

ABSTRACT
This study describes a protocol to assess a novel workflow called Epi-Genomic Newborn Screening (EpiGNs) on 100,000 infants from the state of Victoria, Australia. The workflow uses a first-tier screening approach called methylation-specific quantitative melt analysis (MS-QMA), followed by second and third tier testing including targeted methylation and copy number variation analyzes with droplet digital PCR, EpiTYPER system and low-coverage whole genome sequencing. EpiGNs utilizes only two 3.2 mm newborn blood spot punches to screen for genetic conditions, including fragile X syndrome, Prader-Willi syndrome, Angelman syndrome, Dup15q syndrome and sex chromosome aneuploidies. The program aims to: identify clinically actionable methylation screening thresholds for the first-tier screen and estimate prevalence for the conditions screened.
Keywords: : early detection, epigenomics, EpiGNs, newborn screening, population-wide study
Plain language summary
Article highlights.
Epi-genomic newborn screening (EpiGNs) protocol highlights
Novel Workflow: EpiGNs is a new approach to newborn screening using a combination of blood spot analysis and genetic testing.
Large-Scale Trial: The study will involve 100,000 infants from Victoria, Australia.
Multi-Tier Testing: A first-tier screen methylation-specific quantitative melt analysis (MS-QMA) will be followed by confirmatory tests (real-time PCR, droplet digital PCR, EpiTYPER, whole genome sequencing) for positive cases.
Target conditions: EpiGNs utilizes only two 3.2 mm newborn blood spot punches to screen for various genetic conditions, including Fragile X, Prader-Willi, Angelman, Dup15q syndromes and sex chromosome aneuploidies (Turner, XXY, XXXY, XXXXY and XXYY syndromes).
Clinically actionable thresholds: The study aims to define thresholds for the first-tier methylation screen that lead to further diagnostic or intervention options.
Prevalence estimation: The research will estimate the prevalence of the targeted conditions in the newborn population.
Long-term follow-up: Children identified will be followed for development and healthcare utilization.
Empowering parents: Early detection may provide parents with more informed reproductive choices.
Overall goal: Reduce diagnostic delays, improve early treatment access and empower parents through genetic information and reduce costs for the families and the health system.
1. Introduction
There are over 7000 rare diseases with the average age of diagnosis around 4–5 years [1]. From birth to time of diagnosis, families consult an average of five doctors, receiving an average of three misdiagnoses. This not only adds to the substantial health burden but also places an economic strain on the affected children, their families and healthcare system [2]. Genomic newborn screening for rare diseases holds the potential to reduce diagnostic delay, provide timely family planning advice and allow earlier access to new therapies, potentially altering developmental and health trajectories. However, a key limitation lies in the availability of accurate and affordable genomic workflows that align with current newborn screening program requirements [3,4].
Government-supported newborn bloodspot screening programs in Australia, Europe and the United States routinely screen all infants for over 25 rare diseases. The primary objective is to reduce mortality and morbidity by diagnosing and treating these diseases early in the first year after birth. Over the past four decades, the classic screening criteria outlined by Wilson and Jungner (1968) for selecting conditions have evolved in response to advancements in genomic and epigenomic testing [5]. Applying new genomic and epigenomic technologies, screening now presents an avenue to identify additional rare diseases where early detection and intervention could provide benefits to affected newborns and their families. Further expansion of newborn genomic screening programs awaits confirmation that their benefits at population scale exceed their costs and potential harms [6,7].
Current standard-of-care (SOC) genomic testing approaches are challenging for newborn screening as first-tier tests due to several constraints, particularly for detection of pathogenic events within repetitive regions [8,9] and imprinting disorders [10]. A notable example is the complexity of SOC genomic testing to effectively identify trinucleotide expansion disorders, such as fragile X syndrome (FXS), which is the most prevalent single gene cause of intellectual disability (ID) in males [11,12]. FXS results from FMR1 full mutation (FM) expansion, which comprises 200 or more CGG repeats, and is the leading cause of inherited ID. This expansion leads to the silencing of FMR1 through DNA methylation and may account for 0.6 to 6.5% of cases of developmental delay referral for genetic testing [13,14]. While recent advances in bioinformatics have shown that detection of a proportion of expanded FMR1 alleles is possible by whole genome sequencing (WGS)[15], this is not yet adopted in clinical practice. The common diagnostic testing for children with developmental delay (DD) and ID involves targeted CGG repeat long-range PCR and Southern blot testing. Early detection of FM alleles is important not only for affected infants but also for their mothers, who face a high chance of having additional affected children before a diagnosis is made (on average around age 3 years) [16].
Other limitations include affordability and DNA requirements, which pose a challenge for the use of chromosomal microarray analysis (CMA), WGS and whole exome sequencing (WES), because they necessitate high-quality and unfragmented DNA. The high costs of testing and DNA quantity required (four to ten 3 mm punches per baby) may also make these options undesirable, especially considering the limited blood spot material available after standard testing has been completed. Consent and follow-up costs and related ethical issues are additional concerns. The disclosure of common variants associated with late-onset adult conditions of incomplete penetrance, such as FMR1 premutation (PM) alleles of 55 to 199 CGG repeats (linked to late-onset disorders) identified as part of newborn screening, could be detrimental to families [11,17]. This may negatively impact on the relationship of the mother with the infant with little to no immediate benefit for the infant [17] and considerably increasing the overall costs for the program. These costs may be very difficult to justify from an infra-structure perspective, with a need for pre-test counselling, informed consent and post-test follow-up, especially considering prevalence of FMR1 premutation alleles and intermediate alleles (45 to 54 CGG repeats) is 1 in 120 and 1 in 70 individuals in the general population [11], respectively.
For imprinting disorders, such as Angelman syndrome (AS), for which current antisense oligonucleotides (ASO) clinical trials provide strong rationale for newborn screening, genomic testing will not identify epimutations which can cause this condition without changes to DNA sequence and will struggle to identify a considerable proportion of individuals with maternal uniparental disomy (UPD) [18], largely due to the lack of widespread adoption of suitable bioinformatic analysis tools.
Availability of only one 3 mm punch of NBS material of poor quality, may prevent SOC testing technologies such as CMA, multiplex ligation dependent probe amplification [19] and methylation-specific high-resolution melt [20] for use in newborn screening for chromosome 15 imprinting disorders. These may require DNA extraction with DNA of much higher concentration and quality than that available for newborn screening. In terms of cost, DNA extraction in itself of over $10 per sample. Together with further costs associated with downstream analyses (accounting to hundreds to thousands of dollars per sample) these would be too expensive for newborn screening.
