

Abstract
Basal forebrain cholinergic neurons (BFCNs) extend long projections to multiple regions in the brain to regulate cognitive functions. Degeneration of BFCNs is seen with aging, after brain injury, and in neurodegenerative disorders. An increase in the amount of the immature proform of nerve growth factor (proNGF) in the cerebral cortex results in retrograde degeneration of BFCNs through activation of proNGF receptor p75NTR. Here, we investigated the signaling cascades initiated at the axon terminal that mediate proNGF-induced retrograde degeneration. We found that local axonal protein synthesis and retrograde transport mediated proNGF-induced degeneration initiated from the axon terminal. Analysis of the nascent axonal proteome revealed that proNGF stimulation of axonal terminals triggered the synthesis of numerous proteins within the axon, and pathway analysis showed that amyloid precursor protein (APP) was a key upstream regulator in cultured BFCNs and in mice. Our findings reveal a functional role for APP in mediating BFCN axonal degeneration and cell death induced by proNGF.
Introduction
Almost three decades of research has shown that cognitive decline and neurodegenerative disorders such as Alzheimer’s disease (AD) are associated with loss and degeneration of basal forebrain cholinergic neurons (BFCNs) (1, 2). BFCNs are highly polarized cells and send long axonal projections to multiple targets in the brain, through which they help regulate cognitive processes such as memory formation, learning and attention (3, 4, 5). BFCNs require neurotrophic factors for their survival, differentiation, maintenance, and function (6) which may be produced by their neuronal target regions, and signal through cognate receptor complexes through the projecting axon terminals. BFCNs are unique in their life-long expression of all the neurotrophin receptors, including the p75 neurotrophin receptor (p75NTR) and the receptor tyrosine kinases TrkA, TrkB, and TrkC. Mature neurotrophins promote a pro-survival response through Trk signaling (7, 8, 9, 10), whereas precursor neurotrophins (pro-NTs) bind p75NTR and sortilin to promote apoptosis in BFCNs (11, 12). p75NTR mediated BFCN degeneration has been studied in mass cultures and after seizure conditions in vivo (12). p75NTR is known to promote secondary neurodegeneration in the penumbra after cortical traumatic brain injury (TBI) (13, 14, 15). Our previous study further established that cortical TBI in mice promotes a retrograde loss of BFCNs through p75NTR, stimulated by proneurotrophins induced in the cortical area of impact, to which a subpopulation of BFCNs project (16). Moreover, direct axonal stimulation with proNGF or proBDNF promotes retrograde axon degeneration and cell death of mouse BFCNs in vitro (16). However, the mechanisms that promote proNT-p75NTR-mediated retrograde degeneration of BFCNs remain to be elucidated.
Studies in the peripheral nervous system have shown that under conditions of axonal injury, the interactions between the axon and soma which promote a degenerative response involve unique pathways and key players that include early and pre-catastrophic calcium waves, retrograde transport via dynein, c-Jun N-terminal kinase (JNK) activity, and local axonal protein synthesis (17, 18, 19). Among mechanisms underlying p75NTR-mediated apoptotic signaling, proneurotrophins activate JNK (12) and the intrinsic caspase signaling pathway (20, 21). Studies have also established an integral role for local protein synthesis in promoting nervous system pathologies at the sub-cellular site of disease or insult due to the extensive axonal and dendritic morphology of neurons (22). Axon degeneration in peripheral dorsal root ganglia (DRG) sensory neurons due to NGF deprivation is regulated by amyloid precursor protein (APP) through modulation of calcium homeostasis (23). Although APP cleavage has been extensively studied in conjunction with several central nervous system (CNS) disorders involving axonopathies, such as AD as well as traumatic brain injury (24, 25), the role of full-length APP in the developing brain as well as in CNS degeneration is unclear.
In our study, microfluidic and filter chambers were used to segregate BFCN soma and axons in vitro to study the pathways underlying the retrograde degenerative effect of proNGF in BFCNs. Our results showed that axon specific stimulation of BFCNs with proNGF led to p75NTR-mediated retrograde axon degeneration and cell death. JNK regulated proNGF-mediated retrograde cell death but not axon fragmentation. We also demonstrate a functional role for local axonal protein synthesis, specifically of APP, in promoting BFCN axon degeneration and cell death in response to proNGF treatment at the axon terminal. In this study, a functional role for APP mediated pre-catastrophic Ca2+ increase in the axons was also established. Overall, this investigation elaborates on the signaling mechanisms that lead to p75NTR-mediated retrograde degeneration of BFCNs following axonal proneurotrophin stimulation.
Results
proNGF promotes retrograde degeneration of basal forebrain cholinergic neurons (BFCNs)
To study the mechanisms underlying the degenerative effect of axonal proNGF stimulation, compartmental cultures of rat BFCNs were established using microfluidic chambers. BFCNs projected to the axon compartment by 5 days in vitro (5-DIV), and co-expressed p75NTR, TrkB, and Tuj1 (Fig. 1, A and B). BFCNs that successfully projected their axons to the distal compartment were identified by pretreatment of the axons with the retrograde tracer choleratoxin B labeled with Alexa Fluor 488 (CTB), whereas propidium iodide (PI) was added to the soma chamber to identify dying cells (Fig. 1, A to C). This was followed by the proNGF treatment in the somas or axons. CTB+/PI+ double–positive neurons indicated neurons degenerating (Fig. 1D). Consistent with our previous study (12), proNGF applied to the somas promoted BFCN cell death and axon degeneration (Fig 1, E to H). As we have previously shown for mouse neurons (16), axonal stimulation of rat BFCNs with proNGF promoted retrograde cell death (Fig. 1E), which was confirmed by TUNEL labeling (Fig. 1, F and G). Axonal stimulation with proNGF also elicited axon degeneration after 24 hours of treatment (Fig 1, H and I). Moreover, live imaging showed that axon fragmentation was observed at around 16 hours, whereas cell death occurred around 24 hours, suggesting that axon degeneration preceded cell death (Fig. 1D). ProNGF is a high-affinity ligand for p75NTR and has been shown to promote cell death through p75NTR in BFCNs in vivo after seizures and cortical injury (12, 16). To investigate whether p75NTR mediated the retrograde, proNGF-induced cell death in rat BFCNs, we pretreated the axons with a p75NTR ligand–blocking antibody then stimulated the axons with proNGF. By 24 hours, the number of degenerating BFCNs induced by axonal proNGF was significantly reduced in the presence of the blocking antibody in the axons (Fig. 1J). In contrast, blocking TrkA activity with K252a did not protect BFCN degeneration (fig. S1, A to D), although as a control it did prevent ERK activation by NGF (fig. S1E), suggesting that TrkA does not play a role in proNGF-induced retrograde degeneration. Additionally, BFCN cultures prepared from p75NTR KO rats were protected from axonal proNGF-induced cell death as well as axon degeneration (Fig. 1, K and L). These results show that p75NTR is necessary for proNGF-induced retrograde degeneration of BFCNs.
Figure 1. proNGF promotes retrograde degeneration of basal forebrain cholinergic neurons (BFCNs) in microfluidic cultures through p75NTR.
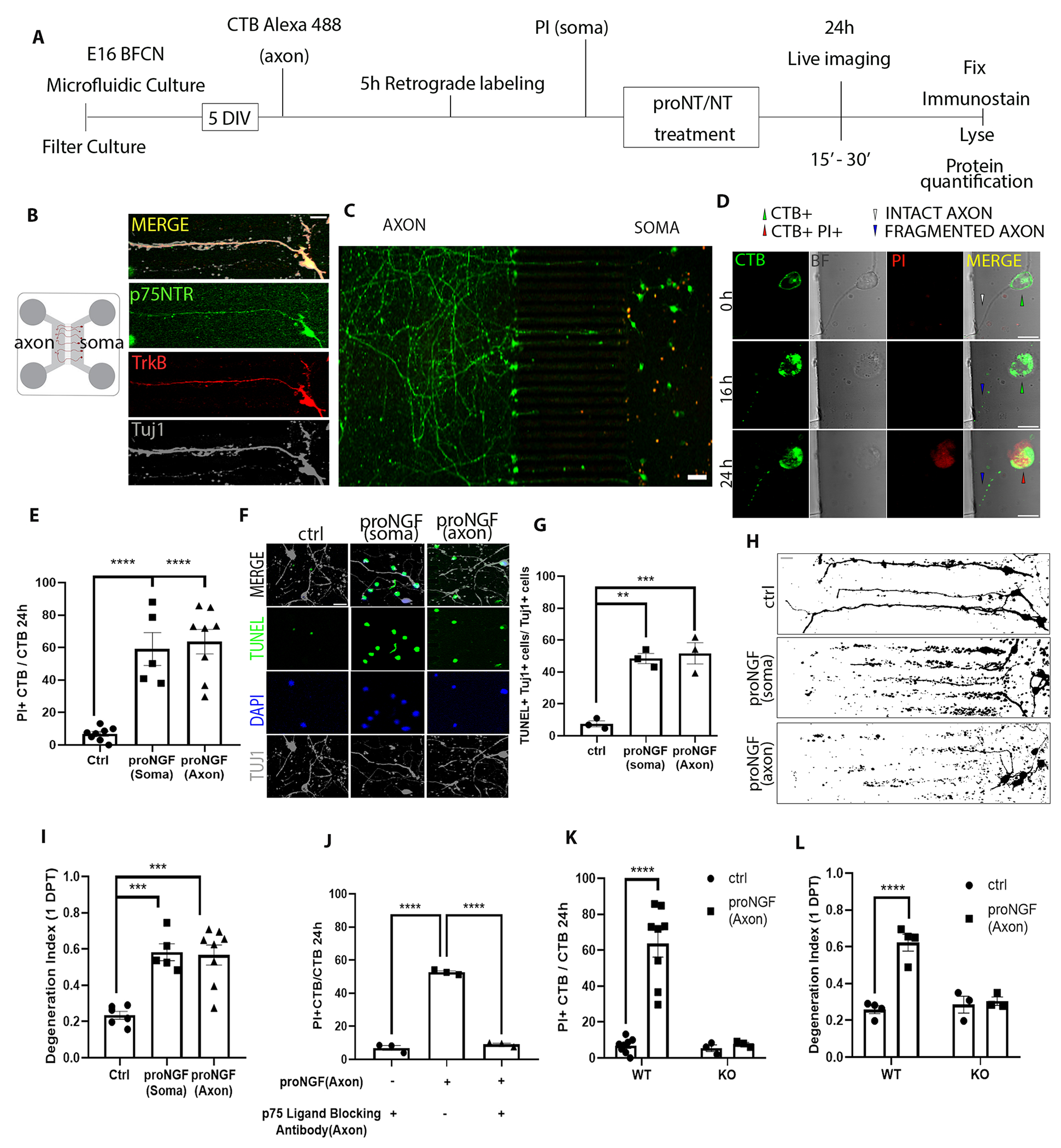
(A to C) Schematic of the experiment in (A), in which E16 rat basal forebrain neurons were cultured in microfluidic chambers for 5 DIV and traced with CTB Alexa-488 retrogradely, from the axon. Propidium iodide (PI) was added to the soma compartment to identify dying neurons following treatment. ProNGF was applied to either soma or axon, as indicated in each figure or legend. Imaging in (B and C) are representative images of p75NTR (green), TrkB (red), and β-tubulin III (Tuj1) in wild-type BFCN axons and somas cultured and treated as described in (A), from at least 5 experiments. Scale bars = 20 μm (B), 50 μm (C). (D) Live-cell imaging for axon fragmentation (CTB) and PI uptake in the nucleus 16 and 24 hours after axonal proNGF stimulation. BF, brightfield. Scale bar = 10μm. Images are representative of 5 experiments in wild-type BFCNs. (E) From imaging described in (D), quantification of dying BFCNs, as the proportion of CTB-positive soma that were PI positive, 24 hours after either soma or axonal treatment with proNGF. Data are mean ± SEM of n = 5 or 8 independent experiments from separate litters, each experiment with technical duplicates and analysis of at least 50 cells per treatment. ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparison tests. (F and G) Wild-type BFCN somas in microfluidic chambers were co-labeled for TUNEL (green), DAPI (blue) and Tuj1 (grey) and treated with proNGF at the soma or the axon. Images were taken 24 hours later (scale bar = 20μm) and analyzed for the proportion of TUNEL-positive Tuj1-positive cells. Data are mean ± SEM of n = 3 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. **p < 0.01, ***p < 0.001 by one-way ANOVA with Tukey’s multiple comparison tests. (H and I) Binary images of Tuj1-immunostained BFCNs (scale bar = 20μm) and quantification of axon fragmentation represented as a degeneration index comparing BFCNs treated with proNGF in the soma or axons and control untreated neurons. Data are mean ± SEM of n = 5 or 8 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. ***p < 0.001 by one-way ANOVA with Tukey’s multiple comparison tests. (J) Analysis of cell death in wild-type BFCNs, assessed by live-cell imaging as described in (D and E), 24 hours after axonal treatment with proNGF with or without axonal pretreatment with a p75NTR ligand-blocking antibody (1 μg/ml for 20 min). Data are mean ± SEM of n = 3 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparison tests. (K) Analysis of cell death in wild-type (WT) and p75NTR-deficient (KO) BFCNs, assessed by live-cell imaging as described in (D and E), 24 hours after axonal proNGF treatment. Data are mean ± SEM of n= 3 or 8 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. ****p < 0.0001 by two-way ANOVA with Sidak’s multiple comparison tests. (L) Degeneration index, assessed by Tuj1 immunostaining as described in (H), in p75NTR wild-type (WT) and knockout (KO) BFCNs after axonal treatment with proNGF. Data are mean ± SEM of n= 3 and 4 independent experiments, each with technical duplicates and at least 50 cells analyzed per treatment. ****p < 0.0001 by Sidak’s multiple comparison tests.
