

Abstract
Mesial temporal lobe epilepsy (mTLE) is the most prevalent type of epilepsy in adults. First and subsequent generations of anti-epileptic therapy regimens fail to decrease seizures in a large number of patients suffering from mTLE, leaving surgical ablation of part of the hippocampus as the only therapeutic option to potentially reach seizure freedom. GluK2 has recently been identified as a promising target for the treatment of mTLE using gene therapy. Here, we engineered an adeno-associated virus serotype 9 vector expressing a cluster of two synthetic microRNAs (miRNAs), expressed from the human synapsin promoter, that target GRIK2 mRNA. Intra-hippocampal delivery of this vector in a mouse model of mTLE significantly reduced GRIK2 expression and daily seizure frequency. This treatment also improved the animals' health, reduced their anxiety, and restored working memory. Focal administration of the vector to the hippocampus of cynomolgus monkeys in GLP toxicology studies led to the selective transduction of hippocampal neurons with little exposure elsewhere in the brain and no transduction outside the central nervous system. Expression of miRNAs in hippocampal neurons resulted in substantially decreased GRIK2 mRNA expression. These data suggest that the intra-hippocampal delivery of a GMP-grade AAV9 encoding a synthetic miRNAs targeting GRIK2 is a promising treatment strategy for mTLE.
Keywords: gene therapy, epilepsy, GRIK2, AAV9, microRNA
Graphical abstract
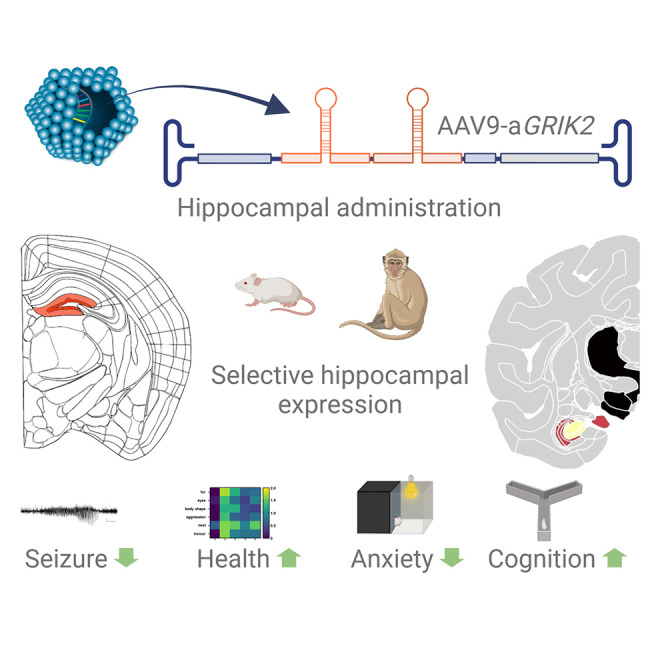
A gene therapy is developed for the treatment of epileptic and non-epiletpic symptoms of mesial temporal lobe epilepsy (mTLE). A GMP-grade AAV9 coding for synthetic miRNAs targeting the kainate receptor GRIK2 showed efficacy and safety in animal models for mTLE epilepsy and non-human primate.
Introduction
Epilepsy is a group of neurological disorders characterized by the occurrence of unpredictable and recurrent seizures. It affects more than 6 million people in Europe1 and 3.4 million in the United States. Focal mesial temporal lobe epilepsy (mTLE) is the most prevalent type of epilepsy in adults, representing over 60% of all diagnosed focal epilepsies.2 In the United States, more than 600,000 people are reported to suffer from mTLE.3
Mesial temporal lobe epilepsy (TLE) is diagnosed by a history of characteristic partial seizure symptoms. In addition to epilepsy, patients with mTLE often suffer from impaired cognitive function and have a high prevalence of mood disorders.4,5 Treatment of mTLE generally begins with anti-epileptic drugs (AEDs), such as Levetiracetam, Lacosamide or Perampanel.6 However, these therapies lead to seizure freedom for only about 25% of patients suffering from mTLE.6 Patients with mTLE who fail to respond to treatment with two AEDs from different classes are considered refractory. For patients with refractory mTLE, and with a clear epileptic focus, surgical resection of the epileptic focus within the hippocampus is the treatment of choice to potentially reach seizure freedom. Resection surgery is an irreversible and invasive procedure involving removal of parts of the hippocampus and the cortex. Surgical resection of the hippocampus is associated with a high risk of post-surgical cognitive decline (e.g., memory loss) and further severe irreversible functional problems (e.g., peripheral vision disturbances, mood swings, loss of motor skills, and speech difficulties) in about 37% of patients.7 Long-term studies investigating post-surgical seizure freedom reveal that a maximum of 70% of patients remain seizure-free after 1 year, decreasing to only 50% remaining seizure free after 5 to 10 years.8 Minimally invasive approaches have been developed as alternatives to surgical resection, but these techniques are only moderately effective for achieving seizure freedom in refractory mTLE patients.9,10,11 Consequently, the demand is high for novel therapeutic approaches in refractory mTLE.
Kainate receptors (KARs) are a family of ionotropic glutamate receptors composed of the tetrameric assembly of GluK1 to GluK5 subunits encoded by the GRIK1 to GRIK5 genes.12 GluK1-GluK3 KAR subunits can assemble into homomeric glutamate receptors or co-assemble into heteromeric receptors with GluK4 or GluK5 subunits to form glutamate-gated cation channels. KARs are expressed in both pre- and postsynaptic compartments in neurons. Activation of postsynaptic KARs by glutamate contributes to the summation of synaptic signals and participates in the regulation of neuronal excitation through both ionotropic and metabotropic mechanisms.13,14 Presynaptic KARs participate in the regulation of neurotransmitter release and in presynaptic forms of synaptic plasticity.13
KARs, in particular the GluK2 subunit, play a key role in the pathophysiology of mTLE. In humans and in rodent models of mTLE, degeneration of pyramidal cells and interneurons in the hippocampus is frequently associated with the emergence of reactive sprouting of mossy fibers.15,16,17 This sprouting is linked to an increase of kainate binding sites across the different subregions of the hippocampus.15 However, most research efforts have focused on the specific recruitment of GluK2/GluK5 at ectopic synapses formed by dentate granule cells (DGCs) onto themselves.18,19 Excitatory synaptic transmission at ectopic synapses on DGCs is meditated in large part by KARs18 following the aberrant synaptic recruitment of GluK2/GluK5 receptors.19,20 The pharmacological inhibition of KARs at these synapses or the constitutive knockout of GluK2 reduces chronic seizures in a pilocarpine mouse model of mTLE.19 The genetic deletion of C1ql2/3, a protein important for the synaptic recruitment of KARs, also leads to decreased spontaneous seizures in a mouse model of mTLE.20 KARs are also associated with other forms of epilepsy. In rodents, for example, GluK2-containing KARs have been implicated in epileptic neonatal mice following a hypoxic insult.21 Gain-of-function mutations in GRIK2 are associated with chronic epilepsy in humans.22 Finally, a rare de novo mutation in NACC1, which interacts with GluK2, is associated with severe neurological symptoms, including epilepsy.23
Selective knockdown of GluK2 in the hippocampus via RNA interference (RNAi) using a proof-of-concept vector was recently shown to efficiently reduce chronic epileptic seizures in a mouse model of mTLE.24 Furthermore, treatment of human hippocampal organotypic slices originating from mTLE patients using this vector decreased the amount of GluK2 protein in the human hippocampus and the frequency of interictal-like epileptiform discharges (IEDs).24 This proof-of-concept study suggests that a gene therapy designed to knock down GluK2 via RNAi represents a potential treatment for mTLE. However, this early construct was suitable only for research use due to its mature miRNA expression profile (which favored the passenger strand), vector genome length (full-length genome of 1.5 kb enabled multiple packaging events per capsid), and use of an shRNA scaffold (which presented a risk to vector genome integrity). The aim of this study was to engineer an optimized AAV-RNAi construct for potential clinical implementation.
Results
Design of AAV9-aGRIK2
Overall, the aim was to identify and optimize up to two miRNAs specific for GRIK2 and express them from an AAV9 vector, under the control of the synapsin 1 promoter (hSyn1). Three construct structures were considered for testing (Figure 1A), a single miRNA (Gen1.1 and 1.2): a single miRNA with a stuffer sequence, placed after the polyA sequence, to fill the size of the AAV genome (Gen1.3) and two miRNA with a stuffer sequence placed after the polyA sequence (Gen2.1 and 2.2). To identify guide sequences capable of efficiently targeting the human GRIK2 mRNA, 100 guide sequences were designed, synthesized as siRNAs, and evaluated in a dual luciferase reporter system to determine knockdown efficiency. Guides were designed and selected based on the following characteristics: (1) full complementarity to the human transcript; (2) degree of complementarity of the total guide sequence or the seed region to rhesus macaque, mouse, and/or rat GRIK2 transcripts; (3) location of the target sequence within the GRIK2 mRNA transcript, favoring those targeting regions predicted to be in an accessible “loop” structure in the full-length human, macaque, and/or mouse transcript, and (4) selectivity for GRIK2 by in silico predicted off-target activity based on cross-referencing known miRNA target sites.
Figure 1.

Design, selection, and optimization of synthetic miRNA constructs capable of efficient GluK2 knockdown with favorable miRNA processing profiles
(A) Schematics depicting the vector genome formats of Gen1.1-.3 and Gen2.1-.2 constructs. (B) Quantification of firefly luciferase(ffluc)-GluK2 reporter knockdown mediated by siRNA and dual reporter plasmid transfection in 293 cells. Data are reported as ffluc RLU from co-transfection of experimental siRNAs with ffluc-GluK2 reporter relative to ffluc RLU from co-transfection of negative control siRNA with ffluc-GluK2 reporter, after normalization to control renilla luciferase RLU (reported as mean ± SEM). (C) Quantification of GRIK2 mRNA levels following miRNA plasmid construct transfection of Gen1.1 constructs into iCell GlutaNeurons, relative to lipid only transfection control (NT), as measured by real-time qPCR and reported as mean ± SD. (D) Quantification of relative levels of mature guide and passenger strand expression from top GRIK2-targeting Gen1.1 constructs as measured by smRNA-seq following plasmid transfection of iCell GlutaNeurons. (E) Assessment of the impact of optimization efforts on G:P by smRNA-seq following Gen1.1 and Gen1.2 plasmid transfection of N2A cells. (F) Assessment of the impact of miRNA concatemerization and addition of a stuffer sequence on G:P by smRNA-seq following plasmid transfection of N2A cells. (G) Quantification of GRIK2 mRNA levels by real-time qPCR (normalized to GAPDH) following transduction of C57Bl6/J mouse cortical neurons with AAV9 vectors packaging Gen1.3, Gen2.1, and Gen2.2 constructs; data are reported as mean ± SEM, NT = non-transduced. (H) Quantification of GluK2 protein levels by western blot (normalized to β-actin) following transduction of C57Bl6/J mouse cortical neurons with AAV9 vectors packaging Gen1.3, Gen2.1, and Gen2.2 constructs. ∗∗p < 0.01, ∗∗∗∗p < 0.0001 one-way ANOVA, followed by Dunnett’s multiple comparison. Data are reported as mean ± SEM, NT = non-transduced.
