

Abstract
Background
Spontaneous supratentorial intracerebral hemorrhage is the deadliest form of stroke with mortality rates over 50%. Currently, no sufficiently effective treatment to improve both mortality and functional outcome rates exists. However, it seems that minimally invasive surgery, especially endoscopic surgery, might be beneficial in improving survival and functional outcome rates, yet large confirmatory studies thereof are lacking. The aim of this trial is to compare whether early minimally invasive endoscopic surgery leads to improved functional outcome rates compared to the best medical treatment.
Methods
This is a prospective, parallel-arm, outcome assessor blinded multicenter trial across Switzerland. Endoscopic surgery will be compared to the best medical treatment in a 1:1 randomization over a total time of 12 months. The primary outcome is defined as improved functional outcome (mRS < 3) after 6 months; secondary outcomes include mortality and morbidity rates as well as patient reported outcomes and the temporal evolution of serum biomarkers for brain damage.
Discussion
Currently, large, randomized trials assessing the role and potential effect of early endoscopic surgery in intracerebral hemorrhage are lacking. Potential practical and methodological issues faced in this trial are patient enrollment, adherence to the hematoma evacuation technique used, potential patient cross-over, and the adaptive Bayesian statistical design. Nonetheless, this trial would be among the first to research the effects of early minimally invasive endoscopic surgery for SSICH and can provide class I evidence for future treatment options in intracerebral hemorrhage.
Trial registration
ClinicalTrials.gov NCT05681988. Registered on January 3, 2023.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13063-024-08534-7.
Keywords: Minimally invasive surgery, Endoscopic surgery, Intracerebral hemorrhage, Functional outcome, Patient reported outcome measures, Bayesian design, Study protocol
Administrative information
Note: the numbers in curly brackets in this protocol refer to SPIRIT checklist item numbers. The order of the items has been modified to group similar items (see http://www.equator-network.org/reporting-guidelines/spirit-2013-statement-defining-standard-protocol-items-for-clinical-trials/).
| Title {1} | Early minimally invasive image-guided endoscopic evacuation of intracerebral hemorrhage (EMINENT-ICH): a randomized controlled trial |
| Trial registration {2a and 2b} |
Registered in ClinicalTrials.gov, identifier NCT05681988 SNCTP registration number: SNCTP000005643 |
| Protocol version {3} | Version 2.0, dated 05.12.2023 |
| Funding {4} | Swiss National Science Foundation (SNSF); Project Number 213616 |
| Author details {5a} |
Tim Jonas Hallenberger1,2, M.D., Urs Fischer2,3, M.D., Leo Hermann Bonati4, M.D., Gilles Dutilh5, Ph.D., Rosine Mucklow6*, MSc, Andrea Sarti Vogt1*, Claudia Boeni-Eckstein1*, Andrea Cardia7, M.D., Gerrit A. Schubert8, M.D., Phillipe Bijlenga9, M.D. Ph.D., Mahmoud Messerer10, M.D., Andreas Raabe11, M.D., Kevin Akeret12, M.D. Ph.D., Christian Zweifel2,13, M.D., Jens Kuhle3,14, M.D. Ph.D., Alex Alfieri15,16, M.D., Jean-Yves Fournier17, M.D. Ph.D., Javier Fandino18, M.D., Isabel Charlotte Hostettler19, M.D. Ph.D., Ulf Christoph Schneider20, M.D., Raphael Guzman1,2,14, M.D. and Jehuda Soleman1,2, M.D 1Department of Neurosurgery, University Hospital Basel, Spitalstrasse 21, CH-4031 Basel, Switzerland 2Faculty of Medicine, University of Basel, Klingelbergstrasse 61, CH-4056 Basel, Switzerland 3Department of Neurology, University Hospital Basel and University of Basel, Petersgraben 4, CH-4031 Basel, Switzerland 4Reha Rheinfelden, Salinenstrasse 98, CH-4310 Rheinfelden, Switzerland 5Department of Clinical Research, Division of Statistics, University Hospital Basel, Spitalstrasse 12, CH-4031 Basel, Switzerland 6Buxtorf Quality Services, Traubenweg 4; CH-4123 Allschwil, Switzerland 7Service of Neurosurgery, Neurocenter of the Southern Switzerland, Regional Hospital of Lugano, Ente Ospedaliero Cantonale (EOC), Via Tesserete 46, CH-6900 Lugano, Switzerland 8Department of Neurosurgery, Kantonsspital Aarau, Tellstrasse 25, CH-5001 Aarau, Switzerland 9Department of Neurosurgery, University Hospital Geneva, Rue Gabrielle-Perret-Gentil 4, CH-1211 Geneva, Switzerland 10Department of Neurosurgery, University Hospital Lausanne (CHUV), Rue du Bugnon 46, CH-1011 Lausanne, Switzerland 11Department of Neurosurgery, University Hospital Bern, Freiburgerstrasse 10, CH-3010 Bern, Switzerland 12Department of Neurosurgery, Clinical Neuroscience Center University Hospital and University of Zürich, Raemistrasse 100, CH-8091 Zürich, Switzerland 13Neurosurgical Unit, Kantonsspital Graubünden, Loestrasse 170, CH-7000 Chur, Switzerland 14Multiple Sclerosis Centre and Research Center for Clinical Neuroimmunology and Neuroscience (RC2NB), Departments of Biomedicine and Clinical Research, University Hospital Basel and University of Basel, Hebelstrasse 4, CH-4031 Basel, Switzerland 15Department of Neurosurgery, Kantonsspital Winterthur, Brunngasse 30, CH-8400 Winterthur, Switzerland 16Faculty of Biomedical Sciences, Università della Svizzera Italiana (USI), Via Giuseppe Buffi 13, CH-6900 Lugano, Switzerland 17Department of Neurosurgery, Hospital of Valais, Avenue Grand-Champsec 80, CH-1951 Sion, Switzerland 18Department of Neurosurgery, Hirslanden Klinik Zürich, CH-8008 Zürich, Switzerland 19Department of Neurosurgery, Kantonsspital St. Gallen, CH-9007 St. Gallen, Switzerland 20Department of Neurosurgery, Kantonsspital Luzern, Spitalstrasse 16, CH-6000 Lucerne, Switzerland *Contributed PRO Content |
| Name and contact information for the trial sponsor {5b} |
Prof. Dr. med. Raphael Guzman Professor and Chair of Neurosurgery Department of Neurosurgery, University Hospital Basel Spitalstrasse 21, 4031 Basel Raphael.guzman@usb.ch |
| Role of sponsor {5c} |
The sponsor was involved in the conception, design, and organization of the present study and will be involved in the study conduct and the decision for publication. The funding body had no role in the design of the study, in collection, analysis, interpretation, or writing and publishing of the results. |
Introduction
Background and rationale {6a}
Spontaneous supratentorial intracerebral hemorrhages (SSICH) are the second most common form of stroke, accounting for roughly 9–27% of all strokes and affecting more than 5 million people worldwide annually (approx. 2500 cases in Switzerland annually) [1]. Mortality rates are high, ranging from 40 to 45% [2]. Patients surviving a SSICH often have poor health-related quality of life (HRQoL) and serious neurological deficits, resulting in great burden for them, their relatives, and the social system [3–5].
Primary brain injury in SSICH occurs due to intra-axial bleeding, causing mass effect and direct destruction of brain tissue [6]. Secondary mechanisms of brain injury are accredited to the local decay of hemoglobin, causing further brain damage due to its toxicity, and to delayed brain edema causing mass effect [7]. As such, the hematoma volume plays a vital role, as larger hematoma volumes contribute to more brain damage and mass effect and consequently lead to poorer outcome [8].
Treatment options for SSICH primarily aim to either stop enlargement of the hematoma volume or remove it altogether [9]. Current treatment consists of either best medical treatment (a combination of medical blood pressure control, intensive care, and prevention of secondary complications, short BMT), considered to be the current gold standard, or open surgical hematoma evacuation. Both treatment options showed similarly low rates of both good functional outcome and patient survival (Class of Recommendation 2a, Level of Evidence B) [9, 10].
Surgical treatment options for SSICH can be divided into conventional craniotomy (CC) and minimally invasive surgery (MIS), including endoscopic surgery (ES) and stereotactic aspiration (SA) [9, 11]. The Surgical Treatment for Intracerebral Haemorrhage (STICH) I and II trials failed to show significant superiority of open surgical removal of SSICH with CC compared to BMT for improved functional outcome or survival rates [12, 13]. MIS on the other hand seems to be a promising alternative compared to BMT and CC. Systematic reviews with meta-analysis suggested that MIS leads to markedly improved survival and favorable outcome rates compared to BMT, yet the most promising MIS technique remains elusive [14, 15].
The MISTIE (minimally invasive surgery plus rt-PA [alteplase] in intracerebral haemorrhage evacuation) I, II, and III trials assessed the potential superiority of minimally invasive hematoma removal using SA (stereotactic insertion of a catheter into the hematoma cavity, repetitive irrigation of the blood clot using thrombolytic agents and subsequent hematoma drainage) compared to BMT [16–18]. MISTIE II demonstrated efficacy in reducing hematoma volume, clinical safety, and feasibility. MISTIE III evaluated good functional outcome after 1 year (modified Rankin Scale (mRS) ≤ 3 points) showing no significant difference between SA and BMT, while SA significantly reduced the all-cause mortality throughout the study period [17].
Recently, ES emerged as a safe and effective treatment option of SSICH [14, 19–23]. Meta-analyses showed improved survival and favorable outcome rates after ES, compared to BMT. Further, compared to SA, more rapid hematoma evacuation and compared to CC, lower morbidity rates were seen [14, 19, 20, 22]. These results could be confirmed in a meta-analysis, where ES was compared to BMT as a single comparator, which showed significantly improved favorable outcome and survival rates (p = 0.02 and 0.01 respectively) [23]. Despite these promising findings, to date, large confirmatory randomized controlled trials assessing the superiority of ES over BMT in regards of functional outcome improvement in SSICH are largely lacking [10, 24].