Retrograde transport is necessary for axonal proNGF treatment to induce BFCN axon degeneration and cell death.
To investigate whether retrograde transport is required for the degenerative signal initiated by proNGF at the axon terminal, BFCN axons were pre-treated with ciliobrevin D, an inhibitor of the retrograde motor dynein, for 20 mins before stimulation with proNGF. Live images obtained 24 hours after treatment were analyzed to quantify the number of degenerating neurons as a ratio of CTB+ PI+ cells over total CTB+ cells (Fig. 2A). The presence of ciliobrevin D in the axons rescued the BFCNs from axonal proNGF-induced degeneration and cell death (Fig. 2, B to D), suggesting that retrograde transport is necessary for proNGF-p75NTR mediated signaling from the axons to elicit BFCN retrograde cell death and axon degeneration. Moreover, blocking dynein function in the somas prior to axonal proNGF stimulation also rescued BFCNs from cell death (Fig. 2, E and F) as well as axon degeneration (Fig. 2, G and H) suggesting that following proNGF stimulation transport of cargo to and from the soma is necessary for the degenerative activity of proNGF.
Figure 2. Retrograde transport is necessary for BFCN axon degeneration and cell death induced by axonal proNGF.
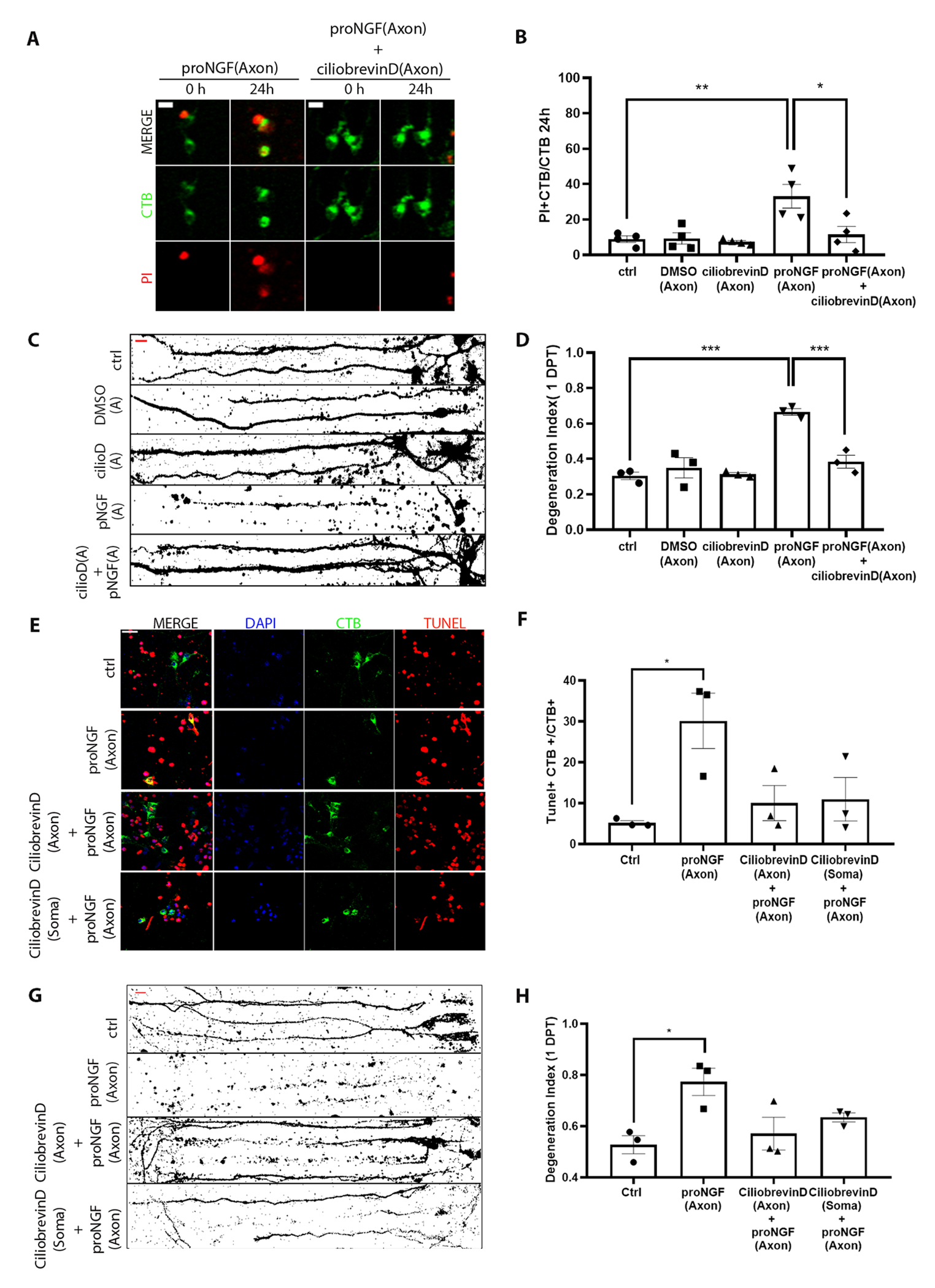
(A and B) Representative images (A) and analysis (B) of CTB and PI labeling in live BFCNs in microfluidic cultures stimulated with proNGF with or without ciliobrevin D in the axons before (0h) and 24 hours after axonal proNGF treatment. Scale = 10 μm. Quantification of dying BFCNs (PI+CTB/CTB) after 24-hour axonal treatment with proNGF with or without ciliobrevin D in the axons are mean ± SEM from n= 4 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. **p = 0.01, *p < 0.05 by one-way ANOVA with Tukey’s multiple comparison tests. (C and D) Binary images of Tuj1-immunostained BFCNs and quantification of axon fragmentation represented as a degeneration index comparing BFCNs treated with proNGF in the axons, with or without ciliobrevin D pretreatment in the axons. Scale = 20μm. Data are mean ± SEM, n = 3 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. ***p < 0.001 by one-way ANOVA with Tukey’s multiple comparison tests. (E and F) BFCN somas in microfluidic chambers co-labeled for TUNEL (red), DAPI (blue) and CTB-488 (green) after 24 hours treatment with proNGF (to the axon), ciliobrevin D+ proNGF (both axon) or ciliobrevin D (to the soma) + proNGF (to the axon). Quantifications of TUNEL+ cells, normalized to CTB, are mean ± SEM of n = 3 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparison tests. Scale = 20μm. (G and H) Binary images of Tuj1-immunostained BFCNs, and quantification of axon fragmentation represented as a degeneration index comparing BFCNs treated with proNGF in the axons, with ciliobrevin D pretreatment in the axons or somas. Scale = 20μm. Data are mean ±SEM, n = 3 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparison tests.
JNK activity is necessary for proNGF induced retrograde BFCN cell death but not axon fragmentation.
To investigate whether JNK, a known downstream target of proNGF-p75NTR–mediated death signaling in hippocampal neurons (26), was activated by proNGF treatment of the axons, BFCNs were grown on filter cultures for Western blot analysis (fig. S2A). Axons grew through the 1-μm pores of the filters separating axons from the somas expressing MAP2 and NeuN (fig. S2, B and C). Diffusion of 10 kDa Dextran beads across the filters was assessed to determine the window of time for specific treatments of axons or somas with proneurotrophins (32-37 kDa). Diffusion from the top to the bottom compartment was observed after 45 min (fig. S2D), whereas diffusion from the bottom to the top compartment was not observed until the 2-hour mark (fig. S2E), thus allowing a 2-hour window for axon-specific treatments. Filter cultures were treated with proNGF in the axons for 15 and 30 minutes, then axon and soma lysates were analyzed for phosphorylation of JNK. Axonal stimulation with proNGF resulted in a significant increase in p-JNK levels over total JNK (Fig. 3, A and B), which was prevented by JNK inhibitor II (Fig. 3, C and D). To assess the role of JNK in mediating retrograde degeneration and cell death, microfluidic cultures were used to test whether blocking JNK activity protected BFCNs from proNGF induced retrograde degeneration. Notably, blocking JNK activation in BFCN axons protected BFCNs from cell death (Fig. 3E) but did not protect from axon degeneration (Fig. 3, F and G) after 24 hours of proNGF axonal stimulation. These results showed that JNK is activated in the axons by proNGF and may be involved in retrograde cell death signaling but not axon degeneration in response to axonal proNGF stimulation, suggesting that different mechanisms may mediate BFCN axon degeneration versus cell death.
Figure 3. JNK activity is necessary for proNGF induced retrograde BFCN cell death but not axon degeneration.
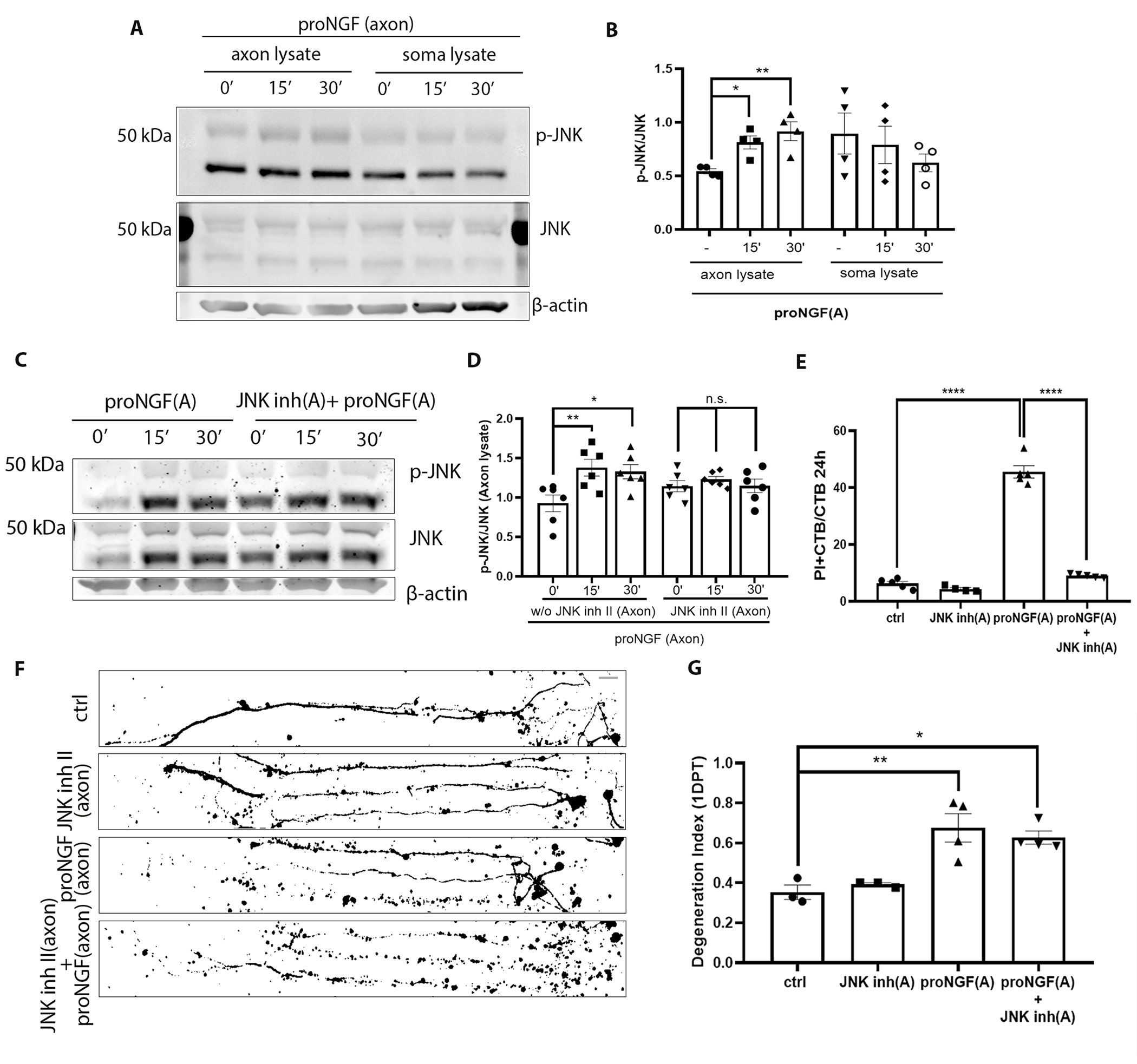
(A and B) Western blot images of total and phosphorylated JNK levels in the BFCN axon and soma lysates after axon stimulation with proNGF for 15 and 30 mins. Quantification of p-JNK is relative to JNK in the axon and soma lysates. Data are mean ± SEM, n = 4 independent experiments. *p < 0.05, **p < 0.01. (C and D) Western blot images and analysis of total and phosphorylated JNK with β-actin in the BFCN axon and soma lysates after axon stimulation with proNGF for 15 and 30 mins with or without JNK inhibitor II in the axons. Data are mean ± SEM, n = 6 independent experiments. **p < 0.01, *p < 0.05. (E) BFCNs plated in microfluidic chambers were pre-treated with JNK inhibitor II in the axons before axonal proNGF treatment [“(A)” in axis labels = axonal application]. Quantifications of the proportion of PI+ CTB BFCNs after 24 hours treatment are from n = 4 or 5 independent experiments. ****p < 0.0001. (F and G) Binary images of Tuj1-labeled BFCNs 24 hours after axonal application of JNK inhibitor II, proNGF, or both, and control untreated BFCNs, with quantification of a degeneration Index. Data are mean ± SEM from n = 3 or 4 independent experiments per treatment. Scale bar = 20μm. p** < 0.01, p* <0.05. All statistical analyses (B to G) were performed using ordinary one-way ANOVA with Tukey’s multiple comparisons test.
