

Abstract

Darunavir, a frontline treatment for HIV infection, faces limitations due to emerging multidrug resistant (MDR) HIV strains, necessitating the development of analogs with improved activity. In this study, a combinatorial in silico approach was used to initially design a series of HIV-1 PI analogs with modifications at key sites, P1′ and P2′, to enhance interactions with HIV-1 PR. Fifteen analogs with promising binding scores were selected for synthesis and evaluated for the HIV-1 PR inhibition activity. The variation of P2′ substitution was found to be effective, as seen in 5aa (1.54 nM), 5ad (0.71 nM), 5ac (0.31 nM), 5ae (0.28 nM), and 5af (1.12 nM), featuring halogen, aliphatic, and alkoxy functionalities on the phenyl sulfoxide motif exhibited superior inhibition against HIV-1 PR compared to DRV, with minimal cytotoxicity observed in Vero and 293T cell lines. Moreover, computational studies demonstrated the potential of selected analogs to inhibit various HIV-1 PR mutations, including I54M and I84V. Further structural dynamics and energetic analyses confirmed the stability and binding affinity of promising analogs, particularly 5ae, which showed strong interactions with key residues in HIV-1 PR. Overall, this study underscores the importance of flexible moieties and interaction enhancement at the S2′ subsite of HIV-1 PR in developing effective DRV analogs to combat HIV and other global health issues.
Keywords: human immunodeficiency virus, HIV-1 protease inhibitors, combinatorial drug design, darunavir, inhibition
Introduction
Acquired immunodeficiency syndrome (AIDS), resulting from human immunodeficiency virus (HIV) infection, remains a significant global health challenge.1,2 The enzyme HIV type-1 protease (HIV-1 PR), crucial in the virus’s life cycle, has been found to be a key target for inhibiting HIV.3−7 To date, the FDA has approved ten HIV-1 protease inhibitors (PIs),8−12 ushering in a new era of highly active antiretroviral therapy (HAART) combining reverse transcriptase inhibitors (RTIs) with these PIs.13,14 Despite these advancements, the persistent threat of multidrug resistance (MDR) undermines long-term treatment efficacy.15−17 Notably, darunavir (DRV), a potent second-generation drug, is also affected by reduced effectiveness against mutant strains of HIV.18−22 The urgent need for potent PIs to address challenges in HIV patient management is therefore undeniable. Enhancing the interaction between inhibitors and the PR emerges as a pivotal strategy for overcoming drug resistance. Subtle mutations not only disrupt the conformation of the active site backbone but also reduce the affinity for inhibitor binding.23−26 The elucidation of the cocrystal structure of HIV-1 PR and inhibitors through X-ray analysis has provided valuable insights into the mechanisms underlying drug resistance.27−29 Mutations within the enzyme pockets, such as I84V, V32I, and I50V, among others, have been extensively documented for their role in conferring resistance to antiretroviral therapies.30−32 Despite efforts to develop effective PIs based on DRV analogs, none have proven successful in efficiently inhibiting DRV-resistant HIV-1 strains, as depicted in Figure 1.19,33−39
Figure 1.
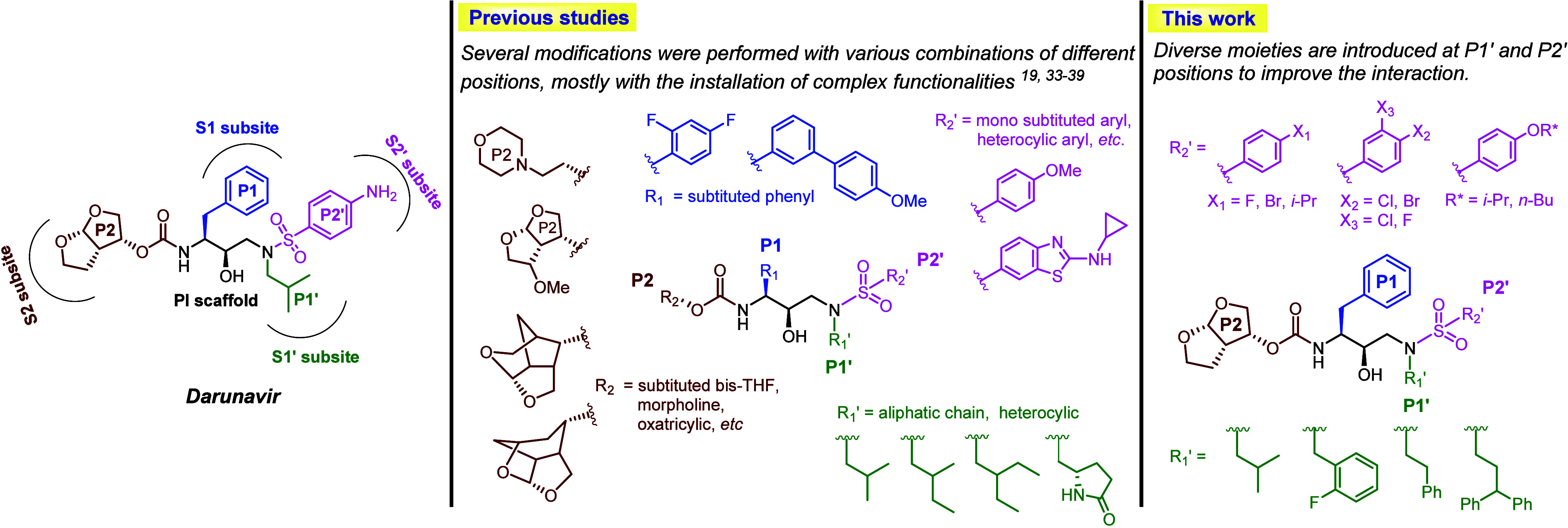
Overview of designs of darunavir analogs previously reported and in the present study.
Structure–activity relationship (SAR) studies have confirmed the necessity for modifications in DRV, particularly at positions P1, P1′, P2, and P2′, as depicted in Figure 1. Key areas of alteration include the heterocyclic functions at P2′ and mono- or disubstituted bis-THF at the P2 location of DRV. The P2′ position, serving as backbone binders, plays a crucial role in enhancing hydrogen bonding interactions between the drug and PR. Additionally, the incorporation of modified moieties at P1′ significantly influences the interaction between the drug and its target, highlighting the intricate interdependence between HIV-1 PR and therapeutic regimens. In our recent study focusing on the FMO-guided generation of darunavir analogs for HIV-1 PR, several DRV analogs were designed and subjected to virtual screening for HIV-1 PR inhibition.39 Analysis revealed that the P1′ and P2′ positions exhibit weaker interactions with the target viral enzyme compared to P1 and P2 positions. This suggests that adding a more flexible moiety at these sites could improve the interaction between modified DRV and PR. In this study, we introduce the expansion of various moieties at the P2′ position alongside the P1′ ligand through combinatorial in silico drug design for HIV-1 PIs, and a selection of promising analogs was synthesized (Figure 1). The in vitro biological activity of these novel DRV analogs against wild-type (WT) HIV-PR was evaluated, and their activity against various HIV-1 PR mutants (MTs) was assessed computationally.
Result and Discussion
Analog Design and Preliminary Screening
A combinatorial in silico drug design approach was employed to enhance the diversity of P1′ and P2′ ligands, aiming to develop inhibitors that retain the DRV core scaffold while improving interactions at the S1′ and S2′ subsites of HIV-1 PR (Figure 2). The interaction profile of DRV in complex with HIV-1 PR indicates that the P1′ position accommodates various hydrophobic moieties, whereas the P2′ region can accommodate a broad range of functionalities.39 Previous studies have primarily focused on modifications at the P1′ position involving long-chain aliphatic groups and heterocycles.35,40−42 In this study, we explored a wider range of aliphatic and aromatic groups with various functionalities to assess their steric and electronic effects on the P1′ region. For P2′ modifications, the S2′ subsite of PR is highly adaptable due to van der Waals forces, hydrophobic interactions, and hydrogen bonding. Literature reviews have identified several promising analogs with halogen substituents, including fluoro,43 chloro,44,45 and occasionally the bromo46 moieties, which were incorporated into some of the P2′ modification in this study (e.g., 2, 3, 4, 9, 14, 18, 19, and 20). Additionally, the introduction of a methoxy group at the P2′ position was found to enhance activity compared to the original DRV.47 Hence, we also varied the length of alkyl (e.g., 16, 17, 22) and alkoxy groups (e.g., 6, 7, 10, 21), to better understand the steric and hydrophobic effects. Various aromatic and heterocyclic moieties (e.g., 1, 4, 5, 11, 12, 13, and 15) were also included to investigate potential π-interactions and hydrogen bonding.
Figure 2.
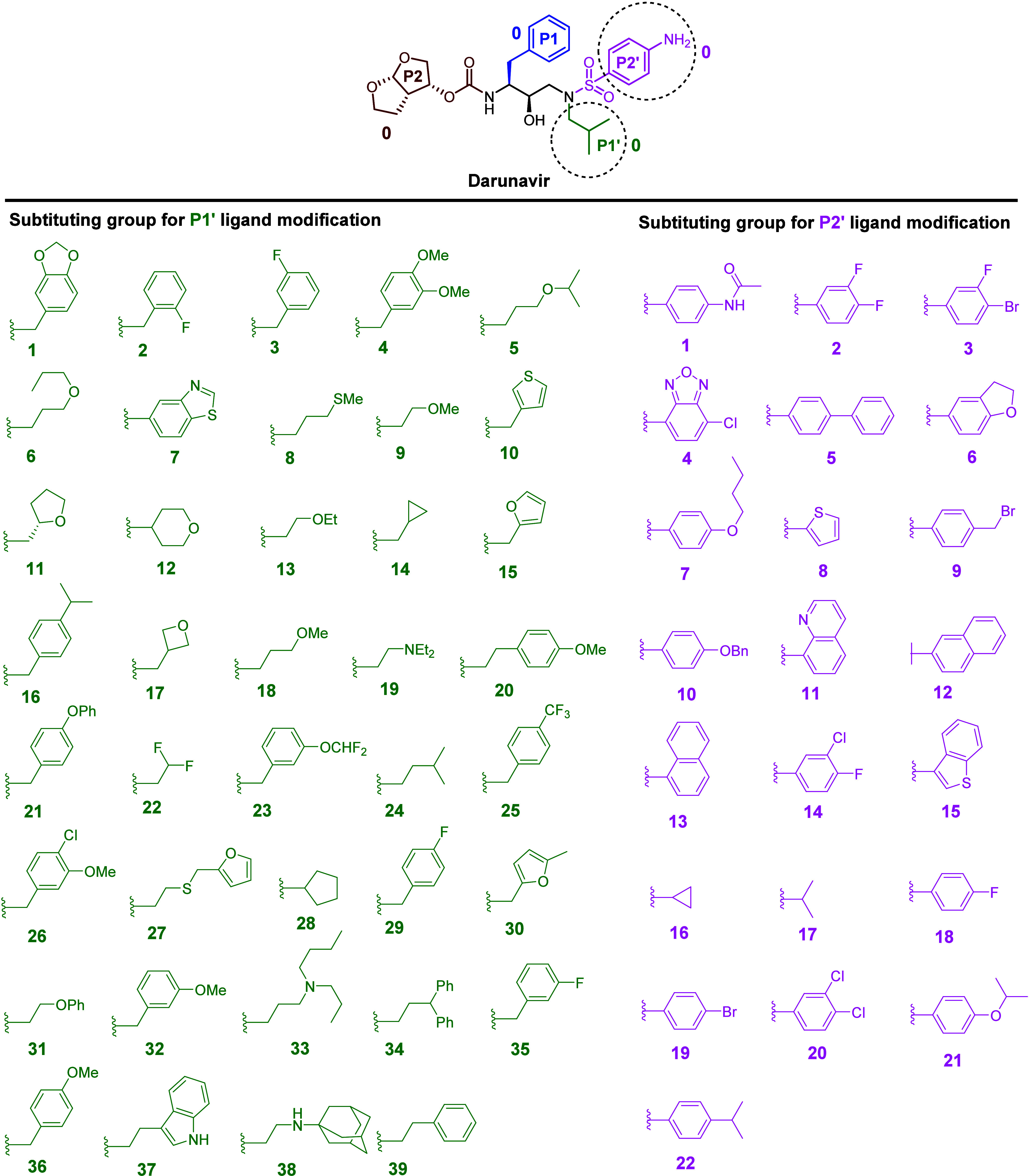
Structures of the DRV scaffold and different substituting groups for the P1′ and P2′ modifications.
These modifications of P1′ and P2′ were combined using Combined Analog Generator Tool (CAT), leading to the generation of 858 novel DRV analogs, which were then docked into the active site of HIV-1 PR as a preliminary virtual screening. Docking scores for the DRV analogs ranged from 65 to 103 (Figure S1), with the reference DRV showing a score of 87.3. Analogs with high docking scores (highlighted in green in Figure S1) were prioritized for synthesis. Prominent candidates for P1′ position included hydrocarbon substituents with different sizes and lengths, such as isobutyl (24), diphenyl ethyl (34), 2-fluorophenyl methyl (35), and phenyl ethyl (39). For the P2′ position, the most promising groups featured phenyl rings with diverse substituents, including aliphatic chains, alkoxy groups, and halogen groups such as 3-F, 4-BrC6H3 (3), 4-On-BuC6H4 (7), 4-FC6H4 (18), 4-BrC6H4 (19), 4-Oi-PrC6H4 (21), and 4-i-PrC6H4 (22). The combinations of long and flexible chains, such as aliphatic and alkoxy groups, were anticipated to augment their interaction with the S2′ subsite by extending deeper into the pocket of HIV-1 PR.