Furthermore, it remains unclear when to best evacuate the hematoma. A subgroup analysis of MISTIE III showed that patients receiving treatment within 36 h after symptom onset profited more than those with later treatment onset (> 36 h) [18]. Likewise, a meta-analysis reports a 2.8 times greater likelihood of achieving functional independence if patients received hematoma evacuation within 24 h after symptom onset [19]. Delayed treatment onset reduces the rate of favorable outcome by 5% per hour lost [25]. These findings suggest that early surgical hematoma evacuation is vital for the improvement of functional outcome and survival in SSICH. Ultra-early hematoma evacuation (< 7 h), however, was associated with higher mortality rates and therefore, it seems that the optimal treatment window lies within the first 6–24 h after bleeding onset [26, 27]. To this date, confirmatory trials analyzing this specific treatment window with ES are lacking.
Lastly, patient reported outcome measures (PROMs) and cognitive outcomes in ES compared to BMT are currently underreported (if reported at all) [28]. Within the frame of the EMINENT-ICH trial, these aspects will be evaluated in a standardized fashion.
The present study thus aims to demonstrate efficacy of ES as add-on therapy to BMT (henceforth simply referred to as ES) versus BMT alone in improving functional outcome and reducing death and dependency among patients with SSICH in a randomized controlled fashion. We further aim to contribute to the ongoing understanding of secondary neuronal damage involved in SSICH and their response to early hematoma evacuation, which could lead to novel insights and possibly novel treatment modalities.
Objectives {7}
The null hypothesis (H0) describes no difference in functional outcome rates of early minimally invasive image-guided endoscopic evacuation additionally to BMT for SSICH compared to BMT alone.
The alternative hypothesis (H1) describes a difference (either improved or worsened [two-sided]) in functional outcome rates of early minimally invasive image-guided endoscopic evacuation additionally to BMT for SSICH compared to BMT alone.
The primary objective is to show superiority of early minimally invasive image-guided hematoma evacuation additionally to BMT compared to BMT alone in functional outcome rates at 6 months after intervention in patients with SSICH.
Secondary objectives are:
To show superior survival rates of patients in the ES arm
To study patient reported quality of life after treatment for SSICH at different time points (3 and 6 months after intervention)
To study patient satisfaction with the outcome after treatment for SSICH at different time points (3 months and 6 months after intervention)
To study cognitive outcome in patients after treatment for SSICH at different time points (3 months and 6 months after intervention)
To study the morbidity rates of patients in both treatment arms
To study the efficacy of ES in reducing the hematoma volume
To study the change in focal neurological deficits exhibited by the patients after treatment
To study the temporal evolution of serum biomarkers (neurofilament light-chain subunit (NfL), glial fibrillary acidic protein (GFAP), S100 calcium-binding protein B (S100B), IL-1α and β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, and TNF-α) and their change in relation to early changes of hematoma volume
Trial design {8}
This is a national, multicenter, parallel-arm, outcome assessor blinded, randomized controlled trial within the stroke units and stroke centers of the Swiss Stroke Registry in a superiority framework. The allocation rate is 1:1.
Methods: participants, interventions, and outcomes
Study setting {9}
The EMINENT-ICH trial will be conducted in 13 stroke centers and stroke units across all regions of Switzerland. Academic hospitals involved are the University Hospitals of Basel, Bern, Zurich, Geneva, and Lausanne as well as cantonal clinics of Aarau, Lucerne, Winterthur, Sion, Lugano, St. Gallen, Zurich (Hirslanden), and Chur.
Eligibility criteria {10}
The study population consists of patients with SSICH defined by the following eligibility criteria.
Inclusion criteria:
Patient age ≥ 18 and < 85
SSICH, defined as the sudden occurrence of bleeding into the lobar parenchyma and/or into the basal ganglia and/or thalamus that may extend into the ventricles confirmed by imaging
SSICH volume ≥ 20 mL < 100 mL (measured using the formula )
- A focal neurological deficit consisting of either:
- ◦ Clinically relevant hemiparesis (≥ 4 motor points on the NIHSS for facial palsy, motoric upper and lower extremities combined)
- ◦ Clinically relevant motor or sensory aphasia (≥ 2 points on the NIHSS)
- ◦ Clinically relevant hemi-inattention (formerly neglect, 2 points on the NIHSS)
- ◦ Decreased level of consciousness (GCS ≤ 13)
Presenting GCS 5–15 (in intubated patients GCS assessment will be performed after Rutledge et al.) [29]. If deemed impossible, the last pre-intubation GCS will be used
Endoscopic hematoma evacuation can be initiated within 24 h after the patient was last seen well/symptom onset
Informed consent of patient (only for patients able to consent)
Exclusion criteria:
SSICH due to known or suspected structural abnormality in the brain (e.g., vascular malformation, aneurysm, AVM, brain tumor) and/or brain trauma and/or hemorrhagic conversion of an ischemic infarction
Multiple simultaneous intracranial hemorrhages (e.g., multifocal ICH, cSDH, aSDH, SAH)
Infratentorial hemorrhage or midbrain extension/involvement of the hemorrhage
Coagulation disorder (including anticoagulation) with an INR of > 1.5 which cannot be pharmacologically reverted until the planned time of evacuation
Positive history of current pregnancy or breast-feeding in premenopausal women
-
Relevant disability prior to SSICH (mRS > 2)
Any comorbid disease or condition expected to compromise survival or ability to complete follow-up assessments through 180 days (e.g., end-stage tumor disease)
Who will take informed consent? {26a}
A trained member of the study team or a trained resident/attending neurosurgeon, in case no member of the study team is available, will explain to each participant the nature of the study, its purpose, the procedures involved, the expected duration, the potential risks and benefits, and any discomfort the study may entail. Each participant will be informed that the participation in the study is voluntary, that the participant may withdraw from the study at any time, and that withdrawal of consent will not affect the participant’s subsequent medical treatment.
The participants will be informed that their medical records may be examined by authorized individuals other than their treating physician.
All participants will be provided with an informed consent form describing the study and providing sufficient information for the participants to make an informed decision about the participation in the study. Enough time needs to be given to the participant to decide whether to participate or not, even in the setting of an emergency. However, since rapid evacuation is a vital asset of this study, we will limit this timeframe depending on the acuteness of the individual situation. The formal consent of a participant using the approved consent form will be obtained before the participant is submitted to any study procedure aside necessary routine examinations aiding to evaluate eligibility.
The consent form will be signed, and a copy of the signed informed consent will be given to the study participants. The consent form will be retained as part of the study records and the process will be documented in the electronic patient file. All further study procedures will be commenced after the participants consented to participate in this study.
In case the patient is unable to consent due to impaired consciousness (i.e., due to the hematoma), an independent physician (who is not participating in/trained for the trial) will be asked to confirm whether the interests of the patient are preserved by participating in the study or not. If the interests of the patient are found to be preserved, the patient will be included in the study and an informed consent form for independent physicians will be completed and filed. As soon as the patient regains the ability to consent, she/he will be retrospectively informed about the study. The patient will then retrospectively be asked to provide informed consent to further participate in the study. In case a patient remains unable to consent or becomes permanently unable to consent, a legal guardian or relative will be informed about the study and will be asked to provide retrospective consent as proxy for the patient.
Additional consent provisions for collection and use of participant data and biological specimens {26b}
A separate consent is asked for potential subsequent use of collected data for further studies.
Interventions
Explanation for the choice of comparators {6b}
Best medical treatment [10], which is considered the current gold standard treatment of SSICH, acts as comparator in this randomized controlled trial. Thus, the intervention will be compared to the current gold standard treatment.
Intervention description {11a}
The intervention group will first receive BMT (as described below) upon admission and early minimally invasive image-guided endoscopic hematoma evacuation as an add-on therapy to BMT. Surgery will be performed within 6–24 h after SSICH symptom onset.
The initial cCT scan will be used for planning of the neuro-navigation. The entry point and trajectory to the hematoma will be determined on a routine presurgical BrainLab® neuro-navigation planning station (BrainLab®, Munich, Germany) or an equivalent neuro-navigation planning station (e.g., Medtronic Stealth planning station, Fig. 1A). The planned trajectory represents the shortest approach from the surface of the brain to the hematoma, ideally in adequate distance from functional (eloquent) areas of the brain. Surgery will be performed in an emergency operating theater or a hybrid operation theater equipped with intraoperative CT (in hybrid OR), neuro-navigation, and neuro-endoscopy. Neuro-navigation will be used to mark the skin incision and the exact location of the entry burr hole (Fig. 1B). The burr hole will be drilled and a transparent trocar (ViewSite Brain Access System®, VycorMedical™, USA or equivalent) will be used as a working channel for the endoscope and the suction device (regular suction device, Artemis®, Apollo®, or other suction devices, Fig. 1C and D). The position and progress of the trocar towards the hematoma cavity will be monitored with neuro-navigation. The endoscope (LOTTA® system, Karl Storz Endoscopes, Germany; Minop®, BBraun, Tuttlingen, Germany or equivalent) will be inserted into the trocar and tracked using neuro-navigation. Using the pre-planned trajectory, the hematoma will be entered. Using continuous suction and irrigation, the hematoma will be aspirated and wash out (Fig. 1E). Under visual control using the endoscope, the hematoma cavity will be continuously monitored and active bleeding areas will be irrigated or coagulated using the endoscopic coagulation device and Floseal®. After the hematoma cavity has been cleared, a final inspection under endoscopic visualization will be carried out (Fig. 1F). Thereafter, the wound is closed in standard neurosurgical fashion [30]. Directly after surgery, while still intubated and sedated, patients will be transferred for a cCT in order to assess adequate hematoma evacuation. In a hybrid OR setting, the cCT will be performed directly in the operating theater. If hematoma removal is found to be insufficient by the treating neurosurgeon, the patient will return to the operating theater for further hematoma aspiration (using the same approach). After surgery, patients will be monitored and cared for in an intensive care/stroke unit. The patient will then be treated, according to the current guidelines for BMT in SSICH [10].
Fig. 1.
