

Abstract
As a traceless, bioreversible modification, the esterification of carboxyl groups in peptides and proteins has the potential to increase their clinical utility. An impediment is the lack of strategies to quantify esterase-catalyzed hydrolysis rates for esters in esterified biologics. We have developed a continuous Förster resonance energy transfer (FRET) assay for esterase activity based on a peptidic substrate and a protease, Glu-C, that cleaves a glutamyl peptide bond only if the glutamyl side chain is a free acid. Using pig liver esterase (PLE) and human carboxylesterases, we validated the assay with substrates containing simple esters (e.g., ethyl) and esters designed to be released by self-immolation upon quinone methide elimination. We found that simple esters were not cleaved by esterases, likely for steric reasons. To account for the relatively low rate of quinone methide elimination, we extended the mathematics of the traditional Michaelis–Menten model to conclude with a first-order intermediate decay step. By exploring two regimes of our substrate → intermediate → product (SIP) model, we evaluated the rate constants for the PLE-catalyzed cleavage of an ester on a glutamyl side chain and subsequent spontaneous quinone methide elimination to regenerate the unmodified peptide (; ). The detection of esterase activity was also feasible in the human intestinal S9 fraction. Our assay and SIP model increase the understanding of the release kinetics of esterified biologics and facilitate the rational design of efficacious peptide prodrugs.
Keywords: Protein Accession Codes, CES1, P23141, CES2, O00748, Glu-C, Q6GI34, PLE, Q29550
Graphical Abstract
INTRODUCTION
Esterases, abundant in organisms from bacteria to mammals, play a key role in the metabolism of xenobiotics and endogenous compounds.1–4 These enzymes catalyze the hydrolysis of esters and other functional groups,4 including some amides and carbamates, to form component acids. In humans, esterases are found in many tissues,2,4 and their overexpression is often associated with cancer.4,5
Among esterases, carboxylesterases (CESs; EC 3.1.1.1) from the serine hydrolase superfamily are known for their ability to activate ester-based prodrugs.4,6 This strategy improves the cell permeability of compounds by masking their anionic carboxylate groups as esters, which can be cleaved tracelessly by intracellular esterases.7 Of the five known human CESs, CES1 and CES2 are the most well-studied.6 CES1 favors substrates (e.g., benazepril) with a small alcohol and a large acyl group, whereas CES2 exhibits the opposite preference (e.g., aspirin). Notably, the specificity of these enzymes is not rigid, allowing them to act on the same substrates (e.g., heroin). Most insights into the biology and scope of esterases come from traditional prodrugs and assays employing small-molecule probes. A broad arsenal of such assays has been developed,6,8–11 providing a platform for the discovery of narrow-spectrum antibiotics,12,13 proximity-based labeling reagents,14 and voltage-sensitive dyes.15
In recent years, the market for biologics has been expanding more rapidly than the market of small molecules, driving innovation across multiple therapeutic sectors.16 Yet, because most biologics do not naturally cross membranes, their use has been largely restricted to the extracellular space. The desire to engage intracellular targets with biologics spurred efforts to apply ester-based prodrug concepts toward peptides,17–22 proteins,23–28 and RNA.29,30 For example, several groups have installed esters in cyclic peptides,17–19,21 often to enhance cell permeability and pharmacokinetics. Others developed strategies for C‑terminal esterification,20,22 advancing methods for peptide labeling in a reversible manner. To unlock the potential of protein prodrugs, our laboratory optimized α-aryl-α‑diazoamides for the chemoselective esterification of carboxyl groups in proteins under mild aqueous conditions.23 With the aid of these compounds, we24–26,28 and others27 have delivered various proteins into live cells.
Research on esterified biologics has outpaced assay development for measuring their cleavage. To the best of our knowledge, no continuous esterase assay based on a carboxyl group within a biologic has been reported to date. This absence contrasts with the abundance of such assays based on probes designed to mimic small molecules6,8–11 and nanomaterials.31,32 Extant probes exhibit different steric constraints and structural complexity relative to biologics, which could result in differences in their interactions with esterases. The cleavage of esterified biologics is typically assessed through mass spectrometry, high-performance liquid chromatography, gel electrophoresis, or readouts based on changes in activity upon de-esterification. These methods rarely provide quantitative kinetic data such as , , or rate changes in activity upon de-esterification. These methods rarely provide quantitative kinetic data such as , or rate constants associated with the decay of short-lived intermediates. Further, the presence of several esters installed in different locations (e.g., multiple esters on a protein) complicates comparative studies.
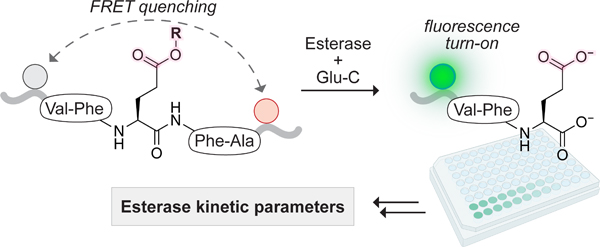
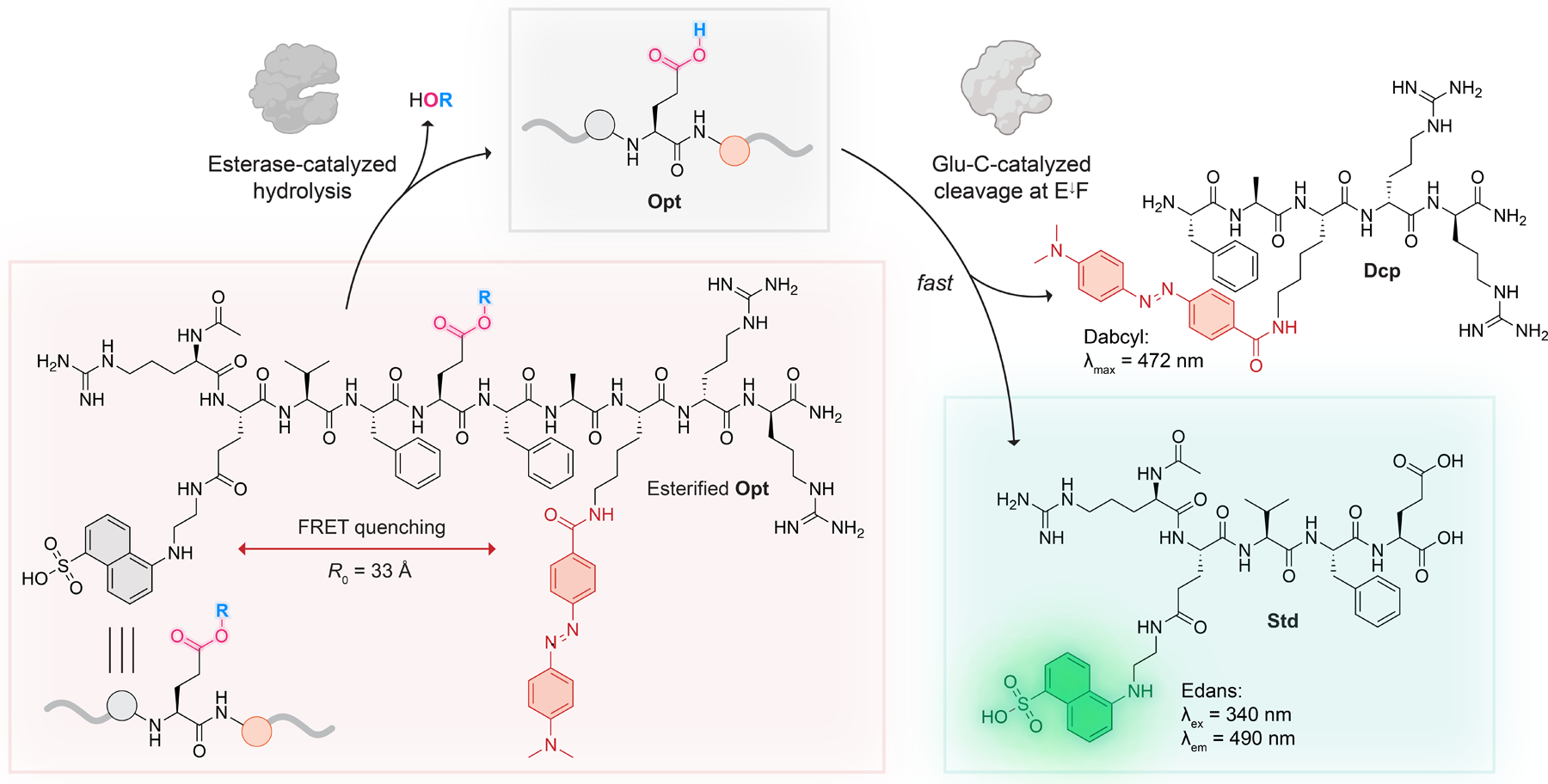
Here, we fill a gap. Specifically, we develop a continuous assay for esterase activity with peptide substrates having aliphatic carboxyl groups (Figure 1). Our assay deploys the Staphylococcus aureus V8 protease (Glu-C33; EC 3.4.21.19) in a realm distinct from its conventional use in proteomic analyses.34 Esterase-mediated hydrolysis of a glutamyl ester in the side chain of our optimal substrate results in the generation of a sequence (Opt) that is cleaved by Glu-C into a fluorescent product (standard, Std) and a dark cleavage product (Dcp), generating fluorescence due to the loss of Förster resonance energy transfer (FRET). Our esterase substrate is an esterified peptide rather than a small molecule, contains a single Glu residue for “one-hit” kinetics, and permits variation of the alcohol portion of the ester.
Figure 1.
Basis of a continuous FRET assay for esterase activity coupled to enzyme Glu-C. R0 refers to the Förster radius between Edans (fluorescence donor) and Dabcyl (fluorescence quencher). The R group refers to either an ester formed directly with the Glu residue or a self-immolative, esterase-sensitive moiety distal from the peptide main chain (for examples of R, see Figure 3).
Using a panel of four peptides with esters of varying sizes and release mechanisms, we validated the assay with pig liver esterase (PLE), CES1, and CES2. We find that esters formed directly with the Glu residue are not cleaved by any tested esterase. In contrast, esters and carbonates that are distal from the peptide main chain (which are designed to be tracelessly released via quinone methide elimination35) are cleaved efficiently, showcasing the importance of self-immolative linkers. Similar findings were obtained when esterified peptides were screened for cleavage in a human intestinal S9 fraction. To account for the rate of the relatively slow stop of intermediate decay, we created a novel substrate → intermediate → product (SIP) kinetic model. By exploring two regimes of SIP, we measured the rate constant of quinone methide elimination, and, for the first time, the associated with esterase-catalyzed hydrolysis of an esterified peptide. Our assay is poised to enable the rational design of esterified peptide prodrugs for intracellular delivery.
RESULTS AND DISCUSSION
Assay Design.
Several considerations were taken into account in the assay design (Figure 1). To endow the assay with high sensitivity and low interference, we focused on the gain of fluorescence due to the loss of FRET. We installed donor and acceptor fluorophores at opposite ends of the peptide backbone and coupled proteolysis to ester hydrolysis. We selected Glu-C, which is a commercially available protease,33 as the detection enzyme due to its specificity for cleaving on the C‑terminal side of glutamate (and occasionally aspartate) residues.36 Previous modeling suggested that the specificity of Glu-C relies on the formation of a hydrogen bond/ion pair between the anionic carboxylate of a glutamate side chain in a substrate and the cationic α‑ammonium group of Val1 at the enzymic N‑terminus.37 That carboxylate–ammonium interaction locates the scissile peptide bond near the active-site nucleophile, the side-chain hydroxy group of Ser169.37 This mechanism served as the foundation for our hypothesis that peptides esterified at the Glu residue would be stable in the presence of Glu-C until esterase-mediated “unmasking.” To maximize the catalytic efficiency of Glu-C, we chose an optimal sequence,36 Val, Phe, Glu, Phe, and Ala in the P3, P2, P1, P1′, and P2′ positions, respectively, as the basis for the substrate peptide, esterified Opt (Figure 1). Our computational model of the complex between Glu-C and a truncated peptide (Ac-Phe-Glu-Phe-NH2) showed the expected salt bridge as well as a hydrogen bond between the glutamyl carboxyl group of the substrate and Thr164 (Figure 2).
Figure 2.