Guide targets spanned the 4.6-kb GRIK2 transcript; the majority were located in the coding sequence, with a minority targeting either the 5′ or 3′ UTRs. Following co-transfection of the synthetic siRNAs and dual luciferase reporter plasmid, up to 87% knockdown was achieved, with 53% of constructs achieving an average knockdown of at least 50% (Figure 1B). Of these 53 constructs, 32% targeted predicted accessible regions of the full-length GRIK2 mRNA, as compared with 19% of constructs that resulted in <50% knockdown, suggesting a correlation between target accessibility and knockdown efficiency. Sixteen guides were selected for further analysis based on knockdown efficiency in the reporter assay, a lack of non-specific activity (as determined by a lack of luciferase knockdown following co-transfection with the non-targeted reporter plasmid, data not shown), and species sequence complementarity with necessary model systems.
Guide sequences were then grafted into one or more miRNA scaffolds (scaffolds A-E) within an AAV cis plasmid under the control of the human hSyn1 promoter to target expression specifically in neurons.25 The ability of each synthetic miRNA construct to target endogenously expressed GRIK2 mRNA following transfection into iCell GlutaNeurons® was determined (Figure 1C). Two versions of guide 19 and scaffold B were designed and evaluated, labeled 19-B.1 and 19-B.2. Construct 10-E achieved the highest knockdown efficiency in this system (33%), and knockdown efficiency was both guide- and scaffold-dependent. In addition to 10-E, three more constructs achieved high relative knockdown and were selected for further evaluation and optimization: 19-B.2 (32% knockdown), 100-D (27% knockdown), and 100-E (30% knockdown). Small RNA (smRNA) sequencing was performed following transfection of iCell GlutaNeurons to assess the guide:passenger ratios (G:P) of mature synthetic miRNA molecules produced by each of these top four constructs (Figure 1D). Only 100-E displayed a G:P that favored guide production (9:1), with the other three constructs displaying G:P favoring passenger production: 0.001:1 (10-E), 1.6:1 (19-B.2), and 0.33:1 (100-D). Since expression of mature passenger strands is undesirable due to potential off-target activity, the sequence of each construct was optimized to favor guide strand selection by RISC over passenger selection through targeted introduction of U-A pair(s) or U-G wobble(s) at the 5′ end of the guide strand (to destabilize base pairing) and/or introduction of G-C pair(s) at the 5′ end of the passenger strand (to stabilize base pairing).26,27,28 A mismatch was introduced in the seed region of miRNAs (g2:p2) between guide strand and passenger strand to promote unwinding of passenger strand during RISC loading, thus promoting RISC maturation.29,30 Multiple mutations to optimize G:P were evaluated per plasmid construct (i.e., three optimized constructs were designed for 10-E, designated 10-E-1, -2, and -3) and optimized constructs are referred to as Gen1.2 constructs (Figure 1A). The impact of these targeted mutations on G:P was assessed by small RNA sequencing following plasmid transfection of N2A cells (Figure 1E). G:P was dramatically improved for all constructs by up to 12,800-fold (100-D to 100-D-2). Two constructs, 19-B.2-3 and 100-E-2, were selected for further development based on Gen1.2 G:P as well as knockdown efficiency of endogenous GRIK2 mRNA following transfection of representative synthetic stem-loop miRNAs in SH-SY5Y cells (data not shown).
To diversify the sites targeted by a single miRNA-expressing construct, Gen2 constructs (Gen2.1 = non-G:P optimized, Gen2.2 = G:P optimized) were designed (Figure 1A), in which two synthetic miRNA constructs were encoded as a concatemer and expressed from a single promoter (hSyn1). To bring the expected AAV genome length to 3.4 kb, which limits packaging to a single genome, an inert stuffer sequence was added (Figures 1A and S1). The impact of concatemerization on mature miRNA expression levels (data not shown) and G:P (Figure 1F) was evaluated by smRNA-seq following plasmid transfection in N2A cells, and concatemerization, including addition of a stuffer sequence, did not negatively impact individual mature miRNA expression levels or G:P of either Gen2.1 or Gen2.2 component miRNAs.
The Gen2.1 and Gen2.2 concatemerized constructs were packaged into AAV9 vectors, alongside equivalent Gen1.3 constructs (single miRNA with inert stuffer); 19-B.2-3 and 100-E-2 are subsequently referred to as miR1 and miR2, respectively, and AAV9 packaging the Gen2.2 construct is subsequently referred to as AAV9-aGRIK2 (Figure 1A). These vectors were then used to transduce mouse cortical neurons in vitro to demonstrate their ability to knock down endogenous GRIK2 mRNA (Figure 1G) and GluK2 protein (Figure 1H) in mouse cells and therefore establish their potential utility in a mouse model of disease. The greatest average GRIK2 mRNA and GluK2 protein knock down was observed following transduction with the Gen2.2 vector, confirming their ability to reduce endogenously produced mouse GRIK2 mRNA and GluK2 protein. Results of RNA-seq analysis performed in iCell GlutaNeurons® treated with AAV9-aGRIK2 and a control vector at 3.0E+05 multiplicity of infection, identified 77 genes with significantly altered expression levels (Table S1). Among these genes two were predicted by in silico analysis (Table S2), GRIK2 and PUM2. Pathway analysis performed on this list of genes retrieved no known enrichment and revealed that these genes have not been described as functionally related.
AAV9-aGRIK2 administration in vivo in the dorsal hippocampus leads to stable miR1/2 expression and long-lasting reduction in GluK2 protein
Following intra-hippocampal delivery of 5.0E+09 genome copies (gc)/hippocampus of AAV9-aGRIK2, in situ hybridization staining for miR1 confirmed that the treatment of control non-epileptic mice led to miR1 expression specifically in the hippocampus (Figure 2A). Analysis of the distribution of miR1 at the caudal site of injection revealed good coverage of the dorsal hippocampus with minimal expression in the ventral hippocampus (Figure 2A). In situ hybridization staining for miR2 showed a similar pattern of expression (data not shown). Intra-hippocampal administration of increasing doses of AAV9-aGRIK2 in pilocarpine-treated epileptic mice led to increased levels of vector DNA in the hippocampus in a dose-dependent manner (Figure 2B). Similarly, real-time-quantitative polymerase chain reaction (RT-qPCR) analysis of mature guide miRNA showed that the administration of increasing doses of AAV9-aGRIK2 corresponded with greater levels of miR1 and miR2 expression (Figures 2C and 2D) also in a dose-dependent manner, and that the expression of both miR1 and miR2 correlated with levels of vector DNA (Figure 2E).
Figure 2.

Administration of AAV9-aGRIK2 decreases GluK2 protein expression in hippocampal neurons in vivo
(A) Schematic showing the two hippocampal injection sites for diluent, AAV9-control (5.0E+9 gc/hippocampus), or AAV9-aGRIK (at different doses) (orange); the corresponding in situ hybridization for miR1, at the sites of injection, is on the right. (B) Quantification of the vector genome copies in the hippocampus. (C) Quantification of miR1 in the hippocampus. (D) Quantification of miR2 in the hippocampus. (E) Correlation between vector copies and miRNA expression. (F) GluK2 protein expression up to 6 months after administration of AAV9-aGRIK2 or diluent in pilocarpine-treated epileptic mice. ∗∗p < 0.01, ∗∗∗∗p < 0.0001 one-way ANOVA, followed by Dunnett’s multiple comparison. Data are reported as mean ± SEM.
The administration of AAV9-aGRIK2 at 5.0E+09 gc/hippocampus in the pilocarpine mouse model of mTLE (also referred to as “epileptic mice”) significantly reduced GluK2 protein levels compared with vehicle-injected epileptic mice up to 3 months after injection (Figure 2F, diluent group N = 7, 7, and 6 mice for 1, 3, and 6 months respectively, treated group N = 7, 9, and 9 mice for 1, 3, and 6 months, respectively). The low level of GluK2 protein was maintained at 6 months, although a decrease in GluK2 protein levels was also found in epileptic mice treated with diluent, which may be due to hippocampal sclerosis.31 Administration of AAV9-aGRIK2 at 5.0E+09 gc/hippocampus in non-epileptic mice significantly reduced GluK2 protein levels compared with vehicle 6 months after injection (Figure S2), indicating that the effect of AAV9-aGRIK2 can last at least 6 months in vivo in mice. Epileptic mice administered AAV9-aGRIK2 5.0E+09 gc/hippocampus showed a consistent correlation between miR1 or miR2 expression and vector DNA levels in the hippocampus at 1, 3, and 6 months post-injection (Figure S2). A consistent correlation between miR1 or miR2 and vector DNA expression was observed, indicating that the promoter activity remained stable over time (Figure S2).
AAV9-aGRIK2 administration reduces the frequency of seizures in a mouse model of mTLE and of IED in hippocampal slices from patients with refractory mTLE
The efficacy of AAV9-aGRIK2 in reducing chronic epileptic activity in vivo was determined in the pilocarpine mouse model of mTLE24,32 and compared with the control vector, AAV9-control. Three weeks after a single injection of AAV9-aGRIK2 (see materials and methods), mice were monitored up to 19 days with telemetric electroencephalogram (EEG) recordings in the DG. We observed that administration of AAV9-aGRIK2 5.0E+09 and 5.0E+10 gc/hippocampus in epileptic mice led to significant reductions in the number of seizures per day compared with those injected with control vector (5.0E+09 gc/hippocampus) (Figures 3A and 3B, AAV9-control N = 11 mice, AAV9-aGRIK2 5.0E+8 N = 17 mice, 5.0E+9 N = 32 mice, and 5.0E+10 N = 8 mice). The seizure reduction was associated with a significant decrease of GRIK2 mRNA expression levels and not PUM2 (Figure S3), indicating that the biological effect of AAV9-aGRIK2 was mediated by GRIK2 and not PUM2.
