

ABSTRACT
Human CD200R1 (hCD200R1), an immune inhibitory receptor expressed predominantly on T cells and myeloid cells, was identified as a promising immuno-oncology target by the 23andMe database. Blockade of CD200R1-dependent signaling enhances T cell-mediated antitumor activity in vitro and in vivo. 23ME–00610 is a potential first-in-class, humanized IgG1 investigational antibody that binds hCD200R1 with high affinity. We have previously shown that 23ME–00610 inhibits the hCD200R1 immune checkpoint function. Herein, we dissect the molecular mechanism of 23ME–00610 blockade of hCD200R1 by solving the crystal structure of 23ME–00610 Fab in complex with hCD200R1 and performing mutational studies, which show 23ME–00610 blocks the interaction between hCD200 and hCD200R1 through steric hindrance. However, 23ME–00610 does not bind CD200R1 of preclinical species such as cynomolgus monkey MfCD200R1. To enable preclinical toxicology studies of CD200R1 blockade in a pharmacologically relevant non-clinical species, we engineered a surrogate antibody with high affinity toward MfCD200R1. We used phage display libraries of 23ME–00610 variants with individual CDR residues randomized to all 20 amino acids, from which we identified mutations that switched on MfCD200R1 binding. Structural analysis suggests how the surrogate, named 23ME–00611, acquires the ortholog binding ability at the equivalent epitope of 23ME–00610. This engineering approach does not require a priori knowledge of structural and functional mapping of antibody–antigen interaction and thus is generally applicable for therapeutic antibody development when desired ortholog binding is lacking. These findings provide foundational insights as 23ME–00610 advances in clinical studies to gain understanding of the hCD200R1 immune checkpoint as a target in immuno-oncology.
KEYWORDS: 23ME-00610, cancer immunotherapy, CD200, CD200R1, deep mutational scan, immune checkpoint
Introduction
CD200R1 is an immune inhibitory receptor and its expression is restricted to immune cells, including myeloid cell subtypes (including neutrophils, eosinophils, basophils, mast cells, microglia, macrophages, and dendritic cells) and subsets of CD4+ and CD8+ T cells.1 Both CD200R1 and its only known ligand, CD200, are type 1 transmembrane proteins with two extracellular immunoglobulin superfamily domains. Structural studies of the murine ligand-receptor complex have confirmed the N-terminus domain of the two proteins as the site of interaction.2
Previous studies have shown that elevated expression of CD200/CD200R1 on tumor cells and tumor-infiltrating immune cells may downregulate the production of proinflammatory cytokines by activated myeloid and/or T cells, including interferon (IFN)α, IFNγ, tumor necrosis factor, macrophage inflammatory protein (MIP)1b, interleukin (IL)-1b, IL-2, IL-5, IL-6, and IL-13.3–11 CD200/CD200R1 signaling may also contribute to an immunosuppressive tumor microenvironment based on its ability to suppress mobilization of CD107a, which is a cytolytic granule component that is indicative of cytotoxic capacity, by antigen-specific T cells.12 In addition, CD200/CD200R1 signaling may activate myeloid-derived suppressive cells and upregulate expression of immunosuppressive proteins, including TGF-β, arginase 1,13 and indoleamine-pyrrole 2,3-dioxygenase.4 Thus, blocking the CD200R1/CD200 immune checkpoint within an immunosuppressive environment has the potential to prevent or reverse immune cell tolerance. To understand the potential of therapeutically targeting this pathway for cancer treatment, we set out to develop an antibody to block CD200R1 interaction with its ligand CD200.
There are five predicted splice isoforms of human CD200R1 (hCD200R1), iso1 to iso5, but only iso1 and iso4 give rise to transmembrane proteins. hCD200R1-iso4 encodes the longest isoform (348 amino acids) and is the most abundant isoform in multiple cancer types (The Cancer Genome Atlas, 2021, http://www.cancer.gov/tcga). The less abundant isoform iso1 has a 23-amino acid truncation at N-terminus due to missing the exon2. In addition to splice variants, the hCD200R1 transcript has four common missense single nucleotide polymorphisms on two haplotypes, reference (Ref) and alternative (Alt). The prevalence of Ref and Alt haplotypes is similar (1000 Genomes Project).14
23ME-00610 is a humanized, monoclonal, effector function-attenuated immunoglobulin G subclass 1 (IgG1) antibody that binds specifically to the hCD200R1 immune inhibitory receptor and inhibits the hCD200R1 immune checkpoint. We previously demonstrated that 23ME–00610 potently binds to all functionally relevant human isoforms and haplotypes of CD200R1 with picomolar affinity and blocks the interaction of hCD200R1 and its cognate ligand hCD200.14 In this work, we determined the molecular mechanism of 23ME–00610 engagement of hCD200R1 using a combination of high-resolution x-ray crystallography and mutagenesis studies. Additionally, we found that 23ME–00610 is highly specific for hCD200R1, but unable to bind CD200R1 from cynomolgus monkey or other potential preclinical species with any measurable affinity. To advance this clinical candidate, we developed a cross-reactive surrogate antibody of 23ME–00610, named 23ME–00611, which targets the cynomolgus monkey’s CD200R1 ortholog (MfCD200R1). This engineered tool antibody enables non-clinical safety evaluations in a pharmacologically relevant species.
Results
Crystal structure of 23ME–00610 bound to human CD200R1
To gain insights into the molecular mechanism for 23ME–00610 blockade of hCD200R1, we solved the crystal structure of 23ME–00610 Fab bound to hCD200R1-iso4-Ref ECD refined to a resolution of 2.89 Å (Figure 1, Supplementary Table S1, S2), which is sufficient to define the binding pose of the antibody and identify specific side chain interactions in the interface. The model comprises residues E1 to D217 of the Fab heavy chain (HC), E1 to C214 of the light chain (LC), and N37 to L232 of hCD200R1-iso4-Ref. One short loop in the CH1 region of the HC is not fully defined by electron density and is not included in the model. We observed one complex in each asymmetric unit.
Figure 1.
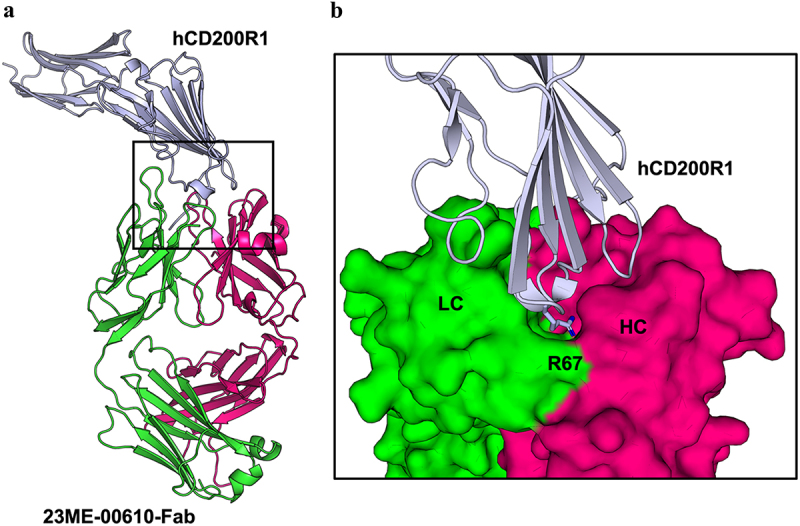
Crystal structure of 23ME-00610-Fab bound to hCD200R1-iso4-ref reveals binding interface. (a) Structure of 23ME-00610-Fab:hCD200R1 complex shown in ribbon. hCD200R1 is in light blue color. 23ME-00610 heavy chain is in magenta, and light chain in green. (b) Zoom-in view of the binding interface with 23ME-00610 is represented as a surface. R67 in hCD200R1, shown as a stick, protrudes into a pocket cavity created at the intersection of the heavy (HC) and light (LC) chains of 23ME-00610.
