

Abstract
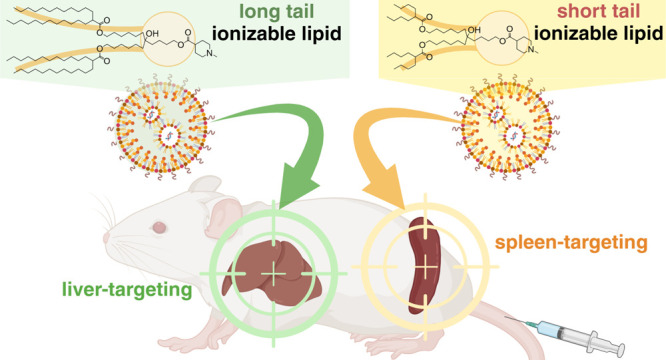
The delivery of mRNA molecules to organs beyond the liver is valuable for therapeutic applications. Functionalized lipid nanoparticles (LNPs) using exogenous mechanisms can regulate in vivo mRNA expression profiles from hepatocytes to extrahepatic tissues but lead to process complexity and cost escalation. Here, we report that mRNA expression gradually shifts from the liver to the spleen in an ionizable lipid tail length-dependent manner. Remarkably, this simple chemical strategy held true even when different ionizable lipid head structures were employed. As a potential mechanism underlying this discovery, our data suggest that 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) is enriched on the surface of mRNA/LNPs with short-tail lipids. This feature limits their interaction with biological components, avoiding their rapid hepatic clearance. We also show that spleen-targeting LNPs loaded with SARS-CoV-2 receptor-binding domain (RBD) mRNA can efficiently induce immune responses and neutralize activity following intramuscular vaccination priming and boosting.
Keywords: mRNA delivery, lipid nanoparticles, ionizable lipids, endogenous targeting, immune cell, vaccine
The successfully developed mRNA (mRNA) vaccines against SARS-CoV-2 highlight the impact of lipid nanoparticle (LNP) technology.1,2 LNPs were first developed for small interfering RNA (siRNA) delivery in the liver, leading to the success of Onpattro, the first approved siRNA drug.3 The liver is a crucial target for various therapeutics because of its role in metabolic and disease processes. Additionally, mRNA-encapsulated liver-targeting LNPs have been used in promising clinical trials for protein replacement and genome editing therapies in liver-related disorders.4
Interest is growing in mRNA delivery beyond the liver. Potential applications include antigen-encoded mRNA delivery to dendritic cells (DCs) for cancer vaccines,5,6 chimeric antigen receptor (CAR) mRNA delivery to T cells for B cell malignancies,7 and protein replacement mRNA therapy to the lung for primary ciliary dyskinesia and cystic fibrosis.8,9 However, commonly used LNPs reach the liver through intravenous administration, primarily via the apolipoprotein E (ApoE)-low-density lipoprotein receptor (LDLR) pathway.10 Moreover, we have demonstrated that interactions between ApoE-bound LNPs and hepatic heparan sulfate proteoglycans contribute to the rapid clearance and accumulation of LNPs in the liver space of Disse.11
To achieve extrahepatic mRNA delivery, researchers have developed functionalized LNPs using exogenous or endogenous mechanisms. Passive targeting platforms with endogenous mechanisms are also promising because of their potential to improve cost and complexity issues in scaled-up processes. As an example of formulation engineering, adding selective organ-targeting (SORT) molecules, such as permanently cationic or anionic lipids, can change the apparent pKa, serum protein interactions, and biodistribution of LNPs.12 Using exogenous ligand-tethered nanoparticles to target specific cell surface receptors is also a promising strategy.13−15 Other studies have reported that engineering the ionizable lipid chemical structure can alter the organ and cellular distribution of LNPs.16−20 However, predicting the LNP organ tropism from lipid structure remains unclear.
Here, we report the relationships between lipid structure and organ tropism. Among the various ionizable lipids reported,21 lipids with a monoamine headgroup and symmetrically branched tails were investigated because they are one of the most promising structures for mRNA delivery. Remarkably, lipid tail lengths could significantly change the mRNA expression profiles between the liver and spleen despite having the same lipid compositions and formulation processes. Importantly, these results were consistent across several different heads, suggesting the wide application potential of this strategy.