Axonal protein synthesis is necessary for proNGF induced retrograde BFCN cell death and axon degeneration.
Recent studies have demonstrated that axonal protein synthesis occurs in response to a variety of environmental cues. A low concentration of puromycin, which incorporates into the C-terminus of elongating nascent chains, was provided to proNGF-treated axons, and showed incorporation 1 hour after treatment, confirming an increase in local protein synthesis in the axons after axonal stimulation with proNGF (Fig. 4, A and B). To determine whether local protein synthesis in the axon was required for proNGF-induced retrograde BFCN degeneration, microfluidic cultures were pre-treated with cycloheximide in the axons to locally inhibit protein synthesis before stimulating axons with proNGF. Inhibition of local axonal protein synthesis protected BFCNs from cell death as well as axon fragmentation caused by proNGF at the axons (Fig. 4, C to E). These results suggest that after axonal stimulation with proNGF, newly synthesized proteins in the axons promote retrograde axon degeneration and cell death. To determine the time window of increased protein synthesis required for degeneration after axonal proNGF stimulation, BFCNs were treated with proNGF in the axons, followed by delayed cycloheximide treatment in the axons 0.5, 1, and 2 hours after exposure to proNGF. BFCNs were not protected from cell death if axonal protein synthesis was blocked after 2 hours of proNGF treatment (Fig. 4E), suggesting that nascent proteins required for proNGF induced BFCN degeneration are synthesized in the axons within 1 to 2 hours after axonal proNGF treatment. These results suggest that proNGF induced new protein synthesis in BFCN axons within 1 to 2 hours of treatment, which promote axon degeneration and retrograde cell death of BFCNs.
Figure 4. Nascent protein synthesis in the axons is necessary for proNGF induced retrograde BFCN cell death and axon degeneration.
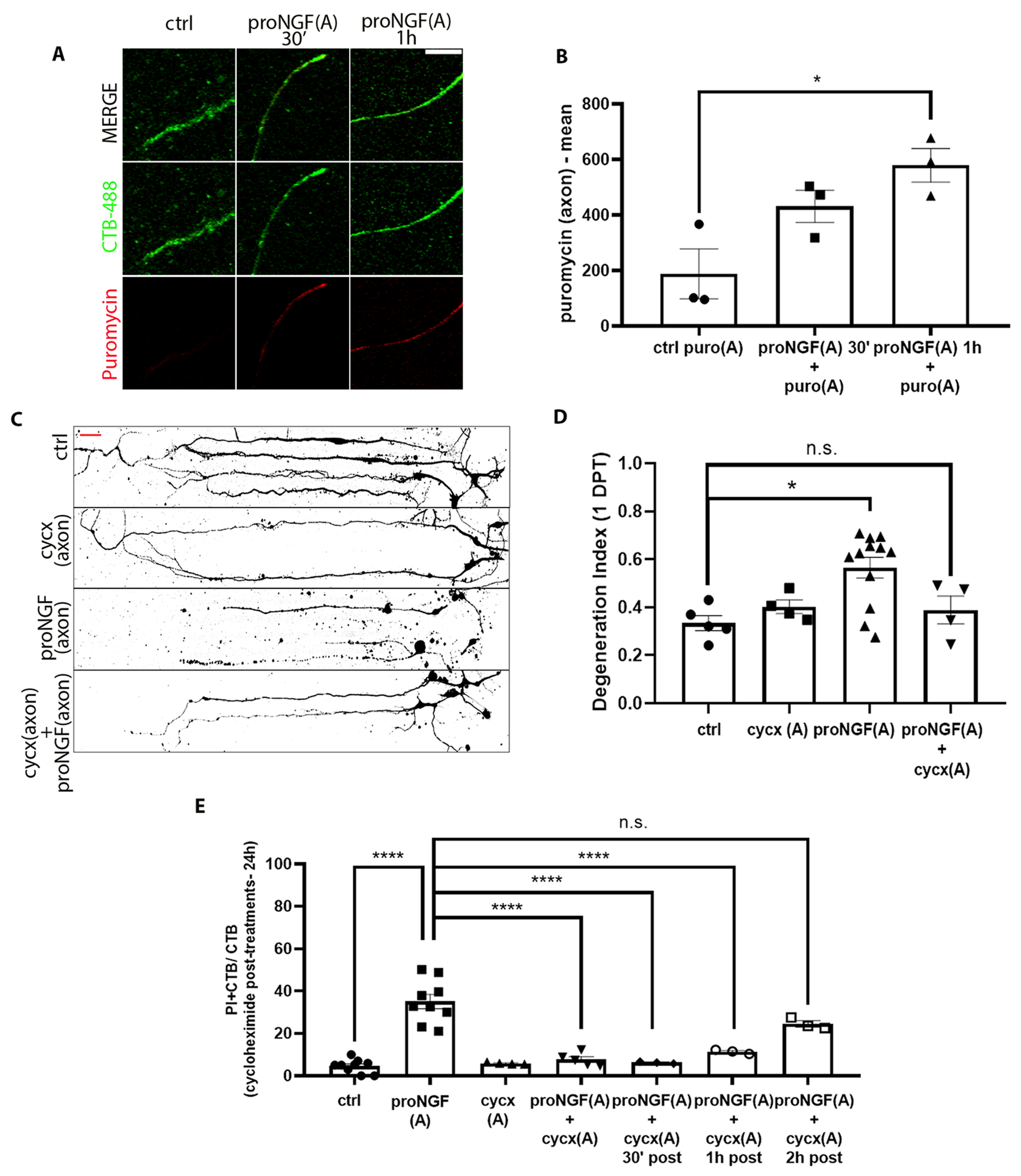
(A and B) Imaging for CTB-488 (green) and puromycin (red) in axons of untreated (ctrl) BFCNs and those treated at the axon with proNGF and puromycin (puro) for 30 min and 1 hour in microfluidic chambers. Quantification of mean intensity of puromycin in CTB+ proNGF-treated vs. untreated axons. Data are mean ± SEM of n = 3 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. In image and graph labels, “(A)” indicates axonal treatment. *p < 0.05. Scale bar = 10μm. (C and D) BFCNs plated in microfluidic chambers were pre-treated with cycloheximide (cycx) in the axons before proNGF treatment at the axon. Binary images of β-tubulin (Tuj1) staining of microfluidic cultures (C), and quantification of a degeneration index in the axons 24 hours after treatment (D). Scale bar = 20μm. Data are mean ± SEM of 4, 5, or 12 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. *p < 0.05, n.s. = not significant/p > 0.05. (E) Quantification of PI+ CTB+ relative to CTB+ BFCNs 24 hours after the indicated treatment, assessed by live imaging. Data are mean ± SEM, n = 3 to 9 independent experiments, each with technical duplicates and analysis of at least 50 cells per treatment. ****p<0.0001. All statistical analysis (B to E) was performed using ordinary one-way ANOVA with Tukey’s multiple comparison tests.
Analysis of axonal protein synthesis induced by proNGF
To analyze the newly synthesized axonal proteins induced by proNGF, the axon compartments of filter cultures were treated for 2 hours with O-propagryl-puromycin (OPP) after 30 min of proNGF treatment in the axons. Axon lysates were processed for mass spectrometry, and the data were analyzed using Ingenuity Pathway Analysis (IPA), a bioinformatic tool that allows prediction of upstream and downstream target proteins as well as cellular pathways likely to be associated with the dataset provided. IPA analysis of top upstream regulators predicted amyloid precursor protein (APP) as one of the major upstream activators of the pathways induced by proNGF axonal stimulation compared to control untreated BFCN axons (Fig. 5, A to C). Moreover, IPA analysis of Diseases and Functions comparing nascent proteins in axon lysates predicted increased probability of cell death, and a decreased probability of cell viability induced by proNGF (Fig. 5B), consistent with the degenerative response of axonal proNGF in BFCNs we observed (Fig. 1). Confirming the results from the mass spec analysis, axonal proNGF stimulation showed a statistically significant increase in the level of APP over 2 hours specifically in the BFCN axons analyzed by Western blot (Fig. 5, D and E). In contrast to axonal proNGF stimulation, BFCN soma stimulation with proNGF did not result in induction of APP in the axons or somas (Fig. 5, F and G), suggesting that the regulation of APP in the BFCN axons is specific to axonally sourced proNGF. Overall, these results showed that the nascent proteome synthesized as a response to proNGF in the axons is predicted to promote BFCN degeneration. Moreover, among upstream regulators, induction of APP, which has been studied in several CNS disorders, (24, 25) was validated as a response to the axonal proNGF stimulation.
Figure 5. proNGF induces new protein synthesis in the axons, associated with cell death and degeneration.