Synthesis of DRV Analogs
The novel DRV analogs in this study were synthesized based on a modified protocol from literature (Figure 3).48 Initially, commercially available Boc-protected epoxide 1 underwent epoxide ring-opening with a corresponding amine, representing the substituents for the P1′ modification. Given that the Boc-protected epoxide functions as a component of the primary scaffold of DRV during interaction with HIV-1 PR. Next, the resulting intermediates 2a–d were then treated with a diverse range of 4-substituted aryl sulfonyl chlorides to introduce the sulfonamide functionality, serving as the P2′ modification. Subsequent deprotection of the Boc-protected carbamate, followed by coupling with carboxylate, yielded the desired analogs 5aa–dg, which were fully characterized through NMR and HRMS analysis. The purity of these analogs was assessed by HPLC analysis. In total, 15 novel DRV analogs were synthesized and used for bioactivity evaluation.
Figure 3.
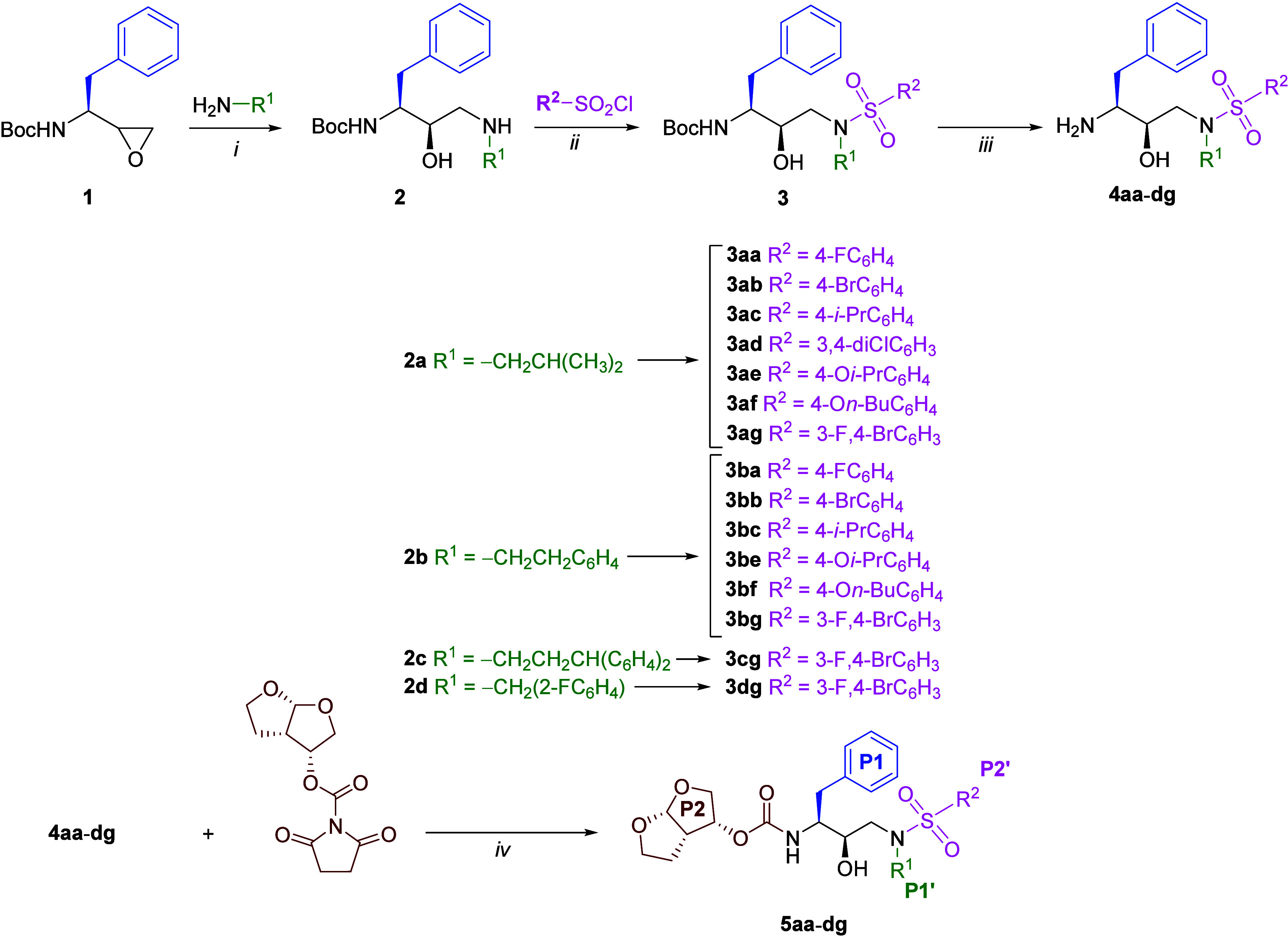
Synthesis of DRV analogs; reagents and conditions: (i) EtOH, 80 °C, 3 h, 60–85%; (ii) TEA, CH2Cl2, 0–25 °C, o/n, 23–83%; (iii) TFA-DCM (1:1), 0–25 °C, o/n, 95–100%; (iv) TEA, CH2Cl2, 25 °C, o/n, 13–50%.
HIV-1 PR Inhibition and Cytotoxicity Analysis
The synthesized DRV analogs underwent initial evaluation for their inhibition activity against WT HIV-1 PR using a fluorogenic assay. The Ki values of each analog are presented in Table 1.49 For the P1’s substitution, compounds featuring larger substituents, such as phenyl ethyl (5ba–bf) or diphenyl propyl (5cg), exhibited reduced activity. This finding is consistent with existing computational based literature, which suggests that the introduction of a large hydrophobic ligand in the P1′ position disrupts the shape of the enzyme's P2′ pocket.38 Regarding P2′ modification, analogs bearing fluoro and dichloro substituents on the phenyl ring, such as compounds 5aa and 5ad demonstrated slightly improved activity (Ki = 1.54 and 0.71 nM, respectively), compared to DRV (Ki = 1.87 nM). This enhancement could be attributed to halogens favorably interacting with amino acid residues D30, V32, V82, and P81 in the S2′ subsite of HIV-1 PR (Figures 5 and S2 in Supporting Information). Furthermore, compounds 5ac, 5ae, and 5af, containing flexible aliphatic or alkoxy groups, exhibited even greater inhibition activity (Ki = 0.31, 0.28, and 1.11 nM, respectively), potentially due to enhanced hydrophobic contact within the S2′ subsite of PR. The structure–activity relationship (SAR) analysis is summarized in Figure 4.
Table 1. HIV-1 PR Inhibition and Cytotoxicity of Darunavir Analogs.

Reported as Mean ± SD; the inhibition values (Ki) of darunavir analog were determined in duplicate.
Reported as Mean ± SD; all darunavir derivatives were screened in both cell lines for 48 h. 1% DMSO was used for the % viability in cytotoxicity assay analysis. n.i. = no inhibition, n.t. = not tested.
Figure 5.

3D diagram of ligand–protein interactions for the five selected analogs with WT HIV-1 PR compared to DRV.
Figure 4.

SAR analysis of DRV analog.
Most compounds showed a moderate to good correlation between docking scores and Ki values. For instance, compounds like 5ac (docking score = 89, Ki = 0.31 nM), 5ad (docking score = 92.9, Ki = 0.71 nM), 5ae (docking score = 101.2, Ki = 0.28 nM), and 5af (docking score = 95, Ki = 1.12 nM) exhibited both high protease inhibitory activity and favorable docking scores. Conversely, compounds such as 5ba (docking score = 77.5, Ki = 18.40 nM) and 5be (docking score = 74.9, Ki = 35.84 nM) showed poorer inhibitory activity and docking scores. There were a few exceptions where the docking scores did not correlate well with inhibitory activity, such as 5bc (docking score = 96, Ki = 1080 nM) and 5cg (docking score = 101.8, Ki = 189 nM). Despite these exceptions, the overall trend indicates a general correlation between docking scores and Ki values for most compounds.
Following an initial assessment against the WT HIV-1 PR, the analogs that showed comparable or lower Ki to the WT HIV-1 PR were tested with the R41T HIV-1 PR mutant (Table 2). Although 5ac, 5ad, and 5ae showed lower Ki against the WT HIV-1 PR (Table 1), they gave higher Ki toward the R41T mutant than darunavir. Among these 5 analogs, only 5aa showed similar Ki to darunavir against R41T mutant. Among the tested analogs, only 5aa exhibited a Ki value similar to that of darunavir against the R41T mutant. This observation suggests that the fluoro functionality at the P2′ position of 5aa may interact favorably with the R41T mutant through multipolar interactions, distinguishing it from the other DRV analogs. Further studies on these analogs against other HIV-1 PR mutant variants could provide additional insights.
Table 2. Darunavir Analogs Inhibition against R41T Mutant.
DRV was used as a reference for DRV analogs investigation against the R41T mutant.
Reported as Mean ± SD; the inhibition values (Ki) of darunavir analog were determined in duplicate.
To evaluate the potential cytotoxicity of DRV analogs, a preliminary cytotoxic study was performed using Vero and 293T cell lines under the conventional mitochondrial toxicity assay.50 Vero is one of the most responsive cell lines for evaluating cytopathic effects, as it lacks cellular defense mechanism to chemical and biological stresses.51 Moreover, 293T is commonly used in antiretroviral drug testing platforms because it predisposed to retroviral infection.52 Remarkably, all DRV analogs examined at 10 μM exhibited >90% cell viability following 2 days of incubation (Table 1), suggesting their potential safety profile for further investigation.
Binding of DRV Analogs to HIV-1 PR WT and MTs
To refine virtual screening and explore the inhibition activity for both WT and various MTs HIV-1 PR, a molecular docking study was applied to investigate the binding of five selected DRV analogs using GOLD program. Among these analogs, 5aa, 5ac, 5ad, 5ae, and 5af demonstrated superior performance in vitro study against WT HIV-1 PR. Also, these analogs showed superior docking scores to DRV (87.3), ranging from 89.0 to 101.2 (Table 3). These compounds were promising and effective in inhibiting most MTs HIV-1 PR, particularly those with higher docking scores than DRV, as indicated by the green color in Table 3.
Table 3. Docking Results of DRV and Its Analogs with WT and Mutants of HIV-1 PR.

Focusing on the binding interaction between promising DRV analogs and WT HIV-1 PR, the binding interaction showed that hydrophobic interactions could be the major contributing interaction for these analogs, interacting with several common key residues in WT HIV-1 PR such as I47, I50, P81, V82, I84, A28′, V32′, I47′, P81′, V82′, and I84′. Additionally, these analogs could form hydrogen bonds with D25, G27, D29, D30, D25′, and D30′. In the 5ad, we could observe a halogen bonding interaction between Cl and D30′ (Figures 5 and S2 in Supporting Information).
Based on our experimental and computational studies, 5ae demonstrated exceptional effectiveness in inhibition activity against HIV-1 PR in the experimental results. It showed adequate effectiveness against other variants of HIV-1 PR in terms of broad-spectrum inhibitors. Thus, 5ae was consequently chosen for further investigation using molecular dynamics (MD) simulations.
Structural Dynamics and Energetic Properties of New Potent Compound
Ligand-binding stability was assessed through the calculation of root-mean-square deviation (RMSD), the number of intermolecular hydrogen bonds (# H-bonds), and the number of atom contacts (# Atom contacts), as shown in Figure 6a. Although both DRV and 5ae complexes with WT HIV-1 PR systems reveal similar RMSD profiles, the main contributing interaction to maintain these ligands in the binding pocket differs. Specifically, hydrogen bonding interactions predominantly contribute to DRV stability, whereas hydrophobic interactions, as indicated by the # Atom contacts, play a significant role in stabilizing 5ae (Figure 6a). The complex structures taken from the last 20 ns simulations, which had reached an equilibrium state, were chosen for structural dynamics and energetic analyses. Although the number of hydrogen bonds in the DRV/WT HIV-1 PR systems (∼4) is higher than in the 5ae/WT HIV-1 PR system (∼3) due to the P2′ modification, the hydrogen bonding stability in the DRV/WT HIV-1 PR is slightly lower than in the 5ae/WT HIV-1 PR. Notably, DRV forms hydrogen bonding interactions with D25 (90 and 88%), D25′ (25%), and D30′ (99%). In contrast, the 5ae system shows slightly higher stability of hydrogen bonding interactions with D25 (97 and 96%), D25′ (52%), and I50′ (79%) (Figure 6a,b). Corresponding with Figure 6b, 5ae also demonstrated potential for hydrophobic interactions with residues L23 (99%), I50 (66%), V82 (7%), I84 (8%), L23′ (20%), F53′ (2%), V82′ (5%), and I84′ (99%) are essential for maintaining the PR inhibitor.53,54 Particularly, L23, which is involved in hydrophobic interactions55,56 and van der Waals forces,38 was considered a critical residue in the majority of PIs.
Figure 6.

(a) All-atom RMSD, # H-bonds, and # Atom contacts of DRV (left) and 5ae (right) in complex with WT HIV-1 PR plotted along the 100 ns MD simulation. (b) Crucial interactions of DRV and 5ae in complex with WT HIV-1 PR are depicted in 2D and 3D pharmacophore models, along with the representative pharmacophore models (RPMs) analyzed from the last 20 ns of MD simulations. Green arrow, red arrow, and yellow circle represent the pharmacophore features of hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), and hydrophobic interaction, respectively.
Furthermore, the hydrophobic interactions between flap residues, such as I50–I84′, play an essential role in retaining the closed conformation of HIV-1 PR.57,58 However, DRV only interacted with I50 (2%), V82 (9%), A28′ (99%), and V82′ (7%) via hydrophobic interaction, consistent with findings from previous studies.39,59
For predicting the binding affinity between the considered compound and WT HIV-1 PR, 100 snapshots of each complex extracted from 80 to 100 ns were also used to calculate for the Molecular mechanics with generalized Born and surface area solvation (MM/GBSA) binding free energy (ΔGbind) and its energy components (Table 4). The ΔGbind values for complexes with DRV and 5ae were −15.9 ± 1.0 and −18.5 ± 0.6 kcal/mol, respectively. This result and the obtained ligand–protein interactions suggest that 5ae could be more susceptible to WT HIV-1 PR than DRV.