We investigated the impact of lipid tail chemistry on the in vivo fate of mRNA/LNPs. The lipid composition was fixed as ionizable lipid, cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2k) at a 50:38.5:10:1.5 ratio. Initially, we used the N-methylpiperidine lipid library as ionizable lipids (Figure 1a).22 We previously demonstrated that a branched tail could enhance the stability, fusogenicity, and functional delivery of mRNAs.23 Thus, we selected symmetrically branched tail lipids, such as CL15F 6–4, CL15F 7–5, CL15F 8–6, CL15F 9–7, CL15F 10–8, CL15F 11–9, CL15F 12–10, and CL15F 14–12 (Figure 1a). To examine the in vivo mRNA delivery profile of each LNP, firefly luciferase (FLuc) mRNA-encapsulated LNPs were intravenously administered in mice. Then, luminescence was captured from each organ using in vivo imaging systems (IVIS) (Figure 1b). Interestingly, LNPs containing short-tail CL15F lipids enhanced mRNA delivery to the spleen despite having the same lipid composition and formulation process (Figure 1c). We compared the relative luminescence values between the liver and spleen, demonstrating that organ tropism was changed significantly in a lipid tail length-dependent manner (Figure 1d). Importantly, this trend was also observed using the previously developed CL4F LNPs (Supplementary Figure S1).23 To determine if these observations were universal, we synthesized additional ionizable lipids containing a short or long tail with different heads, such as piperidine (CL6F), N-methyl pyrrolidine (CL16F), and N-isopropyl piperidine (CL17F) (Figure 1e). The data revealed that ionizable lipids with shorter tails could promote spleen delivery across various head structures (Figure 1f/g). The addition of a SORT lipid, such as 18PA, to LNPs is a well-known spleen-targeted mRNA delivery technique.24 The addition of 18PA to MC3-LNPs and CL15F14–12-LNPs resulted in selective mRNA delivery to the spleen, as expected. LNPs using CL15F 6–4 as a short-tail ionizable lipid, without using 18PA, enhanced mRNA delivery to the spleen both relatively and absolutely, suggesting the potential value of the proposed method (Supplementary Figure S2).
Figure 1.

Ionizable lipid tail length dynamically changed the organ specificity of mRNA delivery. (a) Chemical structures of the CL15F lipid library (termed CL15F m–n, where “m” is the main chain length and “n” is the side chain length). (b) A simple illustration of mRNA/LNP distribution to the liver and spleen following intravenous (i.v.) administration. (c) Effect of CL15F lipid tail length on in vivo mRNA delivery to the liver and spleen. Here, 0.1 mg/kg FLuc mRNA was i.v. injected into mice (n = 3 Balb/c mice per group). (d) Relative luciferase expression in the liver and spleen gradually shifted in an ionizable lipid tail length-dependent manner. (e) Ionizable lipid variants with different head structures (CL6F, CL16F, and CL17F). (f) Effect of lipid tail length with different head structures on in vivo mRNA delivery to the liver, spleen, lung, kidney, and heart. Here, 0.1 mg/kg FLuc mRNA was i.v. injected into mice (n = 3 Balb/c mice per group). (g) Relative luciferase expression in each organ demonstrated that the in vivo fate of mRNA/LNPs can be controlled by strategically adjusting the tail length.
Recent reports of endogenous targeting LNPs with the protein corona have suggested that the LNP surface characteristics, such as charge, are critical for organ tropism and that interactions with (apo)lipoproteins are important for liver tropism.12,25 We therefore hypothesized that the lipid tail chemistry changed the LNP surface state and its interactions with biological components, such as serum (apo)lipoproteins, thus shifting the preference from the liver to the spleen. Lipid-anchored PEG2000 is distributed on the LNP surface and can suppress LNP aggregation during storage.26 The short C14 anchor PEG-lipid is commonly used in mRNA/LNPs, including in this study. Unlike the long C18 anchor PEG-lipid, the C14 anchor PEG-lipid is designed to desorb quickly from LNPs in vivo because of its weak hydrophobic interactions.27,28 Thus, our focus shifted from the PEG layer to the LNP outer shell. Each lipid should be nonuniformly distributed in LNPs. Experimental analysis and molecular dynamics (MD) stimulation have demonstrated that polar lipids, such as DSPC, are preferentially located in the LNP outer shell because of their cylindrical molecular shape.29−31
To theoretically calculate the DSPC density on the surface of each LNP, we used an LNP structural model30 designed from experimental data (Figure 2a). Although controversial, the LNP structure has been interpreted as a densely packed oil droplet in which lipids interact with RNA. This structure differs from classical liposomes, which have an inner water phase surrounded by a lipid bilayer.29 In this model, the LNP core includes the ionizable lipid, cholesterol, mRNA, and water. The core is surrounded by an ∼2.4 nm-thick lipid monolayer, which contains DSPC and a fraction of the ionizable lipid, cholesterol, and lipid-anchoring portion of the PEG lipid. The top PEG layer is approximately 4 nm in thickness. First, the ionizable lipid tail length directly alters its molecular volume, which changes the volume contribution of each lipid in the formulation, even though each LNP is prepared with the same molar ratio. Each lipid’s volume contribution was calculated using both the molecular volume and molar ratio. For example, the DSPC volume contribution was 12.8% and 9.0% in the CL15F 6–4 LNPs and CL15F 14–12 LNPs, respectively, despite having the same molar ratio (10% DSPC) (Figure 2b). Second, short-tail ionizable lipids yielded larger particles across the lipids with different head groups used in this study (Supplementary Figures S3a, S4, S5a). This observation is consistent with data from a recent publication.23 Larger particles are assumed to exhibit a smaller surface area per volume, resulting in a higher DSPC concentration on the LNP surface (Figure 2c). To adequately support this line of reasoning, experimental data on particle size in CL15F LNPs with different tail lengths (Supplementary Figure S3a) were applied to the LNP structure model to calculate the surface area (Supplementary Figure S3b) and number of DSPC molecules (Supplementary Figure S3c). The surface DSPC density of each LNP was then estimated (Figure 2d, Supplementary Figure S3). The results indicated ionizable lipid tail length-dependent changes to the surface DSPC density. The lower and stable polydispersity index (PDI) values in particle size observed throughout this study support the reliability of the estimations.