To address some of these challenges we have developed Epi-Genomic Newborn screening (EpiGNs) workflow. EpiGNs first-tier DNA methylation testing uses only one 3 mm NBS punch followed by confirmatory genomic and epigenetic testing on another 3 mm NBS punch, with no requirements for costly DNA extraction using commercial kits. Moreover, sample requirements for first-tier testing are in line with those for spinal muscular atrophy (SMA) and severe combined immunodeficiency (SCID) newborn screening now implemented internationally [21,22,23]. Importantly, EpiGNs first-tier screen uses the same equipment as that used for SMA/SCID screening. This makes EpiGNs workflow a viable option for integration into existing newborn screening programs upon completion of this study.
Here we describe the protocol and rationale of the EpiGNs program [24] to assess the positive predictive values (PPV) of the first-tier methylation screening test utilized and prevalence estimates for the conditions screened in 100,000 Victorian infants. Additionally, the program uses repeated phenotypic measures from the GenV, Australia's largest study of children and parents [25], to define clinical trajectories of children identified as being affected with each syndrome and compare these to the entire GenV population sample.
1.1. Conditions included in the EpiGNs program
EpiGNs was designed to meet the cost and newborn blood spot material requirements in line with standard of care newborn screening to screen for nine rare genetic conditions including their mosaic and non-mosaic forms: FXS, Prader Willi (PWS), AS, Turner (TS), Dup15q, XXY, XXXY, XXXXY and XXYY syndromes (Supplementary Table S1). These conditions are not included in the current NBS program. Most infants with these conditions do not receive a diagnosis within the first year after birth, which can lead to a ‘diagnostic odyssey’ and increased medical costs and stress for families [10,26,27]. Furthermore, PWS and TS have treatments available from soon after birth [28,29,30], and AS has a molecular therapy currently undergoing clinical trials [31]. New therapies are also being specifically tailored for FXS and are undergoing late-stage clinical trials [32]. These interventions, along with existing treatment options, have the potential to mitigate the impact of severe co-morbidities such as ID, autism and obesity. However, any recommendations for expending the Australian newborn screening program to include some or all of these conditions will be considered only after sufficient data is collected to justify informed decisions on the suitability of the EpiGNs workflow for newborn screening for these conditions. Many individuals with the typical presentation of the nine conditions screened by the EpiGNs program are potentially identifiable through diagnostic testing of children with DD between birth and 4 years of age. Despite significant progress in next-generation sequencing technologies that has greatly enhanced diagnostic outcomes for children with DD referred for genomic testing, a definitive cause remains unidentified in approximately 50% of these cases [33]. This is in part due to the presence of mutations in known ID genes that may be undetected because they are only present in a small proportion of cells. This phenomenon, known as low-level mosaicism, can occur in the brain but may not be easily identified in the tissues typically used for genetic testing, such as blood and saliva. These changes may involve alterations in allele copy number or DNA sequence, and DNA methylation impacting gene regulation. Although CMA and WES are frequently used for DD referrals, they have limitations in detecting clinically significant low-level mosaicism in as little as 5% of the affected tissue [34], as well as changes in large repetitive DNA sequences. Therefore, the FMR1 CGG repeat expansion PCR test is usually conducted separately from CMA and WES to screen for FXS in cases of DD with unknown causes.
The current SOC diagnostic process for FXS starts with determining the CGG repeat size using PCR-based method, such as repeat-primed PCR. After that, the FMR1 promoter methylation status is examined through methylation specific PCR, sometimes accompanied by methylation sensitive Southern blot analysis targeting the FMR1 CpG island, with an analytical sensitivity of 5–20%. In 2014, we demonstrated a cost-effective method known as methylation specific quantitative melt-analysis (MS-QMA), capable of detecting abnormally methylated alleles in just 1% of cells. This approach greatly improved the diagnostic yield for FXS in male probands who had previously tested negative with standard methods [35,36]. Moreover, when used in combination with real-time PCR SRY CNV analysis, the assay was effective in detecting mosaicism for 45X with SRY and 48XXYY/47XXY in individuals with sex chromosome aneuploidies [37]. MS-QMA was also effective in detecting mosaicism for abnormally methylated SNRPN alleles associated with PWS and AS, identifying mosaicism levels as low as 5% of cells with mosaicism between 12 and 19% associated with atypical presentation of AS [10,18]. For these reasons MS-QMA was included as the assay utilized for first-tier newborn screening by the EpiGNs program in this study.
For sex chromosome aneuploidies (SCA), testing involves non-invasive prenatal testing (NIPT) of cell-free DNA in maternal blood [38]. While many companies that offer the sex chromosome assessment report an accuracy rate above 99%, up to 91% of high-risk NIPT SCA results have been reported to be false positives [38,39]. Despite this limitation, prenatal SCA diagnoses are predicted to increase as prenatal NIPT becomes more widely used, with systems now developed to support families of children with SCA diagnoses made prenatally from birth [40]. Similar systems could be utilized for SCAs identified as part of newborn screening, which would have a much wider reach and could be performed at a much lower cost with far fewer false positive results. For those not identified through NIPT, a proportion of SCA diagnoses are made at a later age usually through CMA testing of DD referrals of unknown genetic cause [41].
The current SOC diagnostic testing protocol for PWS or AS usually utilizes analysis of SNRPN promoter methylation at the 15q11-q13 locus as a first-tier diagnostic test on referrals with clinical features suggestive of these disorders. The methylation status of the SNRPN promoter varies depending on its parental origin, with the maternal allele is methylated, while the paternal allele remains unmethylated. In Angelman Syndrome, the SNRPN promoter is generally unmethylated due to the deletion of the maternal allele, paternal UPD of chromosome 15, or a defect in maternal imprinting [42]. In contrast, the SNRPN promoter is fully methylated in PWS, because of paternal deletion, maternal UPD of chromosome 15, or a defect in paternal imprinting [43]. On the other hand, Dup15q results from duplications or triplications of the PWS or AS imprinted region. Triplications occur due to a supernumerary chromosome (isodicentric 15), while duplications are caused by interstitial tandem duplication [42]. Dup15q is often detected through CMA testing in cases of DD with unknown genetic causes and is associated with varying levels of SNRPN promoter methylation depending on the parent of origin and the number of additional copies [10].
2. Materials & methods
2.1. Pilot phase
In a recent pilot studies we have demonstrated feasibility of the EpiGNs workflow targeting FXS [36] and imprinting disorders [10,18]. The EpiGNs workflow uses a single 3.2 mm punch from an NBS for a low-cost, first-tier automated MS-QMA screen to test for DNA methylation changes associated with specific imprinting and X-chromosome aberrations [27,36,37,44]. More costly second- and third-tier epigenetic and genomic testing follows on another 3.2 mm punch to confirm the etiology for a small sample subset with first-tier positive methylation results.