Figure 3.

AAV9-aGRIK2 treatment decreases seizure in epileptic mice and inter-ictal epileptiform discharges in human organotypic hippocampal slices
(A) Timeline of the experimental procedure in epileptic mice (mTLE, top) injected bilaterally in the hippocampus with either AAV9-control (5.0E+9 gc/hippocampus) or AAV9-GRIK2 (at different doses) and example of a typical electrographic seizure (ictal event, bottom). (B) Number of seizures per day in the presence of either AAV9-control or AAV9-GRIK2. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001, one-way ANOVA followed by Tukey’s multiple comparisons test. (C) Photomicrographs showing a representative example of dentate gyrus granule cells (DGCs) transduced with AAV9-GFP (AAV9.hSyn1.GFP) in hippocampal organotypic slices from a patient with temporal lobe epilepsy (TLE), using double immunostaining with anti-Prox1 (left), and anti-GFP (middle) antibodies; Molecular layer (ML), hilus (H), scale bars, top, 50 μm, middle, 100 μm, and bottom, 200 μm. (D) Quantification of miR1 expression level. (E) Quantification of mi2R expression level. (F) Quantification of GRIK2 mRNA in tissue treated with AAV9-aGRIK2 normalized to treatment with the AAV9-control. Student’s t test, ∗∗p < 0.01. (G) Representative traces of LFPs recorded in human hippocampal organotypic slices transduced with either control AAV9-control (top) or AAV9-aGRIK2 (bottom, 2.0E+10 gc per slices); enlarged interictal-like epileptiform discharges (IEDs) shown as upper and lower traces (scale bar, 50 ms); note the decrease of IEDs in slices treated with AAV9-aGRIK2. (H) Quantification of the frequency of IEDs in human organotypic hippocampal slices from five patients with mTLE treated with either AAV9-control or AAV9-aGRIK2; control and treated slices from the same patient are paired (lines) for Student’s t test, ∗p < 0.05. Data are reported as mean ± SEM.
To evaluate the therapeutic potential of AAV9-aGRIK2 in human neural tissue, organotypic hippocampal slices from patients with drug-resistant mTLE were used as an ex vivo translational disease model.24 The pathological features of hippocampal organotypic slices taken from patients with refractory mTLE and the respective donor demographics are shown in Tables S3 and S4. As previously shown,24 we observed that approximately half of the DGCs (Prox1-positive cells) were transduced with AAV9-GFP (2.0E+10 gc per slices) in organotypic hippocampal slices from a patient with TLE (Figure 3C).
Treatment with 2.0E+10 gc AAV9-aGRIK2 per slice led to the expression of miR1 or miR2 at about 5.0E+04 copies/ng of host RNA (Figures 3D and 3E). In these slices originating from four patients with mTLE, the treatment with AAV9-aGRIK2 led to a significant decrease in GRIK2 mRNA expression (Figure 3F, Control N = 9 and AAv9-aGRIK2 N = 9). Additional data from organotypic slices resected from nine mTLE patients showed a significant decrease in GRIK2 mRNA levels following treatment with AAV9-aGRIK2 (Table S3).
In a distinct set of organotypic slices from five mTLE patients (Table S4), the treatment with AAV9-aGRIK2 led to a significant decrease in IED frequency (Figures 3G and 3H). These data demonstrate that downregulation of GRIK2 by a viral-mediated RNAi strategy is effective in suppressing seizures in an in vivo mouse model of mTLE and IEDs in hippocampal tissues from patients with drug-resistant mTLE.
AAV9-aGRIK2 improved general health, reduced anxiety, and restored working memory in a mouse model of mTLE
Chronic epileptic rodents, including mice with TLE, exhibit anxiety-related behavior and impairment of learning and memory,33,34 as well as aggressiveness,35 suggesting that TLE mice reflect several of the behavioral and cognitive comorbidities that are associated with TLE in humans.36,37
Epileptic mice showed a significant deterioration in general health, as determined by grooming/fur quality, protruding eyes, body shape, nesting behavior, tremor, and aggressivity (Figures 4A–4C).35 The administration of AAV9-aGRIK2 at 5.0E+09 gc/hippocampus significantly improved the health severity score (the sum of the score for each health category) for all mice 1 month after administration, whereas administration with the diluent had no effect (Figures 4B and 4C, naive non-epileptic mice N = 6, epileptic mice with diluent N = 7, and epileptic mice with AAV9-aGRIK2 N = 7). As previously observed,24 pilocarpine-treated epileptic mice exhibited pathological hyperlocomotion (Figure S4), a well-described behavioral alteration associated with epilepsy.33 Consistent with our previous research construct,24 epileptic mice receiving AAV9-aGRIK2 showed significantly reduced levels of hyperactivity (demonstrated by the distance traveled in the open-field test) compared with mice receiving diluent (Figure S4). The hyperactivity of epileptic mice could result from increased anxiety.33 This hypothesis was assessed using the standalone light-dark box test, which is used to quantify anxiety induced by light.38 Epileptic mice treated with the diluent displayed a reduced number of entries into the light zone and less time spent in the light zone (Figures 4D–4F, naive non-epileptic mice N = 20 mice, epileptic mice with diluent N = 16, and epileptic mice with AAV9-aGRIK2 N = 16). The administration of AAV9-aGRIK2 at a dose of 5.0E+09 gc/hippocampus significantly increased both the number of entries into the light zone and the time spent in the light compartment indicative of a reduced light-induced anxiety (Figures 4D–4F). Working and spatial memory was also assessed using the Y-Maze and the Y-Maze Novel Arm tasks.39 Epileptic mice treated with diluent exhibited a reduced number of spontaneous alternations (Figures 4G and 4H, for H naive non-epileptic mice N = 21, epileptic mice with diluent N = 20 mice, and epileptic mice with AAV9-aGRIK2 N = 19) and spent significantly less time in the novel arm (Figures 4I and 4J, for J naive non-epileptic mice N = 26, epileptic mice with diluent N = 27, and epileptic mice with AAV9-aGRIK2 N = 24). Administration of AAV9-aGRIK2 at 5.0E+09 gc/hippocampus significantly increased both the number of alternations (Figures 4G and 4H) and the time spent in the novel arm (Figures 4I and 4J), approaching the levels observed in naive non-epileptic mice. Taken together, these results indicate an improvement in general health status, including reduced anxiety, and a restoration of working memory following administration of AAV9-aGRIK2.
Figure 4.

AAV9-aGRIK2 treatment improves comorbidities in epileptic mice
(A) Representative images of pilocarpine mice treated with either diluent (top) or AAV9-aGRIK2 (bottom). (B) Health severity score categories measured in naive non-epileptic and epileptic mice treated with either diluent or AAV9-aGRIK2; yellow is low health and dark blue normal health. (C) Health severity score of naive non-epileptic and epileptic mice with either diluent or AAV9-aGRIK2. ∗∗∗∗p < 0.0001, two-way ANOVA repeated measure followed by Sidak’s multiple comparison test. (D) Schematic representation of the light-dark box. (E) Numbers of entry into the light zone. (F) Time spent in the light zone. (G) Schematic representation of the Y-Maze. (H) Spontaneous correct alternation in the Y-Maze (%). (I) Schematic representation of the novel arm maze. (J) Time spent in the novel arm (%). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, one-way ANOVA Tukey’s multiple comparisons test in (E)–(J). Data are reported as mean ± SEM.
miR1/2 expression and GRIK2 knockdown are largely restricted to the hippocampus following AAV9-aGRIK2 administration in non-human primates (NHPs)
The extent of GRIK2 knockdown in different regions of the brain and body following intra-hippocampal administration of AAV9-aGRIK2 was further assessed in GLP toxicology studies in cynomolgus monkeys receiving bilateral injections with either increasing doses of AAV9-aGRIK2 (60 μL dosed per hippocampus, N = 6 animals) or diluent (control, N = 6 animals). AAV9-aGRIK2 was well tolerated in cynomolgus monkeys at all tested doses with no adverse findings observed in the anticipated human dose range. In all animals including controls, minimal to mild microgliosis, gliosis, and astrocytosis were observed in the hippocampus, adjacent entorhinal cortex, and fornix, especially associated with the injection tract. In the groups treated with AAV9-aGRIK2, increased severity of findings was observed together with mononuclear cell infiltration. At the highest dose tested (1.2E+12 gc/hippocampus) at 3 months, minimal neuronal necrosis was observed in the hippocampus and entorhinal cortex, but by 6 months, these findings resolved and no adverse findings were noted.
Magnetic resonance images (MRIs) acquired during administration revealed gadoteridol coverage of approximatively 35% of the hippocampus (Figure 5A). After 6 months, vector DNA was predominantly detected in the hippocampus, with the highest levels observed in brain sections associated with the injection site (Figure 5B). For each dose, vector DNA, estimated from pooled punch concentrations, was overwhelmingly detected in the hippocampus, accounting for up to 99.8% of the total detected vector DNA in some animals. Low but significant amounts were also found in the entorhinal cortex (EC) (average 1.5% of total vector DNA from punches across all doses) (Figures 5C–5E). In other brain regions, vector DNA levels represented less than 0.1% of the total amount of detected DNA. Outside the brain, vector DNA was not detected in peripheral organs at any dose up to 1.2E+12 gc/hippocampus at 6 months. Vector DNA levels up to 5E+06 gc/μg of host DNA were detected in blood serum immediately after AAV9-aGRIK2 administration, with levels rapidly declining over the course of a week (Figure 5F). Large amounts of vector DNA were detected in cerebrospinal fluid (CSF) immediately after administration with levels approaching 1E+10 gc/μg of host DNA (Figure 5G). By day 29, vector DNA was detected in serum and CSF from animals receiving the highest dose of AAV9-aGRIK2 only, with no vector DNA detected in either fluid thereafter.
Figure 5.

Expression of AAV9-aGRIK2 vector DNA is restricted to the hippocampus in non-human primates
(A) Volume of distribution of gadoteridol inside and outside of the hippocampus. (B) Expression level of the vector DNA in tissue punches sampled in relevant brain regions, after intra-hippocampal administration of AAV9-aGRIK2 at 1.2E+12 gc/hippocampus (black: below detection level and gray: not evaluated, CA: cornu ammonis). (C) Expression of the vector DNA in punches from different brain regions (expressed as percentage of the total amount of vector DNA quantified) after administration AAV9-aGRIK2 at 6.0E+10 gc/hippocampus, (D) 2.4E+11 gc/hippocampus and (E) 1.2E+12 gc/hippocampus. (F) Expression of the vector DNA in the blood serum and (G) in the CSF after administration of AAV9-aGRIK2 at 6.0E+10 gc/hippocampus, 2.4E+11 gc/hippocampus and 1.2E+12 gc/hippocampus. Data are reported as mean ± SEM.