Figure 2.

Surface DSPC density is significant for the spleen-to-liver mRNA delivery ratio. (a) Simple illustration of the LNPs. In this model, the LNP contains a core, lipid monolayer, and PEG layer. (b) The volume contribution differs with varying molecular weight, even with the same molar ratio. (c) The surface-to-volume ratio depends on the particle size. (d) The surface-to-volume ratio and volume contribution of DSPC affect the area per DSPC value. (e) Correlation matrix of the CL15F LNPs between liver mRNA delivery, spleen mRNA delivery, the spleen-to-liver mRNA delivery ratio, size, PDI, encapsulation efficiency, pKa, ζ-potential, and DSPC density created using JMP 16 software. The Pearson correlation coefficients are shown in the heatmap, and the P-values can be visualized by the circle size. (f) Relationships between the DSPC density and spleen-to-liver mRNA delivery ratio. (g) Effect of the DSPC ratio on in vivo mRNA delivery to the liver and spleen. Here, 0.1 mg/kg FLuc mRNA was intravenously injected into mice (n = 3 Balb/c mice per group). The spleen-to-liver ratio values were analyzed using one-way ANOVA with Dunnett’s test for multiple comparisons with DSPC 0% LNPs.
To determine the important factor for organ tropism, we performed a correlation coefficient analysis between liver and spleen mRNA delivery efficiency and particle characteristics, including size, PDI, encapsulation efficiency, pKa value, ζ-potential, and DSPC density (Figure 2e). Our analysis strongly indicated the significance of DSPC density in determining organ-specific mRNA delivery efficiency. The correlation matrix indicated that an enhanced particle size also contributes to mRNA delivery to the spleen. We next graphically displayed the relationship between the theoretical DSPC density and spleen/liver mRNA delivery ratio for each CL15F lipid (Figure 2f).
To further evaluate the importance of DSPC density on LNP organ tropism, we prepared LNPs with different DSPC ratios, while keeping the same ionizable lipid/cholesterol ratio. The in vivo luminescence data revealed that the mRNA expression profiles between the liver and spleen directly correlated with the DSPC ratio, providing further evidence of the significance of DSPC density (Figure 2g, Supplementary Figure S3).
To understand the mechanism controlling how DSPC density can change organ tropism, we specifically focused on the DSPC density in subsequent experiments. We first confirmed the uniformity and spherical morphology of four different LNPs using both transmission electron microscopy (TEM) and dynamic light scattering (DLS) (Figure 3a–d, Supplementary Table S1). These included CL15F 6–4, CL15F 10–8, and CL15F 14–12 LNPs containing 10% DSPC and CL15F 10–8 LNPs containing 30% DSPC. The calculated LNP surface DSPC concentrations were 0.63, 1.18, 1.32, and 0.69, respectively (Figure 3e).
Figure 3.

LNPs with higher DSPC surface densities have fewer interactions with cells in vitro and in vivo. (a–d) TEM images and particle size distribution of each LNP. CL15F 6–4 LNPs with 10% DSPC (a), CL15F 10–8 LNPs with 10% DSPC (b), CL15F 14–12 LNPs with 10% DSPC (c), and CL15F 10–8 LNPs with 30% DSPC (d). Scale bars = 100 nm. (e–g) The in vitro cellular uptake of LNPs carrying DiO-labeled Cy5 FLuc mRNA/FLuc mRNA (= 1:1) (n = 3). Flow cytometry analysis was performed 3 h after the HepG2 cells were transfected with 500 ng mRNA/well. The histogram (e) indicates the Cy5 fluorescence intensity. The graphs in (f) and (g) indicate the Geo mean value of Cy5 and DiO, respectively. The data were analyzed by ordinary one-way ANOVA with Dunnett’s multiple comparisons test of each lipid compared with the CL15F 10–8 DSPC 10% formulation. (h) Luciferase assays were performed 24 h after the HepG2 cells were transfected with LNPs carrying DiO-labeled Cy5 FLuc mRNA/FLuc mRNA (= 1:1). (i) The LNP concentration in blood samples was determined with Cy5-labeled FLuc mRNA-carrying LNPs. Here, 0.4 mg/kg mRNA/LNPs was intravenously (i.v.) injected into mice; then blood was collected; and the Cy5 intensity was measured (n = 3 Balb/c mice per group). (j) Confocal laser scanning microscopic images of the liver were acquired 6 h after administration of DiD-labeled LNPs and FITC conjugate tomato lectin staining. Here, 0.5 mol % DiD-labeled FLuc mRNA was i.v. injected into mice (0.5 mg/kg; n = 3 Balb/c mice per group). The blood vessels are shown in green, and DiD-positive areas are shown in red. Scale bars = 100 μm. (k) Here, 0.1 mg/kg FLuc mRNA/LNPs was i.v. injected into ApoE-deficient mice. Ex vivo luminescence from the liver was captured by in vivo imaging systems (IVIS; n = 3 C.KOR/StmSlc-Apoeshl mice per group). (l) Simple illustration of LNP cellular uptake. We hypothesized that higher DSPC LNPs have fewer interactions with ApoE, resulting in the LNPs not being taken up by the cells. (m) The fluorescence intensity was measured 5 h after incubation of the LNPs with Laurdan at 25 °C. The GP value indicates the membrane hydration level (n = 3).