In our study on chromosome 15 imprinting disorders, we validated MS-QMA for first-tier screening within the EpiGNs workflow. This study involved 1356 samples and demonstrated high specificity, sensitivity and accurate predictive values for distinguishing between newborn blood spots and DNA from blood, saliva and buccal samples of 109 Prader-Willi, 48 Angelman and 9 Dup15q syndrome patient samples compared with neurotypical control samples. We then applied this test to NBS (a single 3.2 mm punch per infant) from 16,579 infants in the general population, who consented to de-identified research as part of the Victorian newborn screening [10]. After conducting second-tier epigenetic and genomic confirmatory testing, we identified two individuals with PWS, two with AS and one with Dup15q syndrome [27]. For FXS FM screening, we performed MS-QMA validation on a single 3.2 mm punch from 89 males and 95 female infants from the general population, 6 males and 10 females with PM alleles and 37 males and 21 females with FXS FM alleles, 0.54 to 18.27 years of age. Interestingly, newborn blood spots of males with FM, FMR1 methylation ratio from MS-QMA testing also strongly correlated with intellectual functioning and autism features [27]. Because FMR1 is X-linked and the CpG sites targeted by MS-QMA are affected by X-chromosome inactivation, the method could also affectively identify individual with different types of SCAs when used in combination with a Y chromosome marker [37].
2.2. Study design
EpiGNs will screen a total of 100,000 infants using the workflow described in the pilot phase. There are two sources for children screened. This includes 50,000 NBS from the GenV cohort and 50,000 NBS from the Victorian newborn screening program. GenV is a state-wide research initiative that aims to give a detailed picture of the health and wellbeing of babies born in Victoria between October 2021 and October 2023, and their parents. On the other hand, the Victorian newborn bloodspot screening is funded by the Department of Health and is offered to all babies in Victoria. VCGS has implemented a consent process for NBS collection, with over 95% parents consenting to the use of blood spots. We plan to utilize 50,000 de-identified, research consented NBS samples from the VCGS collected between 2023 and 2024 and combine them with the retrospectively collected NBS from the GenV cohort. On these samples from the total of 100,000 infants we will perform SNRPN (small nuclear ribonucleoprotein polypeptide N) and FMR1 methylation analysis as a first tier screen. This will enable us to collect and store an additional NBS punch per baby, linking them to data on physical and developmental milestones, parent-reported outcome measures (PROMS), healthcare utilization and resource data for the first 3 years of life and beyond (Figure 1). EpiGNs is projected to identify more than 200 samples from children with those nine genetic conditions. This estimate is derived from the prevalence of each genetic condition, including FXS [45], PWS, AS, Dup15q [10], TS [46], XXY [47], XXXY, XXXXY and XXYY syndromes [48,49]. These data will be used to retrospectively calculate PPV for the first tier testing and prevalence for each condition tested from the combined NBS and GenV cohort of 100,000 infants. Approximately 100 of the 200 samples with confirmed diagnoses are expected to be from the GenV cohort (Figure 1). Outcome measures for these children will be compared with the same outcome measures from the GenV negative cases by the EpiGNs workflow, taking into account non-genetic risk factors such as lifestyle factors and socioeconomic status. Risk factor and outcomes measures will be collected at birth, 1, 2 and 3 years of age to assess trajectories of infants. These comparisons will define screening cut-off for the first tier testing and evaluate cost per additional case identified.
Figure 1.
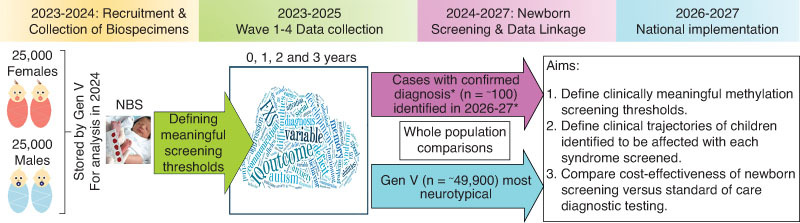
Overview of the GenV component for 50,000 participants. Note: *Fragile X, Prader-Willi, Angelman, Dup15q, Turner, XXY, XXXY and XXYY syndromes.
The study will apply EpiGNs to the NBS of the entire population sample, with EpiGNs molecular results including confirmatory testing to identify the expected 200 samples with FXS, SCAs and chromosome 15 imprinting disorders. For GenV participants, data linkage and phenotype analysis for screen-positive children will be conducted upon completion of first tier testing. GenV provides a unique opportunity to assess the workflow's population-level costs and benefits.
2.3. Study population & eligibility criteria
This study will involve 100,000 infants from Victoria, Australia. To be enrolled in this study, each participant must be a newborn whose parents or guardians have provided consent for either de-identified research within the Victorian newborn screening program or genetic research within the GenV study from October 2021 to October 2023. Additionally, adequate sample of two 3.2 mm punches must be available from the material remaining after completion of SOC newborn screening to be used by the EpiGNs program.
2.4. Ethics approval
This study has received ethics approval from the Royal Children's Hospital Melbourne Research Ethics Committee (reference number: HREC/92777/RCHM-2023(v2)). EpiGNs program does not directly recruit participants or obtain consent. Consent has already been obtained through established and ethically approved processes as part of the Victorian newborn screening program and GenV. EpiGNs follows two independent pipelines for accessing and testing NBS samples:
-
▪
Retesting of NBS samples previously consented for de-identified research at VCGS. This protocol (ethics approval: HREC33066) was utilized in the collection and analysis during the pilot phase of the EpiGNs workflow on NBS consented for de-identified research at VCGS [10].
-
▪
Retesting of newborn blood samples consented for the GenV program (HREC2019.011), which includes optional consent for the use of samples for genetic research. The essential criteria for valid informed consent at GenV include disclosing relevant information to prospective research participants and/or their legally authorized representatives, ensuring voluntary agreement from the participant and having the parent or primary caretaker provide consent for themselves and their children.
2.5. Samples processing
For the VCGS cohort, all NBS materials will be prospectively punched out from the remaining blood spot materials two weeks after the completion of the standard VCGS NBS laboratory test. In the case of GenV infants with consent for genetic analysis, VCGS will retrieve NBS cards and take two further punches from blood spots not used for GenV for EpiGNs. These punches, identified with the EpiGNs ID and including date of birth and sex, will be transferred to EpiGNs to by analyzed following the EpiGNs process. VCGS will punch all materials from both VCGS and GenV cohorts into two replicate 96-well barcoded plates with a single 3.2 mm punch per well. Barcodes on plates and their respective locations will represent the unique NBS ID specific to the EpiGNs program.