The binding site of 23ME–00610-Fab covers a solvent-accessible surface area of ~1044.6 Å2 of hCD200R1-iso4-Ref, of which approximately 642.6 Å2 comes from 23ME–00610 hC interactions and 453.8 Å2 from LC interactions, including a small overlapping area interacting with both chains. The buried surface area of this interaction is similar to that of the known antibody–antigen complexes (1068 ± 314 Å2) in the PDB from a non-redundant set of 1,425 complexes, suggesting that the interface is in the range for potent antibody binding.15 The hCD200R1 extracellular domain (ECD) sequence polymorphisms at positions 89, 121 and 177 in various isoforms and haplotypes are distant from the structural epitope for the antibody, consistent with the finding that 23ME–00610 binds equally to all hCD200R1 variants.14
Since this is the first description of a hCD200R1 structure, we superimposed the previously solved crystal structure of murine CD200R1 (PDB code: 4BFG)2 onto the hCD200R1 ECD and observed that they are highly similar, with a root mean square deviation of 2.16 Å2 over 1205 atoms (Supplementary Figure S1). Noticeable differences were seen in the BC and DE loops of the Fab-bound human CD200R1 structure in comparison to the unbound murine CD200R1 (PDB 4BFG), possibly due to sequence differences in the region (Supplementary Figure S2), different packing of the molecules within the crystal, and/or consequences of 23ME–00610 binding. These two loops exhibited different conformations between free and mCD200-bound mCD200R1 while not involved in the interaction with mCD200 (PDF 4BFI).2
The crystal structure of 23ME–00610-Fab:hCD200R1-iso4-Ref qualitatively defined a set of residues that comprise the epitope of hCD200R1 or the paratope of 23ME–00610 and provides molecular details of the interface (Figure 1b, Supplementary Table S3). As shown in Figure 1b, R67 is part of the BC loop at the center of the structural epitope of hCD200R1, which protrudes deeply into a pocket formed by several 23ME–00610 residues at the junction of heavy and light chain variable domains, namely W52, N58, E95 and G100 of HC and S91, N92, E93, D94 and P96 of LC.
23ME-00610 blocks human CD200R1/CD200 interaction through steric hindrance
To examine how 23ME–00610 blocks hCD200R1 from engaging its ligand hCD200, we generated a homology model of the hCD200:hCD200R1 complex by aligning AlphaFold predicted structures of the individual proteins from UniProt with the known crystal structure of the mouse complex (Figure 2). Comparison of the hCD200:hCD200R1 model with the high-resolution crystal structure of 23ME–00610-Fab:hCD200R1 revealed that hCD200 and 23ME–00610 engage hCD200R1 from opposite directions and the two epitopes have a small overlap (Figure 2a,b; Supplementary Figure S2). The crystal structure shows a set of 25 epitope residues on hCD200R1 for 23ME–00610. Compared to the predicted 24 interface residues with human hCD200, there is an overlap of four residues: T136, P137, D138, and F141. Based on this, 23ME–00610 inhibits hCD200R1 interaction with hCD200 through steric hindrance.
Figure 2.
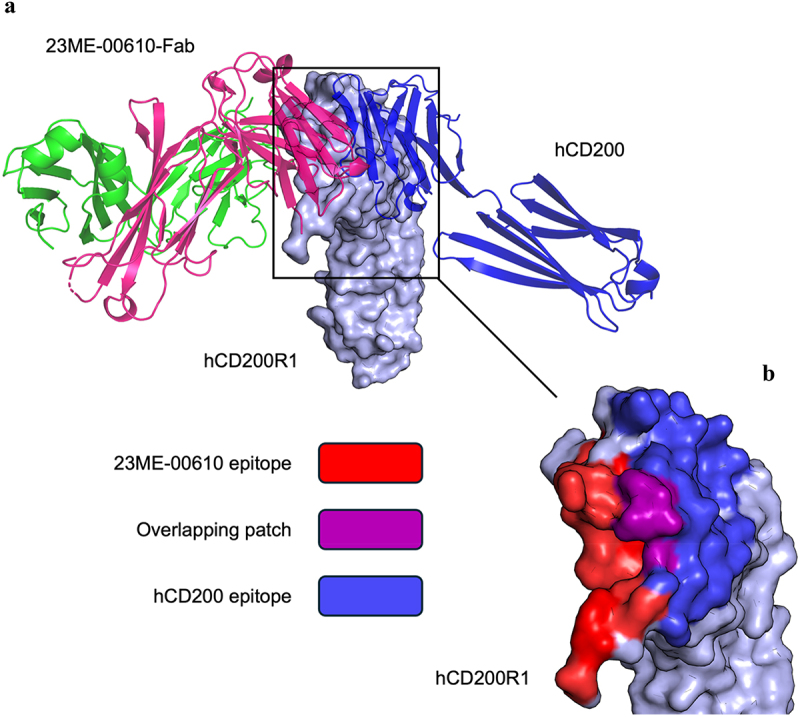
23ME-00610 epitope overlaps with the hCD200 binding site. (a) Structure of 23ME-00610-Fab:hCD200R1 complex is superposed with hCd200:hcd200r1 complex model. 23ME-00610-Fab is shown as ribbons (HC in magenta; LC in green). Surface representation of hCD200R1 is shown in light blue; hCD200 depicted in blue ribbons. Homology model of the hCd200:hcd200r1 complex was generated by aligning AlphaFold predicted structures of hCD200 and hCD200R1 to the known mouse structure (PDB ID 4BFI). (b) Surface representation of hCD200R1 shows structural epitope residues (<5 Å) for 23ME-00610 (red) and hCD200 (blue) and the overlap between the two epitopes (purple).
Given the relatively small structural overlap between the epitopes of 23ME–00610 and hCD200, we examined them further with alanine scanning mutagenesis studies.16 Using the hCD200:hCD200R1 AlphaFold homology model, we selected 24 hCD200R1 residues that are within 5 Å of hCD200 to mutate to alanine as potential epitope sites for 23ME–00610 interaction (Supplementary Figure S2). These hCD200R1 variants were expressed on the surface of HEK293 cells and the binding of 23ME–00610 Fab measured by flow cytometry (Figure 3a). Fab binding potency to cell-surface variants was normalized by the expression level of these variants measured by antibody against the N-terminus hemagglutinin (HA) tag (see Methods). Mutations at positions that significantly reduced binding (p < 0.0001) relative to wild type (WT) included E75, Y88, T95, T136, G139, and F141 (Figure 3a,b). These mutants were then generated as recombinant ECD protein in sufficient quantities for surface plasmon resonance (SPR) analysis, except for Y88A, which formed aggregates and therefore was not tested by SPR. As anticipated, binding of the five alanine mutants of hCD200R1-iso4-Ref exhibited attenuated affinity by 2.9- to 10.4-fold relative to WT from the measured equilibrium dissociation constant (KD) (Supplementary Table S4). Alanine substitution at positions T136, G139, F141 of CD200R1 resulted in the greatest reduction in affinity compared to WT. These three residues are a part of or adjacent to the CD200R1 structural epitope for 23ME–00610 that overlap with the epitope for CD200 (T136, P137, D138, and F141). The mutational studies therefore confirm the functional importance of these CD200R1 epitope residues for 23ME–00610, which can sterically block CD200 engagement.
Figure 3.
