

Abstract
When gastrointestinal stromal tumour (GIST), the most common form of sarcoma, was first recognized as a distinct pathological entity in the 1990s, patients with advanced-stage disease had a very poor prognosis owing to a lack of effective medical therapies. The discovery of KIT mutations as the first and most prevalent drivers of GIST and the subsequent development of the first KIT tyrosine kinase inhibitor (TKI), imatinib, revolutionized the treatment of patients with this disease. We can now identify the driver mutation in 99% of patients with GIST via molecular diagnostic testing, and therapies have been developed to treat many, but not all, molecular subtypes of the disease. Currently, seven drugs are approved by the FDA for the treatment of advanced-stage GIST (imatinib, sunitinib, regorafenib, ripretinib, avapritinib, larotrectinib and entrectinib), all of which are TKIs. While these agents can be very effective for treating certain GIST subtypes, challenges remain that necessitate new therapeutic approaches. In this Review, we discuss the molecular subtypes of GIST and the evolution of the current treatments, as well as their therapeutic limitations. We also highlight emerging therapeutics that might overcome clinical challenges through novel strategies predicated on the biological features of the distinct GIST molecular subtypes.
ToC Blurb
Gastrointestinal stromal tumour (GIST) is the most common form of sarcoma and has become a paradigm of precision medicine owing to fact that almost all patients harbour one of several known molecule drivers, most of which can be targeted therapeutically. Nevertheless, novel therapeutic strategies are required to overcome the intrinsic resistance of certain subtypes of GIST to existing treatments as well as the acquired resistance that eventually arises in initially sensitive subtypes. This Review describes the biology of GIST, the evolution of the current treatments for this cancer, and the emerging therapeutic agents and approaches that might overcome the remaining clinical challenges.
Introduction
Gastrointestinal stromal tumour (GIST) is the most common type of soft-tissue sarcoma. The annual incidence of GIST ranges from 6–22 cases per million individuals worldwide, with an estimated 3,000–6,000 new diagnoses each year in the USA1,2. GIST has become a paradigm for the development of precision medicine treatment approaches owing to the elucidation of the oncogenic drivers that define different molecular subtypes of the disease3. Nearly 99% of GISTs have an identifiable driver alteration, and the presence of a particular driver imparts distinct molecular and biological features and guides treatment strategies using different approved targeted therapies4 (FIGS. 1 and 2). The vast majority of these driver alterations (~85%) are activating mutations in either one of two closely related members of the type III receptor tyrosine kinase (RTK) family, KIT and PDGFRA (also known as PDGFRα)5–7. Other molecular drivers of GIST include rare gene fusions involving different RTKs, mutations in components of oncogenic signalling pathways that are activated downstream of these RTKs, and loss-of-function alterations affecting subunits of the mitochondrial respiratory complex II, succinate dehydrogenase (SDH)1,8,9. We now have therapeutic approaches to treat the majority of advanced-stage GISTs (FIGS. 1 and 2). However, some of the rarer molecular subtypes still lack effective treatments. Even with the available treatments, important clinical challenges remain. In this Review, we discuss the molecular subtypes and current treatment landscape of GIST, as well as the emerging novel therapies that exploit distinct strategies to target the unique biology and/or molecular features of the different GIST subtypes.
Fig. 1 │. Summary of GIST molecular subtypes.
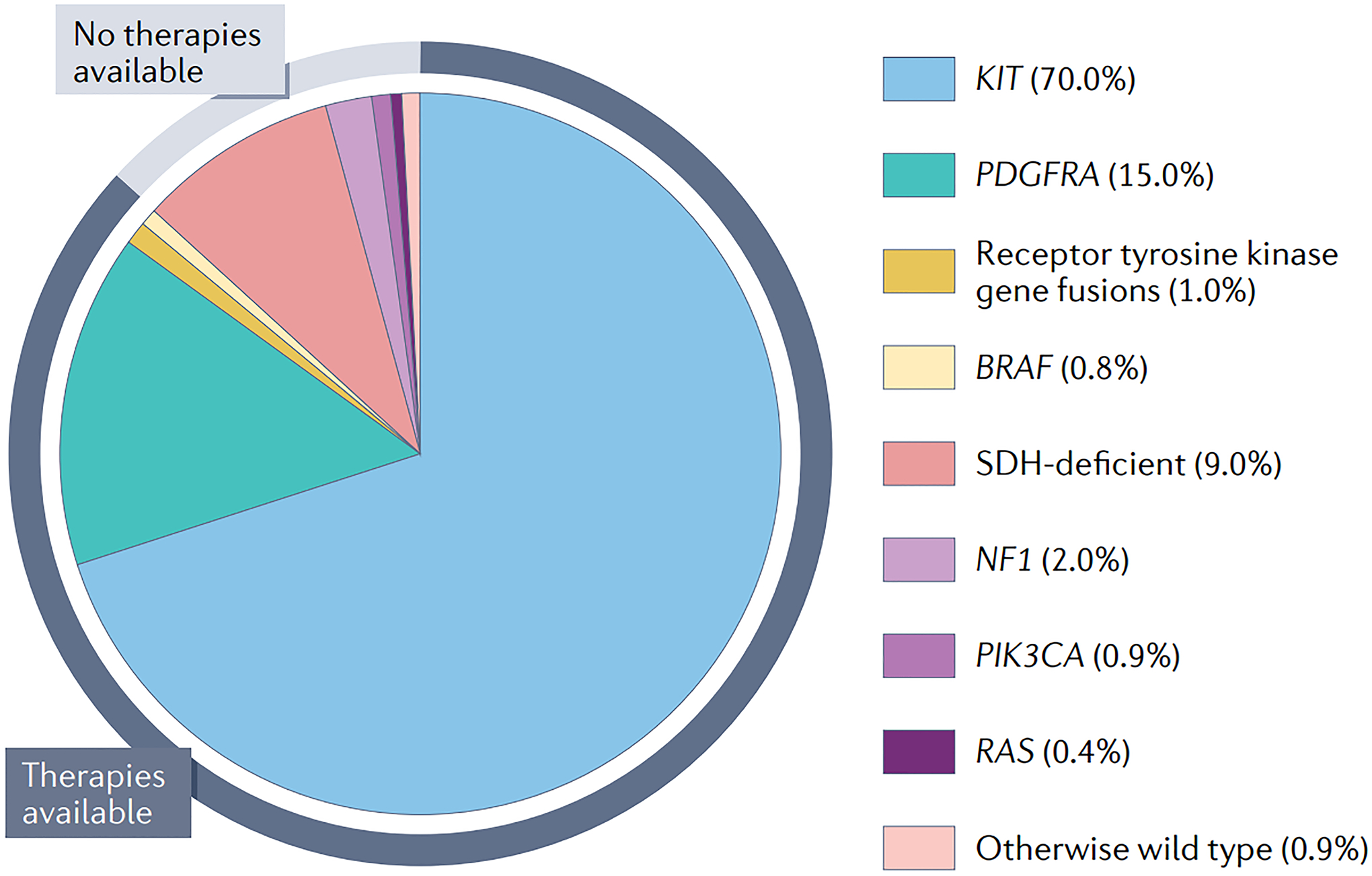
The pie chart indicates the proportion of gastrointestinal stromal tumour (GIST) cases that is driven by each recurrent driver alteration associated with this disease. KIT and PDGFRA mutations account for approximately 85% of GISTs. Effective targeted therapies are now available for patients with GIST harbouring such alterations, as well as those with BRAF mutations or receptor tyrosine kinase gene fusions (predominantly involving FGFR1 or NTRK3), encompassing ~88% of all advanced-stage GISTs (indicated by the black segment of the outer ring). The remaining 12% of GISTs are SDH deficient, NF1, PIK3CA or RAS mutant, or otherwise wild type, and lack effective therapies (indicated by the blue segment of the outer ring).
Fig. 2 │. GIST signalling pathways, drug targets and current systemic therapies.
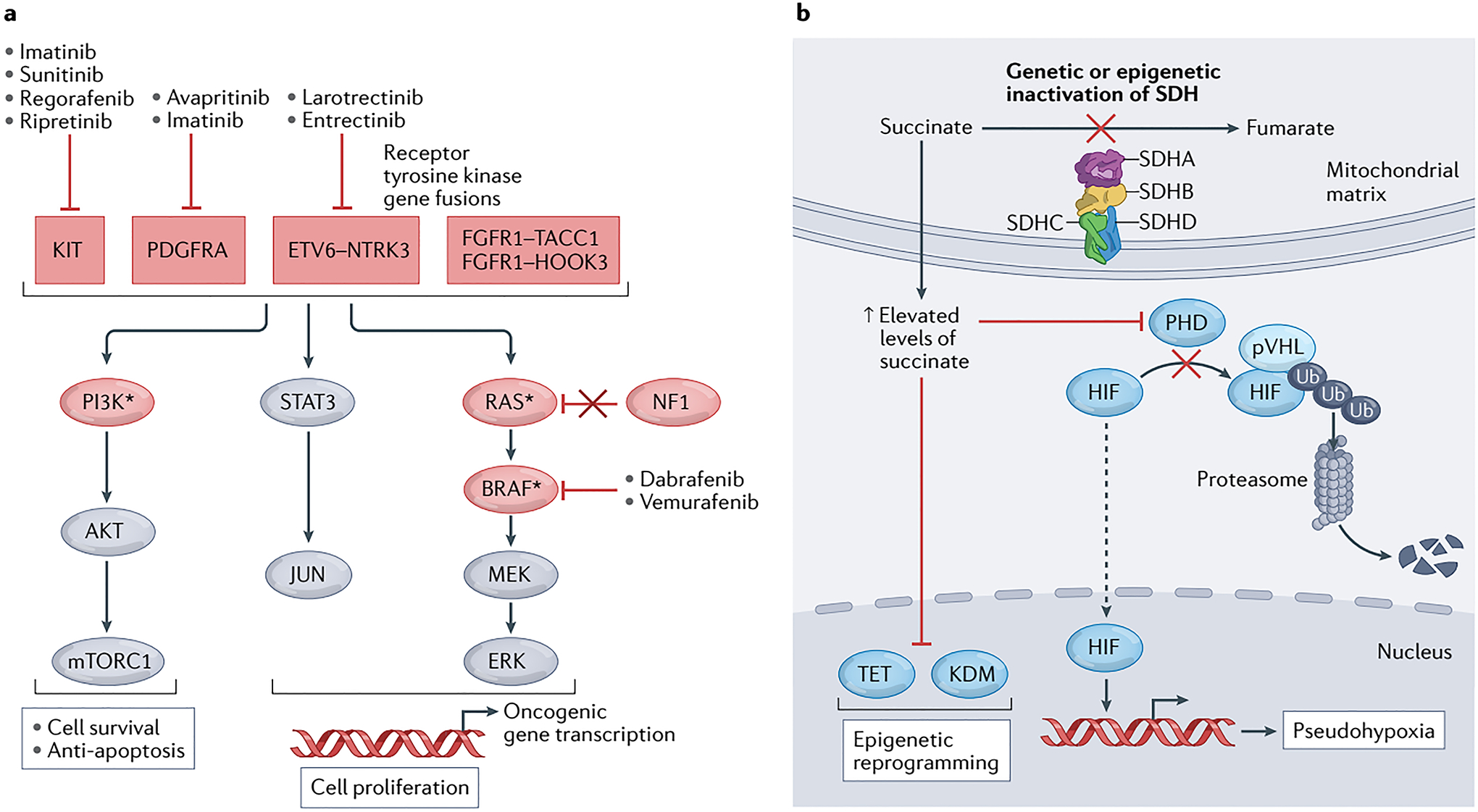
a │ The genetic alterations that drive gastrointestinal stromal tumours (GISTs) generally result in activation of signalling through the MEK–ERK (MAPK), JAK–STAT and PI3K–AKT pathways to prevent apoptosis and drive cell survival and proliferation. The components of these pathways and upstream receptor tyrosine kinases that are recurrently mutated in GIST are shown in red ovals and boxes, respectively; those indicated with an asterisk can also arise as secondary mutations after therapy. Gain-of-function (activating) mutations are found in positive signalling effectors, including KIT, PDGFRA, fusion proteins involving NTRK3 (TRKC) or FGFR1, as well as RAS, PI3Kα or BRAF. Loss-of-function (inactivating) mutations are found in tumour suppressors, such as neurofibromatosis-related protein NF1 (also known as neurofibromin). b │ GIST can also be driven by deficiency of the mitochondrial respiratory complex II, succinate dehydrogenase (SDH), resulting from a genetic mutation in any one of the four SDH subunit genes (SDHA, SDHB, SDHC or SDHD), or more rarely from epigenetic inactivation of SHDC via promoter hypermethylation. Inactivation of the SDH complex results in an accumulation of succinate, which leads to competitive inhibition of α-ketoglutarate-dependent dioxygenases, including those of the hypoxia-inducible factor (HIF)-prolyl hydroxylase domain (PHD), ten-eleven translocation methylcytosine dioxygenase (TET), and lysine-specific histone demethylase (KDM) families. In turn, inhibition of PHDs leads to pseudohypoxia by preventing von Hippel-Lindau disease tumour suppressor (pVHL)-mediated ubiquitination and subsequent proteasomal degradation of HIF, while inhibition of TET and KDM proteins results in increased methylation of DNA and histones, respectively, and thus broad epigenetic reprogramming. Ub, ubiquitin.
