

Abstract
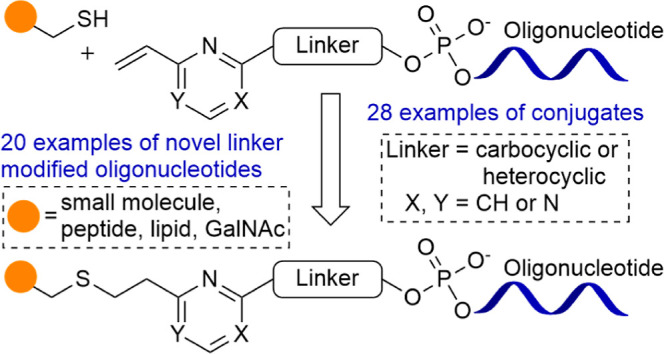
Chemical conjugation of oligonucleotides is widely used to improve their delivery and therapeutic potential. A variety of strategies are implemented to efficiently modify oligonucleotides with conjugating partners. The linkers typically used for oligonucleotide conjugation have limitations in terms of stability or ease of synthesis, which generates the need for providing new improved linkers for oligonucleotide conjugation. Herein, we report the synthesis of novel vinylpyrimidine phosphoramidite building blocks, which can be incorporated into an oligonucleotide by standard solid-phase synthesis in an automated synthesizer. These linker-bearing oligonucleotides can be easily conjugated in a biocompatible manner with thiol-functionalized molecules leading to the efficient generation of oligonucleotide conjugates.
Introduction
One of the major challenges limiting oligonucleotide-based therapeutics from reaching their full potential is the limited ability of these relatively large and often highly charged molecules to traverse cellular membranes, making it difficult to attain therapeutic concentrations at the site of action (in the cytosol or nucleus).1,2 Chemical conjugation of oligonucleotides with transporter molecules is one of the most convenient approaches employed to improve the intracellular delivery and therapeutic potential of these agents.3−5 For these advantages to be realized, it is necessary for the linker joining the oligonucleotide and conjugation partner to be biologically stable as well as able to attach in conditions suitable for both, the oligonucleotide and partner compound.6 A traditional conjugation method is via disulfide bonds7 using cysteine residues. However, disulfides are not stable in vivo and are easily cleaved, which is why they are classified as cleavable linkers. Another widely applied conjugation strategy is to use a triazole linker.8,9 This requires additional modification of the conjugating partners with either an azide or alkyne functionality and, therefore, frequently needs postsynthetic modification of the oligonucleotide. These linkers also typically require the presence of a metal catalyst, such as copper, in order to afford the desired molecule.10 The presence of metals can be detrimental to drug molecules and toxic to biological systems and can also lead to degradation of oligonucleotides. On the other hand, metal-free triazole formation reactions must deal with the bulky reagents and the compatibility of strained alkynes with the conditions necessary for oligonucleotide synthesis. Moreover, triazole linkers are frequently not stable during extended storage and triazole derivatives can display unwanted biological activity, which may lead to off-target effects in vivo.11 Another class of linkers widely used in oligonucleotide conjugation reactions are the maleimide linkers.12−14 However, maleimide linkers present a number of challenges: maleimide-modified oligonucleotides can be difficult to synthesize as the maleimide group is generally not stable under the conditions used in oligonucleotide synthesis. At present, two approaches are employed to mitigate this; often the oligonucleotide needs to be synthesized with an amine modification and it is only after the oligonucleotide has been cleaved from the solid support and purified that this amine is reacted with a bifunctional small-molecule linker containing both, an N-hydroxysuccinimide ester and maleimide group.15 The alternative approach is to make use of commercially available maleimide modifiers, which are both expensive and require additional processing steps. Aside from the difficulties in their preparation, maleimide-functionalized oligonucleotides were reported to display a relatively short half-life in mice plasma due to the ring opening and subsequent cleavage of the amide.16 They also show a narrow pH range for selective reaction with thiols over amines and a lack of selectivity between thiols and disulfide bonds. Accordingly, there is a need to develop improved linkers for oligonucleotide conjugation.
We report the synthesis of a library of novel vinylpyrimidine phosphoramidite linkers (Figure 1) and their successful incorporation at the 5′ end of the oligonucleotide sequences by automated solid-phase synthesis (SPS) using standard phosphoramidite chemistry. After cleavage from the controlled pore glass (CPG) solid support and deprotection, we successfully obtained a panel of novel linker-containing oligonucleotides. Furthermore, we demonstrated the applicability of these linkers by conjugating the linker-containing oligonucleotides to thiol-bearing small molecules, peptide, lipid, and carbohydrate to generate oligonucleotide conjugates via thiol–ene click reactions.17 The rationale behind choosing the vinylpyrimidine scaffold was their successful utilization in preparing protein–drug conjugates by Spring et al.18 However, to the best of our knowledge, vinylpyrimidine phosphoramidite linkers have neither been reported before nor have these linkers been used for preparing oligonucleotide conjugates. As part of our phosphoramidite design, we chose to have heterocyclic and carbocyclic scaffolds attached to the reactive vinyl pyrimidine systems, for their potential rigidity, which we expected would be stable enough to withstand the conditions for automated SPS of oligonucleotides.
Figure 1.

Library of novel linkers synthesized and incorporated into oligonucleotides.
The methods reported can be used as generic techniques for preparing thiol-reactive oligonucleotides that are compatible with conventional solid-phase oligonucleotide synthesis protocols. Factors influencing the reactivity of these linkers such as the position of the double bond, substitution on the double bond, and type of ring (purine/pyrimidine) have also been investigated, which may have an effect on the stability and binding/therapeutic efficiency.
Results and Discussion
The novel phosphoramidite linkers containing the vinylpyrimidine scaffold were synthesized following Schemes 1 and 2. SnAr reaction was carried out with commercially available 2,4-dichloropyrimidine 1 and the corresponding amino alcohols R-OH (commercially sourced cis/trans mixture, unless stated/drawn otherwise) or R′-OH (commercially available) to afford major products 2–4 from R-OH and major products 5–6 from R′-OH in 57–78% yields and their regioisomers 2′–4′ and 5′–6′, respectively, in 4–10% yields as the minor products, which could be easily separated by flash column chromatography. Compounds 2–4 and 5–6 were then subjected to Suzuki coupling to afford the vinylic compounds 7–9 and 13–14, respectively, in 30–96% yields.
Scheme 1. Synthesis of Phosphoramidites 10–12 and 15–16.

Scheme 2. Synthesis of Phosphoramidites 23–27.

Note: 20* was synthesized from 4′ by Suzuki reaction (see the Supporting Information for the detailed procedure). 21# was synthesized from commercially available (R)-1-(4-chloropyrimidin-2-yl)pyrrolidine-3-ol by Suzuki reaction (see the Supporting Information for the detailed procedure).