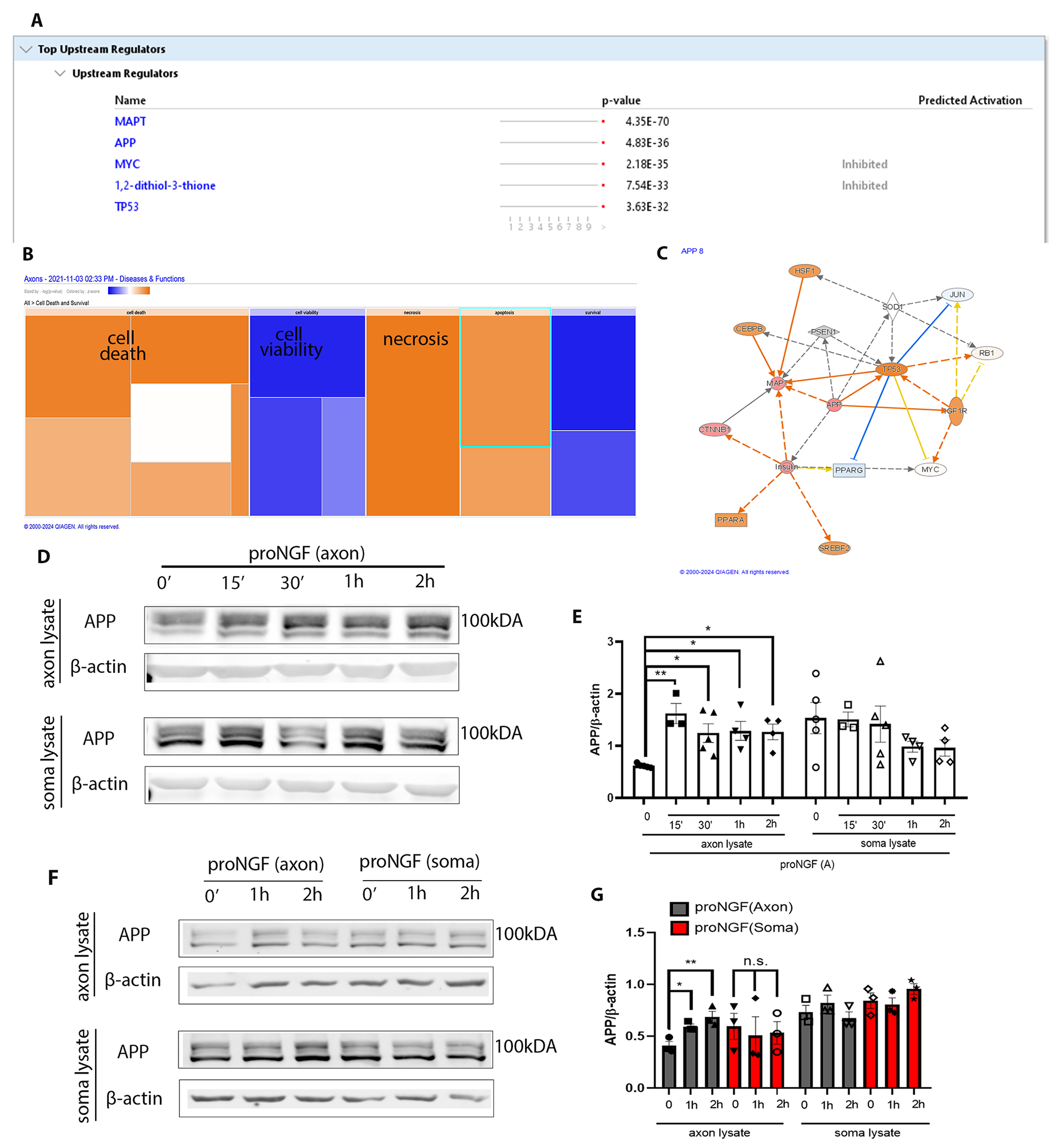
(A and B) Axons of BFCNs plated in filter cultures were treated with proNGF for 30 min, followed by capture of nascent axonal proteins using O-propargyl-puromycin (OPP), which were subjected to mass spectrometry (A) and IPA analysis (B). Samples from 3 independent experiments were combined for analysis. Predicted increases are represented with a scale of orange, predicted decreases represented with a scale of blue, and no significant change in prediction is represented with white. (C) Functional network of significantly regulated proteins from (A) created using IPA analysis with APP as a central regulator. Predicted increase in measurement is represented in red; activation in orange; inhibition in blue. (D and E) Western blotting and analysis of APP levels in the BFCN axons and somas after axon stimulation with proNGF for 15 min to 2 hours. Density was quantified relative to that of β-actin. Data are means ± SEM of n = 3 to 5 independent experiments. *p<0.05, **p<0.001 by ordinary one-way ANOVA with Tukey’s multiple comparison tests. (F and G) Western blot analysis of APP and β-actin levels in the BFCN axons and somas after axon or soma stimulation with proNGF for 1 or 2 hours. Quantification and statistical analysis as in (E), from n = 3 independent experiments per treatment.
APP regulates JNK activation in response to axonal proNGF stimulation of BFCNs
To investigate whether JNK and APP regulate each other in the axons following BFCN exposure to proNGF, axons in filter cultures were pre-treated for 20 mins with combinations of JNK inhibitor II and siRNA for APP. Notably JNK inhibitor II alone increased axonal APP levels, though not to a statistically significant extent (Fig. 6, A to C). However, despite inhibition of JNK activation, axonal APP levels were significantly increased at 30 minutes in comparison to untreated controls (Fig. 6, A and C). In comparison, when local APP induction was blocked, JNK activation was impaired (Fig. 6, D to F). These results suggest that in the axonal proNGF induced degeneration cascade, either APP is activated upstream of JNK, or APP and JNK are components of parallel pathways.
Figure 6. APP regulates JNK activation in response to proNGF in the axons.
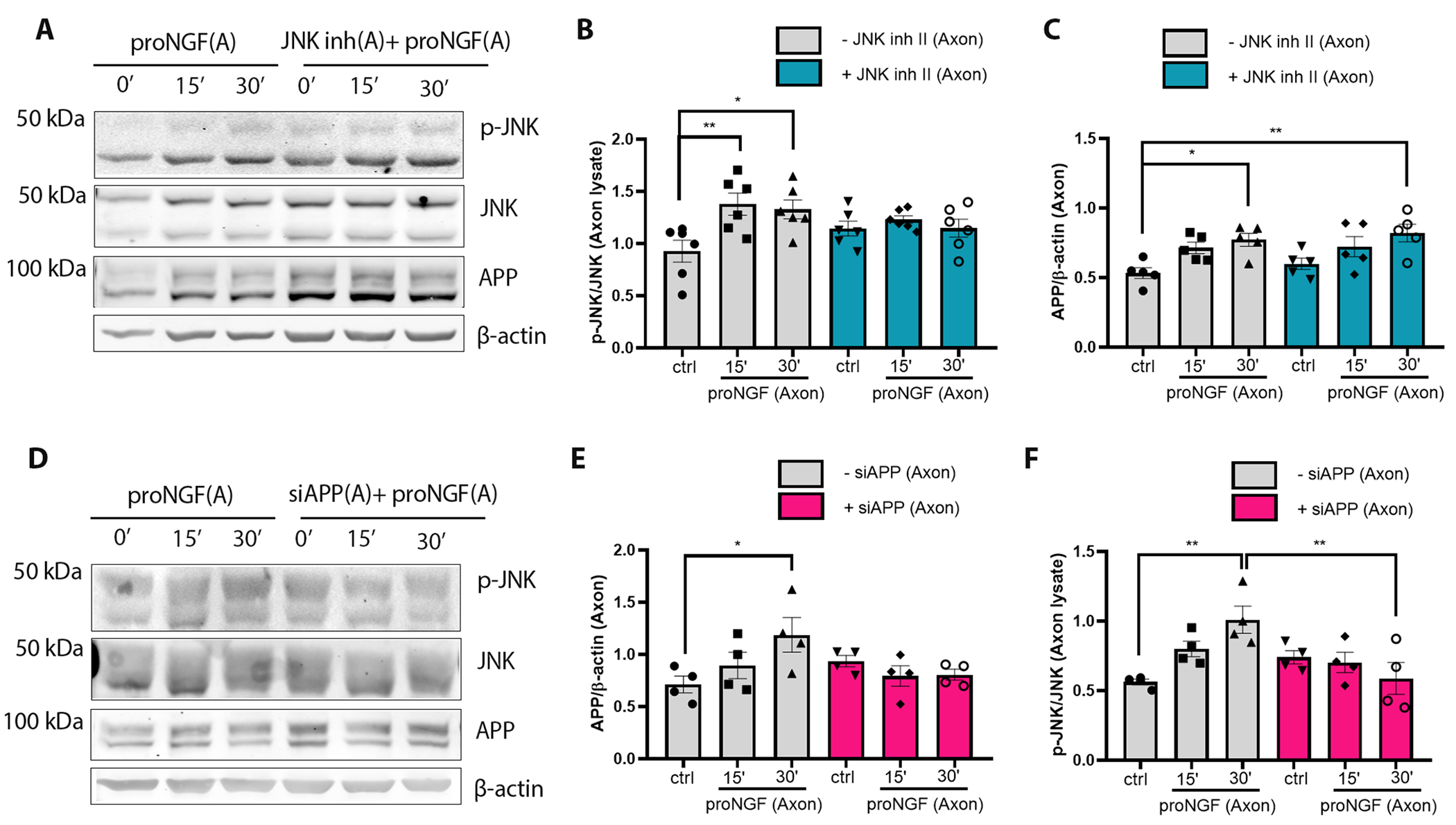
(A to F) Western blot analysis of total and phosphorylated (p-) JNK, APP and β-actin in BFCN axon lysates after axon stimulation with proNGF for 15 and 30 mins with or without (A to C) JNK inhibitor II or (D to F) APP siRNA application in the axons. Data are mean ± SEM, n = 6 (A to C) or 4 (D to F) independent experiments per treatment; *p < 0.05, p** < 0.01, ANOVA with Tukey’s multiple comparison tests.
APP mediates proNGF-induced retrograde axon degeneration and cell death in BFCNs.
A study by de Leon et al. (23), showed that APP has a pro-degenerative function in DRG axons following NGF deprivation. To assess the role of axonal APP in proNGF-induced BFCN degeneration , the increase in APP was prevented with an siRNA specific to APP linked with penetratin-1 (APP siRNA) to promote cell permeability (Fig. 7A). Knockdown of APP in BFCN mass cultures significantly reduced basal APP levels by 2 hours of treatment (Fig. 7, A and B). Additionally, pre-treatment of BFCN axons in filter cultures with two distinct siRNAs targeted to APP for 30 minutes was able to block APP induction in the axons even after 2 hours of proNGF axonal treatment, in contrast to proNGF alone (Fig. 7, C to F). A reduction in basal APP levels was not observed upon the axon-specific application of APP siRNA , with the short pre-treatment time of 30 minutes prior to proNGF addition, as the aim was to restrict the local APP induction that was observed with axonal proNGF treatment (Fig. 7, C and E). Inhibition of axonal protein synthesis using cycxloheximide pre-treatment for 30 minutes in the axons also blocked local APP induction in response to proNGF (Fig. 7, C and D), suggesting that the increase in axonal APP is due to local protein synthesis. Inhibition of APP synthesis in BFCN axons protected BFCNs from proNGF-induced retrograde cell death, as well as axon degeneration (Fig. 7, G to J). These results indicate that APP is locally synthesized in BFCN axons in response to proNGF stimulation in the axons, and plays a critial role in retrograde BFCN cell death and axon degeneration.
Figure 7. APP mediates proNGF-induced retrograde axon degeneration and cell death in BFCNs.
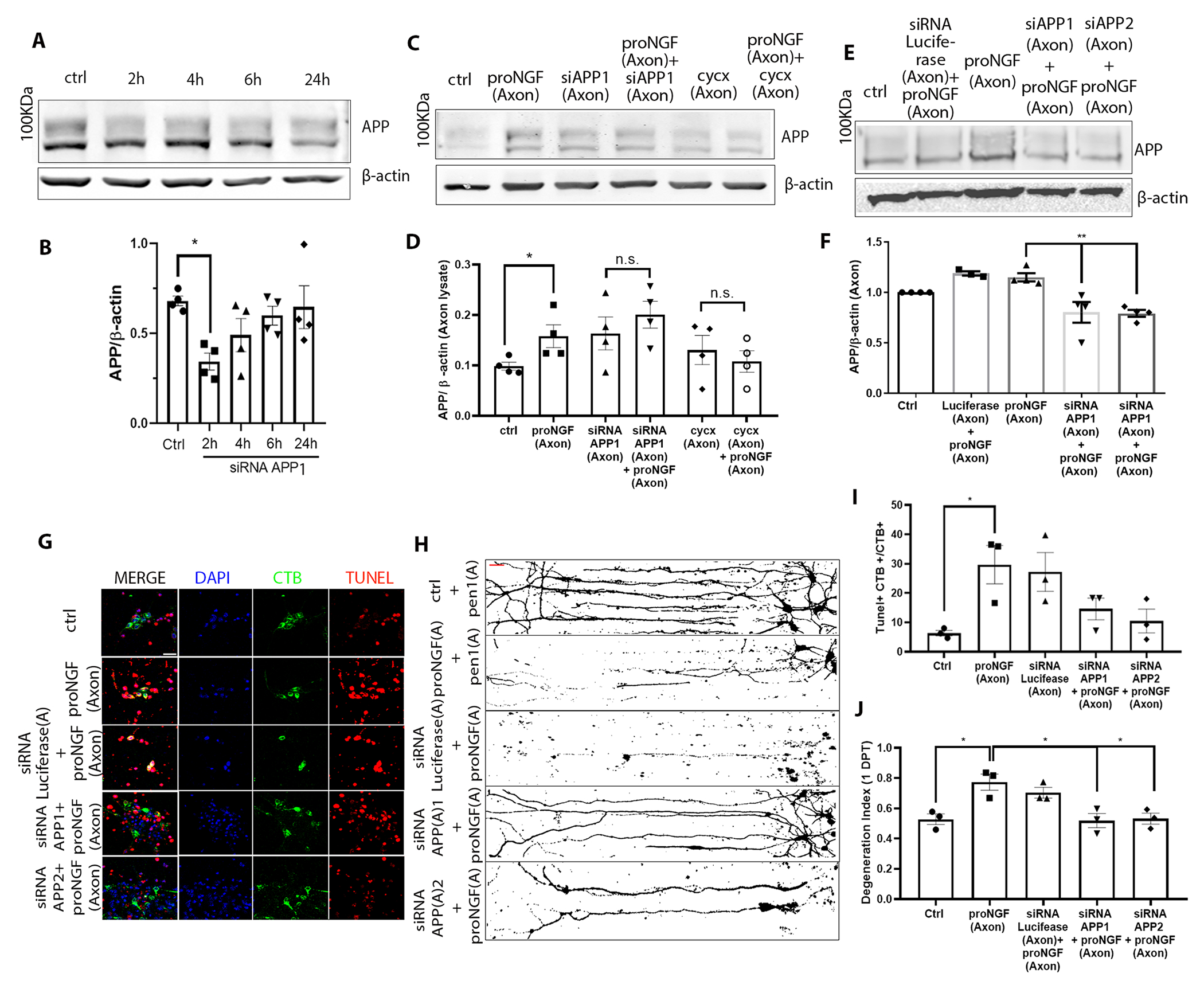
(A to F) Western blotting and analysis of APP expression (100 kDa) in (A and B) whole BFCN cultures when treated with the first of two Pen1-linked siRNAs for APP (siRNA APP1) for 2 to 24 hours, or (C to F) in axon lysates from BFCNs treated at the axons with proNGF after a 30-min pretreatment at the axons with either of two siRNAs to APP [siAPP1 (as in A) or siAPP2] or cycxloheximide. Controls were untreated (ctrl) and application of siRNA against luciferase. Density of APP was calculated relative to that of β-actin. Data are mean ± SEM from n= 4 independent experiments; *p < 0.05, **p < 0.01 by one-way ANOVA with Tukey’s multiple comparison tests. (G to J) Analysis of degeneration in BFCNs treated with proNGF with or without deficiency in APP. In (G), BFCN somas in microfluidic chambers were co-labeled for TUNEL (red), DAPI (blue) and CTB-488 (green) after 24 hours treatment as indicated, with quantification of the proportion of TUNEL-positive CTB+ soma in (I). Scale bar = 20 μm. In (H), BFCNs treated as indicated were Tuj1-immunostained; binary images are representative, with quantification of axon fragmentation in (J). In both graphs, data are mean ± SEM from n= 3 independent experiments, each with technical duplicates and at least 50 cells per treatment; *p < 0.05 by one-way ANOVA with Tukey’s multiple comparison tests.
