

Abstract
Past research suggests that resilience to health hazards increases with age, potentially because less resilient individuals die at earlier ages, leaving behind their more resilient peers. Using lifetime cigarette smoking as a model health hazard, we examined whether accelerated epigenetic aging (indicating differences in the speed of individuals’ underlying aging process) helps explain age-related resilience in a nationally representative sample of 3,783 older U.S. adults from the Health and Retirement Study. Results of mediation moderation analyses indicated that participants aged 86 or older showed a weaker association between lifetime cigarette smoking and mortality relative to participants aged 76–85 and a weaker association between smoking and multimorbidity relative to all younger cohorts. This moderation effect was mediated by a reduced association between smoking pack-years and epigenetic aging. This research helps identify subpopulations of particularly resilient individuals and identifies epigenetic aging as a potential mechanism explaining this process. Interventions in younger adults could utilize epigenetic aging estimates to identify the most vulnerable individuals and intervene before adverse health outcomes, such as chronic disease morbidity or mortality, manifest.
Keywords: Resilience, Mortality, Frailty, Epigenetic aging, Aging
Introduction
Cognitive and physical functioning tend to decline at later ages, leading to chronic disease morbidity, physical limitations and disability, and ultimately mortality (Crimmins 2020; Gladyshev et al. 2021; Khan et al. 2017; López-Otín et al. 2013). Research has found that as individuals age, they become less able to respond to stressors, physical insults, and other health risks (Angevaare et al. 2020; Cosco et al. 2019; Ukraintseva et al. 2021). When individuals cannot protect against or repair damage from these hazards, damage accumulates, leading to physiological dysfunction, morbidity, and mortality. According to the geroscience hypothesis, this damage accumulation represents the aging process, and the most effective way to avoid the frailty, morbidity, and mortality associated with chronological age is to slow the aging process itself (Kennedy et al. 2014; Levine and Crimmins 2018). Epigenetic aging measures, indices of deoxyribonucleic acid methylation (DNAm) sites associated with phenotypic aging, represent useful tools for measuring this underlying aging process (Binder and Horvath 2022; Horvath and Raj 2018).
The aging process is substantially heterogeneous in human populations. Some individuals live to old age in relatively good health despite experiencing stress, engaging in adverse behaviors, and being exposed to other health hazards. Thus, some individuals might be resilient to these hazards, whereas others are particularly vulnerable (Hadley et al. 2017; Pyrkov et al. 2021; Whitson et al. 2016). For example, cigarette smoking is a relatively well-understood health hazard associated with hundreds of thousands of excess deaths per year in the United States (Fenelon and Preston 2012; Lariscy et al. 2018), but individuals vary substantially in the actual health risks associated with smoking (Levine and Crimmins 2014). Understanding vulnerability and resilience in aging is a critical issue for understanding the aging process generally and for developing interventions to support longer, healthier lives.
What Is Resilience?
Resilience here refers to more positive responses to adversity or other health risks than might be expected (Cosco et al. 2019). Typically, research has focused on factors that could buffer or amplify the effect of a health risk (Janssen et al. 2022; Poole et al. 2017; Puterman and Epel 2012). Increasing evidence suggests that differences in resilience might explain why some people live longer, healthier lives. That is, as individuals age, those most vulnerable to health hazards experience the greatest accumulation of damage and greatest acceleration of aging (Carroll et al. 2017; Schutte et al. 2016). These more vulnerable, less resilient individuals tend to die earlier, leaving behind a population of relatively more resilient individuals (Behrman et al. 1990; Rogers 1992; Wrigley-Field 2020). Thus, the oldest-old represent individuals from their cohort who were the most resilient to various health hazards. Research has recognized this heterogeneity in resilience and, conversely, frailty (e.g., Crimmins et al. 2009; Vaupel et al. 1979; Vaupel and Yashin 1985). In the current study, we focus on epigenetic aging as a potential biological mechanism explaining age-associated resilience. That is, we build on this classical demographic research by identifying a biological pathway underlying this process. We focus on lifetime cigarette smoking as a model exposure.
Epigenetic Aging
DNAm is an epigenetic mechanism—a process that affects gene expression in response to environmental factors without altering the underlying genetic code—that has been widely studied in research on health and aging (Andrasfay and Crimmins 2023; Sugden et al. 2023). DNAm, is a processes whereby methyl groups are attached to phosphate groups between cytosine and guanine leading to lowered expression of a given gene (especially when in a gene’s promoter region) (Horvath and Raj 2018). DNAm has been shown to change in response to environmental, social, and behavioral exposures (e.g., socioeconomic status, adverse childhood events, diet) (Sugden et al. 2019, 2023; Waziry et al. 2023) and has been linked to health outcomes (e.g., coronary artery disease, depressive symptoms, mortality) (Klopack, Crimmins et al. 2022; McCrory et al. 2021; Si et al. 2023). Research in animal models has found that anti-aging interventions (e.g., calorie restriction, rapamycin) are associated with changes in age-related DNAm (Unnikrishnan et al. 2019). Indeed, DNAm is considered a hallmark of aging, a basic process underlying the aging process (López-Otín et al. 2013).
Many large, population-based studies have collected DNAm data using the EPIC platform from Illumina. This assay includes more than 850,000 sites from across the genome. Recently, several measures have been produced with these DNAm data to measure aging using machine learning techniques. These epigenetic aging measures are weighted indices of DNAm sites associated with age and health outcomes related to age (Horvath and Raj 2018; Raffington et al. 2021). So-called first-generation epigenetic clocks based on DNAm (e.g., HorvathAge and HannumAge) were designed to identify methylation sites associated with chronological age. These measures have now been shown to have only relatively weak associations with morbidity and mortality (Horvath and Raj 2018).
Alternatively, second-generation DNAm aging measures (e.g., GrimAge and PhenoAge) were trained on chronological age, biomarkers of aging, and aging-related health behaviors (Levine et al. 2018; Lu et al. 2019). More specifically, PhenoAge was produced in two steps: (1) a phenotypic age measure was created using a machine learning approach with 42 biomarkers (reduced to 9 and chronological age via penalized regression) predicting mortality in the National Health and Nutrition Examination Survey; and (2) DNAm PhenoAge (what we use here) was trained using the InCHIANTI data to predict this phenotypic age measure in a penalized regression (Levine et al. 2018). GrimAge was also developed using a two-step approach: (1) DNAm surrogates of 88 plasma proteins and smoking pack-years (indices of weighted DNAm sites that are predictive of each marker and pack-years) were developed using penalized regression in the Framingham Heart Study; and (2) 12 DNAm surrogates that were correlated above r = .35 with their measured value in testing data were used in an elastic net with age and gender predicting time to death (eight surrogates, age, and gender were selected by this model). Thus, GrimAge is an index combining weighted DNAm surrogates of these blood-based biomarkers and pack-years, along with actual age and gender (Lu et al. 2019). Third-generation DNAm aging measures designed to address the rate of aging (e.g., Dunedin Pace of Aging Calculated from the Epigenome [DunedinPACE]) were trained on changes in biomarkers associated with aging over time using the Dunedin Study (Belsky et al. 2020).
As with much of the past research on epigenetic aging, we focus on accelerated epigenetic aging: how much one’s epigenetic age differs from one’s chronological age. Later generation measures of accelerated DNAm aging are predictive of important aging-related health outcomes, including physical frailty, chronic disease morbidity, and mortality (Fiorito et al. 2017; Föhr et al. 2021; Horvath and Raj 2018; McCrory et al. 2021). These measures are also strongly associated with health risk exposures, such as smoking (Horvath and Raj 2018; Lei et al. 2020; Sugden et al. 2019), drinking (Jung et al. 2022; Kim et al. 2022; Zindler et al. 2022), and social stress (Beydoun et al. 2022; Klopack, Crimmins et al. 2022; Oblak et al. 2021; Simons et al. 2021).
