

Abstract
Riboswitches regulate gene expression by modulating their structure upon metabolite binding. These RNA orchestrate several layers of regulation to achieve genetic control. Although Escherichia coli riboswitches modulate translation initiation, several cases have been reported where riboswitches also modulate mRNA levels. Here, we characterize the regulation mechanisms of the thiamin pyrophosphate (TPP) tbpA riboswitch in E. coli. Our results indicate that the tbpA riboswitch modulates both levels of translation and transcription and that TPP sensing is achieved more efficiently cotranscriptionally than post-transcriptionally. The preference for cotranscriptional binding is also observed when monitoring the TPP-dependent inhibition of translation initiation. Using single-molecule approaches, we observe that the aptamer domain freely fluctuates between two main structures involved in TPP recognition. Our results suggest that translation initiation is controlled through the ligand-dependent stabilization of the riboswitch structure. This study demonstrates that riboswitch cotranscriptional sensing is the primary determinant in controlling translation and mRNA levels.
Subject terms: Structural biology, Single-molecule biophysics
In vivo and in vitro studies of an Escherichia coli riboswitch highlights the role of cotranscriptional metabolite sensing and the mechanism of gene regulation.
Introduction
Riboswitches are highly structured regulatory elements located in mRNA untranslated regions1–8. These non-coding RNA structures regulate gene expression by undergoing structural changes upon the binding of cellular metabolites9. There are more than 55 distinct classes of natural riboswitches that have been discovered to date and even more are predicted to exist3, suggesting that riboswitches are widespread regulatory elements. In general, riboswitches exhibit one highly conserved element, the aptamer domain, that is directly involved in metabolite sensing2,10,11. Due to the large variety of detected metabolites3, aptamer domains show diverse structures and exhibit high affinity toward sensed ligands9. In addition to the aptamer, riboswitches contain the expression platform domain that is modulating the expression of downstream gene(s). The structure of the expression platform is not well conserved and may regulate various processes such as transcription termination, translation initiation, mRNA decay and splicing11.
While riboswitches characterized in Bacillus subtilis primarily control premature transcription termination11,12, there is a greater diversity of regulation mechanisms observed for riboswitches in Escherichia coli. For example, in silico structure prediction and in vivo reporter gene assays indicate that the E. coli thiamin pyrophosphate (TPP)-sensing thiM riboswitch strictly modulates translation initiation13. However, transcriptional fusions containing at least 34 thiM codons revealed a TPP-dependent repression of mRNA levels, consistent with Rho-dependent transcription termination modulated by the efficiency of translation13. Similarly, it was found for the TPP-sensing thiC riboswitch that both translation initiation and Rho-dependent transcription termination are modulated upon metabolite binding14. Importantly, in contrast to thiM, it was determined that the Rho binding site is located within the thiC riboswitch domain, thereby suggesting that the thiC riboswitch encodes the regulatory sequences to control both translation and transcription processes14. Lastly, the E. coli tbpA TPP-sensing riboswitch, aka thiB, is also primarily involved in controlling translation initiation15–17. Although it is less studied than the thiC and thiM riboswitches, the tbpA riboswitch was also proposed to regulate Rho transcription termination13. A recent study reported that the primary regulatory control used by the tbpA riboswitch is at the translational level17, suggesting a similar mechanism than used by the thiM riboswitch.
Several experimental evidence suggest a central role for the cotranscriptional process during riboswitch regulation. For instance, it was demonstrated that the transcriptionally-regulating B. subtilis adenine riboswitch poorly achieves ligand binding in the context of the full-length transcript18,19. However, efficient adenine sensing was obtained when performed cotranscriptionally, i.e., while the native RNA polymerase (RNAP) is synthesizing the riboswitch sequence20. Similar conclusions were obtained for the B. subtilis flavin mononucleotide (FMN)-sensing riboswitch, where cotranscriptional FMN sensing relies on several elements such as transcriptional pause sites and the NusA transcription factor21. Interestingly, the preference for cotranscriptional metabolite sensing is preserved in translationally-controlling riboswitches, such as the E. coli TPP-sensing thiC and tbpA riboswitches14,16,17. In both cases, the presence of pause sites and the formation of nascent riboswitch structures contribute to ensure efficient cotranscriptional TPP sensing. While the regulatory mechanism of the thiC riboswitch has been characterized, less is currently known regarding how cotranscriptional sensing of the tbpA riboswitch is harnessed to modulate the initiation of translation.
Here, using a combination of in vivo and in vitro assays, we show that the tbpA riboswitch exerts a control at the translational level. Our data further indicate that the tbpA mRNA levels are also decreased in the presence of TPP, consistent with Rho-dependent transcription termination13. In agreement with tbpA controlling translation initiation, toeprint assays show that the binding of the 30S ribosomal subunit is prevented upon TPP recognition. Our data revealed that TPP sensing is more efficient when occurring cotranscriptionally than post-transcriptionally and that the speed of transcription is important for ligand sensing, in agreement with previous data16. Lastly, smFRET experiments show that structural dynamics is observed at the level of the aptamer domain, consistent with both the aptamer and expression platform being structurally coordinated in the response to TPP.
Results
The tbpA riboswitch regulates both translation and mRNA levels
The tbpA riboswitch controls the expression of the thiBPQ operon that encodes an ABC transporter system involved in thiamin and TPP transport (Fig. 1a)15. The tbpA riboswitch is predicted to regulate at the level of translation initiation, which is accomplished by selectively sequestering the Shine-Dalgarno (SD) sequence upon TPP binding (Fig. 1b). In the absence of TPP, the structure of the riboswitch folds the anti-P1 stem, which disrupts the aptamer domain and exposes the SD sequence to promote translation initiation (Fig. 1a).
Fig. 1. The regulation mechanism of the E. coli tbpA riboswitch.

a The tbpA riboswitch controls the expression of the tbpA-thiP-thiQ operon that is involved in the transport of thiamin and related metabolites. The riboswitch is located in the 5’ untranslated region and adopts the ON state in the absence of TPP in which the anti-P1 stem is formed, thereby allowing the initiation of translation. However, in the presence of TPP, the OFF state is adopted in which the P1 stem of the aptamer domain is stabilized, therefore repressing translation initiation. The GUG start codon is shown in blue. b Secondary structure of the tbpA riboswitch in the TPP-bound OFF structure. The anti-P1 stem is highlighted in yellow and the P1 stem is circled in blue. The Shine-Dalgarno and GUG start codon are shown in blue. c Schematics depicting the constructs used for ß-galactosidase assays. All fusions contain the natural promoter. The “- fusion” represents the construct in which the riboswitch domain is not present. The translational fusion (TrL) contains 11 codons of tbpA and is directly fused to the lacZ reporter. The transcriptional fusion (TrX) contains an additional Shine-Dalgarno (SD) sequence that allows the translation of the lacZ reporter without being affected by the structure of the riboswitch. d ß-galactosidase assays for the promoter-only (-), translational (TrL) and transcriptional (TrX) fusions. The average values of three independent experiments with standard deviations are shown. ß-galactosidase assays for constructs expressed from an arabinose-inducible promoter (pBAD). Gene expression was assessed for the wild-type (WT), G35C, G121C and U130A constructs in the context of both the translational (TrL) (e) and transcriptional (TrX) (f) fusions. The average values of three independent experiments with standard deviations are shown.