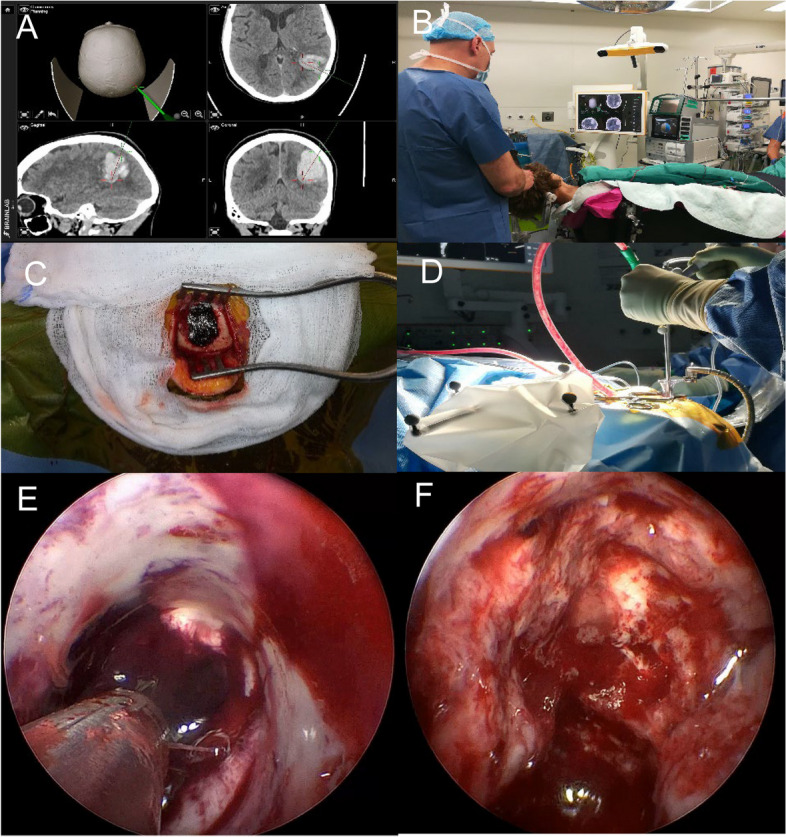
Depiction of the key aspects of the surgical techniques. A Navigation of a lobar hemorrhage; B OR set-up for the proposed intervention; C double burr hole approach; D hematoma evacuation trough the Vycor trocar; E view inside the Vycor trocar showing residual blood trough the transparent sheath; F evacuated hematoma cavity
The control group will receive the current gold standard treatment for SSICH according to the guidelines (BMT) [10]. This involves strict blood pressure control (SBP < 140 mmHg), if needed with intravenous or intraarterial blood pressure lowering agents, reversal of anticoagulation if applicable, intensive care surveillance and nursing on a ICU or stroke unit, control of seizures as well as glucose levels as needed and neurointensive monitoring if deemed necessary [10].
Criteria for discontinuing or modifying allocated interventions {11b}
There are no criteria foreseeing a change in treatment allocation or discontinuation.
Strategies to improve adherence to interventions {11c}
All involved personnel will be trained for the procedures and conduct of the protocol, its interventions, and visits. A detailed standardized presentation of the technique and all critical steps of the surgery including imaging, setup of image guidance and endoscopic equipment, as well as the exact process of aspirating the clot will be provided to all involved surgeons by a proctored workshop (wet lab) and by standard operating procedures (SOPs). Additionally, we published a technical note describing the technique with a video illustrating the steps of the surgery [30]. Regular training will be conducted to train new surgeons and to reinforce the skills needed in the already trained surgeons. Videos and illustrative materials including SOPs for all critical processes of the study will be provided to the involved personnel.
Furthermore, with the help of patient and public involvement (PPI), we designed this trial as pragmatic as possible together with affected patients, caregivers, and patient organization representatives (EUPATI-CH), meaning that all visits and procedures were meticulously evaluated for factors decreasing adherence of both patients and physicians (PPI activity plan can be found in the supplemental material). With this, we are convinced that we will be able to maintain high adherence to the study procedures with all involved study personnel.
Relevant concomitant care permitted or prohibited during the trial {11d}
All additional treatment deemed necessary by the treating physician to address potential complications of SSICH are allowed. This includes antibiotics for any kind of infection (pulmonary, wound infection, etc.), antipsychotics in case of delirium, pain medication when needed, gastric acid inhibitors as addendum to pain medication, reversal of anticoagulation but also re-surgery or secondary surgery if deemed necessary. All occurrences of such treatment will be documented within the case report form (CRF)/electronic CRF (eCRF).
Prohibited concomitant care includes any other primary surgical hematoma removal other than endoscopic surgery.
Oral anticoagulation at admission will be pharmacologically reverted according to the respective center guidelines and according to the current guidelines for ICH management.
Heparin and low molecular weight heparin will be reverted with protamine.
Vitamin K antagonists (VKA) will be reverted with 4-factor prothrombin complex concentrate and/or vitamin K.
Rivaroxaban, apixaban, edoxaban, and fondaparinux (DOACs) will be reverted with 4-factor prothrombin complex concentrate.
Dabigatran (DOAC) will be reverted with idarucizumab.
Anticoagulation is restarted if no new bleeding is demonstrated in the postoperative CT scan at the 3rd postoperative day. DOACs and VKA will be resumed on the 3rd or 4th postoperative day (depending on the patients GFR, if the GFR is > 30 we restart on the 2nd day after surgery or if the GFR is < 30 we restart on the 3rd day after surgery) if the patient is clinically stable and no new bleeding is seen on the postoperative CT scan.
Antiplatelet drugs (aspirin/clopidogrel/prasugrel or ticagrelor) will be handled as follows:
Aspirin will be discontinued if used as primary prophylaxis and reinstated at the 3rd postoperative day. If aspirin is used as secondary prophylaxis, no discontinuation is required.
Clopidogrel will be discontinued before surgery and pharmacologically reverted, if necessary, with platelet concentrate or desmopressin and then reinstated at the 3rd postoperative day.
Prasugrel will be discontinued before surgery and pharmacologically reverted, if necessary, with platelet concentrate and then reinstated at the 3rd postoperative day.
Ticagrelor will be discontinued before surgery and then reinstated at the 3rd postoperative day.
Provisions for post-trial care {30}
An insurance compensating for any harm directly caused by the intervention was instated. No further compensation is foreseen; post-trial care will be conducted according to the current treatment guidelines [10].
Outcomes {12}
Primary outcome:
Good functional outcome 6 months after treatment, measured by the mRS [31]. Good functional outcome is defined as a mRS of ≤ 3 points and will be assessed as binary outcome (yes/no, final value) at 6 months after treatment. The cut-off for good functional outcome is chosen at a mRS score of 3 points, as this reflects the turning point for a patient being able to live a partially self-dependent life or to live a severely disabled life. In this context, a mRS score of 3 points reflects the ability to walk unassisted and care for one’s own bodily needs despite being moderately dependent on assistance, while a mRS score of 4 points describes a patient who is not able to walk and needs assistance with all daily activities and thus marks a severe loss of patient autonomy.
Secondary outcomes:
The mortality rate as measured by death of a participant (binary outcome (yes/no), final value) at 6 and 12 months after intervention.
- Patient reported outcome measures at 3 and 6 months after intervention, those being:
- ◦ Patient and caregiver quality of life as assessed by the PROMIS® Scale v1.2—Global Health questionnaire (continuous variable, final value)
- ◦ Patient satisfaction as assessed by a short survey on a scale of 1–5 (continuous variable, final value, supplemental material)
- ◦ Patient cognitive outcome as assessed by the MOCA® Test (continuous variable, final value)
- The morbidity rate, meaning occurrence of:
- ◦ Ischemic stroke
- ◦ Recurrent SSICH (defined as any radiologically confirmed increase in hematoma volume postoperative/follow-up in the same hematoma cavity that is either asymptomatic or associated with a worsening of the focal-neurological deficit by ≥ 4 points on the NIHSS and/or a decrease in consciousness by ≥ 2 points on the GCS occurring within 30 days after the treatment onset)
- ◦ Epileptic seizures requiring antiepileptic treatment
- ◦ Surgical site infection (intervention group only)
- ◦ Any need for open neurosurgical procedures
- ◦ Life-threatening infections (i.e., pneumonia, urinary tract infection)
- ◦ Any other not defined complication that prolongs the hospital stay and/or leads to further treatment not envisaged in the original treatment plan
The occurrence of any of these events 6 and 12 months after intervention (binary variable (occurrence/no occurrence), final value, proportion).
The change of focal neurological deficit measured by the NIHSS, from baseline to 6 months after intervention as a continuous variable (continuous variable, change from baseline).
The change of disability measured by the mRS, from baseline to 6 and 12 months after intervention as a continuous variable (change from baseline, so-called mRS shift analysis).
The time to intervention, defined as the period from symptom onset/last seen well to start of surgery (start surgical measures, i.e., positioning of patient) or start of medical treatment (admission of first treatment of BMT) (continuous variable, time to event).
The temporal evolution of serum levels of the prespecified biomarkers as continuous variable from start to 6 months after intervention (continuous variable, change from baseline).
The total time spent on the intensive care unit (ICU)/stroke unit as a continuous variable from the first admission to the ICU/stroke unit to discharge from ICU/stroke unit at 7 days after intervention/discharge (continuous variable, final value).
The total time spent in intubation measured in minutes from the start of intubation to extubation as specified in the anesthesiology report at 7 days after intervention/discharge (continuous variable, final value).
Outcomes/measurements applying to the intervention group only:
The proportion of satisfactory hematoma volume reduced to the target volume (goal ≤ 15% of its initial volume). The hematoma volume will be measured on serial cranial computer tomography (cCT) and the difference between the volume of the cCT used for surgery and the cCT directly after surgery will be calculated. The hematoma volume on the pre-operative cCT will be calculated using the formula during screening and secondarily validated using the volumetric function of the navigation software [32]. Hematoma on directly postoperative images will be calculated using the volumetric function of the navigation software. The hematoma volume reduction rate will be a binary variable (achieved reduction < 15%/did not achieve reduction < 15%, final value).
The relative (percentage) reduction or increase of hematoma volume from baseline admission cCT to postoperative cCT directly after surgery as a continuous variable (final value).
Participant timeline {13}
The study schedule consists of 7 study visits (V1–V7), which follow the regular in- and outpatient visit schedule for patients with SSICH. There will be one follow-up telephone interview (visit 7), where the patients are contacted to see whether complications have arisen and whether the reintegration into daily has been possible. All other imaging studies, study visits, the neurological and the clinical examination are part of the daily clinical routine for SSICH, while the study specific examinations will consist of assessing the quality of life, patient satisfaction, and acquiring blood samples. Blood samples will consist of collecting an aliquot of approximately 10 ml blood serum at each indicated visit. The PROMs quality of life and patient satisfaction will be assessed by providing a paper-form questionnaire to the patients and their relatives. If patients are not able to complete the questionnaire, relatives will be asked to provide data. Figure 2 summarizes the participant timeline. Hereafter follows a short description of the procedures per visit.
Fig. 2.