Biology of GIST
GISTs can occur throughout the gastrointestinal tract, most frequently arising in the stomach (60–65%) and small intestine (20–35%); however, their specific molecular distribution and biology can differ between anatomical sites1,3,4,8–13 (TABLE 1). Distinct molecular subtypes of GIST are defined broadly by individual driver alterations. The three major molecular subtypes, KIT mutant, PDGFRA mutant or SDH deficient, constitute nearly 95% of all GISTs. Various other rare molecular drivers account for the remaining cases of GIST, with only ~1% of GISTs lacking a known driver (FIG. 1).
Table 1 │.
GIST subtypes and their cellular and molecular features by anatomical location
| Proportion of GIST cases | Anatomical location | GIST subtype | Cell morphology | Cell of origin |
|---|---|---|---|---|
| 60–65% | Proximal stomach | KIT exon 9 or 11 mutant | Spindle | ICC-IM or ICC-MY |
| Distal stomach | KIT exon 8, 9, 11, 13 or 17 mutant | Spindle | ICC-IM or ICC-MY | |
| PDGFRA exon 12, 14 or 18 mutant | Epithelioid | Telocytes | ||
| SDH deficient | Epithelioid | Unknown | ||
| 20–35% | Small intestine | KIT exon 8, 9, 11, 13 or 17 mutant | Spindle | ICC-MY |
| FGFR1 or NTRK3 RTK fusion | Spindle | Unknown | ||
| BRAF mutant | Spindle | ICC or SM | ||
| NF1 mutant | Spindle | ICC-MY | ||
| 3–5% | Colon or rectum | KIT exon 9 or 11 mutant | Spindle | ICC-IM or ICC-MY |
| FGFR1 or NTRK3 RTK fusion | Spindle | Unknown |
GIST, gastrointestinal stromal tumour; ICC, interstitial cells of Cajal; ICC-IM, intramuscular ICC; ICC-MY: myenteric ICC; SDH, succinate dehydrogenase; SM, smooth muscle precursor; RTK, receptor tyrosine kinase.
Molecular subtypes
KIT-mutant GIST.
In 1998, KIT mutations were the first driver mutation to be discovered in GISTs7,14. Occurring in ~70% of GISTs, these gain-of-function mutations are found at only a few locations in the protein, the membrane-proximal extracellular domain (mutations involving exons 8 or 9), the intracellular juxtramembrane domain (JMD; encoded by exon 11) or the kinase domain (exons 13 or 17), and cause constitutive, ligand-independent kinase activity by disrupting auto-inhibitory regions of the RTK15. The most common primary KIT mutations affect the JMD; point mutations or indels (insertions and/or deletions) within KIT exon 11 drive ~60% of all GISTs1,3,4,8–12,16,17. KIT exon 11-mutant GISTs can occur throughout the gastrointestinal tract, from the oesophagus to the rectum, but account for almost all cases arising in the proximal stomach18 (TABLE 1). KIT exon 11-mutant GISTs, especially those with deletion mutations, typically have high mitotic rates and are associated with a high risk of recurrence and metastasis19,20. As discussed further below, GISTs with KIT exon 11 mutations are extremely sensitive to the KIT tyrosine kinase inhibitor (TKI) imatinib and account for the vast majority of patients with metastatic GIST who respond to this agent. The next most common primary KIT mutations involve the membrane-proximal extracellular region encoded by exon 9 (accounting for 9–10% of all GISTs). Almost all KIT exon 9-mutant GISTs arise in the small intestine, colon or rectum, with rare reports of such GISTs arising in the stomach21 (TABLE 1). Finally, mutations in KIT exons 8, 13 or 17 are rare primary drivers, each accounting for ≤1% of GISTs1,3,4,8–12,16,17. These rare subtypes of KIT-mutant GIST most commonly arise in the intestines, approximately two-fold more often than in the stomach22 (TABLE 1).
PDGFRA-mutant GIST.
PDGFRA is an RTK that is highly homologous to KIT, both functionally and structurally. Accordingly, PDGFRA mutations occur in ~15% of all GISTs (FIG. 1) and are similar to those in KIT; however, distinctions between the two have important treatment implications. Like KIT mutations, PDGFRA mutations in GISTs are gain of function, disrupting auto-inhibitory regions of the RTK and thereby resulting in ligand-independent activation5,6. PDGFRA mutations can also be either point mutations or indels, but the mutational frequencies at the various hot spots in PDGFRA are the opposite of those in KIT1,3,4,8–12,16,17. Whereas the majority of KIT mutations affect the JMD, mutations affecting this region of PDGFRA (encoded by PDGFRA exon 12) are rarely seen in GISTs, with a prevalence of approximately 1–2%. Conversely, the most common PDGFRA mutation, found in 9–10% of all primary GISTs, is a D842V point mutation within the kinase domain activation loop (encoded by exon 18)5,6,23. Notably, the PDGFRA D842V mutation is homologous to the KIT exon 17 D816V mutation found in the vast majority of patients with mastocytosis, although this particular KIT mutation has not been reliably reported as a primary mutation in GIST. Other primary PDGFRA alterations include diverse indels and point mutations in exon 18 (in ~5% of GISTs) and mutations affecting the ATP-binding pocket encoded by exon 14 (in 1%), which is homologous to KIT exon 135. Historically, the survival outcomes of patients with advanced-stage PDGFRA-mutant GIST have been poor because PDGFRAD842V-mutant GIST is highly resistant to imatinib and other type II PDGFRA/KIT TKIs5,24–26. However, the development of avapritinib, a type I PDGFRA/KIT TKI, has now improved the outcomes of many patients with PDGFRAD842V-mutant GIST, as discussed in more detail below. The majority of PDGFRA-mutant GISTs arise in the stomach or the omentum, with rare cases originating in the intestines or mesentery (TABLE 1)27.
SDH-deficient GIST.
SDH-deficient GIST constitutes the third-largest molecular subset, accounting for ~9% of all GISTs1,3,4,8–13,16,17,28–30 (FIG. 1). These tumours have unique clinical and pathological features compared with most other GISTs, given that they most commonly occur in young adults, are almost exclusively gastric in origin (specifically arising in the distal stomach), often have an epithelioid rather than spindle cell morphology and frequently give rise to lymph node metastases13,28,30 (TABLE 1). Across different series, 82–100% of patients with SDH-deficient GIST had an associated germline pathogenic mutation in any one of the four genes encoding the SDH subunits: SDHA, SDHB, SDHC and SDHD13,31,32. Each of the four SDH complex genes are considered classical tumour suppressors, with tumours arising owing to inheritance of a germline loss-of-function (LOF) allele followed by spontaneous somatic loss of heterozygosity of an SDH gene or, less commonly, a second independent somatic LOF mutation in the originally wild-type allele of the same SDH subunit affected by a germline mutation33,34. LOF mutations in any of the SDH subunits lead to dysfunction and degradation of the whole complex, resulting in a loss of SDHB expression as assessed using immunohistochemistry (IHC), which can be used as a diagnostic marker35. The majority of GIST-associated SDH mutations occur in SDHA13,36. In addition to LOF mutations, a minority of SDH-deficient GISTs arise through somatic hypermethylation of the SDHC promoter (~0.5% of all GISTs), leading to loss of both SDHC expression and SDH enzymatic function13,37,38. Most SDH-deficient GISTs behave in an indolent fashion, with a clinical course measured in decades, although some are aggressive and are associated with rapidly progressive disease13. No therapies are specifically approved for SDH-deficient GIST.
Other molecular drivers of GIST.
Beyond KIT, PDGFRA and SDH, a few other molecular drivers account for small subsets of GISTs. Loss of neurofibromin (NF1) accounts for ~2% of GISTs, and activating BRAF V600E mutations for ~0.8%39–42 (FIG. 1). Both of these subtypes of GIST predominately arise in the small intestine via excessive activation of the MEK–ERK signalling pathway (TABLE 1 and FIG. 2a). Treatment with BRAF inhibitors can be effective for patients with BRAFV600E-mutant GIST43. On the basis of the experience in treating BRAF-mutant melanoma, we speculate that combining a MEK inhibitor with a BRAF inhibitor might be even more effective for treating BRAF-mutant GIST44. GISTs with NF1 loss most commonly arise as a manifestation of classical neurofibromatosis type I, owing to a germline mutation in one NF1 allele and subsequent somatic loss or inactivation of the remaining functional allele. Such GIST might also arise spontaneously as a result of somatic homozygous/biallelic or hemizygous LOF mutations in NF1, although such tumours have actually been suggested to constitute unrecognized cases of neurofibromatosis type I45,46. Indeed, malignant GIST occurs in 5–10% of patients with neurofibromatosis type I, with up to 33% of patients having one or more occult GIST found during autopsy47. Neurofibromatosis-associated GISTs occur on average a decade or so earlier than sporadic GISTs, and affected individuals can have multiple, and in some cases, numerous, clinically occult primary GISTs42. Fortunately, many of these tumours have an indolent phenotype and clinical course38,64, and in patients with multiple small tumours located throughout the small intestine, observation rather than radical resection might be indicated39,42,48. Currently, no known effective therapy exists for NF1-mutant GIST. In 2020, the MEK inhibitor selumetinib was approved for treatment of paediatric patients with neurofibromatosis type I who have symptomatic, inoperable plexiform neurofibromas49. This drug is now being evaluated in a phase II study involving patients with NF1-mutant GIST (NCT03109301). Of note, some NF1-mutant GISTs also express mutant forms of KIT, possibly as a secondary mutational event that further augments cell proliferation50,51. This hypothesis is supported by the observation that, in certain tumours, the mutant form of KIT seems to be expressed in some, but not all, cancer cells.
Most recently, gene-fusion proteins involving the RTKs NTRK3 or FGFR1 were discovered as the drivers of up to 1% of all GISTs (FIG. 1), most commonly among those arising in the small intestine or rectum8 (TABLE 1). The fusion partners and nature of these translocations (including NTRK3–ETV6 and TACC1–FGFR1) are similar to those found in other human cancers and result in constitutive signalling by the RTK component52.
Other rare driver mutations involve KRAS or PIK3CA, and are each found in <1% of GISTs8,17,53–55 (FIG. 1). Additionally, some GISTs harbouring KRAS or PIK3CA mutations also have an activating KIT mutation54. In these tumours, the KIT mutation is hypothesized to be the original oncogenic driver, with secondary subclonal mutation of KRAS or PIK3CA, sometimes in the setting of acquired resistance to KIT inhibitors56–58. This observation suggests that activation of RAS or PI3K downstream of mutant KIT provides a proliferative advantage.
Finally, overexpression of FGF4 owing to gene duplication has been reported as a potential cause of a minority of GISTs lacking any known driver mutations59. However, further studies are needed to validate this mechanism of GIST oncogenesis.
Cells of origin
Most GISTs arise from transformation of interstitial cells of Cajal (ICCs), located in the wall of the gastrointestinal tract, which function as pacemakers for peristaltic contractions60. At least four different classes of ICC have been identified, including myenteric ICCs (ICC-MY), intramuscular ICCs (ICC-IM), submucosal plexus ICCs (ICC-SMP) and deep mucosal plexus ICCs (ICC-DMP)61,62. Notably, the distribution of these ICC classes varies throughout the gut. For example, the stomach contains only ICC-MY and ICC-IM, while the large intestine contains these classes as well as ICC-SMP. By contrast, the small intestine lacks ICC-IM and ICC-SMP, but contains ICC-MY and ICC-DMP62. All four ICC subtypes express KIT, but only ICC-MY and ICC-IM can be transformed by KIT mutations63.