All tissue samples with detectable levels of vector DNA were assessed for miRNA expression. No miRNA expression was observed outside the brain. After 6 months, miR1 and miR2 expression was predominantly detected in the hippocampus, with the highest levels observed in brain sections associated with the injection site (Figures 6A and 6B). In general, levels of miR2 expression were higher than for miR1, with levels reaching 1.7E+06 gc in the dentate gyrus up to 6 months post-treatment (Figures 6C and 6D). Both miRNAs showed a good correlation with vector DNA levels (Figures 6E and 6F), indicating no major bias toward the expression of a specific miRNA. Considerably lower levels of miRNA expression were observed in adjacent tissues, such as the EC (Figures 6C and 6D). Interestingly, miR2 was detected in CSF immediately after AAV9-aGRIK2 administration (within approximately 90 min) in all dose groups (Figure 6G). By day 29 and 92, miR2 was detected in the CSF in the mid- and high-dose group and levels were similar to those observed immediately after dosing. However, miR2 was no longer detectable by the end of the study (day 183).
Figure 6.

Knockdown of GRIK2 following administration of AAV9-aGRIK2 is restricted to the hippocampus
(A) Expression level of miR1 and (B) miR2 after intra-hippocampal administration of AAV9-aGRIK2 at 1.2E+12 gc/hippocampus. (C) Expression levels of miR1 and (D) miR2 in different brain regions after administration AAV9-aGRIK2 at 6.0E+10 gc/hippocampus, 2.4E+11 gc/hippocampus and 1.2E+12 gc/hippocampus. (E) Correlation between vector copies and miR1 or (F) miR2 expression. (G) Detection of miR2 in the CSF after administration of AAV9-aGRIK2 at 6.0E+10 gc/hippocampus, 2.4E+11 gc/hippocampus and 1.2E+12 gc/hippocampus. (H) Expression levels of GRIK2 mRNA in the hippocampus and EC after administration AAV9-aGRIK2 at 6.0E+10 gc/hippocampus, 2.4E+11 gc/hippocampus, and 1.2E+12 gc/hippocampus compared with the diluent. (I) Correlation between combined expression of the two miRNAs and the vector DNA. Data are reported as mean ± SEM.
All tissues with detectable levels of miRNA expression (i.e., brain tissues) showed a clear decrease in GRIK2 mRNA in the presence of miR1 and miR2 across all AAV9-aGRIK2 doses (Figure 6H). In addition, brain punches from the hippocampus showed significant reductions in GRIK2 mRNA that varied depending on the level of miRNA expression and the distance from the injection site (Figure 6I) with maximal knockdown at >98%.
Discussion
The aim of this study was to engineer and validate a GMP-grade AAV9-based construct that expresses two miRNAs under the control of a neuronal promoter to selectively knock down GRIK2 in neurons of the hippocampus, thereby providing a potential treatment option for patients with mTLE. Compared with the previously reported research vector,24 the newly engineered, clinically translatable construct showed ideal vector genome occupancy (Figure S1), optimal selectivity for the guide strand, and cross-species reactivity resulting in efficient and sustained downregulation of GRIK2 both in multiple in vitro systems and in vivo in two relevant model species, mice and NHPs. In the two species, the expression of the selected miRNAs (miR1 and miR2) was mainly restricted to the hippocampus following intra-hippocampal administration of AAV9-aGRIK2. The treatment led to a sustained decrease in levels of GluK2 protein and GRIK2 mRNA up to 6 months after administration in mice and NHP, respectively.
More than 30% of people with epilepsy experience persistent seizures despite anti-epileptic pharmacological treatment, and are classified as having drug-resistant epilepsy. Therefore, it is crucial to explore and develop new treatments and strategies for epilepsy. One promising new avenue is the use of gene therapy.3 Gene therapy for focal epilepsy currently focuses on reducing neuronal activity and restoring the balance between inhibition and excitation in seizure-prone brain regions. Previous reports have suggested various approaches, such as overexpressing inhibitory peptides like NPY40 or dynorphin,41 enhancing potassium channels like the engineered voltage-gated potassium channel,42,43 or using optogenetic and chemogenetic tools44,45 to reduce neuronal excitability and suppress seizures. Our proposed gene therapy strategy is based on a novel approach. Rather than reducing neuronal excitability in general by overexpressing an exogenous protein, we propose to selectively downregulate the expression of GluK2, a glutamate receptor that has been shown to play a key role in recurrent seizures in mTLE.19
Similar to our previous research tool vector,24 this study shows that intra-hippocampal administration of GMP-grade AAV9-aGRIK2 decreased the frequency of seizures in the pilocarpine mouse model of mTLE. However, our studies indicate that the benefits of AAV9-aGRIK2 extend beyond seizure control to also improve mTLE-related behavior and general health status, including reduced anxiety and a restoration of working memory. In humans and rodent models with mTLE, increased IED frequency is associated with cognitive decline.34,46 The reduction in IED frequency following AAV9-aGRIK2 administration suggests that the treatment can potentially halt cognitive decline. Indeed, it is anticipated that suppressing paroxysmal activity in dysfunctional epileptic circuits will promote normalization of physiological hippocampal function. These results demonstrate that decreasing neuronal network hyperexcitability in the hippocampus to prevent seizures may have broader positive effects on behavioral symptoms in mTLE patients, with the potential to improve quality of life.
Our studies in cynomolgus monkeys validate the proposed clinical administration of AAV9-aGRIK2 and demonstrate the ability to focally knockdown GRIK2 using a cluster of miRNAs. Magnetic resonance image- (MRI-) guided convection-enhanced delivery of the therapy using a ClearPoint cannula successfully led to the focal distribution of AAV9-aGRIK2 within the hippocampus. Combined miRNA expression was largely restricted to the hippocampus with only the EC showing biologically relevant expression, possibly achieved by retrograde transport through the EC-dentate gyrus circuit.32 Although the hippocampus has a clear and defined role in mTLE, the EC is also implicated in the development of mTLE47,48 likely due to its contribution to the initiation of ictal discharges.49 The focal delivery of miRNAs in the hippocampus achieved up to a 98% reduction in GRIK2 mRNA in brain punches from tissue near the injection site and an overall estimated 50% reduction in the whole hippocampus at a dose of 6.0E+10 gc/hippocampus. All doses of AAV9-aGRIK2 achieved a significant level of miRNA expression and GRIK2 knockdown, and it is anticipated that this level of knockdown would be sufficient to disrupt the aberrant epileptic network in the hippocampus of patients with mTLE.
Based on our findings in mice, the lowest dose of AAV9-aGRIK2 administered to cynomolgus monkeys (6.0E+10 gc/hippocampus) would achieve a predicted minimal efficacy and was not associated with adverse macro or microscopic findings in toxicology studies (Table S5). At higher doses (≥2.4E+11 gc/hippocampus) with very high cell transduction (1E+08 gc per mg; Figure 4B) and miRNA expression (1.7E+06 gc/ng, Figure 5E), minimal neuronal necrosis was observed with associated microgliosis, gliosis, and astrocytosis up to 3 months after treatment. This minimal loss of neurons, described as sparse and uncommon, fully resolved by 6 months.
The hippocampal volume varies between 20 mm3 in mice50 to 200 mm3 in cynomolgus monkeys (based on our MRI data and data from Pardoe et al.51) and 3,000 mm3 in humans. By extrapolation, the volume of low-dose AAV9-aGRIK2 needed to achieve an estimated 50% knockdown of GRIK2 in humans should be 15-fold higher than that needed in monkeys. This suggests that a dose of 1.0E+12 gc/hippocampus would achieve minimum efficacy. Extrapolating directly from mouse data, the volume of AAV9-aGRIK2 needed in humans should be increased by a factor of 150, indicating a slightly lower human dose of 0.75E+12 gc/hippocampus. At these doses, no adverse findings are predicted.
Quantification of transgene product in the CSF and its utility as a surrogate measure of transduction efficiency following intraparenchymal delivery of AAV gene therapies has been previously demonstrated in studies of Huntington disease.52 Both vector DNA and expressed miRNA are released from tissues such as the hippocampus or striatum, directly into the ventricular system and CSF. The presence of vector DNA in CSF (up to 7E+07 gc/mL on day 1 in high-dose animals) is often considered spillage from administration and is quickly eliminated. For AAV9-aGRIK2, miR2 was detected in significant quantities as soon as 90 min after vector delivery. This suggests that in vivo miRNA expression following cell transduction is rapid, abundant, and could be interpreted as a clinical success criteria. However, miR2 in monkeys was detected in the CSF at least 3 months after treatment with AAV9-aGRIK2 1.2E+12 gc/hippocampus (1.5E+05 copies per mL), an observation not previously reported of unknown origin. The observation that miR2 in the CSF was only observed in animals with reported neuronal necrosis suggests its utility as a safety biomarker for AAV-miRNA intracerebral delivery, especially in tissues close to the ventricular system. We therefore propose the close monitoring of CSF miR2 levels during dose evaluation in patients receiving AAV9-aGRIK2 in clinical trials.
A limitation of our study is that AAV9-aGRIK2 treatment did not lead to complete electrographic seizure freedom recorded in the hippocampus in the mouse model of mTLE. However, the systemic injection of pilocarpine results in a severe form of mTLE that affects different brain regions, and not just the hippocampus.53 This suggests that further studies are required to determine whether the residual seizures observed in pilocarpine-treated epileptic mice could be due to extra-hippocampal lesions that would not have been treated by GRIK2 knockdown exclusively in the hippocampus. Our study is further limited by our decision to focus on mTLE because we have identified in past studies a clear mechanistic involvement of aberrantly expressed GluK2 in the hippocampus.19 However, human studies show that gain-of-function mutations in GRIK2 can lead to non-mTLE epilepsy22 and it is well known that GluK2 is expressed in various areas of the brain. The role of GluK2 in the pathophysiology of other forms of focal epilepsy remains to be investigated.
In conclusion, we have developed a promising gene therapy for patients with refractory mTLE that potentially improves the frequency of seizures, mTLE-related behavior, and general health. Results support the development of AAV9-aGRIK2 for the treatment of mTLE and, possibly, for other forms of focal epilepsy.