Over time, GenV will in parallel collect/access by data linkage phenotypic outcomes and healthcare and resource use measures for its participants. The EpiGNs team will have access to GenV phenotypic and other outcomes data to establish neurotypical FMR1 and SNRPN methylation ranges and neurotypical trajectories for phenotypic outcome measures, in order to determine first tier MS-QMA screening thresholds. These thresholds, tailored to each specific condition being screened, represent the cut-off values used in first-tier screening tests to determine whether further diagnostic evaluation or intervention is warranted, ensuring that infants with clinically significant findings receive prompt and appropriate care.
2.6. GenV cohort follow-up & outcome measures
GenV will collect or access risk factors, phenotypic outcomes and measures of healthcare and resource use for all recruited participants. GenV participants will reach age milestones as follows: newborns: Oct 2021–Sept 2023; 1-year-old: Oct 2022–Sept 2024; 2-year-old: Oct 2023–Sep 2025; 3-year-old: Oct 2024–Sept 2026. Outcome data collection will adhere to these timelines, guided by participant response and timing of data linkage. First-tier methylation screening will be conducted on NBS samples collected shortly after birth. Confirmatory testing on shortlisted NBS will align with the ages of the youngest and oldest participants in both the VCGS and GenV cohorts. Centralized assessment of the numbers recruited, screened and short-listed for confirmatory testing will be performed annually. As per GenV protocol and consent, genetic results will not be returned to participants.
Outcome measures will include a mix of data sources, including information directly collected by GenV's ‘ePhenome’ digital tool, data linkage facilitated through extensive partnerships with custodians and extraction of clinical data from hospital records. Data Linkage activities will be undertaken by GenV's data team to ensure that EpiGNs has access to the necessary data within GenV's secure analytical environment.
Specifically, for EpiGNs, GenV will digitally collect ethnic and demographic data at birth and then administer repeated (3–6 month) brief, validated child and parent health economic measures (health-related quality of life, functioning and participation questionnaires), health events and diagnoses, developmental milestones, health, growth, physical activity/function and other parameters such as facial images to assess dysmorphism.
Additionally, establish links to federal and state-curated datasets in collaboration with organisations such as Australian Institute of Health and Welfare, Australian Bureau of Statistics and the Centre for Victorian Data Linkage. These datasets may include Medicare (MBS/PBS), hospital and emergency department admissions, education, social records and the National Disability Insurance Agency (NDIA). The Victorian Consultative Council on Obstetric and Paediatric Mortality and Morbidity database provides important perinatal data on parity, birth order, gestational age, birth weight, length and head circumference at birth. GenV has also formed partnerships with several Victorian pathology providers for laboratory test results. Extract data from state-wide hospital clinical records, with an advanced pilot now testing this for EpiGNs-relevant data on special care nursery admissions and hospital perinatal prescribing.
Ultimately GenV aims to access the following measures via data linkage: Medicare records, hospital records, digital scans like ultrasounds, information collected about pregnancy and education. This includes national literacy and numeracy assessments at school, social support/welfare, housing and general information about environmental exposures such as childcare locations and air pollution. These data will be analyzed for neurodevelopmental trajectories within secure environments as required by state and federal custodians and analyzed without personal identifiers. All results will be stored using Five Safes principles for safe use of data, including ‘Safe Outputs’.
3. Laboratory protocol
3.1. First-tier testing
First-tier MS-QMA analysis targeting FMR1 and SNRPN methylation will be performed on NBS samples punched into 96 well plates, with one 3.2 mm punch per well. We will use an automated high throughput protocol previously validated on NBS from approximately 17,000 infants [10,18] and over 5,000 diagnostic samples [27,35,36,37,50]. Over the course of 5 years, we anticipate processing approximately 100,000 NBS (20,000 per year). The determination of FMR1 and SNRPN methylation positive and negative will rely on reference data from prior studies. The methylation ratio (MR) will be determined using the MS-QMA to quantify the level of methylation at specific CpG sites of FMR1 and SNRPN promoter regions as previously described [10,18,27].
3.2. Second-tier testing
We will retrieve samples (3.2 mm punch per infant from the second plate) that have been shortlisted using above methylation thresholds for second tier testing. For samples suspected of AS, PWS, or Dup15q, this testing will involve SNRPN copy number variation (CNV) and methylation analysis using the real-time PCR relative standard curve method and Competitive Priming Initiated Nested Quantification (CINQ) droplet digital PCR (ddPCR), as previously described [10]. For FMR1 positive calls, this will involve X & Y marker CNV analysis to confirm sex, and the presence or absence of different sex chromosome aneuploidies (SCAs), as previously published [35,50]. All NBS samples with MR <0.17 or >0.39 will undergo this second-tier testing. Those confirmed not to have SCAs with MS-QMA MR >0.49 will be tested using AmplideX CGG sizing PCR, and the EpiTYPER systems, as previously detailed [35,50] to identify infant with FXS FM expansions and abnormal FMR1 methylation. It is important to note that UBE3A mutation reported to cause AS in approximately 9% of cases [51] will not be picked up by EpiGNs, as these individuals have normal SNRPN promoter methylation [10,18,27].
3.3. Third-tier testing
Low-coverage whole genome sequencing (LC-WGS) will be performed to confirm etiology of the first and second tier testing positive results by focusing on Chromosome 15 and X-chromosome for individuals suspected to have deletions, duplication or aneuploidy, consistent with our earlier studies [10,18,27,37]. This will be done using DNA extracted from a single 3.2 mm NBS punch per participant, following established protocols [10] using NextEra DNA Flex Library Prep as per manufacturer's instructions (Illumina, CA, USA), with sequencing performed on the Illumina Novaseq (Illumina, CA, USA) at 2 x 150bp reads, aiming for a minimum of 50 million reads per sample. Reads will be aligned to the human hg19 reference genome using BWA-mem and duplicate reads will be removed using Picard MarkDuplicates. CNV analysis will be performed using WisecondorX [52], where test samples will be compared with 50 controls extracted and sequenced in parallel. As part of second-tier testing to confirm abnormal X, Y and chromosome 15 CNV results, 50kb bin size will be used for WisecondorX analysis and variants greater than 300kb with a ratio mean of less than 0.15 or greater than 0.15 [10]. Previously, this has offered an optimal balance between sensitivity and specificity while minimizing false positives in our previous studies [10]. We expect that 12 PWS/AS samples with deletions within 15q11–13, and 7 duplications and 121 SCAs identified from second tier CNV testing to be characterized by LC-WGS [52].
