DNA Mini Kit (Qiagen, 51304) was used for DNA cleanup. For Columbia samples, 50 mg of tissue was homogenized using a Buffer TE/Rnase A Solution (Maxwell Cat.# A7973). DNA was extracted using a Promega Maxwell kit (AS1610) and cleaned using a Maxwell RSC Tissue DNA Kit (Maxwell, TM476). For all samples, DNA quality was analyzed using a Fragment Analyzer (Advanced Analytics) or BioAnalyzer (Agilent Technologies). Libraries were generated using the Illumina Tru‐Seq PCR‐Free protocol, a short‐read sequencing technology with mean read length of 150 base pairs, and WGS was performed by the NYGC.
2.7. Whole genome sequencing
NYGC performed quality control (QC) on the raw WGS reads and provided the following metrics: Total Reads, passing filter (PF) Reads, % PF Reads, PF Aligned Reads, % PF Aligned, PF Aligned Pairs, % PF Aligned Pairs, Mean Read Length, Strand Balance, Estimated Library Size, Mean Coverage, % Sequence Contamination, Median Insert Size, Mean Insert Size, AT Dropout, GC Dropout, and % Total Duplication. These metrics were generated using Picard tools (v2.4.1, http://picard.sourceforge.net) following paired‐end read alignment to the GRCh38 human reference using the Burrows‐Wheeler Aligner (BWA‐MEM v0.7.15). Sequence contamination was estimated on a per‐sample basis using VerifyBamID (https://genome.sph.umich.edu/wiki/VerifyBamID).
2.8. RNA extraction
RNA extractions were done from frozen tissue from the DLPFC, anterior caudate nucleus, and STG (or temporal pole; Figure 1) for subsequent RNA sequencing. Most donors had tissue from all three regions, but no donors were excluded for lacking samples from any brain regions. Brain tissue from Emory, Banner, and the UFL was sent to Mayo Clinic Jacksonville in Florida for RNA isolation and sequencing. Brain tissue samples for the Mayo cohort were obtained from the Mayo Clinic Brain Bank. RNA was isolated using a Trizol/chloroform protocol, followed by two‐step RNA purification (Qiagen Rneasy Mini Kit) and concentration incorporating on‐column (Qiagen Cat#74106 or 74104 and Cat#79254) and liquid (Zymo Cat# R1014 or R1013) Dnase steps, respectively. The quantity and quality of all RNA samples were determined by the NanoDrop 2000 Spectrophotometer and Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano Chip (Cat# 5067‐1511 from Agilent Technologies).
For all Rush samples, 50 mg of frozen brain tissue was dissected and homogenized in DNA/RNA shield buffer (Zymo, R1100) with 3 mm beads using a bead homogenizer. RNA was subsequently extracted using a Chemagic RNA tissue kit (Perkin Elmer, CMG‐1212) on a Chemagic 360 instrument. RNA was concentrated (Zymo, R1080), and RQN values were calculated with a Fragment Analyzer total RNA assay (Agilent, DNF‐471).
Tissue samples from MSSM and Columbia were prepared for RNA sequencing at the NYGC. Tissue was homogenized using TRIzol (needles), and RNA was extracted using Cloroform. A Qiagen Rneasy Mini Kit was used for RNA cleanup, and quality was analyzed with Fragment Analyzer (Advanced Analytics) or BioAnalyzer (Agilent Technologies).
2.9. RNA sequencing
Brain samples from the Mayo Clinic, Banner Sun Health, UFL, and Emory University were randomized with respect to race, ethnicity, diagnosis (AD, control, other), contributing institution, RNA Integrity Number (RIN), APOE genotypes, sex, and age, prior to transfer to the Mayo Clinic Genome Analysis Core for library preparation and sequencing across 13 flow cells. Total RNA concentration and quality were determined using Qubit fluorometry (ThermoFisher Scientific) and the Agilent Fragment Analyzer. Using Illumina's TruSeq Stranded Total RNA reagent kit (Cat #20020597) and the Illumina Ribo‐Zero Plus rRNA Depletion kit (Cat #20037135), libraries were prepared according to the manufacturer's instructions with 200 ng of total RNA. The concentration and size distribution of the completed libraries were determined using Qubit fluorometry and the Agilent TapeStation D1000. Libraries were sequenced at an average of 200 M total reads, following the standard protocol for the Illumina NovaSeq 6000. The flow cell was sequenced as 100 × 2 paired‐end reads using the NovaSeq S4 sequencing kit and NovaSeq Control Software v1.7.5. Base‐calling was performed using Illumina's RTA version 3.4.4. All RNA samples isolated from tissue samples of the same donor were sequenced together in the same flow cell.
For all Rush samples, after RNA extraction, concentration was determined using Qubit broad‐range RNA assay (Invitrogen, Q10211) according to the manufacturer's instructions; 500 ng total RNA was used as input for sequencing library generation, and rRNA was depleted with RiboGold (Illumina, 20020599). A Zephyr G3 NGS workstation (Perkin Elmer) was used to generate TruSeq stranded sequencing libraries (Illumina, 20020599) with custom unique dual indexes (IDT) according to the manufacturer's instructions with the following modifications. RNA was fragmented for 4 minutes at 85°C. The first strand synthesis was extended to 50 minutes. Size selection post adapter ligation was modified to select for larger fragments. Library size and concentrations were determined using an NGS fragment assay (Agilent, DNF‐473) and Qubit ds DNA assay (Invitrogen, Q10211), respectively, according to the manufacturer's instructions. The modified protocol yielded libraries with an average insert size of ≈ 330 to 370 bp. Libraries were normalized for molarity and sequenced on a NovaSeq 6000 (Illumina) at 40 to 50 M reads, 2 × 150 bp paired end.
Columbia and MSSM samples were sequenced at the NYGC. After rRNA depletion using RiboErase, libraries were prepared using 500 ng of RNA with the KAPA Stranded Total RNA (HMR) RiboErase Kit (kapabiosystems). RNA was fragmented for 5 minutes at 85°C, and first‐strand synthesis was extended to 10 minutes at 25°C, 15 minutes at 42°C, and 15 minutes at 70°C. Size selection post adapter ligation was modified to select larger fragments, which resulted in 480 to 550 bp fragments. Sequencing was performed using an Illumina NovaSeq 6000 to generate 100 bp paired‐end reads. Sequencing quality control was performed using Picard version 1.83 and RseQC version 2.6.1. STAR version 2.5.2a was used to align reads to the GRCh38 genome using Gencode v25 annotation. Bowtie2 version 2.1.0 was used to measure rRNA abundance. Annotated genes were quantified with featureCounts version 1.4.3‐p1. Sequence contamination was estimated on a per‐sample basis using VerifyBamID (https://genome.sph.umich.edu/wiki/VerifyBamID). The identity of the RNA sample is confirmed by evaluating concordance with WGS data using Conpair, a tool that uses a set of single nucleotide polymorphisms common in the human population to determine sample identity.
To maximize the number of brain samples included in the AMP‐AD Diversity Initiative, RIN was measured but not used to filter out samples. The DV200 is an assessment of the proportion of RNA fragments > 200 nucleotides and is considered a more accurate measure of RNA quality when the RIN value is low. 72 For Columbia and RUSH, at least 85% of the low RIN samples (RIN < 5) have a DV200 > 70%; for Mayo, 90% of samples meet this metric, and for MSSM, 95% of the sample pass. Given the high proportion of samples with a DV200 > 70%, samples were not removed based on these metrics but rather assessed carefully at the QC stage.