Phosphitylation of compounds 7–9 and 13–14 afforded the novel phosphoramidites 10–12 and 15–16, respectively, as diastereomeric mixtures in 50–78% yields (Scheme 1). The regioisomers of these phosphoramidites were synthesized following Scheme 2. SnAr reaction was performed with reported compound 2-chloro-4-vinylpyrimidine 17 prepared by the literature procedure19 from 1 and the corresponding amino alcohols R-OH and R′-OH (commercially sourced cis/trans mixture, unless stated/drawn otherwise in the Supporting Information) to afford products 18–19 and 22, respectively, in 34–61% yields, which on phosphitylation afforded accordingly the phosphoramidites 23–24 and 27 as diastereomeric mixtures in 67–72% yields. Compound 20 was synthesized in 96% yield by Suzuki reaction of 4′ (Suzuki conditions described in the Supporting Information) and 21 was synthesized in 45% yield by Suzuki reaction of commercially available material (R)-1-(4-chloropyrimidin-2-yl)pyrrolidin-3-ol under the same reaction conditions. Phosphitylation of 20 and 21 afforded phosphoramidites 25 and 26, respectively, as diastereomeric mixtures in 68–74% yields.
In order to check the feasibility of incorporating the synthesized linkers at the 5′ end of oligonucleotides, we proceeded toward the synthesis of oligonucleotides (ONs) containing the novel linkers via a standard automated solid-phase oligonucleotide synthesis protocol. Initially, we chose to incorporate these linkers into two DNA sequences: ON1 and ON2 (Table 1).
Table 1. Oligonucleotide Sequences Used in This Studya.
| oligonucleotide | 5′ to 3′ sequence |
|---|---|
| ON1 | dCdGdAdCdGdCdTdTdGdCdAdGdCdT |
| ON2 | dCdTdAdCdAdCdTdTdCdCdAdTdCdT |
| ON3 | dGdmCdAdTdTdmCdTdAdAdTdAdGdmCdAdGdmC |
| ON4 | lGlmClAdTdTdmCdTdAdAdTdAdGdmClAlGlmC |
Capital letter is base code: G = guanine, A = adenine, C = cytosine, mC = 5-methylcytosine, T = thymine. Small letters are sugar codes: l = locked nucleic acid (LNA) sugar, d = deoxyribose sugar.
The oligonucleotide synthesis was performed using a standard SPS on a universal solid support (see the Supporting Information for the details). For the deprotection step, which includes cleavage from the solid support and removal of the protecting groups, we used two conventional methods: either treatment with 0.4 M NaOH in MeOH/water (4:1) solution or 32% aqueous ammonia solution. We successfully obtained linker-bearing oligonucleotides ON1-a to l, ON2-a to e, and ON2-j (Scheme 3) in 84–97% purity after HPLC purification (see the Supporting Information for details).
Scheme 3. Library of Novel Linker-Bearing Oligonucleotides Synthesized.

Subsequently, we conjugated our oligonucleotides with thiol-containing molecules via the thiol–ene click reaction. In the first instance, we chose to use 2-mercaptoethanol or glutathione (GSH) as the thiol-bearing partner for conjugation. Oligonucleotides ON1-a to l and ON2-a to e on reaction with 2-mercaptoethanol showed 80–100% conversion [based on liquid chromatography mass spectrometry (LC–MS)] to the corresponding conjugated oligonucleotides ON1-a′ to l′ and ON2-a′ to e′, respectively (see the Supporting Information for details). Oligonucleotides ON1-a to c and ON2-d to e on reaction with GSH resulted in 80–100% conversion to the conjugated oligonucleotides ON1-a″ to c″ and ON2-d″ to e″, respectively (Scheme 4).
Scheme 4. Synthesis of Oligonucleotide Conjugates by Thiol–Ene Click Reaction with 2-Mercaptoethanol and Reduced l-Glutathione.

*Conversion estimated based on LC–MS chromatograms.
Based on the successful synthesis of the linker-bearing oligonucleotides and the promising results of their conjugation with thiols, we broadened the scope to purine-containing linkers and linkers with a substituted double bond. For this purpose, we synthesized phosphoramidite 30 with a purine base (Scheme 5a) and phosphoramidite 35 with fluorine substitutions on the double bond (Scheme 6a).
Scheme 5. (a) Synthesis of Phosphoramidite 30 and (b) Oligonucleotide ON1-i Containing Linker i.

Scheme 6. (a) Synthesis of Phosphoramidite 35 and (b) ON1-j and ON2-j Containing Linker j.

The rationale behind choosing commercially available starting material 28 for the synthesis of 30 was that we wanted to build a directly attached vinylic system for the purine ring, similar to that of our pyrimidine systems. Suzuki coupling of 28 resulted in the vinylic system 29 in 52% yield which on phosphitylation afforded the novel phosphoramidite with the purine scaffold 30 in 76% yield as a diastereomeric mixture (Scheme 5a). For the synthesis of 35, we first performed a SnAr reaction between commercially available 2-chloropyrimidine-5-carbaldehyde 31 and commercially available trans-4-(methylamino)cyclohexanol 32 to obtain aldehyde 33 in 65% yield. The fluorinated vinyl group was introduced by a Wittig-type reaction using triphenylphosphine and sodium 2-chloro-2,2-difluoroacetate20 to afford 34 in 41% yield, which was then phosphorylated to obtain the difluorinated phosphoramidite 35 in 75% yield as a diastereomeric mixture (Scheme 6a).
We could then successfully incorporate bespoke linkers i and j into oligonucleotides to obtain ON1-i (Scheme 5b) and ON1-j and ON2-j (Scheme 6b), respectively.
Our next aim was to determine the reactivity of these oligonucleotides toward the thiol conjugation reactions. We wanted to ascertain whether the substitutions on the alkene, the position of the vinylic double bond, and the type of ring, i.e., purine or pyrimidine, played any role on the reactivity. Hence, we performed a comparison of the reactivity profile of four compounds: ON1-b, ON1-c, ON1-i, and ON1-j owing to their structural differences, in terms of the location of double bond, purine/pyrimidine ring, and fluorine substitution on the double bond. Based on our conjugation studies, oligonucleotide ON1 with linker b and c showed the highest reactivity toward the thiol–ene click reaction. The head-to-head comparison of ON1-b vs ON1-c for the thiol–ene conjugation reaction with 2-mercaptoethanol at time points 0, 1, and 2 h (Figure 2) indicated that they exhibited similar reactivities in spite of the difference in location of the reactive double bond on the pyrimidine ring. It is worth mentioning that in both cases, the desired product formation was detected immediately after addition of the thiolation reagent (0 h time point).
Figure 2.

Reactivity comparison of (a) ON1-b and (b) ON1-c with 2-mercaptoethanol. Reaction conditions: PBS/H2O, 37 °C. (c) Deconvoluted mass spectrum of ON1-b′, [M]− mass calcd: 4613.20; found, 4613.40. (d) Deconvoluted mass spectrum of ON1-c′, [M]− mass calcd: 4613.20; found, 4613.00.
We observed that the oligonucleotide ON1-i with the purine-containing linker required an elevated temperature of 75 °C to react with the thiol reagent and led to poor conversion (4% conversion) to the desired product ON1-i′, exhibiting its much lower reactivity compared to the pyrimidine linker-bearing oligonucleotides (Scheme 7a).
Scheme 7. (a) Conjugation with Purine Analogue ON1-i and (b) Conjugation with Difluoro Analogue ON1-j.