Next, HepG2 cells were transfected with DiO-labeled LNPs carrying Cy5 mRNA. The cellular uptake of these LNPs was evaluated by flow cytometry. Among the 10% DSPC formulations, short-tail CL15F 6–4 LNPs showed significantly less cellular uptake and long-tail CL15F14–12 showed significantly more cellular uptake compared with CL15F10–8 LNPs (Figure 3f/g). In addition, the 30% DSPC formulation showed significantly less cellular uptake than the 10% DSPC formulation. These observations were further validated by the confocal laser scanning microscopic images (Supplementary Figure S5). The in vitro FLuc mRNA expression levels were also lower with these two LNPs (Figure 3h). These trends were also evident in Huh-7 cells (Supplementary Figure S6). Our data strongly suggest that the DSPC density can regulate interactions with the biological membrane.
We then examined the pharmacokinetic (PK) profiles of each LNP carrying Cy5-labeled mRNA. The data revealed that the CL15F 10–8 LNPs containing 30% DSPC and CL15F 6–4 LNPs containing 10% DSPC exhibited extended blood circulation half-lives (Figure 3i). Following administration, LNPs are generally quickly adsorbed by serum proteins, including ApoE, which transport them to hepatocytes via the ApoE-LDLR pathway.10 Thus, we hypothesized that the LNPs with a higher DSPC density would interact less with ApoE after PEG molecule detachment, resulting in prolonged clearance. To evaluate the relationship between DSPC density and ApoE adsorption, we examined LNP liver accumulation following dosing with fluorescently labeled LNPs. The microscopic images indicated that LNPs with a higher DSPC density displayed lower accumulation in the liver hepatocytes (Figure 3j), suggesting reduced ApoE adsorption. Furthermore, each LNP was then injected into ApoE-deficient mice or wild-type mice for liver mRNA delivery efficiency comparisons. The CL15F 10–8 and CL15F 14–12 LNPs containing 10% DSPC showed decreased efficacy in ApoE-deficient mice (Figure 3k). In contrast, the CL15F 6–4 LNPs containing 10% DSPC and CL15F 10–8 LNPs containing 30% DSPC maintained their efficacy in ApoE-deficient mice. These results indicated that the LNPs with a higher DSPC density had reduced ApoE adsorption (Figure 3l). Recombinant ApoE was incubated with LNPs to directly evaluate the interaction between the LNPs and ApoE. Samples were passed through a size exclusion chromatography (SEC) column to isolate ApoE-bound LNPs from unbound ApoE, then ApoE protein levels were detected by Western blot analysis. The results showed that the LNPs with shorter tail lipids adsorbed less ApoE (Supplementary Figure S7). Additionally, less ApoE adsorbed on the LNPs containing 30% DSPC (Supplementary Figure S7).
To better understand the relationship between the DSPC density and LNP surface microenvironment membrane profile, we used Laurdan as a solvatochromic probe. Laurdan is distributed in lipid membranes from its long fatty acids, with an emission spectrum that shifts depending on the dipolar relaxation of the surrounding water molecules. This makes it possible to sensitively distinguish the polarity of the LNP surface.33,34 After Laurdan was incorporated into the LNPs, a wavelength shift was observed. This shift was quantitatively represented by the generalized polarization (GP) value, with the CL15F 6–4 LNPs containing 10% DSPC and CL15F 10–8 LNPs containing 30% DSPC having higher GP values. This indicated an ordered membrane and reduced membrane fluidity (Figure 3m, Supplementary Figure S8). Because increased membrane fluidity has previously been shown to enhance protein adsorption,35,36 we speculated that a reduced membrane fluidity would inhibit ApoE protein adsorption to the surface of these LNPs. Recently, organ tropism was reportedly altered depending on the tail lengths of multitailed ionizable phospholipids (iPhos).37 The iPhos scaffolds employ well-packed saturated hydrocarbon chains, with longer scaffolds showing spleen-selective mRNA delivery. While these lipid species differ from the ionizable lipids focused on in this study, the enhanced spleen selectivity from the increased surface density of rigid DSPCs in this study and increased rigid iPhos content may have similar LNP surface properties. Clarifying the causal relationship with actual changes in physical properties is significant for achieving a predictable LNP design.
