

Abstract
Using proteomics, we identified nucleoside diphosphate kinase A (NDPKA; also known as NME/NM23 nucleoside diphosphate kinase 1: NME1) to be up-regulated in primary cortical neuronal cultures by erythropoietin (EPO) preconditioning. To investigate a neuroprotective role of NDPKA in neurons, we used a RNAi construct to knock-down and an adenoviral vector to overexpress the protein in cortical neuronal cultures prior to exposure to three ischemia-related injury models; excitotoxicity (l-glutamic acid), oxidative stress (hydrogen peroxide), and in vitro ischemia (oxygen-glucose deprivation). NDPKA down-regulation had no effect on neuronal viability following injury. By contrast, NDPKA up-regulation increased neuronal survival in all three-injury models. Similarly, treatment with NDPKA recombinant protein increased neuronal survival, but only against in vitro ischemia and excitotoxicity. These findings indicate that the NDPKA protein may confer a neuroprotective advantage following injury. Furthermore, as exogenous NDPKA protein was neuroprotective, it suggests that a cell surface receptor may be activated by NDPKA leading to a protective cell-signaling response. Taken together both NDPKAs intracellular and extracellular neuroprotective actions suggest that the protein is a legitimate therapeutic target for the design of drugs to limit neuronal death following stroke and other forms of brain injury.
Keywords: Erythropoietin, NDPKA/NME1, Neuroprotection, Two-dimensional gel electrophoresis, In vitro injury models, Neuronal cultures
Introduction
A major objective in stroke research is the development of clinically effective neuroprotective drugs to inhibit neuronal death, but despite numerous efforts, no such drugs are currently available. One approach to the identification of therapeutic targets for neuroprotective drug discovery is to investigate differential protein expression following neuronal preconditioning or the induction of ischemic tolerance. Preconditioning occurs when a mild non-damaging stimulus or biological stimulants activate endogenous mechanisms that can protect neurons against a subsequent damaging insult. Furthermore, because preconditioning is known to be reliant on new protein synthesis, the identification of proteins involved in this process are potential targets in the development of drugs to inhibit ischemic neuronal death. To this end, erythropoietin (EPO) preconditioning and/or treatment in vivo and in vitro have been shown to protect neuronal tissue or cells in various ischemia-related injury models (Morishita et al. 1997; Sakanaka et al. 1998; Brines et al. 2000; Siren et al. 2001; Ruscher et al. 2002; Meloni et al. 2006). In the present study, following EPO preconditioning and two-dimensional gel electrophoresis analysis of protein expression, we identified the nucleoside diphosphate kinase A (NDPKA) protein to be up-regulated in neuronal cultures.
Nucleoside diphosphate kinase A, a 17 kDa protein, also known as NME1 (Nonmetastatic Cells 1, Protein Expressed In), NM23H1 (Nonmetastatic Protein 23, Homolog 1), NM23 (Nonmetastatic Protein 23), and GAAD (Granzyme A-activated DNase), is expressed mainly in the brain, testis, and liver (Lacombe et al. 2000). It belongs to the nucleoside diphosphate kinase family of proteins, which are predominantly involved in development, regulating cell proliferation and differentiation, and in suppressing the metastatic potential of tumors (Steeg et al. 1988; Lombardi et al. 2000). The phosphotransferase and histidine kinase activities of NDPKA are involved in the suppression of cell motility, supporting its role as a metastasis suppressor gene (Wagner and Vu 1995; Freije et al. 1997). Also, the ability of NDPKA to maintain intracellular concentrations of nucleotide di- and tri-phosphates through its phosphotransferase activity is vital for normal cellular metabolic and regulatory functions (Postel 1998). Moreover, NDPKA possesses 3′–5′ exonuclease activity, which is thought to aid the repair of DNA damage and thereby reduce tumorigenesis (Jarrett et al. 2012). While NDPKA is mainly localized in the cytoplasm, it possesses a mitochondria-targeting sequence and has been shown to bind and cross-link with mitochondrial membranes, suggesting that the protein contributes to the mitochondrial inter-membrane space structuring (Francois-Moutal et al. 2013). In addition, NDPKA is known to be found extracellularly (Okabe-Kado et al. 2012).
To date, no study has investigated the role of NDPKA in neuroprotection following ischemic injury. On the basis that EPO treatment increases the expression of other known neuroprotective proteins (Meloni et al. 2005, 2006), we decided to investigate the potential role of NDPKA in neuroprotection using in vitro ischemia-like insults. To do this, we investigated how knockdown or overexpression of NDPKA impacts on the viability of primary cortical neuronal cultures in three different injury models (viz. oxygen-glucose deprivation, hydrogen peroxide, and l-glutamic acid). In addition, we also determined if pre-exposure of recombinant NDPKA protein impacted on the viability of neuronal cultures in the same three-injury models.
Methods
Primary Neuronal Cortical Cultures
All animal procedures were approved by the University of Western Australia Animal Ethics Committee. Establishment of cortical cultures was as previously described (Meloni et al. 2001). Briefly, cortical tissue from E18-E19 Sprague–Dawley rats was dissociated in Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Australia) supplemented with 1.3 mM l-cysteine, 0.9 mM NaHCO3, 10 units/ml papain (Sigma, USA), and 50 units/ml DNase (Sigma) and washed in cold DMEM/10 % horse serum. Neurons were resuspended in Neurobasal (Invitrogen) containing 2 % B27 supplement (B27; Invitrogen). Before seeding, 96-well sized glass wells (7 mm diameter, Grace Davison Discovery Sciences, Australia) or 96-well plastic plates (Nunc, Australia) were coated with poly-d-lysine overnight (50 μl/well: 50 μg/ml; 70–150 kDa, Sigma) followed by incubation with 60 μl of Neurobasal (containing 2 % B27, 4 % fetal bovine serum, 1 % horse serum, 62.5 μM l-glutamic acid, 25 μM 2-mercaptoethanol, and 30 μg/ml streptomycin and 30 μg/ml penicillin) for 1–2 h at 37 °C in CO2 incubator (5 % CO2/95 % air). Culture wells were plated with neurons in 90 μl of Neurobasal/2 % B27 to obtain ≈10,000 viable neurons for each well on day in vitro 11–12. Neuronal cultures were maintained in CO2 incubator at 37 °C. On day in vitro 4, one third of the culture medium was removed and replaced with fresh Neurobasal/2 % B27 containing the mitotic inhibitor, cytosine arabinofuranoside (1 μM final concentration; Sigma). On day in vitro 8, one half of the culture medium was replaced with Neurobasal/2 % B27. Cultures were used on day in vitro 11 or 12 after which time they routinely consist of >97 % neurons and 1–3 % astrocytes (Meloni et al. 2001).