We first investigated the tbpA regulatory mechanism using lacZ reporter gene assays. To determine whether the expression is regulated at the promoter level, we engineered a fusion in which lacZ was directly fused at the endogenous promoter (Fig. 1c). When growing cells in a minimum media without and with 1 mM TPP, no regulatory effect was observed on the activity of the promoter (Fig. 1d), suggesting that the latter is not regulated by TPP levels. We next engineered a translational construct in which lacZ was fused after the 12th codon of tbpA (Fig. 1c). When using this construct, we observed that the expression of tbpA was reduced by ~6.3-fold compared to the promoter-only construct (Fig. 1d). In the presence of TPP, it was observed that lacZ expression was decreased by ~2.3-fold (Fig. 1d), consistent with a riboswitch regulation mechanism. To further investigate the mechanism, we engineered a transcriptional construct in which an additional SD sequence was located immediately upstream of lacZ, thereby making lacZ translation efficiency independent of the riboswitch structure (Fig. 1c). The ß-galactosidase activity was repressed by ~1.6-fold in the presence of TPP in the context of the transcriptional fusion (Fig. 1d). Together, the reporter gene data suggest that TPP sensing by the tbpA riboswitch results in the modulation of both levels of translation and transcription.
We next prepared additional constructs to confirm that the regulation is obtained through riboswitch sensing. Given the low expression of the translational and transcriptional fusions, the new constructs contained an arabinose-inducible promoter (pBAD), as previously used to study the lysC, thiM and thiC riboswitches13,14,22. As expected, when using the pBAD-driven WT constructs, the ß-galactosidase activity was substantially increased (~10-fold) for both translational and transcriptional fusions (Fig. 1e, f). When these constructs were assessed in the presence of TPP, the expression of lacZ was decreased by ~3.2-fold and ~3.9-fold for the translational and transcriptional constructs (Fig. 1e, f), respectively. To ensure that metabolite binding was required for genetic regulation, we introduced a single-point mutation in the TPP binding site (G35C) that is expected to prevent ligand binding23–25. Similar levels of lacZ expression were obtained with and without TPP for both G35C translational and transcriptional constructs (Fig. 1e, f), consistent with the inability of the riboswitch mutant to regulate gene expression.
Given that the expression of translational fusions is affected by both translational and transcriptional levels, it is difficult to conclude whether the regulatory effects obtained with the translational fusions (Fig. 1e) are primarily due to translational, transcriptional or combined effects. To investigate to which extent the tbpA riboswitch rely on translational control to modulate gene expression, we next prepared two different mutants. While the first mutation corresponds to a G121C substitution disrupting the SD sequence, the second one (U130A) transforms the GUG start codon for a GAG sequence, making it translationally inactive. In the context of translational fusions, the SD mutant (G121C) yielded a low lacZ expression that was not modulated by the presence of TPP (Fig. 1e). The expression level of the GUG mutant (U130A) was barely detectable, as expected from the inability to initiate translation. In the case of transcriptional fusions, a similarly low level of expression was obtained for the SD mutant when compared to the translational construct (Fig. 1f). However, in contrast to the translational fusion, the GUG mutant resulted in a detectable expression that was repressed by a factor of ~2.4-fold in the presence of TPP (Fig. 1f). These results suggest that tbpA mRNA levels can be modulated even in the absence of efficient translational control, similarly to the lysC riboswitch22.
TPP binding inhibits translation initiation
Since TPP binding to the tbpA riboswitch is expected to modulate SD accessibility, we used toeprint assays to determine whether TPP binding inhibits the recruitment of the 30S ribosomal subunit (Fig. 2a). In these assays, nascent tbpA transcripts were produced using the E. coli RNAP and the binding of the 30S subunit was assessed using primer extension analyses. In the presence of tRNA-fMet and 30S subunit, primer extension assays of the tbpA RNA yielded a reversed cDNA product at position C144 (Fig. 2b and Supplementary Fig. 1). This toeprint position agrees well with previous data showing that a P-site tRNA in E. coli ribosomes produces a toeprint at +15/16 (where the +1 position is the first nucleotide of the P-site codon26). However, the toeprint efficiency was found to decrease by ~2.2-fold in the presence of TPP (Fig. 2b), consistent with TPP binding inhibiting 30S the recruitment of the 30S subunit. No efficient toeprint was observed in the absence of the 30S subunit or tRNA-fMet (Fig. 2b), in agreement with the toeprint requiring a functional initiation complex.
Fig. 2. The tbpA riboswitch modulates translation initiation through cotranscriptional TPP sensing.
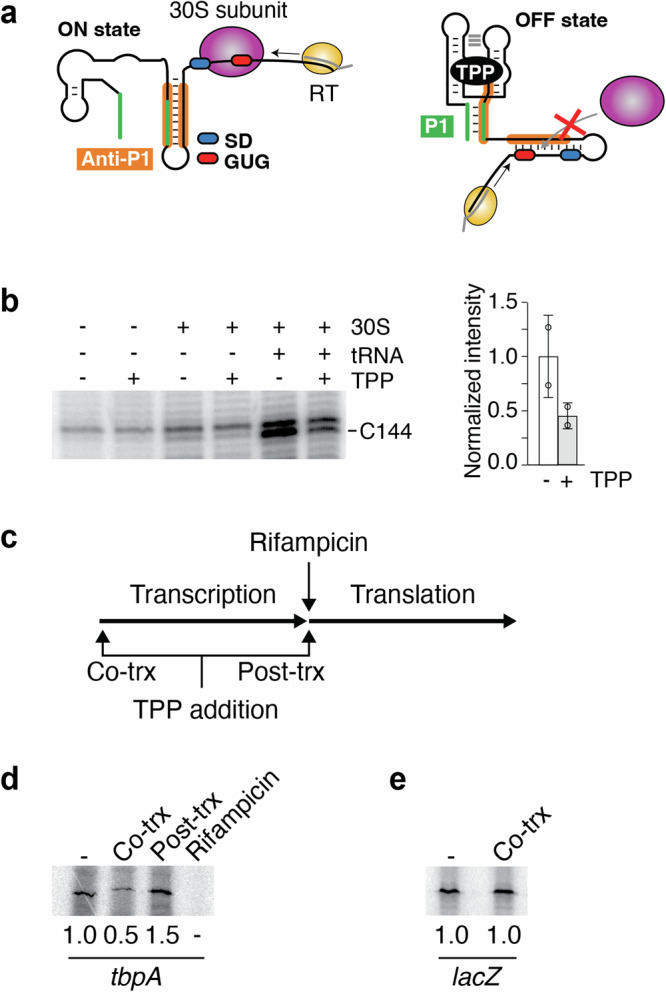
a Schematics representing the toeprint assays. Left, in the absence of TPP, the formation of the ON state allows the binding of the 30S ribosomal subunit. In such a case, the reverse transcriptase (RT) is expected to stop elongating upon reaching the bound 30S subunit. The Shine-Dalgarno (SD) and the GUG start codon are shown in blue and red, respectively. The anti-P1 stem is shown in orange. The DNA oligo used to produce the cDNA is shown in gray. Right, in the presence of TPP, the formation of the OFF state prevents the binding of the 30S subunit, thereby not producing a toeprint at the ribosome binding site. The P1 stem is shown in green. b Left, toeprint assays performed on the tbpA transcript as a function of the 30S subunit, tRNA-fMet and TPP. The toeprint at position C144 is preferentially obtained with the 30S subunit and tRNA-fMet, but is decreased upon TPP binding. Right, quantification of the toeprint efficiency. The experiments have been performed three times and the average and the standard deviations are shown. c Experimental assays monitoring TPP binding using in vitro transcription-translation assays. In these assays, transcription is first allowed to proceed until the addition of rifampicin. The second step involves the addition of amino acids to permit translation. The effect of TPP on the translation efficiency is assessed by adding it during the first (cotranscriptional) or second (post-transcriptional) step. d Transcription-translation assays assessing TPP binding to the riboswitch. Reactions were achieved in the absence (-) or presence of TPP added either cotranscriptionally (Co-trx) or post-transcriptionally (Post-trx) as indicated in c. The ratio of repression is shown below the gels. e Control experiments for transcription-translation assays were performed with lacZ. No translation repression was obtained when adding TPP cotranscriptionally. The ratio of repression is indicated below the gels.