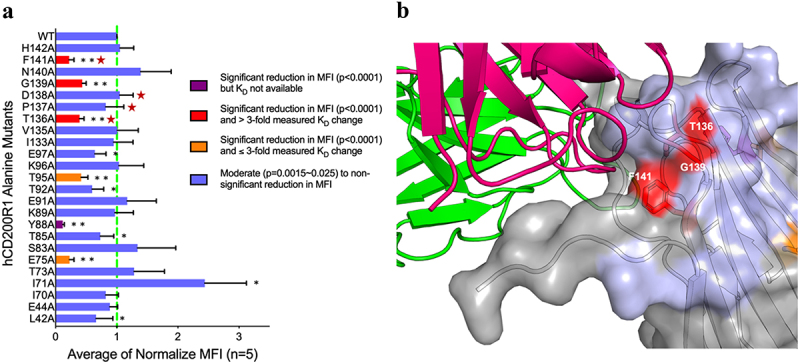
Structural overlaps of hCD200 and 23ME-00610 CD200R1 epitopes are functionally important for 23ME-00610 binding. (a) Relative binding potency of 23ME-00610-Fab to Expi293 cells expressing either wild type (WT) or alanine mutants of hCD200R1-iso4-ref was evaluated by flow cytometry and SPR. Binding MFI (mean fluorescence intensity) was first normalized by the variant expression levels (see methods) and then the ratios to normalized MFI of WT (as 1, green dotted line) were plotted to represent relative binding potency for 23ME-00610. Attenuated binding with p = 0.0015-0.025 (*) or p < 0.0001 (**) relative to WT are shown. Coloring of each bar accords to relative binding potency of variants expressed as full-length protein on Expi293 cells and SPR KD measured as purified ECD protein as labeled. Positions highlighted with a red star are those that overlap between structural epitope of 23ME-00610 and hCD200 as in Figure 2b. (b) With hCD200R1 as surface representation, positions are colored according to the relative binding potency of alanine mutation variants shown in (A) where a significant decrease (**p < 0.0001) in 23ME-00610 binding potency verified by SPR are colored red. 23ME-00610-Fab is shown as ribbons (HC in magenta; LC in green).
23ME-00610 binds with high affinity to human CD200R1 but lacks cross-reactivity to cynomolgus CD200R1 ortholog
We next determined the cross-reactivity of 23ME–00610 to bind cynomolgus CD200R1 ortholog, as we planned to assess the nonclinical safety of blocking hCD200R1 in a pharmacologically relevant species such as a cynomolgus monkey prior to advancement into clinical studies. Human and cynomolgus CD200R1 ortholog (MfCD200R1, UniProt: G7NZT0) were expressed in Expi293 cells and evaluated for binding of 23ME–00610 by flow cytometry. We found that 23ME–00610 binds with high potency to hCD200R1 but shows minimal binding to MfCD200R1 (Figure 4a). MfCD200R1 and hCD200R1 ECD share 90% sequence identity. Functionally important sequence differences are likely present in the interface with 23ME–00610 to prevent binding (Figure 4b).
Figure 4.
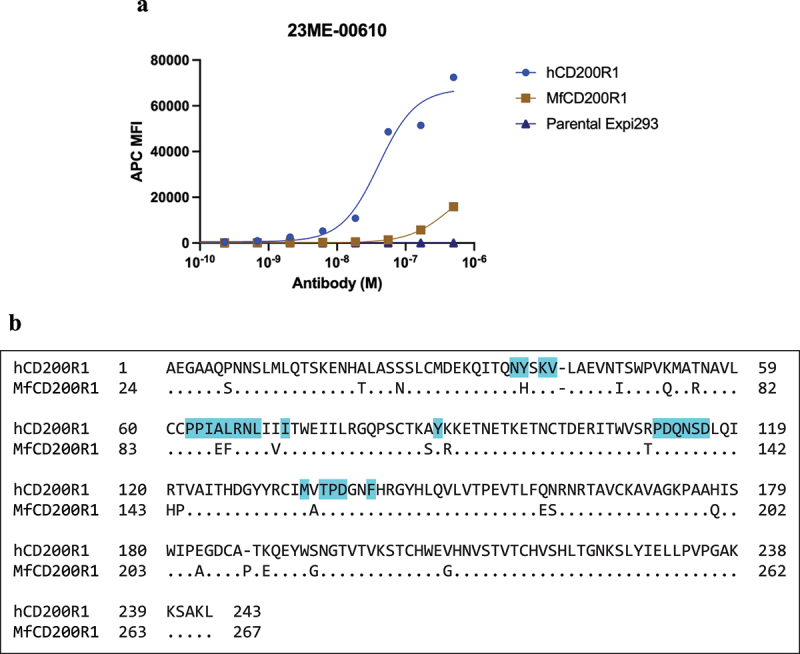
23ME-00610 exhibits minimal cross-reactivity to cynomolgus CD200R1 ortholog (MfCD200R1). (a) Binding of 23ME-00610 to MfCD200R1 or hCD200R1 over-expressed on Expi293 cells was evaluated by flow cytometry. (b) Sequence alignment of the extracellular domains of hCD200R1-iso4-ref and MfCD200R1 (UniProt: G7NZT0) with dots indicating identical sequence. hCD200R1 residues with atoms within 5Å of 23ME-00610 in the solved structure are shaded in cyan.
Engineering 23ME–00611 an MfCD200R1 cross-reactive surrogate antibody
Given the high sequence homology between hCD200R1 and MfCD200R1, we hypothesized that a modest level of mutation in 23ME–00610 may be sufficient to switch on MfCD200R1 binding. We performed deep mutational scanning of the light and heavy chain variable domain complementarity-determining regions (VL or VH-CDR) of the 23ME–00610-Fab as previously described, which is a well-established approach to determine the extent to which each scanned residue tolerates mutation to all other amino acids and thus their functional role for binding interaction.17–19 Our objective was to further understand the high-affinity interaction of 23ME–00610 with hCD200R1. An additional aim was to identify mutations for engineering a surrogate antibody to bind MfCD200R1 with high affinity. Briefly, phage libraries displaying 23ME–00610 Fab with either VL- or VH-CDR randomized to 106-107 degeneracy were generated (see Methods) and characterized by deep sequencing before and after selection for hCD200R1 binding function (Supplementary Figure S3 and Table S5). The enrichment ratios (ER) for each single mutation, i.e., the differential frequency of each mutation in the selected pools over the unselected (pre-sorted) libraries, were tabulated as Log2(ER) in a heat map to indicate each mutation as favorable (red) or unfavorable (blue) for the high-affinity binding interaction (Figure 5a,b). As a metric for an overall tolerance to mutation of each residue, we calculated “mean of Log2ER” for all mutations other than cysteine.
Figure 5.