EPO Preconditioning and Two-Dimensional Gel Electrophoresis Analysis
The EPO preconditioning procedure is described below, while techniques for two-dimensional gel electrophoresis analysis and MALDI-TOF mass spectrometry protein identification have been described in detail in a previous study (Meloni et al. 2006). Neuronal cultures were exposed to EPO preconditioning (0.5 units/ml) on day in vitro 12. EPO preconditioning consisted of adding a 100× concentrated stock of EPO (human recombinant; Janssen Cilag, Australia) prepared in balanced salt solution (BSS: mM 116 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgSO4, 1 NaH2PO4, pH 7.4) directly to neuronal cultures for 12 h before protein isolation for two-dimensional gel electrophoresis. Controls consisted of cortical neuronal cultures treated with an equivalent volume of BSS.
Following electrophoresis, gels were scanned using a Molecular Imager FX (Bio-Rad) equipped with a 488 nm external laser. Differential protein expression profiles were analyzed using Z3 V 2.0 image analysis software (Compugen, Israel). Triplicate images from EPO-treated and control samples were used to compile a raw master reference gel composite. The composite gels generated were then used to compare the protein profiles between EPO and control treatments, in order to determine the level down-/up-regulation of protein spots following EPO treatment. Changes greater than 1.7 fold in protein expression compared to control were considered significant. Differences in protein expression at the 1.7 fold level analyzed by unpaired t test confirmed statistical significance at the 95 % confidence limit.
Short Interfering RNA (siRNA) Treatment of Neuronal Cultures
On day in vitro 6, media were removed from the cortical neuronal cultures and replaced with 50 μl of Neurobasal/2 % B27 media containing 500 nM of a Dharmacon Accell NDPKA siRNA construct (siRNA:NDPKA, Thermo Fisher Scientific, Australia) and incubated at 37 °C for 48 h (Note: no transfection reagent was used). On day in vitro 8, an equal volume of NB/2 % B27 media containing 500 nM siRNA construct was added to the wells and cultures incubated for a further 72 h before being used for protein isolation or being subjected to injury models. The Accell siRNA non-targeting RNAi construct (siRNA:NT) was used as a control in all experiments, while a Bcl-XL RNAi construct (siRNA:Bcl-XL) was used as positive control.
Generation and Amplification of NDPKA Adenoviral Vector and Adenoviral Treatment of Neuronal Cultures
A recombinant adenoviral vector expressing NDPKA was prepared using the AdEasy vector system (Qbiogene, He et al. 1998), with some modifications as described previously (Boulos et al. 2006). The full-length cDNA for rat NDPKA sequence (NCBI reference: NM_138548.2) was subcloned into our modified shuttle plasmid vector designated pRSV/WPRE;CMV:EGFP (Boulos et al. 2006) to create the plasmid pRSV:NDPKA/WPRE;CMV:EGFP (pRSV:NDPKA). Subsequently, pRSV:NDPKA DNA was linearized by PmeI digestion and introduced by electroporation (Gene Pulser II, Biorad) into Escherichia coli strain BJ5183 carrying pAdEasy (Zeng et al. 2001). A recombinant (i.e., a bacterial colony derived from a bacterium that had undergone homologous recombination with the pRSV:NDPKA and pAdEasy plasmids) was selected on media containing 50 μg/ml kanamycin, and plasmid DNA checked by PacI digestion (i.e., homologous recombination). Lipofectamine 2000 (Invitrogen) was used to transfect PacI enzyme linearized pRSV:NDPKA/AdEasy recombinant DNA (3 μg) into HEK293 cells (QBiogene) grown in a 25 cm2 flask. Following the appearance of viral plaques (5 days), viral amplification was performed in a step-wise fashion by infecting HEK293 cells in 175 and 500 cm2 flasks. HEK293 cell lysate-containing adenoviral particles were prepared by three rounds of freezing/thawing before concentration using the Adeno-X virus purification kit (BD Biosciences). The viral titer was determined by the end-point dilution assay as indicated by EGFP reporter expression (Luo et al. 2007). The NDPKA adenoviral vector which also expresses the EGFP reporter under the control of a CMV promoter was designated AdRSV:NDPKA. An empty vector (AdRSV:Empty) and a Bcl-XL vector (AdRSV:Bcl-XL) that also express the EGFP reporter have been described previously (Boulos et al. 2006) and were used as controls.
On day in vitro 9, the media were removed from cortical neuronal cultures and replaced with Neurobasal/2 % B27 media containing purified adenovirus at a multiplicity of infection (MOI) of 75. For dose response experiments, MOIs of 25, 50, 75, and 100 were used. Cultures were incubated for a further 48 h before total protein was isolated for western blot analysis or used for injury models.
