

Abstract
Aims
Inadequate antidepressant response interrupts effective treatment of major depressive disorder (MDD). The BLESS study evaluates the dosage, efficacy, and safety of brexpiprazole adjunctive therapy in Japanese patients with inadequate antidepressant therapy (ADT) response.
Methods
This placebo‐controlled, randomized, multicenter, parallel‐group phase 2/3 study randomized Japanese MDD patients (Hamilton Rating Scale for Depression 17‐item total score ≥ 14; historical inadequate response to 1–3 ADTs) with inadequate response to 8‐week single‐blind, prospective SSRI/SNRI treatment to 6‐week adjunctive treatment with brexpiprazole 1 mg, 2 mg, or placebo. The primary endpoint was change in Montgomery‐Åsberg Depression Rating Scale (MADRS) total score from baseline. Secondary endpoints included MADRS response, remission rate, and Clinical Global Impression‐Improvement score. Safety was comprehensively evaluated, especially regarding antipsychotic adverse events (AEs).
Results
Of 1194 screened patients, 740 were randomized and 736 (1 mg, n = 248; 2 mg, n = 245; placebo, n = 243) had ≥1 baseline/post‐baseline MADRS total score. The LSM (SE) change from baseline in MADRS total score at Week 6 by MMRM analysis was −8.5 (0.47) with brexpiprazole 1 mg, −8.2 (0.47) with brexpiprazole 2 mg, and −6.7 (0.47) with placebo (placebo‐adjusted LSM difference [95% CI]: 1 mg, −1.7 [−3.0, −0.4]; P = 0.0089; 2 mg, −1.4 [−2.7, −0.1]; P = 0.0312). Secondary efficacy results supported the primary endpoint. Brexpiprazole was generally well tolerated.
Conclusion
Brexpiprazole 1 mg daily was an appropriate starting dose and both 1 mg and 2 mg daily were effective and well tolerated as adjunctive therapy for Japanese MDD patients not adequately responsive to ADT.
Keywords: adjunctive therapy, antipsychotic, brexpiprazole, Japanese, major depressive disorder
Major depressive disorder (MDD), a chronic, recurrent psychiatric disorder, causes significant morbidity and mortality related to suicide risk. 1 In Japan, the 12‐month prevalence of MDD (males, 2.2%; females, 3.2%) 2 is lower than the global prevalence compared with other high‐income countries. 3 Despite this, MDD is associated with enormous social and economic costs in Japan, mainly because of reduced workplace productivity and completed suicide. 4
MDD treatment guidelines from the Japanese Society of Mood Disorders (JSMD) 5 are consistent with international guidelines and recommend first‐line antidepressant therapy with one of the following agents: selective serotonin reuptake inhibitors (SSRIs), serotonin‐noradrenaline reuptake inhibitors (SNRIs), or mirtazapine, a noradrenergic and specific serotonergic antidepressant. However, treatment responses are inadequate with antidepressant monotherapy in up to approximately 50% of patients and appear to decrease with further antidepressant treatment lines. 6 Lack of remission leads to ongoing disruption of well‐being, social functioning, quality of life, and broader social and economic effects. 7 , 8 In cases of inadequate response, the JSMD guidelines suggest adjunctive therapy if improvement to dose increase or switching is only partial in terms of the spectrum of depressive symptoms or degree of improvement. 5
Among the options for adjunctive therapy, atypical antipsychotics are associated with the highest evidence levels and are the only option approved by the United States Food and Drug Administration. 9 , 10 In Japan, aripiprazole is currently the only antipsychotic agent approved as adjunctive therapy for MDD. The efficacy of aripiprazole has been confirmed in a Japanese double‐blind, placebo‐controlled trial and international double‐blind trials. 11 , 12 However, results of these trials suggest that a relatively high proportion of patients experience disruptive adverse events (AEs), especially akathisia and insomnia, which may prevent treatment for depressed patients with anxiety and insomnia symptoms. This limitation of aripiprazole justifies the need for other adjunctive therapy options. Patients also report troublesome subjective experiences, especially insomnia, anxiety, and fatigue 13 and addressing these would be of benefit.
Brexpiprazole is a serotonin–dopamine activity modulator that is a partial agonist at the serotonin 5‐HT1A and dopamine D2 receptors and an antagonist at the serotonin 5‐HT2A and noradrenaline α1B/2C receptors, all at similar potency. 14 Unlike aripiprazole, brexpiprazole exhibits subnanomolar potency against the above receptors meaning more potent actions at these receptors. 14 These partial agonist (5‐HT1A/D2) and antagonist (5‐HT2A, α1B/2C) receptor activities modulate the serotonergic, noradrenergic, and dopaminergic systems and are thought to be associated with the antipsychotic and antidepressant effects of brexpiprazole. Further, optimal D2 receptor intrinsic activity (i.e., less partial agonist activity) and potent serotonergic system effects are expected to lower the incidence and severity of akathisia and insomnia compared with aripiprazole. 15 , 16
Brexpiprazole has been approved for the treatment of schizophrenia and MDD in over 60 countries since its first approval in the US in 2015. 17 Internationally, the brexpiprazole clinical trial program for adjunctive treatment in MDD has been robust and comprehensive, including four short‐term trials (Pyxis, 18 Polaris, 19 Syrius, 20 Delphinus 21 ) primarily focused on efficacy. International phase 3 studies have consistently shown a reduction in Montgomery‐Åsberg Depression Rating Scale (MADRS) total score following adjunctive brexpiprazole therapy at doses of 2 mg and 3 mg daily, the recommended approved target and maximum doses, respectively, in the US. 18 , 19 , 20 , 21 A lower incidence of akathisia and insomnia has been reported with brexpiprazole as adjunctive therapy compared with aripiprazole, 16 and is expected to overcome these limitations of aripiprazole. This is believed to be one of the key advantages of brexpiprazole, which may also help improve long‐term adherence. 22 , 23
In Japan, brexpiprazole has been approved for the treatment of schizophrenia since January 2018 based on results of a 6‐week randomized, double‐blind, placebo‐controlled trial in patients with acute relapse, as well as a 52‐week long‐term trial in outpatients. 24 , 25 Brexpiprazole pharmacokinetics are similar between Japanese and non‐Japanese populations. 26 Therefore, brexpiprazole 2 mg daily used in clinical studies outside of Japan and approved in the US should also be effective and tolerable in Japanese patients with MDD. Brexpiprazole 1 mg daily was also chosen to confirm whether it is the lowest effective dose in Japanese patients, although this dose has not been validated in clinical studies in patients with MDD overseas. 19 In short‐term overseas trials, the starting dose was 0.5 mg/day or 1 mg/day, but there was no significant difference in the incidence of adverse events during the titration period with either starting dose. 18 , 19 , 20 , 21 Therefore, a starting dose of 1 mg daily was set, with the objectives of reaching an effective dose quickly, and corroborating if a 1 mg starting dose would represent an effective and tolerable dose in Japanese patients. On this background, the BLESS study aimed to evaluate the efficacy and tolerability of brexpiprazole at doses of 1 mg and 2 mg daily as adjunctive therapy to antidepressant therapies (ADTs) compared with adjunctive placebo, in patients with MDD who showed an inadequate response to antidepressant monotherapy.