2.10. RNA sample exchange
Because RNA sequencing was conducted at three different sequencing centers (Mayo Genome Analysis Core, NYGC, and Rush), a small number of samples were exchanged between the three sequencing centers to evaluate the extent of technical variability between these centers (Figure 3). Mayo Clinic contributed five samples each from the DLPFC and STG to Rush and NYGC. Six DLPFC samples from Columbia were sent to Mayo Clinic and Rush, and four samples each for DLPFC and STG from Mt. Sinai were sent to Mayo and Rush. Rush contributed six samples each from DLPFC and STG to the Mayo Clinic and NYGC. Tissues sent to other sites as part of the swap experiment were also sequenced at each original sequencing site, resulting in three sets of RNAseq data from each participant and brain region for the swapped samples. RNA extraction and sequencing protocol for swap samples at each site is described above (see RNA extraction and RNA sequencing).
FIGURE 3.
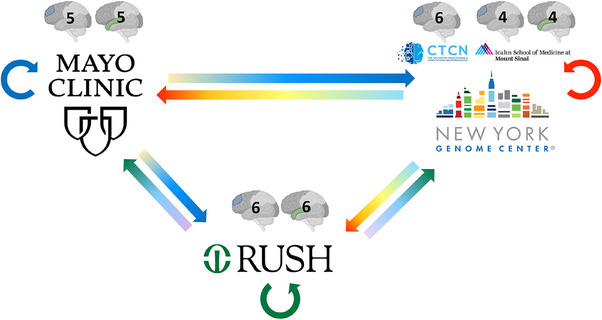
RNAseq sample swaps. To evaluate the technical variability of RNA sequencing among the three sites, RNA tissue from the same brain was sequenced at each site for a small number of samples. The number and region of samples exchanged are illustrated with the grayscale brain image with the exchanged tissue highlighted in color (DLPFC in blue and STG in green). Straight arrows represent tissue exchange; circular arrows represent tissue sequenced at the original site, shown in blue, green, and red circular arrows for Mayo Clinic, Rush, and NYGC, respectively. Samples from MSSM (4 DLPFC, 4 STG) and Columbia (5 DLPFC) were used for the swap experiment at NYGC. DLPFC, dorsolateral prefrontal cortex; MSSM, Mount Sinai School of Medicine; NYGC, New York Genome Center; RNAseq, RNA sequencing.
All samples that were part of the swap study were sequenced in a single batch at Mayo, whereas samples sequenced at NYGC were distributed across five batches, and at Rush, they were distributed across three batches. RIN values for samples sequenced at Mayo ranged between 2.7 and 8.8, whereas those at NYGC ranged between 2.7 and 8.7, and at Rush ranged between 1.3 and 8.0. RNAseq data for swap samples generated across all three sites were consensus processed using MAPRSeq v3 pipeline. 73 Reads were aligned to the reference (GRCh38) using STAR aligner v2.6.1. Sequencing and alignment metrics from FastQC and RseQC were used to evaluate variability across sequencing centers. The median base quality of reads was consistent (Phred ≥ 37) across sites for both DLPFC and STG. Evaluation of base content (percentage of As, Ts, Gs, and Cs at each position in the read) between the 25th and 75th percentile along the read length revealed that the percentage of As and Ts was ≈ 30% and that of Gs and Cs was 20% across all reads and samples. The following summary metrics are summarized by tissue contribution site and sequencing site in Figure S1 in supporting information. Between 104 and 147 M reads were generated for samples sequenced at Mayo, 95% to 98% of which were mapped to the genome and 31% to 54% mapped to genes. For samples sequenced at NYGC, between 58 and 222 M reads were generated, 93% to 98% of which mapped to the genome and 37% to 58% mapped to genes. Similarly, at Rush, between 10 and 125 M reads were generated, 83% to 96% mapped to the genome and 28% to 57% mapped to genes. The median ratio of reads covering the 80th and 20th percentile along the gene body for all genes was between 1 and 1.1, revealing no significant bias toward 3′ or 5′ degradations. Sex deduced from gene expression was consistent with assigned sex based on clinical information. After conditional quantile normalization (CQN) to identify expressed genes, principal component analysis (PCA) was performed to evaluate stratification among samples (Figure S2 in supporting information). When PCs were generated by tissue (one set of PCs each of DLPFC and STG) and plotted together, there was no separation by tissue contribution site (Figure S2A), although there was some separation by sequencing site (Figure S2B), and indeed, sequencing site was the largest source of technical variation. When PCs were generated by tissue contribution site (one set of PCs each for Columbia, Mt. Sinai, Mayo, and Rush) and plotted together, there was no separation by sequencing site but only by tissue (Figure S2C).
2.11. Proteomics
Proteome measurements were conducted in all DLPFC tissue, as well as in STG, for a subset of the samples from the Mayo Clinic to enable joint analyses with other STG proteome data from this Brain Bank. 21 Pre‐ and postprocessing steps for proteomic quantification were performed at Emory University for all samples from all contributing institutions using the following methods. Samples from each individual site were randomized in batches of 15 to 17 and balanced, where possible, with respect to race, ethnicity, diagnosis (AD), sex, and age. 74 The batching schema is included in the proteomics biospecimen metadata file (syn53185805).
2.11.1. Brain tissue homogenization and protein digestion
Procedures for tissue homogenization for all tissues were performed essentially as described. 48 , 75 Approximately 100 mg (wet tissue weight) of brain tissue was homogenized in 8 M urea lysis buffer (8 M urea, 10 mM Tris, 100 mM NaHPO4, pH 8.5) with HALT protease and phosphatase inhibitor cocktail (ThermoFisher) using a Bullet Blender (NextAdvance) essentially as described. 75 Each Rino sample tube (NextAdvance) was supplemented with ≈ 100 µL of stainless steel beads (0.9 to 2.0 mm blend, NextAdvance) and 500 µL of lysis buffer. Tissues were added immediately after excision, and samples were placed into the bullet blender at 4°C. The samples were homogenized for two full 5 minute cycles, and the lysates were transferred to new Eppendorf Lobind tubes. Each sample was then sonicated for three cycles of 5 seconds of active sonication at 30% amplitude, followed by 15 seconds on ice. Samples were centrifuged for 5 minutes at 15,000 × g, and the supernatant was transferred to a new tube. Protein concentration was determined by bicinchoninic acid (BCA) assay (Pierce). For protein digestion, 100 µg of each sample was aliquoted, and volumes were normalized with additional lysis buffer. An equal amount of protein from each sample was aliquoted and digested in parallel to serve as the global pooled internal standard (GIS) in each isobaric tandem mass tag (TMT) batch, as described below. Similarly, GIS pooled standards were generated from all cohorts. Samples were reduced with 1 mM dithiothreitol (DTT) at room temperature for 30 minutes, followed by 5 mM iodoacetamide (IBA) alkylation in the dark for another 30 minutes. Lysyl endopeptidase (Wako) at 1:100 (w/w) was added, and digestion was allowed to proceed overnight. Samples were then 7‐fold diluted with 50 mM ammonium bicarbonate. Trypsin (Promega) was added at 1:50 (w/w), and digestion was carried out for another 16 hours. The peptide solutions were acidified to a final concentration of 1% (vol/vol) formic acid (FA) and 0.1% (vol/vol) trifluoroacetic acid (TFA), and desalted with a 30 mg HLB column (Oasis). Each HLB column was first rinsed with 1 mL of methanol, washed with 1 mL 50% (vol/vol) acetonitrile (ACN), and equilibrated with 2 × 1 mL 0.1% (vol/vol) TFA. The samples were loaded onto the column and washed with 2 × 1 mL 0.1% (vol/vol) TFA. Elution was performed with 2 volumes of 0.5 mL 50% (vol/vol) ACN. The eluates were then dried to completeness using a SpeedVac.