These epigenetic aging measures were trained on different criterion variables, were developed in different populations, and there is little overlap in the DNAm sites that are identified by each measure (Belsky et al. 2018). Past studies utilizing these measures have typically analyzed only one of them (Föhr et al. 2021; Protsenko et al. 2021; Yang and Wu 2021) or analyzed them in separate analyses (Crimmins et al. 2021; Fiorito et al. 2017; Klopack, Crimmins et al. 2022; McCrory et al. 2021; Simons et al. 2022). Because these epigenetic aging measures were developed using different processes, some have argued that they tap into different parts of the same underlying aging process. For example, because GrimAge was designed to capture DNAm variation associated with pack-years, it might be more sensitive to smoking behavior and more predictive of lung disease than other DNAm aging measures (Klopack, Carroll et al. 2022). DunedinPACE measures the current pace of aging and therefore might be more sensitive to changes and recent exposures (Belsky et al. 2020). However, despite their differences, these summary measures of epigenetic methylation tend to covary and were designed to assess the same underlying construct. To the extent that these measures assess acceleration of the underlying aging process, combining information from multiple epigenetic aging measures (e.g., principal components analysis, confirmatory factor analysis) might provide a more accurate estimate of epigenetic aging acceleration. This combination represents an important contribution to the literature on epigenetic aging because it shows how these measures function as indicators of a single underlying aging process and how they differ in their predictive capability.
Lifetime Cigarette Smoking
Cigarette smoking is an ideal exposure to examine resilience in the aging process. Smoking is a widespread health behavior with well-understood health risks (Goodchild et al. 2018; Klopack, Carroll et al. 2022; Preston et al. 2014; U.S. Department of Health and Human Services 2014;) and is robustly associated with epigenetic aging (Beach et al. 2015; Gao et al. 2016; Lei et al. 2020; Yang et al. 2019). In addition, recent evidence suggests that the timing and number of cigarettes smoked across the life course affect epigenetic aging (Klopack, Carroll et al. 2022). Thus, research utilizing life course smoking measures, such as lifetime pack-years (the product of packs of cigarettes smoked per day and years smoking) is particularly vital. However, not all smokers are at equal risk. Emerging evidence suggests that smoking is less robustly associated with mortality and health-relevant biomarkers among older adults than younger adults, suggesting that these older adults might be more resilient members of their cohort who survived to later ages (Levine and Crimmins 2014).
Current Study
Our conceptual model is shown in Figure 1. As noted earlier, past research suggests that resilience reduces, prevents, or helps repair damage from health risk exposures. This damage might manifest as accelerated epigenetic aging. More resilient individuals experience less accelerated aging despite health risk exposure and therefore live longer, healthier lives and make up a larger proportion of older cohorts. To assess vulnerability and resilience in the aging process, we utilized nationally representative data from U.S. adults older than 55. We divided the sample by age into four groups (56–65, 66–75, 76–85, and 85 or older) and assessed the effects of interactions between smoking pack-years and age category on epigenetic aging multimorbidity and mortality. Following emerging research in geroscience, we argue that aging is a general process whereby molecular and cellular processes deteriorate with age, leading to larger systemic dysfunction, morbidity, and ultimately death. Research indicates that the epigenetic aging measures utilized in the current study are useful indicators of this general aging process. Because we expect aging to affect multiple systems in similar ways, we focus on multimorbidity across seven diseases. In the current study, we address two questions. First, does the effect of smoking pack-years on multimorbidity and mortality differ by age group? Second, does epigenetic aging help explain the differential effect of smoking pack-years by age (mediated moderation)?
Figure 1.
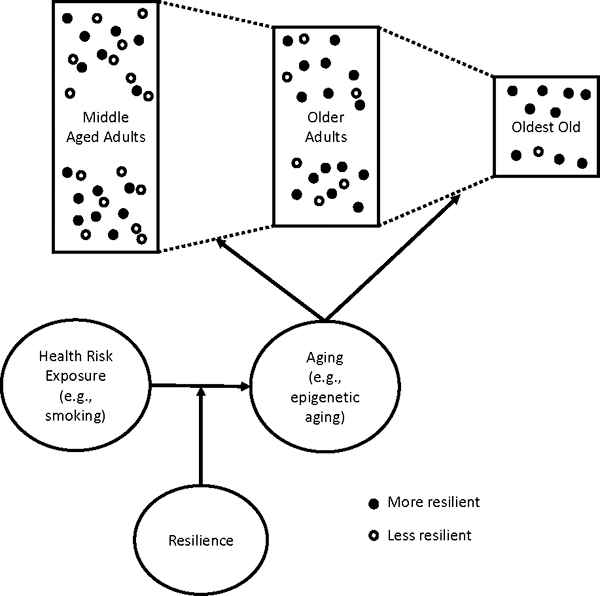
Conceptual model
Methods
Data
We utilized data from the Health and Retirement Study (HRS), a nationally representative sample of community-dwelling older adults in the United States. As part of data collection in 2016, a subsample of participants agreed to participate in the Venous Blood Study (VBS). Roughly 4,000 participants—who, when weighted, are a representative sample of those aged 56 years or older—had epigenome-wide analysis conducted using the Infinium MethylationEPIC BeadChip. More information about the VBS and epigenetic data analysis is available elsewhere (Crimmins et al. 2017; Crimmins et al. 2020).
Measures
Mortality
Mortality was assessed over four years after the venous blood collection as participant deaths known to the HRS as of 2020.
Multimorbidity
We constructed an index of seven chronic conditions by summing whether respondents reported in 2016 that a doctor had ever told them they had “high blood pressure or hypertension;” “diabetes or high blood sugar;” “cancer or a malignant tumor of any kind except skin cancer;” “chronic lung disease except asthma such as chronic bronchitis or emphysema;” “heart attack, coronary heart disease, angina, congestive heart failure, or other heart problems;” “stroke or transient ischemic attack (TIA);” or “arthritis or rheumatism.”
Our multimorbidity measure in this study has been used in past research. Other studies using this measure in the HRS have found that it is highly similar to other instrumentalizations of multimorbidity (Klopack2023). Research also indicates that the number of chronic diseases has predictive qualities similar to other multimorbidity measures (Huntley et al. 2012).
Epigenetic Aging
We utilized second- and third-generation epigenetic aging measures that have established associations with smoking, mortality risk, chronic disease morbidity, and lung functioning. Specifically, we used three epigenetic clocks: GrimAge, PhenoAge, and DunedinPACE. To create GrimAge, researchers created seven DNAm surrogates of proteins associated with mortality and a surrogate marker of smoking pack-years. They then used these surrogate markers, age, and gender to train a measure predictive of time to death (Lu et al. 2019). PhenoAge is a phenotypic age measure created using nine blood-based markers of immune and tissue function predictive of mortality. PhenoAge was then trained in epigenome-wide DNAm data to predict this phenotypic aging measure (Levine et al. 2018). Finally, DunedinPACE was trained using the rate of change in 19 biomarkers of healthy aging over two decades of life (Belsky et al. 2022).
Lifetime Pack-Years
HRS asks respondents to indicate the ages at which they started and quit smoking cigarettes and about how many cigarettes per day they smoked when they were smoking. We used the earliest age a respondent reported smoking and the age at which they quit (if they quit). We then multiplied the number of years smoking by the average of the number of cigarettes a respondent reported smoking per day at each wave and the maximum number smoked per day at the peak of their cigarette consumption divided by 20 (the typical number of cigarettes per pack). This product indicated an individual’s lifetime cigarette smoking pack-years. For more information about this measure, see Haghani et al. (2020).
Age
Participants were divided into four chronological age groups: 56–65 (the youngest participant was 56), 66–75, 76–85, and 86 or older. The first three groups represent decades of life, but all participants over age 85 were combined because few participants were older than 95.
Controls
All models control for race/ethnicity (Black, not Hispanic; Hispanic; other race, not Hispanic; and White, not Hispanic as the reference group), gender (female = 1), educational attainment (less than 12 years, 12 years, 13–15 years, and 16 or more years as the reference group), and body mass index (BMI; underweight, overweight, obese [obese 1], morbidly obese [obese 2], and normal weight as the reference group).