Methods
This placebo‐controlled, randomized, multicenter, parallel‐group phase 2/3 study comprised a screening phase of up to 28 days, an 8‐week single‐blind prospective treatment phase (Phase A), and a 6‐week double‐blind, randomized, placebo‐controlled treatment phase (Phase B), followed by a 30‐day post‐treatment observation phase for patients not enrolled in a subsequent long‐term extension study (Fig. 1).
Fig. 1.
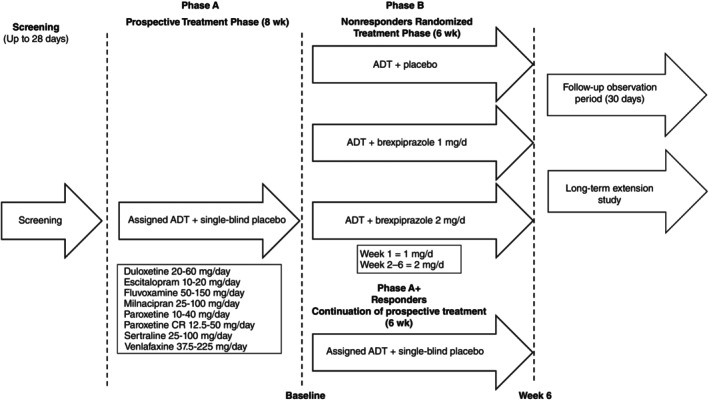
Study design. Antidepressant responders not meeting the criteria for enrolment in the randomized treatment phase were permitted to continue single‐blind antidepressant treatment for an additional 6 weeks at the same regimen and dose as the prospective treatment phase of the study (Phase A+).
The study was approved by the Institutional Review Board (IRB) at the local or central institutional review board at each participating site and complied with Good Clinical Practice (GCP) guidelines, Declaration of Helsinki, and all local laws. All patients provided written informed consent to participate in the study.
This study was registered at ClinicalTrials.gov (NCT03697603).
Patients
Screening eligibility
Japanese adults aged 20 to 64 years were eligible if they had a single or recurrent episode of MDD according to Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM‐5) criteria of ≥8 weeks' duration. Patients were required to be outpatients at the time of informed consent or able to be successfully transferred to outpatient status before enrolment in the antidepressant treatment period (Phase A).
During the current MDD episode, patients were required to have received adequate treatment with 1–3 antidepressants and have had an inadequate response to each agent. Adequate treatment was defined as any antidepressant treatment at an approved dose for ≥6 weeks. Inadequate response was defined as <50% improvement on patient self‐evaluation of their previous ADT improvement, with 100% representing complete improvement and 0% representing no improvement in depressive symptoms.
Prospective, single‐blind treatment (Phase A) eligibility
Adult outpatients who met screening criteria and had a HAM‐D 17‐item total score 27 of ≥18 (indicating moderate to severe depression) at screening and initiation of Phase A were eligible for enrolment into the 8‐week antidepressant treatment period (Phase A).
Randomized, double‐blind treatment (Phase B) eligibility
Patients with a HAM‐D 17‐item total score of ≥14 (mild to severe depression) at Week 8 of Phase A were subsequently randomized into the double‐blind treatment phase (Phase B) if they had an inadequate response to antidepressant treatment, defined as a <50% reduction rate in the HAM‐D 17‐item total score at Week 8 of Phase A compared with initiation of Phase A, and consistently had a Clinical Global Impression‐Improvement (CGI‐I) score 28 of between 3 (minimally improved) and 7 (very much worse) throughout Phase A.
Comprehensive inclusion and exclusion criteria are provided in Table S1 and a listing of prohibited concomitant medications is provided in Table S2.
Treatment and randomization
During the prospective treatment phase, patients received placebo in addition to physician‐determined treatment with an SSRI or SNRI (Table S3) distinct from the previous antidepressant in a single‐blind manner. The antidepressant treatment dose was titrated to within the approved dosage range and then fixed for the last 2 weeks of the 8‐week treatment period. Patients were withdrawn if antidepressant treatment was unable to be administered at a fixed dose and regimen for the last 2 weeks of the treatment period for tolerability reasons.
Patients who met the criteria for enrolment in the randomized treatment phase were subsequently randomized in a 1:1:1 ratio to receive double‐blind brexpiprazole 1, 2 mg, or placebo once daily for 6 weeks. Both patients and investigators were blinded to treatment during the randomized treatment phase. An interactive web response system was used for assigning treatments using a computer‐generated block randomization schedule. The allocation code was not made known to the patient or to the investigator or sub‐investigator. For the brexpiprazole 2 mg group, treatment was started at 1 mg once daily until Week 1, then increased to 2 mg once daily until Week 6. The final dosage of SSRI or SNRI used in the prospective phase was continued without any change in dosage or regimen. Study visits took place weekly during double‐blind treatment.
Antidepressant responders not meeting the criteria for enrolment in the randomized treatment phase were permitted to continue single‐blind antidepressant treatment for an additional 6 weeks at the same regimen and dose as the prospective treatment phase of the study (Phase A+).
Endpoints and assessments
The primary endpoint was the mean change from baseline (Week 8 of Phase A) in MADRS total score at Week 6. 29 Secondary endpoints, assessed at Week 6, included the MADRS response and remission rate, CGI‐I improvement rate and mean change from baseline in CGI‐S, HAM‐D 17‐item total scores, 27 mean Sheehan Disability Scale (SDS) scores, 30 and MADRS Self‐report (MADRS‐S) 31 total scores.
Safety was evaluated by assessment of treatment‐emergent adverse events (TEAEs), laboratory tests, vital signs, physical examinations, waist circumference, body weight, body mass index, 12‐lead ECG, Columbia Suicide Severity Rating Scale (C‐SSRS), drug‐induced extrapyramidal symptoms scale (DIEPSS), Abnormal Involuntary Movement Scale (AIMS), and Barnes Akathisia Rating Scale (BARS).
The incidence and severity of TEAEs (adverse events occurring after initiation of treatment in Phase B) were recorded during each study visit in the double‐blind treatment period.
Efficacy and safety‐related assessment schedule items have been detailed in Table S4.
Statistical analysis
The sample size calculation was based on an expected effect on the primary efficacy endpoint in the brexpiprazole arms compared with placebo. Based on a previous phase 3 double‐blind, placebo‐controlled trials of fixed‐dose brexpiprazole conducted outside of Japan, 18 , 19 , 20 654 evaluable patients (218 patients per group) were required to detect with 90% power a between‐group difference of −2.4 (SD, 7.7) in mean change from baseline to Week 6 in MADRS total score, at a two‐sided significance level of 0.05. Allowing for an attrition rate of 7% due to discontinuations during the double‐blind phase and exclusions from the analysis, a total of 720 randomized patients (240 patients per group) were therefore planned.