Table 4. Comparison of the MM/GBSA Binding Free Energy and Energetic Components (kcal/mol) between DRV and Its Newly Potent Analog 5ae toward WT HIV-1 PR.
| DRV | 5ae | |
|---|---|---|
| ΔEele | –47.5 ± 0.6 | –32.8 ± 0.5 |
| ΔEvdW | –57.7 ± 0.3 | –61.7 ± 0.4 |
| ΔEMM | –105.2 ± 0.9 | –94.5 ± 0.9 |
| ΔGnonpolar,sol | –8.3 ± 0.2 | –8.4 ± 0.1 |
| ΔGele,sol | 66.2 ± 0.4 | 55.9 ± 0.3 |
| ΔGsol | 57.9 ± 0.4 | 47.5 ± 0.3 |
| ΔGele,sol+ ΔEele | 18.7 ± 1.0 | 23.1 ± 0.8 |
| ΔGnonpolar,sol+ ΔEvdW | –66.0 ± 0.6 | –70.1 ± 0.5 |
| –TΔS | –31.4 ± 0.7 | –28.4 ± 1.0 |
| ΔGbind | –15.9 ± 1.0 | –18.5 ± 0.6 |
In detail, the vdW interactions act as the primary force inducing molecular complexation in both DRV (−57.7 ± 0.3 kcal/mol) and 5ae (−61.7 ± 0.4 kcal/mol).
Conclusions
In conclusion, our study focused on enhancing the binding interactions of DRV HIV-1 PI with the PR target through molecular modifications. By synthesizing a series of DRV derivatives with alterations in the P1′ and P2′ ligands, we aimed to improve the efficacy of HIV-1 PR inhibition. Our findings from inhibition studies revealed promising results, with several DRV analogs such as 5aa (1.54 nM), 5ac (0.31 nM), 5ad (0.71 nM), 5ae (0.28 nM), and 5af (1.11 nM) displaying superior activity against WT HIV-1 PR compared to DRV (1.87 nM). Importantly, cytotoxicity assays indicated the safety profile of these analogs, making them promising candidates for further development. Five DRV analogs demonstrated potent inhibition of both WT and MT forms of HIV-1 PR through hydrophobic and H-bond interactions in molecular docking. Notably, 5ae emerged as the most promising analog, exhibiting strong vdW interactions in MD simulations, consistent with experimental findings. Overall, our investigation showed how crucial the linker’s length is in determining the inhibitor’s affinity for the enzyme’s S2′ subsite and how these effectively created DRV analogs might be applied to combat HIV and other global health concerns. These findings hold promise for future research addressing global health challenges and advancing drug discovery efforts in antiretroviral therapy.
Experimental Section
Computational Study
The structure-based drug design approach served as the model for creating the new promising DRV correspondents in this work. The 3D structure of DRV analogs were created using CAT program.39,59 The crystal structures of HIV-1 PR WT in complex with DRV (PDB ID: 4LL3(60)), and several mutation strains, D30N (PDB ID: 7DOZ(61)), V32I (PDB ID: 2HS1(62)), M46L (PDB ID: 2HS2(62)), G48V (PDB ID: 3CYW(63)), I50V (PDB ID: 6DH6(64)), I54M (PDB ID: 3D1Z(63)), I54V (PDB ID: 3D20(63)), L76V (PDB ID: 3PWM(65)), V82A (PDB ID: 2IDW(66)), I84V (PDB ID: 2IEO(66)), N88S (PDB ID: 3LZU(21)), L90M (PDB ID: 6OOS(21)), and I93L (PDB ID: 5T2E(67)), were used as the protein receptor for a molecular docking study using the GOLD program with a genetic algorithm (GA) was employed.68 Note that the structure of mutant R41T was created using the Alphafold 269 via Colabfold.70,71 The protonation state of protein receptors was determined using the PROPKA implementation in the PDB 2PQR Web server.72 The 10 Å sphere centered at DRV in complex with the HIV-1 PR at the active site was defined as the docking site. The Accelrys Discovery Studio 3.0 was used to visualize the interactions between the ligand and protein binding.
MD simulations were conducted using AMBER2273 with periodic conditions, as detailed in prior studies.74 The electrostatic potential (ESP) charges of the optimized compounds were determined at the HF/6-31(d) level of theory and subsequently fitted into restrained ESP (RESP) charges using the ANTECHAMBER module of AmberTools21.73 The AMBER ff19SB force field and GAFF2 were applied for the protein and compound, respectively. Missing hydrogen atoms in both the protein and compound were added using the tLEaP module. The system was neutralized with counterions before immersion in the TIP3P water model in the octahedral box75 by extending at least 10 Å from the protein surface. The systems were performed for the structural minimization with 1500 steps using steepest descent (SD) followed by conjugated gradient (CG) methods to minimize energy for the entire complex. Subsequently, MD simulations with a 2 fs time step were executed. Nonbonded interactions had a short-range limit of 10 Å, and long-range electrostatic interactions76 were treated using the Particle Mesh Ewald (PME) summation approach. Pressure and temperature were controlled, and covalent bonds containing hydrogen atoms were constrained with the SHAKE methodology.77 The simulated models were maintained at a constant temperature of 310 K until 100 ns. The CPPTRAJ module was used to analyses for plot the all-atom root-mean-square deviation (RMSD), the number of intermolecular hydrogen bonds, and the number of contact atoms between the compound and protein over the production phase.78 MM/GBSA79 was calculated to estimate the binding affinity of the compound/protein complex for each simulation using 100 snapshots from the last 20 ns. The 2D interaction was visualized using LigandScout 4.4.8 program.80
Chemistry
All reagents were obtained from TCI Chemicals (Tokyo, Japan) and Fluorochem (Hadfield, United Kingdom). All required solvents purchased from RCI Laboratories (Samsutsakorn, Thailand) were filtered through distillation before being used for both reaction and column chromatography. All experiments were carried out in dried glassware with rubber septa inside an inert system of positive pressure of nitrogen gas. Thin-layer chromatography (TLC) was used to track reactions using aluminum Merck TLC plates coated in silica gel 60 F254. Silica gel 60 was used for the column chromatography (0.063–0.200 mm, 70–230 mesh ASTM, Merck, Darmstadt, Germany). Tetramethyl silane (TMS) was used as an internal reference while recording nuclear magnetic resonance spectra for both 1H and 13C in CDCl3 on the Joel JNM-ECZ500/S1 (500 MHz). The coupling constant J values were reported in Hz, and the chemical shift (δ) was expressed in parts per million (ppm) linked to the corresponding solvent peak (CDCl3: 1H, δ 7.26 ppm, 13C, δ 77.16 ppm). The HRMS mass spectrometer was used to gather data for mass spectra.
The comprehensive synthetic scheme that involved the following steps to synthesize new darunavir analogs have been described in Figure 3.
Epoxide Ring Opening (Step (i))
A mixture of tert-butyl ((S)-1-((S)-oxiran-2-yl)-2-phenylethyl)carbamate (Boc-protected epoxide, 1, 1.0 equiv, commercially purchased from fluorochem) and the corresponding amine (1.0 equiv) was added to the dried EtOH solvent in a round-bottom flask. The reaction was heated for 3 h at 85 °C under N2. The crude mixture was concentrated in vacuo and purified through column chromatography using EtOAc/hexane eluent system. The products 2a–d were obtained in the range of 60–85% yield.
tert-Butyl ((2S,3R)-3-Hydroxy-4-(isobutyl amino)-1-phenyl butan-2-yl)carbamate (2a)
White solid (82.6%); Rf (EtOAc/hexane = 70:30) = 0.1; 1H NMR, (500 MHz, CDCl3): δ 7.31–7.26 (m, 3H), 7.26–7.18 (m, 2H), 4.98 (d, J = 9.5 Hz, 1H), 3.72 (q, J = 8.3 Hz, 1H), 3.57 (dd, J = 9.3, 4.1 Hz, 1H), 2.96–2.87 (m, 2H), 2.66–2.59 (m, 1H), 2.60–2.51 (m, 1H), 2.44–2.31 (m, 2H), 1.73–1.65 (m, 1H), 1.39 (s, 9H), 0.88 (dd, J = 6.8, 1.9 Hz, 6H).
tert-Butyl ((2S,3R)-3-Hydroxy-4-(phenethyl amino)-1-phenyl butan-2-yl)Carbamate (2b)
White solid (73%); Rf (EtOAc/hexane = 70:30) = 0.1; 1H NMR, (500 MHz, CDCl3): δ 7.29–7.24 (m, 6H), 7.23–7.17 (m, 2H), 7.17–7.13 (m, 2H), 5.04 (d, J = 9.6 Hz, 1H), 3.72 (q, J = 8.4 Hz, 1H), 3.51 (ddd, J = 9.5, 4.0, 1.5 Hz, 1H), 2.95–2.76 (m, 5H), 2.73 (dd, J = 7.3, 4.9 Hz, 2H), 2.62–2.53 (m, 2H), 1.40 (s, 9H).
tert-Butyl ((2S,3R)-4-((3,3-Diphenyl propyl)amino)-3-hydroxy-1-phenyl butan-2-yl)Carbamate (2c)
White solid (84.9%); Rf (EtOAc/hexane = 70:30) = 0.1; 1H NMR, (500 MHz, CDCl3): δ 7.34–7.27 (m, 5H), 7.25–7.14 (m, 10H), 4.72 (d, J = 9.1 Hz, 1H), 4.01 (t, J = 7.9 Hz, 1H), 3.53 (q, J = 7.0 Hz, 1H), 3.49–3.43 (m, 1H), 2.96 (dd, J = 14.1, 4.7 Hz, 1H), 2.93–2.75 (m, 2H), 2.65 (d, J = 5.1 Hz, 2H), 2.62–2.52 (m, 2H), 2.27–2.22 (m, 2H), 1.36 (d, J = 11.9 Hz, 9H).
tert-Butyl ((2S,3R)-4-((2-Fluoro benzyl)amino)-3-hydroxy-1-phenylbutan-2-yl)carbamate (2d)
White solid (61.9%); Rf (EtOAc/hexane = 70:30) = 0.1; 1H NMR, (500 MHz, CDCl3): δ 7.32–7.28 (m, 2H), 7.26–7.15 (m, 5H), 7.11–6.98 (m, 2H), 4.74 (d, J = 9.2 Hz, 1H), 4.31 (q, J = 4.2 Hz, 1H), 3.95 (q, J = 6.5 Hz, 1H), 3.86–3.77 (m, 2H), 3.69 (dd, J = 12.6, 3.1 Hz, 1H), 3.56–3.49 (m, 1H), 3.41 (dd, J = 12.7, 4.2 Hz, 1H), 2.94 (dd, J = 14.1, 4.7 Hz, 1H), 2.85 (h, J = 7.4 Hz, 1H), 2.69 (d, J = 5.9 Hz, 1H), 1.32 (s, 9H).
Sulfonamide Synthesis (Step (ii))
To a solution of 2a–d (1.0 equiv) in dried CH2Cl2, was added the triethylamine (1.1 equiv). Then, substituted aryl sulfonyl chloride (1.1 equiv) was added in portions to the reaction mixture at 0 °C, over 30 min, and the reaction mixture was stirred overnight at room temperature of 25 °C. Following the completion of the reaction, the crude was concentrated in vacuo and purified through column chromatography using EtOAc/hexane eluent system. The products 3aa–dg were obtained in the range of 23–83% yield.
tert-Butyl ((2S,3R)-4-((4-Fluoro-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (3aa)
White solid (77.2%); Rf (EtOAc/hexane = 30:70) = 0.6; 1H NMR, (500 MHz, CDCl3): δ 7.77–7.73 (m, 2H), 7.31–7.27 (m, 2H), 7.25–7.13 (m, 5H), 4.98 (d, J = 9.5 Hz, 1H), 3.76–3.71 (m, 1H), 3.67 (q, J = 8.4 Hz, 1H), 3.48 (d, J = 3.2 Hz, 1H), 3.25 (dd, J = 15.2, 9.3 Hz, 1H), 3.01–2.84 (m, 3H), 2.80 (dd, J = 15.3, 2.9 Hz, 1H), 2.69 (dd, J = 13.3, 6.4 Hz, 1H), 1.63–1.47 (m, 1H), 1.40 (s, 9H), 0.79 (dd, J = 9.8, 6.6 Hz, 6H).
tert-Butyl ((2S,3R)-4-((4-Bromo-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (3ab)
White solid (69.5%); Rf (EtOAc/hexane = 30:70) = 0.8; 1H NMR, (500 MHz, CDCl3): δ 7.65–7.56 (m, 4H), 7.29 (dd, J = 8.2, 6.8 Hz, 2H), 7.25–7.18 (m, 3H), 4.96 (d, J = 9.5 Hz, 1H), 3.73 (d, J = 9.2 Hz, 1H), 3.67 (q, J = 8.4 Hz, 1H), 3.43 (d, J = 3.2 Hz, 1H), 3.24 (dd, J = 15.2, 9.2 Hz, 1H), 3.00–2.86 (m, 1H), 2.81 (dd, J = 15.2, 2.9 Hz, 3H), 2.70 (dd, J = 13.3, 6.4 Hz, 2H), 1.58–1.51 (m, 1H), 1.40 (s, 9H), 0.79 (dd, J = 9.2, 6.6 Hz, 6H).