The tbpA riboswitch senses TPP cotranscriptionally
To investigate in more detail the regulation of translation initiation, we next used in vitro transcription-translation assays14. In these assays, the first step consists in initiating transcription reactions by adding the E. coli RNAP (Fig. 2c). In the second step, transcription reactions are stopped by adding rifampicin and translation reactions are initiated by adding amino acids (Fig. 2c). As a result, these assays effectively permit to uncouple transcription and translation and therefore to determine at which level(s) TPP sensing is achieved. We first assessed the riboswitch regulatory activity by adding TPP during the transcription step, thus allowing cotranscriptional TPP sensing. In such conditions, the expression of ThiC was repressed by ~2-fold (Fig. 2d). We next repeated the experiments by adding TPP post-transcriptionally, i.e., during the translation step. In this case, although ThiC expression was increased by ~1.5-fold, suggesting that the presence of TPP may improve translation efficiency, no repression was detected (Fig. 2d). Control experiments showed that when rifampicin was added at the beginning of transcription reactions, no ThiC product was observed (Fig. 2d), consistent with rifampicin efficiently blocking transcription activity. Furthermore, no TPP-dependent modulation was detected when using lacZ (Fig. 2e), consistent with the expression being controlled by the riboswitch. Together, these results indicate that the tbpA riboswitch directly modulates the efficiency of translation initiation via the cotranscriptional sensing of TPP.
To investigate in more detail the cotranscriptional metabolite sensing by the riboswitch, we studied the kinetics of TPP binding to the tbpA riboswitch using RNase H probing assays13,14,16,27. In these experiments, TPP was added either during transcription (cotranscriptionally) or after the addition of heparin (post-transcriptionally)14. Control experiments showed that the probe binding to the aptamer can lead to RNase H cleavage in the absence of TPP (Fig. 3a, Ctrl), indicating that this region is accessible in the ligand-free conformation. However, when TPP was added cotranscriptionally, protection from RNase H cleavage was observed at early time points (15 s and 30 s) (Fig. 3a, Co-trX), suggesting efficient TPP binding. In contrast, when TPP was added post-transcriptionally, stronger RNase H cleavages were detected at all time points (Fig. 3a, Post-trX), in agreement with TPP sensing being less efficient. A fitting analysis revealed a fast-binding rate of 0.17 ± 0.01 s−1 and a slower-binding rate of 0.033 ± 0.001 s−1 for cotranscriptional and post-transcriptional binding assays, respectively (Fig. 3b). These data suggest that TPP cotranscriptional sensing is performed ~5.7-fold more efficiently than post-transcriptionally, similarly to what reported for the thiC riboswitch14.
Fig. 3. TPP sensing is preferentially achieved cotranscriptionally by the tbpA riboswitch.

a RNase H probing experiments monitoring TPP binding when performed cotranscriptionally (Co-trx) or post-transcriptionally (Post-trx). Control reactions (Ctrl) done in the presence of RNase H (RH) show a cleaved product (P) and the uncleaved full-length (FL) transcript without TPP. Cotranscriptional binding was determined by adding TPP before transcription initiation and performing RNase H cleavage during transcription. Post-transcriptional measurements were achieved by performing transcription without TPP, which was followed by the addition of rifampicin, TPP and RNase H cleavage assays. b Quantifications of cotranscriptional and post-transcriptional binding assays. Experiments were fitted to a single-exponential assays and the values of the calculated rates are indicated. c Kswitch measurements for the wild-type tbpA riboswitch done in the presence of increasing TPP concentrations. The full-length (FL) and the cleave product (P) are shown on the left of the gel. d Quantification of the Kswitch experiments done when using 50 µM or 500 µM NTP. The Kswitch value is increasing upon decreasing the NTP concentrations. The average and the standard deviations are shown for each data point.
The rate of transcription is important for TPP sensing
Given that our data suggest that the tbpA riboswitch preferentially binds to TPP cotranscriptionally to regulate translation (Figs. 2d and 3a), we next examined the importance of the rate of transcription for metabolite sensing14,16,20,21. Importantly, due to the presence of three different pause sites located at positions 104, 117 and 13616, it is hypothesized that such pause sites are important for TPP sensing by allowing more time for cotranscriptional sensing. To study the cotranscriptional binding, we transcribed the tbpA riboswitch using various NTP concentrations (500 µM and 50 µM). We reasoned that transcribing at a slower speed would allow more time for TPP binding, thereby decreasing the required TPP concentrations to induce a riboswitch conformational change. When performing RNase H assays as a function of TPP concentration, we found that the cleavage efficiency of RNase H targeting the SD sequence progressively decreased when transcribing with 500 µM NTP (Fig. 3c). Fitting analysis of the experimental data yielded a half TPP concentration (Kswitch) value of 604 ± 110 nM for the structural change (Fig. 3d). When the experiments were performed by using a lower NTP concentration (50 µM), the calculated Kswitch value was 265 ± 63 nM (Fig. 3d). These results indicate that a slower rate of transcription leads to a ~2.3-fold reduction in the required TPP concentration to trigger the riboswitch conformational change, consistent with TPP sensing being performed cotranscriptionally. These data are in agreement with our previous study of the tbpA riboswitch where the rate of RNAP elongation prior to reaching the pause site 136 modulates TPP sensing16.
smFRET analysis of the tbpA aptamer
To determine how the structural dynamics of the riboswitch may be used to control the accessibility to the SD region, we employed single-molecule Förster resonance energy transfer (smFRET) to monitor the TPP-induced conformational changes of the aptamer domain28–30. Fluorescent donor (Cy3) and acceptor (Cy5) dyes were introduced in the tbpA aptamer to follow the RNA conformational changes upon TPP sensing16. While the Cy3 dye is located at the 5’ extremity of the P1 stem, the Cy5 dye is located at position U14 that is at the interface of the P2 and P3 stems. Because the P3-L5 tertiary interaction is formed in the presence of TPP23–25, we reasoned that the TPP-dependent structural change should be detected by the smFRET vector defined by positions 1 and 14. To obtain the Cy3-Cy5 dual-labeled tbpA aptamer, we relied on a strategy in which a 40-nt 5’ synthetic strand containing both Cy3 and Cy5 was ligated to a 67-nt 3’ transcript strand (Fig. 4a)18. Control experiments showed that both 5’ and 3’ RNA molecules were efficiently ligated by the T4 RNA ligase (Fig. 4b), thus making the complete tbpA aptamer.