Plan of Analysis
To assess our arguments, we estimated three structural equation models (SEMs) regressing each of the two outcome variables on epigenetic aging, smoking pack-years, age, and smoking pack-years × age and regressing epigenetic aging on smoking pack-years, age, and smoking pack-years × age. SEM allows us to test epigenetic aging as a mediator of the moderating effect of age on the association between smoking pack-years and health outcomes. Because all three epigenetic aging measures theoretically capture the same underlying construct, we created a latent factor representing epigenetic aging using confirmatory factor analysis (CFA) with the second- and third-generation measures as indicators. CFA is particularly well-suited to our study. A large body of research suggests that different epigenetic aging measures capture part of an underlying but unobserved aging phenomenon (e.g., Belsky et al. 2018). CFA can estimate latent factors representing underlying constructs that are partially measured by multiple indicator variables, making it useful not only for data reduction but also for measuring constructs not directly measured by individual indicators.
We declared mortality as dichotomous using the “CATEGORICAL ARE” command in Mplus. Because mediated moderation effects are likely nonnormally distributed, we utilized 95% confidence intervals (CIs) using a bias-corrected bootstrap procedure for significance tests of moderated mediation.
Both GrimAge and PhenoAge were developed without a focus on the reliability of probes assessing DNAm at individual sites. Research suggests that poor reliability probes introduce noise into the data and therefore might affect the generalizability and replication of results. We therefore utilized so-called principal component (PC) versions of these measures, using PCs from individual site-level data to calculate aging measures. These PC measures have been shown to be more reliable longitudinally and to be more sensitive to the effects of interventions (Higgins-Chen et al. 2022). Because DunedinPACE was developed using only probes shown to be highly reliable, we used this measure without PC adjustment. To assess accelerated epigenetic aging, we then regressed chronological age on these measures. We saved the residuals from these regressions and used them as indicators of accelerated epigenetic aging.
We applied survey weights and strata provided in the HRS tracker file to make the sample population representative. Of the 3,875 participants who were eligible for the study, 23 were missing information on lifetime smoking and 69 were missing information on one or more control variables. Thus, 3,783 participants were included in the current study. We produced descriptive statistics using R and adjusted sample means using HRS sample weights. Significance tests are from bivariate regressions of each variable on age group. All models were fully recursive, so model fit statistics were uninformative. Data were prepared in R 4.1.3 “One Push-up” (R Core Team 2022) using the tidyverse (Wickham et al. 2019) and survey (Lumley 2004) packages. Analyses were conducted in Mplus 8.6 (Muthén and Muthén 2015).
Results
Descriptive Statistics
Descriptive statistics are shown in Table 1. Participants in the oldest category (86 or older) were more likely to die over the next four years relative to every other group. There were 105 deaths in the oldest group, 161 in the 76–85 group, 92 in the 66–75 group, and 63 in the 56–65 group. The oldest group had more chronic illnesses than the youngest two groups (ages 56–65 and 66–75) and greater smoking pack-years than the youngest group. Mean chronic morbidities were 2.9 for the oldest group, 2.8 for the 76–85 group, 2.5 for the 66–75 group, and 1.8 for the youngest group. The oldest group did not significantly differ from the other groups in terms of accelerated epigenetic aging because chronological age has been regressed out of these measures. We also include descriptive statistics for epigenetic aging variables without accelerated aging adjustment. As expected, PhenoAge and GrimAge increase with chronological age. Aging as measured by DunedinPACE is slower for the two youngest age groups than for the oldest group. We used the accelerated aging measures (i.e., with chronological age regressed out) in all models.
Table 1.
Study variable means for each age group
| Ages 56–65 | Ages 66–75 | Ages 76–85 | Ages 86+ | |
|---|---|---|---|---|
| (N = 1,453) | (N = 1,169) | (N = 920) | (N = 241) | |
|
| ||||
| Mortality Over Four Years (proportion) | .039*** | .067*** | .175*** | .496 |
| Multimorbidity (number of conditions) | 1.818*** | 2.480*** | 2.768 | 2.870 |
| DunedinPACE Acceleration (in years of epigenetic aging per chronological year) | −0.002 | 0.001 | 0.005 | −0.006 |
| PhenoAge Acceleration (in years) | −0.071 | 0.181 | −0.053 | −0.310 |
| GrimAge Acceleration (in years) | −0.040 | 0.057 | 0.075 | −0.198 |
| DunedinPACE (in years of epigenetic aging per chronological year) | 0.999*** | 1.024*** | 1.052 | 1.066 |
| PhenoAge (in years) | 58.515*** | 67.390*** | 76.164*** | 85.309 |
| GrimAge (in years) | 71.176*** | 78.424*** | 85.913*** | 93.437 |
| Lifetime Pack-Years | 10.284** | 14.808 | 15.721 | 14.820 |
| Gender (female = 1) | .510*** | .537** | .575** | .668 |
| Education: 0–11 Years | .339*** | .311*** | .230* | .205 |
| Education: 12 Years | .105 | .131 | .194 | .274 |
| Education: 13–15 Years | .269** | .304** | .358 | .352 |
| Education: 16+ Years | .287** | .254* | .218 | .169 |
| Race/Ethnicity: White, Not Hispanic | .744* | .786 | .847 | .829 |
| Race/Ethnicity: Black, Not Hispanic | .113 | .101 | .076 | .084 |
| Race/Ethnicity: Hispanic | .095 | .088 | .062 | .073 |
| Race/Ethnicity: Other, Not Hispanic | .049*** | .025 | .015 | .013 |
| BMI: Underweight | .010* | .015 | .025 | .048 |
| BMI: Normal | .218*** | .235*** | .321** | .449 |
| BMI: Overweight | .355 | .382 | .391 | .377 |
| BMI: Obese 1 | .244*** | .235*** | .191*** | .103 |
| BMI: Obese 2 | .174*** | .133*** | .072*** | .023 |
Note: Significance tests are from a bivariate regression of the variable on the age category, with ages 86+ as the reference group.
p < .05;
p < .01;
p < .001
We also divided participants into tertiles of accelerated aging for each clock (see Table S1, online appendix). Relative to the tertile with relatively less accelerated aging (tertile 1), participants in the most accelerated aging tertile had significantly greater mortality risk, had more morbidities, smoked more, were less likely to be female, were more likely to have less than 12 or 12 years of education, and were more likely to be morbidly obese (obese 2) for all three epigenetic aging measures.
SEM Results
SEM results are shown in Figures 2 and 3 for mortality and multimorbidity, respectively. Because the epigenetic aging measures and health outcomes are on different scales, these figures display standardized results to ease comparison across models. All three epigenetic aging measures loaded significantly on a single latent factor. All factor loadings were highly significant and greater than .6 in both models, suggesting that the latent variable characterizes the underlying latent construct of accelerated epigenetic aging well.
Figure 2.