tert-Butyl ((2S,3R)-3-Hydroxy-4-((N-isobutyl-4-isopropyl phenyl)sulfonamido)-1-phenyl butan-2-yl)carbamate (3ac)
White solid (47%); Rf (EtOAc/hexane = 30:70) = 0.8; 1H NMR, (500 MHz, CDCl3): δ 7.71–7.66 (m, 2H), 7.37–7.32 (m, 2H), 7.29 (dd, J = 7.9, 6.8 Hz, 2H), 7.26–7.18 (m, 3H), 4.65 (d, J = 8.7 Hz, 1H), 3.84–3.68 (m, 1H), 3.14–3.03 (m, 2H), 3.02–2.87 (m, 3H), 2.83 (dd, J = 13.3, 6.7 Hz, 1H), 1.86–1.82 (m, 1H), 1.34 (s, 9H), 1.27 (d, J = 6.9 Hz, 6H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H).
tert-Butyl ((2S,3R)-4-((3,4-Dichloro-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (3ad)
White solid (60%); Rf (EtOAc/hexane = 30:70) = 0.7; 1H NMR, (500 MHz, CDCl3): δ 7.87 (d, J = 1.9 Hz, 1H), 7.60–7.57 (m, 2H), 7.33–7.28 (m, 2H), 7.25–7.21 (m, 3H), 4.65 (d, J = 8.1 Hz, 1H), 3.83–3.74 (m, 2H), 3.14–3.12 (m, 2H), 3.02–2.95 (m, 1H), 2.91 (td, J = 14.2, 12.2, 5.5 Hz, 2H), 1.91–1.81 (m, 1H), 1.36 (s, 9H), 0.88 (dd, J = 9.8, 6.6 Hz, 6H).
tert-Butyl ((2S,3R)-3-Hydroxy-4-((N-isobutyl-4-isopropoxy phenyl)sulfonamido)-1-phenyl butan-2-yl)carbamate (3ae)
Sticky solid (65.5%); Rf (EtOAc/hexane = 30:70) = 0.6; 1H NMR, (500 MHz, CDCl3): δ 7.71–7.65 (m, 2H), 7.32–7.27 (m, 2H), 7.26–7.18 (m, 3H), 6.96–6.90 (m, 2H), 4.63–4.60 (m, 1H), 3.97 (s, 1H), 3.83–3.73 (m, 2H), 3.12–2.98 (m, 3H), 2.92 (td, J = 15.8, 15.0, 8.3 Hz, 1H), 2.80 (dd, J = 13.3, 6.8 Hz, 1H), 1.90–1.78 (m, 1H), 1.67 (s, 1H), 1.36 (d, J = 6.0 Hz, 6H), 1.34 (s, 9H), 0.88 (dd, J = 19.5, 6.6 Hz, 6H).
tert-Butyl ((2S,3R)-4-((4-Butoxy-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3af)
White solid (63.7%); Rf (EtOAc/hexane = 30:70) = 0.8; 1H NMR, (500 MHz, CDCl3): δ 7.70–7.66 (m, 2H), 7.32–07.26 (m, 2H), 7.26–7.19 (m, 3H), 6.97–6.92 (m, 2H), 4.65 (d, J = 8.7 Hz, 1H), 4.06–3.91 (m, 2H), 3.85–3.71 (m, 2H), 3.11–2.98 (m, 3H), 2.96–2.72 (m, 3H), 1.89–1.74 (m, 3H), 1.51 (dt, J = 15.1, 7.4 Hz, 2H), 1.34 (s, 9H), 0.98 (t, J = 7.4 Hz, 3H), 0.88 (dd, J = 19.0, 6.6 Hz, 6H).
tert-Butyl ((2S,3R)-4-((4-Bromo-3-fluoro-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3ag)
White solid (64%); Rf (EtOAc/hexane = 30:70) = 0.5; 1H NMR, (500 MHz, CDCl3): δ 7.72 (dd, J = 8.4, 6.4 Hz, 1H), 7.54 (dd, J = 7.8, 2.1 Hz, 1H), 7.45 (dd, J = 8.4, 2.0 Hz, 1H), 7.32 (t, J = 7.5 Hz, 2H), 7.30–7.22 (m, 3H), 4.69 (d, J = 8.4 Hz, 1H), 3.85–3.76 (m, 3H), 3.16–3.12 (m, 2H), 3.02–2.89 (m, 3H), 1.93–1.85 (m, 1H), 1.38 (s, 9H), 0.90 (dd, J = 9.0, 6.5 Hz, 6H).
tert-Butyl ((2S,3R)-4-((4-Fluoro-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3ba)
White solid (36.3%); Rf (EtOAc/hexane = 30:70) = 0.6; 1H NMR, (500 MHz, CDCl3): δ 7.73 (dd, J = 8.6, 4.9 Hz, 2H), 7.33–7.27 (m, 2H), 7.24 (ddd, J = 11.4, 7.3, 2.9 Hz, 5H), 7.13 (t, J = 8.5 Hz, 2H), 7.10–7.00 (m, 2H), 5.00 (d, J = 9.4 Hz, 1H), 3.69 (h, J = 7.5, 6.2 Hz, 2H), 3.48–3.33 (m, 1H), 3.33–3.25 (m, 1H), 3.19 (d, J = 3.5 Hz, 1H), 2.99–2.90 (m, 2H), 2.90–2.67 (m, 3H), 1.41 (s, 9H).
tert-Butyl ((2S,3R)-4-((4-Bromo-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3bb)
White solid (23.1%); Rf (EtOAc/hexane = 30:70) = 0.7; 1H NMR, (500 MHz, CDCl3): δ 7.58 (s, 4H), 7.33–7.19 (m, 8H), 7.06–7.01 (m, 2H), 5.00 (d, J = 9.4 Hz, 1H), 3.70 (td, J = 10.8, 9.7, 5.8 Hz, 2H), 3.40–3.25 (m, 2H), 3.24–3.15 (m, 1H), 2.99–2.91 (m, 2H), 2.88 (dd, J = 13.6, 8.3 Hz, 1H), 2.75 (td, J = 9.1, 6.0 Hz, 2H), 1.41 (s, 9H).
tert-Butyl ((2S,3R)-3-Hydroxy-4-((4-isopropyl-N-phenethyl phenyl)sulfonamido)-1-phenyl butan-2-yl)carbamate (3bc)
White solid (25%); Rf (EtOAc/hexane = 30:70) = 0.9; 1H NMR, (500 MHz, CDCl3): δ 7.73–7.69 (m, 2H), 7.35 (dd, J = 9.2, 2.8 Hz, 3H), 7.31 (d, J = 7.2 Hz, 2H), 7.30–7.27 (m, 2H), 7.25–7.20 (m, 3H), 7.16 (dd, J = 6.8, 1.8 Hz, 2H), 4.60 (d, J = 8.2 Hz, 1H), 3.86–3.78 (m, 1H), 3.77 (s, 1H), 3.38–3.33 (m, 2H), 3.25–3.07 (m, 2H), 3.01–2.94 (m, 2H), 2.95–2.87 (m, 3H), 1.38 (s, 9H), 1.28 (d, J = 7.1 Hz, 6H).
tert-Butyl ((2S,3R)-3-Hydroxy-4-((4-isopropoxy-N-phenethyl phenyl)sulfonamido)-1-phenyl butan-2-yl)carbamate (3be)
White solid (83%); Rf (EtOAc/hexane = 30:70) = 0.8; 1H NMR, (500 MHz, CDCl3): δ 7.71–7.66 (m, 2H), 7.34–7.28 (m, 3H), 7.28–7.19 (m, 6H), 7.16 (dd, J = 7.0, 1.8 Hz, 2H), 6.96–6.90 (m, 2H), 4.66–4.59 (m, 2H), 3.89–3.65 (m, 2H), 3.41–3.30 (m, 2H), 3.22–3.09 (m, 2H), 3.01–2.97 (m, 1H), 2.93–2.85 (m, 3H), 1.41–1.35 (m, 15H).
tert-Butyl ((2S,3R)-4-((4-Butoxy-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3bf)
Sticky transparent solid (33.1%); Rf (EtOAc/hexane = 30:70) = 0.8; 1H NMR, (500 MHz, CDCl3): δ 7.64 (d, J = 8.6 Hz, 2H), 7.33–7.18 (m, 8H), 7.04 (d, J = 7.4 Hz, 2H), 6.91 (d, J = 8.7 Hz, 2H), 5.00 (d, J = 9.5 Hz, 1H), 3.98 (t, J = 6.5 Hz, 2H), 3.70–3.63 (m, 1H), 3.37–3.26 (m, 1H), 3.20 (d, J = 3.2 Hz, 1H), 3.14–3.08 (m, H), 2.94 (dd, J = 13.5, 7.0 Hz, 1H), 2.91–2.82 (m, 2H), 2.80–2.66 (m, 2H), 1.82–1.73 (m, 1H), 1.48 (h, J = 7.5 Hz, 1H), 1.40 (s, 4H), 0.97 (t, J = 7.4 Hz, 1H).
tert-Butyl ((2S,3R)-4-((4-Bromo-3-fluoro-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3bg)
White solid (54.5%); Rf (EtOAc/hexane = 30:70) = 0.8; 1H NMR, (500 MHz, CDCl3): δ 7.65 (dd, J = 8.4, 6.4 Hz, 1H), 7.45 (dd, J = 7.7, 2.2 Hz, 1H), 7.37 (dd, J = 8.4, 2.1 Hz, 1H), 7.33–7.28 (m, 2H), 7.28–7.20 (m, 6H), 7.06–7.01 (m, 2H), 4.95 (d, J = 9.4 Hz, 1H), 3.71–3.64 (m, 1H), 3.37 (ddd, J = 15.0, 9.3, 6.0 Hz, 1H), 3.34–3.18 (m, 2H), 3.12 (d, J = 4.1 Hz, 1H), 2.99 (dd, J = 14.8, 3.2 Hz, 1H), 2.96–2.83 (m, 2H), 2.81–2.69 (m, 2H), 1.41 (s, 9H).
tert-Butyl ((2S,3R)-4-((4-Bromo-3-fluoro-N-(2-fluoro benzyl)phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (3cg)
White solid (66.1%); Rf (EtOAc/hexane = 30:70) = 0.7; 1H NMR, (500 MHz, CDCl3): δ 7.66 (dd, J = 8.3, 6.5 Hz, 1H), 7.45 (dd, J = 7.7, 2.1 Hz, 1H), 7.38 (ddd, J = 13.5, 8.0, 1.9 Hz, 2H), 7.31–7.19 (m, 4H), 7.17–7.12 (m, 2H), 7.11–6.92 (m, 2H), 4.53–4.40 (m, 2H), 4.35 (d, J = 8.4 Hz, 1H), 3.68 (d, J = 13.7 Hz, 2H), 3.52 (s, 1H), 3.30–3.18 (m, 2H), 2.86 (dd, J = 13.9, 5.3 Hz, 1H), 2.79 (dd, J = 14.0, 7.6 Hz, 1H), 1.35 (s, 8H).
tert-Butyl ((2S,3R)-4-((4-Bromo-N-(3,3-diphenyl propyl)-3-fluoro phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (3dg)
White solid (37.9%); Rf (EtOAc/hexane = 30:70) = 0.9; 1H NMR, (500 MHz, CDCl3): δ 7.63 (dd, J = 8.3, 6.5 Hz, 1H), 7.40 (dd, J = 7.7, 2.1 Hz, 1H), 7.33–7.25 (m, 8H), 7.25–7.17 (m, 8H), 4.62 (d, J = 7.6 Hz, 1H), 3.83 (t, J = 7.8 Hz, 1H), 3.78–3.72 (m, 3H), 3.21 (dd, J = 14.8, 3.2 Hz, 1H), 3.16–3.02 (m, 3H), 2.96 (dd, J = 14.0, 4.8 Hz, 1H), 2.87 (dd, J = 14.1, 8.2 Hz, 1H), 2.35 (q, J = 7.4, 6.9 Hz, 2H), 1.37 (s, 8H).
BOC Deprotection (Step (iii))
To a solution of 3aa–dg (1.0 equiv) in CH2Cl2, trifluoroacetic acid (0.3 equiv) was added at 0 °C. The mixture was stirred overnight at room temperature of 25 °C. The reaction was quenched by sat. NaHCO3 (aq), extracted with EtOAc, washed with brine, and concentrated in vacuo. The corresponding compounds with good yields (95–100%) were used directly for the final step without further purification.