Fig. 4. Single-molecule FRET studies of semi-synthetic tbpA aptamers.

a Schematic representing the procedure to obtain the semi-synthetic tbpA aptamer. A 5’ synthetic RNA strand contains Cy3 and Cy5 dyes at the 5’ extremity and position 14, respectively. The complete aptamer is reconstituted by ligating the 5’ strand and a 3’ T7 RNAP-transcribed strand. The aptamer is attached to a PEG slide using a sequence that is hybridized to a DNA anchor containing a biotin. b Ligation reactions performed in the presence of the 5’ and 3’ RNA strands. While the 3’ strand corresponds to the 67 nt product, the ligated product of both RNA molecules corresponds to the 107 nt product. c smFRET histograms of the semi-synthetic tbpA aptamer obtained in the absence and presence of 1 mM TPP. The folded (F) and unfolded (U) states are indicated. Histograms obtained without and with TPP were built using 264 molecules and 323 molecules, respectively. d smFRET time trajectories obtained in the absence (Left) and presence (Right) of TPP. The anti-correlated Cy3 donor and Cy5 acceptor emission intensities are shown with the resulting FRET trace. Photobleaching events are shown by an asterisk. e smFRET contour plots obtained in the absence (Left) and presence (Right) of TPP. Selected traces have been accumulated for the first 50 s. Histograms representing the percentual occupancy of each state are shown to the right. Contour plots obtained without and with TPP were built using 48 molecules and 51 molecules, respectively.
To perform smFRET assays, we used a biotinylated DNA anchor that hybridized to the 3’ region of the Cy3-Cy5 dual-labeled aptamer allowing to immobilize the molecules on a biotin-functionalized polyethylene glycol surface (Fig. 4a). Single-molecule imaging was performed using wide-field total internal reflection fluorescence microscopy (TIRFM)31. In the absence of TPP, the smFRET analysis of the reconstituted tbpA aptamer revealed a high fraction of molecules (86%) adopting a high-FRET conformation (EFRET ~ 0.75) (Fig. 4c), which corresponds to the previously characterized unfolded (U) state16. The aptamer was also found to fold into a minor population (14%) exhibiting a low-FRET conformation (EFRET ~ 0.3) that is similar to the TPP-bound folded structure (F state) (Fig. 4c)16. In the presence of 1 mM TPP, the proportion of the F state significantly increased (48%), consistent with the formation of a TPP-tbpA aptamer complex (Fig. 4c). The analysis of time traces using hidden Markov modeling without TPP showed long-lived U states (Fig. 4d). Furthermore, a minority of traces revealed that molecules adopted the F state over long period of times or fluctuated between the U and F states (Supplementary Fig. 2a). When the experiments were repeated with TPP, long-lived F states were observed (Fig. 4d) that showed fluctuations between both U and F states (Supplementary Fig. 2b). These results suggest that the U and F conformers show relatively low dynamics during the analyzed timeframe. The distribution of U and F states was obtained by accumulating the FRET time traces from molecules across a time window of ~50 s. While the contour plot obtained in the absence of TPP shows both U (72%) and F (28%) states (Fig. 4e), the contour plot calculated with TPP reveals a significant increase of the F state (46%) across time (Fig. 4e), reflecting the dynamic equilibrium observed in individual traces (Supplementary Fig. 2).
The presence of a substantial fraction (52%) of the unfolded state obtained with the semi-synthetic aptamer in the presence of TPP (Fig. 4c) prompted us to study tbpA structural dynamics using nascent transcripts. Due to the inability of the E. coli RNAP to incorporate fluorescent nucleotides such as Cy3 or Cy516,32, we recently developed an approach to prepare dual-labeled Cy3-Cy5 nascent transcripts obtained with the E. coli RNAP16. The introduction of Cy3 and Cy5 dyes in transcripts is allowed by initiating transcription with a Cy3-labeled trinucleotide and the site-specific incorporation of an azido-uridine analog. The latter permits the subsequent coupling to a Cy5-alkyne through a click reaction, namely strain-promoted azide-alkyne cycloaddition (SPAAC)33. Similarly to the semi-synthetic construct, the aptamer contained a sequence at the 3’ extremity that allowed the hybridization of a DNA anchor to immobilize transcripts on a quartz slide (Fig. 5a). Single-molecule imaging showed that Cy3-Cy5 dual-labeled nascent aptamers adopted in a high proportion the U state (79%) in the absence of TPP (Fig. 5b). However, when the experiments were done with 1 mM TPP, the aptamers mostly folded into the F state (64%) (Fig. 5b), which is in contrast to the semi-synthetic construct (Fig. 4c). These results suggest that the use of nascent transcripts allows to obtain better metabolite-dependent folding efficiency. When analyzing individual time traces recorded in the absence of TPP, long-lived U states were observed (Fig. 5c) that were also found to transit to the F state (Supplementary Fig. 3a). In contrast, when doing the experiments with 1 mM TPP, nascent transcripts were found to transit more efficiently to the F state (Fig. 5c) and to exhibit higher dynamics between the U and F states (Supplementary Fig. 3b). The contour plots obtained for a ~50 s timeframe showed that while nascent transcripts remain relatively static in the U state without TPP, they exhibit a larger degree of the F state (66%) in the presence of TPP (Fig. 5d). Together, these results indicate that nascent tbpA aptamers show a higher conversion of the U to F state in the presence of TPP when compared to the renatured construct.
Fig. 5. Single-molecule FRET studies of nascent tbpA aptamers obtained by stepwise transcription.

a Schematic representing the nascent RNA construct. The nascent RNA has been produced using stepwise transcription reactions and contains a 3’ sequence allowing to hybridize to a DNA anchor coupled to a biotin. b smFRET histograms of the nascent tbpA aptamer obtained in the absence and presence of 1 mM TPP. The folded (F) and unfolded (U) states are indicated. Histograms obtained without and with TPP were built using 210 molecules and 398 molecules, respectively. c smFRET time trajectories obtained in the absence (Left) and presence (Right) of TPP. The anti-correlated Cy3 donor and Cy5 acceptor emission intensities are shown with the resulting FRET trace. Photobleaching events are shown by an asterisk. d smFRET contour plots obtained in the absence (Left) and presence (Right) of TPP. Selected traces have been accumulated for the first 50 s. Histograms representing the percentual occupancy of each state are shown to the right. Contour plots obtained without and with TPP were built using 108 molecules and 117 molecules, respectively.