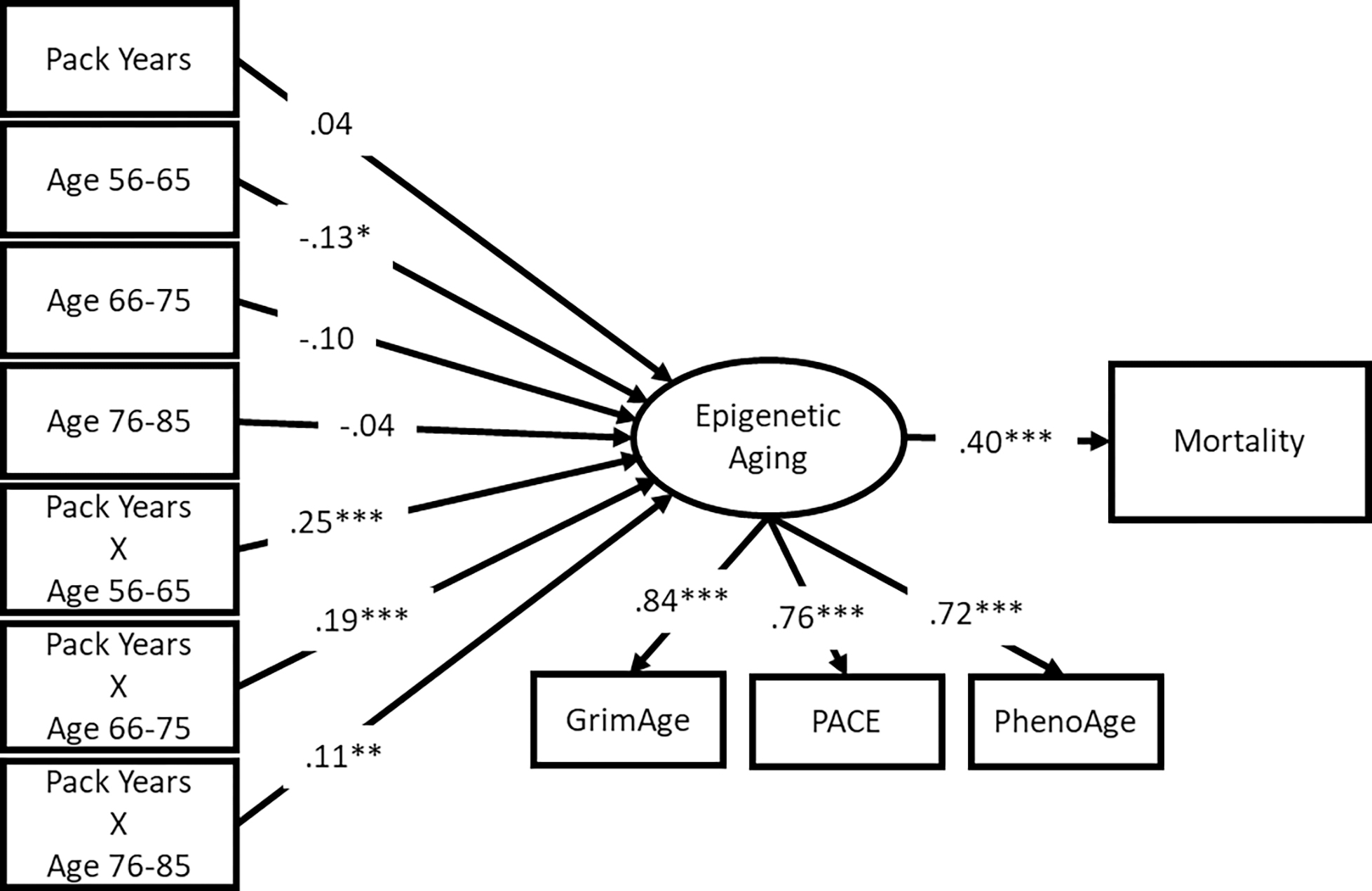
SEM results for Mortality
Note: standardized results shown; *** p < .001, ** p < .01, * p < .05
Figure 3.
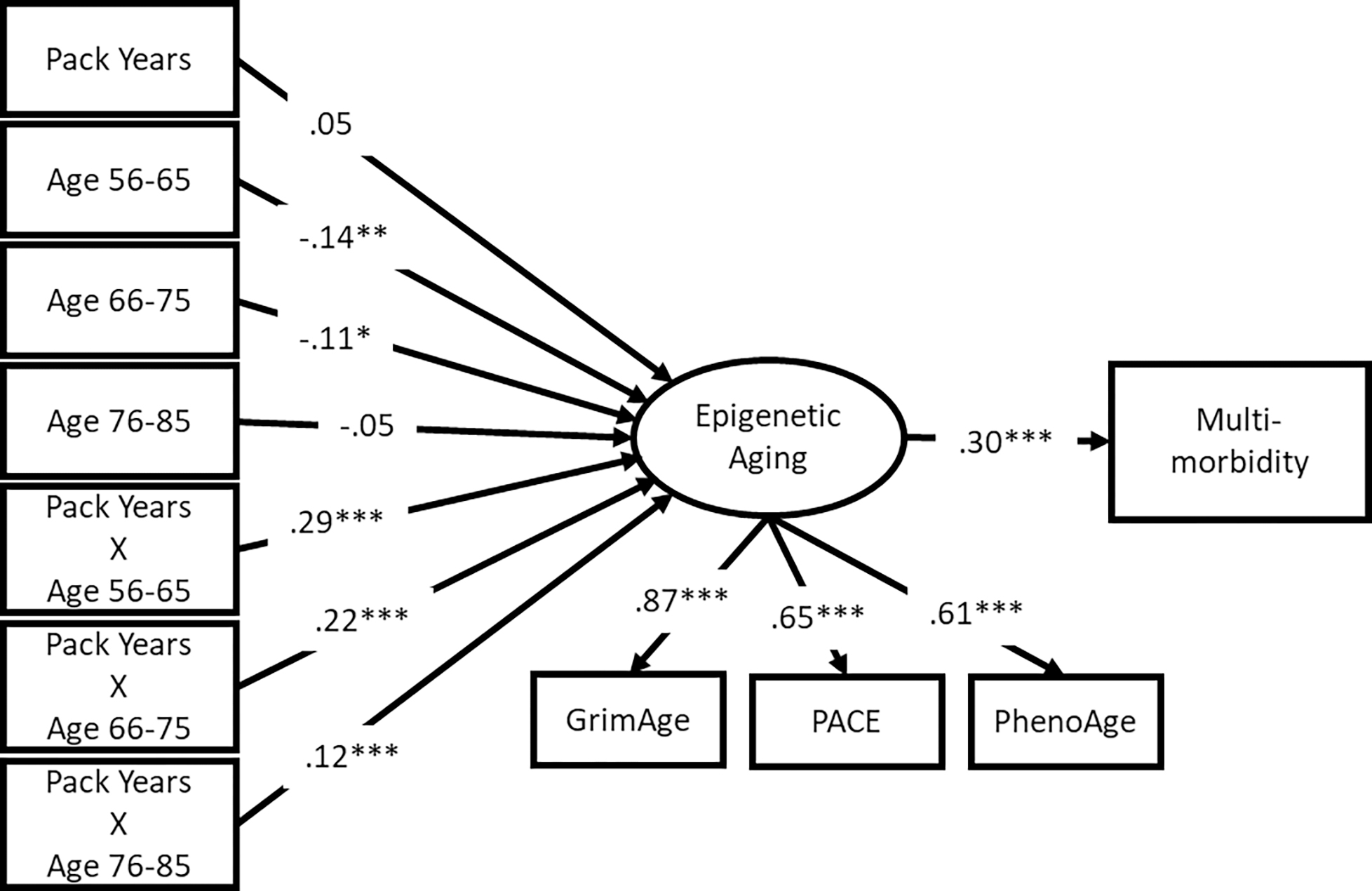
Multi-morbidity results
Note: standardized results shown; *** p < .001, ** p < .01, * p < .05
Mortality risk and multimorbidity were significantly associated with the latent accelerated epigenetic aging factor (β = .40 and .30, respectively; p < .001). Specifically, participants who had more accelerated epigenetic aging also had a higher mortality risk and more chronic illnesses relative to same-aged peers. Across both models, relative to respondents aged 86 or older, respondents aged 56–65 had less accelerated aging if they reported 0 pack-years (β = −.13 and −.14, respectively; p < .05 and p < .01, respectively (because of the inclusion of the interaction terms). Similarly, participants aged 66–75 had less accelerated aging (β = −.11, p < .05) compared with the oldest group of participants in the multimorbidity model. The interaction terms were all significantly associated with accelerated epigenetic aging (all p < .01). These results suggest that the health risk associated with smoking pack-years is greater for younger respondents.