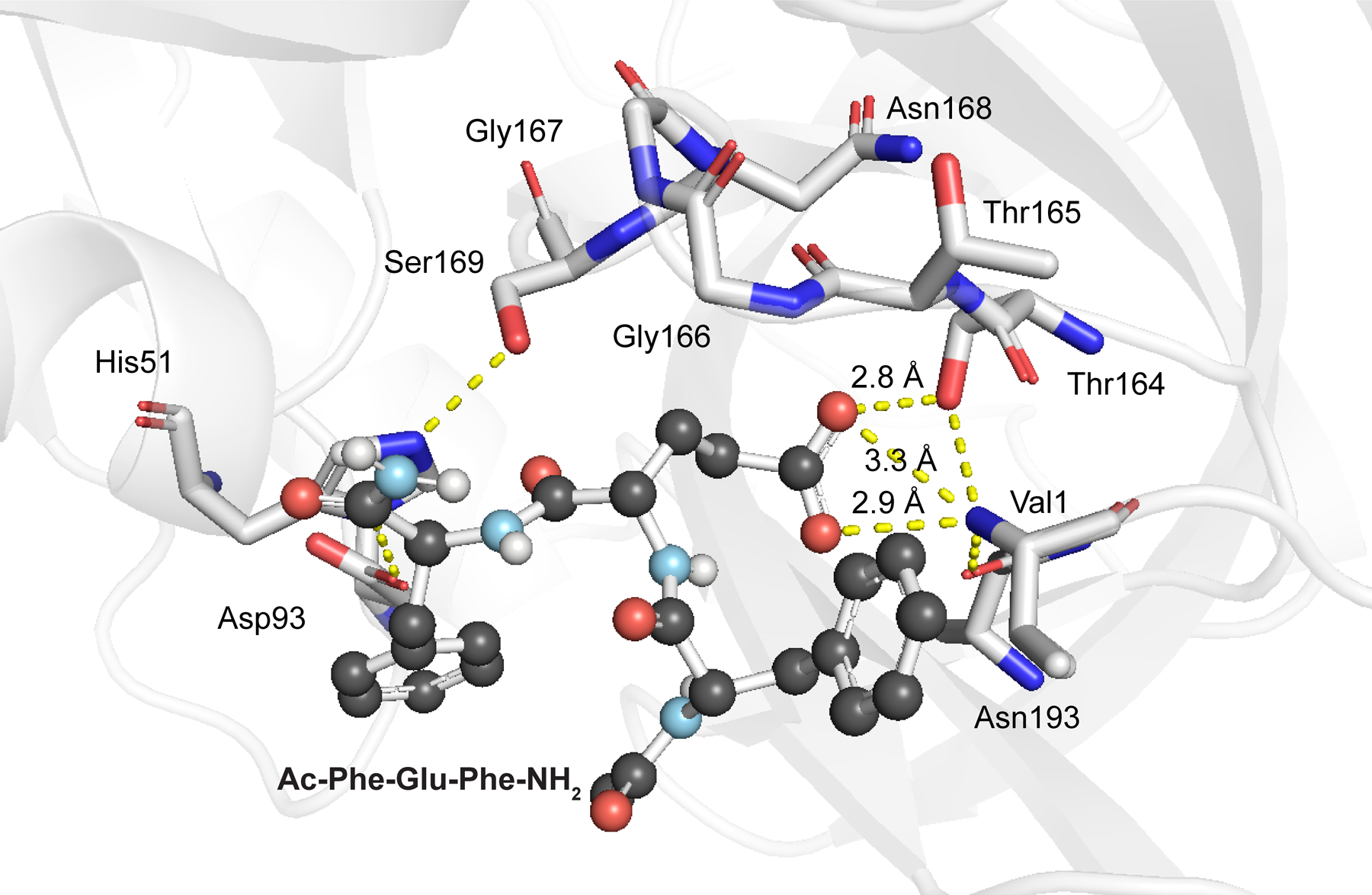
Computational model of a complex between Glu-C (PDB ID 1qy6)37 and Ac-Phe-Glu-Phe-NH2, which is deprotonated at the Glu residue and bound to the active site.
As the FRET pair for the substrate, we selected Dabcyl and Edans due to their permissive Förster radius 38 and the bioorthogonality of Edans fluorescence .39 We also flanked our peptide with d-arginine residues to improve its water solubility and discourage its binding to exopeptidases in complex mixtures. The final design considerations for Opt were the acetylation of its N terminus to prevent intramolecular aminolysis of the ester and amidation of the C terminus to ensure that the sole carboxyl group is on the glutamate side chain.
Synthesis of a Panel of Esterified Peptides for Assay Validation.
To validate our assay, we designed a panel of four esterified peptides (Figure 3). Because we were curious about the impact of steric effects on the ability of esterases to cleave our substrates, we focused on varying the alcohol portion of the ester and on aspects of the traceless release mechanism.
Figure 3.
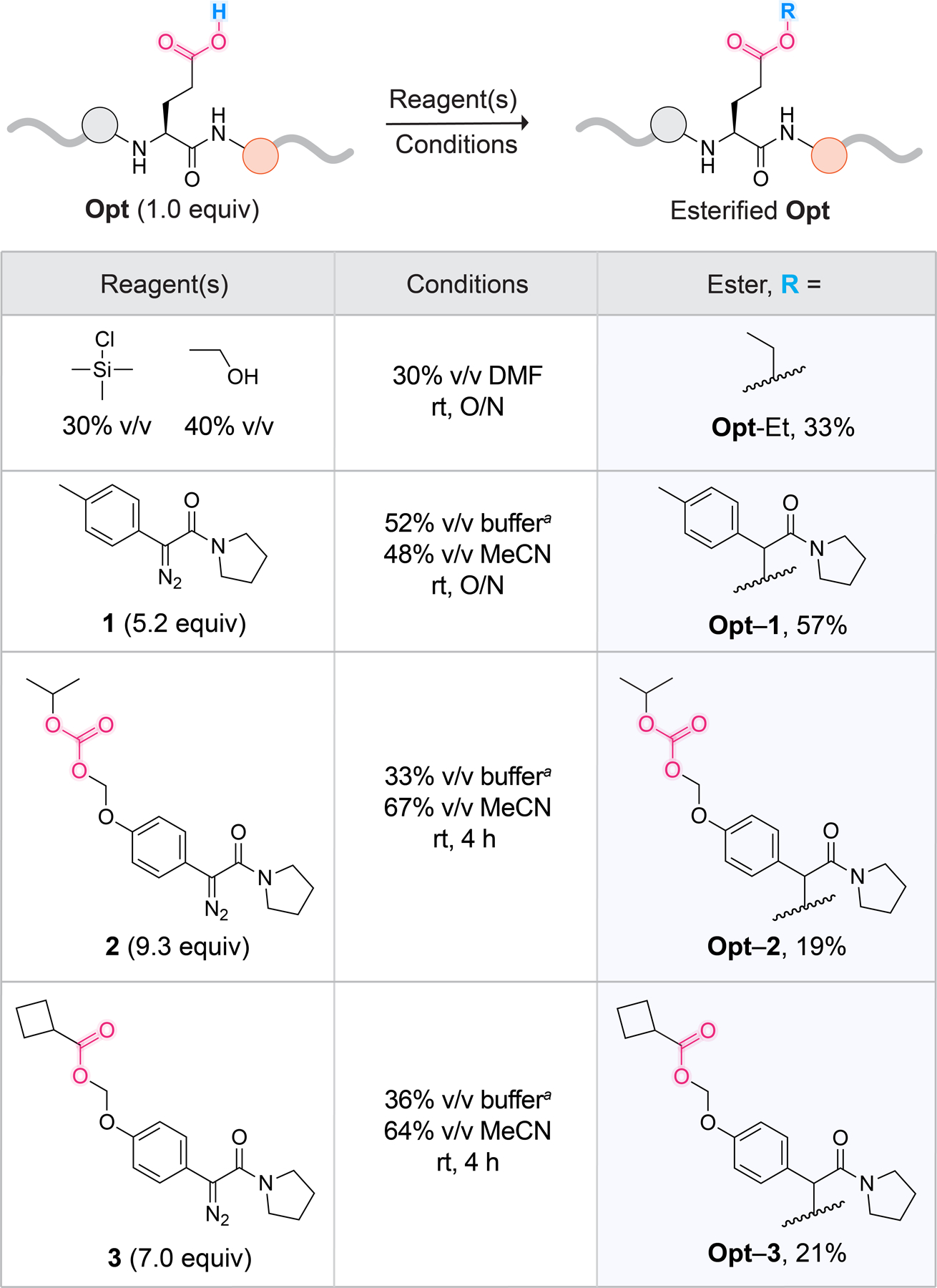
Conditions for the synthesis of a panel of esterified Opt peptides. a10 mM MES–HCl buffer, pH 6.0. For details on reagent concentrations, solid-phase peptide synthesis of Opt, synthesis of diazo compounds 1–3 based on established routes,26,46,55 and portionwise diazo compound addition, see Schemes S1–S8 and Figures S1–S6.
To begin, we chose Opt-Et, which would enable us to explore the hydrolysis of an ethyl ester, which is prevalent in small-molecule prodrugs40–42 but novel in the context of a peptide. Adapting reported routes for amino acid esterification,43–45 we synthesized Opt-Et by adding EtOH and trimethylsilyl chloride to Opt in DMF (Figures 3 and S3 and Scheme S5).
Next, we wanted to compare the release kinetics of esters generated from α-aryl-α-diazoacetamides containing various functionalities in the para position of the aryl ring: a methyl group (Opt–1), an alkyloxycarbonyloxymethyl (Opt–2), and an α-cyclopropyl ester (Opt–3) (Figure 3). In contrast to Opt–1, peptides Opt–2 and Opt–3 feature a second, more sterically accessible site for esterase-mediated cleavage, which enables the traceless release of the carboxyl group through 1,6‑quinone methide elimination. Opt–3 is inspired by the work of Lavis and co-workers,46 who reported that α‑cyclopropyl esters in fluorescein were efficiently cleaved in a variety of endogenous settings. Thus, we were intrigued to see if Opt–3 would be a better esterase substrate than Opt–2. To synthesize Opt–1, Opt–2, and Opt–3, we reacted Opt with diazo compounds 1, 2, and 3, respectively, at pH 6.0 (Figures 3 and S4–S6, Schemes S6–S8). α-Aryl-α‑diazoacetamides, preferentially label acids of higher pKa in aqueous buffers where the pH is close to or larger than the pKa of the target acid.47,48 Hence, under our reaction conditions, we expected diazo compounds to favor the esterification of the carboxylic acid of glutamate (pKa 4) over the sulfonic acid in Edans. (The pKa of the sulfonic acid moiety in p-toluenesulfonic acid is −2.8.49)
Preparation of Peptide Stock Solutions.
Using absorbance, we measured the concentrations of DMF stock solutions of Std (εEdans = 5438 M−1 cm−1 at 336 nm),39 Opt (εDabcyl = 15,100 M−1 cm−1 at 472 nm, where Edans does not absorb; Figure S8),39 and esterified peptides (εDabcyl), by diluting them into a buffer containing Triton X-100 (0.8% w/v). This nonionic detergent is commonly used in esterase assays (at 0.1–1% w/v)8,50,51 and, depending on the type of esterase and substrate, can have varying effects on enzymatic activity.52–54 We noticed that lower concentrations of TritonX-100 resulted in decreased apparent concentrations of all peptides except the more soluble Std. This decrease was more evident at larger dilutions, suggesting that additives prevent peptide loss from the solution due to undesirable peptide adherence to surfaces. Hence, in most subsequent experiments, we supplemented solutions with Triton X-100 at 0.8% w/v.
Optimization of Buffer, Glu-C Concentration, and Substrate Concentration.
To select a buffer compatible with our assay, we considered the pH optima for catalysis by both esterases (pH 6.5–8.0 for carboxylesterases)4 and Glu-C (two optima: pH 4.0 and 7.8).33 We were also aware that monovalent anions inhibit Glu-C, possibly due to competing with substrate for binding to the enzymic active site.56 Taking these constraints into account, we settled on using 10 mM HEPES–NaOH buffer, pH 7.4, for all assays.
Next, we sought to ensure that the signal from the detection step in our coupled assay is directly proportional to product concentration. This assumption holds if the rate of Opt→Std is much faster than that of Opt formation, making the rate of Glu-C-mediated proteolysis negligible. To find a condition in which catalysis by Glu-C is not rate-limiting, we evaluated the time course of 10 µM (Figure 4A) or 1 µM (Figure S7) Opt fluorescence after adding various amounts of Glu-C. In both experiments, the addition of 1 µM of Glu-C resulted in the near-immediate onset of a fluorescence plateau, prompting us to use this concentration in subsequent assays.
Figure 4.