Example template of recommended content for the schedule of enrollment, interventions, and assessments. *mRS assessed blinded by telephone
-
V1: Study inclusion, baseline assessment and treatment: up to 24 h after symptom onset
Study eligibility is confirmed and written informed consent obtained. After informed consent was given, randomization to the intervention or the control group will be conducted by trained personnel through SecuTrial®.
A clinical examination is performed including vital signs (resting blood pressure and heart rate, height, and weight), NIHSS, GCS, and mRS. Blood sampling for biomarkers will be obtained. Patient baseline data is acquired.
Endoscopic hematoma evacuation must be initiated within 24 h according to the procedure described. Patients randomized to the control group will receive standard medical care.
-
V2: Day 1 assessment: 24 ± 6 h after start of treatment
Clinical examination is performed as detailed for V1 except for body weight and height. All patients (control and intervention group) will receive a CT scan to assess the evolution of the hematoma volume (recurrent hemorrhage or postoperative hematoma reduction in case of the intervention group).
-
V3: Day 3 assessment: 72 ± 12 h after treatment
Clinical examination is performed as detailed for V1 except for body weight and height. Blood sampling for biomarkers will be obtained.
-
V4: Day 7 assessment: 7 ± 1 days after treatment or at hospital discharge
Clinical examination is performed as detailed for V1 except for body weight and height.
-
V5: Month 3 assessment: 3 months ± 14 days after treatment
Clinical examination is performed as detailed for V1 except for body weight and height and mRS. A MOCA® Test is performed. A questionnaire regarding patient satisfaction will be filled out whenever possible through a relative or otherwise together with a member of the study team. Patients and their relatives will be asked to fill out a questionnaire together with a member of the study team regarding quality of life. Patients are telephonically contacted by a study-team member (blinded to the allocation) to assess the mRS.
-
V6: Month 6 assessment: 6 months ± 7 weeks after treatment
Clinical examination is performed as detailed for V1 except for body weight and height and mRS. Blood sampling for biomarkers will be obtained. A cCT will be performed for all patients (control and intervention group) to evaluate possible rebleeding, hematoma resorption, and the shape of the hematoma cavity. A MOCA® Test is performed. A questionnaire regarding patient satisfaction will be filled out whenever possible through a relative or otherwise together with a member of the study team. Patients and their relatives will be asked to fill out a questionnaire together with a member of the study team regarding the quality of life. Patients are telephonically contacted by a study-team member (blinded to the allocation) to assess the mRS.
-
V7: Month 12 assessment: 12 months ± 12 weeks after treatment
A telephone interview is conducted assessing potential morbidities of the patients and reintegration into daily life 1 year after intervention. Furthermore, the mRS is assessed as detailed in visits 5 and 6.
Sample size {14}
The sample size estimation is based upon the results from Hallenberger et al., Yao et al., and Sondag et al.’s systematic reviews with meta-analyses [15, 20, 23].
In Hallenberger et al., favorable outcome, defined as mRS 0–3, Barthel Index ≥ 70, Glasgow Outcome Scale 4–5, or an Activity of Daily Living score 1–3, 6 months after treatment was assessed as primary outcome. They found a cumulative relative risk (RR) of having a favorable outcome of 1.93 [1.12;3.33] (p = 0.02) in favor for ES compared to BMT with insignificant heterogeneity (0%, p = 0.92) [23].
Yao et al. reported all-cause mortality as primary outcome while poor functional outcome (mRS 4–6, Glasgow Outcome Scale 1–3, or corresponding clinical presentation) was a secondary outcome. They found a RR of having poor outcome of 0.78 [0.70;0.87] (p < 0.001) with an insignificant heterogeneity (0%, p = 0.60) in favor of ES. The reciprocal value of this would correspond to a RR for favorable outcome of 1.28 in favor of ES [20].
Sondag et al. compared all surgical treatment (craniotomy, craniopuncture, stereotactic aspiration, MISTIE, and ES) to BMT in RCTs regarding favorable functional outcome and death. Favorable functional outcome was defined as good outcome, described as mRS 0–3, Glasgow Outcome Scale 4 and 5, BI ≥ 60 and an extended Glasgow Outcome scale of 5–8 points or, if none of these scores was reported, according to the definition of favorable outcome defined by the authors of the included studies. If available, the outcome assessed at 6-month follow-up was assessed; if not available the 3- or 12-month follow-up was assessed together with the 6-month outcome for the meta-analysis. A subgroup analysis comparing MIS (stereotactic aspiration, MISTIE, and ES) versus BMT alone showed an RR of 1.47 to achieve favorable outcome in favor of MIS [15].
Combined, the RRs result in an overall RR of 1.56 or roughly RR of 1.6 (equaling an odds ratio of 4) as effect size for the intervention. For the effect size of the control group, we analyzed favorable outcome rates (mRS 0–3 after 3 months) based on BMT data from 2020 to 2022 extracted from the Swiss Stroke Registry. This rate of favorable outcome was approximately 65%. However, all patients, even those with minimal bleeding and minimal impairment alongside patients with bleedings of > 100 mL and very poor outcome, are included in the Swiss Stroke Registry, potentially diluting the functional outcome in favor of good outcomes rather than worse outcomes. In consideration of that aspect, a plausible assumption for the proportion of positive outcomes in the control arm would be 50% (0.5).
Based on these assumptions, the sample sizes needed for achieving “compelling evidence,” as described by Schönbrodt and Wagenmakers, was calculated [33]. Compelling evidence is defined as finding a Bayes factor (BF) that points with a certain strength at favoring the null hypothesis or the alternative hypothesis, i.e., hypothetical experiments, expressing how much more likely the data were generated from a model where both study arms have the same probability of a positive outcome, compared to a model where there is a difference in the probability of a positive outcome. In this study, we aim to achieve a BF of 10 in favor of either hypothesis, often interpreted to represent strong evidence. Of all the strengths when using BF as opposed to p values for inference, two stand out:
A BF may quantify evidence against or in favor of the null hypothesis.
BFs may be monitored continuously as the data is generated and collected without the need of correction schemes as needed in a classical null-hypothesis significance testing procedure.
Both strengths described above are exploited in the analysis performed here: data at any moment can be assessed to determine whether enough evidence is present to allow to decide in favor of either the alternative or the null hypothesis. As such, it does not make sense to additionally calculate a p value at the finally achieved n as such a p value would be biased towards “significance.” However, as a sensitivity analysis, we plan to perform one classic/frequentist null-hypothesis significance test at the same time as the first Bayesian analysis of the data (after 40 patients have been included in both arms). The pre-planned hypothesis test is not biased.
We performed a Bayesian logistic regression, following the methods described by Bartos and Wagenmakers to calculate an approximate Bayes factor [34]. In particular, the regression was performed using a so-called moment prior on the beta-weight of the study arms [35]. The moment prior was chosen such that it reflects our prior assumption that the true odds ratio of achieving a positive outcome under treatment vs under control is 4.
The BF is regularly calculated until either a pre-set threshold of evidence is achieved in favor of either of the hypotheses, or when the maximum sample size is reached. For the current study, the chosen total sample size threshold as 200 patients, and the evidence threshold at a Bayes factor of 10. If these thresholds are reached without clear evidence, a new cost–benefit analysis of continuing to collect data will be performed. In our simulations, we accounted for the expected evidence up to 160 patients per arm.
Figure 3 shows the results of the Bayesian logistic analysis. The figure shows results for various assumptions including (1) the true proportion of positive outcomes under BMT (the different panels) and (2) the true odds ratios in favor of ES (different colored lines). The lines indicate how the probability that trial has finished (reaching the threshold of BF = 10) increases as the number of included patients increases (x-axis). Solid lines indicate the proportion of trials that are finished because the BF strongly favors the alternative hypothesis; dashed lines indicate the proportion of trials where the analysis concludes in strong favor of the null hypothesis.
Fig. 3.

Projected sample size estimations assuming different proportions of favorable outcome in the BMT group
Comparing different panels, we see that evidence is collected quicker when the total proportion of favorable outcome for BMT is closer to 50%. Comparing different colored lines, we see that evidence is increasing quicker when the true odds ratio is larger. When inspecting the dashed lines, we see that when the null hypothesis is true (odds ratio = 1), evidence in favor of the null hypothesis increases almost as strongly as the evidence for the alternative increases when the true odds ratio is 4.
Systematic reviews as well as data analyzed from the Swiss Stroke Registry over the last 2 years suggest that a plausible assumption for the proportion of positive outcomes in the control arm is 50% (0.5) (Fig. 3B) with our best estimate of the odds ratio, OR = 4, indicated by the magenta line. Inspecting this line indicates that after collecting about 80 patients per study arm, our trial has a probability (or “Bayesian power”) of 90% to be finished. Note that after 40 patients per arm, there is already a probability of over 50% for the trial to finish. Further, the figure shows that it is very unlikely that the trial would take more than 100 patients per arm. Based on these results, we assume that we will be able to stop collecting data before reaching 80 patients per arm (total of 160 patients) to reach a BF of 10.
Recruitment {15}
The recruitment procedure will take place at either the emergency department or the stroke center of the participating sites. Participants will be recruited by trained personnel in the form of consecutive ongoing enrollment in daily practice as well as recruitment through referring family physicians or peripheral hospitals to the emergency department.
Initial screening according to the eligibility criteria will be performed upon admission. Some of the eligibility criteria are evaluated in daily practice and must be applied before informed consent is given as this is necessary to determine the underlying disease and assess study eligibility.
By close collaboration with the respective departments of neurology in the centers, we anticipate screening almost every patient arriving with an SSICH at one of our centers. Thus, together with trained personnel assessing patients, we are convinced that we will be able to enroll most eligible patients. We tested feasibility of recruitment in our pilot trial (NCT048005177) over a duration of one and a half years, where we screened every patient with any kind of parenchymal hemorrhage at our main center (University Hospital of Basel). Based on this screening process, the inclusion rate of eligible patients was 71.4%.
To estimate the enrollment rates of our collaborating centers, we analyzed the caseload for every stroke unit or stroke center across Switzerland based on reported cases within the Swiss Stroke Registry over 2 years (2020–2022, 838 cases of any size, etiology, and location). However, SSICHs are underreported in most centers as we as insufficiently documented, so the true number of SSICHs is probably much higher for every center than actually registered. We extrapolated our own inclusion rate to the other centers to arrive at their approximate presumed patients per year. On that basis, we contacted the participating centers, who confirmed that based on their own analysis of their caseload per year, the presumed enrollment numbers we estimated were valid.