Discussion
Our study suggests that the E. coli TPP-sensing tbpA riboswitch regulates gene expression both by modulating translation initiation and mRNA levels, similarly to other E. coli riboswitches sensing TPP (thiC and thiM)13,14, lysine (lysC)22,34 and FMN (ribB)35. Importantly, the secondary structure of the tbpA riboswitch is very similar to that of the thiM riboswitch, suggesting that a related regulation mechanism is expected. However, in contrast to the thiM riboswitch in which 34 codons are necessary to downregulate mRNA levels upon TPP sensing13, our data showed that only 12 codons are sufficient to obtain significant regulation (Fig. 1f). These results indicate that a smaller portion of tbpA 5’ UTR is required to modulate mRNA levels compared to thiM. Thus, both tbpA and thiM may rely on part of the coding region to modulate mRNA levels, which is in contrast to thiC and lysC that only require the aptamer domain14,22. Both thiC and lysC riboswitches are larger than tbpA and thiM, suggesting that they embed the structural information to control mRNA levels using Rho or RNase E14,22. More work will be required to understand molecular details of tbpA translational and transcriptional regulatory mechanisms. For instance, it was recently proposed that TPP sensing by the tbpA riboswitch represses gene expression at the translational level, but not at the transcriptional level17. While these experiments ruled out a Rho-dependent transcription termination mechanism using bicyclomycin, it is still possible that mRNA levels could be modulated by other factors, such as RNase E. Furthermore, given that a shorter construct (4 codons) was used in that study, it is conceivable that regulatory elements contained within the downstream 8 codons sequence are missing. If this is the case, the regulation at the RNA level would be triggered because of translation inhibition. Interestingly, our data suggest that the TPP-dependent RNA regulation may be obtained in the absence of active translation elongation (Fig. 1f, TrX construct, GUG mutant). However, since no mRNA regulation was observed when mutating the SD sequence (Fig. 1f, TrX constructs, SD mutant), it indicates that the binding of the ribosome at the SD sequence is required for the mRNA regulation to take place. Thus, although these results indicate that no active translation is needed to control the mRNA levels, they suggest that ribosome binding is still required for the mRNA regulation.
Our toeprint analysis using tbpA nascent transcripts revealed that TPP binding could sterically modulate the access of the 30S ribosomal subunit (Fig. 2b), consistent with the riboswitch regulation mechanism. The in vitro uncoupling of transcription and translation further showed that the riboswitch translation regulation relied on the cotranscriptional TPP sensing (Fig. 2d), similar to the thiC riboswitch14. The preference for ligand sensing during transcription elongation was also observed when monitoring kinetics of RNase H cleavage reactions (Fig. 3a). Using a probe binding to the aptamer domain, the TPP binding rate was found to be ~5.7-fold more efficient when achieved under cotranscriptional conditions than post-transcriptionally (Fig. 3b), a condition similar to the thiC riboswitch14. Furthermore, transcription reactions performed at different elongation rates showed that the Kswitch value decreased by ~2.3-fold when transcription reactions were done lower nucleotide concentrations (Fig. 3d). The preference for TPP binding under cotranscriptional conditions suggest that TPP sensing is performed within a defined transcriptional window, i.e., when the RNAP is transcribing a specific locus of the 5’ UTR. While the upstream boundary for TPP binding should correspond to the formation of the aptamer domain, the downstream boundary is less clear. However, it was previously determined using smFRET assays that transcription complexes located at the pause site 136 are refractory to TPP binding16. These results suggest that at position 136, the formation of the anti-P1 stem most likely occurs (Fig. 1b), which is expected to disrupt the aptamer domain and therefore to perturb ligand binding. Consequently, the formation of the anti-P1 stem is predicted to define the downstream boundary of the TPP sensing window. This situation is similar to the thiC riboswitch for which the formation of the anti-P1 stem is also involved in the formation of a structure precluding TPP sensing14. Since it was recently determined that an intermediate structure embedded within the anti-P1 stem is involved in the cotranscriptional folding of the tbpA riboswitch17, it suggests that different mechanisms of cotranscriptional folding are used by tbpA and thiC riboswitches.
Our single-molecule FRET analysis of the tbpA aptamer was done using two versions of the tbpA riboswitch: semi-synthetic and nascent RNA constructs. The semi-synthetic construct allows to position the Cy3 and Cy5 dyes at any positions during the chemical RNA synthesis. However, due to its inherent nature, it does not allow to study nascent RNA molecules, which are likely representing more biologically relevant structures. The nascent RNA constructs rely on the use of stepwise transcription and click chemistry reaction to introduce a Cy3 dye at the 5’ end of the transcript and a Cy5 dye at an uracil residue. Despite the fundamental differences between these two approaches, both techniques revealed that the tbpA aptamer adopted two FRET states, U and F, and that the F state was favored in the presence of TPP (Figs. 4c and 5b). The main difference observed between both constructs lies in the proportion of F state obtained in the presence of TPP, which is higher for the nascent RNA construct. These results indicate that the use of nascent transcripts allows to ensure better folding efficiency upon metabolite binding. Specifically, the higher efficiency of nascent transcripts is consistent with their folding being adopted cotranscriptionally, which has been shown to be crucial for the adoption of native RNA structures36. In contrast, due to the nature of their preparation, semi-synthetic RNA aptamers are denatured and renatured prior to smFRET assays, thus possibly allowing for partially misfolded structures and hence lower TPP binding efficiency36,37.
Furthermore, our smFRET data show that relatively low structural dynamics of the tbpA aptamer in the absence of TPP for both constructs, which is mostly stabilized in the U state (Figs. 4e and 5d). In the presence of TPP, while a greater proportion of the F state is observed, a relatively low dynamic is occurring between both U and F state (Figs. 4e and 5d). Such a low structural exchange could be due to the absence of the expression platform, which should allow competition between the ON and OFF states14,16. Therefore, these smFRET data are consistent with the tbpA aptamer exhibiting a large structural change in the presence of TPP but in which a low level of dynamics is observed between both FRET states. While more work is required to decipher the molecular mechanism between the U and F conformational states, previous data indicate that higher rates of structural exchanges may be obtained in the context of the full tbpA riboswitch in which the anti-P1 stem could be formed16. Thus, in a cotranscriptional context, it is conceivable that exchange between both states occurs within the transcriptional window in which the anti-P1 stem may partially be completed, most particularly when intermediate structures are formed17.
Overall, our findings are consistent with the tbpA riboswitch performing a dual modulation of gene expression by controlling both translation initiation and mRNA levels, which is similar to other E. coli riboswitches13,14,22,34,35. Such a control of transcriptional levels allows to regulate the expression of several genes embedded within downstream operons, thus permitting to efficiently adjust the production of involved metabolic pathways using additional regulatory mechanisms such as Rho-dependent transcription termination13,35. For example, when sensing TPP, the E. coli tbpA, thiC and thiM riboswitches38 collectively downregulate the mRNA levels of 11 genes involved either in the transport or biosynthesis of TPP. While such an expanded regulatory effect is expected from transcriptionally-regulating riboswitches, such as in B. subtilis, our findings add new data for the tbpA riboswitch supporting the idea that translationally-regulating E. coli riboswitches also modulate whole operon mRNAs upon metabolite sensing. The ability for riboswitches to regulate mRNA levels is possible due to their propensity to sense metabolites cotranscriptionally14,16,17,35,39. The higher efficiency of cotranscriptional sensing for translational riboswitches implies that metabolite recognition is achieved by nascent transcripts, which is supported by our RNase H and smFRET data (Figs. 3d and 5b). Lastly, given that the selective sequestration of the Shine-Dalgarno sequence may be performed post-transcriptionally, such riboswitches may also control outside of the cotranscriptional binding window and benefit from an additional layer of genetic control allowing for fine tuning regulation20. Clearly, the cotranscriptional sensing of cellular metabolites is at the heart of both transcriptional and translational riboswitches and therefore suggests that the transcriptional process is essential for riboswitch regulation.