3.4. Data management & confidentiality
VCGS will provide de-identified biological tissues and certain personal data (including the VCGS ID, two punches per NBS, plate barcode and sex) to the EpiGNs program. The samples must meet the following criteria: flagged as consented for de-identified research or genetic testing as part of GenV.
The results from EpiGNs testing will include genetic data for an estimated 200 individuals with confirmed diagnosis of the conditions being tested, with all data de-identified. Sensitive data will include abnormal results confirming diagnosis of FXS, chromosome 15 imprinting disorder and SCAs, along with their etiology, including genetic subtype and/or presence of mosaicism for the FMR1 and SNRPN loci tested. Given that the EpiGNs workflow analyzes will be restricted to genetic loci associated with the tested conditions, it is highly unlikely to identify any other clinically significant conditions.
The new data generated by the EpiGNs program will include, SNRPN and FMR1 methylation results from across the entire cohort and the estimated 200 cases with confirmed diagnoses. Additionally, results from confirmatory testing from second- and third-tier testing are expected to identify around 200 samples from the combined cohort of 100,000 infants. Genetic data from first-, second- and third-tier testing will be securely stored in a password protected REDCap (Research Electronic Data Capture) database. For the GenV subset, EpiGNs will integrate its methylation data (whole EpiGNs-GenV cohort) and confirmatory data (estimated at ~100 cases in the GenV cohort) into the GenV datasets using secure MCRI-approved upload processes. Following the GenV principle of Open Science, these data will be made available to the broader research after completion of the study. These data will support the descriptive, modelling and case-cohort analyzes of phenotypic and health economic outcomes, according to each individual's EpiGNs methylation and genomic status.
Access to files will be restricted and the data collected from participants will be managed according to the mandatory archive period for future research studies. EpiGNs-specific data will be stored electronically for a minimum of 5 years post study closure. GenV's data are intended to remain in use indefinitely as per its existing ethical approval.
To ensure confidentiality and reduce the risk of identification throughout data collection, analysis and storage, no personally identifiable participant data (e.g., phone number, child's name) will be transferred to the EpiGNs program, except for sex and date of birth. Date of birth is important for interpreting the quality of methylation data, as it allows for calculating the timing of NBS collection (i.e. days from birth to NBS collection).
3.5. Handling of missing data
Missing data may occur due to technical faults during NBS punching, NBS lysis and DNA release steps, as well as automated bisulfite conversion and first, second and third tier testing. These faults, when combined, occur in less than 2% of NBS tested, with automated bisulfite conversion issues being the most common, affecting less than 1% of NBS tested. In the event of such occurrences, quality control procedures embedded in the Q'Max software used for MS-QMA analysis will flag them and the results for the failed analyzes will be discarded from the entire analyzes. There will not be sufficient NBS materials remaining for repeat testing. In GenV, missing data may arise for certain data points, such as the e-phenome or due to failure to link the requested data for a specific phenotypic domain. This is mitigated by extensive data linkage in GenV, which examines overlapping phenotypic domains, where one missing outcome will not mean removal for the genotype-phenotype analyzes planned. In this approach, one missing outcome will not result in removal from the planned genotype-phenotype analyzes.
3.6. Analysis of data
The PPV will be defined as the likelihood that individuals with a positive NBS first-tier MS-QMA result truly have a targeted genetic disorder, which is confirmed using second and third tier testing. Prevalence will be determined as the number of true positive infants for each screened syndrome divided by the total number of NBS analyzed using MS-QMA. To determine clinically meaningful MS-QMA methylation thresholds within the MR ranges used to screen for each syndrome, we will use Receiver Operating Characteristic (ROC) analysis. ROC analysis will assess the diagnostic accuracy of each syndrome across different methylation thresholds. Using ROC, we will determine optimal thresholds to discriminate between individuals with and without a specific syndrome that is expected to be identified. This will be achieved by simultaneously assessing sensitivity, and PPV, either by maximizing the sum [13] or product [53] of these measures. Values below and above the threshold for each syndrome will be classified into a binary variable, to estimate various performance characteristics for each threshold, such as prevalence, sensitivity, likelihood ratio for a positive test and PPV.
For the longitudinal data obtained from the GenV cohort, we will conduct two separate analyzes. First, we will use linear regression to investigate whether the level of methylation at birth (as a predictor) in infants with identified syndromes can predict future clinical outcome measures (e.g. developmental functioning and autism features from e-phenome data) at 1, 2 and 3 years of age. Second, we will use latent class modelling approaches, such as Group-Based Trajectory Modelling (GBTM) [54] or growth mixture modelling (GMM) [55] to identify groups of children following similar growth trajectories, including persistent no change, increase and decrease in health and economic outcome measures. Each outcome (physical and developmental milestones, healthcare use and resource utilization) will be individually examined by this model and the identified groups exhibiting different growth patterns will be compared between the largely neurotypical whole population sample and those with each syndrome defined by EpiGNs. Moreover, the analyzes will consider non-genomic risk for poorer neurodevelopmental and other outcomes, such as demographic and socioeconomic circumstances.
We will model the likely cost-effectiveness of EpiGNs NBS testing by calculating the cost per positive diagnosis. Using decision analytic modelling the incremental cost of EpiGNs NBS testing compared with standard diagnostic testing, per additional early diagnosis of a case will be estimated. Model parameters include the prevalence of each identified condition and the sensitivity of each testing strategy. Where possible we will calculate and compare across strategies the costs/savings and health outcomes associated with early diagnosis.
4. Future perspective & limitations
The outcomes of this study will provide an evidence-based assessment of the feasibility of EpiGNs workflow, to inform rational changes to policy and practice and the implementation of new models for genomic newborn screening nationally and internationally, with the potential to expand the panel of conditions screened at little additional cost. Defining positive predictive values and prevalence estimates for each condition will enhance the robustness of the EpiGNs workflow, allowing for potential expansion to test for additional conditions. Furthermore, the prevalence data of EpiGNs will inform its potential integration into existing SOC newborn screening programs and may provide new information about atypical clinical presentations for the conditions screened which may be driven by socioeconomic, environmental and biological factor including mosaicism (currently not well understood at population level). This new knowledge has the potential to impact recognition of these atypical features for these conditions. This is important because referral for testing for these conditions may not occur due to bias in ascertainment toward a more typical presentation associated with the SOC diagnostic testing.