2.11.2. Isobaric TMT peptide labeling
The Synapse DOI giving sample to batch arrangement is presented Table 4. In preparation for labeling, each brain peptide digest was resuspended in 75 µL of 100 mM triethylammonium bicarbonate (TEAB) buffer; meanwhile, 5 mg of TMT reagent was dissolved into 200 µL of ACN. Each sample (containing 100 µg of peptides) was resuspended in 100 mM TEAB buffer (100 µL). The TMT labeling reagents (5 mg; Tandem Mass Tag [TMTpro] kit [ThermoFisher Scientific, A44520]) were equilibrated to room temperature, and anhydrous ACN (256 µL) was added to each reagent channel. Each channel was gently vortexed for 5 minutes, and then 41 µL from each TMT channel was transferred to the peptide solutions and allowed to incubate for 1 hour at room temperature. The reaction was quenched with 5% (vol/vol) hydroxylamine (8 µL; Pierce). All channels were then combined and dried by SpeedVac (LabConco) to ≈ 150 µL and diluted with 1 mL of 0.1% (vol/vol) TFA, then acidified to a final concentration of 1% (vol/vol) FA and 0.1% (vol/vol) TFA. Labeled peptides were desalted with a 200 mg C18 Sep‐Pak column (Waters). Each Sep‐Pak column was activated with 3 mL of methanol, washed with 3 mL of 50% (vol/vol) ACN, and equilibrated with 2×3 mL of 0.1% TFA. The samples were then loaded and each column was washed with 2×3 mL 0.1% (vol/vol) TFA, followed by 2 mL of 1% (vol/vol) FA. Elution was performed with two volumes of 1.5 mL 50% (vol/vol) ACN. The eluates were then dried to completeness using a SpeedVac.
TABLE 4.
Synapse doi's of data shared on the AD Knowledge Portal for the AMP‐AD Diversity Initiative. a
| Data type | Doi |
|---|---|
| AMP‐AD Diverse Cohorts RNAseq Sample Exchange Data Subset | https://doi.org/10.7303/syn53420676 |
| AMP‐AD Diverse Cohorts Raw TMT Proteomics Data | https://doi.org/10.7303/syn53420674 |
| AMP‐AD Diverse Cohorts Raw WGS Data | https://doi.org/10.7303/syn53420673 |
| AMP‐AD 1.0 Raw WGS Data for Diverse Cohorts Individuals without WGS | https://doi.org/10.7303/syn53420677 |
| AMP‐AD Diverse Cohorts Raw RNAseq Data | https://doi.org/10.7303/syn53420672 |
Abbreviations: AD, Alzheimer's disease; AMP‐AD, Accelerating Medicines Partnership in AD; RNAseq, RNA sequencing; TMT, isobaric tandem mass tag; WGS, whole genome sequencing.
All accompanying individual, biospecimen, and assay metadata are included in the dois; entire study project found at (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage/StudyDetails?Study=syn51732482).
2.11.3. High‐pH off‐line fractionation
High‐pH fractionation was performed essentially as described with slight modification. 75 , 76 Dried samples were re‐suspended in high pH loading buffer (0.07% vol/vol NH4OH, 0.045% vol/vol FA, 2% vol/vol ACN) and loaded onto a Water's BEH 1.7 um 2.1 mm by 150 mm. A Thermo Vanquish or Agilent 1100 HPLC system was used to carry out the fractionation. Solvent A consisted of 0.0175% (vol/vol) NH4OH, 0.01125% (vol/vol) FA, and 2% (vol/vol) ACN; solvent B consisted of 0.0175% (vol/vol) NH4OH, 0.01125% (vol/vol) FA, and 90% (vol/vol) ACN. The sample elution was performed over a 25 minute gradient with a flow rate of 0.6 mL/minute. A total of 192 individual equal‐volume fractions were collected across the gradient and subsequently pooled by concatenation into 96 fractions (Rush, MSSB, and Mayo cohorts) or 48 fractions for the Emory cohort. All peptide fractions were dried to completeness using a SpeedVac. Off‐line fractionation of the Mount Sinai and Emory cohorts was performed as previously described. 75 , 77
2.11.4. TMT mass spectrometry
All fractions were resuspended in an equal volume of loading buffer (0.1% FA, 0.03% TFA, 1% ACN) and analyzed by liquid chromatography coupled to tandem mass spectrometry (MS/MS) essentially as described, 78 with slight modifications. Peptide eluents were separated on a self‐packed C18 (1.9 µm, Dr. Maisch, Germany) fused silica column (25 cm × 75 µM internal diameter [ID]; New Objective) by a Dionex UltiMate 3000 RSLCnano liquid chromatography system (ThermoFisher Scientific) and monitored on a mass spectrometer (ThermoFisher Scientific). Sample elution was performed over a 180 minute gradient with a flow rate of 225 nL/min. The gradient was from 3% to 7% buffer B over 5 minutes, then 7% to 30% over 140 minutes, then 30% to 60% over 5 minutes, then 60% to 99% over 2 minutes, then held constant at 99% solvent B for 8 minutes, and then back to 1% B for an additional 20 minutes to equilibrate the column. The mass spectrometer was set to acquire data in data‐dependent mode using the top‐speed workflow with a cycle time of 3 seconds. Each cycle consisted of one full scan followed by as many MS/MS (MS2) scans that could fit within the time window. The full scan (MS1) was performed with an m/z range of 350 to 1500 at 120,000 resolution (at 200 m/z) with AGC set at 4 × 105 and a maximum injection time of 50 ms. The most intense ions were selected for higher energy collision‐induced dissociation (HCD) at 38% collision energy with an isolation of 0.7 m/z, a resolution of 30,000, an AGC setting of 5 × 104, and a maximum injection time of 100 ms. Of the 72 TMT batches for the DLPFC tissues, 34 were run on an Orbitrap Fusion Lumos mass spectrometer, 24 batches were run on an Orbitrap Fusion Eclipse GC 240 mass spectrometer, and 14 batches were run on an Orbitrap Eclipse mass spectrometer as previously described. 75 Collectively, liquid chromatography MS/MS led to a total of 6479 raw files from frontal cortex, and 1824 raw files from temporal cortex tissue samples (Figure 1A), with the distribution as follows: Emory University Frontal Cortex Cohort: 431; Mayo Clinic Frontal Cortex Cohort: 2304; Mount Sinai Frontal Cortex Cohort: 1344; Rush University Frontal Cortex Cohort: 2400; and Emory University and Mayo Clinic Temporal Cortex Cohort: 1824. Lobes are demarcated by their different colors shown on the brain schematic. The sites that provided samples from the specific brain regions are shown with the colored circles.