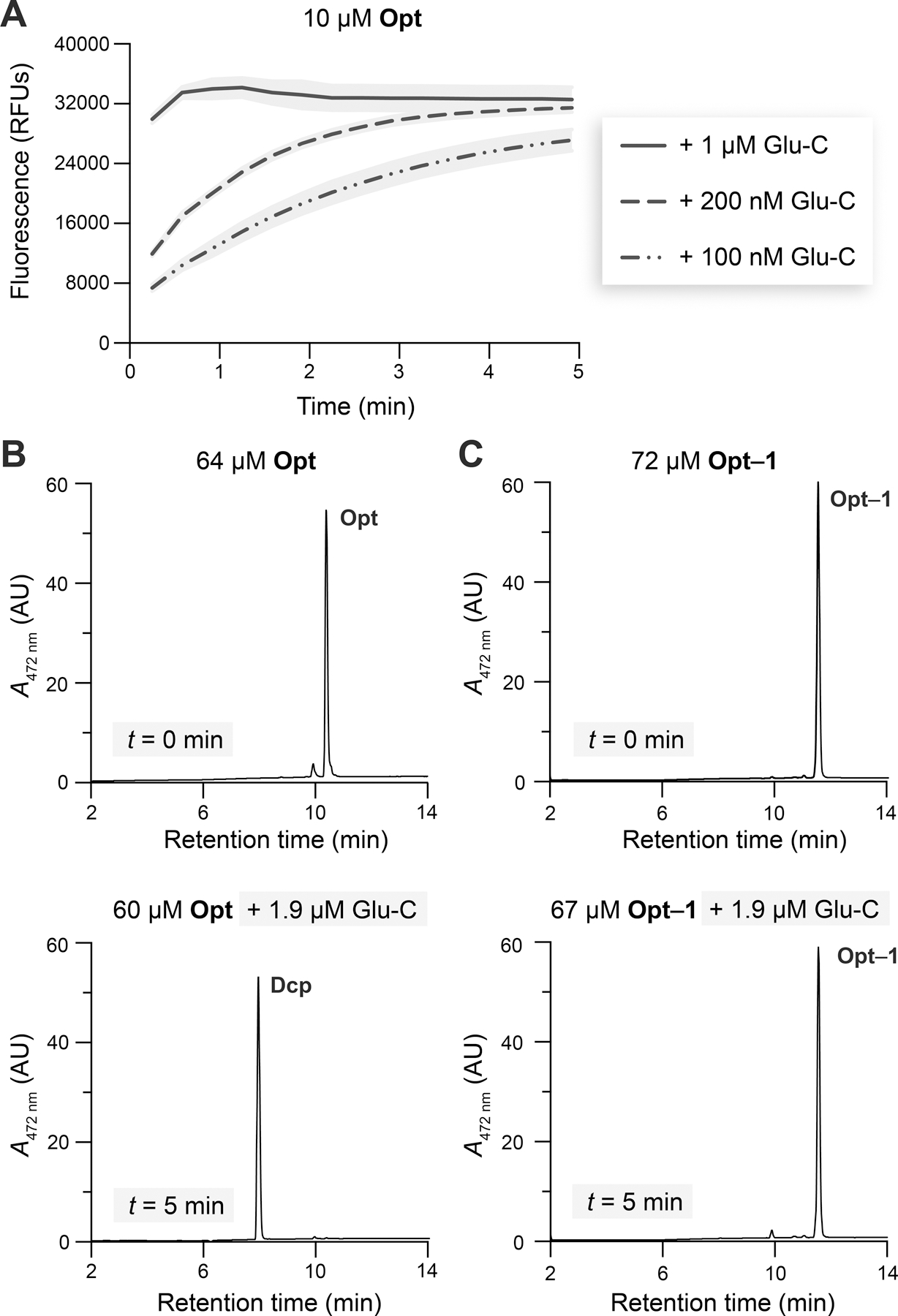
(A) Fluorescence of Opt incubated with Glu-C. Gray areas represent the SD; n = 2 independent experiments, n = 3 technical replicates. (B) LC-MS absorbance traces of Opt and Opt incubated with Glu-C. (C) LC-MS traces of Opt–1 and Opt–1 incubated with Glu-C. Studies were performed in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (1.5% v/v, assay; 5% v/v, LC-MS) and Triton X-100 (0.8% w/v, assay; 0% w/v, LC-MS) at 37 °C. See Figures S9 and S13G,H for LC-MS analyses of Glu-C incubations with all esterified peptides.
To confirm that Glu-C cleaves Opt at its Glu↓Phe bond, we analyzed an early time point of the reaction using quadrupole time-of-flight (Q-TOF) liquid chromatography–mass spectrometry (LC-MS). Indeed, after 5 min of incubation with Glu-C, Opt was fully converted into Dcp (Figure 4B) and Std (Figure S8D).
Our next goal was to validate that esterification protects Opt from cleavage by Glu-C. Previously, glutamate methylation was shown to endow peptides with resistance against Glu-C.57 After 5 min of incubation with Glu-C, Opt-Et (Figure S9B) and Opt–1 (Figure 4C) remained intact, whereas a small amount of Opt–2 (3%, Figure S9F) and Opt–3 (8%, Figure S9H) was cleaved to Dcp. In a separate experiment, after 40 min of Opt–3 incubation with Glu-C, 9% of the esterified peptide was cleaved to Dcp (Figure S13H). Overall, the esterified peptides were more stable to Glu-C than Opt, which was cleaved quickly and completely (cf: Figures 4B,C, S9B, and S13H).
Having validated Glu-C selectivity, we sought to optimize the working range of the assay. We were aware that the magnitude of the inner filter effect—a fluorescence suppression phenomenon arising from light reabsorption—depends on the concentration of quenching groups.58 Upon measuring the fluorescence of Std in the presence of increasing concentrations of Opt (0–10 µM), we found that fluorescence attenuation in this concentration range was <15% (Table S1 and Figure S10). Accordingly, the inner filter effect in the tested regime was minor and did not require any corrections.58–60 The limit of detection of our assay, estimated with eq S1, was ≥240 nM, and Opt cleavage by Glu-C resulted in an 18-fold increase in fluorescence (Figure S11). Taking these data into account, we performed our assays within a 240 nM–10 µM range of Opt-based substrates.
Cleavage of a Panel of Esterified Peptides by PLE.
Having optimized our assay, we proceeded to characterize the cleavage of four esterified peptides (Figure 3) by PLE, which shares a high sequence identity with human CES1.61 To eventually construct a progress curve of the product (Opt) concentration, , we measured the fluorescence associated with four blank-subtracted (blank: buffer alone) conditions (see additional discussion with eqs S3–S5): substrate incubated with both esterase and Glu-C , substrate incubated with esterase , substrate incubated with Glu-C , and Opt incubated with Glu-C . refers to fluorescence turn-on from product formation and other cleavage events that could lead to the loss of FRET. is the turn-on from the background fluorescence of the substrate and any proteolytic activity of the esterase. partially accounts for the loss of FRET due to aqueous ester hydrolysis and proteolysis of the esterified substrate by Glu-C. reflects the maximal turn-on from complete loss of FRET. We tested the cleavage of esterified peptides (10 µM) with a large amount of PLE (2 µM) to maximize conversion to product. The results of the screening are shown in Figure 5A. The two peptides with esters closest to the peptide main chain, Opt-Et and Opt–1, did not appear to get cleaved by PLE. In contrast, the two peptides with secondary release handles, Opt–2 and Opt–3, were cleaved efficiently, with Opt–3 getting cleaved slightly faster than Opt–2.
Figure 5.
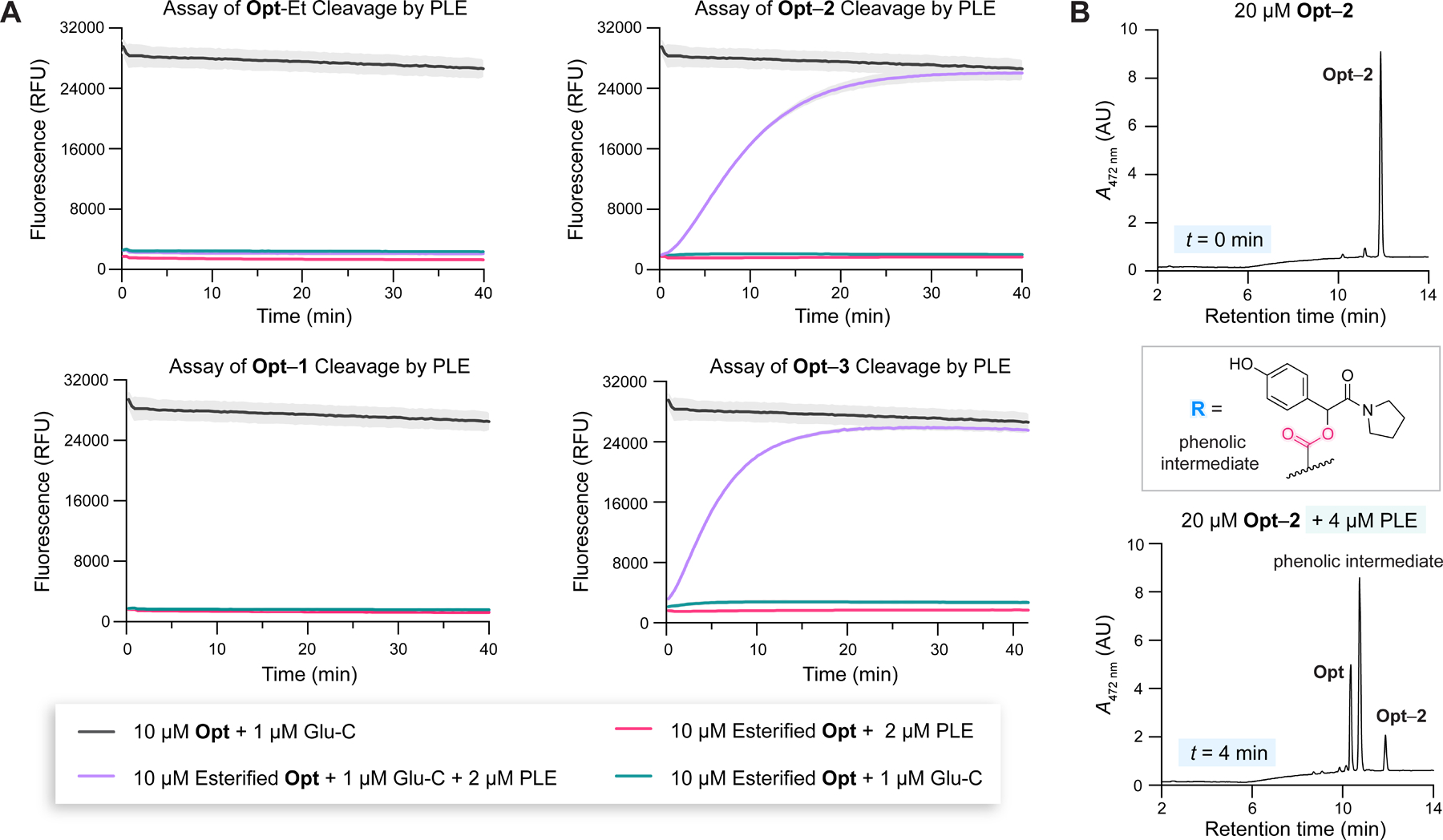
(A) Fluorescence progress curves related to assaying PLE cleavage of esterified peptides. Gray areas represent the SD; n = 1 independent replicate, n = 3 technical replicates. (B) LC-MS absorbance traces of Opt–2 and Opt–2 incubated with PLE for 4 min. The R group refers to an ester formed directly with the Glu residue of Opt. Studies were performed in 10 mM HEPES–NaOH buffer, pH 7.4, with DMF (1.5% v/v, assay; 6% v/v, LC-MS) and Triton X-100 (0.8% w/v, assay; 0% w/v, LC-MS) at 37 °C. For LC-MS analysis of PLE incubation with all esterified peptides, see the Supporting Information.
LC-MS Validation of the Assay and Detection of a Phenolic Intermediate.
To confirm the results of the PLE assay and discern the formation of any intermediates, we incubated the esterified peptides with PLE for 4 min and analyzed the samples via LC-MS. As expected, Opt-Et (Figure S16) and Opt–1 (Figure S15) were intact, whereas Opt–2 (Figures 5B and S14B) and Opt–3 (Figure S13B) were mostly consumed. Intriguingly, Opt was not the major product of PLE-catalyzed hydrolysis of Opt–2 and Opt–3. Instead, a dominant peak corresponding to a phenolic intermediate (Figures 5B, S13B, and S14B) was apparent in the chromatogram that was consistent with incomplete quinone methide elimination.26,48 At longer time scales (Figure S13C,D), most of the phenolic intermediate converted into Opt. From these findings, we inferred that, at high PLE concentrations, the rate-limiting step of the overall reaction was quinone methide elimination, not esterase cleavage.
Next, we explored the products of the reaction of Opt–3 in the presence of both PLE and Glu-C (Figure S13E,F). We detected Dcp and the phenolic intermediate. Surprisingly, we also observed the slow formation of a peptide corresponding to Dcp without a C-terminal Phe residue, phenylalanine cleavage product (Pcp). We attributed this byproduct to proteolytic activity of PLE.4 Because this byproduct did not change the fluorescence readout and formed downstream of the Glu-C step, its formation is inconsequential.
LC-MS Evaluation of the Stability of Esterified Peptides in Aqueous Buffer at pH 7.4.