Based on this calculation, all centers taken together should be able to recruit the intended maximum of 200 patients during a planned recruitment phase of 48 months.
Assignment of interventions: allocation
Sequence generation {16a}
The computer-generated allocation sequence is programmed in our CDMS SecuTrial®, which uses a random-block-size algorithms for a randomization in a 1:1 fashion. Randomization is stratified for center. It will also include a standard minimization algorithm which will ensure that the treatment groups are balanced.
Concealment mechanism {16b}
The allocation to the study arm will be generated by the randomization algorithm implemented into our CDMS (SecuTrial®) and disclosed to the treating team electronically. Thus, the treatment allocation is concealed to the study team until randomization is complete.
Implementation {16c}
The allocation sequence will be implemented in our CDMS (SecuTrial®) by the data manager and the trial statistician. Trained personnel will enroll the patients and implement the randomization procedure within 24 h after symptom onset trough SecuTrial®. Randomization results are directly disclosed to the treating team.
Assignment of interventions: blinding
Who will be blinded {17a}
Since this is a surgical trial, blinding is not possible for primary care providers and patients. However, the laboratory personnel analyzing the biomarkers, the primary outcome assessors as well as the statistician will be blinded as these are not directly involved in the patient care. Laboratory personnel will be blinded as only encoded material will be processed where patients are identified by their patient ID. Likewise, the study statistician responsible for the primary analysis will receive an allocation-blinded copy of the data set. Furthermore, we will conduct blinded outcome assessment at 3- and 6-month follow-up trough study personnel blinded to the allocation of the patient in form of a standardized telephone assessment [36]. This form of assessment is validated, highly comparable with face-to-face evaluation as well as short and easy to implement [37].
Procedure for unblinding if needed {17b}
Since no patient or primary care provider blinding is possible (due to the study design including a surgical treatment arm), no emergency unblinding is foreseen.
Data collection and management
Plans for assessment and collection of outcomes {18a}
All data like scores (NIHSS, GCS, mRS, MOCA®, PROMIS®, patient satisfaction), patient history, and surgical baseline data will be collected or observed in daily clinical routine and transcribed to a paper CRF referring to the patient’s study ID. Information on radiological imaging or laboratory values will be extracted from the respective clinic information system. Data from radiological images will be directly transcribed to the paper CRF. Vital parameters (BP, HR, temperature, weight, height) will be directly transferred from the electronic patient file located in the hospital information system to the paper CRF. Laboratory values (encoded) will be provided per mail to the investigators by our laboratories (Clinical Neuroimmunology and Brain Ischemia) and transcribed to the paper CRF. The blinded outcome assessment will be directly entered into the eCRF by the assessor. All study personnel will be trained in the respective study procedures.
Study data will be transferred from the paper CRF to an eCRF captured via an online Clinical Data Management System (secuTrial®), based at the IT department of the University Hospital Basel. The data collected is entered into the study eCRF. Additional storage capacity can be added as needed. For each enrolled study participant, an individual eCRF is maintained.
Source data for this study will be all data collected within the paper CRF, namely scores of the mRS, NIHSS, GCS, PROMIS®, MOCA®, patient satisfaction, patient and surgical baseline data, morbidities, and findings of the clinical examination except for the vital sings and radiological assessments as they are documented in daily practice in the electronic patient file or radiology program respectively and can be found there for monitoring purposes.
The questionnaires used in this study are the Montreal Cognitive Assessment (MOCA), the PROMIS Scale v1.2 Global Health, and a questionnaire for patient satisfaction derived in our patient and public involvement meetings (all in German, French, Italian, and English). Both, MOCA® and PROMIS® are well established and validated questionnaires to assess cognition and quality of life respectively and were deemed the most effective and patient friendly by our PPI representatives [38, 39]. Further, the time point and method of assessment was discussed and decided upon with the PPI representatives. The questionnaire regarding patient satisfaction was created together with the PPI representatives by first collecting the most important aspects of patient satisfaction, which were then condensed to the 5 most important questions. This questionnaire is not validated in clinical trials, but a reflection of important PROMs and a product of intense collaboration with PPIs (the questionnaire can be found in the supplemental materials). Questionnaires for patient satisfaction will, whenever possible, be assessed by relatives of the patients. The PROMIS® and MOCA® will be assessed by trained study personnel.
Biomarkers
Blood sampling will consist of collecting an aliquot of approximately 10 mL blood serum at visits 1, 3, and 6. The additional blood samples will be taken, if possible, during daily routine while the patient will be in the hospital (V1 and 3). For the follow-up visit (V6), an additional puncture will be necessary to obtain the blood samples. The blood samples will be labeled with the patient study ID and sent for processing, afterwards they will be sent to the Department of Biomedicine at the University Hospital Basel for biomarker analysis. The biomarkers analyzed are validated and can be used to monitor brain injury in SSICH [40–42]. NfL are highly specific structural proteins of neurons released by the disruption of axonal membranes and associated with the severity, activity, and treatment response in neuronal injury [43–45]. S100B is a non-specific marker for neuronal injury and the disruption of the blood–brain barrier which correlates with the infarction volume and clinical outcome in ischemic stroke and SSICH [46, 47]. GFAP is an intermediate filament protein expressed by astrocytes. GFAP levels are specifically higher among patients with SSICH than among patients with ischemic stroke when measured early after symptom onset [48].
We will use a novel assay to measure NfL which was developed in the Laboratory of Clinical Neuroimmunology at the University Hospital Basel. The test is based on single-molecule array (SIMOA) technology for digital immunoassays, using the capture monoclonal antibody (mAB) 47:3 and the biotinylated detector mAB 2:1 from UmanDiagnostics (Umeå, Sweden), transferred onto the SIMOA platform. SIMOA has been shown to be more sensitive than conventional ELISA or ECL based assays to quantify NfL in serum [49].
S100B and GFAP will be measured using a standardized immunoassay from frozen plasma samples via electrochemiluminescence (Roche Cobas Elecsys, Roche, Switzerland).
Interleukins (IL-1α and β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p70, TNF-alpha) will be measured with electrochemiluminescent (ECL) immunosorbent assays (Meso Scale Discovery, MD 20877, USA). This platform allows for testing of multiple biomarkers in limited sample volumes.
The blood samples and aliquots are stored in an appropriate cooling system in a restricted area only accessible to the authorized personnel and handled under appropriate conditions.
Plans to promote participant retention and complete follow-up {18b}
We designed this trial together with affected patients, caregivers, and patient organization representatives in the scope of PPI as pragmatic as possible, meaning that all visits and procedures were meticulously evaluated. With this, we are convinced that patients will remain in the trial, as only little, if any at all, expenditure will be expected, and retention therefore is maintained.
Data management {19}
Every assessor will be trained on how to complete the CRFs and the conduct of the questionnaires. Paper CRFs are transferred by the study personnel into our CDMS; hardcopies are securely stored. An audit trail will maintain a record of initial entries and any changes made. The eCRF will be implemented by the data management group at the Department of Clinical Research (DKF) of the University Hospital Basel using the CDMS secuTrial®. The CDMS runs on a server maintained by the IT department of the University Hospital Basel. Data managers at the DKF Basel will implement validation rules and range checks in the CDMS. When data gets saved in an eCRF, it will be validated for completeness and discrepancies (i.e., using mandatory fields and active missing value handle). Data will be reviewed by the responsible investigator as well as an independent monitor. The monitor will raise queries using the query management system in secuTrial®. Designated investigators must respond to the query and confirm or correct the corresponding data. Thereafter, the monitor can close the query.
For quality assurance, the sponsor, the Ethics Committee, or an independent trial monitor may visit the research site. Direct access to the source data and all study related files is granted on such occasions. All involved parties keep the participant data strictly confidential.
Confidentiality {27}
A unique patient identifier (i.e., patients study ID) will be used to identify patients and a password protected list will be maintained for traceability. The patient ID is generated when the patient is enrolled in the CDMS secuTrial® by consecutive automatic numbering (i.e., USB-NNN with USB referring to the center and NNN being a tree digit number). Only the PI or delegated study personnel will have access to the encoding key. Enrollment and screening logs will be filed to ensure traceability. The principal investigator, the study team, and, if applicable, delegates at the site will be authorized to do eCRF entries. The CDMS is accessible via a standard browser on devices with internet connection. Password protection and user-right management ensures that only authorized study investigators, study team, monitors, data managers, and local authorities (if necessary) will have access to the data during and after the study. User administration and user training are performed by the DKF according to predefined processes. An audit trail will maintain a record of initial entries and any changes made; time and date of entry; and username of person authorizing entry or change. Participant’s identification logs will be stored as a password protected word files and saved on protected servers of the respective study site. On CRFs and other study specific documents, participants are only identified by the patient’s study ID derived by secuTrial®. Completed paper CRFs will be kept locked in a drawer at the respective study site with access only to a very limited number of study team members. ECRFs will be secured in secuTrial®, only accessible by the study teams at the respective sites. The investigators and the sponsor endorse responsibility that nobody else will have access to the confidential data and they guarantee protection against dissemination. Trial and participant data will be handled with uttermost discretion and is only accessible to authorized personnel who require the data to fulfill their duties within the scope of the study.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use {33}
Biological material in this study (i.e., blood samples) is not identified by participant name but by the patient’s study ID. Biological material is stored in an appropriate cooling system in a restricted area only accessible to the authorized personnel and handled under appropriate conditions. The material will be sent coded by the patient’s study ID to the Department of Biomedical Research, University Hospital Basel. The results of laboratory analysis will be provided by mail to the study investigators and will not show in the hospital electronic record system. A back-up copy will be kept at the archives of the hospital’s laboratory. The material’s location is tracked by a laboratory log which is kept in the respective investigator site file (ISF).
Statistical methods
Statistical methods for primary and secondary outcomes {20a}
The primary analyses are performed following the intention-to-treat principle. Additionally, per-protocol analysis will be performed for sensitivity analysis. We study the ratio of positive outcomes in both study arms by performing a Bayesian logistic regression after every 40 additional patients, using the method described by Bartos and Wagenmakers using a moment prior for the regression weight [34, 35]. Data collection (and the periodical analysis of these data) will continue until a Bayes factor of 10 (or 1/10) is achieved. The odds ratio and its 95% credible interval will be reported as based on an uninformed prior. Covariates ICH volume, presence of intraventricular bleeding, do-not-resuscitate orders, location of hematoma, and center will be included in a Bayesian logistic regression model. For continuous variables, a Bayesian regression with normal error term is used, for time-to-event outcomes Bayesian cox-regression is used, and for binary variables Bayesian logistic regression is used. A complete mRS shift analysis will be calculated. Statistical analysis is performed with R version 4.2.1 or higher [50].