Interestingly, the difluorinated linker-containing oligonucleotide ON1-j did not afford any conjugated product under the general thiol–ene click reaction conditions employed for the other pyrimidine linker-bearing oligonucleotides with an unsubstituted double bond. However, when oligonucleotide ON1-j was treated with 2-mercaptoethanol under near-neat conditions (with minimal amount of solvent; in our case, water), the thiol–ene click reaction proceeded at 65 °C to the desired conjugated product ON1-j′ with 33% conversion (based on LC–MS) (Scheme 7b).
Having demonstrated that our methodology could generate oligonucleotide conjugates rapidly and efficiently, and to test the applicability of our linkers on the therapeutic sequences already validated in the field, we decided to incorporate our linker b into the metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) antisense oligonucleotide (ASO) sequences ON3 and ON4 (Table 1).
MALAT1 is a highly abundant, well-conserved, nuclear-retained long noncoding RNA and is a common target used for testing ASOs.21,22 MALAT1 is expressed in a variety of cells and tissues and has been shown to play an important role in regulating genes at both transcriptional and post-transcriptional levels. The ability to incorporate our linker into a MALAT1 sequence would demonstrate the therapeutic application of this method. To our satisfaction, we successfully synthesized linker-bearing MALAT1 ASOs ON3-b and ON4-b following the standard SPS protocol. Next, we attempted the conjugation of the modified MALAT1 sequence ON3-b with various thiol-bearing molecules, namely, 2-mercaptoethanol, a hexapeptide P1 (NH2-SYQGWC-COOH) with a cysteine residue at the C-terminus,23 a lipid moiety, mercaptoethyl-d-α-tocopheryl succinate (Toco-SH) 37, and α-GalNAc-PEG3-Thiol (GalNAc-SH) (Scheme 8).
Scheme 8. Conjugation of MALAT1 ASOs with Thiol-Bearing Moieties.

Successful conjugation to these thiol-modified molecules would considerably enhance the applicability of our methodology. Linker b containing MALAT1 ASOs ON3-b and ON4-b reacted with 2-mercaptoethanol smoothly at 37 °C to afford the conjugated oligonucleotides ON3-b′ and ON4-b′ in 90 and 100% conversion, respectively. To further extend the applicability of our methodology, we embarked onto the conjugation of peptide P1 containing a cysteine to MALAT1 ASOs ON3-b and ON4-b. Peptide conjugation is a frequently utilized approach to improve the delivery of oligonucleotides.24−27 To our delight, peptide P1 reacted smoothly at 37 °C with both linker-bearing MALAT1 sequences, ON3-b and ON4-b, to afford desired peptide-conjugated products ON3-b-P1 and ON4-b-P1, in 79 and 92% conversion, respectively. To better understand the reactivity profile of MALAT1 ASOs ON3-b and ON4-b, we performed a time-course experiment of the peptide conjugation reactions with these sequences at different time points of 0, 1, 2, 3, 4, 6, and 22 h (Figure 3). In both cases, the oligonucleotides were completely consumed after 22 h with the formation of the desired peptide-conjugated oligonucleotides. A closer look at the reaction profile at different time points showed that the LNA-modified MALAT1 ON4-b seemed to be reacting faster than its counterpart, since at the 2 h time point, it already showed ∼50% conversion to the desired product, while the non-LNA sequence ON3-b required 4 h to reach a similar conversion to the expected product. Parallelly, we also carried out a control experiment by replacing the cysteine of the peptide with alanine (peptide P2, NH2-SYQGWA-COOH) and subjecting it to the previously described reaction conditions with ON3-b and ON4-b oligonucleotides. In both cases (and as expected), no reaction occurred, confirming the thiol-specific reactivity of our linker-bearing oligonucleotides (see the Supporting Information for chromatograms). Our initial conjugation was performed with 50 equiv of the peptide, but to enhance the applicability of our methodology, we reduced the equivalents of the peptide to 20 and 3 equiv to see if the conjugation can still be achieved. To our satisfaction, both reactions occurred under the same conditions to afford the desired peptide–oligonucleotide conjugate, albeit the reaction rate was slower and in 25 h, we obtained 22% conversion with 20 equiv peptide and in 27 h, we obtained 27% conversion with 3 equiv peptide (conversions determined by LC–MS). Experimental data of lower equivalence reactions and control experiment with only the peptide have been included in the Supporting Information.
Figure 3.

Conjugation reaction of (a) ON3-b and (b) ON4-b with peptide P1. (c) Deconvoluted mass spectrum of ON3-b-P1, [M]− mass calcd: 5959.42; found, 5959.00. (d) Deconvoluted mass spectrum of ON4-b-P1, [M]− mass calcd: 6127.48; found, 6127.30.
Conjugation of oligonucleotides with lipids is another widely explored strategy to improve site-specific delivery and cellular uptake of the oligonucleotides.28,29 It has been demonstrated that various lipid conjugates supported extra-hepatic delivery of both siRNAs30,31 and ASOs.32 Since tocopherol–small interfering RNA (siRNA) and tocopherol–ASO conjugates have previously demonstrated improved efficacy in vivo,33,34 we chose mercaptoethyl-d-α-tocopheryl succinate (TocoSH, 37) as the coupling partner for our conjugation reaction. Compound 37 was synthesized from commercially available d-α-tocopheryl succinate by N,N′-dicyclohexylcarbodiimide (DCC) coupling with 2-mercaptoethanol following the literature procedure35 (see the Supporting Information for the detailed synthetic protocol and NMR data). We then performed the conjugation reaction between ON3-b and 37. Due to its hydrophobic nature, 37 was dissolved in acetonitrile (ACN) and upon mixing with the oligonucleotide (dissolved in water) formed a slightly opaque solution. Trace amounts of the conjugated product (ON3-b-TF) were detected immediately; however, the overall conversion rate was relatively slow, most likely due to the biphasic nature of the reaction (Figure 4a). Running the reaction overnight did not improve the conversion (data not shown). Instead, undesired product formation was observed, resulting from the decomposition of 37 (visible at 8.4 min in the chromatogram in Figure 4a). As the formation of the desired conjugate ON3-b-TF and the decomposition of the starting material 37 were competitive, we decided to terminate the reaction after 4 h. To confirm that the undesired product was arising from the starting Toco-SH 37, and not the oligonucleotide, we performed a control experiment with 37, mimicking the same reaction conditions but without the oligonucleotide. As expected, this experiment also gave the same peak at 8.4 min by LC–MS (see the Supporting Information for the chromatogram), confirming our initial assumption.
Figure 4.

Conjugation reaction of ON3-b with (a) Toco-SH 37 (product: ON3-b-TF) and (b) GalNAc-SH (product: ON3-b-GalNAc). (c) Deconvoluted mass spectrum of ON3-b-TF, [M]− mass calcd: 5807.52; found, 5807.45. (d) Deconvoluted mass spectrum of ON3-b-GalNAc, [M]− mass calcd: 5586.04; found, 5586.45.