Peptides containing activated side-chain carboxyl groups can undergo spontaneous cyclization reactions and subsequent hydrolytic cleavage via aspartimide,62 glutarimide,63 or pyroglutamyl imide formation.64 The rates of such cyclization reactions are dependent on the amino acid sequence surrounding the ester, as can be gleaned from asparagine and glutamine deamidation rates.65,66 In some peptides, such cyclization reactions are fast on physiological time scales.62 Because cyclization reactions would be problematic for our analysis, we used LC-MS to discern the integrity of our esterified peptides after a 4-h incubation at pH 7.4 and 37 °C. Gratifyingly (Figure S17), Opt-Et and Opt–1 were resistant to hydrolysis, whereas Opt–2 and Opt–3 released only a small amount of Opt, which would be accounted for by measuring . We note that pH–rate profiles67–69 for ester hydrolysis can be used to gain further insights into the stability of our esterified peptides at more acidic or basic conditions.
Mathematical Model for Analysis of Fluorescence Data.
To derive quantitative kinetic information from our FRET assay and controls (, , and ), we developed a strategy to measure progress curves using controls associated with substrate cleavage by esterase (e.g., Figure 5A). We established a simple formula (see eqs S3–S5 for the derivation and Figure S18 as an example):
| (1) |
Equation 1 is valid under the following three conditions: (1) Glu-C-mediated cleavage of Opt is not rate-limiting (see Figure 4A), (2) the esterase does not cleave the substrate (see Figure 5A), and (3) all reaction species upstream of have the same background fluorescence. To validate the use of eq 1, we ensured that neither Glu-C nor PLE is inhibited by Opt–1, which is a structural mimetic (OH→CH3) of the phenolic intermediate, in the working range of the assay (Figures S19 and S20, respectively).
Michaelis–Menten Kinetic Model with an Intermediate.
The observed formation of an intermediate upon esterase cleavage of Opt–2 and Opt–3 (Figures 5B and S13B) raised questions about the speed, significance, and tunability of quinone methide elimination in physiological settings. Knowing its rate constant, , is critical for addressing these questions as well as properly describing the kinetics of esterase-catalyzed hydrolysis.
To find the relevant kinetic constants, we constructed a kinetic model (Figure 6 and eqs S6–S11) of key steps leading to the product in our system: (1) formation of an E∙S complex, (2) esterase catalysis to generate the intermediate (I), and (3) quinone methide elimination to generate Opt. The last step is assumed to follow first-order kinetics. Our model was inspired by a conceptually similar model, RIP, which also relied on the last assumption.70 Though useful for studying the self-immolation of spacers activated by stimuli such as light, the RIP model did not encompass enzymatic catalysis. We call our model “SIP” (substrate → intermediate → product), where “S” implies the presence of enzyme.
Figure 6.
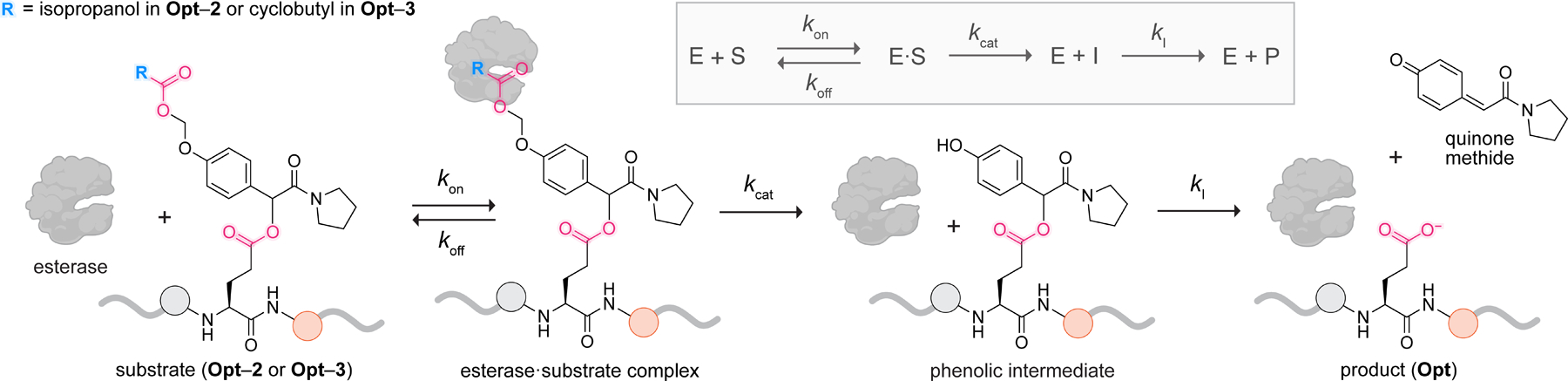
Substrate → intermediate → product (SIP) model that describes our system. This kinetic model is applicable when hydrolysis of a substrate by esterase (E) generates an intermediate that spontaneously converts into a product (Opt) via first-order kinetics (characterized by the rate constant ).
To conceive an approach to measure using our assay, we sought inspiration from a kinetic barrier diagram (Figure 7A). This diagram defines the barrier of a particular step as the presence of 2 µM of PLE, the rate-limiting step of product release was quinone methide elimination. Because all fluxes reciprocal of the flux, invoking a connection with ∆G‡.71,72 From the LC-MS data (Figures 5B), we inferred that, in the presence of 2 µM of PLE, the rate-limiting step of product release was quinone methide elimination. Because all fluxes before the rate-limiting step contribute to the overall observed flux, it was initially unclear how to isolate from other unknown parameters (e.g., , , and ). We realized, however, that a sufficiently large would decrease the barriers associated with the first two steps, enabling us to isolate the rate of the last one. In a regime with sufficiently large , our model would reduce to the first-order process: (Figure 7B and eqs S12–S14). Hence, the formation of would follow the equation:
| (2) |
where is the plateau of the curve at complete conversion, , and is chosen such that but . Equation 2 can then be fitted to experimental data to find the value of .
Figure 7.
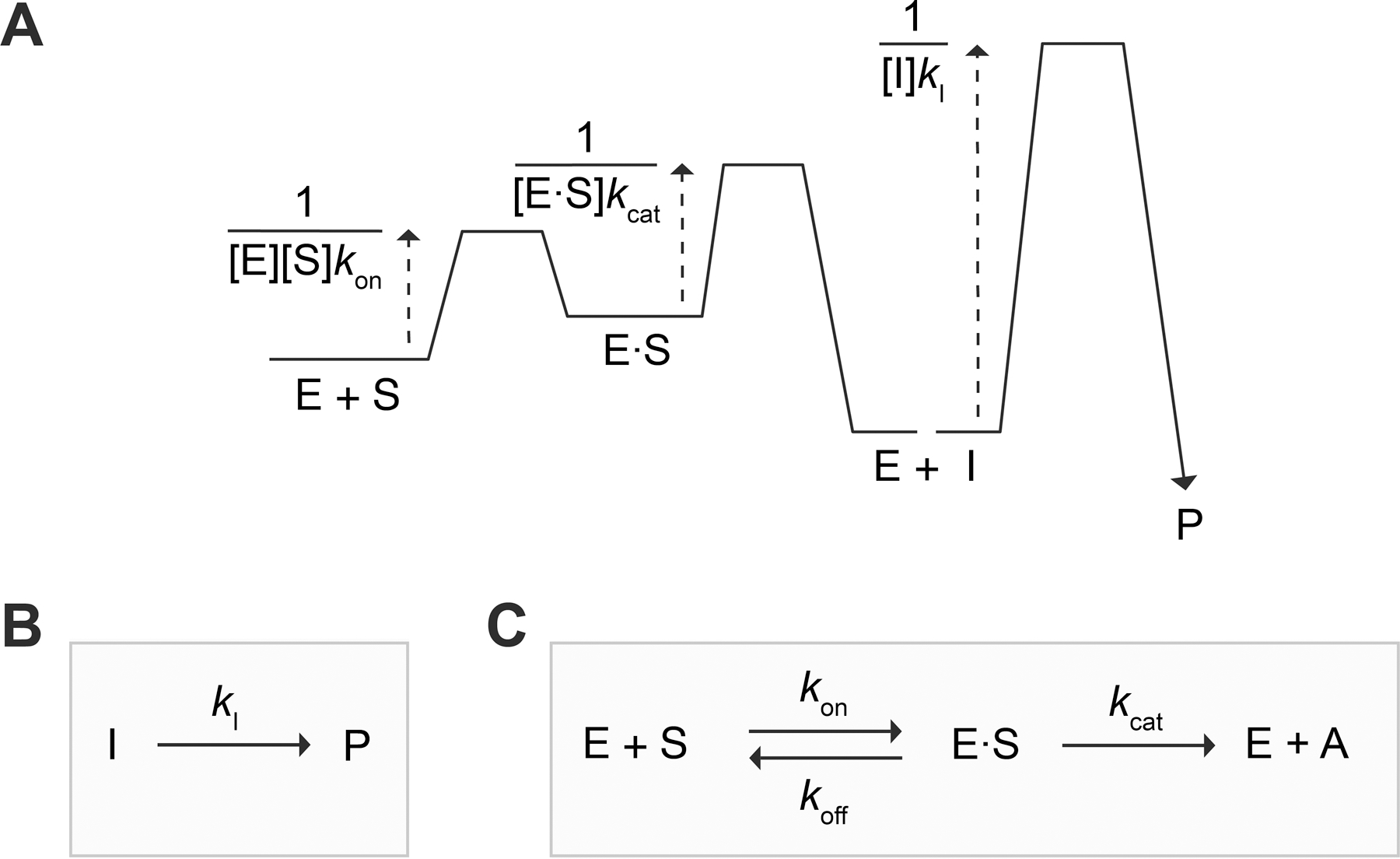
(A) Kinetic barrier diagram71,72 of the SIP model. (B) Large regime, which enables the determination of . (C) Small regime, which enables the determination of and once is known. In panel C, , as in eq 4.
Experimentally, a “sufficiently large” can be found by screening increasing esterase concentrations and identifying when the rate of product formation becomes independent of enzyme. In theory, accessing the first-order regime of the SIP model (Figure 7B) and applying eq 2 to measure demands that because, in contrast to the enzyme-catalyzed step, the spontaneous step cannot be accelerated by increasing . Further, in the particular scenario when , we can show that a “sufficiently large” needs to be
| (3) |
(For proof that satisfying these two conditions, and eq 3, is sufficient, see the discussion following eq S21 and S22 in the Supporting Information.) After and are determined, it should be verified that the experiment did indeed satisfy and eq 3 in the large regime.
Once is determined, it becomes possible to calculate (eq S18) and even (eq S20). Then, and of the esterase can be measured by transforming our model (Figure 7A) into the Michaelis–Menten model73 (Figure 7C). This transformation can be accomplished by defining a new quantity, (see the discussion following eqs S23–S27 in the Supporting Information):
| (4) |
The rate of change of is described by the familiar Michaelis–Menten equation:73
| (5) |
which is solvable for and when the steady-state approximation is valid, i.e., .74,75 In our particular setup, we will work in the initial-rate regime (when is linear with ) such that can be replaced by in eq 5.
Measuring for Opt–2 and Opt–3.
Before proceeding to measure , we first ensured that eq 2 describes our system. We used our assay to evaluate the conversion rates of Opt–2 and Opt–3 into products with PLE concentrations increasing from 2 to 16 µM (Figures S23 and S24). We expected that, if the simplified model in Figure 7B applies, these rates would eventually become indistinguishable (since Opt–2 and Opt–3 share a common intermediate) and stop depending on . Indeed, we found that doubling PLE from 2 to 4 µM increased the rate of Opt–2 conversion into product, whereas the rate associated with Opt–3 did not change appreciably. Thus, already at 2 µM PLE, the rate of esterase-catalyzed hydrolysis of Opt–3 was much faster than the rate of intermediate decay. At larger PLE concentrations, the rates of Opt–2 and Opt–3 conversion into product became even closer, but we noticed some fluorescence artifacts. Overall, 4 µM PLE appeared optimal for finding as this condition allowed for the largest time window for fitting eq 2.
Using the optimal PLE concentration and Opt–3, we were equipped to measure of quinone methide elimination. Upon converting the fluorescence data from four assay controls (Figure S25A) into , we fitted to eq 2 (Figure 8). Our fitted was 0.00325 ± 0.00020 s−1. The corresponding half-life of the phenolic intermediate was 3.55 ± 0.22 min, calculated with eq S15. Then, we plotted (eq S18), (eq S20), and (eq S25) using the determined value of . As a self-consistency check, we ensured that, at the chosen , was close to 0 (Figure 8), and the value of obtained from the fit was close to the one plotted (Table S2). Our measured values of and (Figure 8) for Opt–2 and Opt–3 were comparable to values reported for scaffolds that release CO2 (from carbamate fragmentation) or alcohols via quinone methide elimination.70 Notably, the short-lived nature of this intermediate highlights the advantage of measuring rate constants with our continuous fluorescence assay rather than HPLC.
Figure 8.