The full analysis set (FAS), used for all efficacy analyses was defined as all randomized patients who received ≥1 dose of study medication during the double‐blind phase and had ≥1 baseline and post‐baseline MADRS total score available. The safety analysis set (SAS) used for all safety analyses was defined as all patients who were randomized and received ≥1 dose of study medication during the double‐blind phase.
For the primary analysis, a mixed model repeated‐measures (MMRM) was fitted with an unstructured variance covariance structure using change from baseline during Phase B in MADRS total score as the dependent variable based on the observed cases dataset. The model included fixed effect terms for treatment group, visit (Weeks 1–6), and interaction between treatment group and visit as factors, and baseline and interaction between baseline and time point as covariates.
For each time point, least square means (LSM) for each treatment group and differences in LSM between each brexpiprazole group and the placebo group, as well as the two‐sided 95% CIs were calculated. To control overall type I error rate, a fixed sequence procedure was used. Comparison between brexpiprazole 2 mg and placebo was first performed; comparison between brexpiprazole 1 mg and placebo was performed with a two‐sided significance level of 5% only when a statistically significant difference was observed between brexpiprazole 2 mg and placebo at a two‐sided significance level of 5%.
For the MADRS response rate, MADRS remission rate, and CGI‐I improvement rate, χ2 tests were performed for between‐treatment‐group comparisons using the last observation carried forward (LOCF) data set. CGI‐S, SDS, and MADRS‐S were analyzed by MMRM in a similar way as for the primary endpoint. For the mean change from baseline in HAM‐D 17‐item total score, ANCOVA model with treatment group as factor and baseline as covariate was performed using the LOCF dataset. LSM of each treatment group and differences in LSM between each brexpiprazole group and the placebo group, as well as the two‐sided 95% CIs were determined.
All statistical analyses were performed using SAS® software version 9.4 (SAS Institute, Cary, NC, USA).
Results
A total of 1194 patients were screened from 145 facilities in Japan and were eligible for inclusion in the antidepressant treatment phase (Phase A) (Fig. 2). The trial was held from 30 July 2018 to 4 July 2022. Of these, 1192 patients received antidepressant treatment and 108 patients discontinued treatment during the prospective treatment phase, while 346 patients were considered responders and continued prospective treatment (Phase A+).
Fig. 2.
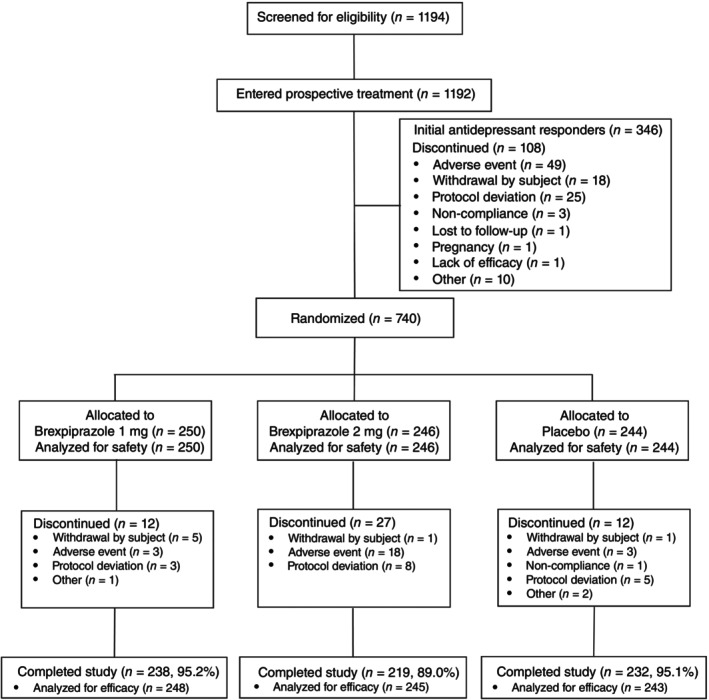
Patient disposition. Percentages are based on the number of subjects in treatment group; Safety analysis set comprise subjects who have received at least one dose of investigational product in Phase B; Full analysis set comprise subjects who have received at least one dose of investigational product in Phase B, and from whom MADRS total scores have been obtained at baseline and at least one post baseline timepoint.
A total of 740 patients were eligible for enrolment in the randomized, double‐blind treatment phase and received ≥1 dose of brexpiprazole (1 mg, n = 250; 2 mg, n = 246) or placebo (n = 244), which comprised the SAS. Of these, 736 patients (1 mg, n = 248; 2 mg, n = 245; placebo, n = 243) had ≥1 baseline and post‐baseline MADRS total score available, which comprised the FAS. Patient disposition is further summarized in Fig. 2. 689 (93.1%) patients completed treatment (brexpiprazole 1 mg, 95.2%; brexpiprazole 2 mg, 89.0%; placebo 95.1%).
Baseline demographics and clinical characteristics were generally similar across treatment groups in the randomized, double‐blind phase (Table 1). Mean (SD) MADRS total scores were similar between brexpiprazole 1 mg (26.7 [6.4]), 2 mg (26.9 [6.9]) and placebo groups (27.3 [6.2]) and indicated that patients generally had moderately severe MDD (20–34). For the overall population, the most commonly used concomitant ADT were escitalopram, sertraline, duloxetine, and venlafaxine. Details of assigned concomitant ADT are included in Table S5.
Table 1.
Patient demographics and baseline characteristics (Phase B, FAS)
| Brexpiprazole | ||||
|---|---|---|---|---|
| 1 mg (n = 248) | 2 mg (n = 245) | Total (n = 493) | Placebo (n = 243) | |
| Age, years, mean (SD) | 40.9 (10.8) | 40.0 (10.7) | 40.4 (10.7) | 39.8 (10.8) |
| Sex, male, n (%) | 132 (53.2) | 143 (58.4) | 275 (55.8) | 138 (56.8) |
| BMI, kg/m2, mean (SD) | 23.5 (4.2) | 23.8 (4.6) | 23.6 (4.4) | 23.4 (4.6) |
| Duration of the current episode (months), mean (SD) | 12.2 (16.5) | 13.0 (19.1) | 12.6 (17.9) | 13.5 (22.4) |
| Number of adequate antidepressant treatments in the current episode, n (%) | ||||
| 1 | 191 (77.0) | 185 (75.5) | 376 (76.3) | 189 (77.8) |
| 2 | 49 (19.8) | 48 (19.6) | 97 (19.7) | 43 (17.7) |
| 3 | 8 (3.2) | 12 (4.9) | 20 (4.1) | 11 (4.5) |
| DSM‐5 diagnosis, n (%) | ||||
| Single episode | 95 (38.3) | 98 (40.0) | 193 (39.1) | 91 (37.4) |
| Recurrent episode | 153 (61.7) | 147 (60.0) | 300 (60.9) | 152 (62.6) |
| MADRS total score, mean (SD) | 26.7 (6.4) | 26.9 (6.9) | 26.8 (6.6) | 27.3 (6.2) |
| HAM‐D 17 total score, mean (SD) | 20.5 (4.0) | 20.8 (3.9) | 20.7 (3.9) | 21.2 (3.7) |
| CGI‐S score, mean (SD) | 4.1 (0.6) | 4.2 (0.6) | 4.1 (0.6) | 4.1 (0.6) |
Abbreviations: BMI, body mass index; CGI‐S, Clinical Global Impression‐Severity of Illness; DSM‐5, Diagnostic and Statistical Manual of Mental Disorders Fifth Edition; FAS, full analysis set; HAM‐D, Hamilton Rating Scale for Depression; MADRS, Montgomery‐Åsberg Depression Rating Scale; SD, standard deviation.