Materials and methods
Ethics
In vivo mouse experiments were performed in accordance with the European community council directives (2010/63/UE) and received approval from the French Ministry for Research, after ethical evaluation by the institutional animal care and use committee of Aix-Marseille University (protocol number: #9896–201605301121497, #38075–202207221022330). For experiments using human tissues, all patients gave their written consent and protocols were approved: for AP-HM by Inserm (N2017–00031), AP-HM (No. M17-06) under the supervision of CRB TBM/AP-HM (AC: 2018–3105)22; and for CHU Bordeaux by CNRS (Codecoh DC-2020-3863). NHP studies were performed at Northern Biomedical Research (Michigan, USA), and all studies complied with the current version of the Final Rules of the Animal Welfare Act Regulations (9 CFR) and the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, 8th edition. In all experiments, to ensure unbiased quantification, the experimenters were blinded to the conditions and identity of the virus injected until quantification was finalized. In the mouse studies, animal weight and unexpected death were recorded throughout the study, and we did not observe any significant correlation between unexpected death and group or unexpected death and procedure (Table S6).
GMP-grade AAV9 production
GMP-grade AAV9 vector was produced in a certified GMP cell culture suite compliant with Food and Drug Administration GMP guidelines. A comprehensive quality assurance system was embedded throughout the manufacturing process, including an array of in-process and release QC assays to ensure that the vector met specifications and regulatory standards for quality and safety.
Dual luciferase reporter assay
The coding sequence and 5′ and 3′ UTRs of the human GRIK2 mRNA, transcript variant 1 (GenBank ID NM_021956) was cloned into the 3′ UTR of firefly luciferase (ffluc) in the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Cat. #E1330). Experimental custom Silencer Select siRNAs were synthesized by Thermo Fisher, and negative control Silencer Select siRNA #1 was purchased off-the-shelf (Thermo Fisher Scientific, Cat. # AM4635). HEK293 cells were seeded in black-walled 96 well plates 24 h prior to transfection, and confluency of 70%–90% was confirmed prior to transfection with Lipofectamine 3000 (Thermo Fisher Scientific, Cat. #L3000001). Cells were transfected according to manufacturer’s instructions for transfection of siRNAs with 10 pmol siRNA and 5 ng pmiRGLO-GluK2 or pmiRGLO-empty. Luciferase signal was quantified 48 h post-transfection using the Dual-Glo Luciferase Assay System (Promega, Cat. #E2920) according to manufacturer’s instructions. Firefly luciferase signal was first normalized to renilla luciferase signal and then reported as relative RLU, or the ratio between the normalized RLU following negative control siRNA transfection and experimental siRNA transfection.
Transfection and GRIK2 mRNA quantification in iCell GlutaNeurons
iCell GlutaNeurons (FUJIFILM Cellular Dynamics, Cat. #R1061) were seeded at a density of 17.5 k cells/well in 384-well plates coated with PEI (pre-coated) and laminin (at seeding). On day 11 post-seeding, cells were transfected with 30 ng plasmid per well with ViaFect (Promega, Cat. #E4981) at a ratio of 4:1 transfection reagent:DNA, and a complete media change with complete GlutaNeuron medium was performed after overnight incubation. On day 16, cells were lysed and GRIK2 mRNA levels quantified relative to GAPDH by real-time qPCR the TaqPath 1-Step system (ThermoFisher Scientific, Cat. #A28525), and fold change in normalized GRIK2 mRNA levels relative to ViaFect-treated only controls is reported.
Transfection of N2A cells
Mouse N2A neuroblast cells (ATCC CCL-131) cultured in DMEM (Gibco, Cat. #11965092) supplemented with 10% FBS were seeded in 24-well plates at 9.0E4 cells/well, and after 24 h the cells were transfected with 250 ng plasmids and 1.2 μL Lipofectamine 3000/well (Thermo Fisher Scientific, Cat. #L3000001) according to manufacturer instructions. After 48 h, cells were lysed, and RNA enriched with small RNAs was isolated using the miRNeasy Mini kit (QIAGEN, Cat. #217004) and was subjected to library preparation and small RNA sequencing.
Small RNA sequencing
Sequencing libraries were prepared from the total RNAs using the NEXTFlex small RNA kit v3 (PerkinElmer/BIOO) to reduce biased ligation by using the adapter with randomized end NNNN. Libraries were pooled to 4 nM, denatured, and diluted to 11 pM with 10% PhiX spike-in according to manufacturer instructions. Sequencing was performed on the MiSeq System (Illumina, San Diego, California) using the MiSeq Reagent Kit v3 (Illumina, Cat. # MS-102-3001) and 80 sequencing cycles. Bioinformatics was performed in Linux operating system (Ubuntu) with cutadapt for adaptor trimming, Bowtie for sequence alignment, and Awk language for data extraction and reporting.
In silico analysis
For both miR1 and miR2, the input sequences corresponding to nucleotide 2 to 17 from the 5′ end (miR1g2-g17 and miR2g2-g17) and nucleotide 2 to 15 from the 5′ end (miR1g2-g15 and miR2g2-g15) were assessed for complementarity to all transcripts on GlutaNeuron transcriptome. The guide nucleotides from g2 to g17 are critical, with g2-g8 as seed region to bind target mRNA, and g9-g17 pairing with mRNA to allow for AGO2 cleavage. From nucleotide g18 and onward, nucleotides insertions or deletions or substitutions have little or no effect on miRNA binding affinity or AGO2 cleavage rate.54,55 Therefore, it is accepted that perfect pairing from nucleotide g2 to g17 gives cleavage of target mRNA as efficiently as full-length pairing and is consequently assessed in the present in silico off-target study and defined as high-risk binding. Because g2-g15 perfect pairing gives cleavage but with compromised efficiency, we also assessed perfect pairing of g2-g15 in this in silico off-target study and classified it as lower risk binding. To further expand the prediction of off-target cleavage hits of low risk, in silico alignment was performed for the sequence from g2-g15 of miR1 and miR2 allowing one nucleotide mismatch with target. Of note, a mismatch located on g16 or g17 with perfect target pairing with g2-g15 gives mRNA cleavage but with compromised efficiency with same risk as a perfect pairing g2-g15. Therefore, the one mismatch allowance prediction is run only on the g2-g15 sequences.
RNA-seq
The RNA-seq library via polyA selection and non-stranded preparation was sequenced at Genewiz. The reads of rRNAs were removed using Bowtie2 alignment on hg19_SILVA rRNA index. All read pairs that did not align onto rRNA were collected for further spliced transcripts alignment using STAR with annotation index generated from ENSEMBL human genome annotation gtf file. The same file created from STAR alignment was processed into bam file using Samtools, which is further used to quantify transcripts by using StringTie. After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis. Using DESeq2 in R Studio, a comparison of gene expression between AAV9-control (AAV9 containing only the stuffer sequence) and AAV9-aGRIK2 was performed. The criteria used for the DE gene analysis were adjusted p value <0.05, Fold Change <0.8 and >1.2.
Transduction of mouse cortical neurons
Cortical neurons from P0-P1 C57Bl6/J mice were dissociated and seeded in six-well plates at a concentration of 5.5e+5 cells per well. Two or 3 days after plating (day-in vivo, DIV2-3), half of the medium was removed, and vectors were added with MOI 7.5E+4.
For RNA collection for GRIK2 mRNA and miRNA quantification by real-time qPCR, mouse cortical cultures were rinsed in ice-cold PBS at DIV 13 and then scraped into 700 μL of Qiazol (QIAGEN). Total RNA was extracted using the miRNeasy mini kit (QIAGEN 217004). Total RNA contents and quality of sample were assessed by NANODROP One Spectrophotometer (Thermo Fischer Scientific) and subjected to real-time qPCR for mouse GRIK2 mRNA (TaqMan assay mm01181234-m1, Thermo Fisher Scientific) and the mouse Gapdh (Taqman Assay mm99999915-g1, Thermo Fischer Scientific) on a Light-Cycler480 real-time PCR system (Roche). Relative expressions for test items were obtained from triplicate, normalized to the GAPDH mRNA level. The same total RNA preparation was used to quantify synthetic miRNAs as described below.
For total protein collection, mouse cortical cultures at DIV 13 were scraped into 100 μL of lysis buffer (50 mM HEPES, 100 mM NaCl, 1% Glycerol, 0.5% n-Dodecyl β-D-maltoside, pH 7.2; protease inhibitor cocktail set III [Calbiochem] and phosphatase cocktail [Pierce]). Total protein contents were quantified by BCA assay (Pierce 23225) At DIV 13, mouse neuronal cultures were lysed and the lysate was used for SDS-PAGE and immunoblotting. Ten micrograms were loaded and separated on 4%–20% gradient precast gels (Bio-Rad) and then transferred to nitrocellulose membranes (Trans-Blot turbo, Bio-Rad) for immunoblotting analysis. The following antibodies were applied: rabbit anti-GluK2/3 (clone NL9 04–921; Merck-Millipore; d1/2000e) and mouse anti-β-actin (A5316; Sigma; d1/5000e) was used as primary and an appropriate 800-nm fluorophore-conjugated secondary antibody produced in goat (IRDye 800 goat anti-mouse [Li-COR 926–32210; Li-COR] or IRDye 800 goat anti-rabbit Li-COR [926–32211; Li-COR]) were used as secondary antibody. Target proteins were detected by reading at 800 nm on Li-COR. Analysis was performed with the Empiria studio software. For quantification, the intensity of the fluorescent signal of each lane was normalized by β-actin expression and then by the control condition.
Animal breeding, mice model of TLE, and AAV9 stereotaxic injection
Swiss male mice (Janvier and Charles River Labs, 30–40 g) were used and housed at room temperature (20°C–22°C) on a 7:00-19:00 light/dark cycle with ad libitum access to food and water. Mice included in the experiments were not littermates but were housed together from weaning. Each cohort contained an equal number of mice treated with either the AAV9-control (or diluent) or AAV9-aGRIK2 to ensure balanced comparisons within the experimental design. Chronic epileptic mice were generated using an adapted pilocarpine-induced status epilepticus (SE) model.28 For treatment with AAV9-aGRIK2, AAV9-control, or diluent, epileptic mice (>2 months post SE) were injected bilaterally into the dorsal and ventral hippocampus using a Hamilton syringe (5E+9 gc/hippocampus, 1 μL/injection site × 4 injection, rate: 0.2 μL/min) (AP –1.8 mm, ML ±1 mm, DV –2 mm, and AP –3.3 mm, ML ± 2.3 mm, DV –2.5 mm) as previously described.24 A subset of age-matched, non-epileptic Swiss male mice (8 weeks old, Janvier and Charles River Labs) were also injected with AAV9-aGRIK2 (5.0E+09 gc/hippocampus) using similar coordinates and techniques.