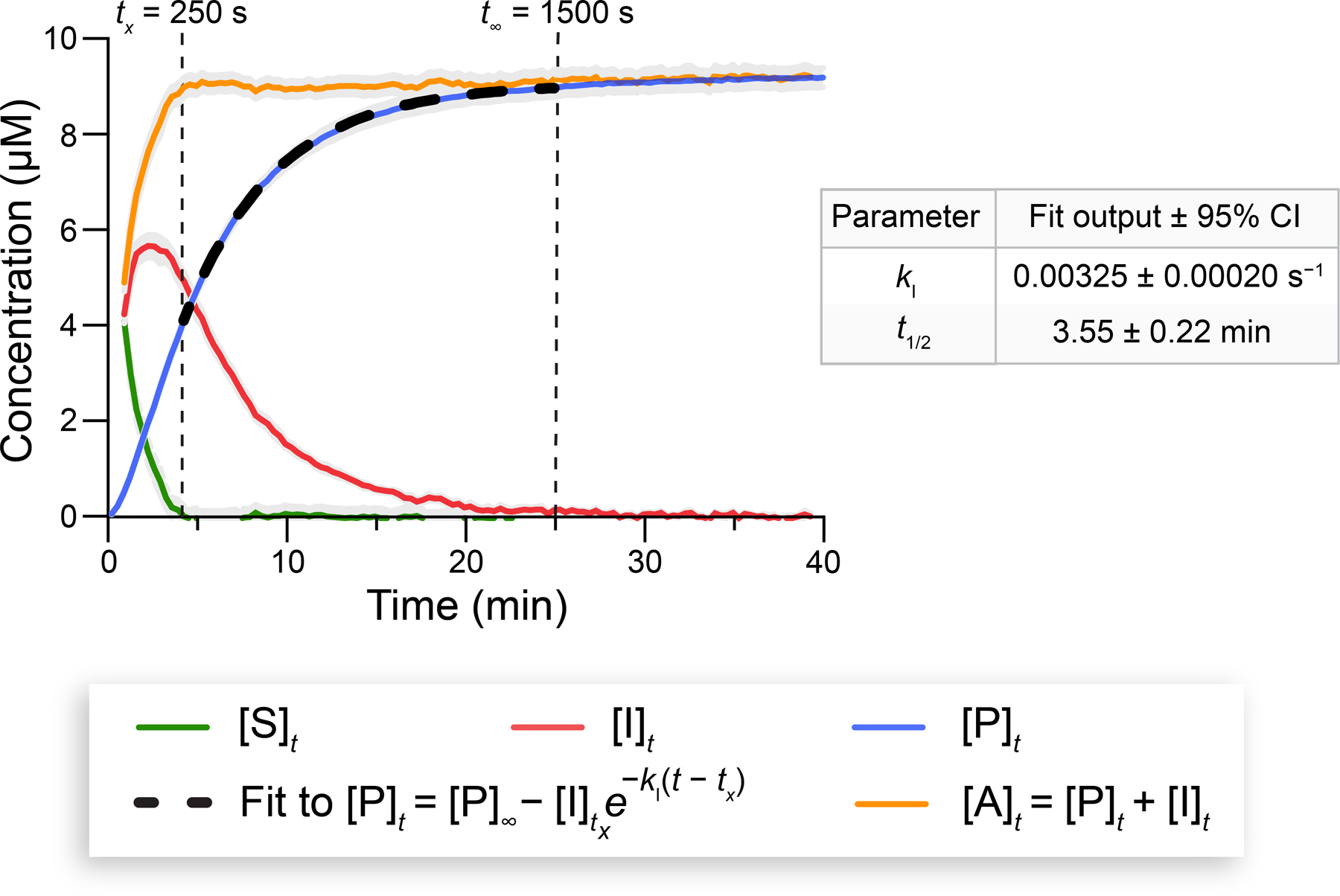
Progress curves of reaction species associated with Opt–3 (10 µM) cleavage by PLE at 37 °C. Fitting to eq 2 enabled the determination of the values of and (±95% CI) for Opt–2 and Opt–3. See Figure S25A for the corresponding fluorescence curves. The Glu-C assay was performed in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (1.5% v/v) and Triton X-100 (0.8% w/v). Gray areas represent the SD; n = 2 independent replicates, n = 3 technical replicates. and fitting parameters are listed in Table S2.
Measuring for PLE and Opt–2.
Once was known, we applied eqs 4 and 5 and the simplified model in Figure 7C to find the of PLE. As a proof of concept, we decided to characterize the cleavage kinetics of Opt–2 quantitatively because this substrate would provide an estimate of the lower limit for the of Opt–3. Qualitatively, it is possible to infer that of of Opt–2 from Figure S18 because (1) Opt–3 and Opt–2 transition through the same intermediate upon esterase cleavage, (2) experiments were performed under the same conditions, and (3) formation in Figure S18 is slightly faster for Opt–3 than for Opt–2. Note that, when intermediates are involved, such qualitative differences are best detected in datasets that utilize values < “sufficiently large” .
Following the traditional Michaelis–Menten model, we measured product formation associated with PLE-mediated cleavage of various concentrations of Opt–2 (0.8–10 µM) (Figure 9A). To apply eq 5 and satisfy the constraint that , we chose a PLE concentration of 25 nM. The initial lag phase in the resultant curves (Figure 9A) was expected because the substrate must first pass through the intermediate before getting converted into a detectable product. Using eq S25 and the measured (Figure 8), we converted into (Figure 9B). Gratifyingly, upon this transformation, the lag phase associated with disappeared, resulting in linear progress curves. Note that this change is mathematically nontrivial and sensitive to the input value of . In fact, as a self-check, we verified that significantly different values of result in the disappearance of the linear profile.
Figure 9.
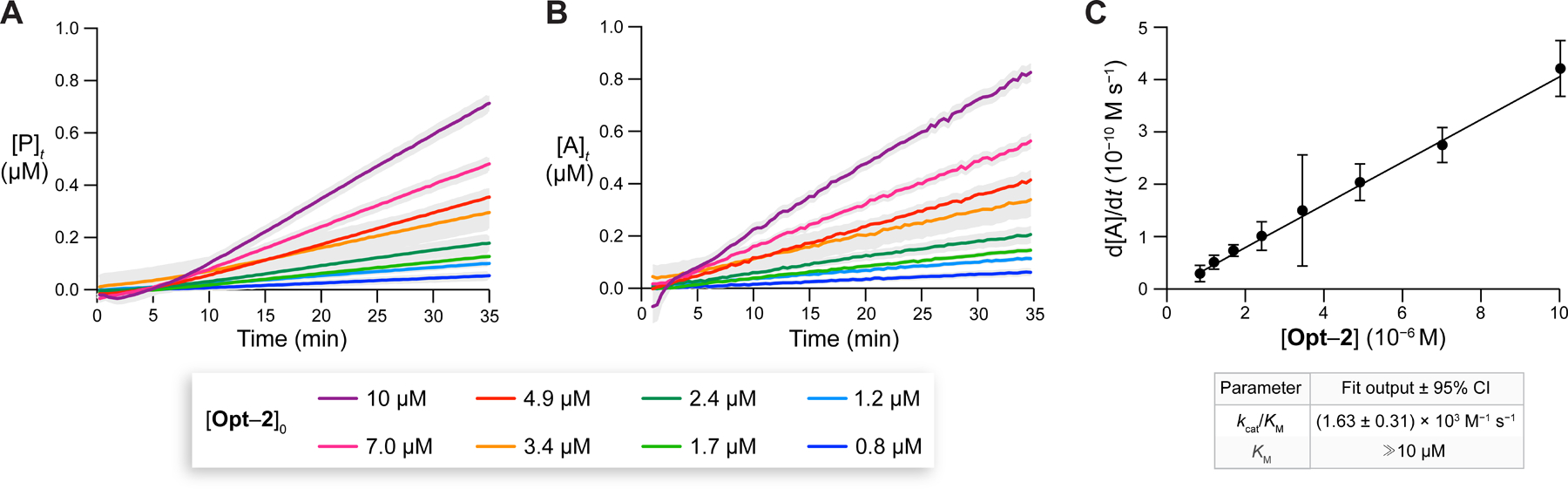
(A) Time-dependence of [P] from Opt–2 cleavage by PLE (25 nM) at 37 °C. (B) Corresponding time-dependence of . (C) Michaelis–Menten plot for the initial rates of formation as a function of [Opt–2]0. A linear fit to eq 5 enabled the determination of the values of (±95% CI) and a lower limit for . Assays were performed in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (1.5% v/v) and Triton X-100 (0.8% w/v). Gray areas represent the SD; n = 2 independent replicates, n = 3 technical replicates.
Finally, we computed the rate of change of for each progress curve and plotted the results as a function of , producing a traditional Michaelis–Menten plot (Figure 9C). Because this plot is linear, we inferred that the of Opt–2 was . The value of of Opt–2, (1.63 ± 0.31) × 103 M−1 s−1, was obtained from the slope of the line using eq S27. Note that, because the working range of our assay is within 240 nM–10 µM of , it is challenging to measure and separately for esterase substrates with using traditional methods. For example, fitting eq 5 to obtain and separately requires reaching ,76 and fitting the Lambert function requires on the order of .77 The more complex Bayesian inference approach can accommodate virtually any combination of and ,78,60 and this approach could be used in future work to measure both and of a substrate with our assay.
Confirming the Validity of Equation 3 and in the Large Regime.
Once we experimentally evaluated (Figure 8, ~0.00325 s−1) of Opt–3 and Opt–2 as well as (Figure 9C, ~1.63 × 103 M−1 s−1) of PLE and Opt–2, we were able to go back and verify that our (4 µM) for determination was indeed “sufficiently large” by checking the validity of the relationship in eq 3 for Opt–2. Because, for Opt–2, for even the largest tested [Opt–2]0 (since ), it was easily verified that
| (6) |
Further, because “sufficiently large” (4 µM) in our experiments, can also be inferred from eq 6. Note that whereas was determined by using the curve of Opt–3, this curve was close to that of Opt–2 at 4 µM (Figure S24).
Importance of a Self-Immolative Linker.
To gain insights into the fate of our esterified peptides in more translationally relevant settings, we assayed their cleavage to Opt by CES1, CES2, and the human intestine S9 fraction. The results of the screening are summarized in Table 1. We found that the selectivity of CES1 mimicked that of its homologue, PLE (Figure 5A). Opt-Et and Opt–1 were not cleaved under any of the tested conditions, whereas both Opt–2 and Opt–3 were efficiently cleaved by CES1 and CES2. Our assay also reported on the traceless cleavage of Opt–3 in the cytosol and microsomes (S9 fraction) of the intestine, validating the utility of our approach in physiological environments.
Table 1.
Summary of Esters as Substrates for Esterases.a
| Esterase | Opt-Et | Opt–1 | Opt–2 | Opt–3 |
|---|---|---|---|---|
| PLE | ❌ | ❌ | ✔ | ✔ |
| CES1 | ❌ | ❌ | ✔ | ✔ |
| CES2 | ❌ | ❌ | ✔ | ✔ |
| Intestine S9 fraction | ❌ | ❌ | ❌ | ✔ |
For fluorescence data, see Figures 5A and S26–S28.
We hypothesize that the esters in Opt-Et and Opt–1 are not cleaved because these peptides are unable to bind to the active site of esterases, possibly due to sterics associated with the esterified Glu residue in the middle of the peptide sequence (Figure 2). In support of our hypothesis, 5 µM Opt–1 does not inhibit PLE (4 µM or 25 nM) from cleaving 5 µM Opt–3 (Figure S20). Altogether, our results suggest that self-immolative linkers connecting an exposed esterase-cleavable handle to a more hindered ester near the backbone are crucial for the design of esterified peptides with enhanced release properties.
Conclusions
We have advanced the development of peptide prodrugs in two ways. First, we reported a continuous fluorescence assay for esterase activity using an esterified peptide as a substrate. Our substrate is unique in the sense that, relative to small-molecule and nanomaterial-based esterase probes, it better recapitulates the interactions of esterases with esterified peptides. Second, we put forward the SIP kinetic model, which incorporates a first-order intermediate decay step into the traditional Michaelis–Menten model. By exploring a high and a low esterase regime of SIP, we were able to calculate, for the first time, the for the esterase-catalyzed cleavage of an esterified peptide.
Our work opens the door for the rational design of esterified peptides with desirable attributes. One future direction would be to potentiate the traceless delivery of therapeutic peptides (perhaps cyclic79–82 or protein domain-mimetic83,84) into the cytosol of live cells by esterifying them with diazo compound 3, as Opt–3 is efficiently cleaved by human CES1, CES2, and the S9 intestinal fraction (Table 1). Another direction would be to tune the rate of quinone methide elimination or another bioreversible (triggerable or spontaneous) self-immolative mechanism that could tracelessly release a peptidic carboxyl group.85 As long as and eq 3 hold, our SIP kinetic model would be applicable to characterizing the decay of any intermediate by a first-order process. Our assay is also applicable to the study of other enzymes with esterase activity, including lipases, cholinesterase, paraoxonase, and even cytochrome P450.2 Although we did not identify a mammalian esterase that can catalyze the hydrolysis of ester moieties in Opt-Et and Opt–1, such enzymes could exist in cells or tissues. Tuning the alcohol portion of the esters to target specific esterases (e.g., intracellular human CES2 versus extracellular mouse CES1b) might be possible.12,13,46 Our assay could also be combined with the Bayesian inference approach60,78 to enable the measurement of both and .