ProNGF promotes axon degeneration in BFCNs through APP-regulated pre-catastrophic Ca2+increase
To investigate whether increased intracellular Ca2+ ion concentration is necessary for axon degeneration in BFCNs after proNGF stimulation, BFCNs grown in microfluidic cultures were loaded with Fluo4-AM, a cell permeant fluorescent calcium indicator prior to proNGF stimulation. Notably, a statistically significant increase in Ca2+ intensity was observed in proNGF-treated axons in comparison to control untreated BFCNs, specifically preceding axon fragmentation (Fig. 8, A to C, fig. S3, and movies S1 and S2). These results suggest that increased Ca2+ in the axons is a pre-catastrophic event in proNGF-induced axon degeneration in BFCNs. Moreover, blocking the intracellular Ca2+ increase by pre-treatment with BAPTA-AM, a cell-permeant Ca2+ chelator, blocked axon degeneration upon proNGF treatment (Fig. 8, A to C, fig. S3, and movies S4 and S5), suggesting that Ca2+ increase is required for proNGF to promote axon degeneration. Additionally, when proNGF-induced APP induction in the axon was blocked using the penetratin-linked siRNA for APP, proNGF stimulation failed to induce an axonal Ca2+ increase, and BFCN axon integrity was rescued (Fig. 8, A, D and E; fig. S3, and movie S3). Intracellular Ca2+ overload has been reported to activate calpains, a group of cysteine proteases which promote apoptosis [reviewed in (47)]. Blocking calpain activation by pre-treatment with ALLN, a cell-permeable calpain inhibitor, specifically in the axons protected BFCNs from proNGF-induced axon degeneration and cell death (Fig. 8, F to H). These results indicate that Ca2+ overload in response to proNGF in BFCN axons activated calpains that promoted BFCN degeneration. Overall, these results suggest that proNGF-mediated retrograde BFCN degeneration involves an APP-mediated, pre-catastrophic Ca2+ increase and calpain activation in the axon.
Figure 8. ProNGF-induced BFCN axon degeneration involves APP mediated Ca2+.
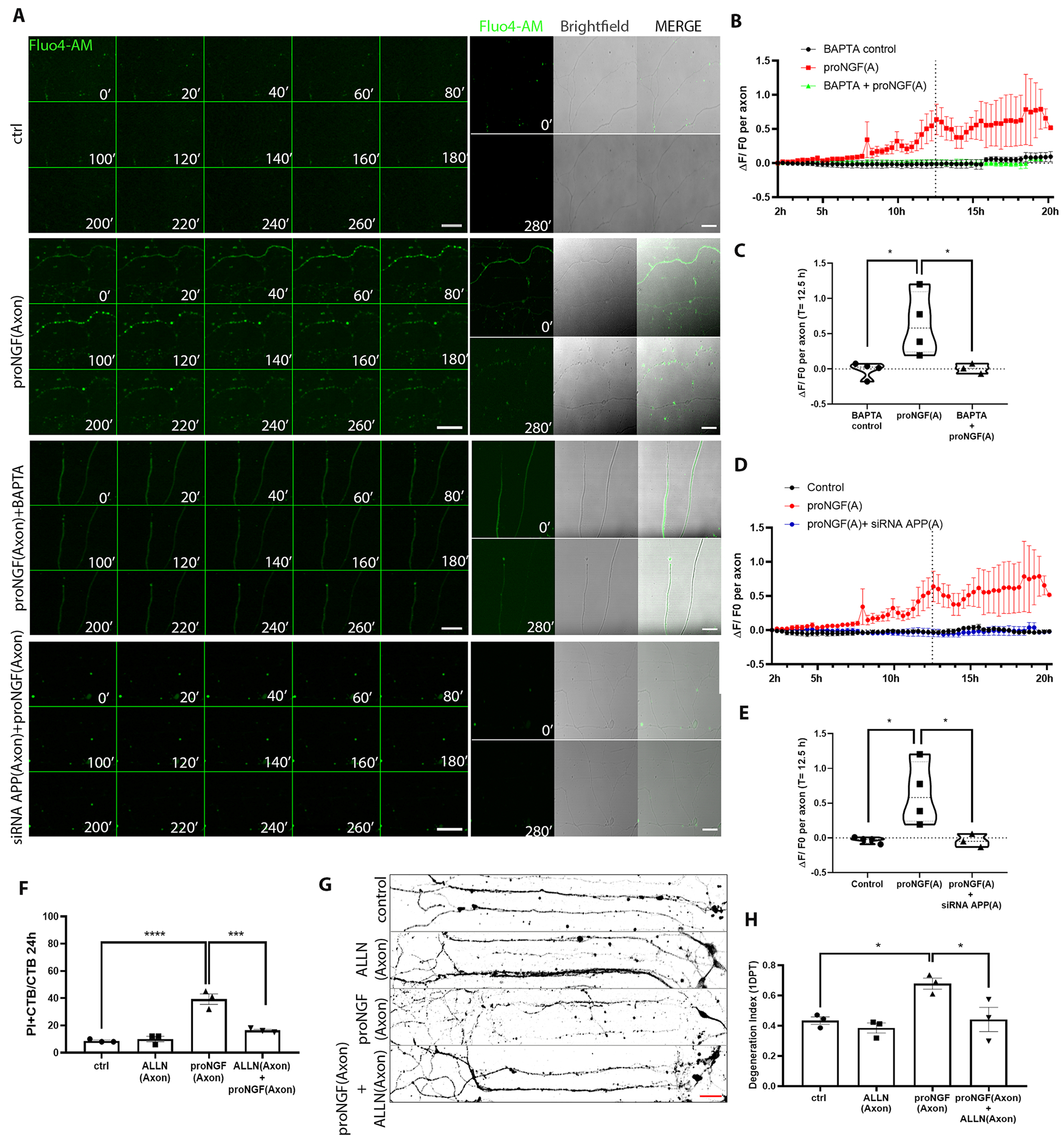
(A) Representative images of BFCN axons loaded with 5 μM Fluo4-AM (green) in control untreated axons, treated as indicated at the axon, from 10 to 14.6 hours (280 mins, 20-min lapses) after treatment, representative of 3 or 4 independent experiments. Corresponding axon images with Fluo4-AM and Brightfield at 0mins and 280mins. Scale bar = 50 μm, 40X. (B to E) Time series of the change in Fluo4-AM intensity (ΔF/F0) 2 hours after the indicated pretreatment, at the axon, and comparison of ΔF/F0 between treatments at 12.5 hours. Data are from n= 3 or 4 independent experiments, each with an analysis of at least 5 axons per treatment. ProNGF data sets are the same, for comparison. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparison tests. (F) Quantification of dying BFCNs (PI+CTB/CTB) after 24 hours axonal treatment with proNGF with or without ALLN in the axons, analyzed by live imaging. Data are mean ± SEM, n= 3 independent experiments and analysis of at least 50 cells per treatment. ****p < 0.0001, ***p < 0.001 by one-way ANOVA with Tukey’s multiple comparison tests. (G and H) Binary images of Tuj1-immunostained BFCNs treated as indicated, with quantification of axon fragmentation, represented as a degeneration index. Scale bar = 50 μm, 20X. Data are mean ± SEM from n = 3 independent experiments and analysis of at least 50 cells per treatment. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparison tests.
Discussion
Traumatic brain injury elicits increased levels of proNGF and proBDNF in the injured cortex, leading to local p75NTR-mediated loss of neurons in the penumbra of the injury (13, 15, 16). BFCNs, which project their axons throughout the cortex, express p75NTR throughout life, which may render these neurons vulnerable to injury-associated increases in proneurotrophins. ProNGF can promote cell death through p75NTR in CNS neurons (27) including BFCNs (12). Our previous study showed that cortical TBI promotes retrograde axonal degeneration of basal forebrain afferents (16). In this study, we investigated mechanisms underlying retrograde degeneration of BFCNs in vitro using compartmental cultures of primary BFCNs subjected to axon-specific stimulation with proNGF. This treatment promoted significant axon degeneration and cell death, establishing a novel retrograde degenerative effect of proNGF in BFCNs, which required p75NTR given that no BFCN loss was seen in p75NTR-deficient neurons, as before (16).
Studies in neurons of the peripheral nervous system have shown that dynein-mediated retrograde transport propagates target-derived neurotrophin signals for neuronal survival (29) and is necessary for p75NTR-mediated cell death after axonal stimulation (28, 30, 46). In our experiments, blocking dynein function in the axons protected BFCNs from proNGF-induced cell death as well as axon degeneration. Moreover, inhibiting dynein in the somas prior to proNGF stimulation in the axons also rescued BFCNs from axon degeneration and cell death, suggesting that proNGF requires axonal cargo to be retrogradely transported to the soma to execute the degeneration cascade. Degeneration of DRG neurons in response to NGF deprivation requires retrograde transport and subsequent anterograde transport of pro-apoptotic factors to the axon (49), indicating that both retrograde and anterograde transport are necessary to promote neurodegeneration in response to axonal stimuli.
Axon stimulation with proNGF resulted in activation of JNK, a well-established component of the proNGF degenerative signal (26). Blocking JNK activity in the BFCN axons protected BFCNs from cell death, but not from axon fragmentation after axonal proNGF stimulation, suggesting that mechanisms governing axon degeneration may be distinct from those governing cell death. JNK plays a role in p75NTR-mediated retrograde death of sympathetic neurons (30), associating with both anterograde and retrograde motors in axons, specifically interacting with p150Glued, a dynactin subunit that promotes p75NTR-mediated degeneration (31, 32).
Injury of peripheral axons evokes novel protein synthesis locally within the axon to regulate the neuronal response (33, 34). CNS neurons that degenerate in response to axonal exposure to Aβ also show intra-axonal protein synthesis (35). Therefore, we investigated the role of axonal protein synthesis as a potential mechanism governing proNGF induced retrograde degeneration. Axonal protein synthesis has been associated with several neurological disorders, including AD and amyotrophic lateral sclerosis [reviewed in (36)]. Blocking axonal protein synthesis within one to two hours of proNGF treatment protected BFCNs from axon degeneration and cell death. Proteomics analysis of nascent axonal proteins induced by proNGF predicted proteins, pathways, diseases, and functions that may be substantially regulated. IPA analysis predicted an increase in cell death and decreased cellular viability, confirming that local protein synthesis in axons stimulated with proNGF promotes a pro-degenerative response. APP was predicted to be a key upstream regulator of the axonal proNGF response; however, Aβ1-40 and Aβ1-42 were not detected in the proteomics analysis, indicating that the pro-degenerative effect was likely mediated by the full-length APP protein. APP levels were significantly increased in the axons after proNGF axonal stimulation, validating the predicted data. The role of APP in axon degeneration (23) has been investigated in the context of several neurodegenerative diseases, including AD [reviewed in (37)]. Excess APP disrupts endocytic pathways, leading to impaired NGF trafficking, promoting neurodegeneration (38). In our study, preventing JNK activation did not block increased axonal APP levels, although APP knockdown led to a loss of axonal JNK activation, suggesting that APP induction is upstream of JNK activation following proNGF stimulation. The JNK scaffolding protein JIP interacts with vesicular cargo through membrane-associated proteins, such as APP, suggesting a link between JNK activation and APP induction in the BFCN axons (39). Further analysis of our proteomics results may lead to additional mechanistic insight and interesting links. For example, the increase in microtubule-associated protein tau (MAPT) levels may suggest a possible role of MAPT in proNGF-mediated axon degeneration. Several neurodegenerative diseases involve tauopathies with axon degeneration observed as an early event (45), suggesting a probable role of MAPT in proNGF-induced axon degeneration.