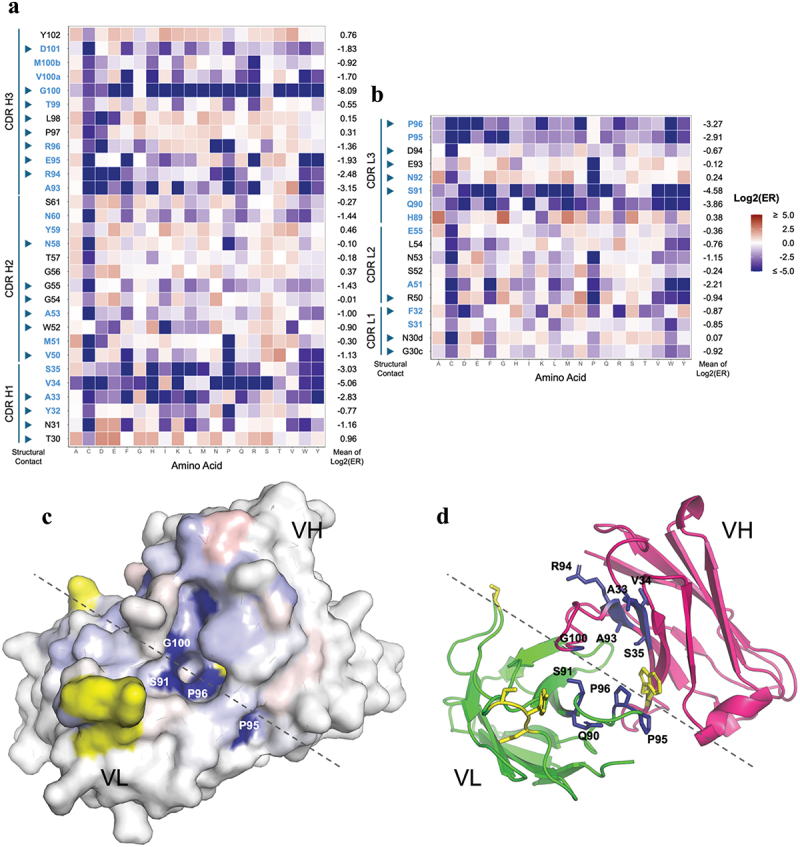
Deep mutational scan of 23ME-00610 for mapping the high affinity interaction with hCD200R1. Single mutation enrichment of all randomized CDR positions of 23ME-00610 VH (a) or VL (b) from hCD200R1 panning is shown with Log2(ER) colored as labeled. Positions with low solvent accessibility are in blue, which is defined as < 25% of the whole residue area20 being solvent accessible as unbound 23ME-00610 fab using FreeSASA.21 CDR positions in the scan that are part of the structural contact sites with hCD200R1 are pointed with arrowheads. For each position, the mean of the Log2(ER) is calculated as the average of Log2(ER) of all mutation except cysteine. (c) The mean of the Log2(ER) is mapped on the structure of 23ME-00610 fab in a top-down view using the same color scale as A and B. Approximate border between VH and VL is denoted with a dotted line. Residues in yellow are part of the structural contact but not included in the mutational scan. (d) Same view of fab as (C) is plotted in ribbon with residues with Log2(ER) < −2 in blue sticks.
Among the 27 scanned CDR residues that make structural contact (<5 Å) with hCD200R1 (indicated with arrowhead), a subset (A33, R94, and G100 of HC; A51, S91, and P96 of LC) were shown to be highly intolerant to mutation with mean Log2ER <-2 (Figure 5a,b). Interestingly, these residues also belong to a set of CDR residues that have low solvent accessibility, characterized as having <25% of the whole residue area being solvent accessible as Fab alone based on the Fab structure in the complex (Figure 5a,b, residue in blue). In fact, all 10 of the residues with mean Log2ER of <-2 belong to this group of residues with low solvent accessibility, suggesting that they play an important role structurally and/or functionally to enable specific and high-affinity interactions with hCD200R1. By mapping the mean Log2ER onto the structure of the antibody (Figure 5c,d), we deduced that the pocket in which the CD200R1 BC loop buries deeply is the central functional hotspot. VH-E95 is one of the residues lining the pocket making a charge interaction with R67 of hCD200R1 buried in the pocket. Consistently, VH-E95 does not tolerate mutation to residues opposite in charge (arginine, lysine) or larger in size (tyrosine, phenylalanine, tryptophan), while accepting mutation to other polar or homologous amino acids, likely to fit into the tight pocket with R67 of hCD200R1.
To engineer MfCD200R1 binding, the VH-CDR and VL-CDR phage libraries were selected against MfCD200R1. In contrast to selection against hCD200R1, more rounds of phage panning were required to enrich the phage clones that potentially possess binding activity toward MfCD200R1. This is expected because the variant clones that can bind MfCD200R1 should be relatively rare. From sequencing the selected library pools that appear to bind MfCD200R, we found four enriched mutations that might confer binding to MfCD200R1. Three mutations were in VH (A33R, W52Y, Y102N) and one in VL (E93W) (Figure 6a). We generated this new variant as full IgG named 23ME–00611 and confirmed that it binds recombinant MfCD200R1 ECD with a KD of 2 nM as evaluated by SPR measurements (Figure 6b). This is in marked contrast to the total lack of measurable binding of 23ME–00610 toward MfCD200R1 (Figure 6b, left). This switch was also reproduced by flow cytometry with cynomolgus PBMC (Figure 6c). Additionally, 23ME–00611 is shown to completely block the interaction of MfCD200R1 with its cognate ligand MfCD200 as evaluated by enzyme-linked immunosorbent assay (ELISA) (Figure 6d).
Figure 6.
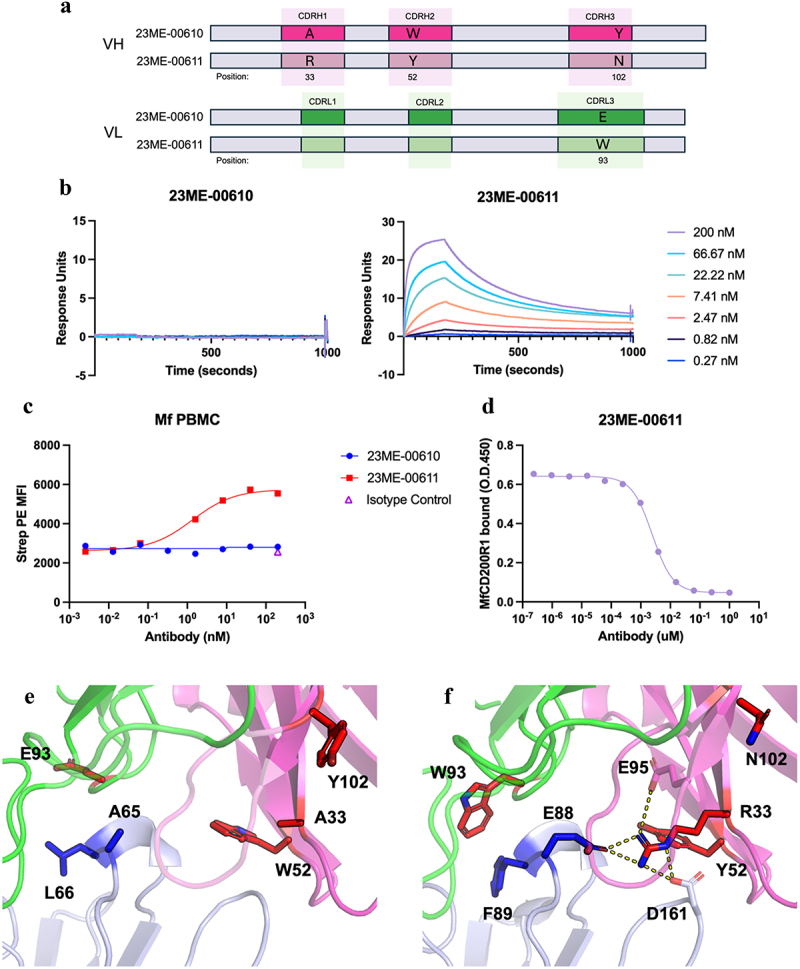
Mutations in 23ME-00611 surrogate antibody confers high affinity binding to MfCD200R1. (a) Schematics that show the VH or VL CDR positions mutated from 23ME-00610 to 23ME-00611; (b) biacore sensorgrams that show lack of 23ME-00610 binding to MfCD200R1 with concentration up to 200 nM (left) and 23ME-00611’s binding to MfCD200R1 at KD = 2 nM at 37°C (right); (c) cynomolgus PBMC FACS study confirms lack of 23ME-00610 binding to MfCD200R1 while 23ME-00611showed good binding potency. (d) 23ME-00611’s potent inhibition of MfCD200R1:MfCD200 interaction as shown by ELISA; (e) zoom-in views of 23ME-00610:hCD200R1 crystal structure and (f) 23ME-00611:MfCD200R1AlphaFold model show the two structurally equivalent residues (blue sticks) in hCD200R1 or MfCD200R1 that can explain the lack of 23ME-0610 binding and the four fab mutations in red sticks that confer 23ME-00611 high affinity binding to MfCD200R1. 23ME-00611 A33R mutation permits a network of charge interaction (dotted line) to accommodate E88 in MfCD200R1. Heavy and light chains indexed using kabat and chothia numbering, respectively.22–24
To understand the gain of MfCD200R specificity by these mutations, we compared the solved crystal structure of 23ME–00610-Fab bound to hCD200R1 with an AlphaFold model of 23ME–00611 bound to MfCD200R1 (Figure 6e,f). Relative to hCD200R1, MfCD200R1 contains two amino acid substitutions E88 and F89 for A65 and L66 in hCD200R1, respectively (Figures 4b, 6e,f). These two residues are a part of the BC loop that buries into the pocket at the junction of VL and VH of 23ME–00610. The A88E substitution in MfCD200R1 would introduce a buried unsatisfied polar group into a hydrophobic interface area in the 23ME–00610:hCD200R1 complex, which likely precludes binding of 23ME–00610. The VH-A33R mutation in 23ME–00611 could compensate for this new buried polar group introduced by the A88E substitution in MfCD200R1 as it forms a new polar interaction network to stabilize the complex (Figure 6f). VH-W52 in 23ME–00610 is substituted for Y52 in 23ME–00611, which could allow for more space to accommodate R33 compared to W52. Position 89 in MfCD200R1 is a phenylalanine residue, which is more hydrophobic than the leucine in hCD200R1. The VL substitution at position 93 replaces a polar glutamic acid with a hydrophobic tryptophan, which could stabilize the more hydrophobic F89 in MfCD200R1. Given that VH-R33 substitution in 23ME–00611 stabilizes E88 in MfCD200R1, it is likely that this substitution contributes most to the gain in MfCD200R1 binding. To confirm this, individual point mutations were made in 23ME–00610 and binding to MfCD200R1 was characterized using Biacore. Among the four mutants, 23ME-00610_A33R indeed displayed the strongest binding (Supplementary Figure S4). 23ME-00610_E93W shows modest binding, with the other single mutations displaying no detectable binding. These data suggest that the A33R substitution contributes most for gaining MfCD200R1 binding. Further, consistent with the mutational scanning of 23ME–610 for hCD200R1 binding (Figure 5a), 23ME–00611 can still bind hCD200R1 well, albeit with weaker affinity (KD = 1 nM) than 23ME–610 (KD = 0.1 nM). Of the four mutations, only A33R appeared unfavorable (Log2ER = −1.84) for 23ME–00610 interaction with hCD200R1, while the others were well tolerated.