To investigate the potential application of CL15F 6–4 LNPs with enhanced functional mRNA delivery to the spleen, we examined the mRNA delivery efficiency at the cellular level using Ai14 transgenic mice (Figure 4a). These Ai14 transgenic mice express tdTomato upon Cre mRNA translation, which facilitates the evaluation of mRNA delivery efficiency with high sensitivity at the cellular level (Figure 4b).38 The microscopic images revealed that the mice treated with the CL15F 14–12 LNPs exhibited significant tdTomato fluorescence levels in the liver hepatocytes (Figure 4c). Low levels of tdTomato fluorescence were detected in the livers of mice treated with the CL15F 6–4 LNPs, consistent with our FLuc mRNA delivery data (Figure 1c). Flow cytometry analysis was performed to evaluate the mRNA delivery in splenocytes. The results indicated that the CL15F 6–4 LNPs preferentially promoted mRNA expression in antigen-presenting cells in the spleen, such as DCs, macrophages, and neutrophils (Figure 4d–f, Supplementary Figure S9). Almost no tdTomato fluorescence was observed in B or T cells (Figure 4g–i). Using DiO-labeled LNPs, the data revealed that the CL15F 6–4 LNPs were significantly distributed in DCs compared with the CL15F 14–12 LNPs (Supplementary Figure S10). Although the mRNA expression levels were very low, the CL15F 6–4 LNPs accumulated in B cells, while the CL15F 14–12 LNPs accumulated in T cells. Therefore, using CL15F 6–4 LNPs was superior for mRNA distribution and expression in DCs, with limited mRNA expression in the liver.
Figure 4.

In vivo expression and distribution profiles of mRNA/LNPs at the cellular level. (a) Experimental schematic for evaluating the in vivo cellular distribution of LNPs using Ai14 transgenic mice. Briefly, liver and spleen tissues were collected from Ai14 mice 24 h following dosing with 0.5 mg/kg Cre mRNA/LNPs (n = 3 mice per group). The splenocytes were disassociated and analyzed by flow cytometry. (b) The Ai14 mice have a loxP-flanked stop codon site to suppress tdTomato expression. With Cre recombinase, tdTomato will have strong transcriptional activation under the CAG promoter. (c) Liver samples were observed by confocal laser scanning microscopy 24 h after administration of mRNA/LNPs and 20 μg/mL FITC-conjugated isolectin B4 and 1 μg/mL Hoechst 33342. The blood vessels are shown in green, tdTomato-positive areas are shown in red, and the nuclei are shown in blue. Scale bars = 100 μm. (d–i) Here, 0.5 mg/kg Cre mRNA/LNPs was injected into mice. The splenocytes were collected and stained, then the tdTomato expression levels were analyzed by flow cytometry. The bar graphs show the percentage of tdTomato-positive cells in DCs (d), macrophages (e), neutrophils (f), B cells (g), CD8+ T cells (h), and CD4+ T cells (i). The data were analyzed using unpaired Student’s t tests. N.S. denotes not significant (P > 0.05).
Viral protein-encoding mRNA/LNPs can serve as immunogens and adjuvants, triggering acquired immunity. This approach was demonstrated during the COVID-19 pandemic. Because DC activation is critical for inducing neutralizing antibodies, cytotoxic T lymphocytes, and immune memory, these cells have become attractive targets for vaccine development.39,40 CL15F short-tail lipid-containing LNPs, which selectively deliver mRNA to antigen-presenting cells in the spleen, have vaccine application potential. In this study, we used CL15F 7–5 because large LNPs over 200 nm, such as CL15F 6–4 LNPs, may not transfer from the administration site into systemic circulation via the lymphatic system.41 As a control, we used CL4F 7–5, a previously developed liver-oriented lipid and the clinically successful SM-102 lipid as a benchmark. First, we evaluated the expression profile following intramuscular administration. FLuc mRNA was encapsulated in each LNP, with the lipid composition maintained as described above. Of the LNPs used, the SM-102 LNPs showed the highest protein expression levels in the muscle and liver. The CL15F 7–5 LNPs showed predominant expression in the muscle and spleen, with limited expression in the liver. The protein expression levels in the spleen were significantly higher for CL15F 7–5 LNPs compared with SM-102 LNPs. Additionally, the CL4F 7–5 LNPs showed higher expression levels in the liver than in the muscle (Figure 5a). Importantly, the expression profiles in the liver and spleen were consistent between intramuscular and i.v. administration (Figure 1d, Supplementary Figure S1). This observation seems reasonable considering previous findings that intramuscularly administered LNPs can enter systemic circulation via the lymphatic system.41−43 We also assessed changes in body weight and performed hematological and histopathological analyses after repeated administration of these LNPs, confirming their minimal toxicity (Figure S11).
Figure 5.