Alternative cells of origin have been proposed for certain GIST subtypes. Telocytes were identified in 2005 as a ICC-like cell type with a CD34+PDGFRA+ immunophenotype in the gastrointestinal tract64. Moreover, hyperplasia of telocytes has been observed in rare families with germline PDGFRA mutations, analogous to ICC hyperplasia that occurs in individuals with germline KIT mutations65. These observations suggest that telocytes are the cell of origin for PDGFRA-mutant GISTs64. Whether transformed ICCs can also give rise to PDGFRA-mutant GISTs is unclear. In the case of BRAF-mutant GIST, findings in different animal models indicate that either ICCs or a smooth muscle-derived cell type can serve as the cell of origin66,67. Currently, the exact cell(s) of origin for SDH-deficient GIST remains unknown.
Differences between these cells of origin are thought to underlie the clinical observation that certain molecular subtypes of GIST commonly arise at specific locations along the gastrointestinal tract (TABLE 1). For example, the vast majority of SDH-deficient and PDGFRA-mutant GISTs occur in the distal stomach; these two subtypes also share a distinctive epithelioid cell morphology18,68. By contrast, KIT-mutant GISTs predominate in the proximal stomach, but can be found throughout the gastrointestinal tract18. NF1-mutant and KIT exon 9-mutant GISTs arise almost exclusively in the small intestine. Histologically, GISTs harbouring KIT, NF1 or BRAF mutations are most commonly composed of spindle cells1,3 (TABLE 1).
Pathobiology
In GISTs, RTK-activating mutations lead to ligand-independent kinase activation and increased signalling through downstream proliferative and survival pathways, including the PI3K–AKT, JAK–STAT and RAS–RAF–MEK–ERK (MAPK) cascades3,6,69,70 (FIG. 2a). Rarely, mutations involving other effectors within these pathways, including PI3K, BRAF or RAS proteins, or their regulators, such as NF1 (a GTPase-activating protein that inactivates RAS), can also drive oncogenic signalling8,40,71,72 (FIG. 2a).
SDH-deficient GIST is one molecular subtype that seems to deviate from this canonical RTK signalling-driven mechanism of tumorigenesis73. Loss of SDH activity owing to LOF mutations in any of the SDH genes results in accumulation of its substrate, succinate, and a decrease in fumarate production74–76 (FIG. 2b). Elevated succinate has been suggested to act as an oncometabolite that drives tumorigenesis in multiple ways, including inhibition of ⍺-ketoglutarate-dependent dioxygenases, such as hypoxia-inducible factor (HIF)-prolyl hydroxylases, TET family methylcytosine dioxygenases and lysine-specific histone demethylases, which induces both pseudohypoxic HIF-1 signalling and DNA and histone hypermethylation phenotypes77–79 (FIG. 2b).
Lessons learned from imatinib
Prior to the year 2000, no effective medical therapies were available for patients with advanced-stage GIST. GISTs have minimal sensitivity to the chemotherapy agents commonly used to treat other sarcomas. Historically, GISTs have also been considered to be resistant to external beam radiotherapy80. Although, more recent studies, including a single prospective phase II trial, have shown that radiotherapy can provide palliative disease stabilization in selected patients81–83. The only known effective GIST therapy at the turn of this century was surgery, which is performed with curative intent for patients with localized disease or to palliate patients with advanced-stage GIST through selective metastasectomy84.
The discovery of activating KIT mutations in GIST in 1998 (REFS.7,14) led to the hypothesis that KIT inhibitors might be effective for the treatment of this disease. Around this same time, imatinib was identified as potent KIT TKI, with activity against mutant forms of KIT, and had already undergone extensive clinical testing for the treatment of patients with chronic myeloid leukaemia (CML)85. Accordingly, imatinib was evaluated as a treatment for GIST in several phase II studies and subsequently in several large-cohort international randomized phase III trials86–89. In these studies, imatinib induced a high rate of clinical benefit (either a complete response, partial response or durable stable disease) ranging from 70–84%, with median progression-free-survival (PFS) durations in the range of 20 months86–89. These results lead to the 2001 FDA approval of imatinib for the treatment of metastatic GIST90. Notably, the median overall survival (OS) duration of patients with advanced-stage GIST is estimated to have increased from 12 months to 4–5 years with the use of imatinib, with 10-year OS estimates of 10–20%84,91,92. Imatinib is generally well tolerated, with many of the common adverse effects being mild or easily managed with standard supportive care measures; however, up to 5% of patients are intolerant of imatinib and require a change in therapy based on toxicity alone93,94.
The success of imatinib had major effects on GIST therapy, drug development and research. Ultimately, the development of all currently approved systemic treatments for GIST, which are all TKIs, was sparked by the limitations of imatinib, particularly those related to primary (intrinsic) or secondary (acquired) resistance to this agent (FIG. 3). These limitations also provide a perspective from which to consider novel therapeutic approaches.
Fig. 3 │. Typical pattern of GIST response and evolution during TKI treatment.
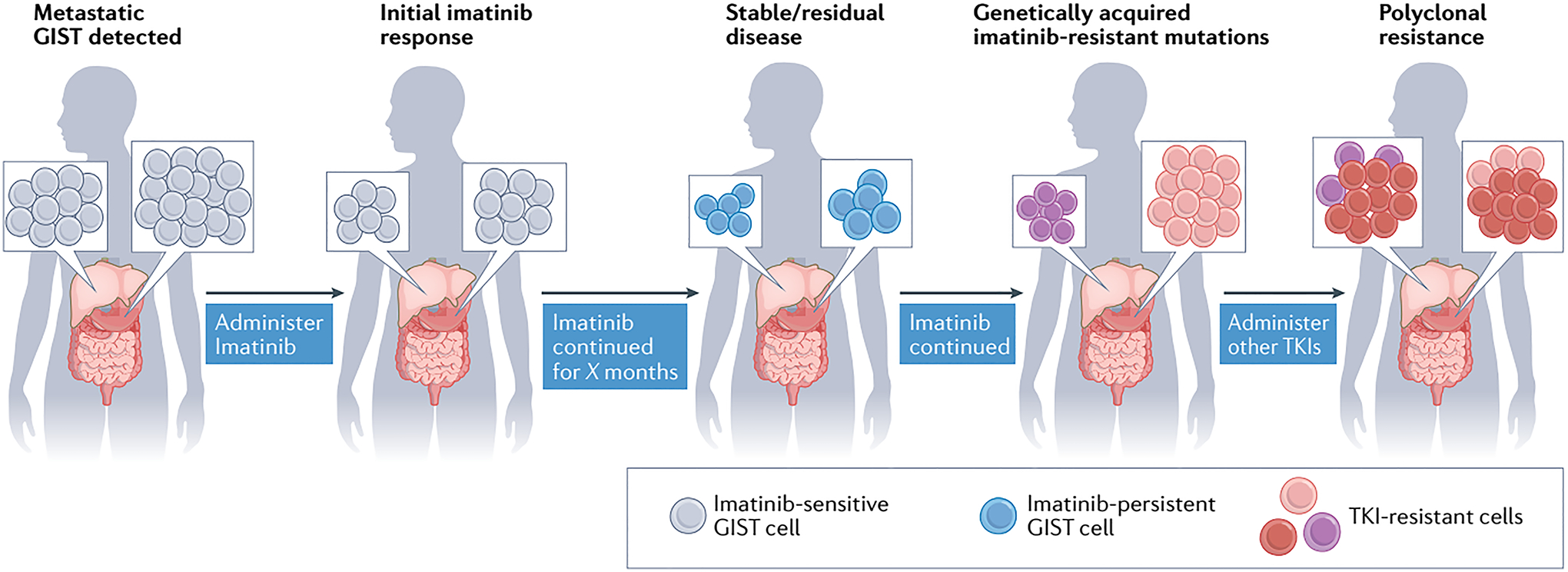
Patients diagnosed with metastatic KIT-mutant gastrointestinal stromal tumour (GIST; lesions indicated by grey cells) are initially treated with the KIT-targeting tyrosine kinase inhibitor (TKI) imatinib. Typically, imatinib induces tumour shrinkage by inducing apoptosis of GIST cells; however, not all GIST cells are eradicated, with a fraction persisting throughout treatment by entering a non-proliferative, quiescent state (imatinib-persistent GIST cells, shown in blue). Some of these persistent GIST cells will eventually acquire genetic mutations that confer resistance to imatinib, leading to tumour outgrowth and disease progression (pink and purple cells). Other KIT TKIs can be administered sequentially to patients with GISTs harbouring resistance mutations. Nevertheless, genetically heterogeneous subclones can arise across tumour lesions (shown as pink, purple and red cells), leading to polyclonal resistance of the tumours to multiple TKIs. Both intertumour and intratumour heterogeneity can be found in patients with TKI-resistant GIST. Intertumour heterogeneity is illustrated by the presence of different imatinib-resistant subclones, either pink or purple, across the two lesions. In the rightmost panel, intratumour heterogeneity is also depicted by the co-existence within a single lesion of newly emergent red subclones together with the pink or purple cell population.
Molecular testing to optimize therapy
A notable finding in the early studies of imatinib was that not all GISTs responded uniformly. We now know that these variable outcomes can be almost entirely understood through molecular testing of the primary tumour. Most patients with GISTs lacking a KIT mutation had minimal to no clinical response to imatinib and generally had markedly inferior PFS and OS relative to patients with KIT-mutant GIST91,95,96. Remarkably, even among patients with KIT-mutant GIST, the likelihood and durability of the response to imatinib can be predicted based on the specific primary KIT mutation; patients with exon 11 mutations have superior outcomes to those with exon 9 mutations, especially when using standard-dose imatinib (400 mg total daily dose)91,95,96. These responses led to different dosing recommendations for patients, depending on the specific KIT mutation detected97.
Further investigation of GISTs with primary resistance to imatinib (that is, those in patients with disease progression <6 months after starting treatment), ultimately led to the identification of the other molecular drivers of this disease, including the PDGFRA D842V mutations (although not all GISTs with PDGFRA mutations are imatinib-resistant). For decades, patients with GISTs harbouring the D842V mutation, the most common PDGFRA mutation, had no therapeutic options because this mutation confers resistance to imatinib. Therefore, the D842V variant of PDGFRA presented a key target for rational drug design5,95. Indeed, the development of the type I PDGFRA/KIT TKI avapritinib has provided patients with PDGFRA -mutant GIST with a promising treatment option98,99. In 2020, avapritinib was approved by the FDA specifically for patients with advanced-stage GIST harbouring a PDGFRA exon 18 mutation, including D842V mutations, based on data from the phase I NAVIGATOR trial showing an objective response rate (ORR) of 84% (7% complete responses), with 61% of responses lasting ≥6 months100.
The remaining molecular subtypes of GIST (SDH deficient, BRAF mutant or RTK translocated) were also identified through studies in the imatinib-refractory, KIT/PDGFRA-wild-type population, and the subsequent development and application of mutation-specific treatments has been shown to benefit some patients. For example, patients with BRAFV600E-mutant GIST have been successfully treated with dabrafenib43, although this agent is not yet formally FDA approved for this indication. Moreover, the TRK TKIs larotrectinib and entrectinib are now FDA approved for the treatment of patients with solid tumours harbouring NTRK fusions, including GISTs, based in part on a 100% ORR and durable responses among patients with GIST in the histology-agnostic registrational studies101,102. Of note, the appropriate diagnosis of GIST with NTRK fusions is challenging, potentially requiring multiple techniques that can include fluorescence in situ hybridization (FISH), IHC and RNA sequencing103. Treatment of SDH-deficient GISTs with imatinib is associated with a very low ORR (<5%), although the second-line TKI sunitinib has been reported to have modest activity in terms of disease stabilization, with a reported partial response rate of approximately 15%13,104. Notwithstanding, the treatment outcomes of patients with advanced-stage SDH-deficient GIST lag far behind those of patients with KIT-mutant GIST, underscoring an unmet need for more effective therapies.
The variable, mutation-dependent responses to imatinib highlight just how crucial molecular testing and patient selection based on the detection of a specific molecular driver is for achieving clinical success. This paradigm has subsequently been applied to other therapies for GIST, beginning with the KIT TKIs sunitinib, regorafenib and ripretinib, but also avapritinib, larotrectinib and entrectinib, and will need to be considered in the development of any future novel therapeutic approaches.