3. DISCUSSION
This is a data descriptor study for the AMP‐AD 20 Diversity Initiative that was launched to generate, analyze, and make available to the research community multi‐omics data in AD and older control brain donors from multi‐ethnic populations enriched for BA and LA participants who are at higher risk 2 for AD but traditionally underrepresented in research. 3 , 4 , 5 , 6 While GWAS in BA and LA participants are orders of magnitude smaller than that for NHW, multi‐omics studies are essentially non‐existent, especially in brain tissue from these populations. This underrepresentation in brain multi‐omics studies is in part due to lower autopsy rates in BA and LA populations, 79 , 80 the causes of which are multi‐factorial but must be comprehensively understood to overcome this barrier in research. There are efforts to increase diversity in autopsy studies for ADRD, 63 , 81 , 82 which have led to the discovery that some but not all neuropathologies have ethnoracial differences. 81 , 83 , 84 , 85
To our knowledge, there are no sizable multi‐omics studies of ADRD including age‐matched control BA and LA donors to uncover the molecular underpinnings of these neuropathologies. In contrast, the AMP‐AD Target Discovery and Preclinical Validation Project generated 21 , 22 , 23 , 24 and broadly shared 25 multi‐omics data on > 2500 brain samples, primarily from NHW donors. These previous studies also pointed to the importance of sampling multiple brain regions. 21 , 23 , 24 Sampling from a homogenized whole brain would likely dilute important signals specific to different disease stages, as it is known that different regions are affected at different stages of disease. 69 The previous and current AMP‐AD projects contain donors whose neuropathologies reflect the full spectrum of AD, from little or no neuropathology to high levels of neuropathology. These multi‐omics data revealed brain molecular alterations in specific biological pathways, including but not limited to innate immunity, synaptic biology, myelination, vascular biology, and mitochondrial energetics, 28 , 29 , 30 , 32 , 33 , 34 , 37 , 39 , 45 , 54 , 86 , 87 , 88 , 89 thereby supporting complex, heterogeneous molecular etiologies, resulting in > 600 therapeutic candidates with a step closer to precision medicine in ADRD.
Recognizing the essential importance of inclusivity in precision medicine, 56 we launched the AMP‐AD Diversity Initiative with the objective of performing multi‐omics profiling and analysis of samples from diverse cohorts to discover the full spectrum of therapeutic targets and biomarkers that will be of utility to all populations affected with AD. In this data descriptor article, we describe the first wave of data generated and shared with the research community, comprising transcriptome from three brain regions, WGS, and proteome measures from 908 multi‐ethnic donors enriched for BA (n = 306) and LA (n = 326). We emphasize that this is the initial set of data currently being expanded to include other ‐omics measures, namely metabolome, single‐cell RNAseq, and epigenome in the AMP‐AD Diverse Cohorts Study.
We must emphasize that multi‐omics studies alone are unlikely to be sufficient to discover all causes of ADRD or explain the disparities in risk observed for BA and LA participants. 4 , 6 , 90 Rather, this requires a full understanding of the role of the exposome, including sex, race, ethnicity, lifetime health measures, co‐morbidities, and additional structural and social determinants of health (SSDoH). 54 , 91 , 92 , 93 , 94 , 95 , 96 Only by capturing the exposome and evaluating its complex interactions with multi‐omics measures and disease‐related outcomes can we have a holistic lens into the etiopathogenesis of ADRD. With this goal in mind, the AMP‐AD Diversity Initiative is in the process of curating and harmonizing exposome data for the donors in the AMP‐AD Diverse Cohorts Study.
Despite the potential utility of this foundational multi‐omics dataset from a multi‐ethnic autopsy cohort, there are shortcomings in the current study. To include the largest possible number of BA and LA donors, brain tissue from both archival brain banks and longitudinal studies was included, resulting in variability in the types of clinical and neuropathologic data available. We strove to overcome this variability by careful harmonization of the neuropathologic data to the extent possible, although we must underscore the need to have more diverse autopsy cohorts with in‐depth and uniform phenotyping, including clinical and neuropathologic variables. For this study, we accepted self‐reported race and ethnicity. We recognize that race and ethnicity are highly complex constructs 6 , 80 , 90 , 97 that must consider SSDoH, cultural, historical, and biological variables, and context. We also recognize that there is an ongoing and shifting conversation about the most appropriate terminology, such as that seen in the recent update by the US Census (https://www.census.gov/newsroom/blogs/random‐samplings/2024/04/updates‐race‐ethnicity‐standards.html), which combines race and ethnicity and adds another category, “Middle Eastern or North African.” We are currently unable to update our data to this new standard, given that the bulk of this data was collected as part of archival brain banks and/or at a time prior to these definitions, using older surveys that did not encompass the full range of possible terminology and that cannot provide more nuanced categories within BA and LA populations (such as country‐specific terms within the LA population). Given the availability of genetic data in this study, genetic ancestry markers could be calculated as well, which future studies will aim to do in the context of accounting for population stratification in statistical genetics studies. However, such adjustments are not appropriate for all statistical analyses, considering the salient social‐cultural impacts of self‐reported race. While we will aim to incorporate as many exposome variables into this study as possible, there is clearly a need for multi‐disciplinary teams to assess all non‐biological and biological variables and context holistically in large‐scale population‐based studies to understand disparities in and causes of disease risk. Finally, though our study is a step in the right direction for inclusivity in precision medicine studies, there are many other underrepresented groups in ADRD research in the United States and globally. 3 , 79 National and global initiatives are required to expand this research to all affected populations.
In summary, we describe transcriptome data from 2224 brain samples, proteome data from 1385 samples, and new whole genome sequencing from 626 samples, primarily from 908 multi‐ethnic donors enriched for BA and LA participants. These data are accompanied by harmonized neuropathologic diagnoses of AD (n = 500), control (n = 211), or other (n = 185). These data made available to the research community are expected to be an initial step to bridge our data and knowledge gap in the understanding of AD in underrepresented and at‐risk populations.