APP has been shown to regulate calcium (Ca2+) release from intracellular stores preceding axon fragmentation in DRG neurons (23, 19). We investigated whether increased Ca2+ plays a role in APP-mediated degeneration in response to axonal proNGF. Blocking intracellular Ca2+ increase with BAPTA-AM protected the axons from degeneration. Moreover, inhibition of APP induction in the axons prevented the increase in Ca2+ levels in the presence of proNGF, suggesting that APP induction leads to increased intracellular Ca2+ and subsequent axon degeneration. Studies have shown that axonal injury induces early calcium waves that prepare axons for local translation events leading to axon degeneration (50). Increased intracellular Ca2+ can also activate calpain, which may promote neurodegeneration (47). Traumatic axonal injury elicits a biphasic calpain activation where transient calpain activity accompanied early structural damage and transport impairment, while delayed activation occurred in the pre-catastrophic phase (48). We showed that calpain activity is necessary for proNGF-induced axonal degeneration; however, it is unclear whether cleavage products from transient calpain activation need to be transported to promote axon degeneration, given that retrograde transport was also necessary for proNGF-induced retrograde degeneration.
This study reveals a mechanism by which p75NTR mediates retrograde degeneration of BFCNs in response to axonal proNGF exposure (Fig. 9). In vivo, mature neurotrophins are expressed in the brain to support many neuronal populations. However, after injury there are increased levels of uncleaved proneurotrophins due to alterations in their processing and maintenance (42). Proneurotrophins induced in the cortical target regions of BFCNs following TBI elicit their retrograde degeneration (16). In this study, we analyzed signaling mechanisms in BFCNs specific to axonal proNGF stimulation. Further knowledge of mechanisms underlying the BFCN response to proneurotrophin stimulation in injured target regions will shape our understanding of brain health in conditions of injury and other pathologies.
Figure 9. Model of proNGF-p75NTR retrograde degeneration and cell death.
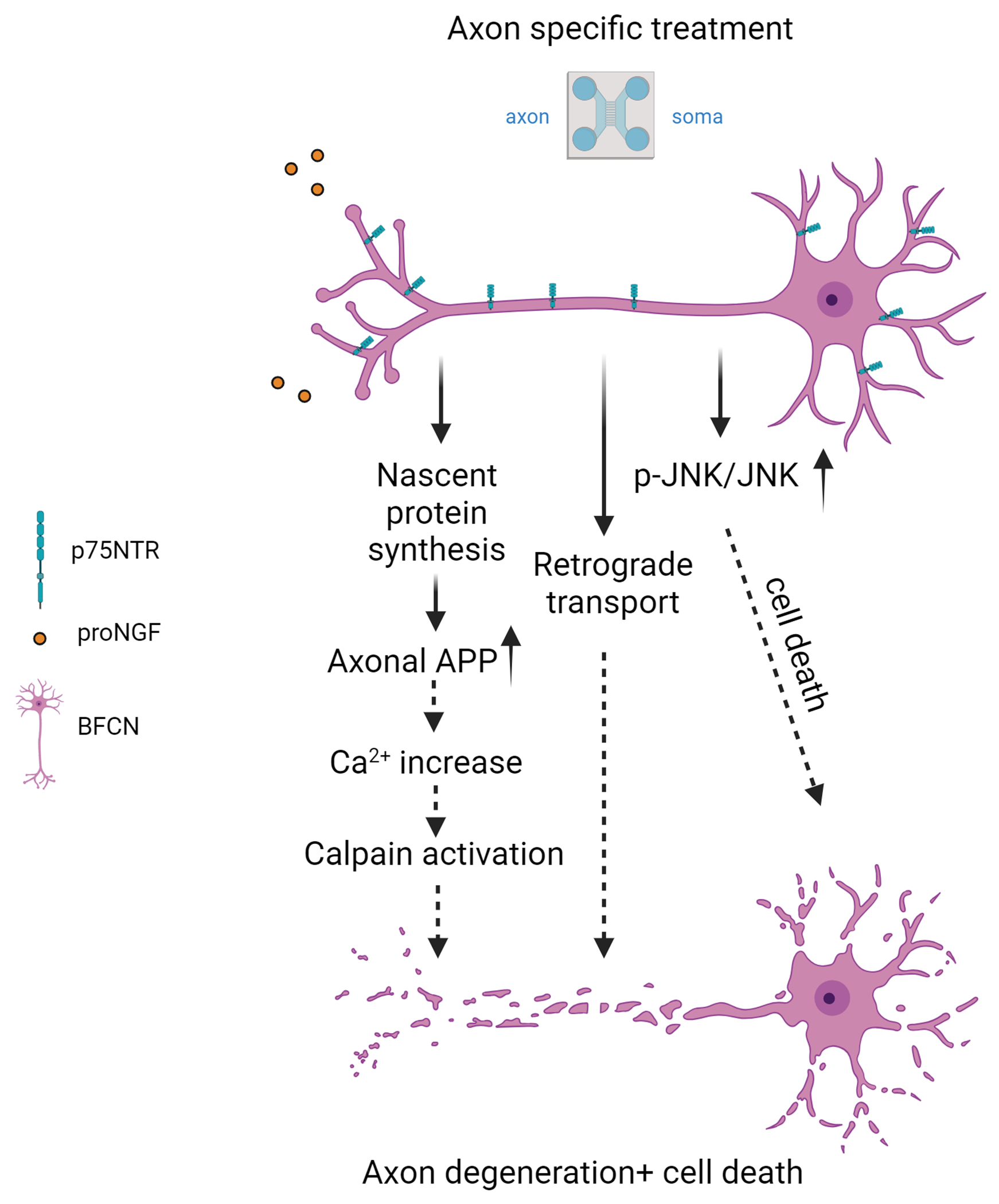
Exposure of axon terminals to proNGF elicits local protein synthesis of APP within the axon, which promotes increased intracellular calcium levels, leading to calpain activation and axon degeneration and cell death. Activation of p-JNK signals retrograde cell death, but not axon fragmentation. Retrograde transport mediated by dynein is necessary for both axon degeneration and cell death, although identification of the transport cargo requires further investigation. Created in BioRender.com.
Materials and Methods
Wild-type and p75NTR knockout rats
All experiments were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) policies and approved by Rutgers University, Newark. Wild-type rats were purchased from Charles River. NGFR global knockout (p75NTR KO) rats were obtained from SAGE/Horizon Laboratories and bred in house. Both males and females were used in all experiments.
Reagents
Recombinant human proNGF protein (cleavage resistant; Cat# N-285) was purchased for Alomone Labs (Tel Aviv, Israel). Poly-D-lysine, glucose, transferrin, insulin, putrescine, selenium, progesterone, penicillin, and streptomycin were purchased from Sigma Aldrich. Minimum Essential Medium (MEM), Ham’s F-12 Media and B-27™ Supplement (50X; # 17504-044) was purchased from Gibco. Microfluidic chambers were made in-house using Microfluidic chamber master molds which were a generous gift from Dr. Eran Perlson, Tel Aviv University, using the protocol described previously (51).
Cholera Toxin Subunit B (Recombinant), Alexa Fluor™ 488 Conjugate (CTB) (Cat# C34775) was purchased from Invitrogen. Propidium iodide (PI; Cat# P1304MP) was obtained from Molecular Probes. The cytoplasmic dynein inhibitor ciliobrevin D (Cat# 250410) and JNK inhibitor II (Cat# 420119) were purchased from Calbiochem. Cycloheximide (Cat# C81040) was purchased from Research Products International. Puromycin dihydrochloride (Cat# P8833) was obtained from Sigma. Fluo4-Am cell permeant was obtained from Invitrogen (Cat# F14201). TUNEL kit was purchased from Invitrogen (# C10617). Penetratin-Arg peptide (Pen1; SKU:11PENA1000) was purchased from MP Biomedicals. siRNA to APP was custom made from Dharmacon. BAPTA-AM (Cat# B1205) was purchased from Invitrogen. ALLN (Cat#208719) was purchased from Sigma-Aldrich.
Antibody to TrkB, rabbit (Cat# 07-225) was purchased from Millipore. Antibody to p75NTR, goat (Cat# AF1157SP) was purchased from R&D Systems. Antibody to puromycin, mouse, clone 12D10 (Cat# 12D10) was obtained from Millipore-Sigma. Antibody to β-tubulin III, mouse (Tuj1; Cat# G712A) was purchased from Promega. Antibody to MAP2, rabbit (Cat# 8707) was obtained from Cell Signaling Technology. Antibody to NeuN, mouse (Cat# MAB377) was obtained from Millipore. Antibody to phosphorylated JNK, rabbit (Thr183/Tyr185, Thr221/Tyr223) was purchased from Upstate Biotechnology. Recombinant antibody to amyloid precursor protein (APP), rabbit (Tyr188; Cat# AB32136) was obtained from Abcam. Antibody to β-actin, mouse (Cat# A2228) was obtained from Sigma. Ligand-blocking antibody to p75NTR (REX) was a generous gift from Dr. Louis Reichardt (University of California at San Francisco).
Alexa Fluor 488 and Alexa Fluor 555 anti-goat and anti-rabbit secondary antibodies and Alexa Fluor 647 anti-mouse secondary antibody were purchased from Invitrogen. LICOR Mouse 800, Rabbit 800, and Mouse 680 secondary antibodies were purchased from LICOR. DAPI Fluoromount-G (Cat# 0100-20) was obtained from Southern Biotech.
O-propargyl-puromycin (OPP; Cat# C10459) was purchased from ThermoFisher. Phenylmethylsulfonyl fluoride (PMSF; Cat# 13559061) was purchased from Boehringer Mannheim. Roche cOmplete, Mini Protease Inhibitor Cocktail EDTA free (Cat# 11836170001) was obtained from Roche. Biotin azide (Cat# B10184) was purchased from Invitrogen. TCEP [tris(2-carboxyethyl) phosphine hydrochloride; Cat# C4706], and TBTA (tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine; Cat# 678937) were purchased from Sigma-Aldrich. Streptavidin magnetic beads (Cat# 88816) and Zeba Spin columns (7K MW cutoff, Cat# 89882) were obtained from Thermo-Fisher.
Neuronal cultures
Wild-type and p75NTR KO pregnant rats were euthanized by exposure to CO2 and soaked in 70% ethanol for 5 min for sterilization. Embryonic day 16 (E16) rat fetuses were removed under sterile conditions and kept in PBS on ice. Basal forebrains were dissected and dissociated in serum-free medium (SFM) (8) composed of a 1:1 mixture of Eagle’s MEM and Ham’s F-12 supplemented with glucose (6 mg/ml), putrescine (60 μm), progesterone (20 nm), transferrin (100 μg/ml), selenium (30 nm), penicillin (0.5 U/ml), and streptomycin (0.5 μg/ml). The cells were then plated in microfluidic chambers (43) attached to glass coverslips in tissue culture dishes that were precoated overnight with poly-D-lysine (0.2 mg/ml). 200 μl of media was added to the soma compartment and 100 μl media was maintained in the axon compartments to maintain a media volume difference that facilitated the growth of the axons of the neurons through the microgrooves towards the axon compartment. The cells were maintained with the volume difference between the soma and axon compartments in SFM supplemented with 1% B-27 for 5 days at 37°C to obtain compartmentalized BFCN cultures which can be treated separately at the axons or somas. For biochemical experiments, BFCNs were plated on the top compartment of filter inserts with a pore size of 1 μm, allowing neurites to grow through the filter to the bottom compartment providing an axon-enriched compartment for axon or soma specific stimulation (44).
Live imaging of BFCNs in microfluidic cultures
BFCN microfluidic cultures were prepared for live imaging at 5-DIV. The axon compartment was treated with recombinant cholera toxin subunit B, Alexa Fluor™ 488 conjugated (CTB, 1 μg/ml; Cat# C34775), a retrograde tracer, for 20 min and washed twice with SFM + 1% B27 to retrogradely label the BFCNs which had extended axons to the distal compartment through the microgrooves. After 5 hours, the CTB from the axons was found to be transported into the cytoplasm of the BFCNs that projected axons to the distal compartment. The soma compartment was treated with propidium iodide (PI, 1 μg/ml; Cat# P1304MP) to label dying neurons. BFCNs were then treated with recombinant human proNGF protein (cleavage resistant; Cat# N-285; 20 ng/ml) in the axon compartment and compared with control untreated compartmentalized BFCNs to assess the effect of axonal stimulation with proNTs on neuronal degeneration. Growth media volume difference was maintained as described in the neuronal culture method to restrict stimulation with proNTs exclusively to the axons. To assess surviving versus dying neurons, live imaging of neurons was performed using a Zeiss LSM 510 confocal microscope maintaining constant temperature and CO2 (37°, 5%) for the duration of the experiment to capture images at 10X, 4 images per chamber. Incorporation of PI in the nucleus of CTB-positive neurons after the 24-hour axonal treatment was assessed.