Together, our esterase assay, kinetic framework, and modular substrate create a platform for studying the release properties of esterified peptides, providing new insights for their rational design.
EXPERIMENTAL PROCEDURES
Proteins and Intestinal S9 Fraction.
Lyophilized bacteria-derived V8 protease Glu-C (product number: IBCTV8LY, 1 mg) was from Innovative Research (Novi, Michigan). Lyophilized PLE (product number: 46058-10MG-F, ≥ 50 U/mg) with lot number BCCJ5320 (lot activity result: 62.6 U/mg) was from Sigma–Aldrich (St. Louis, MO). Lyophilized human (HEK293, C-His) recombinant CES1 (product number: 30105-H08H) with specific activity >10,000 pmol/min/μg was from Sino Biological (Wayne, PA). Lyophilized human (HEK293, C-His) recombinant CES2 (product number: HY-P76192) with specific activity >20000 pmols/min/μg was from MedChemExpress (South Brunswick Township, NJ). Suspension of human intestine S9 fraction (4 mg protein/mL), phenylmethylsulfonyl fluoride-free, mixed gender, pool of 4, was from XenoTech (Kansas City, KS).
Supplies.
Low protein-binding microcentrifuge tubes (product number: 90410) from Thermo Scientific (Waltham, MA) or polymerase chain reaction (PCR) snapsrtip II tubes from GeneMate were used in all manipulations with purified peptides. 96-well, half area, black, flat-bottom polystyrene nonbinding surface microplates (product number: 3993) from Corning (Corning, NY) were used in all fluorescence assays. Advantage polytetrafluoroethylene (PTFE) microspin centrifuge filters with polypropylene housing, pore size 0.2 µm, were from Analytical Sales and Services (Flanders, New Jersey). See the Supporting Information for a more extensive list of supplies.
Computational Modeling of Glu-C in Complex with a Peptide Substrate.
Molecular docking studies were performed with Autodock Vina.86 The model Glu-C substrate (Ac-Phe-Glu-Phe-NH2) was optimized using density functional theory at the M06–2X/6–31+g(d,p) level of theory87,88 with Gaussian 16.89 The resultant atomic coordinates are listed in the Supporting Information. The optimized structure was then subjected to molecular docking into the active site of Glu-C (PDB ID 1qy6),37 which was kept rigid.
Chemical Synthesis of Diazo Compounds and Peptides.
For experimental details on the synthesis and characterization of diazo compounds and peptides, see the Supporting Information.
Preparation of Stock Solutions of Glu-C, PLE, and Peptides.
A stock solution (80 µM based on a molecular weight90 of 27 kDa) of Glu-C was prepared on ice by dissolving 1.0 mg of the enzyme in 463 µL of water. Stock solutions of PLE (typically in assay buffer, see the Supporting Information for more details) were prepared on ice by measuring their absorbance on the DS-11 UV–vis spectrophotometer from DeNovix (Wilmington, DE) and applying the Beer–Lambert law. The extinction coefficient of PLE (ε280 nm = 87,000 M−1 cm−1) was calculated from its amino acid sequence with the ProtParam program from Expasy (Swiss Institute of Bioinformatics).91 Stock solutions of all peptides were prepared in DMF. The concentration of stock solutions of Std (εEdans = 5438 M−1 cm−1 at 336 nm),39 Opt (εDabcyl = 15,100 M−1 cm−1 at 472 nm),39 and esterified peptides (εDabcyl) were determined in 10 mM MES–HCl buffer, pH 6.0, containing Triton X-100 (0.8% w/v) and DMF (3% v/v) by measuring the absorbance of solutions with the DS-11 UV–vis spectrophotometer and applying the Beer–Lambert law. See the Supporting Information for more details.
Plate-Reader Setup for Fluorescence Measurements.
Fluorescence in relative fluorescence units (RFU) was measured with the Spark multimode microplate reader from Tecan (Männedorf, Switzerland) at 37 °C. Plates were shaken for 15 s before the first measurement. Readings were made from the top of the plate at (bandwidth, 20 nm) and (bandwidth, 20 nm) for Edans,39 typically every 20 s for continuous measurements. The sample volume in each well was 100 μL. The gain was set to 63, the z-position to 18547 μm, the settle time to 50 ms, the lag time to 0 μs, the integration time to 40 μs, the number of flashes to 30, and the mirror to “automatic” (50% mirror). See the Supporting Information for more details.
Screen of Glu-C Concentrations for Complete Cleavage of Opt.
2× solutions (20 µM and 2 µM) of Opt were prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (3% v/v) and Triton X-100 (0.8% w/v). 2× solutions of Glu-C (2 µM, 0.4 µM, and 0.2 µM) were prepared in the same buffer without DMF. To wells of a nonbinding 96-well plate were added aliquots (50 µL) of the 2× Opt solution. All solutions were pre-equilibrated to 37 °C for 10 min before measurements. After the indicated time, aliquots (50 µL) of the 2× Glu-C solutions were added to wells containing Opt, and fluorescence was recorded as described in the “Plate Reader Setup for Fluorescence Measurements” section above. See “Statistical Analysis: Computing Errors” for error propagation methods.
Q-TOF LC-MS Setup for Analyzing Peptide Transformations.
Due to the tendency of esterified peptides to stick to Eppendorf tubes in the absence of Triton X-100, all manipulations for LC-MS analysis involving dilutions of the peptide stock solutions in DMF were performed at 37 °C (all reagents were also pre-equilibrated to 37 °C) as this approach minimized analyte loss. Intact masses of peptides were analyzed with a Q-TOF 6530C mass spectrometer in electrospray ionization positive mode, equipped with a Poroshell 120 EC-C18 column from Agilent Technologies (Santa Clara, CA). Before analysis, samples were diluted (see the Supporting Information for details) and passed through PTFE filters. Dabcyl and Edans absorbance39 was monitored at 472 and 336 nm, respectively, with a 1290 Infinity II diode array detector FS (G7117A) from Agilent Technologies. The reference wavelength was set to 0. The following gradient with solvent C (100:0.1 water/formic acid) and solvent D (100:0.1 acetonitrile/formic acid) was applied for elution: 20% v/v D in C for 0–2 min, 20–80% v/v D in C for 2–17 min, and 95% v/v D in C for 17–19 min. See the Supporting Information for more details.
LC-MS Validation of Opt Cleavage by Glu-C at Glu↓Phe.
All manipulations were performed at 37 °C with prewarmed reagents. A 64 µM solution of Opt was prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (5% v/v). To 20 µL of the 64 µM Opt solution was added 16 µL of MeCN followed by 44 µL of solvent C (final MeCN concentration: 20% v/v). The diluted solution was then filtered and analyzed by Q-TOF LC-MS, as described previously. To 30 µL of the 64 µM Opt solution was added a aliquot (2 µL) of Glu-C (30 µM) in 10 mM HEPES–NaOH buffer, pH 7.4, and the resultant reaction mixture was incubated for 5 min on a shaker. The reaction mixture was then diluted, filtered, and analyzed by Q-TOF LC-MS, as described previously. See the Supporting Information for more details.
LC-MS Assessment of the Stability of Esterified Peptides in the Presence of Glu-C.
All manipulations were performed at 37 °C with prewarmed reagents. Solutions of Opt-Et (87 µM), Opt–1 (72 µM), Opt–2 (97 µM), and Opt–3 (58 µM) were prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (5% v/v). To 20 µL of these solutions was added 16 µL of MeCN followed by 44 µL of solvent C (final MeCN concentration: 20% v/v). The diluted solutions were then filtered and analyzed by Q-TOF LC-MS as described previously. Next, to 30 µL of the solutions of esterified peptides were added aliquots (2 µL) of Glu-C (30 µM) in 10 mM HEPES–NaOH buffer, pH 7.4, and the resultant reaction mixtures were incubated for 5 min on a shaker. The reaction mixtures were then diluted with 25.6 µL of MeCN and 70.4 µL of solvent C (final MeCN concentration of 20% v/v), filtered, and analyzed by Q-TOF LC-MS as described previously. See the Supporting Information for more details.
Inner Filter Effect, LOD, and Fold Increase in the Fluorescence Intensity of Opt upon Cleavage by Glu-C.
For respective experimental procedures and discussion, see the Supporting Information.
Assaying Esterified Peptides for Cleavage by PLE.
2× 20 µM solutions of Opt, Opt-Et, Opt–1, Opt–2, and Opt–3 were prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (3% v/v) and Triton X-100 (0.8% w/v). 4× solutions of PLE (8 µM) and Glu-C (4 µM) were prepared in the same buffer without DMF. To wells of a nonbinding 96-well plate were added aliquots (50 µL) of the 20 µM peptide solutions. Then, the following strips of PCR tubes were prepared: (1) strips containing 45 µL of 4× Glu-C solution, (2) strips containing 35 µL of 4× PLE solution, (3) strips containing 45 µL of reaction buffer without DMF, (4) strips containing 35 µL of 4× Glu-C solution, (5) strips containing 45 µL of reaction buffer without DMF, and (6) strips containing 35 µL of 4× PLE solution. All solutions were pre-equilibrated to 37 °C for 10 min. After the indicated time, using a multichannel pipet, 35 µL of PCR strip 1 was added to PCR strip 2, 35 µL of PCR strip 3 was added to PCR strip 4, and 35 µL of PCR strip 5 was added to PCR strip 6. Then, aliquots (50 µL) of resulting solutions of either PLE (4 µM) and Glu-C (2 µM), Glu-C alone (2 µM), or PLE alone (4 µM) were rapidly added to the 50-µL peptide solutions in the 96-well plate using a multichannel pipet. Fluorescence was recorded as described in the “Plate-Reader Setup for Fluorescence Measurements” section above. See the Supporting Information for more experimental details and assumptions for converting fluorescence data into curves using eq 1. Note that , , , and in eq 1 refer to blank (background buffer fluorescence) subtracted values. See “Statistical Analysis: Computing Errors” for error propagation methods.
LC-MS Detection of the Phenolic Intermediate upon Opt–2 and Opt–3 Incubation with PLE.
All manipulations were performed at 37 °C with prewarmed reagents. Solutions of Opt–2 (40 µM) and Opt–3 (40 µM) were prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (6% v/v). To aliquots (30 µL) of these solutions were added either an aliquot (30 µL) of PLE (8 µM) in 10 mM HEPES–NaOH buffer, pH 7.4, or an aliquot (30 µL) of buffer alone. The solutions were incubated for 0 and 4 min on a shaker. After the indicated times, 20 µL of the prepared solutions of esterified peptides were combined with 16 µL of MeCN and 44 µL of solvent C (final MeCN concentration: 20% v/v). The diluted solutions were then filtered and analyzed by Q-TOF LC-MS, as described previously. For LC-MS analysis of all esterified peptides in the presence of PLE, Glu-C, both PLE and Glu-C, or buffer alone (aqueous hydrolysis) at various time points, see the Supporting Information.
Ruling out Glu-C and PLE Inhibition by Opt–1, a Mimetic of the Phenolic Intermediate.
For experimental validation of these negative controls, see the Supporting Information.
Mathematical Primer on Finding and Esterase and .
For all equations describing the SIP model, additional derivations, formulas for computing d[P]/dt, , , and , and proof that and eq 3 are sufficient for accessing the first-order regime of the SIP model, see the Supporting Information.
Identifying a “Sufficiently Large” PLE Concentration for Evaluating .
2× solutions (20 µM) of Opt–2 and Opt–3 were prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing DMF (3% v/v) and Triton X-100 (0.8% w/v). 4× solutions of PLE (64 µM, 32 µM, 16 µM, and 8 µM) and Glu-C (4 µM) were prepared in 10 mM HEPES–NaOH buffer, pH 7.4, containing Triton X-100 (0.8% w/v). Aliquots (50 µL) of the 2× peptide solutions were added to the wells of a nonbinding 96-well plate. The following strips of PCR tubes were prepared: (1) strips containing 45 µL of 4× Glu-C solution, and (2) strips containing 35 µL of 4× PLE solutions. All solutions were pre-equilibrated to 37 °C for 10 min. After the indicated time and using a multichannel pipet, 35 µL of strip (1) was added to strip (2). Then, aliquots (50 µL) of the resultant solutions (32 µM, 16 µM, 8 µM, or 4 µM of PLE and 2 µM of Glu-C) were added to peptide solutions in the 96-well plate. Fluorescence was recorded as described in the “Plate-Reader Setup for Fluorescence Measurements” section above. See “Statistical Analysis: Computing Errors” for error propagation methods.