Requirement for continuous treatment
Researchers and clinicians quickly recognized that continuous, long-term treatment with imatinib is required to achieve disease control because inhibition of KIT does not result in elimination of all GIST cells; some cells persist by entering a nonproliferative, quiescent state105,106 (FIG. 3). These so-called persistent GIST cells can rapidly proliferate again when KIT inhibition is removed, as demonstrated in vitro and also clinically through randomized discontinuation trials in patients with long-term responses to imatinib107,108. Persistent GIST cells typically lack acquired genetic aberrations that might confer more permanent drug resistance, and despite being insensitive to TKIs while quiescent, these cells remain sensitive to imatinib should they re-enter the proliferative state105,109. Nevertheless, the quiescent cells are thought to provide a source of subclones that confer clinical resistance once they acquire secondary mutations.
Several clinical studies have investigated the benefit of adjuvant imatinib for various durations, revealing that longer treatment results in better outcomes110. Indeed, in patients with imatinib-sensitive GIST, the relapse rate is <2% per year during adjuvant imatinib treatment but increases substantially after imatinib therapy is stopped, probably owing to re-entry of quiescent cells into a proliferative state111–113. For example, ~10% of patients had recurrence during 3 years of continuous treatment with imatinib, but an additional 40% of patients relapsed after completing adjuvant therapy111.
Secondary resistance mutations
Similar to what had already been observed for BCR–ABL1 in studies of imatinib-refractory CML, secondary KIT mutations were identified as the main cause of imatinib resistance in KIT-mutant GISTs114. These mutations clustered in two regions of the KIT kinase domain: the ATP/drug-binding pocket (encoded by KIT exons 13 and 14) and the activation loop (exons 17 and 18)115,116,117–119. Rarely, drug resistance can arise owing to mutations in genes encoding downstream effectors, such as PIK3CA or KRAS, which promote activation of the cell proliferation and survival pathways56,58 (FIG. 2a).
Independent of the emerging understanding of imatinib-resistance mechanisms, additional KIT inhibitors were generated, many of them through programmes aiming to develop multi-kinase inhibitors capable of also inhibiting PDGFR and/or VEGFR family members. Large-cohort randomized phase III studies of sunitinib94 and regorafenib120, both with placebo control arms, resulted in approval of these agents for second-line and third-line treatment of advanced-stage GIST, respectively. The median time to tumour progression was 27.3 weeks in patients receiving sunitinib after failure of imatinib, compared with 6.4 weeks in those receiving placebo (HR 0.33; P <0.0001)94. In the case of regorafenib used to treat patients after failure of prior imatinib and sunitinib, the median PFS duration was 4.8 months versus 0.9 months with placebo (HR 0.27; P <0.0001)120. Over time, the basis for the notably lower ORRs and shorter PFS durations with these agents relative to imatinib became known: both sunitinib and regorafenib have activity against some, but not all, secondary, imatinib-resistance mutations in KIT. This knowledge provided a molecular explanation for mixed tumour responses in an individual patient, whereby some lesions regress but at the same time anatomically distinct lesions progress, which defines the clinical entity of complex polyclonal resistance121,122 (FIG. 3). Ultimately, understanding of the mechanisms of drug resistance in KIT-mutant GISTs resulted in the development of broad-spectrum KIT inhibitors, with activity against most, if not all, described resistance mutations. The success of this approach is exemplified by the development of ripretinib, a TKI that binds to a novel region of both the KIT and PDGFRA kinases, referred to as the switch control pocket123. In the randomized, double-blind phase III INVICTUS trial involving patients with advanced-stage GIST that had progression on at least imatinib, sunitinib and regorafenib, ripretinib resulted in an ORR of 9% versus 0% with placebo (P = 0.05), median PFS of 6.3 months versus 1.0 months (HR 0.15; P <0.0001) and median OS of 15.1 months versus 6.6 months (HR 0.36, 95% CI 0.21–0.62; P value not evaluated)124,125. These results formed the basis for the FDA approval of ripretinib for this indication in 2020 (REF.125).
Polyclonal TKI resistance, both within and across tumours, in patients with advanced-stage GIST continues to present a substantial clinical challenge when eventually none of the approved TKIs can control all lesions within a given patient (FIG. 3). Not only is polyclonal resistance recognized for KIT-mutant GIST, but has also been seen in patients with PDGFRA-mutant GIST treated with avapritinib, and will probably be a challenge in patients with NTRK-rearranged GIST treated with larotrectinib or entrectinib126,127.
Novel strategies to treat GIST
The limitations of the therapies discussed above present opportunities for innovation to develop novel treatment strategies including those that utilize TKIs, both newly developed and currently available ones, as well as approaches that are entirely new to the field of GIST therapy (FIG. 4 and TABLE 2). Importantly, although not a novel strategy, developing new KIT and/or PDGFRA inhibitors that are capable of controlling a broader range of resistance mutations when used as single agents remains very clinically relevant for GIST (FIG. 4a). Currently, at least two novel KIT TKIs are entering clinical testing as single agents in patients with advanced-stage GIST: THE-630 (NCT05160168) and NB003 (formally known as AZD3229; NCT04936178) (TABLE 2). Both of these agents have potent in vitro activity against all reported secondary KIT mutations (with the possible exception of certain mutations involving codon 816)128,129. This spectrum of activity could theoretically overcome polyclonal resistance in patients with KIT-mutant GIST that has progressed on multiple prior lines of treatment with KIT TKIs. If promising clinical activity is identified in this population with advanced-stage, multi-drug-resistant disease, additional studies will probably test the new agents in earlier lines of therapy and they could potentially replace the current standard-of-care TKIs. Nevertheless, additional compound mutations will eventually result in clinical drug resistance even to these new agents, similar to that observed with the latest generations of EGFR and ABL1 inhibitors130–132.
Fig. 4 │. New therapeutic approaches exploiting different elements of GIST biology.
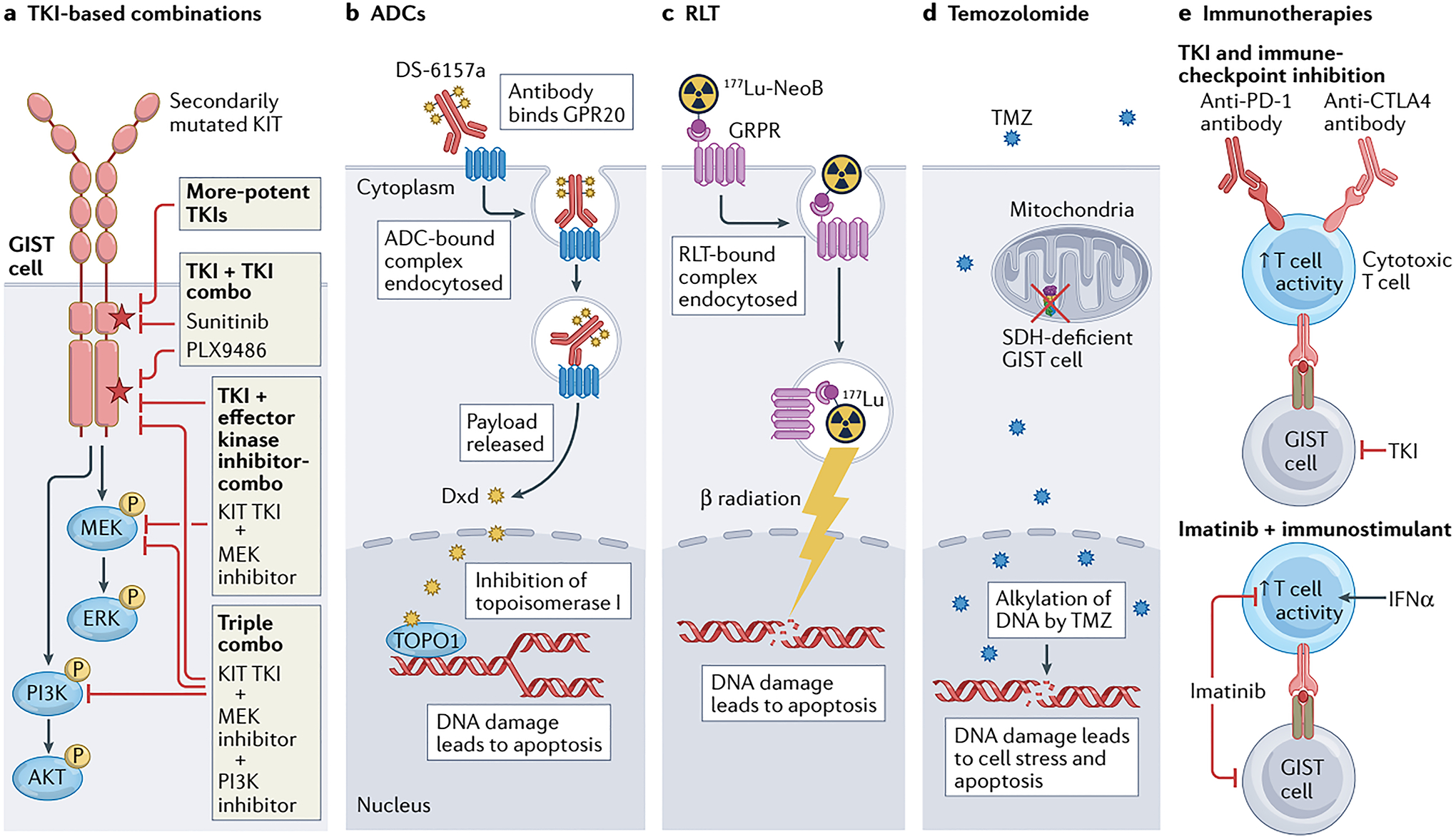
a │ Tyrosine kinase inhibitor (TKI)-based therapy remains relevant for the majority of patients with advanced-stage GIST, particularly those with KIT-mutant disease; however, new strategies are required to overcome treatment resistance and thereby improve outcomes, including the development of more-potent TKIs, or combinations of two TKIs or a TKI plus inhibitors of downstream effector kinases (such as MEK and/or PI3K). Many of these approaches might also be applicable in PDGFRA-mutant GIST if PDGFRA-specific TKIs, such as avapritinib, are utilized. b │ The antibody–drug conjugate (ADC) DS-6157a combines an anti-G protein-coupled receptor 20 (GPR20) antibody and the DNA topoisomerase I (TOPO1) inhibitor deruxtecan (Dxd). Upon binding to GPR20, the receptor–ADC complex is endocytosed, with subsequent lysosomal degradation of the complex resulting in release of the Dxd payload that in turn causes DNA damage and cell death. c │ 177Lu-NeoB is a radioligand therapy (RLT) consisting of the radioisotope 177Lu conjugated via the chelating agent dodecanetetraacetic acid (DOTA) to a peptide antagonist of the gastrin-releasing peptide receptor (GRPR; also known as bombesin receptor subtype 2 (BB2)). Thus, this agent enables specific intracellular delivery of radiation to GRPR-expressing GIST cells, resulting in DNA damage and apoptosis. d │ In an approach specific to SDH-deficient GIST cells, treatment with the alkylating agent temozolomide (TMZ) can cause irreparable DNA damage and cell death. This vulnerability is probably at least partially attributable to epigenetic silencing of 6-O-methylguanine-DNA methyltransferase (MGMT), which is involved in the repair of alkylated DNA, as a consequence of the metabolic alterations resulting from SDH deficiency in these cells. e │ Various immuno-oncology approaches to the treatment of GIST have been proposed, including combining a TKI with PD-1 and/or CTLA4 immune-checkpoint inhibitors to simultaneously suppress GIST cells while stimulating antitumour T cells, or imatinib with an immunostimulant such as IFNα to prevent imatinib-related T cell inactivation.
Table 2 │.
Predicted efficacy of novel treatment strategies against common subtypes of GIST
| Treatment strategy | Novel treatment | Target | Predicted efficacy | ||
|---|---|---|---|---|---|
| KIT-mutant GIST | PDGFRA-mutant GIST | SDH-deficient GIST | |||
| TKI-based | THE-630 (NCT05160168) | KIT driver (all known primary and secondary variants) | √ | x | x |
| NB003 (NCT04936178) formally known as AZD3229 | KIT or PDGFRA driver | √ | √ | x | |
| Bezuclastinib (previously known as CGT9486 and PLX9486) + sunitinib (NCT05208047;134) | KIT driver | √ | √ | x | |
| TKI + MEKi (such as imatinib plus binimetinib; NCT01991379;137,139) | KIT or PDGFRA driver | √ | √ | x | |
| TKI + PI3Ki + MEKi (140) | KIT or PDGFRA driver | √ | √ | x | |
| ADC | DS-6157a (NCT04276415) | GPR20 | √ | √ | √ |
| RLT | 177Lu-NeoB (NCT03872778) | GRPR (expression determined by 68Ga-NeoB uptake) | √ | ? | ? |
| DDR | Temozolomide (NCT03556384;161) | SDH-deficient cells (negative for MGMT expression) | x | x | √ |
| IO | TKI + ICI (such as axitinib and avelumab; NCT04258956) | KIT or PDGFRA driver and PD-1 or PD-L1 | √ | √ | x |
| TKI + immunostimulant (such as IFNα167) | KIT or PDGFRA driver and cytokine receptors | √ | √ | x | |
?, unknown; ADC, antibody–drug conjugate; DDR, DNA damage repair; GIST, gastrointestinal stromal tumour; GPR20, G protein-coupled receptor 20; GRPR, gastrin-releasing peptide receptor (also known as bombesin receptor subtype 2 (BB2)); ICI, immune-checkpoint inhibitor; IO, immuno-oncology; MEKi, MEK inhibitor; PI3K, PI3K inhibitor; RLT, radioligand therapy; SDH, succinate dehydrogenase; TKI, tyrosine kinase inhibitor.