This study highlights the potential of the EpiGNs program for early identification of developmental delays. However, certain limitations must be acknowledged. Methylation analysis may not capture all genetic variations associated with the screened conditions [44]. The inclusion of conditions outside established screening criteria, as outlined by Wilson and Jungner [5], limits the generalizability of the findings to other programs. Moreover, the ethical implications of disclosing genetic information, particularly regarding late-onset conditions and carrier status, necessitate careful consideration. The potential psychological impact on families receiving such information, especially when immediate benefits for the newborn are unclear, must be addressed through comprehensive pre-test counselling and informed consent processes. Ongoing dialogue with stakeholders, including healthcare providers, societies, policymakers and the public, will be essential to navigate these issues. Additionally, the evolving nature of treatments for some conditions, with some still in trial phases, raises questions about the program's long-term cost-effectiveness. Future studies should consider the applicability of the EpiGNs workflow in different demographic and geographic contexts. Research could also refine inclusion criteria to align better with established standards and explore the program's adaptability to emerging knowledge about these conditions and their treatments.
5. Conclusion
The implementation of EpiGNs in a large cohort of 100,000 infants represents a breakthrough in genomic newborn screening, with the potential to enhance early detection and establish prevalence rates for conditions that are often diagnosed late or missed entirely. The study's design incorporates robust methodologies to estimate the prevalence of these conditions, which is essential for understanding the potential impact of the screening program on public health. By utilizing data from the GenV cohort, the EpiGNs program will also allow for longitudinal tracking of developmental outcomes of infants. If successful, the EpiGNs program could serve as a model for other regions considering the integration of epigenomic techniques into their newborn screening protocols. The ability to identify conditions earlier than current practices could lead to reduced healthcare costs associated with delayed diagnoses and prolonged diagnostic odysseys. Furthermore, the findings could inform policy decisions regarding the expansion of newborn screening programs to include additional genetic conditions, ultimately improving health outcomes on a population level.
Moreover, the EpiGNs program the potential to transform the early identification of genetic conditions associated with ID and autism. While the EpiGNs program in its current form only targets a panel of 9 conditions, there are over 120 rare diseases where changes to DNA sequence cause changes to DNA methylation that can be used for screening and diagnostic testing in future studies [44,56]. By integrating low-cost high-throughput DNA methylation screen with second- and third-tier genomic and epigenetic analyses and comprehensive data linkage, this study has the potential to enhance our understanding of the conditions screened and improve outcomes for affected infants and their families. As the field of genomic and epigenomic medicine continues to evolve, the insights gained from the EpiGNs program may pave the way for broader implementation of similar screening initiatives worldwide.
Supplementary Material
Funding Statement
The research conducted at the Murdoch Children's Research Institute was supported by the Victorian Government's Operational Infrastructure Support Program. The salaries were supported by National Health and Medical Research Council project grants (No. 1049299 and No. 1103389 to DE Godler); Murdoch Children's Research Institute, Royal Children's Hospital Foundation (to DE Godler); Next Generation Clinical Researchers Program–Career Development Fellowship, by the Medical Research Future Fund (MRF1141334 to DE Godler); the Genomics Health Futures Mission (MRFF2016199 to DE Godler), the Financial Markets Foundation for Children (Australia; No. 2017-361 to DE Godler and DJ Amor); the Genetics of Learning Disability (GOLD) Service (M Field); the Foundation for Prader-Willi Syndrome Research, USA (No. 43445 and No. 501393 to DE Godler and DJ Amor); the Angelman Syndrome Foundation (to DE Godler); Victorian Medical Research Acceleration Fund (to DE Godler) and joint funding from the Prader-Willi Syndrome Association (Australia) (to DE Godler); Foundation for Angelman Syndrome Therapeutics (Australia) (to DE Godler) and Dup15q Australia Ltd (to DE Godler).
Supplemental material
Supplemental data for this article can be accessed at https://doi.org/10.1080/17501911.2024.2402681
Author contributions
DE Godler is the principal investigator of the EpiGNs program, where all authors participated in contributing to the design of the protocol, contributing to its conceptualization and providing critical feedback leading to the approval of the final manuscript version. M Alshawsh was responsible for drafting the protocol paper, with subsequent review by all authors.
Financial disclosure
The research conducted at the Murdoch Children's Research Institute was supported by the Victorian Government's Operational Infrastructure Support Program. The salaries were supported by National Health and Medical Research Council project grants (No. 1049299 and No. 1103389 to DE Godler); Murdoch Children's Research Institute, Royal Children's Hospital Foundation (to DE Godler); Next Generation Clinical Researchers Program–Career Development Fellowship, by the Medical Research Future Fund (MRF1141334 to DE Godler); the Genomics Health Futures Mission (MRFF2016199 to DE Godler), the Financial Markets Foundation for Children (Australia; No. 2017-361 to DE Godler and DJ Amor); the Genetics of Learning Disability (GOLD) Service (M Field); the Foundation for Prader-Willi Syndrome Research, USA (No. 43445 and No. 501393 to DE Godler and DJ Amor); the Angelman Syndrome Foundation (to DE Godler); Victorian Medical Research Acceleration Fund (to DE Godler) and joint funding from the Prader-Willi Syndrome Association (Australia) (to DE Godler); Foundation for Angelman Syndrome Therapeutics (Australia) (to DE Godler) and Dup15q Australia Ltd (to DE Godler).
Competing interests disclosure
DE Godler reporting being an inventor on patents related to the technologies described in this publication and being an executive director of EDG Innovations & Consulting, which receives funds from this intellectual property. He also has acted as a paid consultant for Bellberry, Ltd and Actinogen Medical, Pty, Ltd. No other disclosures were reported.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
This study has been granted ethics approval by the Royal Children's Hospital Melbourne Research Ethics Committee under the reference number: HREC/92777/RCHM-2023-(v2) and have followed the principles outlined in the Declaration of Helsinki for human experimental investigations.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Marwaha S, Knowles JW, Ashley EA. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med. 2022;14(1):23. doi: 10.1186/s13073-022-01026-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tisdale A, Cutillo CM, Nathan R, et al. . The IDeaS initiative: pilot study to assess the impact of rare diseases on patients and healthcare systems. Orphanet J Rare Dis. 2021;16:1–18. doi: 10.1186/s13023-021-02061-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stark Z, Scott RH. Genomic newborn screening for rare diseases. Nat Rev Genet. 2023;24(11):755–766. doi: 10.1038/s41576-023-00621-w [DOI] [PubMed] [Google Scholar]; • Highlights the potential of incorporating genomic sequencing into newborn screening programs to identify rare diseases early, leading to improved patient outcomes.