Carbamate Formation (Step (iv))
To a solution of 4aa–dg (1.0 equiv) in CH2Cl2, (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl 2,5-dioxopyrrolidine-1-carboxylate (1.0 equiv) was added under an inert system. Then, 1.0 eq of triethylamine was gradually added to the reaction mixture and the mixture was stirred for 24 h at room temperature of 25 °C. The crude was concentrated and purified using column chromatography using 3:2 EtOAc/hexane as the eluent to give 5aa–dg with yields of 13–50%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Fluoro-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5aa)
White solid (41%); Rf (EtOAc/hexane = 40:60) = 0.4; m.p. = 128.1–130.2 °C; 1H NMR, (500 MHz, CDCl3): δ 7.83 (ddd, J = 8.9, 4.9, 2.0 Hz, 2H), 7.35–7.28 (m, 3H), 7.28–7.21 (m, 4H), 5.68 (dd, J = 5.2, 1.8 Hz, 1H), 5.05 (td, J = 8.8, 2.8 Hz, 2H), 4.02–3.95 (m, 1H), 3.94–3.84 (m, 3H), 3.79–3.68 (m, 2H), 3.66 (s, 1H), 3.21 (dd, J = 15.3, 8.4 Hz, 1H), 3.16–2.98 (m, 3H), 2.94 (q, J = 8.1 Hz, 1H), 2.91–2.79 (m, 2H), 1.92–1.84 (m, 1H), 1.70–1.60 (m, 1H), 1.51–1.44 (m, 1H), 0.96 (dd, J = 6.7, 1.9 Hz, 3H), 0.91 (dd, J = 6.6, 1.9 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 165.3 (d, J = 255.7 Hz), 155.6, 137.6, 134.4, 130.1 (d, J = 9.0 Hz), 129.4, 128.7, 126.7, 116.6 (d, J = 22.3 Hz), 109.4, 73.6, 72.9, 70.9, 69.7, 58.7, 55.2, 53.6, 45.5, 35.7, 27.3, 25.9, 20.2, 19.9. HRMS (m/z): [M + H] + calcd for C27H35FN2O7S, 551.2222; found 551.2240. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 95.88%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5ab)
White solid (49%); Rf (EtOAc/hexane = 40:60) = 0.3; m.p. = 131.1–132.4 °C; 1H NMR (500 MHz, CDCl3): δ 7.69–7.60 (m, 4H), 7.31–7.24 (m, 2H), 7.21 (t, J = 5.9 Hz, 3H), 5.64 (d, J = 5.2 Hz, 1H), 5.02 (dt, J = 9.4, 5.1 Hz, 2H), 3.97–3.90 (m, 1H), 3.91–3.84 (m, 2H), 3.82 (dd, J = 8.3, 1.9 Hz, 1H), 3.74–3.62 (m, 2H), 3.59 (d, J = 2.6 Hz, 1H), 3.16 (dd, J = 15.1, 8.6 Hz, 1H), 3.08 (dd, J = 14.1, 4.2 Hz, 1H), 3.05–2.94 (m, 2H), 2.89 (td, J = 9.3, 8.6, 3.7 Hz, 1H), 2.84–2.74 (m, 2H), 1.87–1.79 (m, 1H), 1.68–1.56 (m, 1H), 1.42 (dd, J = 13.2, 5.7 Hz, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 155.6, 137.6, 137.3, 132.6, 129.4, 128.9, 128.7, 128.1, 126.7, 109.4, 73.6, 72.8, 70.9, 69.7, 58.7, 55.3, 53.6, 45.5, 35.7, 27.3, 25.9, 20.2, 19.9. HRMS (m/z): [M + H] + calcd for C27H35BrN2O7S, 611.1421; found 611.1431. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.36%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-3-fluoro-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5ag)
White solid (44%); Rf (EtOAc/hexane = 40:60) = 0.41; m.p. = 134.4–135.1 °C; 1H NMR (500 MHz, CDCl3): δ 7.73 (dd, J = 8.3, 6.5 Hz, 1H), 7.52 (dd, J = 7.7, 2.1 Hz, 1H), 7.44 (dd, J = 8.3, 2.0 Hz, 1H), 7.29 (dd, J = 8.2, 6.6 Hz, 2H), 7.24–7.19 (m, 3H), 5.65 (d, J = 5.2 Hz, 1H), 5.04 (dt, J = 8.2, 6.2 Hz, 1H), 4.94 (d, J = 8.4 Hz, 1H), 3.96 (dd, J = 9.7, 6.1 Hz, 1H), 3.91–3.83 (m, 3H), 3.75–3.64 (m, 2H), 3.47 (d, J = 3.1 Hz, 1H), 3.16 (dd, J = 15.2, 8.4 Hz, 1H), 3.06 (ddd, J = 17.9, 14.5, 3.2 Hz, 2H), 2.98 (dd, J = 13.5, 8.3 Hz, 1H), 2.94–2.90 (m, 1H), 2.87 (dd, J = 13.5, 6.9 Hz, 1H), 2.79 (dd, J = 14.1, 9.0 Hz, 1H), 1.91–1.80 (m, 1H), 1.69–1.61 (m, 1H), 1.49–1.44 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 159.1 (d, J = 253.5 Hz), 155.7, 139.5, 137.5, 134.7, 129.4, 128.8, 126.9, 124.1, 115.7 (d, J = 25.3 Hz), 109.4, 73.7, 72.8, 70.9, 69.7, 58.6, 55.3, 53.6, 45.4, 35.7, 27.3, 25.9, 20.9, 19.9. HRMS (m/z): [M + H] + calcd. for C27H34BrFN2O7S, 629.1327; found 629.1335. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.23%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((3,4-Dichloro-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5ad)
White solid (32%); Rf (EtOAc/hexane = 40:60) = 0.39; m.p. = 132.8–133.1 °C; 1H NMR (500 MHz, CDCl3): δ 7.86 (d, J = 1.5 Hz, 1H), 7.64–7.56 (m, 2H), 7.33–7.26 (m, 2H), 7.21 (d, J = 8.2 Hz, 3H), 5.65 (d, J = 5.2 Hz, 1H), 5.03 (dt, J = 8.2, 6.2 Hz, 1H), 4.91 (d, J = 8.3 Hz, 1H), 3.99–3.93 (m, 1H), 3.91–3.81 (m, 2H), 3.72 (dd, J = 9.7, 6.0 Hz, 1H), 3.68 (dd, J = 9.6, 3.6 Hz, 1H), 3.50 (d, J = 3.1 Hz, 1H), 3.17 (dd, J = 15.2, 8.5 Hz, 1H), 3.11–3.03 (m, 2H), 3.00 (dd, J = 13.5, 8.4 Hz, 1H), 2.95–2.90 (m, 1H), 2.87 (dd, J = 13.5, 6.9 Hz, 1H), 2.80 (dd, J = 14.1, 9.0 Hz, 1H), 1.87–1.80 (m, 1H), 1.69–1.59 (m, 1H), 1.46 (dd, J = 13.2, 5.9 Hz, 1H), 0.93 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 155.7, 138.3, 137.9, 137.5, 134.0, 131.4, 129.4, 129.2, 128.8, 126.9, 126.4, 109.4, 73.7, 72.8, 70.9, 69.7, 58.6, 55.4, 53.5, 45.4, 35.7, 27.3, 25.9, 20.2, 19.9. HRMS (m/z): [M + H] + calcd for C27H34Cl2N2O7S, 601.1537; found 601.1502. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.41%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-Hydroxy-4-((N-isobutyl-4-isopropyl phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (5ac)
Sticky oil (29%); Rf (EtOAc/hexane = 40:60) = 0.43; 1H NMR (500 MHz, CDCl3): δ 7.71–7.66 (m, 2H), 7.39–7.34 (m, 2H), 7.28 (d, J = 7.3 Hz, 2H), 7.24–7.17 (m, 3H), 5.64 (d, J = 5.2 Hz, 1H), 5.01 (dt, J = 8.2, 6.2 Hz, 2H), 3.94 (dd, J = 9.7, 6.2 Hz, 1H), 3.91–3.81 (m, 3H), 3.75 (s, 1H), 3.72–3.64 (m, 2H), 3.17 (dd, J = 15.2, 8.6 Hz, 1H), 3.08 (dd, J = 14.2, 4.3 Hz, 1H), 3.04–2.93 (m, 3H), 2.92–2.86 (m, 1H), 2.84–2.76 (m, 2H), 1.83 (ddd, J = 12.8, 8.3, 6.3 Hz, 1H), 1.72–1.57 (m, 1H), 1.46–1.43 (m, 1H), 1.27 (d, J = 6.9 Hz, 6H), 0.94 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 155.6, 154.6, 137.7, 135.3, 129.5, 128.6, 127.6, 127.4, 126.7, 109.4, 73.5, 73.0, 70.9, 69.7, 59.1, 55.2, 53.9, 45.4, 35.7, 34.3, 27.4, 25.9, 23.7, 20.2, 19.9. HRMS (m/z): [M + H] + calcd for C30H42N2O7S, 575.2785; found 575.2704. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 97.54%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-Hydroxy-4-((N-isobutyl-4-isopropoxy phenyl)sulfonamido)-1-phenylbutan-2-yl)carbamate (5ae)
Sticky oil (28.9%); Rf (EtOAc/hexane = 40:60) = 0.42; 1H NMR (500 MHz, CDCl3): δ 7.70–7.65 (m, 2H), 7.30–7.18 (m, 5H), 6.97–6.91 (m, 2H), 5.63 (d, J = 5.2 Hz, 1H), 5.01 (dt, J = 12.3, 7.6 Hz, 2H), 4.62 (p, J = 6.1 Hz, 1H), 3.96–3.91 (m, 1H), 3.91–3.81 (m, 2H), 3.74 (d, J = 2.6 Hz, 1H), 3.71–3.64 (m, 2H), 3.15 (dd, J = 15.2, 8.6 Hz, 1H), 3.08 (dd, J = 14.1, 4.3 Hz, 1H), 3.02–2.92 (m, 2H), 2.92–2.83 (m, 1H), 2.83–2.73 (m, 2H), 1.87–1.79 (m, 1H), 1.67–1.54 (m, 1H), 1.45–1.41 (, 1H), 1.36 (d, J = 6.0 Hz, 6H), 0.92 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 161.7, 155.5, 137.7, 129.6, 129.4, 129.0, 128.6, 126.6, 115.7, 109.4, 73.5, 72.9, 70.9, 70.5, 69.7, 59.0, 55.2, 53.8, 45.4, 35.7, 27.3, 25.9, 21.9, 20.2, 20.0. HRMS (m/z): [M + H] + calcd for C30H42N2O8S, 591.2735; found 591.2758. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 97.19%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Butoxy-N-isobutyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5af)
White solid (13.7%); Rf (EtOAc/hexane = 40:60) = 0.36; m.p. = 129.8–131.2 °C; 1H NMR (500 MHz, CDCl3): δ 7.69 (d, J = 8.9 Hz, 2H), 7.30–7.24 (m, 3H), 7.21 (d, J = 7.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 5.63 (d, J = 5.2 Hz, 1H), 5.01 (dt, J = 12.3, 7.2 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 3.95 (td, J = 10.4, 9.8, 6.5 Hz, 1H), 3.91–3.84 (m, 2H), 3.82 (dd, J = 8.3, 2.2 Hz, 1H), 3.74 (d, J = 2.7 Hz, 1H), 3.71–3.61 (m, 2H), 3.18–3.04 (m, 2H), 3.01–2.93 (m, 2H), 2.92–2.86 (m, 1H), 2.83–2.76 (m, 2H), 1.88–1.73 (m, 5H), 1.67–1.55 (m, 1H), 1.54–1.45 (m, 2H), 1.46–1.40 (m, 1H), 0.98 (t, J = 7.4 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 162.8, 155.5, 137.7, 129.5, 129.4, 129.3, 128.6, 126.6, 114.8, 109.4, 73.5, 72.9, 70.9, 69.7, 68.2, 58.9, 55.2, 53.8, 45.4, 35.7, 31.1, 27.3, 25.9, 20.2, 20.0, 19.2, 13.9. HRMS (m/z): [M + H] + calcd for C31H44N2O8S, 605.2891; found 605.2783. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.74%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (5ba)
White solid (50%); Rf (EtOAc/hexane = 40:60) = 0.39; m.p. = 141.8–42.3 °C; 1H NMR (500 MHz, CDCl3): δ 7.80–7.74 (m, 2H), 7.29 (td, J = 8.0, 6.6 Hz, 4H), 7.23 (dd, J = 7.3, 1.5 Hz, 2H), 7.22–7.17 (m, 4H), 7.17–7.13 (m, 2H), 5.66 (d, J = 5.2 Hz, 1H), 4.84 (d, J = 8.9 Hz, 1H), 3.97 (dd, J = 9.7, 6.3 Hz, 1H), 3.86 (ddt, J = 9.2, 6.0, 3.2 Hz, 1H), 3.71 (ddd, J = 16.5, 9.0, 5.7 Hz, 2H), 3.41 (dd, J = 8.7, 6.2 Hz, 3H), 3.38–3.30 (m, 1H), 3.29 (d, J = 3.4 Hz, 1H), 3.16 (dd, J = 14.9, 8.2 Hz, 1H), 3.10 (dd, J = 14.8, 3.5 Hz, 1H), 3.04 (dd, J = 14.1, 4.9 Hz, 1H), 2.95–2.91 (m, 1H), 2.90–2.86 (m, 2H), 2.80 (d, J = 9.4 Hz, 1H), 1.72–1.60 (m, 1H), 1.54–1.47 (m, 1H). 13C NMR (126 MHz, CDCl3): δ 165.3 (d, J = 257.04 Hz), 155.8, 138.1, 137.5, 134.5, 130.1 (d, J = 9.5 Hz), 129.4, 129.0, 128.9, 128.7, 126.8, 126.9, 116.7 (d, J = 22.0 Hz), 109.4, 73.6, 72.5, 70.9, 69.7, 55.2, 53.1, 52.2, 45.4, 35.7, 35.6, 25.9. HRMS (m/z): [M + H] + calcd for C31H35FN2O7S, 599.2222; found 599.2242. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 96.92%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)carbamate (5bb)