Efficacy
Primary endpoint
In the FAS, the LSM (SE) change from baseline in MADRS total score at Week 6 by MMRM analysis was −8.5 (0.47) in the brexpiprazole 1 mg group and − 8.2 (0.47) in the brexpiprazole 2 mg group versus −6.7 (0.47) in the placebo group (Table 2, Fig. 3). Placebo‐adjusted LSM treatment differences were statistically significant for both brexpiprazole 1 mg (LSM difference [95% CI]: −1.7 [−3.0, −0.4]; P = 0.0089) and 2 mg (LSM difference [95% CI]: −1.4 [−2.7, −0.1]; P = 0.0312) treatment groups. Improvements in MADRS total score were rapid with both brexpiprazole 1 mg and 2 mg, with separation from placebo observed as early as Week 2 (Fig. 3).
Table 2.
Primary and secondary endpoints at Week 6 of the double‐blind treatment period (Phase B)
| Brexpiprazole | Placebo | ||
|---|---|---|---|
| 1 mg (n = 248) | 2 mg (n = 245) | (n = 243) | |
| MADRS Total Score, MMRM | |||
| Mean (SD) at baseline | 26.7 (6.4) | 26.9 (6.9) | 27.3 (6.2) |
| LSM change at Week 6 (SE) | −8.5 (0.47) | −8.2 (0.47) | −6.7 (0.47) |
| Treatment difference (95% CI) | −1.7 (−3.0, −0.4) | −1.4 (−2.7, −0.1) | |
| P‐value | 0.0089 | 0.0312 | |
| MADRS Response Rate † | |||
| Proportion of responders at Week 6 (LOCF), n (%) | 63 (25.4) | 60 (24.5) | 46 (18.9) |
| Treatment difference (95% CI) | 6.5 (−0.8, 13.8) | 5.6 (−1.7, 12.9) | |
| P‐value | 0.0844 | 0.1364 | |
| MADRS Remission Rate ‡ | |||
| Proportion of patients in remission at Week 6 (LOCF), n (%) | 44 (17.7) | 43 (17.6) | 33 (13.6) |
| Treatment difference (95% CI) | 4.2 (−2.3, 10.6) | 4.0 (−2.5, 10.4) | |
| P‐value | 0.2048 | 0.2265 | |
| CGI‐I Improvement Rate § | |||
| Proportion of responders at Week 6 (LOCF), n (%) | 87 (35.1) | 86 (35.1) | 72 (29.6) |
| Treatment difference (95% CI) | 5.5 (−2.8, 13.7) | 5.5 (−2.8, 13.8) | |
| P value | 0.1969 | 0.1964 | |
| CGI‐S Score, MMRM | |||
| Mean (SD) at baseline | 4.1 (0.6) | 4.2 (0.6) | 4.1 (0.6) |
| LSM change at Week 6 (SE) | −0.7 (0.05) | −0.8 (0.05) | −0.6 (0.05) |
| Treatment difference (95% CI) | −0.1 (−0.3, 0.0) | −0.2 (−0.3, 0.0) | |
| P‐value | 0.0976 | 0.0292 | |
| HAM‐D 17 Total Score, ANCOVA | |||
| Mean (SD) at baseline | 20.5 (4.0) ¶ | 20.8 (3.9) | 21.2 (3.7) |
| LSM change at Week 6 (SE) | −6.3 (0.34) | −5.6 (0.35) | −5.3 (0.35) |
| Treatment difference (95% CI) | −1.0 (−2.0, −0.1) | −0.3 (−1.3, 0.6) | |
| P‐value | 0.0330 | 0.4914 | |
| Mean SDS Score, MMRM | |||
| Mean (SD) at baseline | 5.38 (2.16) †† | 5.62 (2.07) ‡‡ | 5.62 (2.11) §§ |
| LSM change at Week 6 (SE) | −1.13 (0.114) | −1.12 (0.118) | −0.64 (0.115) |
| Treatment difference (95% CI) | −0.49 (−0.81, −0.17) | −0.48 (−0.80, −0.15) | |
| P‐value | 0.0026 | 0.0038 | |
| MADRS‐S Total Score, MMRM | |||
| Mean (SD) at baseline | 10.60 (4.19) | 10.90 (4.59) | 11.01 (4.28) |
| LSM change at Week 6 (SE) | −2.46 (0.199) | −1.87 (0.203) | −1.47 (0.200) |
| Treatment difference (95% CI) | −0.99 (−1.55, −0.44) | −0.41 (−0.97, 0.15) | |
| P‐value | 0.0005 | 0.1543 | |
Abbreviations: ANCOVA, analysis of covariance; CGI‐I, Clinical Global Impression‐Improvement; CGI‐S, Clinical Global Impression‐Severity of Illness; CI, confidence interval; HAM‐D, Hamilton Rating Scale for Depression; LOCF, last observation carried forward; LSM, least squares mean; MADRS, Montgomery‐Åsberg Depression Rating Scale; MADRS‐S, MADRS Self‐report; MMRM, mixed model repeated‐measures; SD, standard deviation; SE, standard error; SDS, Sheehan Disability Scale.
MADRS response rate at Week 6, defined as the proportion of patients with a reduction of ≥50% in MADRS total score from baseline at Week 6.
MADRS remission rate at Week 6, defined as the proportion of patients with a reduction of ≥50% in MADRS total score from baseline at Week 6 and a MADRS total score of ≤10 at Week 6.
CGI‐I improvement rate at Week 6, defined as the proportion of patients with a score of 1 or 2 on the CGI‐I scale at Week 6.
n = 247.
n = 240.
n = 234.
n = 241.
Fig. 3.
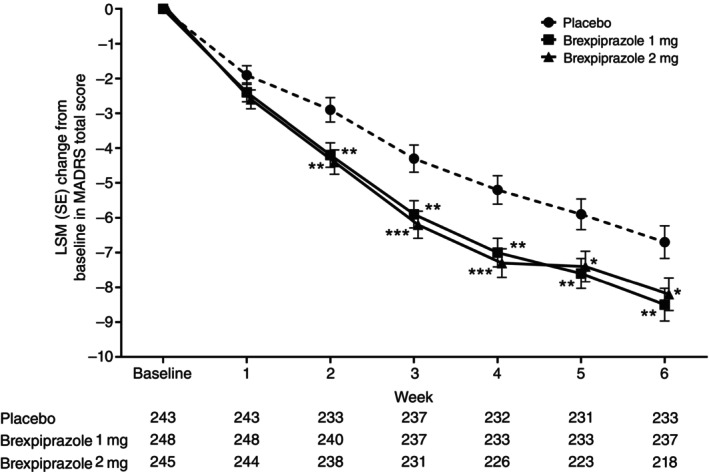
LSM (SE) change from baseline in MADRS total score by MMRM analysis (FAS population). *P < 0.05; **P < 0.01; ***P < 0.001.