Inhibition of axonal transport, protein synthesis, and JNK activity
Prior to treatment with proNGF, the axon compartment of microfluidic BFCN cultures was pretreated with the cytoplasmic dynein inhibitor ciliobrevin D (Calbiochem, Cat# 250410) at 50 μm for 20 mins to inhibit axonal transport; cycloheximide at 1 μg/ml for 20 mins to inhibit protein synthesis; or JNK inhibitor II (Caliochem, Cat#420119) at 40 nm for 20 min to inhibit JNK activity.
Immunocytochemistry (ICC)
Basal forebrain microfluidic cultures were fixed with 4% paraformaldehyde for 20 min, washed with PBS and permeabilized with 0.5% Triton X-100 in PBS for 10 min. The cells were then blocked for 1 hour with 5% normal goat serum and 1% bovine serum albumin (BSA) in PBS and incubated overnight at 4°C with primary antibody prepared in 1%BSA in PBS. Primary antisera were directed against the following: p75NTR (goat, 1:500; RRID:AB_2298561), TrkB (rabbit, 1:500; RRID:AB_310445); β-tubulin III (Tuj1; mouse, 1:1000; RRID:AB_430874), APP (rabbit, 1:500; RRID:AB_2289606). Cells were then washed with PBS, exposed to the appropriate secondary antibodies coupled to different fluorophores, and highly cross-adsorbed against different species (Alexa Fluor 488, Alexa Fluor 594 and Alexa Fluor 647; Invitrogen). Coverslips were mounted on slides using DAPI Fluoromount-G to label the nuclei. Images were obtained using Zeiss LSM 510 META confocal microscope at 20X, 8 images per chamber, and analyzed to measure axon fragmentation using Image J software.
Axon fragmentation analysis
Axon fragmentation was quantified as Degeneration index (DI). DI was calculated as the ratio of the area of fragmented axons over the total area of axons (intact axons + fragmented axons) by using Anti-β-Tubulin III (Tuj1; Cat# G712A)-stained fluorescence images. A total of 8 images were analyzed per chamber. All images were processed using ImageJ software. To analyze the size of axonal fragments, binary masks were created for each image. Particles with a size area equal or lower than 60 μm2 and with a circularity index higher than 0.03 were classified as degenerated neurite fragments.
Western blot analysis
5-DIV filter cultures were treated with proNGF, and axon and soma lysates were obtained in 30- and 60-μl, respectively, of lysis buffer comprised of 10% glycerol (70%), 10% Triton (10%), 10% NP40 (10%) and 10X protease inhibitors (5%) and 10X phosphatase inhibitors (5%). After protein quantification, equal amounts of protein were run on a 12% polyacrylamide gel and transferred to nitrocellulose membrane. Equal protein loading was assessed by Ponceau staining, which was washed out with TBS with 0.05% Tween 20 (TBST) and was confirmed by re-probing the membranes with anti-actin. The membranes were blocked with 5% nonfat milk prepared in TBST for 1 hour and incubated with primary antibody overnight. After washing 3 X 10 min with TBST, the blots were incubated with appropriate secondary antibodies for 1 hour at room temperature. The membrane was washed 3 X 10 min with TBST and then scanned with the Odyssey infrared imaging system (LI-COR Bioscience). All blots shown are representative of at least three independent experiments.
TUNEL assay
BFCNs in microfluidic cultures were fixed with 4% PFA after 24h treatment with proNTs and used to perform TUNEL assay. The assay was performed using Click-iT Plus TUNEL kit from Invitrogen as per manufacturer’s protocol (reference no. C10617) to label fragmented DNA with Alexa Fluor™ 488. The cells were then immunostained for β-tubulin III (Tuj1) and DAPI. Tuj1 and TUNEL double-labeled BFCNs extending their axons to the axon compartment were counted to analyze the number of dying neurons as a result of the respective treatments.
Axonal immunolabeling of newly synthesized proteins
Monitoring of protein synthesis was performed using a modified protocol obtained from (52). To label nascent proteins synthesized in the BFCN axons after axonal proneurotrophin stimulation, 5-DIV BFCNs were labeled with the retrograde tracer CTB-Alexa 488 as described in the Live imaging section above. Microfluidic cultures were then treated with proNGF in the axons for 30 mins or 1 hour, followed by treatment with 10 μg/ml puromycin for 10 mins in the axons. Cells were then fixed with 4% PFA in PBS for 20 mins and stained with anti-puromycin antibody (RRID: AB_2566826, 1:10,000) and anti-mouse secondary antibody labeled with Alexa Fluor 594. Intensity of puromycin in CTB-labeled axons after 30 mins or 1 hour of axonal proNGF treatment was assessed by intensity analysis using Image J software.
Metabolic labeling with OPP
BFCNs grown on filter cultures were treated with proNGF in the axon compartment for 30 mins. 10 μM OPP was added to the axon compartment for 2 hours in proNGF-treated and control untreated filter cultures, washed with PBS and lysed with lysis buffer.
Biotinylation click reaction was performed using 5 mM biotin-PEG3-azide. Proteins were then precipitated with 5 vols of acetone until precipitate was observed and stored overnight at −20°C. Proteins were further prepared for mass-spectrometry analysis by methanol precipitation, followed by desalting using Zeba Spin columns (7K cutoff, Thermo 89882). In new 2-mL tubes eluted samples were pulled down using 80 μL of magnetic streptavidin coated beads (Pierce). Beads were washed with urea and samples were stored in cold PBS.
Fifty microliters of axonal protein lysate from each sample were separated on SDS-PAGE, followed by in-gel trypsin digestion. Alkylation and trypsin digestion steps were performed using 80 μl of 5 mM DTT followed by 10 mM iodoacetamide. Final desalting was performed by activated C-18 OMIX tips (Agilent, A57003100K) with 200 μl 99.9% acetonitrile + 0.1% formic acid (FA dissolved in HPLC grade water) placed in 2 ml tubes with adapters (GL Sciences Japan 5010-2514). Eluates were transferred to 0.6 ml tube, dried by SpeedVac.
Subsequently, equal amounts of peptides from each sample were labeled with one of the 16-plex TMT reagents. These labeled peptides were combined and further fractionated using high pH reversed-phase liquid chromatography (RPLC) on an Acquity UPLC system (Waters). Each fraction obtained was subsequently subjected to analysis via RPLC-MS/MS on an Orbitrap Fusion Lumos Tribrid Mass spectrometer, which was coupled with an Ultimate 3000 nano LC system (Thermo Scientific). The MS/MS spectra obtained were searched against UniProt Rat database (29,918 sequences as downloaded 3/12/2022) using the Sequest search engine within the Proteome Discovery (V2.4, Thermo Scientific) platform for both protein identification and quantitation. Proteins and peptides were identified at 1 % false discovery rate.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium via the PRIDE partner repository with the dataset identifier PXD043846 and 10.6019/PXD043846. Functional analysis of protein expression data was performed using Ingenuity Pathway Analysis (IPA, QIAGEN).
Knockdown of axonal APP
APP siRNA was linked to Penetratin1 (MP Biomedicals) to allow for cell permeability (53). Two different APP siRNAs (APP1 372-391 gtgcctagttggtgagttt; and APP2 1496-1515 tcaacatgctgaagaagta) and a luciferase siRNA as a nonspecific control (cguacgcggaauacuucgauu) were synthesized by Dharmacon with a 5’thiol on the sense strand and mixed with Penetratin1 (Pen1) at a ratio of 1:10 (Pen1 to siRNA APP) by incubating for 1 hour at 37°C. To confirm linkage, Pen1-siRNA APP was run on a 20% non-reducing gel and compared with unlinked Pen1. Cells were treated with 80 nM Pen1-siRNA, while control cells were treated with Pen1 only or with the Pen1-luciferase siRNA. Prior to treatment, Pen1-siRNA was heated at 65°C for 10 min and added to the cultures as a 2X solution.
Calcium imaging
Calcium imaging was performed in BFCNs grown in microfluidic chambers using Fluo-4 AM, a cell permeant labeled calcium indicator (Cat# F14201). Fluo4-AM stock solution was prepared according to the manufacturer’s protocol. Cells (5 DIV) were treated with 5μM Fluo4 AM for 1hr in the incubator. Cells were then washed with indicator-free media to remove unlinked Fluo4-AM. The cells were further incubated for 30 min with the indicator-free media to allow complete de-esterification of intracellular AM esters. Cells treated with BAPTA-AM received 15 μM BAPTA-AM for 1 hour, which was then replaced with fresh media. This was followed by axonal treatment with proNGF. For siRNA knockdown of axonal APP, cells were transfected with Pen1-linked APP siRNA (80 nM) for 30 mins. This was followed by treatment with proNGF (20 ng/ml). Cells with or without proNGF treatment were compared by time-lapse imaging of Fluo4-AM (Excitation/Emission: 494/506 nm) to observe changes in fluorescence as a response to the treatment with images captured every 20 min, over 20 hours. Time-lapse images were obtained using Zeiss LSM 510 META confocal microscope and analyzed to show changes Ca 2+ levels in BFCNs using Image J software by quantifying the ratio of change in Fluo4 intensity, normalized to the initial intensity per axon (ΔF/F0).
Experimental Design and Statistical Analyses
In all experiments “n” refers to the number of independent experiments performed using cell cultures established from separate litters. Statistical analysis was performed in Prism 5.0 software (GraphPad). Image analysis was performed using ImageJ software. All measurements are shown as mean ± SEM. For samples defined by one factor, data were analyzed by one-way ANOVA with Tukey’s post hoc multiple-comparisons test when three or more independent group of samples were compared. For samples defined by two factors, data were compared by two-way ANOVA with Sidak’s post hoc multiple-comparisons test. For in vivo experiments, sample size (n) was defined as the number of mice that were quantified. For in vitro experiments, sample size (n) was defined as the number of independent experiments obtained from separate pregnant rats. P values <0.05 are considered significant. Figure each figure, the statistical test, sample size (n), and the p values are reported in the figure legends specific to each experiment. Epifluorescent images were assembled using Adobe Photoshop.
Supplementary Material
Acknowledgments:
The authors would like to thank Dr. Eran Perlson (Tel Aviv University) for providing the microfluidic templates. We thank Dr. Louis Reichardt (University of California, San Francisco) for the p75NTR ligand blocking antibody, REX.
Funding:
This work was supported by NIH/NINDS 1R01NS127894 (to WJF) and Rutgers Busch Biomedical Grant Program (to HL and WJF).
Footnotes
Competing interests: The authors declare that they have no competing financial interests.