Vector DNA quantification
DNA and RNA were extracted from frozen tissue using the AllPrep DNA/RNA Mini kit from QIAGEN. The AAV9-aGRIK2 cis plasmid was linearized and used to generate a standard curve. The size and concentration of standard DNA were used in the following equation to convert the working stock into copy numbers per microliter where C is concentration of the linearized DNA in ng/μL, N is the size of the plasmid DNA in base pairs (6211 bp), and Avogadro’s number is 6.022 × 1023 molecules per mole, following the equation:
Copy numbers per μL = (C ∗ 6.022 × 1023 molecules / mole) / (N ∗ 1 × 109 ng/g ∗ 650 g / mole of bp).
This calculation assumes that the average molecular weight of a DNA bp is 650 Da. The standard curve has a range of 1.0E+08 gc/reaction to 1.0E+02 gc/reaction (=1E+08 copies/4 μL to 1E+02 copies/4 μL). A qPCR assay targeting the rabbit globin polyA sequence was developed to quantify vector genomes of AAV9-aGRIK2 and control AAV9 in Swiss mice. The linearized AAV9-aGRIK2 plasmid was used as the quantification standard. Based on results from plasmid dilutions, a calibrator curve was established by linear regression. The amount of vector DNA in the samples was calculated by interpolation from the calibrator curve and reported as gc/μg of genomic DNA. The range of the plasmid standard line was 1.0E+02 to 1.0E+08 copies per reaction. The assay has a lower quantification limit (LLOQ) of 1.0E+02 transgene copies/reaction.
Quantification of miR1 and miR2 and GRIK2 mRNA
Total RNA concentrations (from hippocampal samples) were determined by measuring absorbance at an optic density of 260 nm using the NanoDrop2000 (ThermoFisher Scientific, CAT: ND-2000). Total RNA was normalized to 20 ng/μL and a 5-μL input was used for each reaction. miRNAs were reverse transcribed into cDNA using a TaqMan MicroRNA Reverse Transcription (RT) Kit and two gene-specific RT primers to target the miRNAs of interest in duplex and to synthesize the cDNA. As the quantification standard, standard lines were prepared using the following formula, where miRNA copy is the highest point of the standard line, the molecular weight (MW) is the MW of the respective oligonucleotide, 1.0E+9 is the conversion from g to ng and Avogadro’s number is 6.022 × 1023 molecules per mole following the equation:
((miRNA copies ∗ Molecular Weight ∗ 1.0E+9)/6.022E+23) = ssRNA in ng.
This formula gives the number of ng required for the highest point of the standard line, after which a 10-fold serial dilution was performed to prepare a 7-point standard line ranging from 1.25E+09 miRNA copies/5 μL until 1.25E+03 copies/5 μL. From the qPCR results of the standard line miR1-24nt and miR2-22nt, a calibrator curve (for each miRNA) was established by linear regression. The number of molecules of miR1 and miR2 in the samples was calculated by interpolation from the calibrator curve and reported as molecules of miR1 copies and miR2 copies per ng RNA. The range of the standard line was 1.25E+09 to 1.25E+03 molecules per reaction.
After reverse transcription, three different real-time qPCRs were performed for each sample, two different human GRIK2 mRNA assays (Grik2_assay #1: forward primer: TG-For AGCGAGTCCTCACCTCTGAT; reverse primer: TG-Rev GTTTGCCTTCCTCTT GCAGC: probe: 5ʹ-/56-FAM/CCT GAC ACA/ZEN/GAT TGG CGG CC/3IABkFQ/-3ʹ; Grik2_assay #2: forward primer: TG-For AGCATCCAAGAAAGCCTGTGA; reverse primer: TG-Rev GCACGGCTGAGTGAAGAGAA; probe: 5ʹ-/56-FAM/TCG GGC CTT/ZEN/CAC ACA GCT CA/3IABkFQ/-3ʹ) and one multiplex housekeeping genes assay (hsGAPDH-FAM and hsATP5B-VIC). The expression of GRIK2 mRNA levels were determined using the GRIK2 targeting TaqMan assay. To normalize the obtained GRIK2 levels, two different TaqMan assays targeting two different housekeeping genes were used (GAPDH and ATP5B). After quantification, the differential expression was calculated using the 2–ΔΔCt method. The housekeeping gene used for normalization was selected by calculating the standard deviation of all the samples for each housekeeping gene: the housekeeping gene with the lowest standard deviation was selected for normalization, which in this experiment was ATP5B (ΔCt). For the second normalization the average of the ΔCt from a control group was used.
GLUK2 protein quantification by mass spectrometry
All solvents were high-performance liquid chromatography (HPLC) grade from Sigma-Aldrich and all chemicals (unless otherwise stated) were obtained from Sigma-Aldrich. Brain tissue samples were homogenized and denatured using Biognosys’ Denature Buffer and a Precellys Evolution Homogenizer (Bertin Instruments). Samples were reduced and alkylated using Biognosys’ Reduction and Alkylation solution, and digested overnight with sequencing grade trypsin (Promega) at a protein:protease ratio of 50:1. Peptides were desalted using 96-well μHLB plates (Waters) according to the manufacturer’s instructions and dried using a SpeedVac system. Peptides were resuspended in 1% acetonitrile and 0.1% formic acid, and spiked with Biognosys’ iRT kit calibration peptides. Peptide concentrations in mass spectrometry-ready samples were measured using the mBCA assay (Thermo Scientific Pierce). Three stable isotope-labeled reference peptides were spiked into the final peptide samples at known concentrations (Vivitide, the quality grade of the reference peptides was ±10% quantification precision, >95% purity; purity of peptide TVTVVYDDSTGLIR was 93.4%).
For liquid chromatography parallel reaction monitoring (LC-PRM) measurements, 1 μg of peptides per sample was injected into an in-house packed C18 column (PicoFrit emitter with 75 μm inner diameter, 60 cm length, and 10 μm tip from New Objective, packed with 1.7 μm Charged Surface Hybrid C18 particles from Waters) on a Thermo Scientific Q Easy nLC 1200 nano-liquid chromatography system connected to a Thermo ScientificQ Exactive HF-X mass spectrometer equipped with a standard nano-electrospray source. LC solvents were A: 1% acetonitrile in water with 0.1% FA; B: 20% water in acetonitrile with 0.1% FA. The LC gradient was 0%–59% solvent B in 54 min followed by 59%–90% B in 12 s, 90% B for 8 min (total gradient length was 67 min). A scheduled run in PRM mode was performed before data acquisition for retention time calibration using Biognosys’ iRT concept.36 The data acquisition window was 6.7 min per peptide. Signal processing and data analysis were carried out using SpectroDive 11.6 – Biognosys’ software for multiplexed minimal reaction monitoring (MRM)/PRM data analysis based on mProphet. A Q-value filter of 1% was applied.
EEGs electrode implantation and recording
Pilocarpine-treated epileptic mice were implanted with one depth wire DSI electrode 3 weeks after AAV administration (AAV9-control or AAV9-aGRIK2), as described previously.24 Surgery was conducted under isoflurane anesthesia. Electrodes were placed stereotactically into the DG (Paxinos and Watson coordinates from bregma: AP −2.55 mm, ML +1.65 mm, DV −2.25 mm) and an additional screw was placed over the cerebellum, serving as the ground electrode. The electrode and screw were secured on the skull with dental cement. During recovery, animals were given 5 mg/kg subcutaneous carprofen (RIMADYL) at 24 and 48 h. EEGs (amplified (1,000X), filtered at 0.16–97 Hz pass, acquired at 500 Hz) were monitored 9–19 days using a telemetric system (Data Sciences International, St. Paul, MN), 24 h per day. Intra-hippocampal EEG traces represent the difference in potential between the electrode inserted into the DG and an electrode positioned above the cerebellum. EEGs were analyzed using the Ponemah software (DSI, St. Paul, MN). As previously described, seizures were defined as rhythmic (>4 up to 60 Hz) and prolonged spike trains with an amplitude of at least twice the EEG baseline and a duration of at least 8 s.24 The time-frequency spectrogram of EEG was performed using a Fast Fourier Transform (FFT) algorithm with a sliding 1s-Hanning windows (“periodogram” function of DSI).
Locomotion tracking
The locomotion of pilocarpine-treated epileptic mice was evaluated 2 weeks after injection of AAV9-aGRIK2 compared with the mice injected with diluent, as previously described.24 Mice were transferred to the behavior analysis room 1 day prior to experiments for habituation to the environment and kept at room temperature (20°C–22°C) in a 9:00–18:00 light/dark cycle with ad libitum access to food and water. All materials in contact with the animals tested were washed with acetic acid in order to prevent olfactory cues. Spontaneous exploration behavior was tested with the open-field test.33 Briefly, mice were placed into the center of a 50 × 50 × 50-cm blue polyvinyl chloride box for 10 min, and the trajectories were recorded with a video camera connected to tracking software (EthoVision Color; Noldus, The Netherlands). The speed and total distance covered by the mice during 10-min of exploration were analyzed.
Y-Maze spontaneous alternation test, Y-Maze novel arm test, and light-dark box
The Y-Maze and the Y-Maze Novel Arm Tests were used to assess working memory and spatial memory,39 respectively. The light-dark box was used to assess anxiety-like behavior.38 All these tests were done in epileptic mice (treated with pilocarpine) evaluated 3–4 weeks after administration of AAV9-aGRIK2 or diluent; untreated non-epileptic Swiss male mice (naive mice, 11–13 weeks old, Janvier and Charles River Labs) were also evaluated. The mice were first tested in the Y-maze, then the day after in the Y-Maze Novel Arm, and the week after in the light-dark box. The maze consisted of three arms (A, B, C), each measuring 50 × 10 cm, with opaque walls that were 30-cm high. Mice were placed in the entry arm (A), and the sequence of entries into the arms was recorded for 5 min. An entry was considered correct if it differed from the two previous entries, such as A-B-C, B-C-A, or A-C-B, etc. Entries with repeated letters, such as A-A-B, were counted as incorrect alternation. The percentage of alternation was used as an index of spatial working memory performance and was calculated as follows: percentage of alternation = [N-correct/(N-total − 2)]∗100, where N-correct is the number of correct entries into a new arm, and N-total is the total number of entries.