Combination therapy using TKIs
The current KIT TKIs lack activity against all relevant drug-resistance mutations, which limits their effectiveness as single agents. However, each agent has a unique spectrum of activity against the different KIT variants; therefore, combination therapy is one way to potentially overcome the challenges of polyclonal resistance and/or drug-persistent cells in GISTs (FIG. 4a). Combinations of different KIT TKIs have been studied in vitro and in a few clinical studies. In a phase I study, Serrano et al.133 investigated a novel strategy of alternating sunitinib and regorafenib to overcome polyclonal resistance involving different secondary KIT mutations in patients previously treated with at least imatinib, sunitinib and regorafenib. Unfortunately, this approach was unsuccessful, probably owing to overlapping toxicities of the two TKIs (gastrointestinal and hand–foot skin reaction) and difficulties in devising a tolerable and effective dosing scheme133; although, perhaps also because the patients all had tumours that had previously been exposed to, and thus might have developed resistance against, both drugs. More recently, bezuclastinib (previously known as CGT9486 and PLX9486), a type I inhibitor with potent activity against KIT exon 17 and 18 (activation loop) resistance mutations, showed good tolerability and clinical activity when combined with sunitinib, a type II inhibitor that has potent activity against KIT exon 13 and 14 (ATP-binding pocket) resistance mutations, in patients with advanced-stage, TKI-refractory GIST134. On the basis of the promising data from this phase Ib/IIa trial, including a clinical benefit rate of 80% and a median PFS duration of 12.1 months134, a randomized phase III trial comparing the combination treatment versus single-agent sunitinib for the treatment of imatinib-resistant, sunitinib-naive GIST is underway (NCT05208047) (TABLE 2).
As mentioned previously, primary KIT mutations in GISTs result in activation of the downstream MAPK, PI3K–AKT, and JAK–STAT pathways (FIG. 2). MEK–ERK and PI3K–AKT are particularly crucial effectors of mutant KIT signalling, and evidence suggests that both pathways need to be inhibited to optimally decrease the proliferation and/or induce apoptosis of GIST cells135,136. The combination of imatinib with either a MEK inhibitor or a PI3K inhibitor has been tested in patients with advanced-stage GIST, usually in the setting of acquired resistance to imatinib (for example, NCT01735968, NCT01468688 and NCT01991379). To date these studies have not provided any strong signs of efficacy137,138, probably because in the setting of imatinib resistance these treatments no longer function as a combination therapy, but instead as MEK or PI3K inhibitor monotherapy. Given the activation of multiple pathways by unopposed KIT signalling in the setting of imatinib resistance, it is not surprising that inhibiting a single downstream pathway is not effective. More promising activity has been demonstrated in the setting of frontline treatment of advanced-stage GIST by combining imatinib and the MEK inhibitor binimetinib. In a phase II study (NCT01991379), this therapeutic strategy produced an ORR of 69.0%, with a median PFS duration of 29.9 months139; however, in the absence of a randomized control arm, whether this approach is superior to front-line imatinib monotherapy is impossible to determine. Given the long PFS duration associated with single-agent frontline therapy of GIST, along with concerns about increased toxicity and financial costs, a study comparing imatinib with imatinib plus binimetinib in the first line is unlikely.
An alternative and potentially more feasible approach to combining downstream kinase inhibitors with imatinib would involve the use of short-term pulse combination therapy in order to eliminate drug-persistent GIST cells (FIG. 3). The results of an in vitro study by Gupta et al.140 demonstrated that triplet therapy with imatinib plus a MEK inhibitor and a PI3K inhibitor can eliminate cells that persist during imatinib treatment. Triplet therapy would probably also be limited by toxicities, but might be clinically feasible using pulse treatments of limited duration, such as a cyclical treatment schedule. Further clinical studies are needed to determine the safety and potential efficacy of this treatment approach.
Antibody–drug conjugates
Antibody–drug conjugates (ADCs) are one of the most rapidly expanding therapeutic classes in clinical oncology. ADCs entered clinical studies in the 1980s, but initially failed to yield relevant clinical benefits141. However, continued improvements in this technology resulted in the first ADC approval in 2000, of gemtuzumab ozogamicin for acute myeloid leukaemia, followed by a second in 2011, brentuximab vedotin for Hodgkin lymphoma or anaplastic large cell lymphoma141. Currently, a total of 11 ADCs are FDA approved for the treatment of various cancers and dozens of new agents are currently in clinical studies141. ADCs consist of three elements: 1) a tumour-associated antigen-specific monoclonal antibody; 2) a chemical linker; and 3) a potent cytotoxic agent (also known as the ‘payload’)142. The monoclonal antibody element enables tumour-selective targeting, with the therapeutic index optimized through selection of an antigen with high levels of tumoural expression and minimal to no expression by nonmalignant cells141. Advances in the design and synthesis of ADCs have increased the drug-to-antibody ratio, thereby improving payload delivery to cells targeted by the monoclonal antibody141. Theoretically, the payload is only released after the ADC enters a cell by endocytosis and the linker is subsequently cleaved or degraded in the lysosome, thus minimizing systemic toxicity from the payload agent142–144 (FIG. 4b).
In 2021, a report identified GPR20 as a novel GIST-specific target antigen for ADC treatment145. GPR20 is an orphan G protein-coupled receptor that was found to be strongly expressed in the vast majority of GISTs, regardless of molecular subtype, as well as in subsets of ICCs, but not other nonmalignant tissues or types of sarcoma145. On the basis of these findings, DS-6157a was generated using a humanized anti-GPR20 antibody, a protease cleavable maleimide Gly-Gly-Phe-Gly tetrapeptide-based linker and an exatecan-derivative topoisomerase I inhibitor payload (deruxtecan)145 (FIG. 4b). DS-6157a was shown to have antitumour activity against KIT-mutant GIST cells in vitro and in patient-derived xenograft models, regardless of the presence or absence of secondary resistance mutations145. The favourable pharmacokinetic profiles, efficacy and safety and tolerability metrics in animal models have led to advancement of DS-6157a to a first-in-human phase I study in patients with advanced-stage GIST145 (NCT04276415; TABLE 2). The success of this approach will depend in part on GPR20 expression, and thus a diagnostic grade GPR20 IHC assay will probably need to be developed.
Radioligand therapy
Radiolabelled peptides, also known as radioligand therapies (RLTs), have been developed for imaging and/or treatment of various cancers, leading to the birth of the field of theragnostics (or theranostics)146. For example, 68Ga-DOTATATE and 177Lu-DOTATATE are both FDA approved for imaging and treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumours146,147. In addition, 177Lu-PSMA-617 has been granted FDA priority review as a treatment for metastatic castration-resistant prostate cancer148. Several receptors expressed on GIST cells have been proposed as targets for RLT, including somatostatin receptors 1 and 2 (SST1/2) and the gastrin-releasing protein receptor (GRPR, also known as bombesin receptor subtype 2 or BB2)149–151 (FIG. 4c). Follow-up imaging studies have revealed that most GISTs do not express sufficient levels of SST1/2 for effective targeting152,153, although a radiolabelled antagonist of GRPR known as NeoB (previously NeoBOMB1) continues to garner interest as a treatment of GIST. In patient-derived xenograft models of KIT exon 13-mutant GIST, 177Lu-NeoB was found to localize to the tumours, with only minimal accumulation in nonmalignant tissues154. Notably, near complete tumour regression and improved survival was noted in mice treated with a 400 pmol dose of 177Lu-NeoB154. Imaging of patients with GIST using a 68Ga-labelled version of NeoB revealed both interpatient and intrapatient tumour heterogeneity in GRPR expression, with some patients having uptake in 100% of tumours, but others having uptake in only some of the tumours155, suggesting that imaging with this agent could be used for patient selection for treatment with 177Lu-NeoB (TABLE 2). Currently, 177Lu-NeoB is being tested in the phase I/II NeoRay study involving patients with advanced-stage breast or prostate cancer, GIST or glioblastoma (NCT03872778). In this study, patients are being imaged with 68Ga-NeoB PET–CT and those with at least one measurable NeoB-avid lesion will be treated with a putative therapeutic dose of 177Lu-NeoB.
Both ADC and RLT approaches have the potential to overcome several limitations of the current kinase-directed therapies for GIST. First, target expression might be independent of kinase mutation status, as seems to be the case for GPR20145; therefore, these treatments might prove effective for GISTs with no proven effective therapies, including NF1-mutant or SDH-deficient GIST. Second, given that ADCs and RLTs have distinct mechanisms of action and are unlikely to have substantial overlapping toxicity with TKIs, each of these agents could potentially be combined to achieve an additive or synergistic effect. Finally, because these ADCs and RLTs act in a kinase-independent fashion, they might be effective against tumours with secondary resistance mechanisms, including both secondary kinase mutations and kinase-independent mechanisms of drug resistance.
Temozolomide for SDH-deficient GIST
SDH-deficient tumours in general have presented a persistent therapeutic challenge. Technically, the approved indications for imatinib, sunitinib, regorafenib and ripretinib include patients with SDH-deficient GIST, although it is clinically recognized that KIT/PDGFRA TKIs, with the possible exception of sunitinib, provide limited benefit for these patients13,104. Therefore, different therapeutic approaches are needed for SDH-deficient GIST, and exploiting the unique biology of this disease subtype is a possible strategy. Much has been learned about SDH-deficient GIST from the study of other SDH-deficient tumours, owing to their shared pathobiology. For example, SDH-deficient tumours are known to have functional defects in DNA damage repair (DDR) pathways156, prompting investigation of DDR-targeting agents in these cancers. Additional studies in a small number of patients with SDHB-mutant paraganglioma have shown that treatment with the alkylating agent temozolomide often results in disease stabilization157,158 (FIG. 4d). Lack of expression of the DNA dealkylating enzyme 6-O-methylguanine-DNA methyltransferase (MGMT), owing to promoter hypermethylation and resultant transcriptional silencing of MGMT, is predictive of a favourable response to temozolomide in patients with glioblastoma, an approved indication, as well as paraganglioma157–159. SDH-deficient GISTs have been shown to lack MGMT expression160, and in vitro studies using novel patient-derived SDH-deficient GIST models provide evidence of the sensitivity of these tumour to temozolomide161. A phase II study of temozolomide in patients with advanced-stage SDH-deficient GIST is underway (NCT03556384), with promising preliminary results in five initial patients, including two partial responses and disease control in the three other patients161 (TABLE 2).
Immunotherapeutic approaches
Immunotherapy approaches have impressive activity against advanced-stage tumours of certain histologies (such as melanoma, non-small-cell lung cancer and renal cell carcinoma). However, immune-checkpoint inhibitors, including anti-PD-1 antibodies as single agents or in combination with anti-CTLA4 antibodies, have thus far shown only modest activity in patients with GIST. For example, in a randomized phase II study of nivolumab versus nivolumab plus ipilimumab, the median PFS duration in the monotherapy arm was 11.7 weeks, and was only 8.3 weeks in the combination therapy arm162. Nonetheless, 3 of 35 patients across both arms had PFS durations >1.5 years, suggesting the feasibility of an immunotherapy approach for GIST if pre-treatment characteristics of such long-term responders could be identified and used to select patients for treatment162. Similar to these clinical findings, monotherapy with anti-PD-1 or anti-PD-L1 antibodies had no effect on tumour growth in a mouse model of GIST; however, addition of an anti-PD-1 antibody to imatinib markedly decreased tumour growth compared with single-agent imatinib163. Thus, the feasibility of combining TKIs with immunostimulatory agents needs to be tested in clinical studies. Indeed, the combination of the anti-PD-L1 antibody avelumab and KIT/PDGFRA TKI axitinib is currently being tested in the phase II AXAGIST study involving patients with advanced-stage GIST that has progressed after treatment with at least imatinib and sunitinib (NCT04258956) (TABLE 2 and FIG. 4e). One caveat is that these combination therapies depend upon effective KIT/PDGFRA inhibition, indicating the need to partner immunotherapeutic agents with broad-spectrum KIT/PDGFRA TKIs, likely earlier rather than later in the treatment paradigm. Thus, testing such combination therapies as part of first-line treatment of advanced-stage GIST might be warranted.