Evaluating of Opt–2 and Opt–3 Using the Optimal PLE Concentration.
The experiment described in the section “Assaying Esterified Peptides for Cleavage by PLE” was repeated with Opt and Opt–3 except the final [PLE] in the assay was changed to 4 µM. The final [Opt] and [Opt–3] in the assay was 10 µM and the final [Glu-C] was 1 µM. Blank (buffer alone) subtracted fluorescence data was converted into using eq 1. See “Statistical Analysis: Computing Errors” for details on error propagation methods and statistical analysis. Using the method described in the Supporting Information section “Finding as well as Intermediate and Substrate Evolution Curves”, data were fitted with Python’s curve_fit function (scipy library) to eq 2 with three free parameters (, , and ). For fitting, we selected a time window , where and and checked that the obtained value of was not strongly dependent on this choice. As a further check, we ensured that, at the chosen , was close to 0 (Figure 8), and the value of obtained from the fit was close to the one plotted (Table S2). For more details, see the Supporting Information. The raw datasets and Python code used for data plotting, fitting, and statistical analysis are freely available on GitHub (see “Python Code Availability Statement”).
Evaluating of PLE with Opt–2.
The method described in the Supporting Information section “Defining a New Term, “A”, and Finding Esterase and ” was applied to find of PLE. The experiment detailed in the section “Assaying Esterified Peptides for Cleavage by PLE” was repeated with Opt and Opt–2 except: (1) the final [PLE] in the assay was 25 nM and (2) instead of testing a single final 10 µM concentration of both Opt and Opt–2, a broader range of final concentrations was tested (10 × 0.70, 10 × 0.71, 10 × 0.72, 10 × 0.73, 10 × 0.74, 10 × 0.75, 10 × 0.76, and 10 × 0.77 µM). Blank (buffer alone) subtracted fluorescence data was converted into using eq 1. See “Statistical Analysis: Computing Errors” for details on error propagation methods and statistical analysis. Using the previously calculated value of , values were computed with eq S25. Only values <10% of were used for computing . The linearity of progress curves was sensitive to the value of (3× higher and 3× lower values resulted in linearity loss). Using the curve_fit function (scipy library) in Python, was calculated for each versus plot, assuming a linear fit. The resultant values were plotted as a function of . The curve_fit function was once again applied to calculate the slope of the graph in Figure 9C with a linear fit to obtain of Opt–2. For more details, see the Supporting Information. The raw datasets and Python code used for data plotting, fitting, and statistical analysis are freely available on GitHub (see “Python Code Availability Statement”).
Assaying Esterified Peptides for Cleavage by Human Carboxylesterases.
Experimental protocols for assaying esterase activity of CES1, CES2, and the human intestinal S9 fraction can be found in the Supporting Information. Note that, in the case of CES1 and CES2, the final Triton X-100 concentration in the assay buffer was lowered to 0% w/v and 0.7% w/v, respectively.
Statistical Analysis: Computing Errors.
Errors were propagated using standard formulas. See the Supporting Information for additional details and strategy for error propagation on derivates.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Erica L. Debley for assistance with compound synthesis and Drs. Bjarne Silkenath, Jinyi Yang, JoLynn B. Giancola, and Kenton J. Hetrick for advice. They also thank Drs. Mohanraja Kumar and Walt W. Massefski for help with HRMS and NMR analyses of compounds, respectively. Some figures in this manuscript were created with BioRender and PyMOL.
Funding
Y.D.P., E.Z., N.S.A., and E.C.W. were supported by NSF Graduate Research Fellowships. C.S.G. was supported by an NDSEG Fellowship sponsored by the Air Force Research Laboratory. K.R.W. was supported by the MIT Undergraduate Research Opportunities Program. This work used computational resources through allocation BIO230178 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program (NSF). This work was supported by Grants R35 GM148220 and R21 GM135780 (NIH) and the Merkin Institute for Transformative Technologies in Healthcare.
Abbreviations
- AU
absorbance units
- CES
carboxylesterase
- CI
confidence interval
- Dabcyl
4‑(dimethylaminoazo)benzene-4-carboxylic acid
- DMF
dimethylformamide
- Edans
(5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid)
- FRET
Förster resonance energy transfer
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- PCR
polymerase chain reaction, pig liver esterase
- PTFE
polytetrafluoroethylene
- Q-TOF
quadrupole time-of-flight
- SIP
substrate → product → intermediate
- LC-MS
liquid chromatography-mass spectrometry
- HRMS
high resolution mass spectrometry
- RFU
relative fluorescence units
- SD
standard deviation
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/xxxx.xxxxxxx.
General information including safety considerations; instrumentation; atomic coordinates of Ac-Phe-Glu-Phe-NH2 in complex with Glu-C; chemical synthesis and characterization of diazo compounds (Scheme S1–S4) and peptides (Scheme S5–S8, Figure S1–S6); preparation of stock solutions; screen of [Glu-C] for complete cleavage of Opt (Figure S7); validation of Opt cleavage by Glu-C at Glu↓Phe (Figure S8); stability of the esterified peptides in the presence of Glu-C (Figure S9); inner filter effect characterization and LOD (Table S1, Figure S10, eq S1); fold increase in fluorescence intensity upon Opt cleavage by Glu-C (eq S2, Figure S11); detailed experimental protocol for assaying esterified peptides for cleavage by PLE; mass spectrometry analysis of products formed within the PLE assay (Figures S12–S16); evaluation of the stability of esterified peptides to hydrolysis (Figure S17); assumptions and equations for converting fluorescence data into curves (eqs S3–S5, Figure S18); ruling out Glu-C (Figure S19) and PLE (Figure S20) inhibition by Opt–1; primer on finding of intermediate decay and of esterase (Figures S21 and S22, eqs S6–S27) including proof that relationships in eq S21 and eq S22 are sufficient for accessing the first-order kinetics regime of the SIP model; identifying a “sufficiently large” [PLE] for evaluating (Figures S23 and S24); evaluating using optimal [PLE] (Figure S25, Table S2); detailed experimental protocol for measuring of PLE with Opt–2; assaying CES2 (Figure S26), CES1 (Figure S27), and the intestinal S9 fraction (Figure S28); statistical and error analysis (eqs S28–S35); NMR spectra (PDF).
Python Code Availability Statement
Python codes for plotting, computing and , and obtaining error bounds are freely available on GitHub together with respective raw datasets from the Glu-C assay: https://github.com/yana-d-petri/Finding-kI-and-kcat-KM-using-fluorescence-data-from-Glu-C-assay.
The authors declare no competing financial interest.
DEDICATION
This paper is dedicated to the memory of Christopher T. Walsh, who was a lifelong mentor to R.T.R.
REFERENCES
- (1).Panda T; Gowrishankar BS Production and applications of esterases. Appl. Microbiol. Biotechnol 2005, 67, 160–169. [DOI] [PubMed] [Google Scholar]
- (2).Liederer BM; Borchardt RT Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci 2006, 95, 1177–1195. [DOI] [PubMed] [Google Scholar]
- (3).Lian J; Nelson R; Lehner R Carboxylesterases in lipid metabolism: From mouse to human. Protein Cell 2018, 9, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wang D; Zou L; Jin Q; Hou J; Ge G; Yang L Human carboxylesterases: A comprehensive review. Acta Pharm. Sin. B 2018, 8, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xu G; Zhang W; Ma MK; McLeod HL Human carboxylesterase 2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. Clin. Cancer Res 2002, 8, 2605–2611. [PubMed] [Google Scholar]
- (6).Singh A; Gao M; Beck MW Human carboxylesterases and fluorescent probes to image their activity in live cells. RSC Med. Chem 2021, 12, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rautio J; Kumpulainen H; Heimbach T; Oliyai R; Oh D; Järvinen T; Savolainen J Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov 2008, 7, 255–270. [DOI] [PubMed] [Google Scholar]
- (8).Hetrick KJ; Aguilar Ramos MA; Raines RT Terbium(III) luminescence-based assay for esterase activity. Anal. Chem 2019, 91, 8615–8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dai J; Hou Y; Wu J; Shen B A minireview of recent reported carboxylesterase fluorescent probes: Design and biological applications. ChemistrySelect 2020, 5, 11185–11196. [Google Scholar]
- (10).Lan L; Ren X; Yang J; Liu D; Zhang C Detection techniques of carboxylesterase activity: An update review. Bioorg. Chem 2020, 94, 103388. [DOI] [PubMed] [Google Scholar]
- (11).Gil-Rivas A; de Pascual-Teresa B; Ortín I; Ramos A New advances in the exploration of esterases with PET and fluorescent probes. Molecules 2023, 28, 6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hetrick KJ; Aguilar Ramos MA; Raines RT Endogenous enzymes enable antimicrobial activity. ACS Chem. Biol 2021, 16, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hetrick KJ; Raines RT Assessing and utilizing esterase specificity in antimicrobial prodrug development. Methods Enzymol 2022, 664, 199–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pani S; Qiu T; Kentala K; Azizi S-A; Dickinson BC Bioorthogonal masked acylating agents for proximity-dependent RNA labelling. Nat. Chem 2024, 16, 717–726. [DOI] [PubMed] [Google Scholar]
- (15).Liu P; Grenier V; Hong W; Muller VR; Miller EW Fluorogenic targeting of voltage-sensitive dyes to neurons. J. Am. Chem. Soc 2017, 139, 17334–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Deshaies RJ Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. [DOI] [PubMed] [Google Scholar]
- (17).Pauletti GM; Gangwar S; Okumu FW; Siahaan TJ; Stella VJ; Borchardt RT Esterase-sensitive cyclic prodrugs of peptides: Evaluation of an acyloxyalkoxy promoiety in a model hexapeptide. Pharm. Res 1996, 13, 1615–1623. [DOI] [PubMed] [Google Scholar]
- (18).Wang B; Gangwar S; Pauletti GM; Siahaan TJ; Borchardt RT Synthesis of a novel esterase-sensitive cyclic prodrug system for peptides that utilizes a “trimethyl lock”-facilitated lactonization reaction. J. Org. Chem 1997, 62, 1363–1367. [Google Scholar]
- (19).Herfindal L; Kasprzykowski F; Schwede F; L̷ankiewicz L; Fladmark KE; L̷ukomska J; Wahlsten M; Sivonen K; Grzonka Z; Jastorff B; Do̷skeland SO Acyloxymethyl esterification of nodularin-R and microcystin-LA produces inactive protoxins that become reactivated and produce apoptosis inside intact cells. J. Med. Chem 2009, 52, 5758–5762. [DOI] [PubMed] [Google Scholar]
- (20).Arbour CA; Mendoza LG; Stockdill JL Recent advances in the synthesis of C-terminally modified peptides. Org. Biomol. Chem 2020, 18, 7253–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hosono Y; Uchida S; Shinkai M; Townsend CE; Kelly CN; Naylor MR; Lee H-W; Kanamitsu K; Ishii M; Ueki R; Ueda T; Takeuchi K; Sugita M; Akiyama Y; Lokey SR; Morimoto J; Sando S Amide-to-ester substitution as a stable alternative to N-methylation for increasing membrane permeability in cyclic peptides. Nat. Commun 2023, 14, 1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hanssens J; van Dun S; Lokate THG; Reinartz VRAM; van den Bos LJ; Orrù RVA; Saya JM C-Terminal peptide modification: Merging the Passerini reaction with chemo-enzymatic synthesis. chemRxiv 2024, doi: 10.26434/chemrxiv-2024-pt1ln. [DOI] [Google Scholar]
- (23).Mix KA; Raines RT Optimized diazo scaffold for protein esterification. Org. Lett 2015, 17, 2358–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mix KA; Lomax JE; Raines RT Cytosolic delivery of proteins by bioreversible esterification. J. Am. Chem. Soc 2017, 139, 14396–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ressler VT; Mix KA; Raines RT Esterification delivers a functional enzyme into a human cell. ACS Chem. Biol 2019, 14, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jun JV; Petri YD; Erickson LW; Raines RT Modular diazo compound for the bioreversible late-stage modification of proteins. J. Am. Chem. Soc 2023, 145, 6615–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Maynard JRJ; Saidjalolov S; Velluz M-C; Vossio S; Aumeier C; Moreau D; Sakai N; Matile S Toward a traceless tag for the thiol-mediated uptake of proteins. ChemistryEurope 2023, 1, e202300029. [Google Scholar]