White solid (42%); Rf (EtOAc/hexane = 40:60) = 0.47; m.p. = 129.3–30.1 °C; 1H NMR (500 MHz, CDCl3): δ 7.63–7.55 (m, 4H), 7.28–7.22 (m, 4H), 7.19 (dd, J = 13.8, 7.1 Hz, 4H), 7.10 (d, J = 7.0 Hz, 2H), 5.63 (d, J = 5.2 Hz, 1H), 5.06–4.98 (m, 1H), 3.92 (dd, J = 9.7, 6.2 Hz, 1H), 3.88–3.78 (m, 2H), 3.76–3.61 (m, 3H), 3.42–3.31 (m, 3H), 3.13 (d, J = 6.3 Hz, 2H), 3.01 (dd, J = 14.1, 4.7 Hz, 1H), 2.93–2.78 (m, 3H), 2.74 (dd, J = 14.0, 9.7 Hz, 1H), 1.67–1.59 (m, 1H), 1.46–1.40 (m, 1H). 13C NMR (126 MHz, CDCl3): δ 155.7, 138.0, 137.6, 137.5, 132.6, 129.4, 128.9, 128.8, 128.7, 128.1, 126.9, 126.7, 109.4, 73.6, 72.5, 71.0, 69.7, 55.2, 52.9, 51.9, 45.5, 35.6, 25.9. HRMS (m/z): [M + H] + calcd. for C31H35BrN2O7S, 659.1421; found 659.1415. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 98.18%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-3-fluoro-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (5bg)
White solid (30.4%); Rf (EtOAc/hexane = 40:60) = 0.39; m.p. = 133.2–134.8 °C; 1H NMR (500 MHz, CDCl3): δ 7.69 (dd, J = 8.4, 6.4 Hz, 1H), 7.46 (dd, J = 7.7, 2.1 Hz, 1H), 7.41 (dd, J = 8.3, 2.1 Hz, 1H), 7.32–7.27 (m, 4H), 7.25–7.18 (m, 4H), 7.16–7.10 (m, 2H), 5.66 (d, J = 5.2 Hz, 1H), 5.06 (dt, J = 8.2, 6.0 Hz, 1H), 4.95 (d, J = 8.8 Hz, 1H), 3.97 (dd, J = 9.8, 6.1 Hz, 1H), 3.89–3.83 (m, 2H), 3.79–3.71 (m, 2H), 3.71–3.66 (m, 1H), 3.41 (td, J = 7.4, 3.6 Hz, 2H), 3.25 (d, J = 3.7 Hz, 1H), 3.16 (d, J = 5.8 Hz, 2H), 3.05 (dd, J = 14.1, 5.0 Hz, 1H), 2.95–2.86 (m, 3H), 2.77 (dd, J = 14.1, 9.5 Hz, 1H), 1.66–1.62 (m, 1H), 1.51–1.47 (m, 1H). 13C NMR (126 MHz, CDCl3): δ 159.0 (d, J = 253.4 Hz), 155.8, 139.6, 137.7 (d, J = 47.0 Hz), 134.7, 129.4, 128.9, 128.9, 128.7, 127.01, 126.8, 124.0 (d, J = 3.8 Hz), 115.6 (d, J = 25.2 Hz), 109.4, 73.7, 72.5, 70.9, 69.7, 55.3, 52.8, 51.9, 45.5, 35.6, 35.7, 25.9. HRMS (m/z): [M + H] + calcd for C31H35BrFN2O7S, 677.1327; found 677.1295. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 97.93%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-3-Hydroxy-4-((4-isopropyl-N-phenethyl phenyl)sulfonamido)-1-phenyl butan-2-yl)carbamate (5bc)
White solid (40%); Rf (EtOAc/hexane = 40:60) = 0.37; m.p. = 123.5–124.2 °C; 1H NMR (500 MHz, CDCl3): δ 7.69 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.29 (t, J = 7.3 Hz, 4H), 7.23–7.19 (m, 4H), 7.17–7.11 (m, 2H), 5.66 (d, J = 5.2 Hz, 1H), 5.07–5.00 (m, 1H), 4.85 (d, J = 9.1 Hz, 1H), 3.97 (dd, J = 9.6, 6.3 Hz, 1H), 3.86 (td, J = 8.3, 2.3 Hz, 2H), 3.74–3.67 (m, 3H), 3.47–3.38 (m, 2H), 3.35–3.27 (m, 1H), 3.17 (dd, J = 14.9, 8.3 Hz, 1H), 3.11–3.02 (m, 2H), 3.01–2.95 (m, J = 6.9 Hz, 1H), 2.90 (ddt, J = 21.1, 8.9, 6.2 Hz, 3H), 2.79 (dd, J = 14.1, 9.5 Hz, 1H), 1.71–1.60 (m, 1H), 1.49 (dd, J = 13.2, 5.8 Hz, 1H), 1.27 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 155.7, 154.7, 138.3, 137.6, 135.5, 129.5, 129.0, 128.8, 128.7, 127.6, 127.5, 126.8, 126.7, 109.4, 73.6, 72.6, 70.9, 69.7, 55.1, 53.4, 52.5, 45.4, 35.9, 35.6, 34.3, 25.9, 23.7. HRMS (m/z): [M + H]+ calcd for C34H43N2O7S, 623.2785; found 623.2717. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 91.94%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b] furan-3-yl ((2S,3R)-3-Hydroxy-4-((4-isopropoxy-N-phenethyl phenyl)sulfonamido)-1-phenyl butan-2-yl)carbamate (5be)
White solid (19%); Rf (EtOAc/hexane = 40:60) = 0.4; m.p. = 130.3–131.4 °C; 1H NMR (500 MHz, CDCl3): δ 7.67 (d, J = 8.9 Hz, 2H), 7.31–7.25 (m, 4H), 7.21 (t, J = 7.7 Hz, 4H), 7.15 (d, J = 7.0 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 5.65 (d, J = 5.2 Hz, 1H), 5.04 (q, J = 6.2 Hz, 1H), 4.90 (d, J = 9.1 Hz, 1H), 4.66–4.58 (m, 1H), 3.96 (dd, J = 9.6, 6.3 Hz, 1H), 3.85 (dt, J = 8.1, 4.2 Hz, 2H), 3.75–3.65 (m, 3H), 3.43–3.24 (m, 3H), 3.19–3.00 (m, 3H), 2.94–2.84 (m, 3H), 2.81–2.74 (m, 1H), 1.51 (s, 2H), 1.49 (d, J = 7.4 Hz, 1H), 1.36 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 161.8, 155.7, 138.3, 137.7, 129.6, 129.5, 129.2, 129.0, 128.8, 128.7, 126.8, 126.7, 115.8, 109.4, 73.5, 72.6, 70.9, 70.6, 69.7, 55.1, 53.3, 52.4, 45.4, 35.8, 35.6, 25.9, 21.9. HRMS (m/z): [M + H] + calcd for C34H43N2O8S, 639.2735; found 639.2743. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 98.21%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Butoxy-N-phenethyl phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (5bf)
White solid (41.8%); Rf (EtOAc/hexane = 40:60) = 0.39; m.p. = 127.9–128.5 °C; 1H NMR (500 MHz, CDCl3): δ 7.69 (d, J = 8.9 Hz, 2H), 7.31–7.26 (m, 5H), 7.22 (dt, J = 12.0, 4.6 Hz, 3H), 7.15 (dd, J = 6.9, 1.7 Hz, 2H), 6.98–6.92 (m, 2H), 5.66 (d, J = 5.2 Hz, 1H), 5.04 (dt, J = 8.3, 6.3 Hz, 1H), 4.86 (d, J = 9.1 Hz, 1H), 4.01 (t, J = 6.5 Hz, 2H), 3.97 (dd, J = 9.6, 6.2 Hz, 1H), 3.86 (td, J = 8.1, 2.4 Hz, 2H), 3.75–3.65 (m, 2H), 3.45–3.38 (m, 1H), 3.37 (d, J = 3.0 Hz, 1H), 3.32–3.26 (m, 1H), 3.15 (dd, J = 14.9, 8.3 Hz, 1H), 3.11–2.99 (m, 2H), 2.96–2.83 (m, 3H), 2.79 (dd, J = 14.2, 9.5 Hz, 1H), 1.82–1.76 (m, 2H), 1.72–1.60 (m, 1H), 1.55–1.44 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 162.9, 155.7, 138.3, 137.7, 134.8, 129.5, 129.5, 129.0, 128.8, 128.7, 126.8, 126.7, 115.0, 109.4, 73.6, 72.6, 70.9, 69.7, 68.3, 55.1, 53.3, 52.4, 45.4, 35.8, 35.6, 31.1, 25.9, 19.2, 13.9. HRMS (m/z): [M + H] + calcd for C35H45N2O8S, 653.2891; found 653.2812. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.88%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-3-fluoro-N-(2-fluoro benzyl) phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (5dg)
White solid (16.9%); Rf (EtOAc/hexane = 40:60) = 0.43; m.p. = 136.9–137.2 °C; 1H NMR (500 MHz, CDCl3): δ 7.68 (dd, J = 8.4, 6.4 Hz, 1H), 7.44 (dd, J = 7.7, 2.1 Hz, 1H), 7.40 (dd, J = 8.3, 2.1 Hz, 1H), 7.36 (dd, J = 7.6, 1.8 Hz, 1H), 7.32–7.23 (m, 4H), 7.23–7.17 (m, 1H), 7.16–7.12 (m, 2H), 7.09 (d, J = 1.2 Hz, 1H), 6.98–6.94 (m, 1H), 5.64 (d, J = 5.3 Hz, 1H), 5.02 (dt, J = 8.2, 5.9 Hz, 1H), 4.72–4.66 (m, 1H), 4.54–4.36 (m, 2H), 3.94 (dd, J = 9.8, 6.1 Hz, 1H), 3.84 (td, J = 8.2, 2.2 Hz, 1H), 3.80–3.77 (m, 1H), 3.72 (dd, J = 9.7, 5.8 Hz, 1H), 3.70–3.65 (m, 1H), 3.54 (dt, J = 6.5, 3.2 Hz, 1H), 3.32 (d, J = 3.6 Hz, 1H), 3.30–3.18 (m, 2H), 2.96 (dd, J = 14.1, 4.7 Hz, 1H), 2.93–2.88 (m, 1H), 2.70 (dd, J = 14.1, 9.2 Hz, 1H), 1.67–1.58 (m, 1H), 1.47–1.42 (m, 1H). 13C NMR (126 MHz, CDCl3): δ 161.0 (d, J = 247.7 Hz), 156.8 (d, J = 297.4 Hz), 139.9 (d, J = 6.0 Hz), 137.3, 134.6, 131.5 (d, J = 3.5 Hz), 130.5 (d, J = 8.2 Hz), 129.4, 128.7, 126.8, 124.7 (d, J = 3.6 Hz), 123.9 (d, J = 3.8 Hz), 122.2 (d, J = 14.3 Hz), 115.7 (d, J = 12.3 Hz), 115.5 (d, J = 16.0 Hz), 115.1 (d, J = 21.5 Hz), 109.4, 73.6, 72.2, 70.9, 69.7, 54.8, 52.6, 47.1, 45.5, 35.7, 25.9. HRMS (m/z): [M + H] + calcd for C30H32BrFN2O7S, 681.1076; found 6.81.1042. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.19%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3R)-4-((4-Bromo-N-(3,3-diphenyl propyl)-3-fluoro phenyl)sulfonamido)-3-hydroxy-1-phenyl butan-2-yl)carbamate (5cg)
White solid (24.7%); Rf (EtOAc/hexane = 40:60) = 0.35; m.p. = 122.3–124.7 °C; 1H NMR (500 MHz, CDCl3): δ 7.65 (dd, J = 8.3,6.4 Hz, 1H), 7.38 (dd, J = 7.7, 2.0 Hz, 1H), 7.32–7.27 (m, 7H), 7.25–7.14 (m, 9H), 5.65 (d, J = 5.2 Hz, 1H), 5.06–4.99 (m, 1H), 4.87 (d, J = 8.8 Hz, 1H), 3.98–3.89 (m, 1H), 3.91–3.79 (m, 3H), 3.78 (d, J = 4.0 Hz, 1H), 3.74–3.63 (m, 2H), 3.24 (d, J = 3.7 Hz, 1H), 3.21–3.11 (m, 2H), 3.12–2.99 (m, 3H), 2.95–2.89 (m, 1H), 2.77 (dd, J = 14.1, 9.5 Hz, 1H), 2.42–2.27 (m, 2H), 1.71–1.61 (m, 1H), 1.47 (dd, J = 13.0, 6.1 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 159.0 (d, J = 253.1 Hz), 155.8, 143.6 (d, J = 9.6 Hz), 139.5, 137.5, 134.7, 129.4, 128.8 (d, J = 5.9 Hz), 127.7, 126.8 (d, J = 13.6 Hz), 123.9, 115.6 (d, J = 25.2 Hz), 115.1 (d, J = 20.7 Hz), 109.4, 73.7, 72.5, 70.8, 69.7, 55.4, 53.0, 49.4, 48.7, 45.5, 35.6, 34.9, 25.9. HRMS (m/z): [M + H]+ calcd for C38H41BrFN2O7S, 767.1796; found 767.1547. HPLC analytical purity (ACN/H2O: 70/30)190–800 nm = 99.60%.
HIV-1 PR Expression and Purification
Escherichia coli BL21 (DE3) carrying pET28a-HIV-1 PR plasmid were grown in LB medium containing 30 μg/mL kanamycin at 37 °C with shaking speed of 250 rpm. When OD600 reached 0.9–1.0, the expression of recombinant HIV-1 protease was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 0.5 mM and incubating at 16 °C, 160 rpm overnight. Cells were harvested by centrifugation, dissolved with PBS (pH 7.4) containing 1 mM Ethylenediaminetetraacetic acid (EDTA) and lysed by sonication. As both WT and mutated HIV-PR were expressed in inclusion bodies, the pellets were collected by centrifugation at 10,000 × g, 4 °C for 30 min and subsequently resuspended in PBS containing 6 M urea. The pellet was removed by configuration at 30,000 × g, 4 °C for 30 min and the supernatant was subjected to column chromatography.
Crude protein was loaded onto a Ni Sepharose 6 Fast Flow column, pre-equilibrated with PBS containing 6 M urea. After washing the column with Triton-X (1% v/v) and 20 mM imidazole respectively, the HIV-1 PR was eluted by PBS containing 300 mM imidazole and 6 M urea. The HIV-1 PR was then refolded by slowly reducing urea concentration in 100 mM acetate buffer (pH 5.0) containing ethylene glycol (5% v/v) and glycerol (10% v/v). The refolded HIV-1 PR was further purified by Superdex 200 Increase 10/300 GL. The protein was concentrated and stored at–80 °C until use.