Mediated Moderation
Unstandardized results of the mediated moderation analysis are shown in Figure 4. Starting with results for mortality, total effect estimates and the percentage of the total effect explained by epigenetic aging are not easily interpretable because direct effects are in the opposite direction of total effects for ages 56–65. The total effect of smoking pack-years on mortality is significant only for participants aged 66–75 (95% CI = [0.001, 0.012]) and 76–85 (95% CI = [0.002, 0.010]), potentially because of the relatively few mortality events among younger participants. (Only 63 participants aged 56–65 died during the study period.) However, the mediated moderation effect was significant for participants aged 56–65 (95% CI = [0.007, 0.013]), 66–75 (95% CI = [0.005, 0.010]; 100% of the total effect), and 76–85 (95% CI = [0.003, 0.007]; 83.33% of the total effect). That is, for younger groups, the association between smoking pack-years and mortality mediated by epigenetic aging was larger than for older groups. Among the oldest three groups—who had a larger number of mortality events—the total effect of smoking pack-years on mortality was significant for participants aged 66–75 and 76–85 and this effect was entirely or mostly mediated by epigenetic aging; neither the total effect of smoking pack-years on mortality (95% CI = [−0.005, 0.011]) nor the indirect effect through epigenetic aging (95% CI = [−0.002, 0.003]) was significant for participants aged 86 or older. These findings suggest that the mortality risk associated with smoking was reduced for the oldest participants because of their greater resilience to smoking in terms of epigenetic aging.
Figure 4.
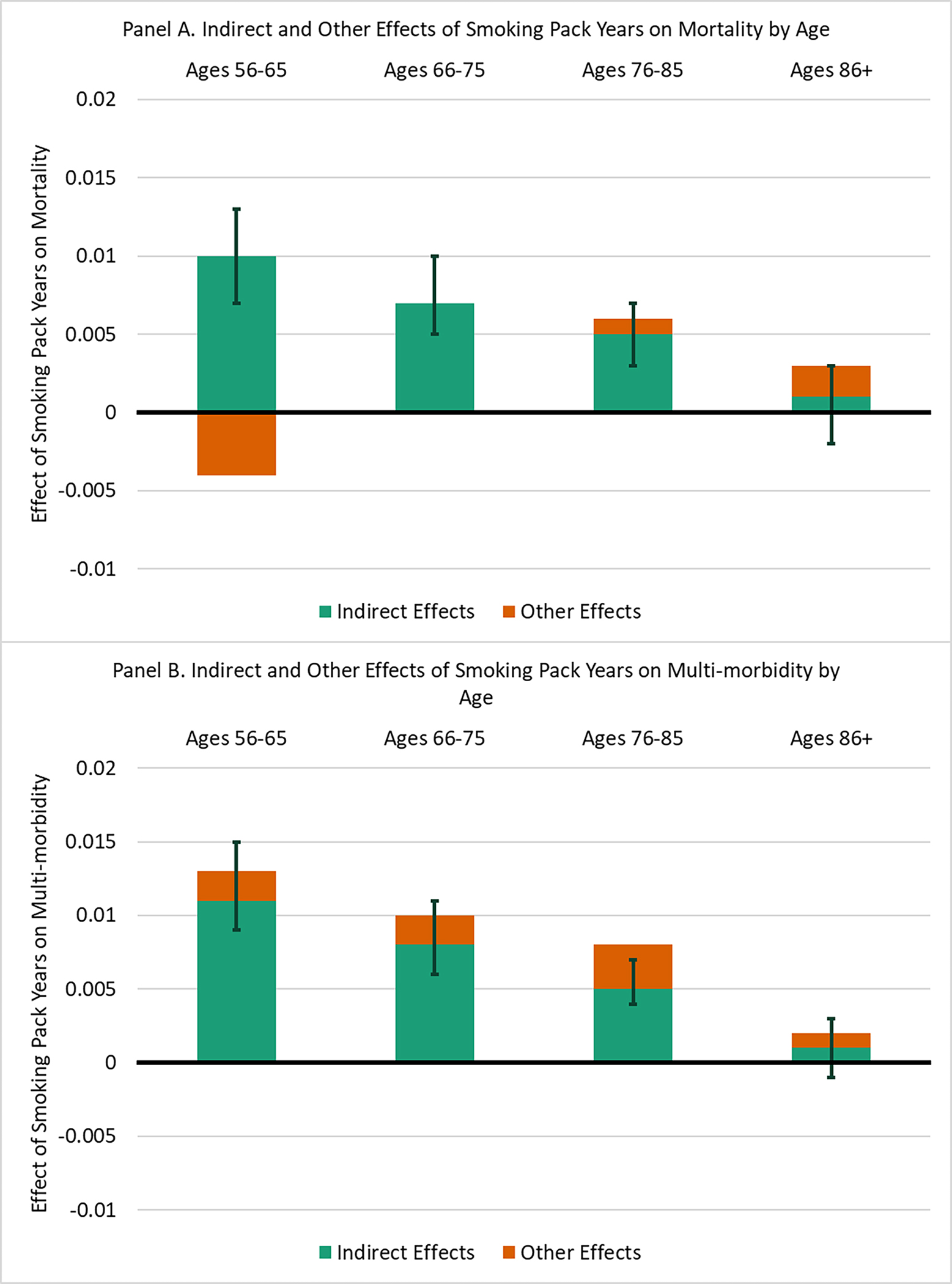
Moderated mediation results.
Note: unstandardized results shown. Estimates and 95% confidence intervals calculated from results of SEMs in Figures 2 and 3. The full bar represents the total effect of lifetime smoking pack years for each age group. The green section represents the indirect effect of pack years through epigenetic aging. The orange section represents all other effects. Some direct effects of pack years on mortality are in the opposite direction of the total effect. 95% confidence intervals for indirect effects from bias-corrected bootstrap procedure.
[EK: I rearranged this section so that information is presented in the same order as for mortality above].
For multimorbidity, the total effect of smoking pack-years on multimorbidity was significant for participants aged 56–65 (95% CI = [0.009, 0.017]), 66–75 (CI = [0.005, 0.014]), and 76–85 (95% CI = [0.004, 0.011]). The moderated mediation effect was significant for participants aged 56–65 (95% CI = [0.009, 0.015]; 84.62% of the total effect), 66–75 (95% CI = [0.006, 0.011]; 80.00% of the total effect), and 76–85 (95% CI = [0.004, 0.007]; 62.50% of the total effect), and the portion of this total effect mediated by epigenetic aging was significant. For respondents aged 86 or older, neither the total (95% CI = [−0.005, 0.011]) nor indirect (95% CI = [−0.001, 0.003]) effects were significant. These findings suggests that the effect of lifetime cigarette smoking on chronic disease morbidity was weaker for the oldest adults because their epigenetic aging was less affected by cigarette smoking.
Additional and Supplemental Analyses
As a sensitivity analysis, we ran the main models with pack-years capped at 55 to determine years of smoking before age 56 (the age of the youngest respondent). We capped this variable at age 55 to ensure that associations involving pack-years were not inflated for older adults as a function of their having been alive longer, giving them more opportunity to smoke. This model was not substantively different from the main model (see Figures S1–S3, online appendix); however, for the mortality model, the total effect of pack-years on mortality risk was only marginally significant for the 66–75 age group. We also ran models without covariates to assess whether conditioning on these variables affected results (see Figures S4–S6, online appendix). The results were highly similar; however, the total effect of pack-years on mortality risk was significant for the 66–75 age group, and the indirect effect of pack-years on multimorbidity mediated by accelerated aging was significant for participants aged 86 or older (although the total effect was still nonsignificant). Because recollection error in the amount smoked in the past could bias the results, we reestimated the main models using the number of years reported smoking (duration) before age 56 (see Figures S7–S9, online appendix). The results were generally greater in magnitude, but the pattern of significant results was similar and led to the same substantive conclusions.