Recombinant NDPKA Protein Production and Purification
To produce recombinant protein, the full-length rat NDPKA sequence (NCBI reference: NM_138548.2) was amplified using PCR. The primers used were rat NDPKA-P1 (GTCGACCCACCATGC, with the SalI restriction enzyme site underlined) and rat NDPKA-P2 (CTCGAGTCACTCATAGATCCAGGTTC, with the XhoI restriction enzyme site underlined). PCR-amplified NDPKA cDNA was inserted into the pET-28 bacterial expression vector (Studier and Moffatt 1986) and transformed into chemically competent KRX E. coli cells. Subsequently, several colonies were inoculated into 2 ml microfuge tubes containing 1.8 ml 2X YT media containing kanamycin (50 μg/ml) and fitted with Eppendorf LidBac membrane filters (PTFE). These were incubated overnight at 37 °C with shaking at 1,400 rpm (Eppendorf thermomixer). Four ml of the overnight cultures was used to inoculate 400 ml of terrific media supplemented with 50 μg/ml kanamycin in a 2 L conical flask. The flask was incubated at 37 °C with shaking (275 rpm) until the cell growth achieved an optical density (OD600) of 0.8–1.0, after which time incubation was at room temperature with shaking (275 rpm) until an OD600 of 1.2–1.5 was obtained. At this point, protein expression was induced by the addition of 4 ml 0.1 M isopropyl-β-D-1-thiogalactopyranoside (IPTG, Promega) and 2 ml 20 % (w/v) rhamnose (Sigma). After overnight incubation at room temperature with shaking (275 rpm), the culture was transferred to a 250 ml centrifuge tube and centrifuged at 10,000 g for 10 min at 4 °C. The cell pellet was resuspended in 25 ml cold lysis buffer (20 mM Tris–HCl, 300 mM NaCl, 20 mM imidazole, 1 % NP-40, pH 8) containing EDTA-free protease inhibitor (Sigma) and DNase (100 units/ml) and subjected to three passages of homogenization using a French press. The lysate was centrifuged at 10,000 rpm for 15 min at 4 °C before for purification using a HIS-tag nickel- nitrilotriacetic acid (NTA) agarose flow cartridge (Qiagen). Briefly, a cartridge was equilibrated with 10 ml lysis buffer before 25 ml of the lysate was applied to the column (1 ml/min), followed by 15 ml of wash buffer (20 mM Tris–HCl, 300 mM NaCl, 30 mM imidazole, pH 8) containing 1 % Triton X-114 (Sigma) as an aid to remove endotoxin. The protein was then eluted by passing 10 ml elution buffer (20 mM Tris–HCl, 300 mM NaCl, 250 mM imidazole, pH 8) through the cartridge and collecting the effluent in 1 ml aliquots. Before pooling 1 ml aliquots, they were checked for purity and yield using SDS-PAGE. Only those fractions showing a distinct single protein band on SDS-PAGE were pooled and subjected to dialysis (Slide-A-Lyzer dialysis cassette; Thermo Fischer Scientific) to remove low molecular weight contaminants and allow equilibration with PBS. Dialysis was performed against 2 L of PBS with three exchanges over a period of 18 h. After the dialysis, the protein solution was filtered through a 0.2 μm Mustang E-membrane filter (endotoxin-binding filter; Pall Corporation, USA) to allow sterilization of the protein solution and removal of any residual endotoxin contaminants. Before aliquoting and storage (500 μl/−80 °C) of purified NDPKA samples, the Bradford assay was used to determine protein concentration.
SDS-PAGE and Western Blotting
Total protein was isolated from neuronal cultures by treatment with lysis buffer [50 mM Tris–HCl pH 7.5, 100 mM NaCl, 20 mM EDTA, 0.1 % SDS, 0.2 % (w/v) deoxycholic acid, 0.25 % Igepal (Sigma), Complete™ protease inhibitor, (Roche)] followed by centrifugation at 10,000 rpm for 1 min at 4 °C. Protein concentration in samples was determined using the Bradford protein assay. Protein samples (5–10 μl; ≈10 μg per lane) were loaded onto 10 % or 4–12 % SDS poly-acrylamide Bis–Tris mini gels (NuPAGE, Invitrogen) and separated via electrophoresis for 50 min at a constant voltage (200 volts). Separated proteins were electrotransferred at 30 volts for 1 h onto polyvinylidinedifluoride or nitrocellulose membranes (Pall Corporation). Membranes were washed with 0.1 % Tween 20 in PBS (PBS/T) for 15 min before being blocked with 5 % skim milk in PBS/T for 1 h at room temperature. Membranes were incubated with primary antibody [rabbit anti-NDPKA (nm23-H1 c-20); Santa Cruz, rabbit anti-Bcl-XL; BD Biosciences] PBS/T containing ovalbumin (1 mg/ml) at 4 °C overnight. After washing with PBS/T for 15 min, the membranes were incubated with a horseradish peroxidase-conjugated secondary antibody in PBS/T containing ovalbumin for 1 h at room temperature. Immuno-reactive bands were visualized using an enhanced chemiluminescence detection system (Amersham, UK) and exposure to X-ray film (Hyperfilm, Amersham). For immuno-detection of the β-tubulin loading control protein, membranes were incubated for 10 min at room temperature with stripping solution (Alpha Diagnostic, USA) prior to re-probing with a mouse anti-β-tubulin antibody (BD Biosciences).
Treatment of Neuronal Cultures with Recombinant NDPKA Protein
On day in vitro 10, media from cortical neuronal cultures were removed and replaced with Neurobasal/2 % B27 media containing purified recombinant NDPKA protein at different concentrations (100, 500, and 1,000 nM). Cultures were incubated at 37 °C for 24 h before being subjected to in vitro injury (viz. oxygen-glucose deprivation, l-glutamic acid, and hydrogen peroxide) with or without incubation with NDPKA-containing medium during the injury phase. In the case of the l-glutamic acid and hydrogen peroxide injury models, the NDPKA-containing medium used for neuronal culture during the injury period was replaced by NDPKA-free medium post-injury. By contrast, in the case of the oxygen-glucose deprivation injury models, the NDPKA-containing medium used during the injury phase was diluted 50 % with fresh NDPKA-free medium post-injury. Qualitative and quantitative measurement of cell viability of neuronal cultures was performed. For all models, three independent experiments were performed and data obtained was pooled for statistical and graphical analysis.
Neuronal In Vitro Injury Models
In Vitro Ischemia Model (Oxygen-Glucose Deprivation)
In order to expose rat cortical neuronal cultures to in vitro ischemia (oxygen-glucose deprivation), media were removed from each well and washed with 315 μl of BSS (pH 6.9), followed by the re-addition of 60 μl of BSS. Culture wells were placed in an anaerobic chamber (Don Whitely Scientific, England) with an atmosphere of 5 % CO2, 10 % H2, and 85 % argon at 98 % humidity and 37 °C for 40 or 45 min (40 min was used for adenoviral vector experiments as it was observed that neurons were slightly more sensitive to injury). Following removal from the anaerobic incubator, an equal volume of Neurobasal media containing 2 % N2 supplement (Invitrogen) was added into each well, and cultures placed in a CO2 incubator at 37 °C. Control cultures were subjected to the same wash procedures only and maintained in a CO2 incubator. Neuronal viability was assessed at 24 h after the initiation of in vitro ischemia.