The Y-Maze Novel Arm Test consisted of two phases separated by a 1-h interval. In the first phase, which trains the rodents for 5 min, each mouse was placed at the end of the start arm and was allowed to explore only two arms of the maze, while the third arm (novel arm) was blocked. After an interval of 15 min, the test trial, also known as the retention test, was performed according to the following protocol: each mouse was placed at the end of the start arm and allowed to freely explore all three arms for 5 min. The maze arms were thoroughly cleaned between trials to avoid olfactory interaction. The percentage of exploration time in each arm was recorded throughout the experimental period.
The light-dark box apparatus consisted of two compartments: one well-lit (400–450 lux) and one dark. These compartments were connected by an opening that allows the mice to move between them. The mice were acclimated to the testing room (10–20 lux) for at least 1 h before the test, and then they were allowed to explore the light-dark box for 10 min.
IED activity measurements in human hippocampal organotypic slices from patients with refractory mTLE
Organotypic slices (also termed “human slice cultures”) were prepared from surgical resections of the hippocampus from patients diagnosed with refractory mTLE (AP-HM, Hôpital de La Timone, Marseille, France; CHU Pellegrin, Bordeaux, France) as previously described.25 All patients gave written consent. Protocols for AP-HM were approved by Inserm (N° 2017-00031), AP-HM (N° M17-06) under the supervision of CRB TBM/AP-HM (AC: 2018–3105)22, and protocols for CHU Bordeaux were approved by CNRS (Codecoh DC-2020-3863). Tissue blocks were transported from the hospital to the laboratory in a cold (2°C–5°C), oxygenated modified artificial cerebro-spinal fluid (mACSF) containing 132 mM choline, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 7 mM MgCl2, 0.5 mM CaCl2, and 8 mM glucose (Sigma-Aldrich). Slices (350-μm thick) were prepared in a biosafety cabinet using a vibratome (Leica VT1200S) in the same solution. After cutting, slice cultures were rinsed for 15 min in oxygenated “washing medium” at room temperature (22°C). Washing medium contained Hanks Balanced Salt Solution (HBSS, Gibco) completed with HEPES (20 mM, Gibco), glucose (17 mM, Sigma-Aldrich) and antibiotics (0.5% Anti-Anti, Gibco). Organotypic slices were placed on individual Minicell culture inserts (PICMORG50, Millipore) in a six-well plate (30 mm Transwell, Merck-Millipore). Culture medium (1 mL) containing 50% MEM, 25% horse or human serum, 15% HBSS, 2% B27, 0.5% antibiotics (Gibco), 11.8 mM glucose, and 20 mM sucrose (Sigma-Aldrich) was applied to each well. Culture plates were maintained in an incubator at 37°C/5% CO2. The culture medium was changed every 2 days. For the transduction of slices with AAV9 vectors, a phosphate-buffered saline medium (2–5 μL) containing AAV9 vectors was applied directly to slices at DIV 1 (2.0E+10 gc/slice). Electrophysiological recordings were performed between DIV10 and DIV14.
For electrophysiological recordings, the organotypic slices were individually transferred to a recording chamber maintained at 30°C–32°C and continuously perfused (2–3 mL/min) with oxygenated (95% O2 and 5% CO2) ACSF containing 126 mM NaCl, 3.5 mM KCl, 1.2 mM NaH2PO4, 26 mM NaHCO3, 1.3 mM MgCl2, 2.0 mM CaCl2, and 10 mM glucose (pH ∼7.4) (Sigma-Aldrich). Before analyzing the effect of AAV9-aGRIK2 treatment, the integrity of the epileptic microcircuit was systematically probed under hyperexcitable conditions (ACSF containing 5 μM gabazine and 50 μM 4-AP). Slices that responded positively with ongoing spontaneous IEDs under hyperexcitable conditions were switched back to the standard medium (ACSF) and washed for 25 min before analysis in a physiological condition.18 Local Field Potential (LFP) were recorded with a monopolar Nichrome wire placed in the DGC layer and a DAM-80 amplifier (low-pass filter, 0.1 Hz; high-pass filter, 3 kHz; World Precision Instruments, Sarasota, FL). Data were digitized (20 kHz) with a Digidata 1440A (Molecular Devices), and acquired using Clampex 10.1 software (PClamp, Molecular Devices). Signals were analyzed offline using Clamp fit 9.2 (PClamp) and MiniAnalysis 6.0.1 (Synaptosoft, Decatur, GA).
Immunostaining of human organotypic slices
Slices fixed in PFA 4% after recording were permeabilized in blocking solution containing 10% horse serum (HS) (human slices) and PBST (PBS +0.5% Triton) for 2 h at room temperature, and were incubated with anti-GFP antibody (Abcam, AB-300798, 1:1000) and with anti-Prox1 antibody (R&D systems AF2727, 1:100) for 48 h at 4°C. Slices were then incubated for 2 h with Alexa Fluor conjugated secondary antibodies (Invitrogen 1:500) and coverslipped in Fluoromount (Thermo Fisher). Fluorescence images were acquired using a LSM800 confocal microscope (Zeiss) using 10X/0.3NA and 20X/0.8NA objectives. Images were processed using NIH ImageJ software. GRIK2 mRNA quantification was performed as described above.
Toxicology and biodistribution studies in NHPs
Mauritian cynomolgus (Macaca fascicularis) monkeys were chosen as they are a natural host for AAV, are the safety and pharmacology species of choice for translation to humans, and have perfect homology in the miRNA binding sequences for GRIK2. The pivotal toxicology and biodistribution studies were performed in accordance with Good Laboratory Practices.
Animals were approximately 2.5 years old at the time of the study and weighed between 2 and 4 kg. Equal numbers of males and females were used, and groups ranged from four to six animals per group with 34 animals total. Anesthetized animals were placed in an MRI-compatible frame, a coronal incision made over the calvarium, and a craniotomy made over the hippocampus (bilateral). After identification of the infusion site by MRI scan, each animal received a single dose of AAV9-aGRIK2 bilaterally to the hippocampus by MRI-guided convection-enhanced delivery using a custom 16 gauge ClearPoint SmartFlow Neuro Ventricular cannula in a volume of 60 mL infused at a rate of 1–3 mL/min. AAV9-aGRIK2 was prepared in a diluent of intrathecal buffer supplemented with ProHance (gadoteridol solution for MRI visualization) and administered at 6.0E+10, 2.4E+11 and 1.2E+12 gc/hippocampus.
Following administration of AAV9-aGRIK2, animals were monitored daily and assessed for clinical tolerability and pathology (blood chemistry, hematology, and CSF). Animals were euthanized 3 or 6 months post-treatment, formalin-fixed tissues were examined microscopically, and tissues were collected for the quantification of vector DNA, miRNA, and GRIK2 expression using validated and qualified qPCR assays. In particular, 4-mm brain punches from the different brain regions were sampled to understand the biodistribution of AAV9-aGRIK2 within the brain.
Cohort management and statistical analysis
In each experimental group, equal numbers of control (AAV9-control or diluent) and AAV9-aGRIK2-treated epileptic mice from cohorts of co-housed mice (after weaning) were used to ensure balanced comparisons within the experimental design. The data analysis was conducted using GraphPad Prism version 8.3.1. The normality of raw data was checked using the Shapiro-Wilk test and the homogeneity of variance was checked using Brown-Forsythe test or visual inspection of a plot of residuals vs. plotted values. If the data were deemed to violate the assumption of normality or homogeneity of variance, an appropriate nonparametric test was used in addition to a parametric test. The detail of the N and p values as well as the statistical test performed is included in the legend of the figure.
Data and code availability
All data associated with this study are present in the paper or the supplemental information.
Acknowledgments
This work was supported by uniQure, Corlieve and a BPI DeepTech grant, the Institut National de la Santé et de la Recherche Médicale (INSERM to V.C. and C.B.), the Aix-Marseille University (AMU to V.C. and C.B.), A∗Midex (Initiative d'Excellence d'Aix-Marseille Université, AMX-19-IET-004) and ANR funding (ANR-17-EURE-0029) (to V.C. and C.B.), the Centre National de la Recherche Scientifique (CNRS to C.M. and S.D.), the Bordeaux University (UB to C.M., S.D., J.G.), the AP-HM La Timone Marseille Hospital (to F.B. and D.S.), the CHU Pellegrin Bordeaux (to G.P.), the Agence Nationale de la Recherche (ANR-18-CE17-0023-01 to C.M. and V.C.), Inserm Transfert (to V.C.). We also thank E. Pallesi, F.J. Michel and S. Pellegrino-Corby for their technical assistance and heading their respective INMED core facilities, J. Teillon and the Bordeaux Imaging Center (BIC) - a service unit of the CNRS-Inserm and Bordeaux University, member of the national infrastructure France BioImaging, Y. Rufin from the Biochemistry and Biophysics platform of Bordeaux Neurocampus, E. Verdier and S. Daburon from the Cell Biology Facility of IINS for providing us respectively mouse primary neurons. Some schematic illustrations have been adapted from templates by BioRender.com. We thank Brad Hollidge from Regenxbio, Inc. (Maryland, USA) for technical support and our collaborators at Northern Biomedical Research and Northern Biomedical Services (Michigan, USA) for providing support for the non-human primate studies.
Author contributions
S.J.B., A.G., N.Pearson, C.H., C.M., V.C., and R.P. contributed to the conception and design of the studies. S.J.B., A.G., N.Pearson, S.D., C.B., N.Partouche, A.B., J.G., M.W., I.B., C.H., J. Sims, and R.P. contributed to the acquisition and analysis of data. D.S., F.B., J.A., and G.P. supported access to human resected material. S.J.B., A.G., N.Pearson, J. Smith, C.M., V.C. and R.P. contributed to drafting the manuscript and preparing the figures.
Declaration of interests
A patent application has been filed relating to this work. S.J.B., N.Pearson, C.H., N.Partouche, J.G., and R.P. declare an association with uniQure/Corlieve Therapeutics. M.W. and I.B. declare an association with uniQure B.V. A.G., J.S., A.M., and O.D. declare association with Regenxbio Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101342.