Notably, the microenvironment of KIT-mutant GIST is altered by KIT TKIs, with an initial augmented immune response owing to activation of CD8+ T cells and dendritic cells164,165. With chronic imatinib therapy, however, the abundance of both intratumoural dendritic cells and CD8+ T cells decreases, which dampens the immune response to tumour cells165. This blunted immune response partly reflects reduced type I interferon (IFN) production, which in turn leads to decreased CD8+ T cell activity166. Restoration of type I IFN signalling through administration of IFNα could potentially reverse the chronic immune-inhibitory effects of imatinib (TABLE 2 and FIG. 4e), a concept previously tested with promising initial results in a small phase II study conducted in the early 2000s167. On the basis of our improved understanding of the GIST microenvironment, this approach should be further tested in a larger study. Another approach to reversing the immune-inhibitory effects of chronic KIT TKI therapy would be to use other cytokines and/or chemical stimulants, such as FLT3 ligand and the Toll-like receptor agonist polyinosinic:polycytidylic acid (poly I:C), to promote dendritic cell expansion and maturation165.
Conclusions
For the past 5 years we have been able to identify the oncogenic driver event in 99% of patients with GIST, but our ability to target all driver mutations has lagged behind. The past few years have, nevertheless, yielded new strategies for targeting the drivers of GIST beyond KIT, providing effective therapies for approximately 80–90% of patients with advanced-stage GIST. However, the limitations of current TKI therapies pose challenges to long-term disease control, and some subtypes of GIST are inherently insensitive to such agents. Thus, alternative approaches will be required to better manage advanced-stage GIST, and as new therapies become available, the optimal treatment sequence will need to be continuously refined.
Key points.
All currently approved systemic therapies for GIST are tyrosine kinase inhibitors (TKIs); these agents can be used to treat the majority of patients with advanced-stage GIST.
TKIs have limitations, particularly in the setting of advanced-stage disease, owing to secondary intra-allelic mutations that confer drug resistance, but also the need for indefinite treatment to control quiescent, drug-persistent tumour cells.
Understanding the underlying biology of the different molecular subtypes of GIST has presented new therapeutic approaches beyond TKIs.
TKIs remain relevant for GISTs harbouring receptor tyrosine kinase mutations or fusions, but applying them in new, more strategic ways will benefit patients.
Acknowledgements
The work of M.C.H. has been supported by grants from the Department of Veterans Affairs (1 I01 BX005358-01A1) and the NIH National Cancer Institute (1 R21 CA263400-01), and by philanthropic donations from the GIST Cancer Research Fund and the Jonathan David Foundation.
Footnotes
Competing interests
M.C.H. has been a consultant for Blueprint Medicines, Deciphera Pharmaceuticals, Novartis and Theseus Pharmaceuticals, and has a patent for the treatment of GIST using imatinib that has been licensed by his institute to Novartis. The other authors declare no competing interests.
References
- 1.Blay JY, Kang YK, Nishida T & von Mehren M Gastrointestinal stromal tumours. Nat Rev Dis Primers 7, 22, doi: 10.1038/s41572-021-00254-5 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Soreide K et al. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol 40, 39–46, doi: 10.1016/j.canep.2015.10.031 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Corless CL, Barnett CM & Heinrich MC Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer 11, 865–878, doi: 10.1038/nrc3143 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Bannon AE, Klug LR, Corless CL & Heinrich MC Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors. Expert Review of Molecular Diagnostics, 1–13, doi: 10.1080/14737159.2017.1308826 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Corless CL et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 23, 5357–5364, doi: 10.1200/JCO.2005.14.068 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299, 708–710, doi: 10.1126/science.1079666 (2003). [DOI] [PubMed] [Google Scholar]; First description of PDGFRA mutations in GIST.
- 7.Hirota S et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279, 577–580 (1998). [DOI] [PubMed] [Google Scholar]; Seminal paper with the first description of KIT mutations in GIST.
- 8.Shi E et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. Journal of translational medicine 14, 339, doi: 10.1186/s12967-016-1075-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive molecular characterization of the mutational landscape of GIST using next-generation sequencing. This paper details FGFR1 and NTRK3 translocations in GIST, and provides one of the first descriptions of the responsiveness of NTRK3-rearranged GIST to larotrectinib.
- 9.Serrano C & George S Gastrointestinal Stromal Tumor: Challenges and Opportunities for a New Decade. Clin Cancer Res 26, 5078–5085, doi: 10.1158/1078-0432.CCR-20-1706 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Cassier PA & Blay JY Molecular response prediction in gastrointestinal stromal tumors. Target Oncol 5, 29–37, doi: 10.1007/s11523-010-0134-9 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Huss S et al. A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del). Mod Pathol 26, 1004–1012, doi: 10.1038/modpathol.2013.47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verschoor AJ et al. The incidence, mutational status, risk classification and referral pattern of gastro-intestinal stromal tumours in the Netherlands: a nationwide pathology registry (PALGA) study. Virchows Arch 472, 221–229, doi: 10.1007/s00428-017-2285-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boikos SA et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA oncology, doi: 10.1001/jamaoncol.2016.0256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive clinical and molecular characterization of GISTs lacking KIT or PDGFRA mutations. This paper provides a detailed characterization of the molecular mechanisms underlying SDH deficiency in GIST and of the clinical responses of SDH-deficient GISTs to conventional TKIs.
- 14.Taniguchi M et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 59, 4297–4300 (1999). [PubMed] [Google Scholar]
- 15.Klug LR, Kent JD & Heinrich MC Structural and clinical consequences of activation loop mutations in class III receptor tyrosine kinases. Pharmacology & therapeutics 191, 123–134, doi: 10.1016/j.pharmthera.2018.06.016 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Nannini M et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer 14, 685, doi: 10.1186/1471-2407-14-685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vanden Bempt I et al. Comprehensive targeted next-generation sequencing approach in the molecular diagnosis of gastrointestinal stromal tumor. Genes, chromosomes & cancer, doi: 10.1002/gcc.22923 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Sharma AK et al. Location of Gastrointestinal Stromal Tumor (GIST) in the Stomach Predicts Tumor Mutation Profile and Drug Sensitivity. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-21-1221 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper comprehensively maps mutational status and primary tumour location of gastric GISTs to provide novel insights on pathogenesis, and suggests differential sensitivity of different ICC subtypes to neoplastic transformation by various mutations.
- 19.Wang HC et al. KIT Exon 11 Codons 557–558 Deletion Mutation Promotes Liver Metastasis Through the CXCL12/CXCR4 Axis in Gastrointestinal Stromal Tumors. Clin Cancer Res, doi: 10.1158/1078-0432.ccr-15-2748 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Dematteo RP et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST). Cancer 112, 608–615, doi: 10.1002/cncr.23199 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Kunstlinger H et al. Gastrointestinal stromal tumors with KIT exon 9 mutations: Update on genotype-phenotype correlation and validation of a high-resolution melting assay for mutational testing. Am J Surg Pathol 37, 1648–1659, doi: 10.1097/PAS.0b013e3182986b88 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Lasota J et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol 21, 476–484, doi: 10.1038/modpathol.2008.2 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Klug LR, Corless CL & Heinrich MC Inhibition of KIT Tyrosine Kinase Activity: Two Decades After the First Approval. J Clin Oncol 39, 1674–1686, doi: 10.1200/JCO.20.03245 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cassier PA et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res 18, 4458–4464, doi: 10.1158/1078-0432.CCR-11-3025 (2012). [DOI] [PubMed] [Google Scholar]
- 25.von Mehren M et al. Clinical efficacy comparison of avapritinib with other tyrosine kinase inhibitors in gastrointestinal stromal tumors with PDGFRA D842V mutation: a retrospective analysis of clinical trial and real-world data. BMC Cancer 21, 291, doi: 10.1186/s12885-021-08013-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; Real-world comparison of outcomes of patients with PDGFRAD842V-mutant GIST treated with conventional TKIs versus avapritinib.
- 26.Yoo C et al. Efficacy of Imatinib in Patients with Platelet-Derived Growth Factor Receptor Alpha-Mutated Gastrointestinal Stromal Tumors. Cancer Res Treat 48, 546–552, doi: 10.4143/crt.2015.015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lasota J & Miettinen M Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 53, 245–266, doi: 10.1111/j.1365-2559.2008.02977.x (2008). [DOI] [PubMed] [Google Scholar]
- 28.Miettinen M et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol 35, 1712–1721, doi: 10.1097/PAS.0b013e3182260752 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed clinicopathological characterization of SDH-deficient GIST.
- 29.Janeway KA et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 108, 314–318, doi: 10.1073/pnas.1009199108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first descriptions of SDH-deficient GIST.
- 30.Doyle LA, Nelson D, Heinrich MC, Corless CL & Hornick JL Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype-phenotype correlation study. Histopathology 61, 801–809, doi: 10.1111/j.1365-2559.2012.04300.x (2012). [DOI] [PubMed] [Google Scholar]
- 31.Pantaleo MA et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 103, 983–987, doi: 10.1093/jnci/djr130 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Pantaleo MA et al. SDHA Germline Variants in Adult Patients With SDHA-Mutant Gastrointestinal Stromal Tumor. Frontiers in oncology 11, 778461, doi: 10.3389/fonc.2021.778461 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evenepoel L et al. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet Med 17, 610–620, doi: 10.1038/gim.2014.162 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Gill AJ Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 72, 106–116, doi: 10.1111/his.13277 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Gaal J et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol 24, 147–151, doi: 10.1038/modpathol.2010.185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pantaleo MA et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. European journal of human genetics : EJHG 22, 32–39, doi: 10.1038/ejhg.2013.80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Killian JK et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Science translational medicine 6, 268ra177, doi: 10.1126/scitranslmed.3009961 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of SDHC promoter hypermethylation as a mechanism causing SDH-deficiency in GIST.
- 38.Casey RT et al. SDHC epi-mutation testing in gastrointestinal stromal tumours and related tumours in clinical practice. Sci Rep 9, 10244, doi: 10.1038/s41598-019-46124-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miettinen M & Lasota J Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 23, 70–83, doi: 10.1053/j.semdp.2006.09.001 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Agaimy A et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. Journal of clinical pathology 62, 613–616, doi: 10.1136/jcp.2009.064550 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Hostein I et al. BRAF mutation status in gastrointestinal stromal tumors. American journal of clinical pathology 133, 141–148, doi: 10.1309/ajcppckga2qgbj1r (2010). [DOI] [PubMed] [Google Scholar]
- 42.Salvi PF et al. Gastrointestinal stromal tumors associated with neurofibromatosis 1: a single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol 2013, 398570, doi: 10.1155/2013/398570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive clinicopathological characterization of GIST in patients with classical neurofibromatosis type I.
- 43.Falchook GS et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 4, 310–315, doi: 10.18632/oncotarget.864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; First description of successful treatment of BRAF-mutant GIST using a BRAF kinase inhibitor.