Data and materials availability:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium via the PRIDE partner repository with the dataset identifier PXD043846 and 10.6019/PXD043846. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Ballinger EC, Ananth M, Talmage DA, Role LW, Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron, 91, 1199–1218 (2016). 10.1016/J.NEURON.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, & DeLong MR (1982). Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science, 215, 1237–1239. 10.1126/SCIENCE.7058341 [DOI] [PubMed] [Google Scholar]
- 3.Hasselmo ME (2006). The Role of Acetylcholine in Learning and Memory. Current Opinion in Neurobiology 16, 710. 10.1016/J.CONB.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada K, Nishizawa K, Kobayashi T, Sakata S, & Kobayashi K (2015). Distinct roles of basal forebrain cholinergic neurons in spatial and object recognition memory. Scientific Reports 5, 1–13. 10.1038/srep13158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitz TW, Cheng FHT, & de Rosa E (2010). Failing to Ignore: Paradoxical Neural Effects of Perceptual Load on Early Attentional Selection in Normal Aging. Journal of Neuroscience 30, 14750–14758. 10.1523/JNEUROSCI.2687-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nonomura T, Nishio C, Lindsay RM, & Hatanaka H (1995). Cultured basal forebrain cholinergic neurons from postnatal rats show both overlapping and non-overlapping responses to the neurotrophins. Brain Research 683, 129–139. 10.1016/0006-8993(95)00357-V [DOI] [PubMed] [Google Scholar]
- 7.Alderson RF, Alterman AL, Barde YA, & Lindsay RM (1990). Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron 5, 297–306. 10.1016/0896-6273(90)90166-D [DOI] [PubMed] [Google Scholar]
- 8.Friedman WJ, Ibáňez CF, Hallböök F, Persson H, Cain LD, Dreyfus CF, & Black IB (1993). Differential Actions of Neurotrophins in the Locus Coeruleus and Basal Forebrain. Experimental Neurology 119, 72–78. 10.1006/EXNR.1993.1007 [DOI] [PubMed] [Google Scholar]
- 9.Hefti F, Hartikka J, Eckenstein F, Gnahn H, Heumann R, & Schwab M (1985). Nerve growth factor increases choline acetyl-transferase but not survival or fiber outgrowth of cultured fetal septal cholinergic neurons. Neuroscience 14, 55–68. 10.1016/0306-4522(85)90163-0 [DOI] [PubMed] [Google Scholar]
- 10.Kromer LF (1987). Nerve Growth Factor Treatment After Brain Injury Prevents Neuronal Death. Science 235, 214–216. 10.1126/SCIENCE.3798108 [DOI] [PubMed] [Google Scholar]
- 11.Nykjaer A, Lee R, Teng KK, & Jansen P (2004). Sortilin is essential for proNGF- induced neuronal cell death. Nature, 427(February), 15–20. 10.1038/nature02289. [DOI] [PubMed] [Google Scholar]
- 12.Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, & Friedman WJ (2006). Interaction of Survival and Death Signaling in Basal Forebrain Neurons: Roles of Neurotrophins and Proneurotrophins. Journal of Neuroscience 26, 7756–7766. 10.1523/JNEUROSCI.1560-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alder J, Fujioka W, Giarratana A, Wissocki J, Thakkar K, Vuong P, Patel B, Chakraborty T, Elsabeh R, Parikh A, Girn HS, Crockett D, & Thakker-Varia S (2015). Genetic and pharmacological intervention of the p75NTR pathway alters morphological and behavioural recovery following traumatic brain injury in mice. Brain Injury. 30, 48–65. 10.3109/02699052.2015.1088963 [DOI] [PubMed] [Google Scholar]
- 14.Delbary-Gossart S, Lee S, Baroni M, Lamarche I, Arnone M, Canolle B, Lin A, Sacramento J, Salegio EA, Castel MN, Delesque-Touchard N, Alam A, Laboudie P, Ferzaz B, Savi P, Herbert JM, Manley GT, Ferguson AR, Bresnahan JC, Bono F, & Beattie MS (2016). A novel inhibitor of p75-neurotrophin receptor improves functional outcomes in two models of traumatic brain injury. Brain 139, 1762–1782. 10.1093/BRAIN/AWW074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montroul LE, Rothbar DE, Kana HD, D’Mell V, Dodso V, Tro CM, Zani JP, Leviso SW, & Friedma WJ (2020). Proneurotrophins Induce Apoptotic Neuronal Death After Controlled Cortical Impact Injury in Adult Mice. ASN Neuro, 12. 10.1177/1759091420930865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dasgupta S, Montroull LE, Pandya MA, Zanin JP, Wang W, Wu Z, & Friedman WJ (2023). Cortical Brain Injury Causes Retrograde Degeneration of Afferent Basal Forebrain Cholinergic Neurons via the p75NTR. Eneuro, 10(8). 10.1523/ENEURO.0067-23.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagendran T, & Taylor AM (2019). Unique Axon-to-Soma Signaling Pathways Mediate Dendritic Spine Loss and Hyper-Excitability Post-axotomy. Frontiers in Cellular Neuroscience, 13, 472505. 10.3389/FNCEL.2019.00431/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rishal I, & Fainzilber M (2013). Axon–soma communication in neuronal injury. Nature Reviews Neuroscience 2013 15:1, 15(1), 32–42. 10.1038/nrn3609 [DOI] [PubMed] [Google Scholar]
- 19.Yong Y, Gamage K, Cheng I, Barford K, Spano A, Winckler B, & Deppmann C (2019). P75NTR and DR6 Regulate Distinct Phases of Axon Degeneration Demarcated by Spheroid Rupture. Journal of Neuroscience, 39(48), 9503–9520. 10.1523/JNEUROSCI.1867-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, & Vincenz C (2001). Characterization of a p75 NTR Apoptotic Signaling Pathway Using a Novel Cellular Model. The Journal of Biological Chemistry. 276(36), 33812–33820. 10.1074/jbc.M010548200 [DOI] [PubMed] [Google Scholar]
- 21.Troy CM, Friedman JE, & Friedman WJ (2002). Mechanisms of p75-mediated Death of Hippocampal Neurons. The Journal of Biological Chemistry 277(37), 34295–34302. 10.1074/jbc.M205167200 [DOI] [PubMed] [Google Scholar]
- 22.Gamarra M, de la Cruz A, Blanco-Urrejola M, & Baleriola J (2021). Local Translation in Nervous System Pathologies. Frontiers in Integrative Neuroscience, 15. 10.3389/FNINT.2021.689208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de León A, Gibon J, & Barker PA (2022). APP Genetic Deficiency Alters Intracellular Ca2+ Homeostasis and Delays Axonal Degeneration in Dorsal Root Ganglion Sensory Neurons. Journal of Neuroscience, 42(35), 6680–6691. 10.1523/JNEUROSCI.0162-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gentleman SM, Graham DI, & Roberts GW (1993). Chapter 16 Molecular pathology of head trauma: altered βAPP metabolism and the aetiology of Alzheimer’s disease. Progress in Brain Research, 96I, 237–246. 10.1016/S0079-6123(08)63270-7 [DOI] [PubMed] [Google Scholar]
- 25.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, & Goldstein LSB (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s diseases. Science, 307(5713), 1282–1288. 10.1126/SCIENCE.1105681/SUPPL_FILE/STOKIN-SOM.PDF [DOI] [PubMed] [Google Scholar]
- 26.Friedman WJ (2000). Neurotrophins Induce Death of Hippocampal Neurons via the p75 Receptor. Journal of Neuroscience, 20(17), 6340–6346. 10.1523/JNEUROSCI.20-17-06340.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee R, Kermani P, Teng K, & Hempsted B (2001). Regulation of Cell Survival by Secreted Proneurotrophins. Science, 294(November), 1945–1948. 10.1126/science.1065057 [DOI] [PubMed] [Google Scholar]
- 28.Kenchappa RS, Chao MV, Barker PA, Teng HK, Hempstead BL, & Carter BD (2006). Ligand-Dependent Cleavage of the P75 Neurotrophin Receptor Is Necessary for NRIF Nuclear Translocation and Apoptosis in Sympathetic Neurons. Neuron, 50, 219–232. 10.1016/j.neuron.2006.03.011 [DOI] [PubMed] [Google Scholar]
- 29.Heerssen HM, Pazyra MF, & Segal RA (2004). Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nature. Neuroscience 7, 596–604. 10.1038/nn1242 [DOI] [PubMed] [Google Scholar]
- 30.Escudero CA, Cabeza C, Moya-Alvarado G, Maloney MT, Flores CM, Wu C, Court FA, Mobley WC, & Bronfman FC (2019). c-Jun N-terminal kinase (JNK)-dependent internalization and Rab5-dependent endocytic sorting mediate long-distance retrograde neuronal death induced by axonal BDNF-p75 signaling. Scientific Reports 9, 6070. 10.1038/s41598-019-42420-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavalli V, Kujala P, Klumperman J, & Goldstein LSB (2005). Sunday Driver links axonal transport to damage signaling. The Journal of Cell Biology, 168(5), 775. 10.1083/JCB.200410136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pathak A, & Carter BD (2017). Retrograde apoptotic signaling by the p75 neurotrophin receptor. Neuronal Signaling, 1(1), 20160007. 10.1042/NS20160007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, Van-Minnen J, Twiss JL, & Fainzilber M (2003). Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron, 40(6), 1095–1104. 10.1016/S0896-6273(03)00770-0 [DOI] [PubMed] [Google Scholar]
- 34.Perry RBT, Doron-Mandel E, Iavnilovitch E, Rishal I, Dagan SY, Tsoory M, Coppola G, McDonald MK, Gomes C, Geschwind DH, Twiss JL, Yaron A, & Fainzilber M (2012). Subcellular knockout of importin β1 perturbs axonal retrograde signaling. Neuron, 75(2), 294–305. 10.1016/J.NEURON.2012.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baleriola J, Walker CA, Jean YY, Crary JF, Troy CM, Nagy PL, & Hengst U (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell, 158(5), 1159–1172. 10.1016/J.CELL.2014.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kar AN, Lee SJ, & Twiss JL (2017). Expanding Axonal Transcriptome Brings New Functions for Axonally Synthesized Proteins in Health and Disease. The Neuroscientist, 24(2), 111–129. https://Doi.Org/10.1177/1073858417712668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kneynsberg A, Collier TJ, Manfredsson FP, & Kanaan M (2017). Quantitative and semi-quantitative measurements of axonal degeneration in tissue and primary neuron cultures. Journal of Neuroscience Methods, 266, 32–41. 10.1016/j.jneumeth.2016.03.004.Quantitative [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu W, Weissmiller AM, White JA, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, Gunawardena S, Ding J, Mobley WC, & Wu C (2016). Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. Journal of Clinical Investigation, 126(5), 1815–1833. 10.1172/JCI82409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, & Nishimoto I (2001). c-Jun N-Terminal Kinase (JNK)-Interacting Protein-1b/Islet-Brain-1 Scaffolds Alzheimer’s Amyloid Precursor Protein with JNK. Journal of Neuroscience, 21(17), 6597–6607. 10.1523/JNEUROSCI.21-17-06597.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fagan AM, Garber M, Barbacid M, Silos-Santiago I, & Holtzman DM (1997). A Role for TrkA during Maturation of Striatal and Basal Forebrain Cholinergic Neurons In Vivo. Journal of Neuroscience, 17(20), 7644–7654. 10.1523/JNEUROSCI.17-20-07644.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward NL, & Hagg T (2000). BDNF is needed for postnatal maturation of basal forebrain and neostriatum cholinergic neurons in vivo. Experimental Neurology, 162(2), 297–310. 10.1006/EXNR.1999.7346 [DOI] [PubMed] [Google Scholar]
- 42.Le AP, & Friedman WJ (2012). Matrix Metalloproteinase-7 Regulates Cleavage of Pro-Nerve Growth Factor and Is Neuroprotective following Kainic Acid-Induced Seizures. Journal of Neuroscience. 32(2), 703–712. 10.1523/JNEUROSCI.4128-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, & Jeon NL (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nature Methods. 2(8), 599–605. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1558906/pdf/nihms-11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Unsain N, Heard KN, Higgins JM, & Barker PA (2014). Production and Isolation of Axons from Sensory Neurons for Biochemical Analysis Using Porous Filters. Journal of Visualized Experiments. JoVE, 89, 51795. 10.3791/51795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kneynsberg Andrew, Combs Benjamin, Christensen Kyle, Morfini Gerardo and Kanaan Nicholas M.. (2017). Axonal Degeneration in Tauopathies: Disease Relevance and Underlying Mechanisms. Frontiers in Neuroscience. 11, 572 https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2017.00572https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2017.00572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pathak A, Stanley EM, Hickman FE, Wallace N, Brewer B, Li D, Gluska S, Perlson E, Fuhrmann S, Akassoglou K, Bronfman F, Casaccia P, Burnette DT, Carter BD. (2018) Retrograde Degenerative Signaling Mediated by the p75 Neurotrophin Receptor Requires p150s Deacetylation by Axonal HDAC1. Developmental Cell. 46(3):376–387.e7. doi: 10.1016/j.devcel.2018.07.001.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metwally E, Al-Abbadi HA, Hussain T, Murtaza G, Abdellatif AM, & Ahmed MF (2023). Calpain signaling: from biology to therapeutic opportunities in neurodegenerative disorders. Frontiers in Veterinary Science. 10. https://www.frontiersin.org/articles/10.3389/fvets.2023.1235163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, & Meaney DF (2003). Traumatic Axonal Injury Results in Biphasic Calpain Activation and Retrograde Transport Impairment in Mice. Journal of Cerebral Blood Flow & Metabolism. 2003;23(1):34–42. doi: 10.1097/01.WCB.0000035040.10031.B0 [DOI] [PubMed] [Google Scholar]
- 49.Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tešić M, Molina H, Tessier-Lavigne M (2016) Axon Degeneration Gated by Retrograde Activation of Somatic Pro-apoptotic Signaling. Cell. 25;164(5):1031–45. doi: 10.1016/j.cell.2016.01.032. Epub 2016 Feb 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rishal I, Fainzilber M Axon–soma communication in neuronal injury. (2014) Nature Reviews Neuroscience 15, 32–42 . 10.1038/nrn3609 [DOI] [PubMed] [Google Scholar]
- 51.Harris J, Lee H, Vahidi B, Tu C, Cribbs D, Jeon NL, & Cotman C (2007). Fabrication of a Microfluidic Device for the Compartmentalization of Neuron Soma and Axons. JoVE (Journal of Visualized Experiments), 7, e261. 10.3791/261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt E, Clavarino G, Ceppi M, Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nature Methods. 6, 275–277. 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- 53.Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, & Troy CM (2004) Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. Journal of Neuroscience. 24(45):10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium via the PRIDE partner repository with the dataset identifier PXD043846 and 10.6019/PXD043846. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.