Discussion
Antibodies targeting immune checkpoints such as CTLA-4 and PD-1 have shown improved overall survival across numerous solid and hematologic cancers.25 Combination therapy can further improve clinical benefit for patients, but so far only a subset of patients have long-term benefits. Thus, there remains a need to expand the immune checkpoint inhibitor toolbox. Analysis of human genetics has the potential to inform more successful drug target discovery.26 A genome-wide association study analysis using 23andMe’s genomic and health survey database identified the novel immune-oncology target, CD200R1.14 Like the well-characterized CTLA-4 checkpoint inhibitor, genetic variants within the CD200R1 pathway associate with opposing effects in cancer and immune diseases. Therefore, we generated a potentially first-in-class antibody, 23ME–00610, as a blockade of the CD200:CD200R1 checkpoint axis to activate an antitumor immune response and potentially combine with other checkpoint inhibitors.14
We used structural analysis and mutational studies to dissect the molecular basis for the interaction of 23ME–00610 with hCD200R1. These studies provide a comprehensive examination of the molecular mechanism underlying the blockade of human CD200R1 (hCD200R1) by 23ME–00610, an investigational immune checkpoint immunotherapy currently under evaluation in a Phase 1/2a clinical study in patients with advanced solid tumors (NCT05199272).
The 23ME–00610-Fab:CD200R1 crystal structure revealed a well-defined binding interface involving a loop of hCD200R1 inserted deep into a pocket formed by antibody residues from both VH and VL of 23ME–00610. Further, homology modeling of the hCD200:hCD200R1 complex indicated that 23ME–00610 and hCD200 engage hCD200R1 from opposite directions with distinct but partially overlapping epitopes at four residues (T136, P137, D138, and F141) Alanine scanning of hCD200R1 confirmed the functional importance of these structurally overlapping epitope residues for antibody binding. Deep mutational scanning on the antibody side was used to show that the functional hotspot for this high-affinity interaction is contained within the pocket formed at the junction of heavy and light chains where the BC loop of hCD200R1 is deeply buried. Together, these studies indicate that 23ME–00610 disrupts hCD200 binding to hCD200R1 through a steric hindrance mechanism.
While these results provide valuable insights into the mechanism of action of 23ME–00610, one hurdle in therapeutic antibody development is evaluation of safety in preclinical studies using non-human primates (NHP) prior to human trials. This is further complicated when the lead candidate has limited cross reactivity with the orthologous target in the animal model as is the case with 23ME–00610 lacking binding activity toward CD200R1 in cynomolgus monkeys (MfCD200R1), which is the preferred model for preclinical safety study for developing a therapeutic antibody. We investigated whether 23ME–00610 might bind orthologs from other animals such as rhesus, rabbit and marmoset that can potentially be used for safety studies but could not identify any (data not shown). For antibody therapeutics, the feasibility of performing a preclinical safety study in NHP is advantageous for progressing into the clinical trials.
To overcome this limitation, we set out to engineer a surrogate antibody that can bind MfCD200R1 with high affinity and ideally to the equivalent epitope of 23ME–00610 so that the surrogate’s action in cynomolgus would closely represent 23ME–00610 in human. We used the deep mutational scanning antibody phage libraries that randomized VH or VL CDR residues, which is typically done to identify residues involved in antibody–antigen interactions, and we directly selected for the rare variants that bind to MfCD200R1. This library approach allowed each CDR residue in the scan to be any of the 20 amino acids (by using NNK codon) and up to three mutations with one from each of the three HC or LC CDRs can be present in each clone. The design is agnostic to the sequence or structural conformation difference between human and cynomolgus CD200R1 because the CDR residues included in the libraries are standards established for mutational scanning. The theoretical degeneracy of the libraries (106-107) is low enough to be well covered by generated phage libraries and large enough to include a solution for gaining binding function to a homologous protein (Table S5). Indeed, we observed gradual enrichment of phage libraries selected on MfCD200R1 specifically and identified mutations in the CDRs that were confirmed to enable MfCD200R1 binding by the recombinant variant antibodies. Notably, four mutations (A33R, W52Y, Y102N in the VH domain and E93W in the VL domain) were identified to achieve high-affinity binding to MfCD200R1.
Structural modeling suggests that the surrogate 23ME–00611 should bind to the exact same site as 23ME–610 and it explains how mutations at four CDR positions compensate for the key differences between human and cynomolgus CD200R1 molecularly (Figure 6). Importantly, 23ME–00611 effectively blocked the interaction between MfCD200R1 and its cognate ligand, MfCD200, demonstrating that it retains the functionality of 23ME–00610. The successful generation of 23ME–00611 that mirrors how 23ME–00610 binds hCD200R1 has facilitated the evaluation of CD200R1 blockade in NHP.