Antibody responses in SARS-CoV-2 RBD mRNA/LNP vaccinated mice. (a) Bioluminescence from each organ 6 h following intramuscular dosing of 5 μg/30 μL/mouse FLuc mRNA/LNPs (n = 5 C57BL/6 mice per group). (b) The binding of the SARS-CoV-2 virus to the human ACE2 receptor can be mimicked in vitro via purified recombinant hACE2 and the RBD of the SARS-CoV-2 spike protein interaction. This interaction can be blocked by neutralizing antibodies present in the test serum as a virus neutralization test. (c, d) Here, 5 μg/30 μL of SARS-CoV2 mRNA-loaded LNPs was injected into the left thigh muscles of mice twice at two-week intervals. Two weeks following the boosting dose, serum samples were collected from the mice (n = 5 C57BL/6 mice per group). (c) The circulating RBD-specific IgG antibody titers were evaluated. The data were analyzed by ordinary one-way ANOVA with Dunnett’s multiple comparisons test of each lipid compared with SM-102. (d) Inhibition of the SARS-CoV-2 RBD and hACE2 interaction by mouse serum.
To evaluate the immunogenicity of each LNP, mRNA molecules designed to produce high expression levels of the SARS-CoV-2 virus spike protein were used as a model. Immunogenicity was assessed by quantifying the antireceptor-binding domain (RBD) IgG titers and neutralizing activity 2 weeks after priming and boost dosing. To assess the neutralizing activity of the RBD mRNA LNP-induced antibodies, we performed the surrogate virus neutralization test (Figure 5b).44 We first confirmed that the mRNA/LNP immunogenicity was induced in a dose-dependent manner (Supplementary Figure S12). Next, we compared the immunogenicity of each LNP. The anti-RBD IgG titers of the treated mice increased in the following order: CL4F 7–5, SM-102, and CL15F 7–5 LNPs (Figure 5c). The neutralizing activity was proportional to the anti-RBD IgG data (Figure 5d). Despite much lower total mRNA expression levels than SM-102 LNPs, the CL15F 7–5 and CL4F 7–5 LNPs showed comparable immune activity. These results suggest that expression intensity, especially at the injection site, and systemic expression profiles are important for efficient immune induction.
Herein, we discovered a fascinating phenomenon: as the lipid tail shortened, a progressive shift in the mRNA expression profile from the liver to the spleen was observed. We proposed that this finding is attributed to LNP surface state changes. In fact, some studies have indicated that LNP surface characteristics are important for controlling the in vivo fate of mRNA/LNPs. For example, LNPs containing positively or negatively charged lipids can change the protein corona composition and mRNA expression profile across organs.24 Additionally, recent reports have demonstrated that LNPs containing higher DSPC or egg sphingomyelin densities can deliver mRNA to the spleen.45 Our theoretical calculations indicated that LNPs containing lipids with shorter tails exhibited higher DSPC densities on the surface. The in vitro and in vivo experiments also confirmed that LNPs containing shorter tails displayed fewer interactions with biological components such as serum proteins. These results strongly indicate the importance of the LNP surface state.
This study has some limitations. In these experiments, the relationship between tail length and organ tropism was demonstrated by focusing on lipids containing monoamine headgroups and symmetrical branching tails. However, further research is needed to better understand whether this relationship is applicable to lipids with multiamine headgroups or asymmetric tails. Additionally, our theoretical calculations of DSPC densities were performed using parameters from a previous study.30 The parameters used in this model, such as the LNP monolayer thickness and amount of water in the LNP, seem valid and general considering the data from another research group.46 However, they can vary depending on the experimental conditions. Along with studies evaluating the affinity of ApoE and LNPs,47,48 it is challenging but expected to accurately understand the LNP surface state and its interactions with biological components. We also evaluated the LNP membrane fluidity, but this is only one feature that describes the physical properties of the LNPs. In the future, ionizable lipid and LNP design techniques will evolve along with advances in nanoscale, single-particle level analysis, such as fluorescent microscopy, electron microscopy, and nanoflow cytometry.49−52
This study proposed new LNP formulation design guidelines that, unlike existing approaches that add specific ligands to optimized LNPs, have advantages of not increasing the associated cost or process complexity.
Acknowledgments
This work was funded by the Nitto Denko Corporation. Support for research expenses was obtained from the Ministry of Education, Culture, Sports, Science and Technology. The authors would also like to thank Takuya Shishido and Masao Murakawa for their critical review, Chisato Noyama for helping with the physicochemical characteristic measurements, and Chisa Okuma and Keiko Kajiwara for their help with the Ai14 mouse study. The synthesized lipids were analyzed using an NMR installed at the Faculty of Pharmaceutical Sciences, Hokkaido University. The NMR is registered in the Open Facility system and is managed by the Global Facility Center, Creative Research Institution, Hokkaido University. We thank J. Iacona, Ph.D., from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Data Availability Statement
All data generated in this study are available from the corresponding author upon reasonable request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.nanolett.4c02566.
Materials and methods, supplementary Figures and Tables, and characterization of synthetic lipids (PDF)
Author Contributions
Conceptualization and Methodology: K.H., Y.S., A.Ok., and H.H.; Investigation: K.H., Y.S., M.T., S.S., A.Ot., and Y.M.; Writing–Original Draft: K.H.; Writing–Review & Editing: Y.S. and H.H.; Supervision: Y.S., H.E., A.Ok., and H.H.