Secondary endpoints
In the FAS, numerical improvements were observed across all secondary endpoints in favor of brexpiprazole 1 mg and 2 mg versus placebo (Table 2), supporting the results of the primary endpoint. Brexpiprazole 1 mg showed greater efficacy than placebo (P < 0.05) in mean change from baseline at Week 6 in HAM‐D 17, SDS and MADRS‐S. Brexpiprazole 2 mg showed greater efficacy than placebo (P < 0.05) in mean change from baseline at Week 6 in CGI‐S and SDS (Table 2). Compared to placebo, brexpiprazole 1 mg did not show significant improvement in MADRS response rate, MADRS remission rate, CGI‐I improvement rate and CGI‐S at Week 6, while brexpiprazole 2 mg did not show significant improvement in MADRS response rate, MADRS remission rate, CGI‐I improvement rate, HAM‐D 17 and MADRS‐S at Week 6 (Table 2).
Safety
TEAEs, including those with incidence ≥5% in either brexpiprazole group, are summarized in Table 3. TEAEs of any severity grade occurred in 62.0% of patients receiving brexpiprazole 1 mg, 74.0% of patients receiving brexpiprazole 2 mg, and 59.0% of patients receiving placebo. The most common (≥5%) reported TEAEs were weight gain, tremor, akathisia, and nasopharyngitis in the brexpiprazole 1 mg group and akathisia, weight gain, increased serum prolactin level, hyperprolactinemia and nasopharyngitis in the brexpiprazole 2 mg group (Table 3). Most TEAEs were mild or moderate in severity. Severe TEAEs reported by ≥2 patients in either brexpiprazole group were akathisia (1 mg: n = 1; 2 mg: n = 2; placebo: n = 0) and malaise (1 mg: n = 0; 2 mg: n = 2, placebo: n = 0). TEAEs leading to treatment discontinuation were noted in 0.8% of patients receiving brexpiprazole 1 mg, 7.3% of patients receiving brexpiprazole 2 mg, and 1.2% of patients receiving placebo with akathisia being the most common individual cause for discontinuation (brexpiprazole 2 mg: n = 6) and a variety of other causes occurring at low individual rates. In total, eight serious TEAEs were noted during the randomized treatment phase (1 mg: 1.2% [n = 3]; 2 mg: 1.2% [n = 3]; placebo: 0.8% [n = 2]) and constituted the following individual events: appendicitis, intentional overdose, oculomotor nerve paralysis, alcoholic pancreatitis, extramammary Paget's disease, epilepsy, cellulitis, and subarachnoid hemorrhage. However, there were no serious drug‐related TEAEs and no deaths occurred in this study. No clinically meaningful changes in laboratory test values, vital signs, or ECG parameters were observed either with brexpiprazole or placebo. When the incidence of TEAEs was examined by CYP2D6 phenotype (IM, EM, PM, Unknown) and by concomitant medication (ADT), there was no significant difference in the incidence of TEAEs in any subgroup, except for some subgroups with fewer subjects. However, plasma brexpiprazole concentrations were higher in IM patients compared to EM patients in all dose groups, except at certain time points.
Table 3.
Summary of TEAEs and occurrence of TEAEs with incidence ≥5% in either brexpiprazole group during the double‐blind treatment period (SAS)
| Brexpiprazole | Placebo | |||
|---|---|---|---|---|
| 1 mg (n = 250) | 2 mg (n = 246) | Total (n = 496) | (n = 244) | |
| TEAEs | 155 (62.0) | 182 (74.0) | 337 (67.9) | 144 (59.0) |
| Serious TEAEs | 3 (1.2) | 3 (1.2) | 6 (1.2) | 2 (0.8) |
| Severe TEAEs | 4 (1.6) | 5 (2.0) | 9 (1.8) | 2 (0.8) |
| Discontinuation due to TEAEs | 2 (0.8) | 18 (7.3) | 20 (4.0) | 3 (1.2) |
| Deaths | 0 | 0 | 0 | 0 |
| Occurrence of TEAEs ≥5% (PT) | ||||
| Akathisia | 15 (6.0) | 60 (24.4) | 75 (15.1) | 3 (1.2) |
| Weight increased | 18 (7.2) | 19 (7.7) | 37 (7.5) | 6 (2.5) |
| Nasopharyngitis | 20 (8.0) | 16 (6.5) | 36 (7.3) | 24 (9.8) |
| Tremor | 16 (6.4) | 12 (4.9) | 28 (5.6) | 9 (3.7) |
| Blood prolactin increased | 6 (2.4) | 15 (6.1) | 21 (4.2) | 6 (2.5) |
| Hyperprolactinemia | 3 (1.2) | 13 (5.3) | 16 (3.2) | 3 (1.2) |
Note: Data are presented as n (%).
Abbreviations: PT, preferred term; TEAE, treatment‐emergent adverse event.
Aside from akathisia, the rates of activating TEAEs were low overall, including insomnia (1 mg: 3.6%; 2 mg: 4.9%; placebo: 3.3%), restlessness (1 mg: 0.0%; 2 mg: 0.4%; placebo: 0.0%), and anxiety (1 mg: 0.0%; 2 mg: 0.8%; placebo: 0.0%), and occurred at a similar rate to placebo. Extrapyramidal disorder events as a MedDRA preferred term occurred in 4.5% of patients receiving brexpiprazole 2 mg, compared with 0.4% of patients receiving brexpiprazole 1 mg and 0.8% of patients receiving placebo. Most such extrapyramidal disorder events were either mild or moderate in severity. Other subjective negative experiences for patients that have been associated with antipsychotics occurred at a low rate with brexpiprazole, including insomnia, anxiety, and fatigue. Most events of this nature were also mild or moderate in severity, and few cases resulted in discontinuation (e.g., one patient each in the brexpiprazole 2 mg group discontinued due to insomnia or anxiety).
In terms of TEAEs of interest known to be related to antipsychotics, the incidence of extrapyramidal symptom‐related TEAEs was higher with brexpiprazole (1 mg: 14.8%; 2 mg: 36.6%), compared with placebo (7.4%) although all such symptoms were mild to moderate in severity and no cases of tardive dyskinesia were noted. The LSM (SE) change in DIEPSS total score from baseline to the worst point after administration was 0.4 ± 0.10 with brexpiprazole 1 mg and 1.2 ± 0.10 with brexpiprazole 2 mg. Regarding tardive dyskinesia and akathisia, the AIMS score and the BARS assessment, respectively, showed small changes from baseline to the 6‐week evaluation in the 1 mg, 2 mg, and placebo groups and no clinically meaningful changes were observed. Prolactin levels increased from baseline at final assessment with brexpiprazole but remained within the reference range at this point for both men and women. As noted previously, weight gain was greater among patients treated with brexpiprazole but all events were mild or moderate in severity and none led to treatment discontinuation.