Our next target was to achieve an N-acetylgalactosamine (GalNAc) conjugation with our linker-containing MALAT1 ASO ON3-b. GalNAc is a carbohydrate moiety which binds to the asialoglycoprotein receptor and facilitates the uptake of oligonucleotides into hepatocytes by receptor-mediated endocytosis.36,37 The fact that 5 out of 6 clinically approved siRNA therapeutics are GalNAc conjugates,38−40 and many more being in preclinical developments, highlights the importance of this modification. For our experiment, we chose commercially available α-GalNAc-PEG3-Thiol (GalNAc-SH) as the conjugating partner. To our satisfaction, ON3-b reacted smoothly with GalNAc-SH at 37 °C in aqueous conditions and underwent quantitative conversion within 4 h to afford the GalNAc-conjugated oligonucleotide ON3-b-GalNAc (Figure 4b). This achievement opens up the avenue for applicability of our conjugation method for future research related to tissue-specific delivery and improved efficacy, which has already proven to be a success with GalNAc-conjugated ASOs.41,42 To demonstrate the wider applicability of our conjugation technology, we repeated the reaction with lower equivalents of GalNAc-SH (20 and 3 equiv). Both reactions occurred smoothly under the same conditions to afford the desired GalNAc–oligonucleotide conjugate. However, in this case, the reaction rate was slower: we obtained 70% conversion in 23 h with 20 equiv GalNAc-SH and 49% conversion in 50 h with 3 equiv GalNAc-SH (conversions determined by LC–MS). Experimental data of lower equivalence reactions have been included in the Supporting Information.
Conclusions
In summary, we have designed and synthesized a set of novel vinylpyrimidine phosphoramidite linkers, which were successfully incorporated at the 5′ end of various oligonucleotide sequences, including MALAT1, to generate unique oligonucleotides by standard SPS. The syntheses of the phosphoramidites themselves are simple, and thus, the resulting systems will be accessible in practical terms. These linker-modified oligonucleotides were successfully conjugated in a biocompatible manner to thiol-containing molecules such as 2-mercaptoethanol, GSH, peptide, lipid, and GalNAc, exhibiting their specificity and powerful potential for therapeutic and diagnostic applications. The present strategy highlights the sustainability, versatility, and efficiency of our linker technology, displaying its utility in the bioconjugation of oligonucleotides with various thiol functionalities. Furthermore, in our future work, we will focus on potential applications of our developed methodology such as in attaching thiol-functionalized molecular transporters to therapeutic oligonucleotides to improve their PK/PD properties and/or tissue specific delivery, attaching fluorescent moieties to oligonucleotides for use as an imaging tool or probe, and in attaching oligonucleotides to thiol-functionalized surfaces, including the surface of nanomaterials, to build spherical nucleic acids.
Experimental Procedures
Material and Methods
All reactions were carried out under an atmosphere of argon. Reactions were monitored by TLC using aluminum-backed silica gel 60 (F254) plates, visualized using UV 254 nm and vanillin dips. Flash column chromatography was carried out with silica gel (60–120 mesh). Reagents were used as received from commercial sources, unless otherwise stated. All commercial amino alcohols were a mixture of cis and trans isomers unless indicated otherwise in structures or write up. Dry solvents were purchased and used as received. 1H NMR spectra were recorded on a Bruker AVANCE 400 spectrometer (400 MHz) at room temperature. 13C NMR spectra were recorded at a frequency of 101 MHz. Chemical shifts were reported in δ units (parts per million), and coupling constants (J) were measured in Hertz (Hz). The residual solvent signals were used as references. 31P NMR spectra (decoupled) were recorded at a frequency of 162 MHz and chemical shifts were reported in ppm. 19F NMR spectra (decoupled) were recorded at a frequency of 376 MHz and chemical shifts were reported in ppm. Mass spectra were recorded on a Waters LC–MS system with an Acquity QDa detector in positive ion detection mode (ESI+) and analyzed as m/z [M + H]+.
General Method for the Synthesis of Linkers
Compounds 2, 2′, 3, 3′, 4, 4′, 5, 5′, and 6, 6′ were synthesized following method A step 1. Compounds 7, 8, 9, 13, 14, and 21# were synthesized following method A step 2. Compounds 10, 11, 12, 15, 16, 25, 26, and 30 were synthesized following method A step 3.
Representative Examples
Method A Step 1
3-((2-Chloropyrimidin-4-yl)(methyl)amino)cyclobutan-1-ol (2)
To a solution of 2,4-dichloro-pyrimidine 1 (1.00 g, 6.7 mmol) in THF (10 mL) were added Et3N (16.75 mmol, 0.43 mL) and commercially available 3-(methylamino)cyclobutan-1-ol (cis/trans 5:1) (1.02 g, 10.05 mmol). The mixture was stirred at ambient temperature for 7 h and concentrated under reduced pressure. The residue was dissolved in DCM (20 mL) and the solution was poured into saturated aqueous sodium bicarbonate solution (5 mL). The two layers were separated, and the aqueous layer was extracted with DCM (2 × 20 mL). The combined organic layer was dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash column chromatography using hexane/EtOAc (0–100%) to afford 3-((2-chloropyrimidin-4-yl)(methyl)amino)cyclobutan-1-ol 2 (major product) (925 mg, 64.6%) as a pale-yellow gum and 3-((4-chloropyrimidin-2-yl)(methyl)amino)cyclobutan-1-ol 2′ (minor product) (140 mg, 9.8%) as a yellow viscous oil. For 3-((2-chloropyrimidin-4-yl)(methyl)amino)cyclobutan-1-ol 2 (major product), 1H NMR (400 MHz, CDCl3) (cis/trans mixture): δ 7.86 (dd, J = 7.4, 5.9 Hz, 1H), 6.32–6.16 (m, 1H), 4.67–4.33 (m, 1H), 4.09–4.01 (m, 1H), 2.97–2.94 (m, 3H), 2.73–2.33 (m, 2H), 2.32–1.92 (m, 2H) (one exchangeable proton of OH was not observed). 13C NMR (101 MHz, CDCl3) (cis/trans mixture): δ 162.95, 162.90, 160.24, 160.19, 156.68, 156.60, 102.04, 63.82, 60.60, 60.54, 42.66, 38.60, 37.16, 30.68, 30.57.
LC–MS (ESI-MS) m/z: calcd for C9H13ClN3O+, 214.074; found, 214.079 [M + H]+.
For 3-((4-chloropyrimidin-2-yl)(methyl)amino)cyclobutan-1-ol 2′ (minor product), 1H NMR (400 MHz, CDCl3) (cis/trans mixture): δ 8.08 (d, J = 5.1 Hz, 1H), 6.43 (dd, J = 5.0, 1.0 Hz, 1H), 4.55 (tt, J = 9.7, 7.5 Hz, 1H), 4.11–3.97 (m, 1H), 3.05 (d, J = 6.7 Hz, 3H), 2.66–2.58 (m, 2H), 2.21–1.97 (m, 3H).
LC–MS (ESI-MS) m/z: calcd for C9H13ClN3O+, 214.074; found, 214.078 [M + H]+.