We found that GrimAge had the largest factor loading. To assess how much GrimAge drove the significant results, we reassessed models using the individual epigenetic aging measures instead of the latent factor based on multiple clocks (see Figures S10–S18, online appendix). Results using only the GrimAge measure were largely similar to those in the main analyses. Results focused on PhenoAge were generally weaker: the indirect effects of smoking on mortality and multimorbidity via PhenoAge were significant only for the 66–75 and 76–85 age groups. Results using DunedinPACE were weaker than the main models, but significance tests from all indirect paths and total effects were similar.
To determine whether one disease drove the results for multimorbidity, we also estimated models using the same procedure as used for mortality, replacing mortality with each chronic illness (see Figures S19–S26, online appendix). The results differed slightly across diseases, mostly driven by different associations between the chronic illness and accelerated aging. For hypertension, the total effects of smoking were nonsignificant for all age groups, but indirect effects were significant for all but the oldest group. For diabetes, the total effect was significant only for the 76–85 age group, and indirect effects were significant for all but the oldest group. For cancer, total effects were significant for the 66–75 and 76–85 age groups, and indirect effects were significant for all but the oldest group. For lung disease, total effects were significant for all but the oldest group, and indirect effects were significant for all but the oldest group. For heart disease, no total effects were significant, but indirect effects were significant for all but the oldest group. For stroke, total effects were significant only for the 66–75 age group, but indirect effects were significant for all but the oldest group. Finally, for arthritis, total effects were significant for the two youngest groups, but the indirect effect was significant only for the youngest group.
Discussion
Past research indicated hidden heterogeneity in vulnerability and resilience to health risks, such as cigarette smoking, and that this resilience is concentrated in older cohorts (Levine and Crimmins 2014). One explanation for this effect is that less resilient individuals tend to age faster than their peers when exposed to health risks. These less resilient individuals then die earlier, leaving behind a pool of increasingly resilient individuals (Behrman et al. 1990; Vaupel et al. 1979). Our results support this hypothesis: participants in older cohorts tended to experience a weaker association between lifetime cigarette smoking and health outcomes. This moderation effect was mediated by a reduced association between smoking pack-years and epigenetic aging. This effect was clearest in results for multimorbidity: the direct effects of smoking on the number of chronic illnesses remained fairly constant, and the reduced total effect was largely attributable to decreasing associations between smoking pack-years and epigenetic aging.
The direct effect of smoking pack-years on mortality risk was negative for participants aged 56–65, leading to nonsignificant total effects. This finding is likely due to the relatively few mortality events in this younger cohort. However, the indirect effect of smoking pack-years on mortality risk mediated by epigenetic aging was significant for this group. Comparisons among participants aged 66–75, 76–85, and 86 or older (for whom there were more mortality events) were in the expected direction. Thus, future research with more follow-up is needed to assess how cigarette smoking pack-years affects aging and mortality in these younger groups. However, these findings suggest that greater accelerated aging among those aged 66–75 and 76–85 helps explain greater mortality risk for smokers in these cohorts.
Supplementary analyses using individual epigenetic aging measures showed that GrimAge is the most consistent mediator of the interaction between smoking pack-years and mortality and multimorbidity. This finding is consistent with past research finding that GrimAge is relatively robustly associated with health risks in HRS data and is strongly associated with health outcomes in multiple data sets (McCrory et al. 2021). GrimAge and DunedinPACE were developed by predicting time to death and pace of aging in middle-aged adults, respectively. Thus, they may be better suited to capturing long-term exposures from earlier in the life course than PhenoAge, which was trained to predict mortality. Because GrimAge also captures DNAm variance associated with smoking pack-years, it is logical that it would be the strongest mediator of the effects in this study. However, results for all the individual aging measures were generally weaker than those for the latent aging factor. This finding is consistent with our argument that these individual measures capture a portion of the larger underlying construct of accelerated aging and that combining them in a latent factor is useful for understanding aging processes. Ours is one of the first studies to use these measures as indicators of an underlying aging process. Because all three measures can be produced using the same underlying DNAm data, we encourage other researchers to utilize factor analysis in future research focused on accelerated aging.
In analyses focused on individual diseases, results for illnesses most clearly linked to smoking (e.g., lung disease) were the strongest and most consistent with the main multimorbidity model. By contrast, we found relatively weak results for diseases more tenuously linked to smoking (especially arthritis). However, all these models produced weaker results than the main multimorbidity model, consistent with our arguments that aging is a general process and that chronic disease onset broadly results from this process. Models focused on individual diseases are less able to capture the general aging process. Future research focused on aging populations and demographic selection might want to focus on multiple diseases at once rather than individual clinical outcomes to better understand the aging process.
This study has limitations. Although the HRS is representative of older U.S. adults, additional research in international contexts is needed to understand how national context might affect the results. Additionally, it is unknown whether similar patterns would be found among younger people. Epigenetic aging was available at only one time point. Additional data could help differentiate potential age, period, and cohort effects. Additionally, a longer follow-up is needed. As noted earlier, few mortality events occurred among younger cohorts in the study period. As HRS collects more data, it will be possible to determine whether individuals whose epigenetic aging was more affected by smoking die earlier, leaving behind a more resilient cohort of survivors. Additionally, we did not investigate the sources of differential resilience in this study. Past research suggests that genetic and social factors might affect resilience to health hazards (Levine and Crimmins 2016). Smoking might be less important at older ages as other mortality risks become more important. Future research should investigate other mortality risk factors (e.g., diet and exercise, air pollution and environmental toxins) to examine whether the pattern we found for smoking can be replicated. Our results might differ by race/ethnicity and by sex/gender, given the different smoking rates among these groups. Future research should investigate differential resilience among these groups. The fact that a DNAm surrogate of pack-years is a part of the GrimAge measure could be considered a limitation of this study, but we argue that it is a strength. GrimAge partly represents the epigenetic processes associated with smoking and is therefore a potential biological pathway linking smoking behaviors to health and longevity. Analyses of cause-specific mortality are beyond the scope of the current study, but we expect that an analysis focused on causes of death relevant to smoking might show stronger results.
Despite these limitations, our study has important implications for demographic and aging research. Our findings help identify subpopulations of particularly resilient individuals who are notable for the low epigenetic age acceleration associated with exposure to a major health hazard: smoking. There has been a long-standing concern with understanding the sources of differential resilience and frailty (Behrman et al. 1990) and how frailty affects selection into mortality at earlier ages (Vaupel et al. 1979). This study identified epigenetic aging as a potential mechanism explaining this process that can be measured relatively early in life, before morbidity and mortality emerge, to provide indicators of resilience and frailty. Interventions for younger adults could utilize epigenetic aging estimates to identify the most vulnerable individuals and intervene before health outcomes such as chronic disease morbidity or mortality manifest. Future research is needed to understand the sources of differential resilience to understand and promote these factors.
Supplementary Material
Acknowledgments
Research reported here was supported by the National Institute on Aging of the National Institutes of Health under Awards T32AG000037, R01AGAG060110 and P30AG017265. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The Health and Retirement Study is sponsored by the National Institute on Aging (grant NIA U01AG009740) and is conducted by the University of Michigan.