Hydrogen Peroxide Model (Oxidative Stress)
Media from neuronal culture wells were removed and replaced with 100 μl of Neurobasal/1 % N2 supplement containing 3 or 3.5 μM of hydrogen peroxide (3 μM was used for adenoviral vector experiments). Cultures were incubated at 37 °C in an incubator (5 % CO2/95 % air) for 1 h, after which the medium from each well was removed and replaced with 100 μl of Neurobasal/1 % N2 supplement. Cultures were incubated at 37 °C in a CO2 incubator for a further 24 h. Control cultures received the same media changes, but were not exposed to hydrogen peroxide. Neuronal viability was assessed at 24 h after hydrogen peroxide exposure.
l-Glutamic Acid Model (Excitotoxicity)
Media in neuronal culture wells were removed and replaced with 50 μl of Neurobasal/2 % B27 supplement media containing 6 or 25 μM l-glutamic acid (6 μM was used for adenoviral vector experiments). Cultures were incubated at 37 °C in a CO2 incubator for 5 min, after which time the medium from each well was removed and replaced with 100 μl of fresh Neurobasal/1 % N2 supplement before being incubated at 37 °C in a CO2 incubator for a further 24 h. Control cultures were treated the same, but without exposure to l-glutamic acid containing media. A positive control consisted of cultures containing the glutamate receptor antagonists, MK801 (5 μM) and 6-cyano-7-nitroquinoxaline (5 μM; CNQX) during l-glutamic acid exposure. Neuronal viability was assessed at 24 h after l-glutamic acid exposure.
Neuronal Viability Assay and Statistical Analysis
Neuronal cell cultures were examined by light microscopy for qualitative assessment of neuronal cell viability 18–24 h after in vitro injury and quantitatively after 24 h by the colorimetric 3-(4,5-dimethyliazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay. MTS absorbance data were converted to reflect proportional cell viability relative to both the untreated (taken as 100 % viable) and treated controls. For all models, three independent experiments were performed with quadruplicate (l-glutamic acid and hydrogen peroxide exposure) or sextuplicate (oxygen-glucose deprivation) replicate samples. Data obtained were pooled for generating graphs and for analysis by ANOVA followed by post hoc Fischer’s PLSD test, with P < 0.05 values being considered as statistically significant.
Results
EPO and Two-dimensional Gel electrophoresis
A 12-hour exposure of cortical neuronal cultures to EPO resulted in NDPKA protein up-regulation, as determined by two-dimensional electrophoresis and mass spectrometry protein identification; a representative gel is shown in Fig. 1. The degree of NDPKA up-regulation based on triplicate independent gels was determined to be 1.8 fold.
Fig. 1.

Two-dimensional gels of proteins from control and EPO-preconditioned cortical neuronal cultures. Proteins separated using first dimension pH 4–7 IPG strips. NDPKA protein spot circled in control and EPO gels
Down-regulation of NDPKA in Cortical Cultures and Effect on Neuronal Viability in Injury Models
Western blot analysis confirmed that the siRNA NDPKA construct decreased NDPKA expression in cortical neuronal cultures (Fig. 2). Experiments using cortical neuronal cultures treated with the siRNA NDPKA construct subsequently subjected to in vitro ischemia, hydrogen peroxide, and l-glutamic acid exposure did not result in any statistically significant increase or decrease in neuronal viability (Fig. 3a–c), however, there was a slight trend of increased survival (in vitro ischemia P = 0.276, hydrogen peroxide P = 0.21, l-glutamic acid P = 0.1056). In contrast, down-regulation of the anti-apoptotic Bcl-XL protein increased neuronal death (P = 0.018) following hydrogen peroxide exposure (Fig. 3b), while l-glutamic acid receptor blockers significantly increased neuronal viability following l-glutamic acid exposure (P = 0.0023; Fig. 3c).
Fig. 2.

Western blot analysis of cortical neuronal cultures treated with Bcl-XL (siRNA:Bcl-XL), NDPKA (siRNA:NDPKA), and the non-targeting (siRNA:NT) RNAi constructs (500 nM) and probed with a anti-Bcl-XL and b anti-NDPKA antibodies. To insure similar protein loading membranes were probed with an anti-β-tubulin antibody
Fig. 3.
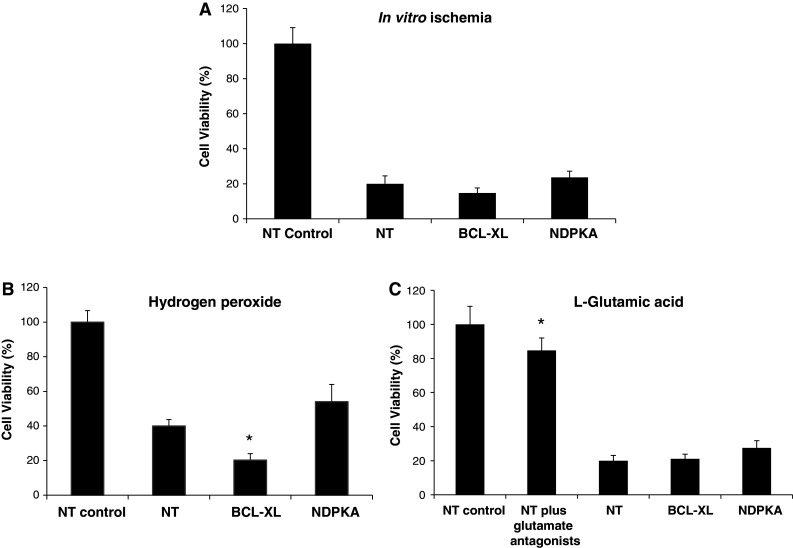
Cell viability of neuronal cultures treated with siRNA constructs (non-targeting control = siRNA:NT, Bcl-XL = siRNA: Bcl-XL, NDPKA = siRNA:NDPKA; 500 nM) at 24 h following; a in vitro ischemia (45 min), b hydrogen peroxide exposure (3.5 μM), and c l-glutamic acid exposure (25 μM). Control siRNA:NT-untreated cultures were taken as 100 % cell survival. The glutamate receptor antagonists used were 5 μM MK801/5 μM CNQX. Values are expressed as means ± SD; n = 12–18. *P < 0.05 compared to NT-treated cultures and subjected to ischemia-like insults
Up-regulation of NDPKA in Cortical Cultures and Effect on Neuronal Viability in Injury Models
Western blot analysis confirmed that the AdRSV:NDPKA adenoviral vector increased NDPKA expression in neuronal cultures (Fig. 4).
Fig. 4.