- 44.Tanda ET et al. Current State of Target Treatment in BRAF Mutated Melanoma. Front Mol Biosci 7, 154, doi: 10.3389/fmolb.2020.00154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belinsky MG et al. Somatic loss of function mutations in neurofibromin 1 and MYC associated factor X genes identified by exome-wide sequencing in a wild-type GIST case. BMC Cancer 15, 887, doi: 10.1186/s12885-015-1872-y (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gasparotto D et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin Cancer Res 23, 273–282, doi: 10.1158/1078-0432.ccr-16-0152 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Zoller ME, Rembeck B, Oden A, Samuelsson M & Angervall L Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer 79, 2125–2131 (1997). [PubMed] [Google Scholar]
- 48.Maertens O et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 15, 1015–1023, doi: 10.1093/hmg/ddl016 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Galvin R et al. Neurofibromatosis in the Era of Precision Medicine: Development of MEK Inhibitors and Recent Successes with Selumetinib. Current oncology reports 23, 45, doi: 10.1007/s11912-021-01032-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burgoyne AM et al. Duodenal-Jejunal Flexure GI Stromal Tumor Frequently Heralds Somatic NF1 and Notch Pathway Mutations. JCO Precis Oncol 2017, doi: 10.1200/PO.17.00014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li K et al. Multiple gastrointestinal stromal tumors: analysis of clinicopathologic characteristics and prognosis of 20 patients. Cancer Manag Res 11, 7031–7038, doi: 10.2147/CMAR.S197560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cocco E, Scaltriti M & Drilon A NTRK fusion-positive cancers and TRK inhibitor therapy. Nature reviews. Clinical oncology 15, 731–747, doi: 10.1038/s41571-018-0113-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miranda C et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res 18, 1769–1776, doi: 10.1158/1078-0432.ccr-11-2230 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Mavroeidis L et al. Comprehensive molecular screening by next generation sequencing reveals a distinctive mutational profile of KIT/PDGFRA genes and novel genomic alterations: results from a 20-year cohort of patients with GIST from north-western Greece. ESMO Open 3, e000335, doi: 10.1136/esmoopen-2018-000335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haefliger S et al. Molecular Profile of Gastrointestinal Stromal Tumors in Sixty-Eight Patients from a Single Swiss Institution. Pathobiology 87, 171–178, doi: 10.1159/000505407 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Serrano C et al. KRAS and KIT Gatekeeper Mutations Confer Polyclonal Primary Imatinib Resistance in GI Stromal Tumors: Relevance of Concomitant Phosphatidylinositol 3-Kinase/AKT Dysregulation. J Clin Oncol 33, e93–96, doi: 10.1200/JCO.2013.48.7488 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lasota J et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Mod Pathol 29, 275–282, doi: 10.1038/modpathol.2015.160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muhlenberg T et al. KIT-Dependent and KIT-Independent Genomic Heterogeneity of Resistance in Gastrointestinal Stromal Tumors - TORC1/2 Inhibition as Salvage Strategy. Mol Cancer Ther 18, 1985–1996, doi: 10.1158/1535-7163.MCT-18-1224 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Urbini M et al. Gene duplication, rather than epigenetic changes, drives FGF4 overexpression in KIT/PDGFRA/SDH/RAS-P WT GIST. Sci Rep 10, 19829, doi: 10.1038/s41598-020-76519-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sircar K et al. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am J Surg Pathol 23, 377–389 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Sanders KM, Kito Y, Hwang SJ & Ward SM Regulation of Gastrointestinal Smooth Muscle Function by Interstitial Cells. Physiology (Bethesda) 31, 316–326, doi: 10.1152/physiol.00006.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Al-Shboul OA The importance of interstitial cells of cajal in the gastrointestinal tract. Saudi J Gastroenterol 19, 3–15, doi: 10.4103/1319-3767.105909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chi P et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467, 849–853, doi: 10.1038/nature09409 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed analysis of the subtypes of ICC that can give rise to GIST and first description of ETV1 as a crucial lineage-specific survival factor.
- 64.Ricci R et al. Telocytes are the physiological counterpart of inflammatory fibroid polyps and PDGFRA-mutant GISTs. Journal of cellular and molecular medicine 22, 4856–4862, doi: 10.1111/jcmm.13748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Report of telocytes as an alternative cell of origin to ICC in PDGFRA-mutant GIST.
- 65.Manley PN et al. Familial PDGFRA-mutation syndrome: somatic and gastrointestinal phenotype. Hum Pathol 76, 52–57, doi: 10.1016/j.humpath.2018.02.014 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Kondo J et al. A smooth muscle-derived, Braf-driven mouse model of gastrointestinal stromal tumor (GIST): evidence for an alternative GIST cell-of-origin. J Pathol 252, 441–450, doi: 10.1002/path.5552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ran L et al. ETV1-Positive Cells Give Rise to BRAF(V600E) -Mutant Gastrointestinal Stromal Tumors. Cancer Res 77, 3758–3765, doi: 10.1158/0008-5472.CAN-16-3510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using different mouse models, the preceding two references describe either ICC or smooth muscle precursor cells as the cell of origin in BRAF-mutant GIST.
- 68.Mei L et al. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 4, 74–91, doi: 10.1016/j.trecan.2017.11.006 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Ronnstrand L Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci 61, 2535–2548, doi: 10.1007/s00018-004-4189-6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; Excellent review of signal transduction from KIT and the role of its cognate ligand (stem cell factor) in normal physiological development and homeostasis.
- 70.Duensing A et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 23, 3999–4006, doi: 10.1038/sj.onc.1207525 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Daniels M et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett 312, 43–54, doi: 10.1016/j.canlet.2011.07.029 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Andersson J et al. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 29, 1170–1176 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Lussey-Lepoutre C et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nature communications 6, 8784, doi: 10.1038/ncomms9784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim E et al. Utility of the succinate: Fumarate ratio for assessing SDH dysfunction in different tumor types. Mol Genet Metab Rep 10, 45–49, doi: 10.1016/j.ymgmr.2016.12.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lussey-Lepoutre C et al. In Vivo Detection of Succinate by Magnetic Resonance Spectroscopy as a Hallmark of SDHx Mutations in Paraganglioma. Clin Cancer Res 22, 1120–1129, doi: 10.1158/1078-0432.CCR-15-1576 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Richter S et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. The Journal of clinical endocrinology and metabolism 99, 3903–3911, doi: 10.1210/jc.2014-2151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiao M et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26, 1326–1338, doi: 10.1101/gad.191056.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Killian JK et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer discovery 3, 648–657, doi: 10.1158/2159-8290.cd-13-0092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Detailed molecular and genomic characterization of epigenetic reprogramming due to genome-wide DNA hypermethylation in SDH-deficient tumours.
- 79.Eijkelenkamp K, Osinga TE, Links TP & van der Horst-Schrivers ANA Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin Genet 97, 39–53, doi: 10.1111/cge.13553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeMatteo RP et al. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Annals of surgery 231, 51–58, doi: 10.1097/00000658-200001000-00008 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gatto L et al. Radiotherapy in the management of gist: state of the art and new potential scenarios. Clin Sarcoma Res 7, 1, doi: 10.1186/s13569-016-0065-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuaron JJ, Goodman KA, Lee N & Wu AJ External beam radiation therapy for locally advanced and metastatic gastrointestinal stromal tumors. Radiat Oncol 8, 274, doi: 10.1186/1748-717X-8-274 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Joensuu H et al. Radiotherapy for GIST progressing during or after tyrosine kinase inhibitor therapy: A prospective study. Radiother Oncol 116, 233–238, doi: 10.1016/j.radonc.2015.07.025 (2015). [DOI] [PubMed] [Google Scholar]; Only prospective study of the role of radiotherapy in the treatment of advanced-stage GIST.
- 84.Dematteo RP, Heinrich MC, El-Rifai WM & Demetri G Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol 33, 466–477 (2002). [DOI] [PubMed] [Google Scholar]; Excellent historical comparison of the clinical outcomes of patients with GIST.
- 85.Heinrich MC et al. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 96, 925–932 (2000). [PubMed] [Google Scholar]; One of the first descriptions of imatinib as an inhibitor of both wild-type and exon 11-mutant KIT.
- 86.Demetri GD et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347, 472–480, doi: 10.1056/NEJMoa020461 (2002). [DOI] [PubMed] [Google Scholar]; Phase II trial that led to FDA approval of imatinib for patients with advanced-stage GIST.
- 87.van Oosterom AT et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 358, 1421–1423 (2001). [DOI] [PubMed] [Google Scholar]
- 88.Verweij J et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364, 1127–1134, doi: 10.1016/s0140-6736(04)17098-0 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Blanke CD et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 26, 620–625, doi: 10.1200/JCO.2007.13.4403 (2008). [DOI] [PubMed] [Google Scholar]
- 90.GLEEVEC (imatinib mesylate) tablets Label, <https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021588s024lbl.pdf> (2001).
- 91.Heinrich M et al. Correlation of Long-term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA oncology, doi: 10.1001/jamaoncol.2016.6728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Casali PG et al. Ten-Year Progression-Free and Overall Survival in Patients With Unresectable or Metastatic GI Stromal Tumors: Long-Term Analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup Phase III Randomized Trial on Imatinib at Two Dose Levels. J Clin Oncol 35, 1713–1720, doi: 10.1200/JCO.2016.71.0228 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Sodergren SC et al. Systematic review of the side effects associated with tyrosine kinase inhibitors used in the treatment of gastrointestinal stromal tumours on behalf of the EORTC Quality of Life Group. Critical reviews in oncology/hematology 91, 35–46, doi: 10.1016/j.critrevonc.2014.01.002 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Demetri GD et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368, 1329–1338, doi: 10.1016/S0140-6736(06)69446-4 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Heinrich MC et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21, 4342–4349, doi: 10.1200/JCO.2003.04.190 (2003). [DOI] [PubMed] [Google Scholar]
- 96.Debiec-Rychter M et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42, 1093–1103, doi: 10.1016/j.ejca.2006.01.030 (2006). [DOI] [PubMed] [Google Scholar]; In the preceding two papers, clinical outcomes with imatinib are correlated with tumour mutational status in patients with advanced-stage GIST, which provided a rationale for using a precision oncology approach to treatment selection.
- 97.Casali PG et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO, doi: 10.1016/j.annonc.2021.09.005 (2021). [DOI] [PubMed] [Google Scholar]
- 98.Heinrich MC et al. in Connective TIssue Oncology Society Annual Meeting (Rome, Italy, 2018). [Google Scholar]
- 99.Heinrich MC et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol 21, 935–946, doi: 10.1016/S1470-2045(20)30269-2 (2020). [DOI] [PubMed] [Google Scholar]; First results from the phase I trial that led to FDA approval of avapritinib for advanced-stage PDGFRA exon 11-mutant GIST.
- 100.AYVAKIT (avapritinib) tablets, for oral use, <https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212608s000lbl.pdf> (2020).
- 101.Drilon A et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 378, 731–739, doi: 10.1056/NEJMoa1714448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hong DS et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 21, 531–540, doi: 10.1016/S1470-2045(19)30856-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Solomon JP & Hechtman JF Detection of NTRK Fusions: Merits and Limitations of Current Diagnostic Platforms. Cancer Res 79, 3163–3168, doi: 10.1158/0008-5472.CAN-19-0372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Janeway KA et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 52, 767–771, doi: 10.1002/pbc.21909 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Gupta A et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci U S A 107, 14333–14338, doi: 10.1073/pnas.1000248107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Y et al. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res 68, 9015–9023, doi: 10.1158/0008-5472.CAN-08-1935 (2008). [DOI] [PubMed] [Google Scholar]
- 107.Le Cesne A et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol 11, 942–949, doi: 10.1016/S1470-2045(10)70222-9 (2010). [DOI] [PubMed] [Google Scholar]
- 108.Patrikidou A et al. Long-term outcome of molecular subgroups of GIST patients treated with standard-dose imatinib in the BFR14 trial of the French Sarcoma Group. Eur J Cancer 52, 173–180, doi: 10.1016/j.ejca.2015.10.069 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Boichuk S et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res 73, 5120–5129, doi: 10.1158/0008-5472.CAN-13-0579 (2013). [DOI] [PubMed] [Google Scholar]
- 110.Joensuu H et al. Adjuvant Imatinib for High-Risk GI Stromal Tumor: Analysis of a Randomized Trial. J Clin Oncol 34, 244–250, doi: 10.1200/jco.2015.62.9170 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Joensuu H et al. Survival Outcomes Associated With 3 Years vs 1 Year of Adjuvant Imatinib for Patients With High-Risk Gastrointestinal Stromal Tumors: An Analysis of a Randomized Clinical Trial After 10-Year Follow-up. JAMA oncology 6, 1241–1246, doi: 10.1001/jamaoncol.2020.2091 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Casali PG et al. Final analysis of the randomized trial on imatinib as an adjuvant in localized gastrointestinal stromal tumors (GIST) from the EORTC Soft Tissue and Bone Sarcoma Group (STBSG), the Australasian Gastro-Intestinal Trials Group (AGITG), UNICANCER, French Sarcoma Group (FSG), Italian Sarcoma Group (ISG), and Spanish Group for Research on Sarcomas (GEIS)(). Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 32, 533–541, doi: 10.1016/j.annonc.2021.01.004 (2021). [DOI] [PubMed] [Google Scholar]
- 113.Raut CP et al. Efficacy and Tolerability of 5-Year Adjuvant Imatinib Treatment for Patients With Resected Intermediate- or High-Risk Primary Gastrointestinal Stromal Tumor: The PERSIST-5 Clinical Trial. JAMA oncology 4, e184060, doi: 10.1001/jamaoncol.2018.4060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Al-Ali HK et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib. Hematol J 5, 55–60, doi: 10.1038/sj.thj.6200319 (2004). [DOI] [PubMed] [Google Scholar]
- 115.Gramza AW, Corless CL & Heinrich MC Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin Cancer Res 15, 7510–7518, doi: 10.1158/1078-0432.CCR-09-0190 (2009). [DOI] [PubMed] [Google Scholar]
- 116.Heinrich MC et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 24, 4764–4774, doi: 10.1200/JCO.2006.06.2265 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Bertucci F et al. Acquired resistance to imatinib and secondary KIT exon 13 mutation in gastrointestinal stromal tumour. Oncol Rep 16, 97–101 (2006). [PubMed] [Google Scholar]
- 118.McLean SR et al. Imatinib binding and cKIT inhibition is abrogated by the cKIT kinase domain I missense mutation Val654Ala. Mol Cancer Ther 4, 2008–2015, doi: 10.1158/1535-7163.mct-05-0070 (2005). [DOI] [PubMed] [Google Scholar]
- 119.Chen LL et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 64, 5913–5919, doi: 10.1158/0008-5472.can-04-0085 (2004). [DOI] [PubMed] [Google Scholar]
- 120.Demetri GD et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 295–302, doi: 10.1016/S0140-6736(12)61857-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Serrano C et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer 120, 612–620, doi: 10.1038/s41416-019-0389-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; The preceding two papers describe the placebo-controlled phase III trial that led to FDA approval of sunitinib and regorafenib, respectively, for advanced-stage GIST.