Regarding the time of onset of TEAEs, the greatest single proportion of events in each group occurred in between day 1 and day 7 of treatment (1 mg: 22.0%; 2 mg: 27.2%; placebo: 23.0%) with no events having a higher incidence with increasing duration of treatment. Further, there were no significant differences in the incidence of TEAEs by time of first onset between the brexpiprazole and placebo groups.
In terms of C‐SSRS results, no suicidal behavior was observed at baseline while one patient in the brexpiprazole 2 mg group exhibited suicidal behavior after administration. After administration of treatment, the rate of suicidal ideation was 15.2% of patients in the brexpiprazole 1 mg group, 19.5% of patients in the brexpiprazole 2 mg group and 17.6% of patients in the placebo group.
Discussion
This placebo‐controlled, randomized, phase 2/3 study is the first to comprehensively evaluate the efficacy and tolerability of brexpiprazole as adjunctive therapy in Japanese patients with MDD who showed an inadequate response to antidepressant monotherapy. Improvements in patients receiving adjunct brexpiprazole were greater than in patients taking adjunct placebo, as shown by statistically significant differences versus placebo in the primary endpoint of change from baseline in MADRS total score at week 6 for both brexpiprazole 1 mg and 2 mg. Numerical improvements in secondary endpoints in favor of brexpiprazole 1 mg and 2 mg versus placebo reflected the results of the primary endpoint. Overall, TEAEs were common in all treatment groups, but there were few serious TEAEs. The incidence of certain TEAEs, including akathisia and elevated serum prolactin levels, tended to be higher in patients who received brexpiprazole 2 mg. However, most TEAEs were mild or moderate in severity. The most common TEAEs associated with brexpiprazole were akathisia, weight gain, nasopharyngitis, tremor, increased serum prolactin level, and hyperprolactinemia. There were no notable differences in the incidence of TEAEs by first onset between brexpiprazole and placebo. As such, there is no suggestion that the incidence of tolerability issues increases over time with brexpiprazole.
Efficacy results in the present study are generally consistent with those reported in similar international phase 3 trials of brexpiprazole for this indication. 18 , 19 , 20 Baseline depression scores in the present study were similar to those in pivotal overseas trials among non‐Japanese patients. It should be noted that the primary endpoint was not achieved with brexpiprazole 1 mg in these overseas trials but, for the first time, was achieved among Japanese patients in this study. Given current prescribing patterns in Japan for MDD, it is also worthwhile to compare these results with those of the ADMIRE study, which was designed to evaluate the efficacy and safety of adjunctive aripiprazole in Japanese patients with MDD. 12 Although it is not possible to make an unambiguous comparison due to differences in patient backgrounds and the time of the trial, the design, patient population, and inclusion criteria of the current study and ADMIRE are sufficiently similar to allow such comparisons. Briefly, patients in the 3‐arm ADMIRE study were randomized to adjunctive aripiprazole either as a fixed 3 mg daily dose or a flexible 3–15 mg daily dose or to placebo and were studied over 6 weeks, with an identical primary endpoint as the current study. Change in mean MADRS total score for the adjunctive fixed dose or flexible dose of aripiprazole were significantly greater than with adjunctive placebo. The differences in mean MADRS total score versus placebo are −2.2 to −3.1 with aripiprazole in the ADMIRE study compared with −1.4 to −1.7 in the current trial. However, some of this difference between aripiprazole and brexpiprazole may relate to differences in patient background. For example, the proportion of patients with recurrent episodes at baseline in the BLESS study (60.0–62.6%) was notably higher than in the ADMIRE study (38.1–47.4%) and recurrent episodes have been identified as more difficult to treat. 32 In Japanese patients, it appears that, brexpiprazole 1 mg is an appropriate starting dose at which the primary endpoint was met thus also making it an effective target dose. Further, 1 mg may also avoid the increased tolerability issues associated with 2 mg. As mentioned above, this represents one of the key novel features of this study.
In terms of tolerability, results of the present study were generally consistent with those of international trials with various points of difference in terms of incidence. In international trials, the incidence of akathisia with brexpiprazole 1 mg (4.4%) and 2 mg (7.4–8.3%) were lower than in patients who received brexpiprazole 1 mg (6.0%) but especially those who received 2 mg (24.4%) in this study. The incidence of akathisia with aripiprazole observed in the ADMIRE study was notably higher than in the present study, especially with the flexible dosing strategy (36.6% for aripiprazole 3–15 mg versus 4.1% for placebo) but also with the fixed dosing strategy (14.2% for aripiprazole 3 mg versus 4.1% for placebo). Although there was no detected bias in patient background factors related to akathisia among the studies, 33 the pathophysiology of akathisia is poorly understood. However, as mentioned previously, a relatively lower level of intrinsic activity at D2 receptors and more potent actions at 5HT1A/2A, than aripiprazole may underlie the lower rates of akathisia as well as agitation and activating symptoms noted in relevant clinical trials. 15 It should be reinforced that most cases of akathisia in the present study were mild or moderate in severity and severe cases were reported in only one patient in the brexpiprazole 1 mg group and two patients in the brexpiprazole 2 mg group (versus no patients in the placebo group). The discontinuation rate from akathisia among patients who received brexpiprazole 2 mg was approximately 2.4% compared with 0.0% among patients who received brexpiprazole 1 mg or placebo. Further, most cases of akathisia resolved either following treatment or upon discontinuation. Results of assessments such as BARS showed small changes from baseline to the final evaluation in all treatment groups, with no clinically meaningful changes at the final assessment.