Interim analyses {21b}
The data may be inspected any time, but we plan to perform the primary analyses after every 40th patient (20 per arm) and will stop data collection if the BF, in favor of either the null or the alternative hypothesis, is over 10 or the total sample size has reached 200 patients.
Methods for additional analyses (e.g., subgroup analyses) {20b}
At the time point of the first Bayesian analysis (after a total of 40 patients have been collected in each study arm), we perform a frequentist logistic regression analysis in parallel, calculating a p value for the null hypothesis. In the ES arm, several explorative analyses are performed, studying the relation between hematoma location, hematoma size, and treatment outcome. For parameters measured over time, figures are created illustrating the development over time at both the patient level and group level.
Methods in analysis to handle protocol non-adherence and any statistical methods to handle missing data {20c}
If missing data occurs, we will try to obtain the respective data needed from either the participant, their next of kin, or their treating physician. If acquiring the respective missing data is not possible, we will mark the data as missing. In case of missing laboratory values, we will aim to achieve them; if this proves to be impossible, the data will be marked as missing. As such, missing data is assumed to be missing at random (MAR). In case more than 5% of patients show missing data for the primary outcome across arms, Bayesian multiple imputation using the whole data set is performed. Intention-to-treat and per-protocol analysis are planned. Outcome data collected for dropouts will consist of all study data collected to the last visit. If substantial deviations of the analysis as outlined in these sections are needed for whatever reason, the protocol will be amended. All deviations of the analysis from the protocol or from the detailed analysis plan will be listed and justified in a separate section of the final statistical report.
Plans to give access to the full protocol, participant-level data, and statistical code {31c}
The full protocol as well as the statistical code is available upon request with the principal investigator. Regarding participant level data, the Swiss Ordinance of 20 September 2013 on Clinical Trials in Human Research (Clinical Trials Ordinance, ClinO) ordains that handling of health-related personal data in connection with a clinical trial must be restricted to those persons who require this data to fulfill their duties. The Federal Act of 30 September 2011 on research involving Human Beings (Human Research Act, HRA) requires an informed consent of the person, the legal representative, or next of kin for reuse of personal data. Researchers who wish to reuse the data will have to obtain authorization of the responsible ethics committee as ordained in the Ordinance of 20 September 2013 on Human Research with the exception of clinical Trials (Human Research Ordinance, HRO). The definition of “further use” in the HRO in particular includes the storage in databases and making accessible or available or transferring of health-related personal data already collected. A transfer of research data to a data repository would therefore violate Swiss national law. Although some repositories allow storing data non-public and restricting data access to specific users, they do not support restricted access to a subset of stored variables. Since the responsible ethics committee can exclude certain variables from reuse in a specific project application, restriction to subsets is a required feature.
The only way to circumvent the HRA would be an anonymization of the data, i.e., the masking or deleting of all items which, when combined, would enable to identify a patient without disproportionate effort. Health-related personal data are considered correctly coded in accordance with the HRA if, from the perspective of a person who lacks access to the key, they are to be considered anonymized (HRO Art. 26). Since the investigator must retain all documents required for the identification and follow-up of participants for at least 10 years after the completion of a clinical trial (ClinO Art. 45), data may appear anonymized to third parties, while in fact the HRA considers them coded and forbids reuse without the persons’ consents. Additionally, an anonymization would make the combination of these data with routine data and data from other clinical trials impossible, which is the most important and most common reason for reuse of data in clinical research. Furthermore, it would counteract the efforts of the government-funded Swiss Personalized Health Network (SPHN), which has given priority to effective exchange of patient data.
Instead of transferring the data on a repository, the DKF of the University of Basel will act as an independent data access committee (DAC) and store the data at time of publication on secure servers, maintained and backed-up by the IT department of the University Hospital Basel. Researchers who wish to reuse data may submit a project synopsis to the DKF.
The DKF as independent DAC will answer formal request of applicants, review and submit the project documents to the responsible ethics committee(s), and (upon approval) securely transfer the requested data to the applicants.
Metadata describing the type, size, and content of the datasets will be shared along with the study protocol and case report forms on the public repository dataverse.harvard.edu. Additionally, the CRFs will be uploaded on medical-data-models.org and all variables will be annotated by their Unified Medical Language System Concept Unique Identifier (UMLS CUI) to improve findability for other clinicians. With the metadata registered on a public repository together with a reference to the DAC, this procedure will adhere to the FAIR principles to the best of the legal limitations for clinical research in Switzerland.
The results of this study will be published in a peer-reviewed medical journal, independent of the results and the statistical code is made available upon request. A data sharing statement referring researchers to the DKF for data access will be contained in the study protocol and publication.
Oversight and monitoring
Composition of the coordinating center and trial steering committee {5d}
No formal trial steering committee was put together; however, the Study Coordinating Center (Clinical Neurosurgical Research Centre, Basel) acts as the Trial Management Committee and has regular meetings (monthly) to discuss problems and/or difficulties within the study and their solutions as well as possible improvements. More urgent questions are discussed as needed, e.g., by mail contact or over the phone by the respective parties.
Data management, quality management, monitoring, and statistical support are provided by the DKF.
Composition of the data monitoring committee, its role and reporting structure {21a}
Monitoring duties will be provided by the DKF at the University Hospital Basel. We will apply a centralized monitoring approach, meaning the study sites will be initiated with a site initiation visit and then routinely visited after the first 2–4 patients before switching to centralized monitoring trough secuTrial® with predefined ranges for further on-site monitoring if deemed necessary. All informed consents, source data, e.g., CRF, eCRF, and laboratory results, the trial master file, and the investigator site files will be monitored. All source data and all documents will be made accessible to monitors and questions will be answered during monitoring through the study staff. A data and safety monitoring board (DSMB), independent from the investigator team and consist of an expert in the field, a statistician, and PPI representative, will be assessing safety, adherence, and efficacy of the study and advise on continuation of the study. The DSMB charter regulates the direct affairs of the DSMB and can be requested with the Study Coordinating Center. An independent Clinical Outcome Event Committee will re-evaluate all SAEs occurring in the study and confirm or correct the assessment of the investigators at the hand of the DSMB. The CEC will send the report of their assessment to the DSMB. An assessment will occur whenever 40 patients have been included.
Adverse event reporting and harms {22}
The reporting and follow-up of serious adverse events (SAEs) is regulated by the law in Switzerland; thus, there are clear guidelines to follow. All SAEs are documented and reported immediately to the sponsor-investigator of the study.
Exemptions from expedited reporting may be possible if the SAE is either a clear result of the underlying disease or well-known. These exemptions for the EMINENT-ICH trial are defined below:
Death
Hemorrhagic transformation to an ischemic stroke
Recurrent ICH
Epileptic seizure
Any serious, life-threatening infection (i.e., surgical site infection, pneumonia, etc.)
Persistent focal neurological impairment
These patients will be followed according to the normal procedures of follow-up in case of (S)AE until the (S)AE stabilizes or resolves. All (S)AEs are evaluated by trained study personnel as soon as the (S)AE occurs. On a regular basis (after every 40th patient included), all (S)AEs are re-evaluated and either confirmed or corrected through a clinical event committee (CEC). The CEC will report its findings at the hands of the DSMB.
If immediate safety and protective measures must be taken during the conduct of the study, the investigator notifies the ethics committee of these measures, and of the circumstances necessitating them, within 7 days.
Frequency and plans for auditing trial conduct {23}
Audits are planned by SwissMedic, the governmental institution overseeing clinical research in Switzerland. No concrete schedule for audit frequencies was proposed or planned. Audits may include verification of all source data, site files, study site inspections, and other study documents.
Plans for communicating important protocol amendments to relevant parties (e.g., trial participants, ethical committees) {25}
Substantial changes to the study setup and study organization, the protocol, and relevant study documents are submitted to the Ethics Committee for approval before implementation. Under emergency circumstances, deviations from the protocol to protect the rights, safety, and well-being of human subjects may proceed without prior approval of the Ethics Committee. Such deviations shall be documented and reported to the Ethics Committee as soon as possible.
Upon regular study termination, the Ethics Committee is notified via BASEC within 90 days. Upon premature study termination or study interruption, the Ethics Committee is notified via BASEC within 15 days.
Dissemination plans {31a}
The documentation accompanying the data will consist out of the Data Record Table (DRT) exported from the CDMS secuTrial®, therefore containing all data collected on paper CRF. It is an Excel file summarizing the questions and variables collected in the eCRF. The Excel file consists out of the following sheets:
Configuration: An overview sheet of the internal project’s name, eCRF version, and which extended features of secuTrial® have been activated.
Form overview: In this sheet, all available forms of the eCRF will be listed and the full visit plan (name, day of visit, type of visit, possible deviations in days) will be shown. For each form, the visits in which it is available (or whether it is visit independent) and the name of the.csv table when exported will be given.
There will be one sheet for each form, containing the names of the form and a.csv table. All variables stored in a table will be listed in separate rows, with the following metadata:
Question and description text as shown in the eCRF, caption of the variable in the eCRF, type of the variable (e.g., text field with maximal number of characters, date, number item with maximal number of digits, radio button or drop down selection with list of available answers, check box, date), name of the variable in the exported.csv tables, and if applicable any additional rules (e.g., mandatory item, optional item, “hide item if” with corresponding conditional)
The metadata variables (e.g., patient ID, visit number, date of last edit, person entering data) and potential details regarding processing and analysis saved with the eCRF data in the exported tables will be specified in an additional Excel file. The patient ID will serve as the persistent identifier. During the conduct of the study, we plan to consult patient organization representatives to discuss possibilities to best disseminate the results of the trial. Thus, the following are only tentative plans. We plan to disseminate the results over (1) our trial website (www.eminent-ich.ch), (2) over the SNSF platform and (3) with the help of a patient organization representatives to the respective patient organizations, (4) by publishing the results open-access in a peer-reviewed journal and (5) social media (linked-in, twitter, etc.).