References
- Andrasfay T, & Crimmins E (2023). Occupational characteristics and epigenetic aging among older adults in the United States. Epigenetics, 18, 2218763. 10.1080/15592294.2023.2218763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevaare MJ, Roberts J, van Hout HPJ, Joling KJ, Smalbrugge M, Schoonmade LJ, . . . Hertogh CMPM (2020). Resilience in older persons: A systematic review of the conceptual literature. Ageing Research Reviews, 63, 101144. 10.1016/j.arr.2020.101144 [DOI] [PubMed] [Google Scholar]
- Beach SRH, Dogan MV, Lei MK, Cutrona C, Gerrard M, Gibbons FX, . . . Philibert RA (2015). Methylomic aging as a window onto the influence of lifestyle: Tobacco and alcohol use alter the rate of biological aging. Journal of the American Geriatrics Society, 63, 2519–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrman JR, Sickles RC, & Taubman P (1990). Age-specific death rates with tobacco smoking and occupational activity: Sensitivity to sample length, functional form, and unobserved frailty. Demography, 27, 267–284. [PubMed] [Google Scholar]
- Belsky DW, Caspi A, Arseneault L, Baccarelli A, Corcoran DL, Gao X, . . . Moffitt TE (2020). Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. eLife, 9, e54870. 10.7554/eLife.54870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Caspi A, Corcoran DL, Sugden K, Poulton R, Arseneault L, . . . Moffitt TE (2022). DunedinPACE, a DNA methylation biomarker of the pace of aging. eLife, 11, e73420. 10.7554/eLife.73420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky DW, Moffitt TE, Cohen AA, Corcoran DL, Levine ME, Prinz JA, . . . Caspi A (2018). Eleven telomere, epigenetic clock, and biomarker-composite quantifications of biological aging: Do they measure the same thing? American Journal of Epidemiology, 187, 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beydoun MA, Beydoun HA, Noren Hooten N, Maldonado AI, Weiss J, Evans MK, & Zonderman AB (2022). Epigenetic clocks and their association with trajectories in perceived discrimination and depressive symptoms among U.S. middle-aged and older adults. Aging, 14, 5311–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder AM, & Horvath S (2022). Epigenetic clocks. In Michels KB (Ed.), Epigenetic epidemiology (2nd ed., pp. 261–276). Cham: Springer Nature Switzerland. 10.1007/978-3-030-94475-9_11 [DOI] [Google Scholar]
- Carroll JE, Irwin MR, Levine ME, Seeman TE, Absher D, Assimes T, & Horvath S (2017). Epigenetic aging and immune senescence in women with insomnia symptoms: Findings from the Women’s Health Initiative Study. Mechanisms of Dementia and Delirium, 81, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosco TD, Kok A, Wister A, & Howse K (2019). Conceptualising and operationalising resilience in older adults. Health Psychology and Behavioral Medicine, 7, 90–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins EM (2020). Social hallmarks of aging: Suggestions for geroscience research. Ageing Research Reviews, 63, 101136. 10.1016/j.arr.2020.101136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins EM, Faul JD, Thyagarajan B, & Weir DR (2017). Venous blood collection and assay protocol in the 2016 Health and Retirement Study: 2016 Venous Blood Study (VBS) (HRS documentation report). Retrieved from https://hrsdata.isr.umich.edu/sites/default/files/documentation/data-descriptions/HRS2016VBSDD.pdf [Google Scholar]
- Crimmins EM, Kim JK, Fisher J, & Faul JD (2020). HRS epigenetic clocks (HRS documentation report). Ann Arbor: Survey Research Center, University of Michigan. Retrieved from https://hrsdata.isr.umich.edu/sites/default/files/documentation/data-descriptions/EPICLOCKS_DD.pdf [Google Scholar]
- Crimmins EM, Kim JK, & Seeman TE (2009). Poverty and biological risk: The earlier “aging” of the poor. Journals of Gerontology, Series A: Biological Sciences and Medical Sciences, 64A, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins EM, Thyagarajan B, Levine ME, Weir DR, & Faul J (2021). Associations of age, sex, race/ethnicity, and education with 13 epigenetic clocks in a nationally representative U.S. sample: The Health and Retirement Study. Journals of Gerontology, Series A: Biological Sciences and Medical Sciences, 76, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenelon A, & Preston SH (2012). Estimating smoking-attributable mortality in the United States. Demography, 49, 797–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorito G, Polidoro S, Dugué P-A, Kivimaki M, Ponzi E, Matullo G, . . . Vineis P (2017). Social adversity and epigenetic aging: A multi-cohort study on socioeconomic differences in peripheral blood DNA methylation. Scientific Reports, 7, 16266. 10.1038/s41598-017-16391-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Föhr T, Waller K, Viljanen A, Sanchez R, Miina M, Rantanen T, . . . Sillanpää E (2021). Does the epigenetic clock GrimAge predict mortality independent of genetic influences: An 18 year follow-up study in older female twin pairs. Clinical Epigenetics, 13, 128. 10.1186/s13148-021-01112-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Breitling LP, & Brenner H (2016). Relationship of tobacco smoking and smoking-related DNA methylation with epigenetic age acceleration. Oncotarget, 7, 46878–46889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev VN, Kritchevsky SB, Clarke SG, Cuervo AM, Fiehn O, de Magalhães JP, . . . Cummings SR (2021). Molecular damage in aging. Nature Aging, 1, 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild M, Nargis N, & d’Espaignet ET (2018). Global economic cost of smoking-attributable diseases. Tobacco Control, 27, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley EC, Kuchel GA, Newman AB, Allore HG, Bartley JM, Bergeman CS, . . . Yung R (2017). Report: NIA workshop on measures of physiologic resiliencies in human aging. Journals of Gerontology, Series A: Biological Sciences and Medical Sciences, 72, 980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghani A, Arpawong TE, Kim JK, Lewinger JP, Finch CE, & Crimmins E (2020). Female vulnerability to the effects of smoking on health outcomes in older people. PLoS One, 15, e0234015. 10.1371/journal.pone.0234015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins-Chen AT, Thrush KL, Wang Y, Minteer CJ, Kuo P-L, Wang M, . . . Levine ME (2022). A computational solution for bolstering reliability of epigenetic clocks: Implications for clinical trials and longitudinal tracking. Nature Aging, 2, 644–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, & Raj K (2018). DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nature Reviews: Genetics, 19, 371–384. [DOI] [PubMed] [Google Scholar]
- Huntley AL, Johnson R, Purdy S, Valderas JM, & Salisbury C (2012). Measures of multimorbidity and morbidity burden for use in primary care and community settings: A systematic review and guide. Annals of Family Medicine, 10, 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen I, Powell LH, Everson-Rose SA, Hollenberg SM, El Khoudary SR, & Matthews KA (2022). Psychosocial well-being and progression of coronary artery calcification in midlife women. Journal of the American Heart Association, 11, e023937. 10.1161/jaha.121.023937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, McCartney DL, Wagner J, Yoo J, Bell AS, Mavromatis LA, . . . Lohoff FW (2022). Additive effects of stress and alcohol exposure on accelerated epigenetic aging in alcohol use disorder. Biological Psychiatry, 93, 331–341. [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, . . . Sierra F (2014). Geroscience: Linking aging to chronic disease. Cell, 159, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SS, Singer BD, & Vaughan DE (2017). Molecular and physiological manifestations and measurement of aging in humans. Aging Cell, 16, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Zheng Y, Joyce BT, Jiang H, Greenland P, Jacobs DR, . . . Hou L (2022). Relative contributions of six lifestyle- and health-related exposures to epigenetic aging: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Clinical Epigenetics, 14, 85. 10.1186/s13148-022-01304-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopack ET (2023). Chronic stress and latent virus reactivation: Effects on immune aging, chronic disease morbidity, and mortality. Journals of Gerontology, Series B: Psychological Sciences and Social Sciences, 78, 1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopack ET, Carroll JE, Cole SW, Seeman TE, & Crimmins EM (2022). Lifetime exposure to smoking, epigenetic aging, and morbidity and mortality in older adults. Clinical Epigenetics, 14, 72. 10.1186/s13148-022-01286-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopack ET, Crimmins EM, Cole SW, Seeman TE, & Carroll JE (2022). Accelerated epigenetic aging mediates link between adverse childhood experiences and depressive symptoms in older adults: Results from the Health and Retirement Study. SSM–Population Health, 17, 101071. 