Western blot analysis of cortical neuronal cultures transfected with the control (AdRSV:Empty), Bcl-XL (AdRSV:Bcl-XL), and NDPKA (AdRSV:NDPKA) adenoviral vectors (MOI of 100) and probed with a anti-Bcl-XL and b anti-NDPKA antibodies. To insure similar protein loading membranes were probed with an anti-β-tubulin antibody
In Vitro Ischemia Model
Cortical neuronal cultures treated with the AdRSV:NDPKA adenoviral vector and subjected to in vitro ischemia increased neuronal viability compared to control adenoviral vector-treated cultures (Fig. 5a; P = 0.0014). Although all AdRSV:NDPKA vector doses (MOI: 25 P = 0.06; 50 P = 0.001; 75 P = 0.02; 100 P = 0.06) were associated with increased neuronal survival, only MOIs of 50 and 75 increased cell survival to statistically significant levels (Fig. 5b). Treatment of cultures with the AdRSV:Bcl-XL vector (MOI: 75) did not increase neuronal viability in the in vitro ischemia model (Fig. 5a).
Fig. 5.

a Cell viability of neuronal cultures treated with adenoviral vectors (AdRSV:Empty, AdRSV:Bcl-XL, and AdRSV:NDPKA; MOI of 75) at 24 h following in vitro ischemia. b Relationship between cell viability and MOI of AdRSV:NDPKA vector. Cortical neuronal cultures were treated with AdRSV:NDPKA adenoviral vector at a MOI of 25, 50, 75, or 100 and subjected to in vitro ischemia (40 min). The empty vector (AdRSV:Empty) was used at a MOI of 75 and taken as 100 % cell survival in untreated control cultures. Values are expressed as means ± SD; n = 18. *P < 0.05 compared to empty vector-treated cultures subjected to in vitro ischemia
Hydrogen Peroxide Model
Cortical neuronal cultures treated with the AdRSV:NDPKA adenoviral vector and exposed to hydrogen peroxide increased neuronal viability compared to control adenoviral vector-treated cultures (Fig. 6a; P = 0.019). Although all AdRSV:NDPKA vector doses (MOI: 25 P = 0.06; 50 P = 0.02; 75 P = 0.15; 100 P = 0.21) were associated with increased neuronal survival, only a MOI of 50 was statistically significant (Fig. 6b). Treatment of cultures with the AdRSV:Bcl-XL vector (MOI: 75) also significantly increased neuronal viability in this model (Fig. 6a; P = 0.01).
Fig. 6.

a Cell viability of neuronal cultures treated with adenoviral vectors (AdRSV:Empty, AdRSV:Bcl-XL, and AdRSV:NDPKA; MOI of 75) at 24 h following hydrogen peroxide exposure (3 μM). b Relationship between cell viability and MOI of AdRSV:NDPKA vector. Cortical neuronal cultures were treated with AdRSV:NDPKA adenoviral vector at a MOI of 25, 50, 75, or 100 and exposed to hydrogen peroxide (3 μM). The empty vector (AdRSV:Empty) was used at a MOI of 75 and taken as 100 % cell survival in untreated control cultures. Values are expressed as means ± SD; n = 12. *P < 0.05 compared to empty vector-treated cultures subjected to hydrogen peroxide
l-Glutamic Acid Model
Cortical neuronal cultures treated with the AdRSV:NDPKA adenoviral vector and exposed to l-glutamic acid increased neuronal viability compared to control adenoviral vector-treated cultures (Fig. 7a; P = 0.009). Although most AdRSV:NDPKA vector doses (MOI: 25 P = 0.01; 50 P = 0.28; 75 P = 0.008) were associated with increased neuronal survival, only MOIs of 25 and 75 reached statistical significance (Fig. 7b). Treatment of cultures with the AdRSV:Bcl-XL vector (MOI: 75 P = 0.004) and glutamate receptor blockers also significantly increased neuronal viability in this model (Fig. 7a).
Fig. 7.

a Cell viability of neuronal cultures treated with adenoviral vectors (AdRSV:Empty, AdRSV:Bcl-XL, and AdRSV:NDPKA; MOI of 75) at 24 h following l-glutamic acid exposure. The glutamate receptor antagonists used were 5 μM MK801/5 μM CNQX. Relationship between cell viability and MOI of AdRSV:NDPKA vector. Cortical neuronal cultures were treated with AdRSV:NDPKA adenoviral vector at a MOI of 25, 50, 75, or 100 and exposed to l-glutamic acid (6 μM). The empty vector (AdRSV:Empty) was used at a MOI of 75 and taken as 100 % cell survival in untreated control cultures. Values are expressed as means ± SD; n = 12. *P < 0.05 compared to empty vector-treated cultures subjected to l-glutamic acid
Addition of Recombinant NDPKA Protein in Cortical Cultures and Effect on Neuronal Viability in Injury Models
In Vitro Ischemia Model
A 24-h pre-incubation of neuronal cultures with recombinant NDPKA protein followed by exposure to NDPKA-containing medium during in vitro ischemia resulted in a significant increase in neuronal survival for all three protein concentrations tested (Fig. 8a: 1000 nM P = 0.049; 500 nM P = 0.004; 100 nM P = 0.02). However, no neuroprotection was observed when cultures were only exposed to NDPKA protein either during the 24-hour pre-incubation period or only during the period of in vitro ischemia (Fig. 8b, c). Shortening the pre-incubation period of NDPKA exposure to 6 h compared to 24 h with or without subsequent exposure of cultures to NDPKA during the injury phase also did not result in any neuroprotective effect (Fig. 8d, e).
Fig. 8.

Cell viability of neuronal cultures treated with recombinant NDPKA protein at different concentrations (100 nM, 500 nM, and 1,000 nM); a during both 24 h before and during in vitro ischemia (50 min); b only during in vitro ischemia; c only during the 24 h before in vitro ischemia; d during both 6 h before and during in vitro ischemia; e only during the 6 h before in vitro ischemia. No protein controls consisted of cultures treated with media without NDPKA. Cell survival in control cultures not subjected to in vitro ischemia was taken as 100 %. Values are expressed as means ± SD; n = 18. *P < 0.05 compared to no protein control cultures subjected to in vitro ischemia
Hydrogen Peroxide Model
Treatment of neuronal cultures with recombinant NDPKA protein during the 24-h pre-incubation period and during hydrogen peroxide exposure did not consistently result in any neuroprotection (Fig. 9a). Similarly, no protective effects were observed when cultures were only pre-incubated with NDPKA protein 24 h prior to injury (Fig. 9b).