Discussion
SSICH is the most severe form of stroke lacking sufficiently effective treatment options. Although minimally invasive endoscopic hematoma evacuation has been described to potentially improve functional outcome and survival rates, no large, well-structured confirmatory studies have been conducted. Furthermore, the ideal timing for hematoma evacuation is presumed to be between 6 and 24 h, yet again, this time window has not been researched in large confirmatory trials. Likewise, patient reported outcome measures were largely absent in prior SSICH trials. Lastly, blood serum biomarkers are known as diagnostic and predictive markers in SSICH; however, their natural course in patients receiving hematoma evacuation has not been addressed before. We herein report the protocol of a pragmatic trial, which addresses abovementioned gaps and has an adaptive statistical design to address early endoscopic surgery additional to BMT as potential new treatment option in SSICH. Based on the pathophysiological considerations, the current literature, and our own data, we are convinced that evacuating SSICH in a minimal invasive endoscopic fashion, within 6–24 h and achieving a fast and significant reduction of hematoma volume will result in superiority of good functional outcome and lower morbidity rates compared to BMT.
Although this is a randomized controlled trial, several potential limitations can be expected. Adequate patient enrollment could be a major limitation to this trial, potentially leading to its premature discontinuation. However, we have taken several precautions to mitigate this risk. First, we have meticulously screened all patients with ICH at our main center to evaluate caseload and enrollment ratio. Further, we have screened the national stroke data base to evaluate the SSICH caseload for our partner centers and then approximated overall projected enrollment numbers. With this information, we have developed worst-case and best-case scenarios for enrollment and are adequately prepared. Secondly, we use an adaptive Bayesian statistical approach for the current trial. As previously described, this approach has the advantage of constant, real-time information about the evidence being generated in this trial and thus allows for potential early completion due to our predefined stopping rules and consequently to fewer patients having to be enrolled. Lastly, by using a centralized monitoring approach, we will be informed early if the overall or center specific enrollment rates are not met and be able to adapt respectively.
Further limitations could be imposed by maladherence to our surgical technique. Currently, several techniques for both, open and minimally invasive hematoma evacuation, exist, including conventional craniotomy, stereotactic aspiration, endoscopic surgery, and, most recently, minimally invasive parafalcine surgery as used in the ENRICH trial [51]. Techniques for endoscopic surgery as used in current ongoing trials like EVACUATE (NCT04434807), DIST (NCT05460793), MIND (NCT03342664), NESICH (NCT05539859), and EMINENT-ICH (NCT05681988) all have the same basic technique as proposed by Kellner et al. [52]. However, especially differences regarding the suction devices make each of these techniques unique, which in turn limits generalizability and leads to potential confusion among neurosurgeons. To improve adherence to our technique, we use a normal suction device, making the intervention more pragmatic for daily clinical use as well as potentially more cost-effective. As described by Sondag et al. in the DIST Pilot trial, the learning curve of endoscopic hematoma evacuation can be steep even after very few patients operated [53]. As such, to further improve adherence, we prepared a proctored workshop with a skull and brain model (Synaptive Medical, Toronto, Canada), filled with jam and CT images thereof for neuro-navigation, to train the surgical evacuation. Lastly, we have published our technique for quick reference and have distributed SOPs detailing the surgical process in detail [30]. With these measures, we are confident that we can maintain high adherence rates in the EMINENT-ICH trial.
Not being able to blind patients and investigators is considered as a major limitation of this trial. Due to the nature of our current surgical setting, blinding of patients and investigators becomes virtually impossible while sham surgeries are considered unethical in such a severe disease. We address this issue by providing allocation-blinded outcome assessment at 6 months with telephone interviews trough trained, blinded study personnel. With this, we can mitigate the bias arising from lack of blinding.
Lastly, the potential for cross-over of BMT patients to the surgical arm (i.e., trough craniotomy if considered lifesaving) can pose a significant limitation for the study. The final analysis, however, will incorporate that fact as we perform a per-protocol and intention-to-treat analysis.
Trial status
The current approved protocol is version number 2.0, dated 5th of December 2023. This trial was registered under the number NCT05681988 in trials.gov. As of January 1st, 2024, recruitment has begun at the main site (Basel) with the other sites currently being initiated. The last patient-first visit is estimated for the beginning of December 2028 while the analysis and trial completion is planned for December 2029.
Authors’ contributions {31b}
Supplementary Information
Acknowledgements
We want to thank our project manager, Mrs. Stephanie Andersson-Riva, our trial coordinator Mrs. Birsel Klein-Reesink, and our study nurses Ms. Severine Dominique Miszak and Ms. Monika Pfeffermann for their enormous support in all administrative and coordinating matters. We further want to thank Mrs. Kai-Marie Schimanski for her support in the website set-up. Lastly, our thanks go to Dr. Attill Saemann, who has taken the time to develop our trial logo.
Abbreviations
- AE
Adverse event
- BASEC
Business Administration System for Ethical Committees
- BMT
Best medical treatment
- CC
Conventional craniotomy
- cCT
Cranial computed tomography
- ClinO
Ordinance on Clinical Trials in Human Research
- CRF
Case report form
- DSMB
Data and safety monitoring board
- DOAC
Direct oral anticoagulants
- eCRF
Electronic case report form
- EDC
Electronic data capture
- ES
Endoscopic surgery
- FADP
Federal Act on Data Protection
- FOPH
Federal Office of Public Health
- GCP
Good Clinical Practice
- GCS
Glasgow Coma Scale
- CDMS
Clinical Data Management System
- GFAP
Glial fibrillary acidic protein
- HRA
Human Research Act
- ICH
International Conference on Harmonisation
- ICH
Intracerebral hemorrhage
- ICU
Intensive care unit
- IL
Interleukin
- ISF
Investigator site file
- MAR
Missing at random
- MIPS
Minimally invasive puncture surgery
- MIPS
Minimal invasive parafalcine surgery
- MIS
Minimally invasive surgery
- MOCA
Montreal Cognitive Assessment
- MRI
Magnet resonance imaging
- mRS
Modified Rankin Scale
- NfL
The light-chain neurofilament subunit
- NIHSS
National Institute of Health Stroke Scale
- NOAK
New oral anticoagulants
- OAC
Oral anticoagulants
- PI
Principal investigator
- PPI
Patient and public involvement
- PROMS
Patient reported outcome measures
- RR
Relative risk
- S100B
S100 calcium-binding protein B
- SA
Stereotactic aspiration
- SAE
Serious adverse event
- SIMOA
Single-molecule array
- SIV
Site initiation visit
- SSICH
Spontaneous supratentorial intracerebral hemorrhage
- SCC
Study Coordinating Center
- VKA
Vitamin K antagonists
Authors’ contributions {31b}
RG, JS, LB, UF, and TJH conceived and drafted the study protocol. RG, TJH, UF, and JS acquired funding. All authors will contribute to the implementation and data collection of the study. TJH, AS, CBE, and RM designed the PPI aspects of this study. RG and JK will provide the laboratory material and locations for biomarker analysis. GD will perform statistical analysis. All authors critically revised the protocol before submission to the local ethics committee and prior to submission.
Funding {4}
This study is funded by the Swiss National Science Foundation (SNSF), Project Number 213616. We further received a preparatory grant for patient and public involvement by the SNSF (213916).
Data availability {29}
The plan for data sharing was outlined under Sect. 31c. The Department of Clinical Research will act as an independent data access committee. Any data required to support the protocol can be supplied on request.
Declarations
Consent for publication {32}
Given by all protocol authors.
Competing interests {28}
Dr. Tim Hallenberger discloses that he receives a MD PhD scholarship jointly by the SNSF and the Swiss Academy of Medical Sciences (Grant ID 207030). The remaining authors declare no conflict of interest for the present study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Andrea Sarti Vogt died during the conduct of the study.