- 4.Lancet T . Genomic newborn screening: current concerns and challenges. 2023;402(10398):265. doi: 10.1016/S0140-6736(23)01513-1 [DOI] [PubMed] [Google Scholar]
- 5.Andermann A, Blancquaert I, Beauchamp S, et al. . Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ. 2008;86(4):317–319. doi: 10.2471/BLT.07.050112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey DB Jr, Porter KA, Andrews SM, et al. . Expert evaluation of strategies to modernize newborn screening in the United States. JAMA Netw Open. 2021;4(12):e2140998. doi: 10.1001/jamanetworkopen.2021.40998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Downie L, Bouffler SE, Amor DJ, et al. . Gene selection for genomic newborn screening: moving towards consensus? Genet Med. 2024;26(5):101077. doi: 10.1016/j.gim.2024.101077 [DOI] [PubMed] [Google Scholar]; •• Proposes a consensus gene list for genomic newborn screening, serving as a foundation for systematic harmonization efforts on a global scale.
- 8.Lee JS, Hwang H, Kim SY, et al. . Chromosomal microarray with clinical diagnostic utility in children with developmental delay or intellectual disability. Ann Lab Med. 2018;38(5):473–480. doi: 10.3343/alm.2018.38.5.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han JY, Lee IG. Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Clin Exp Pediatr. 2020;63(6):195. doi: 10.3345/kjp.2019.00808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godler DE, Ling L, Gamage D, et al. . Feasibility of screening for chromosome 15 imprinting disorders in 16 579 newborns by using a novel genomic workflow. JAMA Netw Open. 2022;5(1):e2141911–e2141911. doi: 10.1001/jamanetworkopen.2021.41911 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrates the feasibility of implementing a novel methylation workflow for screening chromosome 15 imprinting disorders in a large cohort of newborns.
- 11.Kraan CM, Bui QM, Field M, et al. . FMR1 allele size distribution in 35,000 males and females: a comparison of developmental delay and general population cohorts. Genet Med. 2018;20(12):1627–1634. doi: 10.1038/gim.2018.52 [DOI] [PubMed] [Google Scholar]; • Reveals no association between FMR1 premutation and gray zone allele expansions and developmental-delay phenotypes in pediatric settings.
- 12.Kraan CM, Godler DE, Amor DJ. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev Med Child Neurol. 2018;61(2):121–127. doi: 10.1111/dmcn.13985 [DOI] [PubMed] [Google Scholar]; •• Highlights how DNA methylation analysis facilitates improved epigenetic assessment of the Fragile X gene.
- 13.Hagerman RJ, Berry-Kravis E, Kaufmann WE, et al. . Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123(1):378–390. doi: 10.1542/peds.2008-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dean DD, Agarwal S, Muthuswamy S. Fragile X molecular investigation and genetic counseling of intellectual disability/developmental delay patients in an Indian scenario. Expert Rev Mol Diagn. 2019;19(7):641–649. doi: 10.1080/14737159.2019.1622416 [DOI] [PubMed] [Google Scholar]
- 15.Ibañez K, Polke J, Hagelstrom RT, et al. . Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: a retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. 2022;21(3):234–245. doi: 10.1016/S1474-4422(21)00462-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bailey DB Jr, Raspa M, Bishop E, et al. . No change in the age of diagnosis for fragile x syndrome: findings from a national parent survey. Pediatrics. 2009;124(2):527–533. doi: 10.1542/peds.2008-2992 [DOI] [PubMed] [Google Scholar]
- 17.Godler DE, Amor DJ, Slater HR. Methylation analysis in newborn screening for fragile x syndrome. JAMA Neurol. 2014;71(6):800. doi: 10.1001/jamaneurol.2014.142 [DOI] [PubMed] [Google Scholar]
- 18.Baker EK, Merton CF, Tan W-H, et al. . Methylation analysis and developmental profile of two individuals with Angelman syndrome due to mosaic imprinting defects. Eur J Med Genet. 2022;65(4):104456. doi: 10.1016/j.ejmg.2022.104456 [DOI] [PMC free article] [PubMed] [Google Scholar]; • Shows the developmental profile of individuals with Angelman syndrome and investigates the utility of methylation analysis in diagnosis.
- 19.Willis AS, van den Veyver I, Eng CM. Multiplex ligation-dependent probe amplification (MLPA) and prenatal diagnosis. Prenat Diagn. 2012;32(4):315–320. doi: 10.1002/pd.3860 [DOI] [PubMed] [Google Scholar]
- 20.Ferreira IR, Costa RA, Gomes LHF, et al. . A newborn screening pilot study using methylation-sensitive high resolution melting on dried blood spots to detect Prader-Willi and Angelman syndromes. Sci Rep. 2020;10(1):13026. doi: 10.1038/s41598-020-69750-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor JL, Lee FK, Yazdanpanah GK, et al. . Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin Chem. 2015;61(2):412–419. doi: 10.1373/clinchem.2014.231019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rochmah MA, Harahap NIF, Niba ETE, et al. . Genetic screening of spinal muscular atrophy using a real-time modified COP-PCR technique with dried blood-spot DNA. Brain Dev. 2017;39(9):774–782. doi: 10.1016/j.braindev.2017.04.015 [DOI] [PubMed] [Google Scholar]
- 23.Kariyawasam D, Russell JS, Wiley V, et al. . The implementation of newborn screening for spinal muscular atrophy: the Australian experience. Genet Med. 2020;22(3):557–565. doi: 10.1038/s41436-019-0673-0 [DOI] [PubMed] [Google Scholar]
- 24.(MCRI) MCsRI . Epi-Genomic Newborn screening program (EpiGNs). 2024. [cited 2024 16 April 2024]. Available from: https://www.mcri.edu.au/research/strategic-collaborations/centres/epi-genomic-newborn-screening-program
- 25.GenV . GenV. 2024. [cited 2024 16 April 2024]. Available from: https://www.genv.org.au/
- 26.Savendahl L, Davenport ML. Delayed diagnoses of Turner's syndrome: proposed guidelines for change. J Pediatr. 2000;137(4):455–459. doi: 10.1067/mpd.2000.107390 [DOI] [PubMed] [Google Scholar]
- 27.Kraan CM, Baker EK, Arpone M, et al. . DNA methylation at birth predicts intellectual functioning and autism features in children with Fragile X syndrome. Int J Mol Sci. 2020;21(20):7735. doi: 10.3390/ijms21207735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimonis VE, Tamura R, Gold JA, et al. . Early diagnosis in Prader-Willi syndrome reduces obesity and associated co-morbidities. Genes. 2019;10(11):898. doi: 10.3390/genes10110898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfäffle R, Bidlingmaier M, Kreitschmann-Andermahr I, et al. . Safety and effectiveness of Omnitrope®, a biosimilar recombinant human growth hormone: more than 10 Years' experience from the PATRO Children Study. Horm Res Paediatr. 2020;93(3):154–163. doi: 10.1159/000508190 [DOI] [PubMed] [Google Scholar]
- 30.Murdock DR, Donovan FX, Chandrasekharappa SC, et al. . Whole-exome sequencing for diagnosis of turner syndrome: toward next-generation sequencing and newborn screening. J Clin Endocrinol Metab. 2017;102(5):1529–1537. doi: 10.1210/jc.2016-3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elgersma Y, Sonzogni M. UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev Med Child Neurol. 2021;63(7):802–807. doi: 10.1111/dmcn.14831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berry-Kravis EM, Harnett MD, Reines SA, et al. . Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, Phase II clinical trial. Nat Med. 2021;27(5):862–870. doi: 10.1038/s41591-021-01321-w [DOI] [PubMed] [Google Scholar]
- 33.Deciphering Developmental Disorders S. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–228. doi: 10.1038/nature14135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krupp DR, Barnard RA, Duffourd Y, et al. . Exonic mosaic mutations contribute risk for autism spectrum disorder. Am J Hum Genet. 2017;101(3):369–390. doi: 10.1016/j.ajhg.2017.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aliaga SM, Slater HR, Francis D, et al. . Identification of males with cryptic fragile X alleles by methylation-specific quantitative melt analysis. Clin Chem. 2016;62(2):343–352. doi: 10.1373/clinchem.2015.244681 [DOI] [PubMed] [Google Scholar]
- 36.Inaba Y, Schwartz CE, Bui QM, et al. . Early detection of fragile X syndrome: applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin Chem. 2014;60(7):963–973. doi: 10.1373/clinchem.2013.217331 [DOI] [PubMed] [Google Scholar]; •• Introduces a novel methylation specific–quantitative melt analysis (MS-QMA), enabling early detection of Fragile X syndrome and potentially enhancing screening accuracy and efficiency.