- 122.Heinrich MC et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 26, 5352–5359, doi: 10.1200/JCO.2007.15.7461 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Smith BD et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 35, 738–751 e739, doi: 10.1016/j.ccell.2019.04.006 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Blay JY et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 21, 923–934, doi: 10.1016/S1470-2045(20)30168-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Placebo-controlled phase III trial that led to FDA approval of ripretinib for advanced-stage GIST previously treated with imatinib, sunitinib and regorafenib.
- 125.QINLOCK™ (ripretinib) tablets, for oral use, <https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213973s000lbl.pdf> (2020).
- 126.Grunewald S et al. Resistance to avapritinib in PDGFRA-driven GIST is caused by secondary mutations in the PDGFRA kinase domain. Cancer discovery, doi: 10.1158/2159-8290.CD-20-0487 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Somwar R et al. NTRK kinase domain mutations in cancer variably impact sensitivity to type I and type II inhibitors. Commun Biol 3, 776, doi: 10.1038/s42003-020-01508-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Banks E et al. Discovery and pharmacological characterization of AZD3229, a potent KIT/PDGFRalpha inhibitor for treatment of gastrointestinal stromal tumors. Science translational medicine 12, doi: 10.1126/scitranslmed.aaz2481 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Rivera VM, Huang WS, Pritchard JR, Dalgamo DC & Shakespeare WC in AACR (2021). Abstract 1292: Preclinical characterization of THE-630, a next-generation inhibitor for KIT-mutant gastrointestinal stromal tumors (GIST). Cancer Res. 81 (13 Suppl.) 1292 (2021). [Google Scholar]
- 130.Tumbrink HL, Heimsoeth A & Sos ML The next tier of EGFR resistance mutations in lung cancer. Oncogene 40, 1–11, doi: 10.1038/s41388-020-01510-w (2021). [DOI] [PubMed] [Google Scholar]
- 131.Zabriskie MS et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 26, 428–442, doi: 10.1016/j.ccr.2014.07.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kang KH et al. Compound mutations involving T315I and P-loop mutations are the major components of multiple mutations detected in tyrosine kinase inhibitor resistant chronic myeloid leukemia. Leuk Res 76, 87–93, doi: 10.1016/j.leukres.2018.10.019 (2019). [DOI] [PubMed] [Google Scholar]
- 133.Serrano C et al. Phase I Study of Rapid Alternation of Sunitinib and Regorafenib for the Treatment of Tyrosine Kinase Inhibitor Refractory Gastrointestinal Stromal Tumors. Clin Cancer Res 25, 7287–7293, doi: 10.1158/1078-0432.CCR-19-2150 (2019). [DOI] [PubMed] [Google Scholar]
- 134.Wagner AJ et al. Association of Combination of Conformation-Specific KIT Inhibitors With Clinical Benefit in Patients With Refractory Gastrointestinal Stromal Tumors: A Phase 1b/2a Nonrandomized Clinical Trial. JAMA oncology, doi: 10.1001/jamaoncol.2021.2086 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; Description of the combined use of KIT TKIs with complementary activity to overcome drug-resistant GIST.
- 135.Gupta A et al. Ripretinib and MEK Inhibitors Synergize to Induce Apoptosis in Preclinical Models of GIST and Systemic Mastocytosis. Mol Cancer Ther 20, 1234–1245, doi: 10.1158/1535-7163.MCT-20-0824 (2021). [DOI] [PubMed] [Google Scholar]
- 136.Floris G et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res 19, 620–630, doi: 10.1158/1078-0432.ccr-12-2853 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chi P et al. Phase Ib trial of the combination of imatinib and binimetinib in patients with advanced gastrointestinal stromal tumors (GIST). Clin. Cancer Res 10.1158/1078-0432.CCR-21-3909 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gelderblom H et al. Imatinib in combination with phosphoinositol kinase inhibitor buparlisib in patients with gastrointestinal stromal tumour who failed prior therapy with imatinib and sunitinib: a Phase 1b, multicentre study. Br J Cancer 122, 1158–1165, doi: 10.1038/s41416-020-0769-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chi P et al. Phase II trial of imatinib plus binimetinib in patients with treatment-naive advanced gastrointestinal stromal tumor. J. Clin. Oncol 10.1200/JCO.21.02029 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gupta A, Ma S, Che K, Pobbati AV & Rubin BP Inhibition of PI3K and MAPK pathways along with KIT inhibitors as a strategy to overcome drug resistance in gastrointestinal stromal tumors. PLoS One 16, e0252689, doi: 10.1371/journal.pone.0252689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; Intriguing description of triplet therapy targeting the KIT, PI3K and MAPK signalling pathways to overcome in vitro quiescence of TKI-resistant GIST cells.
- 141.Drago JZ, Modi S & Chandarlapaty S Unlocking the potential of antibody-drug conjugates for cancer therapy. Nature reviews. Clinical oncology 18, 327–344, doi: 10.1038/s41571-021-00470-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schumacher D, Hackenberger CP, Leonhardt H & Helma J Current Status: Site-Specific Antibody Drug Conjugates. J Clin Immunol 36 Suppl 1, 100–107, doi: 10.1007/s10875-016-0265-6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Joubert N, Beck A, Dumontet C & Denevault-Sabourin C Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals (Basel) 13, doi: 10.3390/ph13090245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baah S, Laws M & Rahman KM Antibody-Drug Conjugates-A Tutorial Review. Molecules 26, doi: 10.3390/molecules26102943 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Iida K et al. Identification and Therapeutic Targeting of GPR20, Selectively Expressed in Gastrointestinal Stromal Tumors, with DS-6157a, a First-In-Class Antibody-Drug Conjugate. Cancer discovery, doi: 10.1158/2159-8290.CD-20-1434 (2021). [DOI] [PubMed] [Google Scholar]; Description of a novel GPR20-targeted antibody–drug conjugate that is currently being tested in a phase I/II trial involving patients with GIST.
- 146.Vahidfar N et al. An Impressive Approach in Nuclear Medicine: Theranostics. PET Clin 16, 327–340, doi: 10.1016/j.cpet.2021.03.011 (2021). [DOI] [PubMed] [Google Scholar]
- 147.Strosberg J et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med 376, 125–135, doi: 10.1056/NEJMoa1607427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sartor O et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 385, 1091–1103, doi: 10.1056/NEJMoa2107322 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Reubi JC, Korner M, Waser B, Mazzucchelli L & Guillou L High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur J Nucl Med Mol Imaging 31, 803–810, doi: 10.1007/s00259-004-1476-2 (2004). [DOI] [PubMed] [Google Scholar]
- 150.Zhao WY et al. Somatostatin receptors in gastrointestinal stromal tumors: new prognostic biomarker and potential therapeutic strategy. Am J Transl Res 6, 831–840 (2014). [PMC free article] [PubMed] [Google Scholar]
- 151.Loaiza-Bonilla A & Bonilla-Reyes PA Somatostatin Receptor Avidity in Gastrointestinal Stromal Tumors: Theranostic Implications of Gallium-68 Scan and Eligibility for Peptide Receptor Radionuclide Therapy. Cureus 9, e1710, doi: 10.7759/cureus.1710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Arne G et al. Gastrointestinal stromal tumors (GISTs) express somatostatin receptors and bind radiolabeled somatostatin analogs. Acta Oncol 52, 783–792, doi: 10.3109/0284186X.2012.733075 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Paulmichl A et al. Targeting Gastrointestinal Stromal Tumor with (68)Ga-Labeled Peptides: An In Vitro Study on Gastrointestinal Stromal Tumor-Cell Lines. Cancer Biother Radiopharm 31, 302–310, doi: 10.1089/cbr.2016.2092 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Montemagno C et al. In Vivo Biodistribution and Efficacy Evaluation of NeoB, a Radiotracer Targeted to GRPR, in Mice Bearing Gastrointestinal Stromal Tumor. Cancers (Basel) 13, doi: 10.3390/cancers13051051 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gruber L et al. MITIGATE-NeoBOMB1, a Phase I/IIa Study to Evaluate Safety, Pharmacokinetics, and Preliminary Imaging of (68)Ga-NeoBOMB1, a Gastrin-Releasing Peptide Receptor Antagonist, in GIST Patients. J Nucl Med 61, 1749–1755, doi: 10.2967/jnumed.119.238808 (2020). [DOI] [PubMed] [Google Scholar]
- 156.Sulkowski PL et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 50, 1086–1092, doi: 10.1038/s41588-018-0170-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tong A et al. Temozolomide Is a Potential Therapeutic Tool for Patients With Metastatic Pheochromocytoma/Paraganglioma-Case Report and Review of the Literature. Front Endocrinol (Lausanne) 11, 61, doi: 10.3389/fendo.2020.00061 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tena I et al. Successful Second-Line Metronomic Temozolomide in Metastatic Paraganglioma: Case Reports and Review of the Literature. Clin Med Insights Oncol 12, 1179554918763367, doi: 10.1177/1179554918763367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hegi ME et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352, 997–1003, doi: 10.1056/NEJMoa043331 (2005). [DOI] [PubMed] [Google Scholar]
- 160.Ricci R et al. Preferential MGMT methylation could predispose a subset of KIT/PDGFRA-WT GISTs, including SDH-deficient ones, to respond to alkylating agents. Clin Epigenetics 11, 2, doi: 10.1186/s13148-018-0594-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yebra M et al. Establishment of Patient-derived Succinate Dehydrogenase-deficient Gastrointestinal Stromal Tumor Models For Predicting Therapeutic Response. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-21-2092 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript details the rationale for an ongoing phase II trial of temozolomide in patients with SDH-deficient GIST.
- 162.Singh AS et al. A Randomized Phase 2 Study Of Nivolumab Monotherapy Or Nivolumab Combined with Ipilimumab In Patients with Advanced Gastrointestinal Stromal Tumors. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-21-0878 (2021). [DOI] [PubMed] [Google Scholar]
- 163.Seifert AM et al. PD-1/PD-L1 Blockade Enhances T-cell Activity and Antitumor Efficacy of Imatinib in Gastrointestinal Stromal Tumors. Clin Cancer Res 23, 454–465, doi: 10.1158/1078-0432.CCR-16-1163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Balachandran VP et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 17, 1094–1100, doi: 10.1038/nm.2438 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Medina BD et al. Oncogenic kinase inhibition limits Batf3-dependent dendritic cell development and antitumor immunity. The Journal of experimental medicine 216, 1359–1376, doi: 10.1084/jem.20180660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Liu M et al. Oncogenic KIT Modulates Type I IFN-Mediated Antitumor Immunity in GIST. Cancer Immunol Res 9, 542–553, doi: 10.1158/2326-6066.CIR-20-0692 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen LL et al. Exploiting antitumor immunity to overcome relapse and improve remission duration. Cancer Immunol Immunother 61, 1113–1124, doi: 10.1007/s00262-011-1185-1 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; The preceding five references describe the immune microenvironment of GIST and the effects of TKIs on the immune response, as well as potential approaches to augment the immune response as a therapeutic strategy.