References
- 1.Feigin VL, Lawes CMM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2(1):43–53. [DOI] [PubMed] [Google Scholar]
- 2.Krishnamurthi RV, Feigin VL, Forouzanfar MH, Mensah GA, Connor M, Bennett DA, et al. Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet Glob Health. 2013;1(5):e259–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dennis MS. Outcome after brain haemorrhage. Cerebrovasc Dis. 2003;16(Suppl 1):9–13. [DOI] [PubMed] [Google Scholar]
- 4.Qureshi AI, Suri MF, Nasar A, Kirmani JF, Ezzeddine MA, Divani AA, et al. Changes in cost and outcome among US patients with stroke hospitalized in 1990 to 1991 and those hospitalized in 2000 to 2001. Stroke. 2007;38(7):2180–4. [DOI] [PubMed] [Google Scholar]
- 5.Christensen MC, Mayer S, Ferran JM. Quality of life after intracerebral hemorrhage: results of the Factor Seven for Acute Hemorrhagic Stroke (FAST) trial. Stroke. 2009;40(5):1677–82. [DOI] [PubMed] [Google Scholar]
- 6.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5(1):53–63. [DOI] [PubMed] [Google Scholar]
- 7.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96(2):287–93. [DOI] [PubMed] [Google Scholar]
- 8.Hemphill JC 3rd, Bonovich DC, Besmertis L, Manley GT, Johnston SC. The ICH score: a simple, reliable grading scale for intracerebral hemorrhage. Stroke. 2001;32(4):891–7. [DOI] [PubMed] [Google Scholar]
- 9.Cordonnier C, Demchuk A, Ziai W, Anderson CS. Intracerebral haemorrhage: current approaches to acute management. Lancet. 2018;392(10154):1257–68. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg SM, Ziai WC, Cordonnier C, Dowlatshahi D, Francis B, Goldstein JN, et al. 2022 guideline for the management of patients with spontaneous intracerebral hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke. 2022;53(7):e282–361. [DOI] [PubMed] [Google Scholar]
- 11.de Oliveira Manoel AL. Surgery for spontaneous intracerebral hemorrhage. Crit Care. 2020;24(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365(9457):387–97. [DOI] [PubMed] [Google Scholar]
- 13.Mendelow AD, Gregson BA, Rowan EN, Murray GD, Gholkar A, Mitchell PM. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet. 2013;382(9890):397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo G, Pan C, Guo W, Bai S, Nie H, Feng Y, et al. Efficacy and safety of four interventions for spontaneous supratentorial intracerebral hemorrhage: a network meta-analysis. J Neurointerv Surg. 2020;12(6):598–604. [DOI] [PubMed] [Google Scholar]
- 15.Sondag L, Schreuder F, Boogaarts HD, Rovers MM, Vandertop WP, Dammers R, et al. Neurosurgical intervention for supratentorial intracerebral hemorrhage. Ann Neurol. 2020;88(2):239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan T, Zuccarello M, Narayan R, Keyl P, Lane K, Hanley D. Preliminary findings of the minimally-invasive surgery plus rtPA for intracerebral hemorrhage evacuation (MISTIE) clinical trial. Acta Neurochir Suppl. 2008;105:147–51. [DOI] [PubMed] [Google Scholar]
- 17.Hanley DF, Thompson RE, Muschelli J, Rosenblum M, McBee N, Lane K, et al. Safety and efficacy of minimally invasive surgery plus alteplase in intracerebral haemorrhage evacuation (MISTIE): a randomised, controlled, open-label, phase 2 trial. Lancet Neurol. 2016;15(12):1228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanley DF, Thompson RE, Rosenblum M, Yenokyan G, Lane K, McBee N, et al. Efficacy and safety of minimally invasive surgery with thrombolysis in intracerebral haemorrhage evacuation (MISTIE III): a randomised, controlled, open-label, blinded endpoint phase 3 trial. Lancet. 2019;393(10175):1021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scaggiante J, Zhang X, Mocco J, Kellner CP. Minimally invasive surgery for intracerebral hemorrhage. Stroke. 2018;49(11):2612–20. [DOI] [PubMed] [Google Scholar]
- 20.Yao Z, Hu X, You C, He M. Effect and feasibility of endoscopic surgery in spontaneous intracerebral hemorrhage: a systematic review and meta-analysis. World Neurosurg. 2018;113:348-56.e2. [DOI] [PubMed] [Google Scholar]
- 21.Kellner CP, Song R, Pan J, Nistal DA, Scaggiante J, Chartrain AG, et al. Long-term functional outcome following minimally invasive endoscopic intracerebral hemorrhage evacuation. Journal of NeuroInterventional Surgery. 2020;12(5):489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M, Mu F, Su D, Han Q, Guo Z, Chen T. Different surgical interventions for patients with spontaneous supratentorial intracranial hemorrhage: a network meta-analysis. Clin Neurol Neurosurg. 2020;188: 105617. [DOI] [PubMed] [Google Scholar]
- 23.Hallenberger TJ, Guzman R, Bonati LH, Greuter L, Soleman J. Endoscopic surgery for spontaneous supratentorial intracerebral haemorrhage: a systematic review and meta-analysis. Front Neurol. 2022;13:1054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steiner T, Al-Shahi Salman R, Beer R, Christensen H, Cordonnier C, Csiba L, et al. European Stroke Organisation (ESO) guidelines for the management of spontaneous intracerebral hemorrhage. Int J Stroke. 2014;9(7):840–55. [DOI] [PubMed] [Google Scholar]
- 25.Kellner CP, Song R, Ali M, Nistal DA, Samarage M, Dangayach NS, et al. Time to evacuation and functional outcome after minimally invasive endoscopic intracerebral hemorrhage evacuation. Stroke. 2021;52(9):e536–9. [DOI] [PubMed] [Google Scholar]
- 26.Morgenstern LB, Demchuk AM, Kim DH, Frankowski RF, Grotta JC. Rebleeding leads to poor outcome in ultra-early craniotomy for intracerebral hemorrhage. Neurology. 2001;56(10):1294–9. [DOI] [PubMed] [Google Scholar]
- 27.Wang YF, Wu JS, Mao Y, Chen XC, Zhou LF, Zhang Y. The optimal time-window for surgical treatment of spontaneous intracerebral hemorrhage: result of prospective randomized controlled trial of 500 cases. Acta Neurochir Suppl. 2008;105:141–5. [DOI] [PubMed] [Google Scholar]
- 28.Green TL, Newcommon N, Demchuk A. Quality of life and caregiver outcomes following decompressive hemicraniectomy for severe stroke: a narrative literature review. Can J Neurosci Nurs. 2010;32(2):24–33. [PubMed] [Google Scholar]
- 29.Rutledge R, Lentz CW, Fakhry S, Hunt J. Appropriate use of the Glasgow Coma Scale in intubated patients: a linear regression prediction of the Glasgow verbal score from the Glasgow eye and motor scores. J Trauma. 1996;41(3):514–22. [DOI] [PubMed] [Google Scholar]
- 30.Hallenberger TJ, Guzman R, Soleman J. Minimally invasive image-guided endoscopic evacuation of intracerebral haemorrhage: how I do it. Acta Neurochir (Wien). 2022;165(6):1597–602. [DOI] [PubMed] [Google Scholar]
- 31.van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19(5):604–7. [DOI] [PubMed] [Google Scholar]
- 32.Kothari RU, Brott T, Broderick JP, Barsan WG, Sauerbeck LR, Zuccarello M, et al. The ABCs of measuring intracerebral hemorrhage volumes. Stroke. 1996;27(8):1304–5. [DOI] [PubMed] [Google Scholar]
- 33.Schönbrodt FD, Wagenmakers EJ. Bayes factor design analysis: planning for compelling evidence. Psychon Bull Rev. 2018;25(1):128–42. [DOI] [PubMed] [Google Scholar]
- 34.Bartoš F, Wagenmakers E-J. A general approximation to nested Bayes factors with informed priors. Stat. 2023;12(1): e600. [Google Scholar]
- 35.Johnson VE, Rossell D. On the use of non-local prior densities in Bayesian hypothesis tests. Journal of the Royal Statistical Society Series B (Statistical Methodology). 2010;72(2):143–70. [Google Scholar]
- 36.Bruno A, Akinwuntan AE, Lin C, Close B, Davis K, Baute V, et al. Simplified modified Rankin scale questionnaire: reproducibility over the telephone and validation with quality of life. Stroke. 2011;42(8):2276–9. [DOI] [PubMed] [Google Scholar]
- 37.Baggio JA, Santos-Pontelli TE, Cougo-Pinto PT, Camilo M, Silva NF, Antunes P, et al. Validation of a structured interview for telephone assessment of the modified Rankin Scale in Brazilian stroke patients. Cerebrovasc Dis. 2014;38(4):297–301. [DOI] [PubMed] [Google Scholar]
- 38.Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695–9. [DOI] [PubMed] [Google Scholar]
- 39.Salinas J, Sprinkhuizen SM, Ackerson T, Bernhardt J, Davie C, George MG, et al. An international standard set of patient-centered outcome measures after stroke. Stroke. 2016;47(1):180–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunswick AS, Hwang BY, Appelboom G, Hwang RY, Piazza MA, Connolly ES Jr. Serum biomarkers of spontaneous intracerebral hemorrhage induced secondary brain injury. J Neurol Sci. 2012;321(1–2):1–10. [DOI] [PubMed] [Google Scholar]
- 41.Senn R, Elkind MS, Montaner J, Christ-Crain M, Katan M. Potential role of blood biomarkers in the management of nontraumatic intracerebral hemorrhage. Cerebrovasc Dis. 2014;38(6):395–409. [DOI] [PubMed] [Google Scholar]
- 42.Alex Matos Ribeiro J, Fernanda Garcia-Salazar L, Regina Saade-Pacheco C, Shirley Moreira Silva E, Garcia Oliveira S, Flavia Silveira A, et al. Prognostic molecular markers for motor recovery in acute hemorrhagic stroke: a systematic review. Clin Chim Acta. 2021;522:45–60. [DOI] [PubMed] [Google Scholar]
- 43.Byrne LM, Rodrigues FB, Blennow K, Durr A, Leavitt BR, Roos RAC, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: a retrospective cohort analysis. Lancet Neurol. 2017;16(8):601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuhle J, Nourbakhsh B, Grant D, Morant S, Barro C, Yaldizli O, et al. Serum neurofilament is associated with progression of brain atrophy and disability in early MS. Neurology. 2017;88(9):826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barro C, Benkert P, Disanto G, Tsagkas C, Amann M, Naegelin Y, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain. 2018;141(8):2382–91. [DOI] [PubMed] [Google Scholar]
- 46.Foerch C, Singer OC, Neumann-Haefelin T, du Mesnil de Rochemont R, Steinmetz H, Sitzer M. Evaluation of serum S100B as a surrogate marker for long-term outcome and infarct volume in acute middle cerebral artery infarction. Arch Neurol. 2005;62(7):1130–4. [DOI] [PubMed] [Google Scholar]
- 47.Delgado P, Alvarez Sabin J, Santamarina E, Molina CA, Quintana M, Rosell A, et al. Plasma S100B level after acute spontaneous intracerebral hemorrhage. Stroke. 2006;37(11):2837–9. [DOI] [PubMed] [Google Scholar]
- 48.Luger S, Witsch J, Dietz A, Hamann GF, Minnerup J, Schneider H, et al. Glial fibrillary acidic protein serum levels distinguish between intracerebral hemorrhage and cerebral ischemia in the early phase of stroke. Clin Chem. 2017;63(1):377–85. [DOI] [PubMed] [Google Scholar]
- 49.Kuhle J, Barro C, Andreasson U, Derfuss T, Lindberg R, Sandelius A, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med. 2016;54(10):1655–61. [DOI] [PubMed] [Google Scholar]
- 50.RCoreTeam. R: a language and environment for statistical computing. Vienna: R Foundation for artistically Computing; 2022.
- 51.Ratcliff JJ, Hall AJ, Porto E, Saville BR, Lewis RJ, Allen JW, et al. Early Minimally Invasive Removal of Intracerebral Hemorrhage (ENRICH): study protocol for a multi-centered two-arm randomized adaptive trial. Front Neurol. 2023;14:1126958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kellner CP, Chartrain AG, Nistal DA, Scaggiante J, Hom D, Ghatan S, et al. The Stereotactic Intracerebral Hemorrhage Underwater Blood Aspiration (SCUBA) technique for minimally invasive endoscopic intracerebral hemorrhage evacuation. J Neurointerv Surg. 2018;10(8):771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sondag L, Schreuder FHBM, Pegge SAH, Coutinho JM, Dippel DWJ, Janssen PM, et al. Safety and technical efficacy of early minimally invasive endoscopy-guided surgery for intracerebral haemorrhage: the Dutch Intracerebral haemorrhage Surgery Trial pilot study. Acta Neurochir. 2023;165(6):1585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The plan for data sharing was outlined under Sect. 31c. The Department of Clinical Research will act as an independent data access committee. Any data required to support the protocol can be supplied on request.