Fig. 9.

Cell viability of neuronal cultures treated with recombinant NDPKA protein at different concentrations (100 nM, 500 nM, and 1,000 nM); a during both 24 h before and during hydrogen peroxide exposure (3.5 μM); b only during the 24 h before hydrogen peroxide exposure. No protein controls consisted of cultures treated with media without NDPKA. Cell survival in control cultures not subjected to hydrogen peroxide was taken as 100 %. Values are expressed as means ± SD; n = 12. There were no statistically significant differences between cultures subjected to hydrogen peroxide and treatment groups
l-Glutamic Acid Model
Treatment of neuronal cultures with recombinant NDPKA protein during the 24-h pre-incubation period and during l-glutamic acid exposure resulted in a significant increase in neuronal survival at all three concentrations tested (Fig. 10a; 1000 nM P = 0.005; 500 nM P = 0.03; 100 nM P = 0.02). Treatment of neuronal cultures with NDPKA protein only during the 24-h pre-incubation period did not exert any protective effect (Fig. 10b). Shortening the pre-incubation period of NDPKA exposure to 6 h with or without subsequent exposure of cultures to NDPKA during the injury phase was not associated with any neuroprotective effect (Fig. 10c, d). In all experiments involving l-glutamic acid exposure, treatment with glutamate receptor antagonist (MK801: 5 μM/CNQX: 5 μM) consistently increased cell survival (Fig. 10a–d).
Fig. 10.

Cell viability of neuronal cultures treated with recombinant NDPKA protein at different concentrations (100 nM, 500 nM, and 1,000 nM); a during both 24 h before and during l-glutamic acid exposure (12.5 μM); b only during the 24 h before l-glutamic acid exposure; c during both 6 h before and during l-glutamic acid exposure; d only during the 6 h before l-glutamic acid exposure. No protein controls consisted of cultures treated with media without NDPKA. Cell survival in control cultures not exposed to l-glutamic acid was taken as 100 %. The l-glutamic acid antagonists used were 5 μM MK801/5 μM CNQX. Values are expressed as means ± SD; n = 12. *P < 0.05 compared to no protein control cultures subjected to l-glutamic acid
Discussion
In this study, we identified NDPKA protein up-regulation in EPO-treated cortical neuronal cultures and used three in vitro injury models (viz. in vitro ischemia/oxygen-glucose deprivation, l-glutamic acid/excitotoxicity, and hydrogen peroxide/oxidative stress) to assess the effects of NDPKA down-regulation, up-regulation, and NDPKA protein treatment on neuronal viability. The down-regulation of NDPKA did not induce any significant neuroprotection or increase in neuronal death. In contrast, up-regulation of NDPKA was able to increase neuronal survival in all three-injury models, while treatment with NDPKA protein increased neuronal survival in the in vitro ischemia and l-glutamic acid models. The neutral down-regulation findings suggest that NDPKA is not associated with neuronal ischemic-related injury mechanisms, while the positive up-regulation findings indicate that the NDPKA protein can confer a neuroprotective advantage following such injuries. Furthermore, the positive findings of exogenously applied NDPKA protein in the in vitro ischemia and l-glutamic acid models are suggestive that the protein is acting via a cell surface receptor to relay a protective signaling response. An alternative possibility is that NDPKA is increasing the extracellular levels of free nucleotides (e.g., ATP, GTP), resulting in the activation of purinergic receptors (e.g., P2X, P2Y) and a neuroprotective signaling response (Weisman et al. 2012).
It is also of interest to examine the results obtained with anti-apoptotic protein Bcl-XL, as they shed light on the cell death processors occurring in each injury model. Down-regulation of Bcl-XL exacerbated neuronal death in the hydrogen peroxide model, but not in the in vitro ischemia or l-glutamic acid injury models. These findings strongly suggest that apoptotic mechanisms contribute to neuronal death in the hydrogen peroxide injury model to a greater extent than in the in vitro ischemia and l-glutamic acid excitotoxic injury models. In this context, it is noteworthy that while apoptosis has been detected, autophagy has been demonstrated to be the main mechanism of cell death in the in vitro ischemia model (Meloni et al. 2011) and a calpain/necrotic mechanism in the l-glutamic acid model (Meade et al. 2010).
While no experiments have examined NDPKA in stroke or cerebral ischemia, studies on the effects of NDPKA in non-neuronal cells provide insight into possible mechanisms whereby NDPKA exerts neuroprotection. It has been reported that overexpression of NDPKA in HeLa cells protects against hydrogen peroxide oxidative stress by reducing ROS levels, thought to be mediated at least in part, by increasing glutathione peroxidase-1 (GPX-1) and p53 protein levels (An et al. 2008), however, western analysis of these two proteins following NDPKA overexpression in neuronal cultures revealed no change in expression (unpublished observation). Other studies have reported that NDPKA protects human lung cancer and murine bone-marrow cell lines against oxidative stress and ionizing radiation by activating the human apurinic endonuclease-1 protein (APE1), which is involved in the cellular DNA damage response (Arnaud-Dabernat et al. 2004; Zhang et al. 2011). Similarly, NDPKA’s 3′–5′ exonuclease activity is thought to be involved in the repair of DNA damage (Jarrett et al. 2012). Taken together, these findings suggest that the neuroprotective effect of NDKPA may be linked to increases in cellular antioxidant levels and to mitigating DNA damage.
In the protein studies, an important finding was that the extracellular NDPKA-mediated neuroprotection observed in the in vitro ischemia and l-glutamic acid models was the requirement of neuronal cultures to be exposed to NDPKA both before (24 h pre-incubation) and during the injury phase. Even reducing the period of pre-incubation to 6 h (compared to 24 h) in both the in vitro ischemia and l-glutamic acid models was sufficient to prevent NDPKAs’ neuroprotective effect. This suggests that neurons need to be exposed to NDPKA prior to and during the injury for any neuroprotective effect to be exerted. What this means mechanistically remains to be established, however, given that NDPKA can be found extracellularly (Okabe-Kado et al. 2012) the finding is not totally unexpected. A potential mechanism whereby extracellular NDPKA can exert an effect on cells is through the interaction with the type 1 membrane glycoprotein of the mucin family (MUC1) membrane receptor (Hikita et al. 2008). NDPKA is known to bind to MUC1 receptors in tumor cell lines (Mahanta et al. 2008), however, the MUC1 receptor does not appear to be expressed in the brain (Allan Brain Atlas: http://www.brain-map.org).