- 37.Godler DE, Inaba Y, Schwartz CE, et al. . Detection of skewed X-chromosome inactivation in Fragile X syndrome and X chromosome aneuploidy using quantitative melt analysis. Expert Rev Mol Med. 2015;17:e13. doi: 10.1017/erm.2015.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizia K, Townsend L, Karatas J. Sex chromosome aneuploidy screening in a general population. Australas J Ultrasound Med. 2016;19(3):105–108. doi: 10.1002/ajum.12019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reiss RE, Discenza M, Foster J, et al. . Sex chromosome aneuploidy detection by noninvasive prenatal testing: helpful or hazardous? Prenat Diagn. 2017;37(5):515–520. doi: 10.1002/pd.5039 [DOI] [PubMed] [Google Scholar]
- 40.Riggan KA, Gross B, Close S, et al. . Prenatal genetic diagnosis of a sex chromosome aneuploidy: parent experiences. J Genet Couns. 2021;30(5):1407–1417. doi: 10.1002/jgc4.1407 [DOI] [PubMed] [Google Scholar]
- 41.Ozkan E, Lacerda MP. Genetics, cytogenetic testing and conventional karyotype. StatPearls: Treasure Island (FL); 2020. [PubMed] [Google Scholar]
- 42.Kalsner L, Chamberlain SJ. Prader-Willi, Angelman, and 15q11–q13 duplication syndromes. Pediatr Clin North Am. 2015;62(3):587–606. doi: 10.1016/j.pcl.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Downie L, Halliday J, Lewis S, et al. . Principles of genomic newborn screening programs. JAMA Netw Open. 2021. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Godler DE, Amor DJ. DNA methylation analysis for screening and diagnostic testing in neurodevelopmental disorders. Essays Biochem. 2019; 63(6): 785–795. doi: 10.1042/EBC20190056 [DOI] [PubMed] [Google Scholar]; •• Explores the potential of genome-wide methylation analysis (GWMA) for diagnosing neurodevelopmental disorders with defined epigenetic signatures.
- 45.Coffee B, Keith K, Albizua I, et al. . Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85(4):503–514. doi: 10.1016/j.ajhg.2009.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gravholt CH, Andersen NH, Conway GS, et al. . Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177(3):G1–G70. doi: 10.1530/EJE-17-0430 [DOI] [PubMed] [Google Scholar]
- 47.Herlihy AS, Halliday JL, Cock ML, et al. . The prevalence and diagnosis rates of Klinefelter syndrome: an Australian comparison. Med J Aust. 2011;194(1):24–28. doi: 10.5694/j.1326-5377.2011.tb04141.x [DOI] [PubMed] [Google Scholar]
- 48.Blumling AA, Martyn K, Talboy A, et al. . Rare sex chromosome variation 48, XXYY: an integrative review. Am J Med Genet C: Seminars in Medical Genetics. 2020;184(2):386–403. doi: 10.1002/ajmg.c.31789 [DOI] [PubMed] [Google Scholar]
- 49.Tartaglia N, Ayari N, Howell S, et al. . 48, XXYY, 48, XXXY and 49, XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011;100(6):851–860. doi: 10.1111/j.1651-2227.2011.02235.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hensel CH, Vanzo RJ, Martin MM, et al. . Abnormally methylated FMR1 in absence of a detectable full mutation in a U.S.A patient cohort referred for fragile X testing. Sci Rep. 2019;9(1):15315. doi: 10.1038/s41598-019-51618-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du X, Wang J, Li S, et al. . An analysis of phenotype and genotype in a large cohort of Chinese children with Angelman syndrome. Genes. 2022;13(8):1447. doi: 10.3390/genes13081447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raman L, Dheedene A, De Smet M, et al. . WisecondorX: improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 2019;47(4):1605–1614. doi: 10.1093/nar/gky1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berry-Kravis EM, Harnett MD, Reines SA, et al. . Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, Phase II clinical trial. Nat Med. 2021;27(5):862–870. doi: 10.1038/s41591-021-01321-w [DOI] [PubMed] [Google Scholar]
- 54.Freriks K, Timmermans J, Beerendonk CC, et al. . Standardized multidisciplinary evaluation yields significant previously undiagnosed morbidity in adult women with Turner syndrome. J Clin Endocrinol Metab. 2011;96(9):E1517–E1526. doi: 10.1210/jc.2011-0346 [DOI] [PubMed] [Google Scholar]
- 55.Nguena Nguefack HL, Page MG, Katz J, et al. . Trajectory Modelling Techniques Useful to Epidemiological Research: A Comparative Narrative Review of Approaches. Clin Epidemiol. 2020;12:1205–1222. doi: 10.2147/CLEP.S265287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadikovic B, Levy MA, Kerkhof J, et al. . Clinical epigenomics: genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet Med. 2021;23(6):1065–1074. doi: 10.1038/s41436-020-01096-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.