Method A Step 2
3-(Methyl(2-vinylpyrimidin-4-yl)amino)cyclobutan-1-ol (7)
To a solution of 3-((2-chloropyrimidin-4-yl)(methyl)amino)cyclobutan-1-ol 2 (141 mg, 0.66 mmol) and vinylboronic acid pinacol ester (0.34 mL, 1.98 mmol) in dioxane (4 mL) and water (1 mL) under an argon atmosphere, Na2CO3 (245 mg, 2.31 mmol) was added. The mixture was degassed for 5 min. Then, catalyst Pd(dppf)Cl2·DCM (54 mg, 0.66 mmol) was added to the mixture and further degassed for 5 min. The reaction mixture was warmed up to 85 °C and stirred at this temperature for 18 h. The reaction mixture was cooled to room temperature, diluted with DCM, and filtered through a bed of Celite, washing several times with DCM (3 × 15 mL). The solvent was removed under vacuum. The residue was subjected to flash column chromatography using heptane/ethyl acetate (0–100%) to afford the product 3-(methyl(2-vinylpyrimidin-4-yl)amino)cyclobutan-1-ol 7 (130 mg, 96.7%) as a pale-yellow gum. 1H NMR (400 MHz, CDCl3) (cis/trans mixture): δ 8.09 (t, J = 6.0 Hz, 1H), 6.67 (dd, J = 17.3, 10.4 Hz, 1H), 6.56–6.40 (m, 1H), 6.32–6.15 (m, 1H), 5.59 (dd, J = 10.4, 2.0 Hz, 1H), 4.55–4.30 (m, 1H), 4.10 (tt, J = 7.6, 6.5 Hz, 1H), 3.17–2.83 (m, 4H), 2.73–2.62 (m, 2H), 2.15–2.06 (m, 2H). 13C NMR (101 MHz, CDCl3) (cis/trans mixture): δ 163.19, 163.16, 161.81, 161.77, 155.36, 155.32, 137.48, 137.45, 122.44, 122.40, 101.96, 101.90, 60.94, 60.55, 42.25, 38.89, 37.39, 30.26, 30.23.
LC–MS (ESI-MS) m/z: calcd for C11H16N3O+, 206.129; found, 206.128 [M + H]+.
Method A Step 3
2-Cyanoethyl (3-(Methyl(2-vinylpyrimidin-4-yl)amino)cyclobutyl) Diisopropylphosphoramidite (10)
A stirred solution of 3-(methyl(2-vinylpyrimidin-4-yl)amino)cyclobutan-1-ol 7 (128 mg, 0.62 mmol) (dried overnight in high vacuum) in DCM (8 mL) was degassed by bubbling with argon for 5 min. The solution was cooled to 0 °C in an ice bath and DIPEA (0.32 mL, 1.86 mmol) was added, followed by dropwise addition of 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (0.21 mL, 0.93 mmol). The reaction mixture was allowed to gradually warm to room temperature and stirred for 6 h. The solvent was removed under vacuum. The residue was subjected to flash column chromatography using heptane/(1% Et3N)/EtOAc (0–100%) to afford product 2-cyanoethyl (3-(methyl(2-vinylpyrimidin-4-yl)amino)cyclobutyl) diisopropylphosphoramidite (diastereomeric mixture) 10 (136 mg, 54.1%) as a colorless gum. 1H NMR (400 MHz, CD2Cl2) (mixture of diastereoisomers): δ 8.14 (d, J = 6.1 Hz, 0.79H), 7.98 (d, J = 6.1 Hz, 0.24H), 6.80–6.63 (m, 1H), 6.60–6.43 (m, 1H), 6.43–6.19 (m, 1H), 5.58 (dd, J = 10.4, 2.3 Hz, 1H), 4.67–4.41 (m, 1H), 4.27–4.09 (m, 1H), 3.92–3.73 (m, 2H), 3.70–3.60 (m, 2H), 3.05 (d, J = 5.6 Hz, 3H), 2.91–2.53 (m, 4H), 2.39–2.16 (m, 2H), 1.27–1.17 (m, 12H). 31P NMR (162 MHz, CD2Cl2) (mixture of diastereoisomers): δ 146.51, 145.59.
Compound 17 was synthesized following method B step 1. Compounds 18, 19, 22, and 33 were synthesized following method B step 2. Compounds 23, 24, 27, and 35 were synthesized following method B step 3 which is the same as method A.
Method B Step 1
1-(4-Vinylpyrimidin-2-yl)piperidin-4-ol (22)
To a solution of 2,4-dichloropyrimidine 1 (519 mg, 3.48 mmol) in anhydrous 2-propanol (10 mL), potassium trifluorovinyl borate (500 mg, 3.83 mmol) and Et3N (0.97 mL, 10.4 mmol) were added under an argon atmosphere in a microwave tube. The mixture was degassed for 5 min. Then, the catalyst Pd(dppf)Cl2·DCM (142 mg, 0.174 mmol) was added to the mixture, and the mixture was further degassed for 5 min. The tube was sealed, and the reaction mixture was warmed up to 90 °C and stirred at this temperature for 5 h. The reaction mixture was then cooled to room temperature, diluted with ethyl acetate (25 mL), filtered through a bed of Celite, and washed several times with ethyl acetate (3 × 10 mL). The solvent was removed under vacuum. Flash column chromatography using heptane/EtOAc (0–100%) afforded product 2-chloro-4-vinylpyrimidine 17 (296 mg, 60.5%) as a pale-yellow oil. 1H NMR data was consistent with the literature data.xvii
1H NMR (400 MHz, CDCl3): δ 8.57 (dd, J = 5.1, 1.3 Hz, 1H), 7.23 (dd, J = 5.1, 0.9 Hz, 1H), 6.71 (ddd, J = 17.4, 10.6, 1.4 Hz, 1H), 6.54 (ddd, J = 17.4, 1.7, 1.0 Hz, 1H), 5.80 (dt, J = 10.5, 1.3 Hz, 1H).
Method B Step 2
1-(4-Vinylpyrimidin-2-yl)piperidin-4-ol (22)
A solution of 2-chloro-4-vinylpyrimidine 17 (100 mg, 0.711 mmol), piperidin-4-ol (86.3 mg, 0.853 mmol), and anhydrous DIPEA (0.35 mL, 1.78 mmol) in anhydrous DMF (1.5 mL) was stirred at 100 °C in a sealed tube for 18 h. The reaction mixture was cooled to room temperature, and solvent was removed under vacuum. Flash column chromatography using heptane/ethyl acetate (0–100%) afforded product 1-(4-vinylpyrimidin-2-yl)piperidin-4-ol 22 (49 mg, 34%) as a pale-brown gum.
1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 5.0 Hz, 1H), 6.59 (dd, J = 17.4, 10.5 Hz, 1H), 6.48 (d, J = 5.0 Hz, 1H), 6.37 (dd, J = 17.4, 1.6 Hz, 1H), 5.57 (dd, J = 10.5, 1.6 Hz, 1H), 4.62–4.40 (m, 2H), 3.94 (tt, J = 8.7, 4.0 Hz, 1H), 3.31 (ddd, J = 13.4, 10.0, 3.3 Hz, 2H), 2.00–1.92 (m, 3H), 1.59–1.50 (m, 2H).
13C NMR (101 MHz, CDCl3): δ 162.89, 161.80, 158.42, 136.27, 121.48, 107.09, 68.57, 41.51, 34.37.
LC–MS (ESI-MS) m/z: calcd for C11H16N3O+, 206.129; found, 206.122 [M + H]+.