In conclusion, our work showed that the E. coli tbpA riboswitch may regulate both at the translational and transcriptional levels. Furthermore, the requirement for cotranscriptional TPP sensing emphasizes the presence of a mRNA regulation mechanism that is also observed in other E. coli riboswitches. Our single-molecule data is consistent with nascent transcripts being more efficient to sense TPP, a situation that is consistent with riboswitches performing efficient genetic response during transcription elongation.
Methods
Bacterial strains and oligonucleotides
Strains used in this study are derived from Escherichia coli MG1655. Strain BL21 (DE3) was used for overproduction of RNA polymerase (RNAP) and sigma70 factor. The E. coli RNAP and sigma70 factors were obtained from the Plateforme de purification des protéines (Université de Sherbrooke). The PM1205 strain (Supplementary Table 1) was used to construct the tbpA translational and natural promoter fusions (Supplementary Table 2)22. The PCR strategies to obtain the various constructs are listed in Supplementary Table 3. DNA oligonucleotides used in this study were purchased from Integrated DNA technologies (IDT) and are listed in Supplementary Table 4.
Beta-galactosidase experiments
Kinetic assays for beta-galactosidase experiments were performed as described previously14,22. Briefly, an overnight bacterial culture grown in M63 0.2% glucose minimal medium was diluted to an OD600 of 0.02 in 50 mL of fresh medium. The various fusions used in this study are described in the Supplementary Table 2. For constructs containing the natural promoter, the culture was incubated at 37°C until an OD600 of 0.1 was obtained, and TPP (500 µg/mL) was then added as indicated. For constructs using a pBAD promoter, arabinose (0.1% final concentration) was added after OD600 of 0.1 was reached, which was followed by the addition of TPP (500 µg/mL). In all cases, the specific ß-galactosidase activities were calculated as previously described22,40.
In vitro transcription-translation assay
The transcription-translation assays were performed using the template pLacUV5-tbpA306cd (Supplementary Table 3) and were done as previously reported14. Briefly, reactions were achieved using the PURExpress Kit from New England Biolabs. To verify that TPP does not generally affect transcription-translation efficiency, a DNA template containing the lacUV5 promoter fused to the first 934 codons of lacZ followed by a T7 terminator was used as an internal control (pLacUV5-lacZ). TPP was added at the final concentration of 25 µM and transcription was carried out with 0.5 U of the RNA polymerase holoenzyme from Epicentre. For tbpA uncoupled transcription-translation assays, reactions were initiated by a transcription step of the pLacUV5-tbpA306cd construct at 37°C for 15 min using a transcription solution without amino acids/transfer RNA. Rifampicin (250 µg/mL) was next added to the reaction and incubated for 1 min to prevent re-initiation of transcription. The translation step was initiated by incubating with a solution comprising amino acids and transfer RNA at 37°C for 4 h. TPP (100 µM) was either added during the transcription step (cotranscriptional) or the translation step (post-transcriptional). Reactions were stopped by placing samples on ice for 10 min and next incubated with 4 volumes of acetone 100% at 4 °C for 15 min. Samples were precipitated, resuspended in denaturing buffer, and resolved on SDS-PAGE.
RNase H probing analysis of transcription reactions
DNA templates corresponding to tbpA EC-248 (pLacUV5-tbpA-EC-248) were incubated with sigma70 factor, the RNAP, and the streptavidin at 37 °C for 5 min. Transcription reactions were performed in 20 mM Tris-HCl pH 8.0, 20 mM MgCl2, 20 mM NaCl, 14 mM 2-mercaptoethanol and 0.1 mM EDTA. Elongation complexes were synchronized at position 9 (EC-9) by incorporating 10 µM GUU trinucleotide, 2.5 µM ATP/CTP, [α-32P] UTP and incubating at 37 °C for 5 min. Sample were passed through G50 columns to remove free nucleotides. Samples were added to a reaction mixture composed of TPP and all four nucleotides, and were incubated at 37 °C for 5 min to elongate at position 248. Transcriptions were completed at various TPP concentrations (1 nM to 500 µM) and NTP concentrations (50 µM and 500 µM). After incubating at 37 °C for 5 min, samples were mixed with 200 µM DNA probe (923AC) and incubated for another 5 min. RNase H cleavage assays were initiated by incorporating a solution of RNase H in 5 mM Tris-HCl, pH 8.0, 20 mM MgCl2, 100 mM KCl and 5 µM EDTA and incubated at 37 °C for 5 min. Reactions were stopped by adding an equal volume of stop solution (95% formamide, 20 mM EDTA and 0.4% SDS).
RNase H probing analysis of TPP kinetics
The kinetics of TPP binding (co- or post-transcriptional binding) was essentially performed as previously described14. Transcription reactions using the tbpA EC-136 DNA template (pLacUV5-tbpA-EC-136) were performed in 20 mM Tris-HCl pH 8.0, 20 mM MgCl2, 20 mM NaCl, 14 mM 2-mercaptoethanol and 0.1 mM EDTA. Radioactively labeled transcripts were prepared as described in the section “RNase H probing analysis of transcription reactions”. TPP (10 µM) was either added cotranscriptionally or post-transcriptionally by adding an excess of heparin prior to TPP14. The use of an excess of heparin was shown to prevent RNAP from initiating transcription41, thus allowing to monitor post-transcriptional TPP binding on transcribed RNA molecules. The obtained transcripts were assessed by RNase H cleavage assays by mixing with 200 µM DNA (24477AC) and RNase H for 15 s at various time points (15 s, 45 s, 90 s, 2 min and 3 min) (see section “RNase H probing analysis of transcription reactions” for RNase cleavage assays). The reported errors for the TPP-binding rates are the standard error in the fitting18, which are assumed to be approximated by the standard deviation.
TPP-dependent toeprint assays
Nascent 180 nt tbpA transcripts were prepared by in vitro transcription reactions using the EC-180 DNA template (pLacUV5-tbpA-EC-180) in 20 mM Tris-HCl pH 8.0, 20 mM MgCl2, 20 mM NaCl, 14 mM 2-mercaptoethanol and 0.1 mM EDTA. Reactions were initiated by incubating the DNA template, sigma70 factor and the RNAP at 37 °C for 5 min. Elongation complexes were synchronized at position 9 (EC-9) by incorporating 25 µM Cy3-GUU trinucleotide, 2.5 µM ATP/CTP/UTP and incubating at 37 °C for 5 min. Sample were passed through G50 columns to remove free nucleotides. Where indicated, 500 µM TPP was added followed by the addition of 500 µM NTPs. Reactions were incubated at 37 °C for 10 min and the obtained RNAs were purified using the ThermoFisher GeneJET RNA purification kit. The obtained transcripts were incubated with the radiolabeled oligonucleotide (3374JG) in 50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2 and 10 mM DTT at 37 °C for 5 min. Next, 300 nM of the 30S subunit, 600 nM tRNA-fMet (MP Biomedicals) and 1 mM TPP and incubated at 37 °C for 5 min. Then, 500 µM dNTPs and 10 U of MulV-RT (New England Biolabs) were added and the reverse transcriptase reaction allowed to proceed at 37 °C for 15 min. Reactions were migrated on an 8% denaturing-PAGE for 2 h and products were quantified by using the Quantity One software. The intensity of the toeprint was normalized to a band that did not show any change across conditions (C138).