10.1016/j.ssmph.2022.101071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lariscy JT, Hummer RA, & Rogers RG (2018). Cigarette smoking and all-cause and cause-specific adult mortality in the United States. Demography, 55, 1855–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M-K, Gibbons FX, Simons RL, Philibert RA, & Beach SRH (2020). The effect of tobacco smoking differs across indices of DNA methylation-based aging in an African American sample: DNA methylation-based indices of smoking capture these effects. Genes, 11, 311. 10.3390/genes11030311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, & Crimmins E (2014). Not all smokers die young: A model for hidden heterogeneity within the human population. PLoS One, 9, e87403. 10.1371/journal.pone.0087403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, & Crimmins EM (2016). A genetic network associated with stress resistance, longevity, and cancer in humans. Journals of Gerontology, Series A: Biological Sciences and Medical Sciences, 71, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, & Crimmins EM (2018). Is 60 the new 50? Examining changes in biological age over the past two decades. Demography, 55, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Quach A, Horvath S, Chen BH, Ferrucci L, . . . Horvath S (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging, 10, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, & Kroemer G (2013). The hallmarks of aging. Cell, 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, . . . Horvath S (2019). DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging, 11, 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumley T (2004). Analysis of complex survey samples. Journal of Statistical Software, 9(8), 1–19. [Google Scholar]
- McCrory C, Fiorito G, Hernandez B, Polidoro S, O’Halloran AM, Hever A, . . . Kenny RA (2021). GrimAge outperforms other epigenetic clocks in the prediction of age-related clinical phenotypes and all-cause mortality. Journals of Gerontology, Series A: Biological Sciences and Medical Sciences, 76, 741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén LK, & Muthén BO (2015). Mplus: Statistical analysis with latent variables—User’s guide (7th ed.). Los Angeles, CA: Muthén & Muthén. Retrieved from https://www.statmodel.com/download/usersguide/MplusUserGuideVer_7.pdf [Google Scholar]
- Oblak L, van der Zaag J, Higgins-Chen AT, Levine ME, & Boks MP (2021). A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Research Reviews, 69, 101348. 10.1016/j.arr.2021.101348 [DOI] [PubMed] [Google Scholar]
- Poole JC, Dobson KS, & Pusch D (2017). Childhood adversity and adult depression: The protective role of psychological resilience. Child Abuse & Neglect, 64, 89–100. [DOI] [PubMed] [Google Scholar]
- Preston SH, Stokes A, Mehta NK, & Cao B (2014). Projecting the effect of changes in smoking and obesity on future life expectancy in the United States. Demography, 51, 27–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protsenko E, Yang R, Nier B, Reus V, Hammamieh R, Rampersaud R, . . . Wolkowitz OM (2021). “GrimAge,” an epigenetic predictor of mortality, is accelerated in major depressive disorder. Translational Psychiatry, 11, 193. 10.1038/s41398-021-01302-0“ 10.1038/s41398-021-01302-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puterman E, & Epel E (2012). An intricate dance: Life experience, multisystem resiliency, and rate of telomere decline throughout the lifespan. Social and Personality Psychology Compass, 6, 807–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrkov TV, Avchaciov K, Tarkhov AE, Menshikov LI, Gudkov AV, & Fedichev PO (2021). Longitudinal analysis of blood markers reveals progressive loss of resilience and predicts human lifespan limit. Nature Communications, 12, 2765. 10.1038/s41467-021-23014-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2022). R: A language and environment for statistical computing [Computer software]. R Foundation for Statistical Computing. Available from https://www.R-project.org/ [Google Scholar]
- Raffington L, Belsky DW, Kothari M, Malanchini M, Tucker-Drob EM, & Harden KP (2021). Socioeconomic disadvantage and the pace of biological aging in children. Pediatrics, 147, e2020024406. 10.1542/peds.2020-024406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers A (1992). Heterogeneity and selection in multistate population analysis. Demography, 29, 31–38. [PubMed] [Google Scholar]
- Schutte NS, Palanisamy SKA, & McFarlane JR (2016). The relationship between positive psychological characteristics and longer telomeres. Psychology & Health, 31, 1466–1480. [DOI] [PubMed] [Google Scholar]
- Si J, Chen L, Yu C, Guo Y, Sun D, Pang Y, . . . Lv J (2023). Healthy lifestyle, DNA methylation age acceleration, and incident risk of coronary heart disease. Clinical Epigenetics, 15, 52. 10.1186/s13148-023-01464-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons RL, Lei M-K, Klopack ET, Berg M, Zhang Y, & Beach SSR (2021). (Re)setting epigenetic clocks: An important avenue whereby social conditions become biologically embedded across the life course. Journal of Health and Social Behavior, 62, 436–453. [DOI] [PubMed] [Google Scholar]
- Simons RL, Ong ML, Lei M-K, Klopack E, Berg M, Zhang Y, . . . Beach SRH (2022). Shifts in lifestyle and socioeconomic circumstances predict change—for better or worse—in speed of epigenetic aging: A study of middle-aged Black women. Social Science & Medicine, 307, 115175. 10.1016/j.socscimed.2022.115175 [DOI] [PubMed] [Google Scholar]
- Sugden K, Hannon EJ, Arseneault L, Belsky DW, Broadbent JM, Corcoran DL, . . . Caspi A (2019). Establishing a generalized polyepigenetic biomarker for tobacco smoking. Translational Psychiatry, 9, 92. 10.1038/s41398-019-0430-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden K, Moffitt TE, Arpawong TE, Arseneault L, Belsky DW, Corcoran DL, . . . Caspi A (2023). Cross-national and cross-generational evidence that educational attainment may slow the pace of aging in European-descent individuals. Journals of Gerontology, Series B: Psychological Sciences and Social Sciences, 78, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukraintseva S, Arbeev K, Duan M, Akushevich I, Kulminski A, Stallard E, & Yashin A (2021). Decline in biological resilience as key manifestation of aging: Potential mechanisms and role in health and longevity. Mechanisms of Ageing and Development, 194, 111418. 10.1016/j.mad.2020.111418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, & Richardson A (2019). The role of DNA methylation in epigenetics of aging. Pharmacology & Therapeutics, 195, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services. (2014). The health consequences of smoking—50 years of progress: A report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. Retrieved from https://stacks.cdc.gov/view/cdc/21569/cdc_21569_ds1.pdf [Google Scholar]
- Vaupel JW, Manton KG, & Stallard E (1979). The impact of heterogeneity in individual frailty on the dynamics of mortality. Demography, 16, 439–454. [PubMed] [Google Scholar]
- Vaupel JW, & Yashin AI (1985). Heterogeneity’s ruses: Some surprising effects of selection on population dynamics. American Statistician, 39, 176–185. [PubMed] [Google Scholar]
- Waziry R, Ryan CP, Corcoran DL, Huffman KM, Kobor MS, Kothari M, . . . Belsky DW (2023). Effect of long-term caloric restriction on DNA methylation measures of biological aging in healthy adults from the CALERIE trial. Nature Aging, 3, 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitson HE, Duan-Porter W, Schmader KE, Morey MC, Cohen HJ, & Colón-Emeric CS (2016). Physical resilience in older adults: Systematic review and development of an emerging construct. Journals of Gerontology, Series A: Biological Sciences and Medical Sciences, 71, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, . . . Yutani H (2019). Welcome to the Tidyverse. Journal of Open Source Software, 4, 1686. 10.21105/joss.01686 [DOI] [Google Scholar]
- Wrigley-Field E (2020). Multidimensional mortality selection: Why individual dimensions of frailty don’t act like frailty. Demography, 57, 747–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, & Wu GWY (2021). DNA methylation clock associated with age-related illnesses is accelerated in PTSD. Neuropsychopharmacology, 46, 225–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gao X, Just AC, Colicino E, Wang C, Coull BA, . . . Baccarelli AA (2019). Smoking-related DNA methylation is associated with DNA methylation phenotypic age acceleration: The Veterans Affairs Normative Aging Study. International Journal of Environmental Research and Public Health, 16, 2356. 10.3390/ijerph16132356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindler T, Frieling H, Fliedner L, Veer IM, Neyazi A, Awasthi S, . . . Friedel E (2022). How alcohol makes the epigenetic clock tick faster and the clock reversing effect of abstinence. Addiction Biology, 27, e13198. 10.1111/adb.13198 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.