The authors declare the following competing financial interest(s): K.H., M.T., S.S., A.Ot., Y.M., H.H., and Y.S. are the authors of patents WO2022/071582 (A1) and WO2023191050 (A1) in relation to this publication. K.H., M.T., S.S., A.Ot., Y.M., H.E., and A.Ok. were employees of the Nitto Denko Corporation at the time of this study.
This paper published ASAP on October 7, 2024 with a duplicate reference (ref 32). The reference was removed and the corrected paper was reposted on October 8, 2024.
Supplementary Material
References
- Polack F. P.; et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden L. R.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019 2019, 14 (12), 1084–1087. 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- Qin S.; et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 71 (7), 1–35. 10.1038/s41392-022-01007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz L. M.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nat. 2016 2016, 534, 396–401. 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- Rojas L. A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nat. 2023 2023, 618, 144–150. 10.1038/s41586-023-06063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Benedicto E.; et al. Spleen SORT LNP Generated in situ CAR T Cells Extend Survival in a Mouse Model of Lymphoreplete B Cell Lymphoma. Angew. Chemie Int. Ed. 2023, 62, e202310395 10.1002/anie.202310395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C. J.; et al. Inhaled delivery of a lipid nanoparticle encapsulated messenger RNA encoding a ciliary protein for the treatment of primary ciliary dyskinesia. Pulm. Pharmacol. Ther. 2022, 75, 102134. 10.1016/j.pupt.2022.102134. [DOI] [PubMed] [Google Scholar]
- Wei T.; et al. Lung SORT LNPs enable precise homology-directed repair mediated CRISPR/Cas genome correction in cystic fibrosis models. Nat. Commun. 2023 2023, 14, 1–14. 10.1038/s41467-023-42948-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; et al. Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y.; Kinami Y.; Hashiba K.; Harashima H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J. Controlled Release 2020, 322, 217–226. 10.1016/j.jconrel.2020.03.006. [DOI] [PubMed] [Google Scholar]
- Dilliard S. A.; Cheng Q.; Siegwart D. J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2109256118 10.1073/pnas.2109256118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rurik J. G.; et al. CAR T cells produced in vivo to treat cardiac injury. Science (80-.) 2022, 375, 91–96. 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; et al. Ligand-tethered lipid nanoparticles for targeted RNA delivery to treat liver fibrosis. Nat. Commun. 2023 2023, 14, 1–12. 10.1038/s41467-022-35637-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. B.; et al. A Glu-urea-Lys Ligand-conjugated Lipid Nanoparticle/siRNA System Inhibits Androgen Receptor Expression In Vivo. Mol. Ther. - Nucleic Acids 2016, 5, e348 10.1038/mtna.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu M.; et al. Lung-selective mRNA delivery of synthetic lipid nanoparticles for the treatment of pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2116271119 10.1073/pnas.2116271119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage M. P.; Sago C. D.; Gan Z.; Krupczak B. R.; Dahlman J. E. Constrained Nanoparticles Deliver siRNA and sgRNA to T Cells In Vivo without Targeting Ligands. Adv. Mater. 2019, 31, 1902251. 10.1002/adma.201902251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H.; et al. Piperazine-derived lipid nanoparticles deliver mRNA to immune cells in vivo. Nat. Commun. 2022, 10.1038/s41467-022-32281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu G. S.; et al. A Combinatorial Library of Lipid Nanoparticles for Cell Type-Specific mRNA Delivery. Adv. Sci. 2023, 10, 2301929. 10.1002/advs.202301929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunovska K.; et al. The Extent to Which Lipid Nanoparticles Require Apolipoprotein e and Low-Density Lipoprotein Receptor for Delivery Changes with Ionizable Lipid Structure. Nano Lett. 2022, 22, 10025–10033. 10.1021/acs.nanolett.2c03741. [DOI] [PubMed] [Google Scholar]
- Han X.; et al. An ionizable lipid toolbox for RNA delivery. Nat. Commun. 2021, 12, 7233. 10.1038/s41467-021-27493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiba K.; et al. Overcoming thermostability challenges in mRNA–lipid nanoparticle systems with piperidine-based ionizable lipids. Commun. Biol. 2024 2024, 71 (7), 1–13. 10.1038/s42003-024-06235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiba K.; et al. Branching ionizable lipids can enhance the stability, fusogenicity, and functional delivery of mRNA. Small Sci. 2023, 3, 2200071. 10.1002/smsc.202300285. [DOI] [Google Scholar]
- Cheng Q.; et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; et al. Multiomics analysis of naturally efficacious lipid nanoparticle coronas reveals high-density lipoprotein is necessary for their function. Nat. Commun. 2023, 14, 1–16. 10.1038/s41467-023-39768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; et al. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 2019, 11, 9023–9031. 10.1039/C9NR02004G. [DOI] [PubMed] [Google Scholar]
- Wilson S. C.; et al. Real time measurement of PEG shedding from lipid nanoparticles in serum via NMR spectroscopy. Mol. Pharmaceutics 2015, 12, 386–392. 10.1021/mp500400k. [DOI] [PubMed] [Google Scholar]