The study highlights the role of antibody engineering in overcoming interspecies reactivity challenges. The deep mutational scanning approach used in the discovery of 23ME–00611 demonstrates the feasibility of generating ortholog cross-reactive antibodies needed for therapeutic development or biological investigation. While we use structural and mutational studies to dissect this change of binding specificity, the approach does not require prior knowledge of 1) the structural or molecular model of the antibody interaction, 2) the key differences in orthologous molecules behind the lack of cross-reactivity, and 3) the antibody residues important for the lack of interspecies cross-reactivity. Another advantage of the approach is that phage display selection allows for an easy way to enrich mutations that confer high-affinity binding by using stringent selection strategy. To the best of our knowledge, this is the first report of engineering antibody cross-species reactivity from non-existent to high affinity (Kd = 2 nM) within one round of engineering. We believe this approach is applicable for similar challenges of engineering interspecies ortholog binding.
In conclusion, our study advances the molecular understanding of 23ME–00610, a potential first-in-class, humanized IgG1 investigational antibody that binds hCD200R1, and provides a framework for engineering species cross reactivity to enable therapeutic development of antibodies.
Material and methods
Purification of CD200R1 and CD200 proteins
Sequences corresponding to the ECD of hCD200R1 or MfCD200R1 were cloned into mammalian expression vectors using a cytomegalovirus (CMV) promoter to drive expression. Sequence encoding a C-terminal hexahistadine (His) purification tag was inserted and CD200R1 was expressed using the Expi293 system according to manufacturer’s instructions and purified using immobilized metal affinity chromatography with a Histrap FF column (Cytiva), followed by size-exclusion chromatography (SEC) with Superdex 200 (Cytiva) using an ÄKTA system. CD200 was similarly expressed except that the vector contained C-terminal human IgG crystallizable fragment (Fc) region with N297G mutation. Recombinant CD200-Fc proteins were purified using affinity chromatography with a MabSelect SuRe (Cytiva) column and followed with a Superdex 200 column.
Measurement of binding affinity using surface plasmon resonance (SPR)
To determine the binding affinity of 23ME–00610 for hCD200R1-iso4-Ref WT or different alanine mutant proteins, and 23ME–00610 and 23ME–00611 for MfCD200R1, SPR measurements were performed using a Biacore 8K instrument at 37°C. The temperature is chosen to reflect physiological conditions. 23ME–00610 (0.5 µg/mL) in HBS-EP buffer (0.01 M 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid [HEPES], pH 7.4; 0.15 M NaCl, 3 mm ethylenediaminetetraacetic acid [EDTA]; 0.005% Tween-20) was injected onto a Protein A capture chip using a flow rate of 30 µL/min. Buffer alone was injected to flow cell 1 (FC1) as a reference. Next, 3-fold serial dilutions of hCD200R1 in 1X HBS-P+ buffer starting from 100 nM were injected at a flow rate of 30 µL/minute. The concentrations of both the antibody and receptor were determined through preliminary experimentation to avoid surface oversaturation, ensure a sufficient binding response, minimize artifacts and capture monovalent binding kinetics. Each sensorgram was subjected to reference and buffer subtraction prior to analysis. Analysis was performed using BIACORE 8K Evaluation Software (version 1.1.1.7442). Association rates (ka) and dissociation rates (kd) were calculated using a one-to-one binding kinetics model. The equilibrium dissociation constant (KD) was calculated as the ratio of kd/ka.
Blocking ELISA
96-well MAXISORP flat-bottom plates were coated overnight at 4°C with MfCD200-Fc at a concentration of 1 µg/mL in phosphate-buffered saline (PBS). Plates were first blocked by addition of 300 μL of 1% bovine serum albumin (BSA) in PBS, pH 7.2. 1:1 mix of 0.04 µg/mL biotinylated MfCD200R1 and 4-fold serial dilutions of 23ME–00611 starting from 1 µM in ELISA buffer (0.1% BSA, 0.05% Tween-20 in PBS, pH 7.2) were pre-incubated for 1 h and transferred to MfCD200-Fc coated plates capture to the unbound biotinylated MfCD200R1 for 15 min which was then detected with poly-horseradish peroxidase (HRP) streptavidin in ELISA buffer and visualized by adding the substrate for HRP (tetramethylbenzidine, TMB). Plates were analyzed by reading the optical density at 450 nm (OD450).
Purification of 23ME–00610-Fab
The sequence encoding the Fab of 23ME–00610 (23ME-00610-Fab) was cloned into a pRK mammalian expression vector as for CD200R1 expression. The vector containing LC and the VH and CH1 domains of HC fused with a histidine purification tag C-terminally. Recombinant 23ME–00610-Fab was expressed and purified using the Expi293 expression system with the immobilized metal affinity chromatography and followed by the SEC Superdex S75 16/600 column (Cytiva).
Protein crystallography, structure modeling, and refinement
To prepare the 23ME–00610-Fab:hCD200R1 complex, 23ME–00610-Fab and hCD200R1 were mixed at a 2:1 mass ratio (10 mg 23ME–00610-Fab, 5 mg hCD200R1) in 1 mL and incubated for 2 h at 4°C, and the mixture was loaded onto a Superdex 200 10/300 to separate unbound protein from the 1:1 complex. Fractions corresponding to the complex were pooled and concentrated, resulting in homogenous stoichiometric protein as evaluated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis. The purified complex was used in crystallization trials using a standard Proteros screen with approximately 1200 different conditions. Crystallization screening of 23ME–00610-Fab:hCD200R1 (45 mg/ml) complex was conducted by sitting-drop vapor diffusion at 20°C. The crystals appeared upon mixing 0.2 µl of protein solution with 0.2 μl of reservoir solution containing sodium cacodylate 0.1 M pH 6.50, glycerol 10.0% (v/v), NaCl 1.0 M, polyethylene glycol (PEG) 600 30.0% weight/volume (w/v). A cryo-protocol was established using Proteros standard protocols. Crystals were flash-frozen and measured at a temperature of 100 K. Diffraction data were collected at the Swiss Light Source (SLS, Villigen, Switzerland) using cryogenic conditions. The crystals obtained belong to space group P 41 21 2. Data was processed using the programs autoPROC, XDS and AIMLESS.27–31 The phase information necessary to determine and analyze the structure was obtained by molecular replacement using a solved structure of 23ME–00610-Fab as a search model. Subsequent model building and refinement was performed according to standard protocols with COOT32 and the software package CCP4,30 respectively. For the calculation of the free R-factor (Rfree), a measure to cross-validate the correctness of the final model, approximately 4.9% of measured reflections were excluded from the refinement procedure (Table S2). TLS refinement (using REFMAC5, CCP4) has been carried out, which resulted in lower R-factors and higher quality of the electron density map. The water model was built with the “Find waters” algorithm of COOT by putting water molecules in peaks of the Fo-Fc map contoured at 3.0 followed by refinement with REFMAC5 and checking all waters with the validation tool of COOT. The criteria for the list of suspicious waters were (1) B-factor greater 80 Å2, (2) 2Fo-Fc map less than 1.2σ, and (3) distance to closest contact <2.3 Å or >3.5 Å. The suspicious water molecules and those in the ligand-binding site (distance to ligand less than 10 Å) were checked manually. The Ramachandran plot of the final model calculated with Molprobity33 showed 96.63% of all residues in the favored region and 3.21% in the allowed region. The residue Arg54 (LC) was an outlier in the Ramachandran plot (data not shown), but it was clearly defined by the electron density. Statistics of the final structure and the refinement process are listed in Tables S1 and S2.
AlphaFold prediction of the 23ME–00611:MfCD200R1 complex
AlphaFold multimer v2.2 was run on AWS development environment using the VH and VL sequences of 23ME–00611 and the ECD of MfCD200R1.