Method B Step 3
2-Cyanoethyl (1-(4-Vinylpyrimidin-2-yl)piperidin-4-yl) Diisopropylphosphoramidite (27)
Starting from 1-(4-vinylpyrimidin-2-yl)piperidin-4-ol 22, compound 2-cyanoethyl (1-(4-vinylpyrimidin-2-yl)piperidin-4-yl) diisopropylphosphoramidite 27 (diastereomeric mixture) was synthesized in 67% yield as a colorless gum, following the same synthetic procedure as mentioned in method A step 3.
1H NMR (400 MHz, CD2Cl2) (mixture of diastereoisomers): δ 8.29 (d, J = 5.0 Hz, 1H), 6.61 (dd, J = 17.3, 10.5 Hz, 1H), 6.50 (d, J = 5.0 Hz, 1H), 6.40 (dd, J = 17.3, 1.7 Hz, 1H), 5.58 (dd, J = 10.5, 1.7 Hz, 1H), 4.24–4.10 (m, 3H), 3.91–3.76 (m, 2H), 3.75–3.60 (m, 4H), 2.67 (t, J = 6.3 Hz, 2H), 2.02–1.87 (m, 2H), 1.76–1.66 (m, 2H), 1.23 (dd, J = 6.8, 1.2 Hz, 12H).
31P NMR (162 MHz, CD2Cl2) (mixture of diastereoisomers): δ 146.14.
General Method for the Synthesis of Oligonucleotides Containing Novel Linkers
Solid-Phase Oligonucleotide Synthesis
Oligonucleotides were synthesized on a 1 μmol scale with the MM12 synthesizer (LGC Biosearch Technologies) using 1000 Å Universal Controlled Pore Glass (CPG) (loading 47.7 μmol/g, LGC Biosearch Technologies). Cleavage of the 4,4′-dimethoxytrityl group was performed with 3% trichloroacetic acid in dichloromethane (DCM) (LGC Biosearch Technologies). DNA and LNA phosphoramidites were prepared as 0.1 M solutions in dry ACN (LGC Biosearch Technologies). Linker phosphoramidites were dissolved in a 50:50 THF/DCM mixture (0.08 M). Solvents were moisture controlled with less than 30 ppm water content. A 0.25 M solution of 5-ethylthio-1H-tetrazole in dry ACN (LGC Biosearch Technologies) was used as an activator. Failed sequences were capped with a 1:1 mixture of Capping Mix A (tetrahydrofuran/lutidine/acetic anhydride, 8:1:1, v/v/v, LGC Biosearch Technologies) and Capping Mix B (16% N-methylimidazole in tetrahydrofuran, LGC Biosearch Technologies). The oxidizing step was performed with 0.02 M iodine in tetrahydrofuran/pyridine/water (7:2:1, v/v/v, LGC Biosearch Technologies).
Oligonucleotide synthesis parameters: Deblock: 2 × 60 s, coupling times: 3 × 180 s (standard DNA phosphoramidites), 3 × 480 s (LNA phosphoramidites), capping: 2 × 60 s, oxidation: 1 × 60 s. For modified phosphoramidites (linker at the 5′ end of the oligonucleotide), the coupling time was 4 × 360 s and the capping step was removed.
General Method for Cleavage and Deprotection of Linker-Containing Oligonucleotides
Method I
5 mg of the CPG containing the linker-modified oligonucleotide was placed in a vial, treated with 100 μL of 0.4 M sodium hydroxide (NaOH) in methanol/water (4:1) solution, and shaken (Eppendorf ThermoMixerC) for 20 min at 80 °C. The sample was left to cool down to room temperature, spun down, and filtered. The filtrate was frozen and freeze-dried. The sample was desalted on a Glen Gel-Pak 0.2 Desalting Column (catalogue no. 61-5002-50, Glen Research) following the manufacturer’s protocol. The collected sample was frozen and freeze-dried.
Method II
5 mg of the CPG containing the linker-modified oligonucleotide was placed in a vial, treated with 100 μL of 32% aqueous ammonia solution, and shaken (Eppendorf ThermoMixerC) overnight at 23 °C. The sample was spun down and filtered. The filtrate was frozen and freeze-dried.
General Method for Oligonucleotide Purification
The sample was purified on a Waters preparative high-performance liquid chromatography (HPLC) system (XBridge BEH C18 OBD Prep Column; 130 Å; 10 mm × 250 mm, 5 μm; Waters) with a flow rate of 4 mL/min. Eluent A: 0.1 M triethylammonium acetate (TEAA) in water; eluent B: 0.1 M TEAA in 50% ACN; gradient 15–45% B in 20 min. Collected fractions were directly desalted on the Glen Gel-Pak 1.0 Desalting Column (catalogue no. 61-5010-50, Glen Research) following the manufacturer’s protocol. The collected sample was frozen and freeze-dried. The sample was dissolved in water and analyzed by LC–MS [Waters LC–MS system with an Acquity QDa detector, ACQUITY PREMIER Oligonucleotide BEH C18 column (130 Å; 2.1 × 50 mm, 1.7 μm; Waters)] at 65 °C with a flow rate of 0.3 mL/min. Eluent A: 7 mM triethylamine (TEA), 80 mM hexafluoroisopropanol (HFIP) in water; eluent B: 3.5 mM TEA, 40 mM HFIP in 50% ACN; gradient 5–30% B in 8 min. Samples were run in the negative mode (ESI–) and analyzed as m/z [M]− or [M – H]−. The purity of the oligonucleotides was auto measured using the integration function on MassLynx software V4.2 (Waters).
General Method for the Analysis of the Oligonucleotide Conjugates
Unless stated otherwise, oligonucleotide conjugates were analyzed on the Waters LC–MS system [with the Acquity QDa detector, ACQUITY PREMIER Oligonucleotide BEH C18 column (130 Å; 2.1 × 50 mm, 1.7 μm; Waters)] at 65 °C with a flow rate of 0.3 mL/min. Eluent A: 7 mM TEA, 80 mM HFIP in water; eluent B: 3.5 mM TEA, 40 mM HFIP in 50% ACN; gradient 5–30% B in 8 min. Samples were run in the negative mode (ESI–) and analyzed as m/z [M]− or [M – H]−.
General Method for Thiol–Ene Click Reactions
Conjugation with β-Mercaptoethanol
A solution of 100 mM β-mercaptoethanol (β-ME) in phosphate-buffered saline (PBS) was freshly prepared: 1.4 μL of β-ME was added to 200 μL of 5× PBS. Then, the oligonucleotide sample (1 nmol dissolved in 50 μL of water) was mixed with 25 μL of conjugation buffer (containing β-ME) and incubated for 2 h at 37 °C. LC–MS analysis (conditions as described above) was performed immediately after mixing (0 h) and then after 2 h (by mixing 5–10 μL of the sample with 10 μL of water) to verify formation of the conjugated product.
Conjugation with GSH
A stock solution of 200 mM reduced GSH in water was freshly prepared: 12.3 mg (40 μmol) of GSH was added to 200 μL of water. Then, the oligonucleotide sample (2 nmol dissolved in 50 μL of water) was mixed with 25 μL of the stock solution (containing GSH) and the mixture was incubated for 6 h at 37 °C. LC–MS analysis (conditions as described above) was performed immediately after mixing (0 h) and then every 2 h (for 6 h in total) to verify formation of the conjugated product (by mixing 5 μL of the sample with 5 μL of water).