The apparent large increase in the incidence of akathisia between 1 mg and 2 mg in the present study also deserves more detailed discussion. In international trials, the incidence of akathisia across the range of 1–3 mg daily has been noted to show a monotonic increasing trend, 34 whereas the incidence in Japanese patients treated with 2 mg seems to deviate from this trend. The lack of apparent difference in BARS scores should be considered in light of the fact that this was only assessed at two observation points, which may be insufficient to detect changes occurring during treatment. Some CYP2D6 phenotypes and the type of concomitant ADT used may have an effect on brexpiprazole blood levels and the CYP2D6*10 allele has also been suggested as a risk factor for extrapyramidal symptoms, including akathisia, in Asian patients with mood disorders. 35 , 36 However, the incidence of AEs examined by CYP2D6 phenotype (IM, EM, PM, Unknown) and by concomitant medication (ADT) showed there were no significant differences, except for some subgroups with fewer patients. This suggests that these genetic factors did not affect akathisia incidence in this study. Differences in pharmacokinetics and pharmacodynamics of brexpiprazole due to possible impact of gene variability requires further exploration. Unfortunately, however, data on the impact of genetic variations on brexpiprazole pharmacodynamics and tolerability are still limited. 37
Despite their widespread use, attention to potential tolerability issues other than akathisia is important when considering using antipsychotic medication in patients with MDD. 38 Extrapyramidal symptoms, metabolic syndrome effects, increased prolactin levels, and subjective negative experiences such as insomnia are particularly important to consider. A systematic review and meta‐analysis of antipsychotics found that brexpiprazole provided a favorable number‐needed‐to‐harm (NNH) value compared with antipyschotics overall (NNH: brexpiprazole, 57; overall, 37). 9 Further, NNH values associated with brexpiprazole for weight gain (NNH = 20) and akathisia (NNH = 17) were more favorable than certain other antipsychotics with available data (NNH for weight gain for olanzapaine, 9; akathisia for aripiprazole, 7). 9 Results from our study found that the incidence of insomnia, which represents a key activating TEAE of brexpiprazole, was relatively similar compared with international trials. Considering the differences with the placebo group, the incidence of insomnia was lower with brexpiprazole in the current trial than with aripiprazole in the ADMIRE study. Weight gain also occurred at a similar rate in both Japanese and non‐Japanese patients although incidences were consistently higher than in placebo‐treated patients in both populations. A recent large international preference study identified insomnia, anxiety, and fatigue as the most important non‐serious AEs troubling to patients treated for depression. 13 The fact that the incidence of these specific AEs was relatively low in the present trial is reassuring.
Limitations of the current study are similar to those of previous studies of brexpiprazole for this indication and are mainly the short treatment duration and lack of an active comparator. Regarding this, a long‐term extension of this study was conducted (NCT03737474) and will be published separately. The generalizability of this study is also limited to Japanese patients and with respect to the eligibility criteria. However, given the primary purpose of this study in evaluating the efficacy and tolerability of brexpiprazole specifically among Japanese patients with MDD and inadequate response to antidepressant therapy, this should not be viewed as a notable limitation. Notwithstanding this, further studies, including real‐world studies and comparative studies among Japanese patients would provide useful additional information for clinicians.
Conclusion
Results of this study found that brexpiprazole 1 mg daily was an appropriate starting dose and brexpiprazole 1 mg and 2 mg daily were effective and well tolerated as adjunctive therapy for Japanese patients with MDD not adequately responsive to ADT monotherapy.
Author contributions
Study concept and design: M Kato, M Shiosakai, K Kuwahara, K Iba, Y Shimada, M Saito, T Higuchi. Acquisition of data: M Kato, M Shiosakai, K Kuwahara, K Iba, Y Shimada, M Saito, T Higuchi. Analysis and interpretation of data, drafting of the manuscript, revising it for intellectual content, and final approval of the completed manuscript: All authors.
Disclosure statement
M.K. reports consulting fees for this study from Otsuka Pharmaceutical Co., Ltd.; consulting fees from outside of this study from Sumitomo Pharma Co., Ltd., Shionogi & Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Lundbeck Japan K.K., Takeda Pharmaceutical Co., Ltd.; payment or honoraria for lectures, presentations, speakers' bureaus, manuscript writing or educational events from Sumitomo Pharma Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Meiji Seika Pharma Co., Ltd., Eli Lilly Japan K.K., MSD K.K., Pfizer Japan Inc., Janssen Pharmaceutical K.K., Shionogi & Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical Co., Ltd., Lundbeck Japan K.K., Viatris Inc., Eisai Co., Ltd., Kyowa Pharmaceutical Industry Co., Ltd., Ono Pharmaceutical Co., Ltd. M.S., K.K., K.I., Y.S., M.S., Y.I., D.S. and K.A. are full‐time employees of Otsuka Pharmaceutical Co., Ltd. N.K. was a full‐time emloyee of Otsuka Pharmaceutical Co., Ltd. T.H. reports consulting fees for this study and honoraria for lectures from Otsuka Pharmaceutical Co., Ltd.
Supporting information
Table S1. Eligibility Criteria.
Table S2. List of prohibited concomitant medications.
Table S3. Commercially available selective serotonin‐reuptake inhibitors and Serotonin‐Norepinephrine Reuptake Inhibitors.
Table S4. Schedule of assessments during double‐blind period (Phase B).
Table S5. Antidepressant therapy at baseline.
Acknowledgements
The authors thank all clinicians for their involvement and contribution to the study. Medical writing support was provided by Jordana Campbell, BSc, CMPP, and Mark Snape, MBBS, CMPP, of inScience Communications, Springer Healthcare. This medical writing assistance was funded by Otsuka Pharmaceutical Co., Ltd.
Data availability statement
Anonymized individual participant data that underlie the results of this study will be shared with researchers to achieve aims prespecified in a methodologically sound research proposal.
References
- 1. Cai H, Xie X‐M, Zhang Q et al. Prevalence of suicidality in major depressive disorder: A systematic review and meta‐analysis of comparative studies. Front. Psych. 2021; 12: 690130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nishi D, Ishikawa H, Kawakami N. Prevalence of mental disorders and mental health service use in Japan. Psychiatry Clin. Neurosci. 2019; 73: 458–465. [DOI] [PubMed] [Google Scholar]
- 3. Ishikawa H, Kawakami N, Kessler RC. Lifetime and 12‐month prevalence, severity and unmet need for treatment of common mental disorders in Japan: Results from the final dataset of world mental health Japan survey. Epidemiol. Psychiatr. Sci. 2016; 25: 217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Okumura Y, Higuchi T. Cost of depression among adults in Japan. Prim. Care Companion CNS Disord. 2011; 13: PCC.10m01082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Committee for Treatment Guidelines of Mood Disorders of the Japanese Society of Mood Disorders . Guidelines for Treatment of Depression, 2nd edn. Igaku‐shoin Ltd., Tokyo, 2017. [Google Scholar]
- 6. Rush AJ, Trivedi MH, Wisniewski SR et al. Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: A star*d report. Am. J. Psychiatry 2006; 163: 1905–1917. [DOI] [PubMed] [Google Scholar]
- 7. Kessler RC. The costs of depression. Psychiatr. Clin. North Am. 2012; 35: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McIntyre RS, O'Donovan C. The human cost of not achieving full remission in depression. Can. J. Psychiatry 2004; 49: 10s–16s. [PubMed] [Google Scholar]
- 9. Kishimoto T, Hagi K, Kurokawa S, Kane JM, Correll CU. Efficacy and safety/tolerability of antipsychotics in the treatment of adult patients with major depressive disorder: A systematic review and meta‐analysis. Psychol. Med. 2023; 53: 4064–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nuñez NA, Joseph B, Pahwa M et al. Augmentation strategies for treatment resistant major depression: A systematic review and network meta‐analysis. J. Affect. Disord. 2022; 302: 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berman RM, Fava M, Thase ME et al. Aripiprazole augmentation in major depressive disorder: A double‐blind, placebo‐controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009; 14: 197–206. [DOI] [PubMed] [Google Scholar]
- 12. Kamijima K, Higuchi T, Ishigooka J et al. Aripiprazole augmentation to antidepressant therapy in japanese patients with major depressive disorder: A randomized, double‐blind, placebo‐controlled study (ADMIRE study). J. Affect. Disord. 2013; 151: 899–905. [DOI] [PubMed] [Google Scholar]
- 13. Chevance A, Tomlinson A, Ravaud P et al. Important adverse events to be evaluated in antidepressant trials and meta‐analyses in depression: A large international preference study including patients and healthcare professionals. Evid. Based Ment. Health 2022; 25: e41–e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maeda K, Sugino H, Akazawa H et al. Brexpiprazole I: In vitro and in vivo characterization of a novel serotonin‐dopamine activity modulator. J. Pharmacol. Exp. Ther. 2014; 350: 589–604. [DOI] [PubMed] [Google Scholar]
- 15. Stahl SM. Mechanism of action of brexpiprazole: Comparison with aripiprazole. CNS Spectr. 2016; 21: 1–6. [DOI] [PubMed] [Google Scholar]
- 16. Citrome L. Activating and sedating adverse effects of second‐generation antipsychotics in the treatment of schizophrenia and major depressive disorder: Absolute risk increase and number needed to harm. J. Clin. Psychopharmacol. 2017; 37: 138–147. [DOI] [PubMed] [Google Scholar]
- 17. US Food And Drug Administration . Rexulti (brexpiprazole) tablets, for oral use. 2015. Available from: https://www.Accessdata.Fda.Gov/drugsatfda_docs/label/2023/205422s009lbl.Pdf [Cited 21 August 2023].