Fluorescent Assay for HIV-1 PR Inhibition
The fluorescence resonance energy transfer (FRET) assay was used to evaluate the HIV-1 PR inhibitory capabilities of all newly synthesized DRV analogs in vitro. The commercially available peptide (Arg-Glu-(EDANS)-Ser-Gly-Ile-Phe-Leu-Glu-Thr-Ser-Lys-(DABCYL)-Arg) was opted as the substrate (10 μM). The wavelengths of excitation and emission were observed at 340 and 490 nm, respectively. To carry out assay analysis, the peptide is tagged at both ends with the energy transfer donor (EDANS) and acceptor (DABCYL) dyes. Dimethyl sulfoxide (DMSO) was used to dissolve the inhibitors and dilute them to the proper amounts. The reaction was carried out in 50 mM sodium acetate, pH 5.0, 0.1 M sodium chloride, and 2% V/V DMF at 27 °C for 30 min. The inhibition assay was performed using 100 nM HIV-1 protease. The relative fluorescence units (RFU) were recorded in real-time using Synergy TM H1/BioTek microplate reader and used for the calculation of initial velocities. Data were fitted by nonlinear regression to Morrison’s equation according to Windsor & Raines, 2015 for Ki determination.49
Cytotoxicity Assay
The cytotoxicity of DRV analogs was tested with Vero (CCL-81) and 293T (CRL3216) cell lines (ATCC, Manassas, VA, USA). 293T was courtesy of Asst. Prof. Pokrath Hansasuta, Division of Virology, Department of Microbiology, Faculty of Medicine, Chulalongkorn University. Each cell line was seeded at 1 × 104 cells per well into 96-well plates and incubated at 37 °C under 5% CO2 overnight. The DRV analogs were added at the indicated concentrations in the final concentrations of 1% DMSO in the maintenance medium. Cells were incubated for 2 days before analyzing the cell viability using CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA) according to manufacturer’s protocol. The plate was read at the A490 nm by VICTORTM X3 microplate reader (PerkinElmer, Waltham, MA, USA). Each sample was analyzed in triplicated, and results were reported as means and standard deviation (SD).50,52
Acknowledgments
We would like to thank all the facilities at Chulalongkorn University that provided support in the completion of the experiments detailed in this manuscript. For computational studies, we would like to thank all the facilities at the Research Program in the Centre for Computational Science, University of Tsukuba, Japan, who provided support in the completion of the computational studies detailed in this manuscript. For the cytotoxicity analysis of 293T cell lines, we would like to thank Dr. Pokrath Hansasuta, Faculty of Medicine, Chulalongkorn University, for providing their assistance. Author, Muhammad Asad Ur Rehman would like to thank the Scholarship Program for ASEAN and Non-ASEAN Countries, and Overseas Research Experience Scholarship, Graduate School, Chulalongkorn University, Thailand.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.4c00040.
Details of characterization analysis of newly synthesized compound (PDF)
Author Contributions
CRediT: Muhammad Asad Ur Rehman data curation, formal analysis, investigation, methodology, visualization, writing - original draft; Hathaichanok Chuntakaruk data curation, investigation, software, visualization; Soraat Amphan data curation, investigation; Aphinya Suroengrit data curation, investigation; Kowit Hengphasatporn formal analysis, software, validation, writing - review & editing; Yasuteru Shigeta funding acquisition, resources, supervision; Thanyada Rungrotmongkol conceptualization, formal analysis, methodology, software, supervision, validation, writing - review & editing; Kuakarun Krusong conceptualization, formal analysis, methodology, supervision, validation, writing - review & editing; Siwaporn Boonyasuppayakorn methodology, validation, writing - review & editing; Chanat Aonbangkhen conceptualization, methodology, resources, validation, writing - review & editing; Tanatorn Khotavivattana conceptualization, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, writing - original draft, writing - review & editing.
This Research is funded by Thailand Science Research and Innovation Fund and Chulalongkorn University (HEA662300079) to Dr. Thanyada Rungrotmongkol, Dr. Kuakarun Krusong, Dr. Chanat Aonbangkhen, and Dr. Tanatorn Khotavivattana. Authors, Dr. Yasuteru Shigeta and Dr. Kowit Hengphasatporn would like to thank for the Grant-in-aid for Scientific Researches (23H04879, 23H02427, 21H05269, and 24K20888) from JSPS. This research in part used the computational resources of Cygnus provided by the Multidisciplinary Cooperative Research Program in the Centre for Computational Sciences, University of Tsukuba, Japan.
The authors declare no competing financial interest.
Supplementary Material
References
- Human immunodeficiency virus (HIV). Transfus. Med. Hemother. 2016, 43 (3), 203–222. 10.1159/000445852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joint United Nations Programme on HIV/AIDS ; 2004: Report on the global AIDS epidemic: 4th global report; UNAIDS, 2004. [Google Scholar]
- Fanales-Belasio E.; Raimondo M.; Suligoi B.; Buttò S. HIV virology and pathogenetic mechanisms of infection: a brief overview. Ann. Ist. Super. 2010, 46, 5–14. 10.4415/ANN_10_01_02. [DOI] [PubMed] [Google Scholar]
- Brik A.; Wong C.-H. HIV-1 protease: mechanism and drug discovery. Org. Biomol. Chem. 2003, 1 (1), 5–14. 10.1039/b208248a. [DOI] [PubMed] [Google Scholar]
- Mitsuya H.; Broder S. Strategies for antiviral therapy in AIDS. Nature. 1987, 325 (6107), 773–778. 10.1038/325773a0. [DOI] [PubMed] [Google Scholar]
- Deeks E. D. Darunavir: a review of its use in the management of HIV-1 infection. Drugs. 2014, 74 (1), 99–125. 10.1007/s40265-013-0159-3. [DOI] [PubMed] [Google Scholar]
- Kohl N. E.; Emini E. A.; Schleif W. A.; Davis L. J.; Heimbach J. C.; Dixon R.; Scolnick E. M.; Sigal I. S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U. S. A. 1988, 85 (13), 4686–4690. 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgil S. C. First-Generation HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. Aspartic acid proteases as therapeutic targets. 2010, 45, 139–168. 10.1002/9783527630943.ch6. [DOI] [Google Scholar]
- Ali A.; Bandaranayake R. M.; Cai Y.; King N. M.; Kolli M.; Mittal S.; Murzycki J. F.; Nalam M. N.; Nalivaika E. A.; Özen A.; Prabu-Jeyabalan M. M.; Thayer K.; Schiffer C. A. Molecular basis for drug resistance in HIV-1 protease. Viruses 2010, 2 (11), 2509–2535. 10.3390/v2112509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Anderson D. D.; Mitsuya H. The FDA Approved HIV-1 Protease Inhibitors for Treatment of HIV/AIDS. Burger's Med. Chem. Drug Dis. 2003, 1–74. 10.1002/0471266949.bmc224. [DOI] [Google Scholar]
- Lv Z.; Chu Y.; Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV/AIDS-Res. Palliat. Care. 2015, 95–104. 10.2147/HIV.S79956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z. Second generation HIV protease inhibitors against resistant virus. Expert Opin. Drug Dis. 2008, 3 (7), 775–786. 10.1517/17460441.3.7.775. [DOI] [PubMed] [Google Scholar]
- Murphy E. L.; Collier A. C.; Kalish L. A.; Assmann S. F.; Para M. F.; Flanigan T. P.; Kumar P. N.; Mintz L.; Wallach F. R.; Nemo G. J. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann. Int. Med. 2001, 135 (1), 17–26. 10.7326/0003-4819-135-1-200107030-00005. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Osswald H. L.; Prato G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59 (11), 5172–5208. 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Sridhar P. R.; Leshchenko S.; Hussain A. K.; Li J.; Kovalevsky A. Y.; Walters D. E.; Wedekind J. E.; Grum-Tokars V.; Das D.; Koh Y.; Maeda K.; Gatanaga H.; Weber I. T.; Mitsuya H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J. Med. Chem. 2006, 49 (17), 5252–5261. 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- Staszewski S.; Morales-Ramirez J.; Tashima K. T.; Rachlis A.; Skiest D.; Stanford J.; Stryker R.; Johnson P.; Labriola D. F.; Farina D.; Manion D. J.; Ruiz N. M. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. N. Engl. J. Med. 1999, 341 (25), 1865–1873. 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- Wainberg M. A.; Friedland G. Public health implications of antiretroviral therapy and HIV drug resistance. JAMA 1998, 279 (24), 1977–1983. 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- Aoki M.; Das D.; Hayashi H.; Aoki-Ogata H.; Takamatsu Y.; Ghosh A. K.; Mitsuya H. Mechanism of darunavir (DRV)’s high genetic barrier to HIV-1 resistance: a key V32I substitution in protease rarely occurs, but once it occurs, it predisposes HIV-1 to develop DRV resistance. MBio. 2018, 9 (2), e0242517 10.1128/mBio.02425-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Rao K. V.; Nyalapatla P. R.; Osswald H. L.; Martyr C. D.; Aoki M.; Hayashi H.; Agniswamy J.; Wang Y.-F.; Bulut H.; Das D.; Weber I. T.; Mitsuya H. Design and development of highly potent HIV-1 protease inhibitors with a crown-like oxotricyclic core as the P2-ligand to combat multidrug-resistant HIV variants. J. Med. Chem. 2017, 60 (10), 4267–4278. 10.1021/acs.jmedchem.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Dawson Z. L.; Mitsuya H. Darunavir, a conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV. Bioorg. Med. Chem. 2007, 15 (24), 7576–7580. 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaranayake R. M.; Kolli M.; King N. M.; Nalivaika E. A.; Heroux A.; Kakizawa J.; Sugiura W.; Schiffer C. A. The effect of clade-specific sequence polymorphisms on HIV-1 protease activity and inhibitor resistance pathways. J. Virol. 2010, 84 (19), 9995–10003. 10.1128/JVI.00505-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M.; Zhou H.; Ma L.; Dong B.; Zhou J.; Zhang G.; Wang M.; Wang J.; Cen S.; Wang Y. Design and evaluation of novel piperidine HIV-1 protease inhibitors with potency against DRV-resistant variants. Eur. J. Med. Chem. 2021, 220, 113450 10.1016/j.ejmech.2021.113450. [DOI] [PubMed] [Google Scholar]
- Tie Y.; Kovalevsky A. Y.; Boross P.; Wang Y. F.; Ghosh A. K.; Tozser J.; Harrison R. W.; Weber I. T. Atomic resolution crystal structures of HIV-1 protease and mutants V82A and I84V with saquinavir. Proteins: Structure, Function, and Bioinformatics. 2007, 67 (1), 232–242. 10.1002/prot.21304. [DOI] [PubMed] [Google Scholar]
- Kovalevsky A. Y.; Tie Y.; Liu F.; Boross P. I.; Wang Y.-F.; Leshchenko S.; Ghosh A. K.; Harrison R. W.; Weber I. T. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J. Med. Chem. 2006, 49 (4), 1379–1387. 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Ramu Sridhar P.; Kumaragurubaran N.; Koh Y.; Weber I. T.; Mitsuya H. Bis-tetrahydrofuran: a privileged ligand for darunavir and a new generation of HIV protease inhibitors that combat drug resistance. ChemMedChem: Chem. Enabling Drug Dis. 2006, 1 (9), 939–950. 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- Agniswamy J.; Shen C.-H.; Wang Y.-F.; Ghosh A. K.; Rao K. V.; Xu C.-X.; Sayer J. M.; Louis J. M.; Weber I. T. Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with P1′-pyrrolidinone or P2-tris-tetrahydrofuran. J. Med. Chem. 2013, 56 (10), 4017–4027. 10.1021/jm400231v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A.; Miller M.; Jaskólski M.; Sathyanarayana B. K.; Baldwin E.; Weber I. T.; Selk L. M.; Clawson L.; Schneider J.; Kent S. B. Conserved folding in retroviral proteases: crystal structure of synthetic HIV-1 protease. Sci. 1989, 245 (4918), 616–621. 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- Lapatto R.; Blundell T.; Hemmings A.; Overington J.; Wilderspin A.; Wood S.; Merson J. R.; Whittle P. J.; Danley D. E.; Geoghegan K. F.; Hawrylik S. J.; Lee S. E.; Scheld K. G.; Hobart P. M. X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature 1989, 342 (6247), 299–302. 10.1038/342299a0. [DOI] [PubMed] [Google Scholar]