AUTHOR CONTRIBUTIONS
Joseph S. Reddy, Laura Heath, Nilüfer Ertekin‐Taner wrote the initial draft of the manuscript. Joseph S. Reddy, Laura Heath, Abby Vander Linden, Mariet Allen, Anna Greenwood, Nilüfer Ertekin‐Taner collated and oversaw the organization of data and samples for the AMP‐AD Diversity Initiative. Joseph S. Reddy, Mariet Allen, Katia de Paiva Lopes, Edward J. Fox, Erming Wang, Yiyi Ma, Stefan Prokop, Thomas G. Beach, Andrew F. Teich, Varham Haroutunian, Marla Gearing, Dennis W. Dickson, and Edward B. Lee provided and organized brain samples from the Mayo Clinic, Rush, Emory, Upenn, Mount Sinai, Columbia, Banner, and the University of Florida Brain Banks. Fatemeh Seifar, Luming Yin, Kaiming Xu, Lingyan Ping, Erica S. Modeste, Eric B. Dammer, Anantharaman Shantaraman, Lingyan Ping, Zachary S. Quicksall, Joseph S. Reddy, Edward J. Fox, Aliza Wingo, Thomas Wingo, William L. Poehlman, Zachary S. Quicksall, Alexi Runnels, Yanling Wang, Duc M. Duong, Erica S. Modeste, and Stephanie R. Oatman analyzed the transcriptome, genome, and proteome data. Laura Heath, Abby Vander Linden, Mariet Allen, Jo Scanlan, Charlotte Ho, Minerva M. Carrasquillo, Merve Atik., Geovanna Yepez, Adriana O. Mitchell, Thuy T. Nguyen, Stefan Prokop, Thomas G. Beach, Andrew F. Teich, Varham Haroutunian, Marla Gearing, and Dennis W. Dickson provided data and performed analyses for phenotype harmonization. Hasini Reddy, Harrison Xiao, Stefan Prokop, Thomas G. Beach, Andrew F. Teich, Varham Haroutunian, Marla Gearing, and Dennis W. Dickson provided neuropathology measures. Sudha Seshadri, Richard Mayeux, Lisa L. Barnes, Philip De Jager, Bin Zhang, David Bennett, James J. Lah, Allan I. Levey, David X. Marquez, Nicholas T. Seyfried, and Nilüfer Ertekin‐Taner led the cohort studies from which donor tissue and data are obtained. Philip De Jager, Bin Zhang, David Bennett, Nicholas T. Seyfried, Anna K. Greenwood, and Nilüfer Ertekin‐Taner obtained funding for and designed the AMP‐AD Diversity Initiative and provided supervision. All authors reviewed and provided feedback for the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
This study was approved by the institutional review board at the respective institutions. All participants or next of kin provided consent.
The AD Knowledge Portal hosts data from multiple cohorts that were generated as part of or used in support of the AMP‐AD Diverse Cohorts Study conducted under the AMP‐AD Diversity Initiative. The portal uses the Synapse software platform for backend support, providing users with web‐based and programmatic access to data files. All data files in the portal are annotated using a standard vocabulary to enable users to search for relevant content across the AMP‐AD datasets using programmatic queries. Data are stored in a cloud‐based manner hosted by Amazon Web Services (AWS), which enables users to execute cloud‐based computing or copy the data to local infrastructure. Detailed descriptions, including data processing, QC metrics, and assay and cohort‐specific variables, are provided for each file as applicable (as noted in Table 4).
Access to the data described herein is controlled in a manner set forth by the IRBs at the Mayo Clinic, MSSM, Rush, Emory, and Columbia. All data use terms include (1) maintenance of data in a secure and confidential manner, (2) respect for the privacy of study participants, (3) including the following in any published text: “The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org/). Data generation was supported by the following NIH grants: U01AG046139, U01AG046170, U01AG061357, U01AG061356, U01AG061359, and R01AG067025. We thank the participants of participants of the Religious Order Study, Memory and Aging Project, the Minority Aging Research Study, Rush Alzheimer's Disease Research Center, Mount Sinai/JJ Peters VA Medical Center NIH Brain and Tissue Repository, National Institute of Mental Health Human Brain Collection Core (NIMH HBCC), Mayo Clinic Brain Bank, Sun Health Research Institute Brain and Body Donation Program, Goizueta Alzheimer's Disease Research Center, New York Brain Bank at Columbia University, New York Genome Center and the Biggs Institute Brain Bank for their generous donations. Data and analysis contributing investigators include Nilüfer Ertekin‐Taner, Minerva Carrasquillo, Mariet Allen, Dennis Dickson (Mayo Clinic, Jacksonville, FL), David Bennett, Lisa Barnes (Rush University), Philip De Jager, Vilas Menon (Columbia University), Bin Zhang, Vahram Haroutanian (Icahn School of Medicine at Mount Sinai), Allan Levey, Nick Seyfried (Emory University), Rima Kaddurah‐Daouk (Duke University), Steve Finkbeiner (University of California‐San Francisco/Gladstone Institutes), Daifeng Wang (University of Wisconsin‐Madison), Stefano Marenco (NIMH HBCC), Anna Greenwood, Abby Vander Linden, Laura Heath, William Poehlman (Sage Bionetworks).” For access to content described in this article see: https://doi.org/10.7303/syn53420672, https://doi.org/10.7303/syn53420673, https://doi.org/10.7303/syn53420674, https://doi.org/10.7303/syn53420676, https://doi.org/10.7303/syn53420677 (also listed in Table 4). To download data, users must register for a Synapse account, provide electronic agreement to the Terms of Use outlined above, and complete a Data Use Certificate. User approvals are managed by the Synapse Access and Compliance Team (ACT). Additional variables will be shared for subsets of the study participants (depending on availability) in future data releases, including other markers of neuropathology (such as markers of Lewy body disease and cerebrovascular disease), and clinical and social information collected ante mortem (such as years of education, cognitive test scores, Area Deprivation Index, comorbidities, and medications). The AD Knowledge Portal does not have the ability to share all available data on every patient, per data use agreements. Readers interested in obtaining additional variables or tissue samples may reach out to individual cohort studies and brain banks to determine availability and access options through individual data use agreement requirements.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We would like to thank the patients and their families for their participation; without them, these studies would not have been possible. The results published here are based on data available in the AD Knowledge Portal (https://adknowledgeportal.org). The Mayo RNAseq study data was led by Dr. Nilüfer Ertekin‐Taner, Mayo Clinic, Jacksonville, FL, as part of the multi‐PI U01 AG046139 (MPIs Golde, Ertekin‐Taner, Younkin, Price) using samples from The Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, P30AG072979, P01AG066597, U19AG062418, U01AG061357, RF1AG062181, P30AG066511 CurePSP Foundation, and support from the Mayo Foundation. Study data included samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona, USA. The Brain and Body Donation Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the National Institute on Aging (P30 AG019610 and P30AG072980, Arizona Alzheimer's Disease Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05‐901 and 1001 to the Arizona Parkinson's Disease Consortium), and the Michael J. Fox Foundation for Parkinson's Research. We would like to thank John Q. Trojanowski (deceased) for his leadership at the Center for Neurodegenerative Disease Research, which helped make acquiring samples from University of Pennsylvania Integrated Neurodegenerative Disease Brain Bank possible. Additional support for these studies was provided by the NINDS grant R01‐NS080820 (NET), NIA grant R01‐AG061796 (NET), NIA grant U19‐AG074879 (NET), and Alzheimer's Association Zenith Fellows Award (NET). We thank the Mayo Clinic Genome Analysis Core (GAC), Co‐Directors Julie M. Cunningham, PhD, and Eric Wieben, PhD, and supervisor Julie Lau, for their collaboration in the collection of ‐omics data.
Reddy JS, Heath L, Linden AV, et al. Bridging the gap: Multi‐omics profiling of brain tissue in Alzheimer's disease and older controls in multi‐ethnic populations. Alzheimer's Dement. 2024;20:7174–7192. 10.1002/alz.14208
Joseph S. Reddy and Laura Heath are the co‐first authors.
Anna K. Greenwood and Nilüfer Ertekin‐Taner are co‐corresponding authors.
Contributor Information
Anna K. Greenwood, Email: anna.greenwood@sagebase.org.