Conjugation with Peptides
Peptide P1 sequence (P1): NH2-SYQGWC-COOH.
Peptide P2 sequence (P2): NH2-SYQGWA-COOH.
100 nmol of P1/P2 was dissolved in 50 μL of a 5× PBS buffer. Then, the oligonucleotide sample (1 nmol dissolved in 50 μL of water) was mixed with 25 μL of the buffer solution (containing P1 or P2) and incubated at 37 °C. LC–MS analysis (conditions as described above) was performed immediately after mixing (0 h) and then after 1, 2, 3, 4, 6, and 22 h (for conjugation with P1) or 1, 2, and 4 h (for conjugation with P2) to verify formation of the conjugated product (by mixing 5–10 μL of the sample with 10 μL of water).
Conjugation with Lipid
A 4 mM stock solution of mercaptoethyl-d-α-tocopheryl succinate (37) in ACN was freshly prepared: 23.6 mg (40 μmol) of 37 was dissolved in 10 mL of ACN. Oligonucleotide sample (1 nmol dissolved in 50 μL of water) was mixed with 25 μL (100 nmol) of the stock solution containing lipid-thiol and incubated at 37 °C. LC–MS analysis (standard conditions) was performed immediately after mixing (0 h) and then after 2 and 4 h to verify formation of the conjugated product (by mixing 5 μL of the sample with 5 μL of water).
Conjugation with GalNAc
A 4 mM stock solution of commercially available α-GalNAc-PEG3-thiol in water was freshly prepared: 1.48 mg (4 μmol) of α-GalNAc-PEG3-thiol was dissolved in 1 mL of water. Oligonucleotide sample (1 nmol dissolved in 50 μL of water) was mixed with 25 μL (100 nmol) of the GalNAc-thiol-containing stock solution and incubated at 37 °C for 4 h. LC–MS analysis (standard conditions) was performed immediately after mixing (0 h) and then after 2 and 4 h to verify formation of the conjugated product (by mixing 5 μL of the sample with 5 μL of water).
Acknowledgments
We would like to thank Professor Nick Lench for his support and critical reading of the manuscript and Mark Cunningham for his overall support. The Nucleic Acid Therapy Accelerator (NATA) is funded by the Medical Research Council UK (MRC): grant reference no. MC_PC_20061.
Glossary
Abbreviations
- EtOAc
ethyl acetate
- DIPEA
N,N-diisopropylethylamine
- ClP(OCE)N(iPr)2
2-cyanoethyl N,N-diisopropylchlorophosphoramidite
- CH2=CHBF3K
potassium vinyltrifluoroborate
- Pd(dppf)Cl2·DCM
[1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-(dimethylamino)pyridine
- DMF
N,N-dimethylformamide
- THF
tetrahydrofuran
- DCM
dichloromethane
- rt
room temperature
- eq
equivalents
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.4c00336.
Additional experimental procedures, NMR spectra, and LC–MS chromatograms (PDF)
The authors declare the following competing financial interest(s): The phosphoramidites, a few of the oligonucleotides, and their conjugates reported in this manuscript are the subject of a patent application from our organisation where the corresponding author (SB) is an inventor (UK Patent Application Number - GB2311271.7).
Special Issue
Published as part of Bioconjugate Chemistryspecial issue “Early Career Innovators in Bioconjugate Chemistry”.
Supplementary Material
References
- Roberts T. C.; Langer R.; Wood M. J. A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discovery 2020, 19, 673–694. 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; Witzigmann D.; Thomson S. B.; Chen S.; Leavitt R.; Cullis P. R.; van der Meel R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- Juliano R. L.; Ming X.; Nakagawa O. The Chemistry and Biology of Oligonucleotide Conjugates. Acc. Chem. Res. 2012, 45 (7), 1067–1076. 10.1021/ar2002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S.; Oliver P. L. The chemistry and biology of oligonucleotide conjugation. Nucleic Acid Insights 2024, 1 (3), 127–138. 10.18609/nai.2024.018. [DOI] [Google Scholar]
- Ming X.; Laing B. Bioconjugates for targeted delivery of therapeutic oligonucleotides. Adv. Drug Delivery Rev. 2015, 87, 81–89. 10.1016/j.addr.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. Oligonucleotide conjugates for therapeutic applications. Ther. Deliv. 2013, 4 (7), 791–809. 10.4155/tde.13.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial M.; Postma A. Disulfide conjugation chemistry: a mixed blessing for therapeutic drug delivery. Ther. Deliv. 2017, 8 (6), 359–362. 10.4155/tde-2017-0003. [DOI] [PubMed] [Google Scholar]
- Astakhova K.; Ray R.; Taskova M.; Uhd J.; Carstens A.; Morris K. Clicking” Gene Therapeutics: A Successful Union of Chemistry and Biomedicine for New Solutions. Mol. Pharm. 2018, 15 (8), 2892–2899. 10.1021/acs.molpharmaceut.7b00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantoni N.; El-Sagheer A. H.; Brown T. A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev. 2021, 121, 7122–7154. 10.1021/acs.chemrev.0c00928. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Matin M. M.; Matin P.; Rahman M. R.; Ben Hadda T.; Almalki F. A.; Mahmud S.; Ghoneim M. M.; Alruwaily M.; Alshehri S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 864286. 10.3389/fmolb.2022.864286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A.; Pedroso E.; Grandas A. Maleimide-Dimethylfuran exo Adducts: Effective Maleimide Protection in the Synthesis of Oligonucleotide Conjugates. Org. Lett. 2011, 13 (16), 4364–4367. 10.1021/ol201690b. [DOI] [PubMed] [Google Scholar]
- Sanchez A.; Pedroso E.; Grandas A. Easy introduction of maleimides at different positions of oligonucleotide chains for conjugation purposes. Org. Biomol. Chem. 2012, 10, 8478–8483. 10.1039/c2ob26514a. [DOI] [PubMed] [Google Scholar]
- Mangla P.; Vicentini Q.; Biscans A. Therapeutic Oligonucleotides: An Outlook on Chemical Strategies to Improve Endosomal Trafficking. Cells 2023, 12, 2253. 10.3390/cells12182253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjærsgaard N. L.; Hansen R. A.; Gothelf K. V. Preparation of Maleimide-Modified Oligonucleotides from the Corresponding Amines Using N-Methoxycarbonylmaleimide. Bioconjugate Chem. 2022, 33 (7), 1254–1260. 10.1021/acs.bioconjchem.2c00144. [DOI] [PubMed] [Google Scholar]