Importantly, exposure of recombinant NDPKA has been shown to have several effects on leukemia cells, and if similar effects were to occur in neurons or the brain may have implications in terms of the protein’s neuroprotective actions. For example, exposure of leukemia cells to NDPKA protein stimulates cytokine secretion, including granulocyte macrophage colony-stimulating factor (GS-CSF), interleukin-1 beta (IL-1β), and interleukin 6 (IL-6) (Okabe-Kado et al. 2009). In particular, GS-CSF is a known growth-inducing cytokine that is reported to exert neuroprotective effects in animal models of cerebral ischemia via the up-regulation of anti-apoptotic proteins (Bcl-2 and Bcl-XL) and by modulating the expression of pro-apoptotic elements, such as Bax, caspase 3, and p53 (Nakagawa et al. 2006; Yata et al. 2007; Kong et al. 2009; Choi et al. 2011). In addition, extracellular NDPKA protein activates mitogen-activated protein kinase (MAPK) pathways in myeloid leukemia cells, which involves the signaling molecules extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 (Okabe-Kado et al. 2009). It has been reported that activation of these pathways might be involved in neuroprotection following in vitro models of l-glutamic acid excitotoxicity and both in vitro and in vivo models of cerebral ischemia (Huang et al. 2010; Karmarkar et al. 2011; Wang et al. 2011; Jiang et al. 2012).
While the current study has provided strong evidence for the beneficial effect of NDPKA in ischemia-like injuries, additional work is required to identify NDPKA-interacting proteins and the survival pathways involved. To this end, a previous study employing a yeast-two hybrid screen using human NDPKA as bait has identified eight proteins that physically interact with NDPKA (Seong et al. 2007). Another avenue for future research are studies investigating the neuroprotective actions of recombinant NDPKA protein in vivo using animal stroke models, although there is the issue of NDPKA’s ability to cross the blood–brain barrier. Alternatively, if available, determining the effects of NDPKA overexpression in transgenic mice following cerebral ischemia would also be of interest.
It is interesting to note that in NDPKA adenoviral experiments not all viral MOIs resulted in significant neuroprotective outcomes. For example, in the in vitro ischemia, oxidative stress, and excitotoxicity models, only MOIs of 50/75, 50, and 25/75, respectively, produced a statistically significant effect. We believe the most likely explanation for MOIs having different efficacies in the different models reflects variability in neuronal cultures (e.g., cell density, level of maturation, and astrocyte population) and the balance between overexpressing sufficient NDPKA protein to induce a protective effect and adenoviral load, which appears to increase neuronal susceptibility to injury.
In conclusion, the current study describes the novel finding that NDPKA is up-regulated by EPO in neuronal cultures and that intracellular and extracellular NDPKA-mediated neuroprotective pathways can be activated. Therefore, based on these findings it is possible that NDPKA and its down-stream neuroprotective effects on neurons provide potential therapeutic target for the design of drugs to limit neuronal death following cerebral ischemia and other forms of brain injury.
Acknowledgments
This study was supported by the Neuromuscular Foundation of Western Australia, the Sir Charles Gairdner Hospital Research Fund, and by a University of Western Australia postgraduate research scholarship to Jonathan Teoh.
Conflict of interest
The authors have no competing financial interests.
References
- An R, Chu YL, Tian C, Dai XX, Chen JH, Shi Q, Han J, Dong XP (2008) Over-expression of nm23-H1 in HeLa cells provides cells with higher resistance to oxidative stress possibly due to raising intracellular p53 and GPX1. Acta Pharmacol Sin 29:1451–1458 [DOI] [PubMed] [Google Scholar]
- Arnaud-Dabernat S, Masse K, Smani M, Peuchant E, Landry M, Bourbon PM, Le Floch R, Daniel JY, Larou M (2004) Nm23-M2/NDP kinase B induces endogenous c-myc and nm23-M1/NDP kinase A overexpression in BAF3 cells. Both NDP kinases protect the cells from oxidative stress-induced death. Exp Cell Res 301:293–304 [DOI] [PubMed] [Google Scholar]
- Boulos S, Meloni BP, Arthur PG, Bojarski C, Knuckey NW (2006) Assessment of CMV, RSV and SYN1 promoters and the woodchuck post-transcriptional regulatory element in adenovirus vectors for transgene expression in cortical neuronal cultures. Brain Res 1102:27–38 [DOI] [PubMed] [Google Scholar]
- Brines ML, Ghezzi P, Keenan S, Agello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A (2000) Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 97:10526–10531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Kim KH, Park H, Park SR, Choi BH (2011) Granulocyte macrophage-colony stimulating factor shows anti-apoptotic activity in neural progenitor cells via JAK/STAT5-Bcl-2 pathway. Apoptosis 16:127–134 [DOI] [PubMed] [Google Scholar]
- Francois-Moutal L, Maniti O, Marcillat O, Granjon T (2013) New insights into lipid-Nucleoside Diphosphate Kinase-D interaction mechanism: protein structural changes and membrane reorganisation. Biochim Biophys Acta 1828:906–915 [DOI] [PubMed] [Google Scholar]
- Freije JM, Blay P, MacDonald NJ, Manrow RE, Steeg PS (1997) Site-directed mutation of Nm23-H1. Mutations lacking motility suppressive capacity upon transfection are deficient in histidine-dependent protein phosphotransferase pathways in vitro. J Biol Chem 272:5525–5532 [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikita ST, Kosik KS, Clegg DO, Bamdad C (2008) MUC1* mediates the growth of human pluripotent stem cells. PLoS ONE 3:e3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Liou YF, Chung SY, Lin WY, Jong GP, Kuo CH, Tsai FJ, Cheng YC, Cheng FC, Lin JY (2010) Role of ERK signaling in the neuroprotective efficacy of magnesium sulfate treatment during focal cerebral ischemia in the gerbil cortex. Chin J Physiol 53:299–309 [DOI] [PubMed] [Google Scholar]