Measurement of binding to alanine hCD200R1 mutants expressed on Expi293 cells by flow cytometry through mammalian display
Structural model of the hCD200:hCD200R1 complex was produced by retrieving individual AlphaFold structure predictions of each protein from UniProt (hCD200: P41217, hCD200R1: Q8TD46) and then a homology model of the human complex based on the x-ray crystal structure of the mouse mCD200:mCD200R1 complex (PDB:4BFI) was generated. Residues on hCD200R1 within 5 Å of hCD200 were selected for alanine mutation as a potential epitope for 23ME–00610. The sequences corresponding to the extracellular and transmembrane domains of WT hCD200R1-iso4-Ref or each alanine mutant were cloned into pRK mammalian expression vectors using a CMV promoter to drive expression. The vectors also contained sequences encoding an N-terminal HA tag. Each hCD200R1 mutant was transfected into Expi293 cells using standard transfection protocols. Transfected cells were harvested and washed with PBS and resuspended in cold fluorescence-activated cell sorting (FACS) buffer (1X PBS + 1% BSA) to obtain a cell concentration of 1 × 106 cells/0.1 mL. Cell suspension was added to each well in a 96-well round-bottom plate and incubated with serially diluted 23ME–00610-Fab for 1 h on ice in the dark. Following incubation, plates were washed 3 times with FACS buffer and incubated with allophycocyanin (APC)-conjugated anti-human Fab antibody (1:100 dilution) for 1 h. A fluorescein isothiocyanate (FITC)-conjugated anti-HA secondary antibody was also added to measure cell surface expression of the hCD200R1 WT and mutants. Cells were analyzed by flow cytometry, gating on fsc (forward scatter) and ssc (side scatter), single cells, on FITC-positive population, and finally on APC-positive cells population, collecting 10,000 events for APC-positive gate.
Measurement of 23ME–00610 binding to cynomolgus CD200R1 ortholog
The vectors containing full-length MfCD200R1 as above for hCD200R1-iso4-Ref were transiently transfected to Expi293. To determine binding of antibodies, cells were washed and resuspended to reach a cell concentration of 10,000 cells/µL. 3X serial dilution of 23ME–00610 started at 500 nM in FACS buffer were added and incubated for 1 h on ice. Cells were then washed and the secondary anti-human IgG antibody (Jackson ImmunoResearch 016-130-084) diluted at 1:100 in FACS buffer was then added to the plate. The plate was again incubated for 1 h on ice in the dark. Another three washes were performed, and a final resuspension of the cells was performed with 150 µL/well of FACS buffer. Plates were then read with the flow cytometer and mean fluorescence intensity (MFI) was plotted.
Binding of 23ME–00611 to peripheral immune cells from cynomolgus monkey
The binding of 23ME–00611 or 23ME–00610 to cynomolgus immune cells was determined by flow cytometry using cynomolgus monkey peripheral blood mononuclear cells (PBMC). Cryopreserved PBMC (BioIVT) were thawed in a 37°C water bath and resuspended in PBMC buffer (PBS +2% heat-inactivated FBS +2 mm EDTA). Cells were centrifuged at 300 × g and washed once with PBMC buffer. Cell suspensions were counted on a Beckman Coulter Vi-Cell instrument to assess viability and resuspended to 2 × 106 cells/mL. A 2X concentration (400 nM) of biotinylated 23ME–00610, 23ME–00611, or isotype control was 5-fold serial diluted, and then an equal volume of the PBMC cell suspension was added to each well, mixed and incubated at 4°C for 20 min. Unbound biotinylated antibody was removed by centrifugation and washed prior to incubation with live/dead viability stain (Thermo Fisher Scientific) for 5 min in the dark at RT. Samples were stained for 20 min at 4°C in the dark with streptavidin-PE (diluted 1:300). Cells were washed twice and collected by centrifugation and resuspended in PBMC buffer before acquiring data on a Beckman Coulter Cytoflex LX system. Single-color stained UltraComp eBeads™ and ArC™ Amine Reactive Compensation Beads were used to perform compensation at the time of acquisition. FlowJo software analyzed the potency of binding of 23ME–00611 to live PBMC. Binding of 23ME–00611 was evaluated using Prism software 3-parameter, log10[23ME-00611] versus response and non-linear curve fit. Streptavidin-PE MFI was plotted.
Fab displaying phage display libraries generation, selection and analysis
Methods for NNK library generation and phage panning selection were performed as described previously.18 Briefly, two phage libraries of 23ME–00610 Fab were constructed for deep mutational scanning by allowing either VH or VL CDR residues to mutate to all 20 amino acids, one residue per CDR at a time using a Kunkel mutagenesis protocol.17 CDR residues selected for mutation are as listed in Figure 5 and Supplementary Figure S3 and as previously described.18 A group of degenerate oligonucleotides carrying NNK degenerate codon was used for each CDR loop with each oligonucleotide mutating a single CDR residue with NNK, which allowed randomization to 20 amino acids. The libraries thus were to allow up to three mutations for each variant, one in each of the three CDRs of a VH or VL CDR library. Mutagenized DNA was electroporated into Escherichia coli XL1-Blue cells yielding 2–4 × 109 transformants, which should cover the designed degeneracy of each library (106-107) well, as described in Supplementary Table S5. Libraries were panned against hCD200R1 ECD coated on ELISA plate and then against biotinylated hCD200R1 in solution panning conditions. The selection stringency was increased in the successive solution panning rounds by incubating the phage libraries with decreasing antigen concentrations (Supplementary Table S5). The affinity-based phage library selection strategies are as previously described.34 The same protocol is used for MfCD200R1 panning.
The presorted libraries and the panning tracks that showed significant enrichment of hCD200R1 binders were subjected to deep sequencing for analysis. After isolating the phagemid double-stranded DNA from the selected rounds, VH and VL segments from each sample were amplified using Illumina 16s library preparation protocol (Illumina). Sequencing adapters and dual‐index barcodes were then added to the amplicons using Illumina Nextera XT Index Kit (Illumina). In preparation for sequencing on Illumina MiSeq, adapter-ligated amplicons were subjected to standard Illumina library denaturing and sample loading protocol using MiSeq Reagent Kit v3 (600 cycles) (Illumina). Paired-end sequencing was performed to cover the entire length of the amplicon with an insert size of 200 bp to 300 bp.
Obtained sequencing data were parsed and analyzed by in-house software. Quality control (QC) is performed on identified amplicon sequences, where each CDR sequence is first checked for the correct length. Reads with stop codons, unexpected insertions/deletions, >1 NNK mutation per CDR or non-NNK mutations were filtered out before further analysis. Mutagenesis efficiency and sequencing quality control results of the libraries are detailed in Supplementary Table S5. All mutations at each mutagenized CDR position were accounted for (Supplementary Figure S3). Weighted frequencies of all mutations for each randomized CDR position were generated before and after phage sorting, and the Log2 of the enrichment ratios for all mutations (Log2(ER)) was calculated by dividing the weighted frequency of a given mutation at a given position in the sorted sample by the weighted frequency of the same mutation in the unsorted sample.35
Abbreviations
- CD
cluster of differentiation
- CDR
complementarity determining region
- ECD
extracellular domain
- ELISA
Enzyme-linked immunosorbent assay
- ER
enrichment ratio
- FACS
fluorescence-activated cell sorting
- Fc
c-terminal human IgG fragment crystallizable
- FC1
flow cell 1
- HC
antibody heavy chain
- hCD200R1
human CD200 receptor 1
- HRP
horseradish peroxidase
- IgG1
immunoglobulin G subclass 1 antibody
- LC
antibody light chain
- MfCD200R1
cynomolgus CD200R1
- MFI
mean fluorescence intensity
- PBMC
peripheral blood mononuclear cells
- SPR
surface plasmon resonance
- VH
variable domain of antibody heavy chain
- VL
variable domain of antibody light chain
Supplementary Material
Acknowledgments
This study was funded by 23andMe. We thank Dylan Glatt, Bill Richards and Amy Kimzey for their input and review of the manuscript. We also thank past and present 23andMe Therapeutics scientists who contributed to P006, especially Chingwei V. Lee and Patrick Koenig. In addition, we thank Rony Nehme from Proteros for technical support. Finally, 23andMe is grateful to the millions of participants who consented to using their genetic and health survey data for research.