- Wang Y.-F.; Tie Y.; Boross P. I.; Tozser J.; Ghosh A. K.; Harrison R. W.; Weber I. T. Potent new antiviral compound shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J. Med. Chem. 2007, 50 (18), 4509–4515. 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockbaum G. J.; Leidner F.; Rusere L. N.; Henes M.; Kosovrasti K.; Nachum G. S.; Nalivaika E. A.; Ali A.; Kurt Yilmaz N.; Schiffer C. A. Structural adaptation of darunavir analogues against primary mutations in HIV-1 protease. ACS Infect. Dis. 2019, 5 (2), 316–325. 10.1021/acsinfecdis.8b00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M.; Ode H.; Suzuki K.; Fujino M.; Maejima M.; Kimura Y.; Masaoka T.; Hattori J.; Matsuda M.; Hachiya A.; Yokomaku Y.; Suzuki A.; Watanabe N.; Sugiura W.; Iwatani Y. Unique flap conformation in an HIV-1 protease with high-level darunavir resistance. Front. Microbiol. 2016, 7, 61. 10.3389/fmicb.2016.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulnik S. V.; Suvorov L. I.; Liu B.; Yu B.; Anderson B.; Mitsuya H.; Erickson J. W. Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochem. 1995, 34 (29), 9282–9287. 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Dou Y.; Ma L.; Dong B.; Zhang F.; Zhang G.; Wang J.; Zhou J.; Cen S.; Wang Y. Novel HIV-1 protease inhibitors with morpholine as the P2 ligand to enhance activity against DRV-resistant variants. ACS Med. Chem. Lett. 2020, 11 (6), 1196–1204. 10.1021/acsmedchemlett.0c00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Kovela S.; Sharma A.; Shahabi D.; Ghosh A. K.; Hopkins D. R.; Yadav M.; Johnson M. E.; Agniswamy J.; Wang Y.; Hattori S.; Higashi-Kuwata N.; Aoki M.; Amano M.; Weber I. T.; Mitsuya H. Design, Synthesis and X-Ray Structural Studies of Potent HIV-1 Protease Inhibitors Containing C-4 Substituted Tricyclic Hexahydro-Furofuran Derivatives as P2 Ligands. ChemMedChem 2022, 17, e202200058 10.1002/cmdc.202200058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Leshchenko-Yashchuk S.; Anderson D. D.; Baldridge A.; Noetzel M.; Miller H. B.; Tie Y.; Wang Y.-F.; Koh Y.; Weber I. T.; Mitsuya H. Design of HIV-1 protease inhibitors with pyrrolidinones and oxazolidinones as novel P1′-Ligands to enhance backbone-binding interactions with protease: synthesis, biological evaluation, and protein–ligand X-ray studies. J. Med. Chem. 2009, 52 (13), 3902–3914. 10.1021/jm900303m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Yu X.; Osswald H. L.; Agniswamy J.; Wang Y.-F.; Amano M.; Weber I. T.; Mitsuya H. Structure-based design of potent HIV-1 protease inhibitors with modified P1-biphenyl ligands: Synthesis, biological evaluation, and enzyme–inhibitor X-ray structural studies. J. Med. Chem. 2015, 58 (13), 5334–5343. 10.1021/acs.jmedchem.5b00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Martyr C. D.; Steffey M.; Wang Y.-F.; Agniswamy J.; Amano M.; Weber I. T.; Mitsuya H. Design, synthesis, and X-ray structure of substituted bis-tetrahydrofuran (bis-THF)-derived potent HIV-1 protease inhibitors. ACS Med. Chem. Lett. 2011, 2 (4), 298–302. 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen J. L.; Leidner F.; Ragland D. A.; Kurt Yilmaz N.; Schiffer C. A. Interdependence of inhibitor recognition in HIV-1 protease. J. Chem. Theory Comput. 2017, 13 (5), 2300–2309. 10.1021/acs.jctc.6b01262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuntakaruk H.; Hengphasatporn K.; Shigeta Y.; Aonbangkhen C.; Lee V. S.; Khotavivattana T.; Rungrotmongkol T.; Hannongbua S. FMO-guided design of darunavir analogs as HIV-1 protease inhibitors. Sci. Rep. 2024, 14 (1), 3639. 10.1038/s41598-024-53940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Fyvie W. S.; Brindisi M.; Steffey M.; Agniswamy J.; Wang Y.-F.; Aoki M.; Amano M.; Weber I. T.; Mitsuya H. Design, synthesis, X-ray studies, and biological evaluation of novel macrocyclic HIV-1 protease inhibitors involving the P1′-P2′ ligands. Bioorg. Med. Chem. Lett. 2017, 27 (21), 4925–4931. 10.1016/j.bmcl.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M.; Ma L.; Dong B.; Zhang G.; Wang J.; Zhou J.; Cen S.; Wang Y. Synthesis and evaluation of potent human immunodeficiency virus 1 protease inhibitors with epimeric isopropanol as novel P1′ ligands. Future Med. Chem. 2020, 12 (9), 775–794. 10.4155/fmc-2019-0331. [DOI] [PubMed] [Google Scholar]
- Lockbaum G. J.; Rusere L. N.; Henes M.; Kosovrasti K.; Rao D. N.; Spielvogel E.; Lee S. K.; Nalivaika E. A.; Swanstrom R.; Yilmaz N. K.; Schiffer C. A.; Ali A. HIV-1 protease inhibitors with a P1 phosphonate modification maintain potency against drug-resistant variants by increased interactions with flap residues. Eur. J. Med. Chem. 2023, 257, 115501 10.1016/j.ejmech.2023.115501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Li Z.; Dhawan G.; Zhang W.; Sorochinsky A. E.; Butler G.; Soloshonok V. A.; Han J. Fluorine-containing drugs approved by the FDA in 2021. Chin. Chem. Lett. 2023, 34 (1), 107578. 10.1016/j.cclet.2022.06.001. [DOI] [Google Scholar]
- Fang W.-Y.; Ravindar L.; Rakesh K.; Manukumar H.; Shantharam C.; Alharbi N. S.; Qin H.-L. Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 173, 117–153. 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodi D.; Ishihara Y. Magic chloro”: profound effects of the chlorine atom in drug discovery. J. Med. Chem. 2023, 66 (8), 5305–5331. 10.1021/acs.jmedchem.2c02015. [DOI] [PubMed] [Google Scholar]
- Faleye O. S.; Boya B. R.; Lee J.-H.; Choi I.; Lee J. Halogenated antimicrobial agents to combat drug-resistant pathogens. Pharmacol. Rev. 2024, 76 (1), 90–141. 10.1124/pharmrev.123.000863. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Jadhav R. D.; Simpson H.; Kovela S.; Osswald H.; Agniswamy J.; Wang Y.-F.; Hattori S.-I.; Weber I. T.; Mitsuya H. Design, synthesis, and X-ray studies of potent HIV-1 protease inhibitors incorporating aminothiochromane and aminotetrahydronaphthalene carboxamide derivatives as the P2 ligands. Eur. J. Med. Chem. 2018, 160, 171–182. 10.1016/j.ejmech.2018.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-H.; Bai X.-G.; Zhou L.; Wang J.-X.; Liu H.-T.; Wang Y.-C. Synthesis and biological evaluation of novel HIV-1 protease inhibitors using tertiary amine as P2-ligands. Bioorg. Med. Chem. Lett. 2015, 25 (9), 1880–1883. 10.1016/j.bmcl.2015.03.047. [DOI] [PubMed] [Google Scholar]
- Windsor I. W.; Raines R. T. Fluorogenic assay for inhibitors of HIV-1 protease with sub-picomolar affinity. Sci. Rep. 2015, 5 (1), 11286. 10.1038/srep11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeanurit N.; Tuong T. L.; Nguyen V.-K.; Vibulakhaophan V.; Hengphasatporn K.; Shigeta Y.; Ho S. X.; Chu J. J. H.; Rungrotmongkol T.; Chavasiri W.; Boonyasuppayakorn S. Lichen-Derived Diffractaic Acid Inhibited Dengue Virus Replication in a Cell-Based System. Molecules 2023, 28 (3), 974. 10.3390/molecules28030974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emeny J. M.; Morgan M. J. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 1979, 43 (1), 247–252. 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- Wansri R.; Lin A. C. K.; Pengon J.; Kamchonwongpaisan S.; Srimongkolpithak N.; Rattanajak R.; Wilasluck P.; Deetanya P.; Wangkanont K.; Hengphasatporn K.; Shigeta Y.; Liangsakul J.; Suroengrit A.; Boonyasuppayakorn S.; Chuanasa T.; De-eknamkul W.; Hannongbua S.; Rungrotmongkol T.; Chamni S. Semi-synthesis of N-aryl amide analogs of piperine from Piper nigrum and evaluation of their antitrypanosomal, antimalarial, and anti-SARS-CoV-2 main protease activities. Molecules 2022, 27 (9), 2841. 10.3390/molecules27092841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armentano M. F.; Lupattelli P.; Bisaccia F.; D'Orsi R.; Miglionico R.; Nigro I.; Santarsiere A.; Berti F.; Funicello M.; Chiummiento L. Novel wild type and mutate HIV-1 protease inhibitors containing heteroaryl carboxamides in P2: Synthesis, biological evaluations and in silico ADME prediction. Res. Chem. 2023, 6, 101165 10.1016/j.rechem.2023.101165. [DOI] [Google Scholar]
- Weber I. T.; Kneller D. W.; Wong-Sam A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med. Chem. 2015, 7 (8), 1023–1038. 10.4155/fmc.15.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedidi R. S.; Garimella H.; Aoki M.; Aoki-Ogata H.; Desai D. V.; Chang S. B.; Davis D. A.; Fyvie W. S.; Kaufman J. D.; Smith D. W.; Das D.; Wingfield P. T.; Maeda K.; Ghosh A. K.; Mitsuya H. A conserved hydrogen-bonding network of P2 bis-tetrahydrofuran-containing HIV-1 protease inhibitors (PIs) with a protease active-site amino acid backbone aids in their activity against PI-resistant HIV. Antimicrob. Agents Chemother. 2014, 58 (7), 3679–3688. 10.1128/AAC.00107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide K.; Aoki M.; Amano M.; Koh Y.; Yedidi R. S.; Das D.; Leschenko S.; Chapsal B.; Ghosh A. K.; Mitsuya H. Novel HIV-1 protease inhibitors (PIs) containing a bicyclic P2 functional moiety, tetrahydropyrano-tetrahydrofuran, that are potent against multi-PI-resistant HIV-1 variants. Antimicrob. Agents Chemother. 2011, 55 (4), 1717–1727. 10.1128/AAC.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheik Ismail Z.; Worth R.; Mosebi S.; Sayed Y. HIV protease hinge region insertions at codon 38 affect enzyme kinetics, conformational stability and dynamics. Protein J. 2023, 42 (5), 490–501. 10.1007/s10930-023-10132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Wang J.; Shao Q.; Shi J.; Zhu W. Effects of drug-resistant mutations on the dynamic properties of HIV-1 protease and inhibition by Amprenavir and Darunavir. Sci. Rep. 2015, 5 (1), 10517. 10.1038/srep10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuntakaruk H.; Boonpalit K.; Kinchagawat J.; Nakarin F.; Khotavivattana T.; Aonbangkhen C.; Shigeta Y.; Hengphasatporn K.; Nutanong S.; Rungrotmongkol T.; Hannongbua S. Machine learning-guided design of potent darunavir analogs targeting HIV-1 proteases: A computational approach for antiretroviral drug discovery. J. Comput. Chem. 2024, 45 (13), 953–968. 10.1002/jcc.27298. [DOI] [PubMed] [Google Scholar]
- Dwipayana I.; Syah Y. M.; Aditama R.; Feraliana F.; Fibriani A. Development of a dimer-based screening system for dimerization inhibitor of HIV-1 protease. J. Microbiol. Biotechnol. 2020, 2, 1–11. 10.37604/jmsb.v2i2.42. [DOI] [Google Scholar]
- Bihani S. C.; Gupta G. D.; Hosur M. V. Molecular basis for reduced cleavage activity and drug resistance in D30N HIV-1 protease. J. Biomol. Struct. Dyn. 2022, 40 (23), 13127–13135. 10.1080/07391102.2021.1982007. [DOI] [PubMed] [Google Scholar]
- Kovalevsky A. Y.; Liu F.; Leshchenko S.; Ghosh A. K.; Louis J. M.; Harrison R. W.; Weber I. T. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J. Mol. Biol. 2006, 363, 161–173. 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Kovalevsky A. Y.; Tie Y.; Ghosh A. K.; Harrison R. W.; Weber I. T. Effect of flap mutations on structure of HIV-1 protease and inhibition by saquinavir and darunavir. J. Mol. Biol. 2008, 381 (1), 102–115. 10.1016/j.jmb.2008.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockbaum G. J.; Leidner F.; Rusere L. N.; Henes M.; Kosovrasti K.; Nachum G. S.; Nalivaika E. A.; Bolon D. N.; Ali A.; Yilmaz N. K.; Schiffer C. A. Correction to Structural Adaptation of Darunavir Analogues against Primary Mutations in HIV-1 Protease. ACS Infect. Dis. 2019, 5 (6), 1044. 10.1021/acsinfecdis.9b00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis J. M.; Zhang Y.; Sayer J. M.; Wang Y.-F.; Harrison R. W.; Weber I. T. The L76V drug resistance mutation decreases the dimer stability and rate of autoprocessing of HIV-1 protease by reducing internal hydrophobic contacts. Biochem. 2011, 50 (21), 4786–4795. 10.1021/bi200033z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Y.; Boross P. I.; Wang Y.-F.; Gaddis L.; Hussain A. K.; Leshchenko S.; Ghosh A. K.; Louis J. M.; Harrison R. W.; Weber I. T. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol. 2004, 338 (2), 341–352. 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- Agniswamy J.; Louis J. M.; Roche J.; Harrison R. W.; Weber I. T. Structural studies of a rationally selected multi-drug resistant HIV-1 protease reveal synergistic effect of distal mutations on flap dynamics. PloS one. 2016, 11 (12), e0168616 10.1371/journal.pone.0168616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267 (3), 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Tourlet S.; Radjasandirane R.; Diharce J.; de Brevern A. G. Alphafold2 update and perspectives. Bio Med. Inform. 2023, 3 (2), 378–390. 10.3390/biomedinformatics3020025. [DOI] [Google Scholar]
- Mirdita M.; Schütze K.; Moriwaki Y.; Heo L.; Ovchinnikov S.; Steinegger M. ColabFold: making protein folding accessible to all. Nat. Methods. 2022, 19 (6), 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer S.; Azijn H.; Surleraux D.; Jochmans D.; Tahri A.; Pauwels R.; Wigerinck P.; de Béthune M.-P. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob. Agents Chemother. 2005, 49 (6), 2314–2321. 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic acids Res. 2007, 35 (Suppl_2), W522–W525. 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D.; Aktulga H.; Belfon K.; Ben-Shalom I.; Berryman J.; Brozell S.; Cerutti D.; Cheatham T.; Cisneros G.; Cruzeiro V.. Amber; 2022. [Google Scholar]
- Somboon T.; Mahalapbutr P.; Sanachai K.; Maitarad P.; Lee V. S.; Hannongbua S.; Rungrotmongkol T. Computational study on peptidomimetic inhibitors against SARS-CoV-2 main protease. J. Mol. Liq. 2021, 322, 114999 10.1016/j.molliq.2020.114999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark P.; Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105 (43), 9954–9960. 10.1021/jp003020w. [DOI] [Google Scholar]
- Chari R.; Jerath K.; Badkar A. V.; Kalonia D. S. Long-and short-range electrostatic interactions affect the rheology of highly concentrated antibody solutions. Pharm. Res. 2009, 26, 2607–2618. 10.1007/s11095-009-9975-2. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9 (7), 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Dis. 2015, 10 (5), 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45 (1), 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.