- (28).Giancola JB; Grimm JB; Jun JV; Petri YD; Lavis LD; Raines RT Evaluation of the cytosolic uptake of HaloTag using a pH-sensitive dye. ACS Chem. Biol 2024, 19, 908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Fang L; Xiao L; Jun YW; Onishi Y; Kool ET Reversible 2′-OH acylation enhances RNA stability. Nat. Chem 2023, 15, 1296–1305. [DOI] [PubMed] [Google Scholar]
- (30).Guo J; Chen S; Onishi Y; Shi Q; Song Y; Mei H; Chen L; Kool ET; Zhu R-Y RNA control via redox-responsive acylation. Angew. Chem., Int. Ed 2024, 63, e202402178. [DOI] [PubMed] [Google Scholar]
- (31).Leung FC-M; Tam AY-Y; Au VK-M; Li M-J; Yam VW-W Förster resonance energy transfer studies of luminescent gold nanoparticles functionalized with ruthenium(II) and rhenium(I) complexes: Modulation via esterase hydrolysis. ACS Appl. Mater. Interfaces 2014, 6, 6644–6653. [DOI] [PubMed] [Google Scholar]
- (32).Breger JC; Susumu K; Lasarte-Aragonés G; Díaz SA; Brask J; Medintz IL Quantum dot lipase biosensor utilizing a custom-synthesized peptidyl-ester substrate. ACS Sens 2020, 5, 1295–1304. [DOI] [PubMed] [Google Scholar]
- (33).Drapeau GR; Boily Y; Houmard J Purification and properties of an extracellular protease of Staphylococcus aureus. J. Biol. Chem 1972, 247, 6720–6726. [PubMed] [Google Scholar]
- (34).Giansanti P; Tsiatsiani L; Low TY; Heck AJR Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc 2016, 11, 993–1006. [DOI] [PubMed] [Google Scholar]
- (35).Rokita SE, Quinone Methides. John Wiley & Sons: Hoboken, NJ, 2009. [Google Scholar]
- (36).Breddam K; Meldal M Substrate preferences of glutamic-acid-specific endopeptidases assessed by synthetic peptide substrates based on intramolecular fluorescence quenching. Eur. J. Biochem 1992, 206, 103–107. [DOI] [PubMed] [Google Scholar]
- (37).Prasad L; Leduc Y; Hayakawa K; Delbaere LTJ The structure of a universally employed enzyme: V8 protease from Staphylococcus aureus. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 256–259. [DOI] [PubMed] [Google Scholar]
- (38).Fang B; Shen Y; Peng B; Bai H; Wang L; Zhang J; Hu W; Fu L; Zhang W; Li L; Huang W Small-molecule quenchers for Förster resonance energy transfer: Structure, mechanism, and applications. Angew. Chem., Int. Ed 2022, 61, e202207188. [DOI] [PubMed] [Google Scholar]
- (39).Matayoshi ED; Wang GT; Krafft GA; Erickson J Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science 1990, 247, 954–958. [DOI] [PubMed] [Google Scholar]
- (40).Testa B; Mayer JM, Hydrolysis in Drug and Prodrug Metabolism. Wiley–VCH: Weinheim, Germany, 2003. [Google Scholar]
- (41).Huttunen KM; Raunio H; Rautio J Prodrugs—from serendipity to rational design. Pharmacol. Rev 2011, 63, 750–771. [DOI] [PubMed] [Google Scholar]
- (42).Redasani VK; Bari SB, Prodrug Design: Perspectives, Approaches and Applications in Medicinal Chemistry. Academic Press: New York, NY, 2015. [Google Scholar]
- (43).Chen B-C; Skoumbourdis AP; Guo P; Bednarz MS; Kocy OR; Sundeen JE; Vite GD A facile method for the transformation of N-(tert-butoxycarbonyl) α-amino acids to N-unprotected α-amino methyl esters. J. Org. Chem 1999, 64, 9294–9296. [Google Scholar]
- (44).Li J; Sha Y A convenient synthesis of amino acid methyl esters. Molecules 2008, 13, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Takaishi T; Izumi M; Ota R; Inoue C; Kiyota H; Fukase K Product selectivity of esterification of L-aspartic acid and L-glutamic acid using chlorotrimethylsilane. Nat. Prod. Commun 2017, 12, 247–249. [PubMed] [Google Scholar]
- (46).Tian L; Yang Y; Wysocki LM; Arnold AC; Hu A; Ravichandran B; Sternson SM; Looger LL; Lavis LD Selective esterase–ester pair for targeting small molecules with cellular specificity. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 4756–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Fei N; Sauter B; Gillingham D The pKa of Brønsted acids controls their reactivity with diazo compounds. Chem. Commun 2016, 52, 7501–7504. [DOI] [PubMed] [Google Scholar]
- (48).Petri YD; Gutierrez CS; Raines RT Chemoselective caging of carboxyl groups for on-demand protein activation with small molecules. Angew. Chem., Int. Ed 2023, 62, e202215614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Guthrie JP Hydrolysis of esters of oxy acids: pKa values for strong acids; Brønsted relationship for attack of water at methyl; free energies of hydrolysis of esters of oxy acids; and a linear relationship between free energy of hydrolysis and pKa holding over a range of 20 pK units. Can. J. Chem 1978, 56, 2342–2354. [Google Scholar]
- (50).Asoodeh A; Ghanbari T Characterization of an extracellular thermophilic alkaline esterase produced by Bacillus subtilis DR8806. J. Mol. Catal. B Enzym 2013, 85–86, 49–55. [Google Scholar]
- (51).Ohgane K; Yoshioka H; Hashimoto Y Multiplexing fluorogenic esterase-based viability assay with luciferase assays. MethodsX 2019, 6, 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Allen SL; Allen JM; Licht BM Effects of Triton X-100 upon the activity of some electrophoretically separated acid phosphatases and esterases. J. Histochem. Cytochem 1965, 13, 434–440. [DOI] [PubMed] [Google Scholar]
- (53).Sakamoto T; Okuda H; Fujii S Studies on sterol-ester hydrolase in rat liver homogenates. J. Biochem 1974, 75, 1073–1079. [DOI] [PubMed] [Google Scholar]
- (54).Zhang C; Xu Y; Zhong Q; Li X; Gao P; Feng C; Chu Q; Chen Y; Liu D In vitro evaluation of the inhibitory potential of pharmaceutical excipients on human carboxylesterase 1A and 2. PLoS One 2014, 9, e93819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jun JV; Raines RT Two-step synthesis of α-aryl-α-diazoamides as modular bioreversible labels. Org. Lett 2021, 23, 3110–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Houmard J Kinetic investigation of the Staphylococcal protease-catalyzed hydrolysis of synthetic substrates. Eur. J. Biochem 1976, 68, 621–627. [DOI] [PubMed] [Google Scholar]
- (57).Terwilliger TC; Koshland DE Sites of methyl esterification and deamination on the aspartate receptor involved in chemotaxis. J. Biol. Chem 1984, 259, 7719–7725. [PubMed] [Google Scholar]
- (58).Liu Y; Kati W; Chen C-M; Tripathi R; Molla A; Kohlbrenner W Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal. Biochem 1999, 267, 331–335. [DOI] [PubMed] [Google Scholar]
- (59).Palmier MO; Van Doren SR Rapid determination of enzyme kinetics from fluorescence: Overcoming the inner filter effect. Anal. Biochem 2007, 371, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wralstad EC; Sayers J; Raines RT Bayesian inference elucidates the catalytic competency of the SARS-CoV-2 main protease 3CLpro. Anal. Chem 2023, 95, 14981–14989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhou Q; Yan B; Sun W; Chen Q; Xiao Q; Xiao Y; Wang X; Shi D Pig liver esterases hydrolyze endocannabinoids and promote inflammatory response. Front. Immunol 2021, 12, 670427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).McFadden PN; Clarke S Chemical conversion of aspartyl peptides to isoaspartyl peptides. A method for generating new methyl-accepting substrates for the erythrocyte D-aspartyl/L-isoaspartyl protein methyltransferase. J. Biol. Chem 1986, 261, 11503–11511. [PubMed] [Google Scholar]
- (63).Zhu J; Marchant RE Solid-phase synthesis of tailed cyclic RGD peptides using glutamic acid: Unexpected glutarimide formation. J. Pept. Sci 2008, 14, 690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Nalbone JM; Lahankar N; Buissereth L; Raj M Glutamic acid selective chemical cleavage of peptide bonds. Org. Lett 2016, 18, 1186–1189. [DOI] [PubMed] [Google Scholar]
- (65).Robinson NE; Robinson ZW; Robinson BR; Robinson AL; Robinson JA; Robinson ML; Robinson AB Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Sci 2004, 63, 426–436. [DOI] [PubMed] [Google Scholar]
- (66).Robinson NE; Robinson AB Prediction of primary structure deamidation rates of asparaginyl and glutaminyl peptides through steric and catalytic effects. J. Pept. Sci 2004, 63, 437–448. [DOI] [PubMed] [Google Scholar]
- (67).Fort JJ; Mitra AK Solubility and stability characteristics of a series of methotrexate dialkyl esters. Int. J. Pharm 1990, 59, 271–279. [Google Scholar]
- (68).Jensen E; Bundgaard H Peptide esters as water-soluble prodrugs for hydroxyl containing agents: Chemical stability and enzymatic hydrolysis of benzyl esters of glycine, diglycine and triglycine. Int. J. Pharm 1991, 71, 117–125. [Google Scholar]
- (69).Kahns AH; Buur A; Bundgaard H Prodrugs of peptides. 18. Synthesis and evaluation of various esters of desmopressin (dDAVP). Pharm. Res 1993, 10, 68–74. [DOI] [PubMed] [Google Scholar]
- (70).Alouane A; Labruère R; Le Saux T; Schmidt F; Jullien L Self-immolative spacers: Kinetic aspects, structure–property relationships, and applications. Angew. Chem., Int. Ed 2015, 54, 7492–7509. [DOI] [PubMed] [Google Scholar]
- (71).Burbaum JJ; Raines RT; Albery WJ; Knowles JR Evolutionary optimization of the catalytic effectiveness of an enzyme. Biochemistry 1989, 28, 9293–9305. [DOI] [PubMed] [Google Scholar]
- (72).Südi J How to draw kinetic barrier diagrams for enzyme-catalysed reactions. Biochem. J 1991, 276, 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Johnson KA; Goody RS The original Michaelis constant: Translation of the 1913 Michaelis–Menten paper. Biochemistry 2011, 50, 8264–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Segel LA; Slemrod M The quasi-steady-state assumption: A case study in perturbation. SIAM Rev 1989, 31, 446–477. [Google Scholar]
- (75).Schnell S; Mendoza C Closed form solution for time-dependent enzyme kinetics. J. Theor. Biol 1997, 187, 207–212. [Google Scholar]
- (76).Bisswanger H Enzyme assays. Perspect. Sci 2014, 1, 41–55. [Google Scholar]
- (77).Goličnik M On the Lambert W function and its utility in biochemical kinetics. Biochem. Eng. J 2012, 63, 116–123. [Google Scholar]
- (78).Hong H; Choi B; Kim JK, Beyond the Michaelis–Menten: Bayesian inference for enzyme kinetic analysis. In Computational Methods for Estimating the Kinetic Parameters of Biological Systems, Vanhaelen Q, Ed. Springer: New York, NY, 2022; pp 47–64. [DOI] [PubMed] [Google Scholar]
- (79).Dougherty PG; Sahni A; Pei D Understanding cell penetration of cyclic peptides. Chem. Rev 2019, 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Goto Y; Suga H The RaPID platform for the discovery of pseudo-natural macrocyclic peptides. Acc. Chem. Res 2021, 54, 3604–3617. [DOI] [PubMed] [Google Scholar]
- (81).Tyler TJ; Durek T; Craik DJ Native and engineered cyclic disulfide-rich peptides as drug leads. Molecules 2023, 28, 3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Ji X; Nielsen AL; Heinis C Cyclic peptides for drug development. Angew. Chem., Int. Ed 2024, 63, e202308251. [DOI] [PubMed] [Google Scholar]
- (83).Merritt HI; Sawyer N; Arora PS Protein domain mimics as modulators of protein−protein interactions. Pept. Sci 2020, 112, e24145. [Google Scholar]
- (84).Dongrui Z; Miyamoto M; Yokoo H; Demizu Y Innovative peptide architectures: Advancements in foldamers and stapled peptides for drug discovery. Expert Opin. Drug Discovery 2024, 19, 699–723. [DOI] [PubMed] [Google Scholar]
- (85).Petri YD; FitzGerald FG Chemoselective reagents for the traceless bioreversible modification of native proteins. Bioconjugate Chem 2024, 35, 1300–1308. [DOI] [PubMed] [Google Scholar]
- (86).Trott O; Olson AJ AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Zhao Y; Truhlar DG Density functionals with broad applicability in chemistry. Acc. Chem. Res 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- (88).Zhao Y; Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- (89).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, rev. C.01; Gaussian, Inc.:Wallingford, CT, 2016. [Google Scholar]
- (90).Drapeau GR The primary structure of staphylococcal protease. Can. J. Biochem 1978, 56, 534–544. [DOI] [PubMed] [Google Scholar]
- (91).Gasteiger E; Hoogland C; Gattiker A; Duvaud S; Wilkins MR; Appel RD; Bairoch A, Protein identification and analysis tools on the Expasy server. In The Proteomics Handbook, Walker J, Ed. Humana Press: Totowa, NJ, 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.