Funding Statement
This work was funded by 23andMe.
Disclosure statement
AD, LL, GF, YH: Employees of 23andMe. OG: Employees of Proteros Biostructures. BC, CM, AJH, and SJB were employees of 23andMe at the time this work was performed.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2024.2410316.
References
- 1.Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, Song Y, Jenmalm M, Gorman D, McClanahan T, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171(6):3034–12. doi: 10.4049/jimmunol.171.6.3034. [DOI] [PubMed] [Google Scholar]
- 2.Hatherley D, Lea SM, Johnson S, Barclay AN.. Structures of CD200/CD200 receptor family and implications for topology, regulation, and evolution. Structure. 2013;21(5):820–832. doi: 10.1016/j.str.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foster-Cuevas M, Wright GJ, Puklavec MJ, Brown MH, Barclay AN. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J Virol. 2004;78(14):7667–7676. doi: 10.1128/JVI.78.14.7667-7676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang S, Cherwinski H, Sedgwick JD, Phillips JH. Molecular mechanisms of CD200 inhibition of mast cell activation. J Immunol. 2004;173(11):6786–6793. doi: 10.4049/jimmunol.173.11.6786. [DOI] [PubMed] [Google Scholar]
- 5.McWhirter JR, Kretz-Rommel A, Saven A, Maruyama T, Potter KN, Mockridge CI, Ravey EP, Qin F, Bowdish KS. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc Natl Acad Sci USA. 2006;103(4):1041–1046. doi: 10.1073/pnas.0510081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, Frederickson S, Maruyama T, Wild MA, Nolan M-J, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol. 2007;178(9):5595–5605. doi: 10.4049/jimmunol.178.9.5595. [DOI] [PubMed] [Google Scholar]
- 7.Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat Immunol. 2008;9(9):1074–1083. doi: 10.1038/ni.1637. [DOI] [PubMed] [Google Scholar]
- 8.Gorczynski RM, Chen Z, Diao J, Khatri I, Wong K, Yu K, Behnke J. Breast cancer cell CD200 expression regulates immune response to EMT6 tumor cells in mice. Breast Cancer Res Treat. 2010;123(2):405–415. doi: 10.1007/s10549-009-0667-8. [DOI] [PubMed] [Google Scholar]
- 9.Mukhopadhyay S, Pluddemann A, Hoe JC, Williams KJ, Varin A, Makepeace K, Aknin M-L, Bowdish DME, Smale ST, Barclay AN, et al. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host & Microbe. 2010;8(3):236–247. doi: 10.1016/j.chom.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Akkaya M, Aknin ML, Akkaya B, Barclay AN, Thomas PG. Dissection of agonistic and blocking effects of CD200 receptor antibodies. PLOS ONE. 2013;8(5):e63325. doi: 10.1371/journal.pone.0063325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salek-Ardakani S, Bell T, Jagger CP, Snelgrove RJ, Hussell T. CD200R1 regulates eosinophilia during pulmonary fungal infection in mice. Eur J Immunol. 2019;49(9):1380–1390. doi: 10.1002/eji.201847861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misstear K, Chanas SA, Rezaee SA. Suppression of antigen-specific T cell responses by the Kaposi’s sarcoma-associated herpesvirus viral OX2 protein and its cellular orthologue, CD200. J Virol. 2012;86(11):6246–6257. doi: 10.1128/JVI.07168-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayakawa K, Wang X, Lo EH. CD200 increases alternatively activated macrophages through cAMP-response element binding protein – C/EBP-beta signaling. J Neurochem. 2016;136(5):900–906. doi: 10.1111/jnc.13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenaux J, Fang X, Huang YM, Melero C, Bonnans C, Lowe EL, Palumbo T, Lay C, Yi Z, Zhou A, et al. 23ME-00610, a genetically informed, first-in-class antibody targeting CD200R1 to enhance antitumor T cell function. Oncoimmunol. 2023. June 5;12(1):2217737. doi: 10.1080/2162402X.2023.2217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reis PBPS, Barletta GP, Gagliardi L, Fortuna S, Soler MA, Rocchia W. Antibody-antigen binding interface analysis in the big data era. Front Mol Biosci. 2022. Jul 14;9:945808. doi: 10.3389/fmolb.2022.945808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244(4908):1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 17.Huang R, Fang P, Kay BK. Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods. 2012;58(1):10–17. doi: 10.1016/j.ymeth.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koenig P, Lee CV, Sanowar S, Wu P, Stinson J, Harris SF, Fuh G. Deep sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular degeneration. J Biol Chem. 2015;290(36):21773–21786. doi: 10.1074/jbc.M115.662783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koenig P, Lee CV, Walters BT, Janakiraman V, Stinson J, Patapoff TW, Fuh G. Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc Natl Acad Sci U S A. 2017;114(4):E486–E495. doi: 10.1073/pnas.1613231114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tien MZ, Meyer AG, Sydykova DK, Spielman SJ, Wilke CO, Porollo A. Maximum allowed solvent accessibilites of residues in proteins. PLOS ONE. 2013;8(11):e80635. doi: 10.1371/journal.pone.0080635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitternacht S. FreeSASA: an open source C library for solvent accessible surface area calculations. F1000Research. 2016;5:189. doi: 10.12688/f1000research.7931.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabat EA, Wu T, Bilofsky H. (U.S.) NI of H. Sequences of immunoglobulin chains: tabulation analysis of amino acid sequences of precursors, V-regions, C-regions, J-Chain BP-Microglobulins, 1979. Department Of Health, Educ, Welf, Public Health Service, Natl Institutes Of Health. 1979. [Google Scholar]
- 23.Kabat EA, Wu T, Foeller C, Perry HM, Gottesman KS. Sequences of proteins of immunological interest. Diane Publishing Company; 1992. [Google Scholar]
- 24.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196(4):901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 25.Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37(4):443–10. doi: 10.1016/j.ccell.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minikel EV, Painter JL, Dong CC, Nelson MR. Refining the impact of genetic evidence on clinical success. Nature. 2024. May;629(8012):624–629. doi: 10.1038/s41586-024-07316-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67(4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sparta KM, Krug M, Heinemann U, Mueller U, Weiss MS. Xdsapp2.0. J Appl Crystallogr. 2016;49(3):1085–1092. doi: 10.1107/S1600576716004416. [DOI] [Google Scholar]
- 30.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(12):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Chen VB, Wedell JR, Wenger RK, Ulrich EL, Markley JL. MolProbity for the masses–of data. J Biomol NMR. 2015;63(1):77–83. doi: 10.1007/s10858-015-9969-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bostrom J, Lee CV, Haber L, Fuh G. Improving antibody binding affinity and specificity for therapeutic development. Methods Mol Biol. 2009;525:353–376. xiii. [DOI] [PubMed] [Google Scholar]
- 35.Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S. High resolution mapping of protein sequence–function relationships. Nat Methods. 2010;7(9):741–746; 10.1038/nmeth.1492. PMID:20711194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.