- Jarrett SG, Novak M, Dabernat S, Daniel JY, Mellon I, Zhang Q, Harris N, Ciesielski MJ, Fenstermaker RA, Kovacic D, Slominski A, Kaetzel DM (2012) Metastasis Suppressor NM23-H1 Promotes Repair of UV-Induced DNA Damage and Suppresses UV-Induced Melanomagenesis. Cancer Res 72:133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang SY, Zou YY, Wang JT (2012) p38 mitogen-activated protein kinase-induced nuclear factor kappa-light-chain-enhancer of activated B cell activity is required for neuroprotection in retinal ischemia/reperfusion injury. Mol Vis 18:2096–2106 [PMC free article] [PubMed] [Google Scholar]
- Karmarkar SW, Bottum KM, Krager SL, Tischkau SA (2011) ERK/MAPK is essential for endogenous neuroprotection in SCN2.2 cells. PLoS ONE 6:e23493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong T, Choi JK, Park H, Choi BH, Snyder BJ, Bukhari S, Kim NK, Huang X, Park SR, Park HC, Ha Y (2009) Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte-macrophage colony-stimulating factor in rats Laboratory investigation. J Neurosurg 111:155–163 [DOI] [PubMed] [Google Scholar]
- Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO (2000) The human Nm23/nucleoside diphosphate kinases. J Bioenerg Biomembr 32:247–258 [DOI] [PubMed] [Google Scholar]
- Lombardi D, Lacombe ML, Paggi MG (2000) nm23: unraveling its biological function in cell differentiation. J Cell Physiol 182:144–149 [DOI] [PubMed] [Google Scholar]
- Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC (2007) A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc 2:1236–1247 [DOI] [PubMed] [Google Scholar]
- Mahanta S, Fessler SP, Park J, Bamdad C (2008) A minimal fragment of MUC1 mediates growth of cancer cells. PLoS ONE 3:e2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade AJ, Meloni BP, Cross J, Bakker AJ, Fear MW, Mastaglia FL, Watt PM, Knuckey NW (2010) AP-1 inhibitory peptides are neuroprotective following acute glutamate excitotoxicity in primary cortical neuronal cultures. J Neurochem 112:258–270 [DOI] [PubMed] [Google Scholar]
- Meloni BP, Majda BT, Knuckey NW (2001) Establishment of neuronal in vitro models of ischemia in 96-well microtiter strip-plates that result in acute, progressive and delayed neuronal death. Neuroscience 108:17–26 [DOI] [PubMed] [Google Scholar]
- Meloni BP, Van Dyk D, Cole R, Knuckey NW (2005) Proteome analysis of cortical neuronal cultures following cycloheximide, heat stress and MK801 preconditioning. Proteomics 5:4743–4753 [DOI] [PubMed] [Google Scholar]
- Meloni BP, Tilbrook PA, Boulos S, Arthur PG, Knuckey NW (2006) Erythropoietin preconditioning in neuronal cultures: signaling, protection from in vitro ischemia, and proteomic analysis. J Neurosci Res 83:584–593 [DOI] [PubMed] [Google Scholar]
- Meloni BP, Meade AJ, Kitikomolsuk D, Knuckey NW (2011) Characterisation of neuronal cell death in acute and delayed in vitro ischemia (oxygen-glucose deprivation) models. J Neurosci Methods 195:67–74 [DOI] [PubMed] [Google Scholar]
- Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R (1997) Erythropoietin receptor is expressed in rat hipocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 76:105–116 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Suga S, Kawase T, Toda M (2006) Intracarotid injection of granulocyte-macrophage colony-stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res 1089:179–185 [DOI] [PubMed] [Google Scholar]
- Okabe-Kado J, Kasukabe T, Honma Y, Kobayashi H, Maseki N, Kaneko Y (2009) Extracellular NM23 protein promotes the growth and survival of primary cultured human acute myelogenous leukemia cells. Cancer Sci 100:1885–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe-Kado J, Kasukabe T, Kaneko Y (2012) Extracellular NM23 Protein as a Therapeutic Target for Hematologic Malignancies. Adv Hematol 2012:879368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postel EH (1998) NM23-NDP kinase. Int J Biochem Cell Biol 30:1291–1295 [DOI] [PubMed] [Google Scholar]
- Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U, Meisel A (2002) Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci 22:10291–10301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakanaka M, Wen T-C, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R (1998) In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 95:4635–4640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong HA, Jung H, Ha H (2007) NM23-H1 tumor suppressor physically interacts with serine-threonine kinase receptor-associated protein, a transforming growth factor-beta (TGF-beta) receptor-interacting protein, and negatively regulates TGF-beta signaling. J Biol Chem 282:12075–12096 [DOI] [PubMed] [Google Scholar]
- Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P (2001) Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 98:4044–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA, Sobel ME (1988) Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst 80:200–204 [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130 [DOI] [PubMed] [Google Scholar]
- Wagner PD, Vu ND (1995) Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J Biol Chem 270:21758–21764 [DOI] [PubMed] [Google Scholar]
- Wang S, Guo SX, Dai ZG, Dong XW, Liu Y, Jiang S, Wang ZP (2011) Dual isoflurane-induced preconditioning improves neuroprotection in rat brain in vitro and the role of extracellular signal-regulated protein kinase. Chin Med Sci J 26:36–42 [DOI] [PubMed] [Google Scholar]
- Weisman GA, Ajit D, Garrad R, Peterson TS, Woods LT, Thebeau C, Camden JM, Erb L (2012) Neuroprotective roles of the P2Y2 receptor. Purinergic Signal 8:559–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yata K, Matchett GA, Tsubokawa T, Tang J, Kanamaru K, Zhang JH (2007) Granulocyte-colony stimulating factor inhibits apoptotic neuron loss after neonatal hypoxia-ischemia in rats. Brain Res 1145:227–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Smith SK, Siegel F, Shi Z, Van Kampen KR, Elmets CA, Tang DC (2001) AdEasy system made easier by selecting the viral backbone plasmid preceding homologous recombination. Biotechniques 31:260–262 [DOI] [PubMed] [Google Scholar]
- Zhang ZM, Yang XQ, Wang D, Wang G, Yang ZZ, Qing Y, Yang ZX, Li MX, Xiang DB (2011) Nm23-H1 protein binds to APE1 at AP sites and stimulates AP endonuclease activity following ionizing radiation of the human lung cancer A549 cells. Cell Biochem Biophys 61:561–572 [DOI] [PubMed] [Google Scholar]