Nilüfer Ertekin‐Taner, Email: taner.nilufer@mayo.edu.
DATA AVAILABILITY STATEMENT
The data described herein is available for use by the research community and has been deposited in the AD Knowledge Portal, with all publicly available data found under The Accelerating Medicines Partnership Alzheimer's Disease Diverse Cohorts Study (AMP‐AD Diverse Cohorts Study): (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage/StudyDetails?Study = syn51732482). Table 4 provides a list of the files and folders containing all data, their specific Synapse identifiers (IDs), DOIs, and brief descriptions of the file or folder contents. These files and their assigned DOIs will be maintained in perpetuity in the AMP‐AD Knowledge Portal. Access to all these files is enabled through the Sage Bionetworks, Synapse repository.
REFERENCES
- 1. GBD 2019 Dementia Forecasting Collaborators . Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7:e105‐e125. doi: 10.1016/S2468-2667(21)00249-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19:1598‐1695. doi: 10.1002/alz.13016 [DOI] [PubMed] [Google Scholar]
- 3. Reitz C, Pericak‐Vance MA, Foroud T, Mayeux R. A global view of the genetic basis of Alzheimer disease. Nat Rev Neurol. 2023;19:261‐277. doi: 10.1038/s41582-023-00789-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Logue MW, Dasgupta S, Farrer LA. Genetics of Alzheimer's disease in the African American population. J Clin Med. 2023;12:5189. doi: 10.3390/jcm12165189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shin J, Doraiswamy PM. Underrepresentation of African‐Americans in Alzheimer's Trials: a call for affirmative action. Front Aging Neurosci. 2016;8:123. doi: 10.3389/fnagi.2016.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chin AL, Negash S, Hamilton R. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25:187‐195. doi: 10.1097/wad.0b013e318211c6c9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53:1276‐1282. doi: 10.1038/s41588-021-00921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reitz C, Jun G, Naj A, et al. Variants in the ATP‐binding cassette transporter (ABCA7), apolipoprotein E ϵ4,and the risk of late‐onset Alzheimer disease in African Americans. JAMA. 2013;309:1483‐1492. doi: 10.1001/jama.2013.2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JH, Barral S, Cheng R, et al. Age‐at‐onset linkage analysis in Caribbean Hispanics with familial late‐onset Alzheimer's disease. Neurogenetics. 2008;9:51‐60. doi: 10.1007/s10048-007-0103-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghani M, Sato C, Lee JH, et al. Evidence of recessive Alzheimer disease loci in a Caribbean Hispanic data set: genome‐wide survey of runs of homozygosity. JAMA Neurol. 2013;70(10):1261‐1267. doi: 10.1001/jamaneurol.2013.3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andrews SJ, Renton AE, Fulton‐Howard B, Podlesny‐Drabiniok A, Marcora E, Goate AM. The complex genetic architecture of Alzheimer's disease: novel insights and future directions. EBioMedicine. 2023;90:104511. doi: 10.1016/j.ebiom.2023.104511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kunkle BW, Schmidt M, Klein H‐U, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta‐analysis. JAMA Neurol. 2021;78:102‐113. doi: 10.1001/jamaneurol.2020.3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sherva R, Zhang R, Sahelijo N, et al. African ancestry GWAS of dementia in a large military cohort identifies significant risk loci. Mol Psychiatry. 2023;28:1293‐1302. doi: 10.1038/s41380-022-01890-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Logue MW, Schu M, Vardarajan BN, et al. Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimers Dement. 2014;10:609‐618.ell. doi: 10.1016/j.jalz.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jin SC, Carrasquillo MM, Benitez BA, et al. TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener. 2015;10:19. doi: 10.1186/s13024-015-0016-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. N'Songo A, Carrasquillo MM, Wang X, et al. African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol Genet. 2017;3:e141. doi: 10.1212/NXG.0000000000000141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olivier M, Asmis R, Hawkins GA, Howard TD, Cox LA. The need for multi‐omics biomarker signatures in precision medicine. Int J Mol Sci. 2019;20:4781. doi: 10.3390/ijms20194781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin J, Dong K, Bai Y, et al. Precision oncology for gallbladder cancer: insights from genetic alterations and clinical practice. Ann Transl Med. 2019;7:467. doi: 10.21037/atm.2019.08.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hodes RJ, Buckholtz N. Accelerating Medicines Partnership: Alzheimer's Disease (AMP‐AD) Knowledge Portal Aids Alzheimer's Drug Discovery through Open Data Sharing. Expert Opin Ther Targets. 2016;20:389‐391. doi: 10.1517/14728222.2016.1135132 [DOI] [PubMed] [Google Scholar]
- 21. Allen M, Carrasquillo MM, Funk C, et al. Human whole genome genotype and transcriptome data for Alzheimer's and other neurodegenerative diseases. Sci Data. 2016;3:160089. doi: 10.1038/sdata.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. St John‐Williams L, Blach C, Toledo JB, et al. Targeted metabolomics and medication classification data from participants in the ADNI1 cohort. Sci Data. 2017;4:170140. doi: 10.1038/sdata.2017.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang M, Beckmann ND, Roussos P, et al. The Mount Sinai cohort of large‐scale genomic, transcriptomic and proteomic data in Alzheimer's disease. Sci Data. 2018;5:180185. doi: 10.1038/sdata.2018.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Jager PL, Ma Y, McCabe C, et al. A multi‐omic atlas of the human frontal cortex for aging and Alzheimer's disease research. Sci Data. 2018;5:180142. doi: 10.1038/sdata.2018.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greenwood AK, Montgomery KS, Kauer N, et al. The AD knowledge portal: a repository for multi‐omic data on Alzheimer's disease and aging. Curr Protoc Hum Genet. 2020;108(1):e105. doi: 10.1002/cphg.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156‐1163. doi: 10.1038/nn.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allen M, Burgess JD, Ballard T, et al. Gene expression, methylation and neuropathology correlations at progressive supranuclear palsy risk loci. Acta Neuropathol. 2016;132:197‐211. doi: 10.1007/s00401-016-1576-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carrasquillo MM, Allen M, Burgess JD, et al. A candidate regulatory variant at the TREM gene cluster associates with decreased Alzheimer's disease risk and increased TREML1 and TREM2 brain gene expression. Alzheimers Dement. 2017;13:663‐673. doi: 10.1016/j.jalz.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen M, Wang X, Burgess JD, et al. Conserved brain myelination networks are altered in Alzheimer's and other neurodegenerative diseases. Alzheimers Dement. 2018;14:352‐366. doi: 10.1016/j.jalz.2017.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Allen M, Wang X, Serie DJ, et al. Divergent brain gene expression patterns associate with distinct cell‐specific tau neuropathology traits in progressive supranuclear palsy. Acta Neuropathol. 2018;136:709‐727. doi: 10.1007/s00401-018-1900-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nho K, Nudelman K, Allen M, et al. Genome‐wide transcriptome analysis identifies novel dysregulated genes implicated in Alzheimer's pathology. Alzheimers Dement. 2020;16:1213‐1223. doi: 10.1002/alz.12092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wan Y‐W, Al‐Ouran R, Mangleburg CG, et al. Meta‐analysis of the Alzheimer's disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 2020;32:107908. doi: 10.1016/j.celrep.2020.107908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X, Allen M, Li S, et al. Deciphering cellular transcriptional alterations in Alzheimer's disease brains. Mol Neurodegener. 