- Gallud A. Time evolution of PEG-shedding and serum protein coronation determines the cell uptake kinetics and delivery of lipid nanoparticle formulated mRNA. bioRxiv 2021, 10.1101/2021.08.20.457104. [DOI] [Google Scholar]
- Kulkarni J. A.; et al. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. 10.1021/acsnano.8b01516. [DOI] [PubMed] [Google Scholar]
- Arteta M. Y.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E3351–E3360. 10.1073/pnas.1720542115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehghani-Ghahnaviyeh S.; et al. Ionizable Amino Lipids Distribution and Effects on DSPC/Cholesterol Membranes: Implications for Lipid Nanoparticle Structure. J. Phys. Chem. B 2023, 127, 6928–6939. 10.1021/acs.jpcb.3c01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasassi T.; De Stasio G.; Ravagnan G.; Rusch R. M.; Gratton E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys. J. 1991, 60, 179–189. 10.1016/S0006-3495(91)82041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlikowska-Rzeznik H.; Krok E.; Chattopadhyay M.; Lester A.; Piatkowski L. Laurdan Discerns Lipid Membrane Hydration and Cholesterol Content. J. Phys. Chem. B 2023, 127, 3382–3391. 10.1021/acs.jpcb.3c00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; et al. Helper lipid structure influences protein adsorption and delivery of lipid nanoparticles to spleen and liver. Biomater. Sci. 2021, 9, 1449–1463. 10.1039/D0BM01609H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande A. H.; Qin S.; Tatulian S. A. Membrane Fluidity Is a Key Modulator of Membrane Binding, Insertion, and Activity of 5-Lipoxygenase. Biophys. J. 2005, 88, 4084–4094. 10.1529/biophysj.104.056788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing. Nat. Mater. 2021 2021, 20, 701–710. 10.1038/s41563-020-00886-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman K. J.; et al. Rapid, Single-Cell Analysis and Discovery of Vectored mRNA Transfection In Vivo with a loxP-Flanked tdTomato Reporter Mouse. Mol. Ther. - Nucleic Acids 2018, 10, 55–63. 10.1016/j.omtn.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galati D.; Zanotta S.; Capitelli L.; Bocchino M. A bird’s eye view on the role of dendritic cells in SARS-CoV-2 infection: Perspectives for immune-based vaccines. Allergy 2022, 77, 100–110. 10.1111/all.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro J. R.; Farber D. L. COVID-19 vaccines: modes of immune activation and future challenges. Nat. Rev. Immunol. 2021 2021, 21, 195–197. 10.1038/s41577-021-00526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di J.; et al. Biodistribution and Non-linear Gene Expression of mRNA LNPs Affected by Delivery Route and Particle Size. Pharm. Res. 2022, 39, 105–114. 10.1007/s11095-022-03166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl K.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. 10.1016/j.ymthe.2017.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardi N.; et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controlled Release 2015, 217, 345–351. 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C. W.; et al. A SARS-CoV-2 surrogate virus neutralization test based on antibody-mediated blockage of ACE2–spike protein–protein interaction. Nat. Biotechnol. 2020 2020, 38, 1073–1078. 10.1038/s41587-020-0631-z. [DOI] [PubMed] [Google Scholar]
- Chander N.; Basha G.; Yan Cheng M. H.; Witzigmann D.; Cullis P. R. Lipid nanoparticle mRNA systems containing high levels of sphingomyelin engender higher protein expression in hepatic and extra-hepatic tissues. Mol. Ther. - Methods Clin. Dev. 2023, 30, 235–245. 10.1016/j.omtm.2023.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; et al. Acidification-Induced Structure Evolution of Lipid Nanoparticles Correlates with Their In Vitro Gene Transfections. ACS Nano 2023, 17, 979–990. 10.1021/acsnano.2c06213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; et al. Helper lipid structure influences protein adsorption and delivery of lipid nanoparticles to spleen and liver. Biomater. Sci. 2021, 9, 1449–1463. 10.1039/D0BM01609H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani F.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of mRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. 10.1021/acsnano.0c10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamanzi A.; et al. Simultaneous, Single-Particle Measurements of Size and Loading Give Insights into the Structure of Drug-Delivery Nanoparticles. ACS Nano 2021, 15, 19244–19255. 10.1021/acsnano.1c04862. [DOI] [PubMed] [Google Scholar]
- Cheng M. H. Y.; et al. Induction of Bleb Structures in Lipid Nanoparticle Formulations of mRNA Leads to Improved Transfection Potency. Adv. Mater. 2023, 35, 2303370. 10.1002/adma.202303370. [DOI] [PubMed] [Google Scholar]
- Chen C.; Chen C.; Li Y.; Gu R.; Yan X. Characterization of lipid-based nanomedicines at the single-particle level. Fundam. Res. 2023, 3, 488–504. 10.1016/j.fmre.2022.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zhou Y.; Chen C.; Zhu S.; Yan X. Quantification of Available Ligand Density on the Surface of Targeted Liposomal Nanomedicines at the Single-Particle Level. ACS Nano 2022, 16, 6886–6897. 10.1021/acsnano.2c02084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in this study are available from the corresponding author upon reasonable request.