Sequencing reactions used for the analysis of reverse transcription products were done using RNA produced using T7 RNAP transcription reactions42 using the pT7-tbpA-EC-180 template. Briefly, 100 nM of RNA was incubated with 250 µM ddNTPs in TE 1X buffer (10 mM Tris-HCl and 1 mM EDTA). The radiolabeled oligonucleotide (3374JG) was incubated with the RNA at 65 °C for 3 min and cooled down at room temperature for 5 min. A mix of SS first strand buffer (Invitrogen), 5 mM DTT and 500 µM dNTPs were added to the mixture following the addition of the Superscript II RT enzyme (Invitrogen) and incubated at 52 °C for 10 min.
Preparation of semi-synthetic tbpA RNA for smFRET assays
The Cy3-Cy5 dual-labeled tbpA aptamer was prepared using ligation assays18. Briefly, the aptamer was reconstituted using a synthetic 5’ strand (IDT) (residues 1–38) and a transcribed 3’ strand (residues 39 to 89). The 5’ RNA strand was ordered as a 5’-labeled Cy3 strand containing an azide-modified U at position 14 (2558JG). The 5’ RNA strand was incubated at 37 °C for 1 h with 63 µM µM DBCO-Cy5 (Jean Bioscience) in 20 mM Tris-HCl pH 8.0, 20 mM MgCl2, 20 mM NaCl, 14 mM 2-mercaptoethanol and 0.1 mM EDTA to obtain a Cy3-Cy5 labeled strand, which was purified by denaturing gel electrophoresis and ethanol precipitation. The 3’ strand was produced using T7 RNAP transcription reactions42 using the 3’ tbpA-89 template. The 3’ strand was obtained through self-cleavage of a larger transcript containing a hammerhead ribozyme located upstream of the aptamer RNA sequence. Transcription reactions were resolved using denaturing polyacrylamide gel electrophoresis and the band corresponding to the 3’ strand was excised from the gel and purified using ethanol precipitation18. The obtained 3’ strand was subsequently phosphorylated using T4 Polynucleotide Kinase (NEB) using the manufacturer’s protocol. Next, the Cy3-Cy5 dual-labeled 5’ strand and the 5’-PO4 3’ transcribed strand were annealed by heating a mixture (molar ratio 1:1) to 80 °C in 50 mM Tris-HCl pH 7.5 and 50 mM NaCl and slowly cooling it to room temperature. The mixture was transferred in a tube containing 50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT and 1 mM ATP and T4 RNA ligase 1 (New England Biolabs), which was incubated at 37 °C for 4 h. Full-length ligated RNA molecules were purified by denaturing polyacrylamide gel electrophoresis and ethanol precipitation.
Preparation of nascent tbpA RNA for smFRET assays
The Cy3-Cy5 dual-labeled nascent tbpA aptamers were prepared using native transcription reactions16. Briefly, DNA templates allowing the transcription of the tbpA aptamer + anchoring sequence (pLacUV5-tbpA-88-extBio) were incubated with sigma70 factor and the RNAP at 37 °C for 5 min. Transcription reactions were done in 20 mM Tris-HCl pH 8.0, 20 mM MgCl2, 20 mM NaCl, 14 mM 2-mercaptoethanol and 0.1 mM EDTA. Elongation complexes were synchronized at position 9 (EC-9) by incorporating 25 µM Cy3-GUU trinucleotide, 1.25 µM ATP/CTP/UTP and incubating at 37˚C for 5 min. Sample were passed through G50 columns to remove free nucleotides. Samples were reacted with 2.5 µM CTP/GTP and 2.5 µM azide-modified UTP analog at 37 °C for 5 min to allow formation of EC-17. The samples were next passed through G50 columns to remove free nucleotides. The full-length transcripts were obtained by adding 1 mm NTP at 37 °C for 5 min, thus releasing the RNA from transcription complexes. The incorporation of Cy5 at the U14 azide group was performed by incubating transcripts with 63 µM DBCO-Cy5 in the transcription buffer at 37 °C for 1 h. The transcripts were passed through G50 columns to remove unreacted DBCO-Cy5 dyes. Samples were collected in the transcription buffer and were incubated 5 min with or without TPP before smFRET measurements.
Single-molecule FRET imaging
Single-molecule FRET experiments were performed as previously described16,18. smFRET traces were recorded from immobilized single tbpA aptamers using a prism-type total-internal reflection setup including an inverted microscope (Olympus IX71) coupled to a 532 nm laser (Crystalaser) and a back illuminated Ixon EMCCD camera (Andor). Microscope quartz slides were passivated with a 30:1 mixture of PEG (Laysan Biosciences, USA) and biotinylated PEG. A concentration of 0.2 mg/mL streptavidin was added to the PEG slide to allow immobilization of the tbpA aptamer hybridized with the biotinylated DNA anchor. A concentration of ~250 pM dual-labeled nascent or semi-synthetic tbpA aptamers was added to the slide and attached to the slide using DNA anchors 2433AC or 2559JG, respectively. Data were acquired using a 50 ms integration time on surface-immobilized molecules in the transcription buffer to which was added 2 mM Trolox, 5 mM 3,4-protocatechuic acid (PCA, Sigma) and 100 nM protocatechuate dioxygenase (PCD, Sigma), pH 7.5, which was used to enhance the photostability of the dye. The setup allowed to record Cy3 and Cy5 signals simultaneously16,18. Left and right images were corrected for optical aberrations and inhomogeneous evanescence-wave illumination using custom-built routines using IDL software (Exelis, USA). Subsequently, an IDL mapping algorithm was used to correlate the position of Cy3 and Cy5 signals from the same molecule and a time trace was built for each molecule. To calculate the percentual contribution of the U and F states at each experimental condition, the single-molecule FRET histogram at each condition was fitted to a two-Gaussian distribution. The area under each Gaussian was extracted and its percentual contribution calculated with respect to the total area under the two-Gaussian curve.
Statistics and reproducibility
All the experiments were repeated for two or three times. For reporter gene assays, independent batches of culture were included in each experiment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We thank members of the Lafontaine and Penedo laboratory for discussion. This work was supported by the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Fonds de recherche du Québec en Natures et Technologies (FRQNT). J.C.P. thanks the School of Biology at the University of St Andrews for financial support.
Author contributions
Conceived, designed, and performed experiments: J.P.G, M.G., M.S.-R., A.C., P.T., P.S.-P., A.D., J.M. Analyzed the data: J.P.G, M.G., M.S.-R., A.C., P.T., P.S.-P., A.D., J.M., E.M., J.C.P. and D.A.L. Participated in writing the manuscript: J.P.G, P.S.-P., J.C.P. and D.A.L.