2020;15:38. doi: 10.1186/s13024-020-00392-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strickland SL, Reddy JS, Allen M, et al. MAPT haplotype–stratified GWAS reveals differential association for AD risk variants. Alzheimers Dement. 2020;16:983‐1002. doi: 10.1002/alz.12099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma Y, Dammer EB, Felsky D, et al. Atlas of RNA editing events affecting protein expression in aged and Alzheimer's disease human brain tissue. Nat Commun. 2021;12(1):7035. doi: 10.1038/s41467-021-27204-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ma Y, Yu L, Olah M, Smith RG, Pishva E, Menon V, et al. Epigenomic features related to microglia are associated with attenuated effect of APOE ε4 on Alzheimer's disease risk in humans: human neuropathology: AD neuropathology. Alzheimers Dement. 2023;18(4):688‐699. doi: 10.1002/alz.043533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Allen M, İş Ö, et al. Alzheimer's disease and progressive supranuclear palsy share similar transcriptomic changes in distinct brain regions. J Clin Invest. 2022;132(2):e149904. doi: 10.1172/jci149904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Batra R, Arnold M, Wörheide MA, et al. The landscape of metabolic brain alterations in Alzheimer's disease. Alzheimers Dement. 2022;19:980‐998. doi: 10.1002/alz.12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oatman SR, Reddy JS, Quicksall Z, et al. Genome‐wide association study of brain biochemical phenotypes reveals distinct genetic architecture of Alzheimer's disease related proteins. Mol Neurodegener. 2023;18(1):2. doi: 10.1186/s13024-022-00592-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Min Y, Wang X, İş Ö, et al. Cross species systems biology discovers glial DDR2, STOM, and KANK2 as therapeutic targets in progressive supranuclear palsy. Nat Commun. 2023;14:6801. doi: 10.1038/s41467-023-42626-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McKenzie AT, Moyon S, Wang M, et al. Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer's disease. Mol Neurodegener. 2017;12(1):82. doi: 10.1186/s13024-017-0219-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beckmann ND, Lin W‐J, Wang M, et al. Multiscale causal networks identify VGF as a key regulator of Alzheimer's disease. Nat Commun. 2020;11(1):3942. doi: 10.1038/s41467-020-17405-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang M, Li A, Sekiya M, et al. Transformative network modeling of multi‐omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer's disease. Neuron. 2021;109:257‐272.e14. doi: 10.1016/j.neuron.2020.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horgusluoglu E, Neff R, Song WM, et al. Integrative metabolomics‐genomics approach reveals key metabolic pathways and regulators of Alzheimer's disease. Alzheimers Dement. 2021;18:1260‐1278. doi: 10.1002/alz.12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnson ECB, Carter EK, Dammer EB, et al. Large‐scale deep multi‐layer analysis of Alzheimer's disease brain reveals strong proteomic disease‐related changes not observed at the RNA level. Nat Neurosci. 2022;25:213‐225. doi: 10.1038/s41593-021-00999-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci. 2018;21:811‐819. doi: 10.1038/s41593-018-0154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. MahmoudianDehkordi S, Arnold M, Nho K, et al. Altered bile acid profile associates with cognitive impairment in Alzheimer's disease‐An emerging role for gut microbiome. Alzheimers Dement. 2019;15:76‐92. doi: 10.1016/j.jalz.2018.07.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seyfried NT, Dammer EB, Swarup V, et al. A multi‐network approach identifies protein‐specific co‐expression in asymptomatic and symptomatic Alzheimer's disease. Cell Syst. 2017;4:60‐72.e4. doi: 10.1016/j.cels.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wingo AP, Dammer EB, Breen MS, et al. Large‐scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun. 2019;10(1):1619. doi: 10.1038/s41467-019-09613-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sung YJ, Yang C, Norton J, et al. Proteomics of brain, CSF, and plasma identifies molecular signatures for distinguishing sporadic and genetic Alzheimer's disease. Sci Transl Med. 2023;15(703):eabq5923. doi: 10.1126/scitranslmed.abq5923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toledo JB, Arnold M, Kastenmüller G, et al. Metabolic network failures in Alzheimer's disease: a biochemical road map. Alzheimers Dement. 2017;13:965‐984. doi: 10.1016/j.jalz.2017.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nho K, Kueider‐Paisley A, MahmoudianDehkordi S, et al. Altered bile acid profile in mild cognitive impairment and Alzheimer's disease: relationship to neuroimaging and CSF biomarkers. Alzheimers Dement. 2018;15:232‐244. doi: 10.1016/j.jalz.2018.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baloni P, Arnold M, Moreno H, et al. Multi‐omic analyses characterize the ceramide/sphingomyelin pathway as a therapeutic target in Alzheimer's disease 2021. medRxiv. 2021. doi: 10.1101/2021.07.16.21260601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reddy JS, Jin J, Lincoln SJ, et al. Transcript levels in plasma contribute substantial predictive value as potential Alzheimer's disease biomarkers in African Americans. EBioMedicine. 2022;78:103929. doi: 10.1016/j.ebiom.2022.103929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Modeste ES, Ping L, Watson CM, et al. Quantitative proteomics of cerebrospinal fluid from African Americans and Caucasians reveals shared and divergent changes in Alzheimer's disease. Mol Neurodegener. 2023;18(1):48. doi: 10.1186/s13024-023-00638-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ginsburg GS, Denny JC, Schully SD. Data‐driven science and diversity in the All of Us Research Program. Sci Transl Med. 2023;15(726):eade9214. doi: 10.1126/scitranslmed.ade9214 [DOI] [PubMed] [Google Scholar]
- 57. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 58. Beach TG, Adler CH, Sue LI, et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology. 2015;35:354‐389. doi: 10.1111/neup.12189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging—Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging—Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1‐13. doi: 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18(Suppl 4):S1‐S2. [PubMed] [Google Scholar]
- 62. Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement. 2014;10:477‐484.e1. doi: 10.1016/j.jalz.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barnes LL, Shah RC, Aggarwal NT, Bennett DA, Schneider JA. The Minority Aging Research Study: ongoing efforts to obtain brain donation in African Americans without dementia. Curr Alzheimer Res. 2012;9:734‐745. doi: 10.2174/156720512801322627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community‐based studies. Neurology. 2006;66:1837‐1844. doi: 10.1212/01.wnl.0000219668.47116.e6 [DOI] [PubMed] [Google Scholar]
- 65. Coleman C, Wang M, Wang E, et al. Multi‐omic atlas of the parahippocampal gyrus in Alzheimer's disease. Sci Data. 2023;10:602. doi: 10.1038/s41597-023-02507-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li K, Rashid T, Li J, et al. Postmortem Brain Imaging in Alzheimer's Disease and Related Dementias: the South Texas Alzheimer's Disease Research Center Repository. J Alzheimers Dis. 2023;96:1267‐1283. doi: 10.3233/jad-230389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐1800. doi: 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 68. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479‐486. doi: 10.1212/wnl.41.4.479 [DOI] [PubMed] [Google Scholar]