- Halloy F.; Iyer P. S.; Ćwiek P.; Ghidini A.; Barman-Aksözen J.; Wildner-Verhey van Wijk N.; Theocharides A. P. A.; Minder E. I.; Schneider-Yin X.; Schümperli D.; Hall J. Delivery of oligonucleotides to bone marrow to modulate ferrochelatase splicing in a mouse model of erythropoietic protoporphyria. Nucleic Acids Res. 2020, 48 (9), 4658–4671. 10.1093/nar/gkaa229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyle C. E.; Bowman C. N. Thiol–Ene Click Chemistry. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- Seki H.; Walsh S. J.; Bargh J. D.; Parker J. S.; Carroll J.; Spring D. R. Rapid and robust cysteine bioconjugation with vinylheteroarenes. Chem. Sci. 2021, 12, 9060–9068. 10.1039/D1SC02722K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raux E.; Klenc J.; Blake A.; Sączewski J.; Strekowski L. Conjugate Addition of Nucleophiles to the Vinyl Function of 2-Chloro-4-vinylpyrimidine Derivatives. Molecules 2010, 15 (3), 1973–1984. 10.3390/molecules15031973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Zhu C.; Cai Y.; Jiang H. Solvent-Switched Oxidation Selectivities with O2: Controlled Synthesis of α-Difluoro(thio)methylated Alcohols and Ketones. Angew. Chem., Int. Ed. 2021, 60, 12038–12045. 10.1002/anie.202017271. [DOI] [PubMed] [Google Scholar]
- Amodio N.; Stamato M. A.; Juli G.; Morelli E.; Fulciniti M.; Manzoni M.; Taiana E.; Agnelli L.; Cantafio M. E. G.; Romeo E.; Raimondi L.; Caracciolo D.; Zuccala V.; Rossi M.; Neri A.; Munshi N. C.; Tagliaferri P.; Tassone P. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. 10.1038/s41375-018-0067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Hamblin M. H.; Yin K. J. The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 2017, 14, 1705–1714. 10.1080/15476286.2017.1358347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Long T.; Zheng J.; Sheng W.; Sun S.; Wei W.; Zhao J.; Wang H. Cysteine-Specific Bioconjugation and Stapling of Unprotected Peptides by Chlorooximes. CCS Chem. 2022, 4, 3355–3363. 10.31635/ccschem.021.202101386. [DOI] [Google Scholar]
- Klabenkova K.; Fokina A.; Stetsenko D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. 10.3390/molecules26175420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fàbrega C.; Aviñó A.; Navarro N.; Jorge A. F.; Grijalvo S.; Eritja R. Lipid and Peptide-Oligonucleotide Conjugates for Therapeutic Purposes: From Simple Hybrids to Complex Multifunctional Assemblies. Pharmaceutics 2023, 15, 320. 10.3390/pharmaceutics15020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClorey G.; Banerjee S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. 10.3390/biomedicines6020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knerr L.; Prakash T. P.; Lee R.; Drury III W. J.; Nikan M.; Fu W.; Pirie E.; Maria L. D.; Valeur E.; Hayen A.; Ölwegård-Halvarsson M.; Broddefalk J.; Ämmälä C.; Østergaard M. E.; Meuller J.; Sundström L.; Andersson P.; Janzén D.; Jansson-Löfmark R.; Seth P. P.; Andersson S. Glucagon Like Peptide 1 Receptor Agonists for Targeted Delivery of Antisense Oligonucleotides to Pancreatic Beta Cell. J. Am. Chem. Soc. 2021, 143 (9), 3416–3429. 10.1021/jacs.0c12043. [DOI] [PubMed] [Google Scholar]
- Tran P.; Weldemichael T.; Liu Z.; Li H. Delivery of Oligonucleotides: Efficiency with Lipid Conjugation and Clinical Outcome. Pharmaceutics 2022, 14, 342. 10.3390/pharmaceutics14020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Feng K.; Li L.; Yang L.; Pan X.; Yazd H. S.; Cui C.; Li J.; Moroz L.; Sun Y.; Wang B.; Li X.; Huang T.; Tan W. Lipid–oligonucleotide conjugates for bioapplications. Natl. Sci. Rev. 2020, 7, 1933–1953. 10.1093/nsr/nwaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscans A.; Coles A.; Haraszti R.; Echeverria D.; Hassler M.; Osborn M.; Khvorova A.; Osborn M.; Khvorova A. Diverse lipid conjugates for functional extra-hepatic siRNA delivery in vivo. Nucleic Acids Res. 2019, 47 (3), 1082–1096. 10.1093/nar/gky1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscans A.; Caiazzi J.; McHugh N.; Hariharan V.; Muhuri M.; Khvorova A. Docosanoic acid conjugation to siRNA enables functional and safe delivery to skeletal and cardiac muscles. Mol. Ther. 2021, 29 (4), 1382–1394. 10.1016/j.ymthe.2020.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash T. P.; Mullick A. E.; Lee R. G.; Yu J.; Yeh S. T.; Low A.; Chappell A. E.; Østergaard M. E.; Murray S.; Gaus H. J.; Swayze E. E.; Seth P. P. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47 (12), 6029–6044. 10.1093/nar/gkz354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina K.; Unno T.; Uno Y.; Kubodera T.; Kanouchi T.; Mizusawa H.; Yokota T. Efficient In Vivo Delivery of siRNA to the Liver by Conjugation of α-Tocopherol. Mol. Ther. 2008, 16 (4), 734–740. 10.1038/mt.2008.14. [DOI] [PubMed] [Google Scholar]
- Nishina T.; Numata J.; Nishina K.; Yoshida-Tanaka K.; Nitta K.; Piao W.; Iwata R.; Ito S.; Kuwahara H.; Wada T.; Mizusawa H.; Yokota T. Chimeric Antisense Oligonucleotide Conjugated to α-Tocopherol. Mol. Ther. Nucleic Acids 2015, 4 (1), e220 10.1038/mtna.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanslambrouck S.; Riva R.; Ucakar B.; Préat V.; Gagliardi M.; Molin D. G. M.; Lecomte P.; Jérôme C. Thiol-ene Reaction: An Efficient Tool to Design Lipophilic Polyphosphoesters for Drug Delivery Systems. Molecules 2021, 26, 1750. 10.3390/molecules26061750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza A. A.; Devarajan P. V. Asialoglycoprotein receptor mediated hepatocyte targeting Strategies and applications. J. Controlled Release 2015, 203, 126–139. 10.1016/j.jconrel.2015.02.022. [DOI] [PubMed] [Google Scholar]
- Tanowitz M.; Hettrick L.; Revenko A.; Kinberger G. A.; Prakash T. P.; Seth P. P. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017, 45 (21), 12388–12400. 10.1093/nar/gkx960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli M.; Manoharan M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51 (6), 2529–2573. 10.1093/nar/gkad067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer A. D.; Dowdy S. F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28 (3), 109–118. 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal I.; Eells K.; Hudson I. A Comparison of Currently Approved Small Interfering RNA (siRNA) Medications to Alternative Treatments by Costs, Indications, and Medicaid Coverage. Pharmacy 2024, 12, 58. 10.3390/pharmacy12020058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker B. F.; Xia S.; Partridge W.; Engelhardt J. A.; Tsimikas S.; Crooke S. T.; Bhanot S.; Geary R. S. Safety and Tolerability of GalNAc3-Conjugated Antisense Drugs Compared to the Same-Sequence 2’- O-Methoxyethyl-Modified Antisense Drugs: Results from an Integrated Assessment of Phase 1 Clinical Trial Data. Nucleic Acid Ther. 2024, 34 (1), 18–25. 10.1089/nat.2023.0026. [DOI] [PubMed] [Google Scholar]
- Al Shaer D.; Al Musaimi O.; Albericio F.; de la Torre B. G. 2023 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2024, 17, 243. 10.3390/ph17020243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.