Peer review
Peer review information
Communications Biology thanks Yu Liu, and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data underlying Figs. 1–3 can be found in the Supplementary Data. The non-cropped version of gels are presented in Supplementary Fig. 4 and 5.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-024-07008-5.
References
- 1.Salvail, H. & Breaker, R. R. Riboswitches. Curr. Biol.33, R343–R348 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breaker, R. R. The Biochemical Landscape of Riboswitch Ligands. Biochemistry61, 137–149 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kavita, K. & Breaker, R. R. Discovering riboswitches: the past and the future. Trends Biochem. Sci.48, 119–141 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ariza-Mateos, A., Nuthanakanti, A. & Serganov, A. Riboswitch Mechanisms: New Tricks for an Old Dog. Biochem. Mosc.86, 962–975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards, J. & Belasco, J. G. Riboswitch control of bacterial RNA stability. Mol. Microbiol.116, 361–365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scull, C. E., Dandpat, S. S., Romero, R. A. & Walter, N. G. Transcriptional Riboswitches Integrate Timescales for Bacterial Gene Expression Control. Front. Mol. Biosci.7, 607158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Micura, R. & Höbartner, C. Fundamental studies of functional nucleic acids: aptamers, riboswitches, ribozymes and DNAzymes. Chem. Soc. Rev.49, 7331–7353 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Bédard, A.-S. V., Hien, E. D. M. & Lafontaine, D. A. Riboswitch regulation mechanisms: RNA, metabolites and regulatory proteins. Biochim. Biophys. Acta Gene Regul. Mech.1863, 194501 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Mccown, P. J., Corbino, K. A., Stav, S., Sherlock, M. E. & Breaker, R. R. Riboswitch diversity and distribution. RNA23, 995–1011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garst, A. D., Edwards, A. L. & Batey, R. T. Riboswitches: structures and mechanisms. Cold Spring Harbor Perspect. Biol.3, a003533 (2011). [DOI] [PMC free article] [PubMed]
- 11.Serganov, A. & Nudler, E. A decade of riboswitches. Cell152, 17–24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breaker, R. R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol.4, a003566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastet, L. et al. Translational control and Rho-dependent transcription termination are intimately linked in riboswitch regulation. Nucleic Acids Res.45, 7474–7486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauvier, A. et al. Transcriptional pausing at the translation start site operates as a critical checkpoint for riboswitch regulation. Nat. Commun.8, 13892 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodionov, D. A., Vitreschak, A. G., Mironov, A. A. & Gelfand, M. S. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem.277, 48949–48959 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Chauvier, A. et al. Monitoring RNA dynamics in native transcriptional complexes. Proc. Natl Acad. Sci. USA118, e2106564118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman, K. E., Steans, R., Hertz, L. M. & Lucks, J. B. A transient intermediate RNA structure underlies the regulatory function of the E. coli thiB TPP translational riboswitch. RNA29, 1658–1672 (2023). [DOI] [PMC free article] [PubMed]
- 18.Lemay, J. F., Penedo, J. C., Tremblay, R., Lilley, D. M. J. & Lafontaine, D. A. A. Folding of the Adenine Riboswitch. Chem. Biol.13, 857–868 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Wickiser, J. K., Cheah, M. T., Breaker, R. R. & Crothers, D. M. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry44, 13404–13414 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Lemay, J.-F. et al. Comparative study between transcriptionally- and translationally-acting adenine riboswitches reveals key differences in riboswitch regulatory mechanisms. PLoS Genet7, e1001278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickiser, J. K., Winkler, W. C., Breaker, R. R. & Crothers, D. M. The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol. Cell18, 49–60 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Caron, M.-P. et al. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc. Natl Acad. Sci.109, E3444–E3453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards, T. E. & Ferre-D’Amare, A. R. Crystal Structures of the Thi-Box Riboswitch Bound to Thiamine Pyrophosphate Analogs Reveal Adaptive RNA-Small Molecule Recognition. Structure14, 1459–1468 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Thore, S., Leibundgut, M. & Ban, N. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science312, 1208–1211 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Serganov, A., Polonskaia, A., Phan, A. T., Breaker, R. R. & Patel, D. J. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature441, 1167–1171 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph, S. & Noller, H. F. EF-G-catalyzed translocation of anticodon stem-loop analogs of transfer RNA in the ribosome. EMBO J.17, 3478–3483 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lussier, A., Bastet, L., Chauvier, A. & Lafontaine, D. A. A kissing loop is important for btuB riboswitch ligand sensing and regulatory control. J. Biol. Chem.290, 26739–26751 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.St-Pierre, P., McCluskey, K., Shaw, E., Penedo, J. C. C. & Lafontaine, D. A. A. Fluorescence tools to investigate riboswitch structural dynamics. Biochim. Biophys. Acta Gene Regul. Mech.1839, 1005–1019 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Suddala, K. C. & Walter, N. G. Riboswitch structure and dynamics by smFRET microscopy. Methods Enzymol.549, 343–373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selvin, P. R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol.7, 730–734 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Roy, R., Hohng, S. & Ha, T. A practical guide to single-molecule FRET. Nat. Methods5, 507–516 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vassylyev, D. G. et al. Structural basis for substrate loading in bacterial RNA polymerase. Nature448, 163–168 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Winz, M.-L., Samanta, A., Benzinger, D. & Jäschke, A. Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic Acids Res.40, e78 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh, T. et al. Direct and indirect control of Rho-dependent transcription termination by the Escherichia coli lysC riboswitch. RNA30, 381–391 (2024). [DOI] [PMC free article] [PubMed]
- 35.Hollands, K. et al. Riboswitch control of Rho-dependent transcription termination. Proc. Natl Acad. Sci. USA109, 5376–5381 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan, T. & Sosnick, T. RNA folding during transcription. Annu Rev. Biophys. Biomol. Struct.35, 161–175 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Perdrizet, G. A. 2nd, Artsimovitch, I., Furman, R., Sosnick, T. R. & Pan, T. Transcriptional pausing coordinates folding of the aptamer domain and the expression platform of a riboswitch. Proc. Natl Acad. Sci. USA109, 3323–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler, W., Nahvi, A. & Breaker, R. R. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature419, 952–956 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Chauvier, A. & Walter, N. G. Beyond ligand binding: Single molecule observation reveals how riboswitches integrate multiple signals to balance bacterial gene regulation. Curr. Opin. Struct. Biol.88, 102893 (2024). [DOI] [PubMed] [Google Scholar]
- 40.Bastet, L. et al. Riboswitch and small RNAs modulate btuB translation initiation in Escherichia coli and trigger distinct mRNA regulatory mechanisms. Nucleic Acids Res.52, 5852–5865 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chamberlin, M. J., Nierman, W. C., Wiggs, J. & Neff, N. A quantitative assay for bacterial RNA polymerases. J. Biol. Chem.254, 10061–10069 (1979). [PubMed] [Google Scholar]
- 42.Milligan, J. F., Groebe, D. R., Witherell, G. W. & Uhlenbeck, O. C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res.15, 8783–8798 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data underlying Figs. 1–3 can be found in the Supplementary Data. The non-cropped version of gels are presented in Supplementary Fig. 4 and 5.