- 69. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 70. Lee Y, Jeon S, Park M, et al. Effects of Alzheimer and Lewy body disease pathologies on brain metabolism. Ann Neurol. 2022;91:853‐863. doi: 10.1002/ana.26355 [DOI] [PubMed] [Google Scholar]
- 71. Braak H, Braak E. Alzheimerʼs disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. 1990;49:215‐224. doi: 10.1097/00005072-199005000-00003 [DOI] [PubMed] [Google Scholar]
- 72. Matsubara T, Soh J, Morita M, et al. DV200 index for assessing RNA integrity in next‐generation sequencing. Biomed Res Int. 2020;2020:9349132. doi: 10.1155/2020/9349132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kalari KR, Nair BA, Bhavsar JD, et al. MAP‐RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinf. 2014;15:224. doi: 10.1186/1471-2105-15-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Maienschein‐Cline M, Lei Z, Gardeux V, et al. ARTS: automated randomization of multiple traits for study design. Bioinformatics. 2014;30:1637‐1639. doi: 10.1093/bioinformatics/btu075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ping L, Duong DM, Yin L, et al. Global quantitative analysis of the human brain proteome in Alzheimer's and Parkinson's Disease. Sci Data. 2018;5:180036. doi: 10.1038/sdata.2018.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mertins P, Tang LC, Krug K, et al. Reproducible workflow for multiplexed deep‐scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography‐mass spectrometry. Nat Protoc. 2018;13:1632‐1661. doi: 10.1038/s41596-018-0006-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bai B, Wang X, Li Y, et al. Deep multilayer brain proteomics identifies Molecular Networks in Alzheimer's Disease Progression. Neuron. 2020;105:975‐991.e7. doi: 10.1016/j.neuron.2019.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wingo AP, Liu Y, Gerasimov ES, et al. Integrating human brain proteomes with genome‐wide association data implicates new proteins in Alzheimer's disease pathogenesis. Nat Genet. 2021;53:143‐146. doi: 10.1038/s41588-020-00773-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Glover CM, Shah RC, Bennett DA, Wilson RS, Barnes LL. Perceived impediments to completed brain autopsies among diverse older adults who have signed a Uniform Anatomical Gift Act for brain donation for clinical research. Ethn Dis. 2020;30:709‐718. doi: 10.18865/ed.30.s2.709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ighodaro ET, Nelson PT, Kukull WA, et al. Challenges and considerations related to studying dementia in Blacks/African Americans. J Alzheimers Dis. 2017;60:1‐10. doi: 10.3233/jad-170242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Santos OA, Pedraza O, Lucas JA, et al. Ethnoracial differences in Alzheimer's disease from the FLorida Autopsied Multi‐Ethnic (FLAME) cohort. Alzheimers Dement. 2019;15:635‐643. doi: 10.1016/j.jalz.2018.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Weiner MW, Veitch DP, Miller MJ, et al. Increasing participant diversity in AD research: plans for digital screening, blood testing, and a community‐engaged approach in the Alzheimer's Disease Neuroimaging Initiative 4. Alzheimers Dement. 2023;19:307‐317. doi: 10.1002/alz.12797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nag S, Barnes LL, Yu L, et al. Association of Lewy bodies with age‐related clinical characteristics in Black and White decedents. Neurology. 2021;97(8):e825‐e835. doi: 10.1212/wnl.0000000000012324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barnes LL, Leurgans S, Aggarwal NT, et al. Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology. 2015;85:528‐534. doi: 10.1212/wnl.0000000000001834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Graff‐Radford NR, Besser LM, Crook JE, Kukull WA, Dickson DW. Neuropathologic differences by race from the National Alzheimer's Coordinating Center. Alzheimers Dement. 2016;12:669‐677. doi: 10.1016/j.jalz.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Conway OJ, Carrasquillo MM, Wang X, et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol Neurodegener. 2018;13(1):53. doi: 10.1186/s13024-018-0289-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Patel T, Carnwath TP, Wang X, et al. Transcriptional landscape of human microglia implicates age, sex, and APOE‐related immunometabolic pathway perturbations. Aging Cell. 2022;21(5):e13606. doi: 10.1111/acel.13606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Strickland SL, Morel H, Prusinski C, et al. Association of ABI3 and PLCG2 missense variants with disease risk and neuropathology in Lewy body disease and progressive supranuclear palsy. Acta Neuropathol Commun. 2020;8(1):172. doi: 10.1186/s40478-020-01050-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Johnson ECB, Dammer EB, Duong DM, et al. Large‐scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769‐780. doi: 10.1038/s41591-020-0815-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Adkins‐Jackson PB, George KM, Besser LM, et al. The structural and social determinants of Alzheimer's disease related dementias. Alzheimers Dement. 2023;19:3171‐3185. doi: 10.1002/alz.13027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gomez‐Pinilla F, Zhuang Y, Feng J, Ying Z, Fan G. Exercise impacts brain‐derived neurotrophic factor plasticity by engaging mechanisms of epigenetic regulation. Eur J Neurosci. 2010;33:383‐390. doi: 10.1111/j.1460-9568.2010.07508.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hajjar I, Yang Z, Okafor M, et al. Association of plasma and cerebrospinal fluid alzheimer disease biomarkers with race and the role of genetic ancestry, vascular comorbidities, and neighborhood factors. JAMA Netw Open. 2022;5:e2235068. doi: 10.1001/jamanetworkopen.2022.35068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Avila‐Rieger J, Turney IC, Vonk JMJ, et al. Socioeconomic status, biological aging, and memory in a diverse national sample of older US men and women. Neurology. 2022;99(19):e2114‐e2124. doi: 10.1212/wnl.0000000000201032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Deniz K, Ho CCG, Malphrus KG, et al. Plasma biomarkers of Alzheimer's disease in African Americans. J Alzheimers Dis. 2021;79:323‐334. doi: 10.3233/jad-200828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Stites SD, Midgett S, Mechanic‐Hamilton D, et al. Establishing a framework for gathering structural and social determinants of health in Alzheimer's Disease Research Centers. Gerontologist. 2021;62:694‐703. doi: 10.1093/geront/gnab182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Stites SD, Coe NB. Let's not repeat history's mistakes: two cautions to scientists on the use of race in Alzheimer's disease and Alzheimer's disease related dementias research. J Alzheimers Dis. 2023;92:729‐740. doi: 10.3233/jad-220507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hendricks‐Sturrup RM, Edgar LM, Johnson‐Glover T, Lu CY. Exploring African American community perspectives about genomic medicine research: a literature review. SAGE Open Med. 2020;8:205031212090174. doi: 10.1177/2050312120901740 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data described herein is available for use by the research community and has been deposited in the AD Knowledge Portal, with all publicly available data found under The Accelerating Medicines Partnership Alzheimer's Disease Diverse Cohorts Study (AMP‐AD Diverse Cohorts Study): (https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage/StudyDetails?Study = syn51732482). Table 4 provides a list of the files and folders containing all data, their specific Synapse identifiers (IDs), DOIs, and brief descriptions of the file or folder contents. These files and their assigned DOIs will be maintained in perpetuity in the AMP‐AD Knowledge Portal. Access to all these files is enabled through the Sage Bionetworks, Synapse repository.