- 18. Thase ME, Youakim JM, Skuban A et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: A phase 3, randomized, placebo‐controlled study in patients with inadequate response to antidepressants. J. Clin. Psychiatry 2015; 76: 1224–1231. [DOI] [PubMed] [Google Scholar]
- 19. Thase ME, Youakim JM, Skuban A et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: A phase 3, randomized, double‐blind study. J. Clin. Psychiatry 2015; 76: 1232–1240. [DOI] [PubMed] [Google Scholar]
- 20. Hobart M, Skuban A, Zhang P et al. A randomized, placebo‐controlled study of the efficacy and safety of fixed‐dose brexpiprazole 2 mg/d as adjunctive treatment of adults with major depressive disorder. J. Clin. Psychiatry 2018; 79: 17m12058. [DOI] [PubMed] [Google Scholar]
- 21. Hobart M, Skuban A, Zhang P et al. Efficacy and safety of flexibly dosed brexpiprazole for the adjunctive treatment of major depressive disorder: A randomized, active‐referenced, placebo‐controlled study. Curr. Med. Res. Opin. 2018; 34: 633–642. [DOI] [PubMed] [Google Scholar]
- 22. Poyurovsky M. Acute antipsychotic‐induced akathisia revisited. Br. J. Psychiatry 2010; 196: 89–91. [DOI] [PubMed] [Google Scholar]
- 23. Thippaiah SM, Fargason RE, Birur B. Struggling to find effective pharmacologic options for akathisia? B‐CALM! Psychopharmacol. Bull. 2021; 51: 72–78. [PMC free article] [PubMed] [Google Scholar]
- 24. Ishigooka J, Iwashita S, Tadori Y. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia in Japan: A 6‐week, randomized, double‐blind, placebo‐controlled study. Psychiatry Clin. Neurosci. 2018; 72: 692–700. [DOI] [PubMed] [Google Scholar]
- 25. Ishigooka J, Iwashita S, Tadori Y. Long‐term safety and effectiveness of brexpiprazole in Japanese patients with schizophrenia: A 52‐week, open‐label study. Psychiatry Clin. Neurosci. 2018; 72: 445–453. [DOI] [PubMed] [Google Scholar]
- 26. Ishigooka J, Iwashita S, Higashi K, Liew EL, Tadori Y. Pharmacokinetics and safety of brexpiprazole following multiple‐dose administration to Japanese patients with schizophrenia. J. Clin. Pharmacol. 2018; 58: 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamilton M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960; 23: 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guy W (ed.). ECDEU assessment manual for psychopharmacology. Rockville, md: Us department of heath, education, and welfare public health service alcohol, drug abuse, and mental health administration. 1976.
- 29. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br. J. Psychiatry 1979; 134: 382–389. [DOI] [PubMed] [Google Scholar]
- 30. Leon AC, Olfson M, Portera L, Farber L, Sheehan DV. Assessing psychiatric impairment in primary care with the sheehan disability scale. Int. J. Psychiatry Med. 1997; 27: 93–105. [DOI] [PubMed] [Google Scholar]
- 31. Fantino B, Moore N. The self‐reported Montgomery‐Asberg depression rating scale is a useful evaluative tool in major depressive disorder. BMC Psychiatry 2009; 9: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hardeveld F, Spijker J, De Graaf R, Nolen WA, Beekman AT. Recurrence of major depressive disorder and its predictors in the general population: Results from The Netherlands mental health survey and incidence study (NEMESIS). Psychol. Med. 2013; 43: 39–48. [DOI] [PubMed] [Google Scholar]
- 33. Sachdev P. The epidemiology of drug‐induced akathisia: Part I. Acute akathisia. Schizophr. Bull. 1995; 21: 431–449. [DOI] [PubMed] [Google Scholar]
- 34. Furukawa Y, Oguro S, Obata S, Hamza T, Ostinelli EG, Kasai K. Optimal dose of brexpiprazole for augmentation therapy of antidepressant‐refractory depression: A systematic review and dose‐effect meta‐analysis. Psychiatry Clin. Neurosci. 2022; 76: 416–422. [DOI] [PubMed] [Google Scholar]
- 35. Bai Y, Chen G, Yang H, Gao K. Do Asian and north American patients with bipolar disorder have similar efficacy, tolerability, and safety profile during clinical trials with atypical antipsychotics? J. Affect. Disord. 2020; 261: 259–270. [DOI] [PubMed] [Google Scholar]
- 36. Han C, Pae C‐U. Do we need to consider ethno‐cultural variation in the use of atypical antipsychotics for Asian patients with major depressive disorder? CNS Drugs 2013; 27: 47–51. [DOI] [PubMed] [Google Scholar]
- 37. Vasiliu O. The pharmacogenetics of the new‐generation antipsychotics – A scoping review focused on patients with severe psychiatric disorders. Front Psychiatry 2023; 14: 1124796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang P, Si T. Use of antipsychotics in the treatment of depressive disorders. Shanghai Arch. Psychiatry 2013; 25: 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Eligibility Criteria.
Table S2. List of prohibited concomitant medications.
Table S3. Commercially available selective serotonin‐reuptake inhibitors and Serotonin‐Norepinephrine Reuptake Inhibitors.
Table S4. Schedule of assessments during double‐blind period (Phase B).
Table S5. Antidepressant therapy at baseline.
Data Availability Statement
Anonymized individual participant data that underlie the results of this study will be shared with researchers to achieve aims prespecified in a methodologically sound research proposal.