Supplemental information
References
- 1.Cross J.H. Epilepsy in the WHO European region: Fostering epilepsy care in Europe. Epilepsia. 2011;52:187–188. doi: 10.1111/J.1528-1167.2010.02903.X. [DOI] [PubMed] [Google Scholar]
- 2.Semah F., Picot M.C., Adam C., Broglin D., Arzimanoglou A., Bazin B., Cavalcanti D., Baulac M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology. 1998;51:1256–1262. doi: 10.1212/WNL.51.5.1256. [DOI] [PubMed] [Google Scholar]
- 3.Asadi-Pooya A.A., Stewart G.R., Abrams D.J., Sharan A. Prevalence and Incidence of Drug-Resistant Mesial Temporal Lobe Epilepsy in the United States. World Neurosurg. 2017;99:662–666. doi: 10.1016/J.WNEU.2016.12.074. [DOI] [PubMed] [Google Scholar]
- 4.Phuong T.H., Houot M., Méré M., Denos M., Samson S., Dupont S. Cognitive impairment in temporal lobe epilepsy: contributions of lesion, localization and lateralization. J. Neurol. 2021;268:1443–1452. doi: 10.1007/s00415-020-10307-6. [DOI] [PubMed] [Google Scholar]
- 5.Tai X.Y., Bernhardt B., Thom M., Thompson P., Baxendale S., Koepp M., Bernasconi N. Blackwell Publishing Ltd; 2018. Review: Neurodegenerative Processes in Temporal Lobe Epilepsy with Hippocampal Sclerosis: Clinical, Pathological and Neuroimaging Evidence. [DOI] [PubMed] [Google Scholar]
- 6.Pohlen M.S., Jin J., Tobias R.S., Maheshwari A. Pharmacoresistance with newer anti-epileptic drugs in mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsy Res. 2017;137:56–60. doi: 10.1016/j.eplepsyres.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helmstaedter C., Petzold I., Bien C.G. The cognitive consequence of resecting nonlesional tissues in epilepsy surgery-Results from MRI- and histopathology-negative patients with temporal lobe epilepsy. Epilepsia. 2011;52:1402–1408. doi: 10.1111/j.1528-1167.2011.03157.x. [DOI] [PubMed] [Google Scholar]
- 8.Mohan M., Keller S., Nicolson A., Biswas S., Smith D., Osman Farah J., Eldridge P., Wieshmann U. The long-term outcomes of epilepsy surgery. PLoS One. 2018;13 doi: 10.1371/JOURNAL.PONE.0196274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrell M.J. 2011. Responsive cortical stimulation for the treatment of medically intractable partial epilepsy On behalf of the RNS System in Epilepsy Study Group. [DOI] [PubMed] [Google Scholar]
- 10.Schulze-Bonhage A., Hirsch M., Knake S., Kaufmann E., Kegele J., Rademacher M., Vonck K., Coenen V.A., Glaser M., Jenkner C., et al. Focal Cortex Stimulation with a Novel Implantable Device and Antiseizure Outcomes in 2 Prospective Multicenter Single-Arm Trials. JAMA Neurol. 2023;80:588–596. doi: 10.1001/jamaneurol.2023.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toffa D.H., Touma L., El Meskine T., Bouthillier A., Nguyen D.K. W.B. Saunders Ltd; 2020. Learnings from 30 Years of Reported Efficacy and Safety of Vagus Nerve Stimulation (VNS) for Epilepsy Treatment: A Critical Review. [DOI] [PubMed] [Google Scholar]
- 12.Contractor A., Mulle C., Swanson G.T. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–163. doi: 10.1016/J.TINS.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mulle C., Crépel V. Elsevier Ltd; 2021. Regulation and Dysregulation of Neuronal Circuits by KARs. [DOI] [PubMed] [Google Scholar]
- 14.Pinheiro P.S., Lanore F., Veran J., Artinian J., Blanchet C., Crépel V., Perrais D., Mulle C. Selective Block of Postsynaptic Kainate Receptors Reveals Their Function at Hippocampal Mossy Fiber Synapses. Cerebr. Cortex. 2013;23:323–331. doi: 10.1093/CERCOR/BHS022. [DOI] [PubMed] [Google Scholar]
- 15.Represa A., Robain O., Tremblay E., Ben-Ari Y. Hippocampal plasticity in childhood epilepsy. Neurosci. Lett. 1989;99:351–355. doi: 10.1016/0304-3940(89)90472-2. [DOI] [PubMed] [Google Scholar]
- 16.Gabriel S., Njunting M., Pomper J.K., Merschhemke M., Sanabria E.R.G., Eilers A., Kivi A., Zeller M., Meencke H.J., Cavalheiro E.A., et al. Stimulus and Potassium-Induced Epileptiform Activity in the Human Dentate Gyrus from Patients with and without Hippocampal Sclerosis. J. Neurosci. 2004;24:10416–10430. doi: 10.1523/JNEUROSCI.2074-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sutula T., Cascino G., Cavazos J., Parada I., Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann. Neurol. 1989;26:321–330. doi: 10.1002/ANA.410260303. [DOI] [PubMed] [Google Scholar]
- 18.Epsztein J., Represa A., Jorquera I., Ben-Ari Y., Crépel V. Recurrent mossy fibers establish aberrant kainate receptor-operated synapses on granule cells from epileptic rats. J. Neurosci. 2005;25:8229–8239. doi: 10.1523/JNEUROSCI.1469-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peret A., Christie L.A., Ouedraogo D.W., Gorlewicz A., Epsztein J.Ô., Mulle C., Crépel V. Contribution of Aberrant GluK2-Containing Kainate Receptors to Chronic Seizures in Temporal Lobe Epilepsy. Cell Rep. 2014;8:347–354. doi: 10.1016/j.celrep.2014.06.032. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda K., Budisantoso T., Mitakidis N., Sugaya Y., Miura E., Kakegawa W., Yamasaki M., Konno K., Uchigashima M., Abe M., et al. Transsynaptic Modulation of Kainate Receptor Functions by C1q-like Proteins. Neuron. 2016;90:752–767. doi: 10.1016/j.neuron.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Grosenbaugh D.K., Ross B.M., Wagley P., Zanelli S.A. The Role of Kainate Receptors in the Pathophysiology of Hypoxia-Induced Seizures in the Neonatal Mouse. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-24722-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stolz J.R., Foote K.M., Veenstra-Knol H.E., Pfundt R., ten Broeke S.W., de Leeuw N., Roht L., Pajusalu S., Part R., Rebane I., et al. Clustered mutations in the GRIK2 kainate receptor subunit gene underlie diverse neurodevelopmental disorders. Am. J. Hum. Genet. 2021;108:1692–1709. doi: 10.1016/j.ajhg.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniel J.A., Elizarova S., Shaib A.H., Chouaib A.A., Magnussen H.M., Wang J., Brose N., Rhee J., Tirard M. An intellectual-disability-associated mutation of the transcriptional regulator NACC1 impairs glutamatergic neurotransmission. Front. Mol. Neurosci. 2023;16 doi: 10.3389/fnmol.2023.1115880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boileau C., Deforges S., Peret A., Scavarda D., Bartolomei F., Giles A., Partouche N., Gautron J., Viotti J., Janowitz H., et al. GluK2 Is a Target for Gene Therapy in Drug-Resistant Temporal Lobe Epilepsy. Ann. Neurol. 2023;94:745–761. doi: 10.1002/ANA.26723. [DOI] [PubMed] [Google Scholar]
- 25.Hioki H., Kameda H., Nakamura H., Okunomiya T., Ohira K., Nakamura K., Kuroda M., Furuta T., Kaneko T. Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther. 2007;14:872–882. doi: 10.1038/sj.gt.3302924. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz D.S., Hutvágner G., Du T., Xu Z., Aronin N., Zamore P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/S0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 27.Khvorova A., Reynolds A., Jayasena S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/S0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 28.Medley J.C., Panzade G., Zinovyeva A.Y. microRNA strand selection: Unwinding the rules. Wiley Interdiscip. Rev. RNA. 2021;12 doi: 10.1002/wrna.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomari Y., Du T., Zamore P.D. Sorting of Drosophila Small Silencing RNAs. Cell. 2007;130:299–308. doi: 10.1016/j.cell.2007.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matranga C., Tomari Y., Shin C., Bartel D.P., Zamore P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 31.Mátyás A., Borbély E., Mihály A. Hippocampal Sclerosis in Pilocarpine Epilepsy: Survival of Peptide-Containing Neurons and Learning and Memory Disturbances in the Adult NMRI Strain Mouse. Int. J. Mol. Sci. 2021;23 doi: 10.3390/IJMS23010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vigier A., Partouche N., Michel F.J., Crépel V., Marissal T. Substantial outcome improvement using a refined pilocarpine mouse model of temporal lobe epilepsy. Neurobiol. Dis. 2021;161 doi: 10.1016/J.NBD.2021.105547. [DOI] [PubMed] [Google Scholar]
- 33.Müller C.J., Bankstahl M., Gröticke I., Löscher W. Pilocarpine vs. lithium-pilocarpine for induction of status epilepticus in mice: development of spontaneous seizures, behavioral alterations and neuronal damage. Eur. J. Pharmacol. 2009;619:15–24. doi: 10.1016/J.EJPHAR.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Kleen J.K., Scott R.C., Lenck-Santini P.-P., Holmes G.L. Cognitive and Behavioral Co-Morbidities of Epilepsy. Jasper’s Basic Mech. Epilepsies. 2012:915–929. doi: 10.1093/med/9780199746545.003.0072. [DOI] [PubMed] [Google Scholar]
- 35.Huang X., McMahon J., Huang Y. Rapamycin attenuates aggressive behavior in a rat model of pilocarpine-induced epilepsy. Neuroscience. 2012;215:90–97. doi: 10.1016/J.NEUROSCIENCE.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keezer M.R., Sisodiya S.M., Sander J.W. Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. 2016;15:106–115. doi: 10.1016/S1474-4422(15)00225-2. [DOI] [PubMed] [Google Scholar]
- 37.Hoppe C., Elger C.E., Helmstaedter C. Long-term memory impairment in patients with focal epilepsy. Epilepsia. 2007;48:26–29. doi: 10.1111/J.1528-1167.2007.01397.X. [DOI] [PubMed] [Google Scholar]
- 38.Bourin M., Hascoët M. The mouse light/dark box test. Eur. J. Pharmacol. 2003;463:55–65. doi: 10.1016/S0014-2999(03)01274-3. [DOI] [PubMed] [Google Scholar]
- 39.Prieur E.A.K., Jadavji N.M. Assessing Spatial Working Memory Using the Spontaneous Alternation Y-maze Test in Aged Male Mice. Bio. Protoc. 2019;9 doi: 10.21769/BIOPROTOC.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woldbye D.P.D., Ängehagen M., Gøtzsche C.R., Elbrønd-Bek H., Sørensen A.T., Christiansen S.H., Olesen M.V., Nikitidou L., Hansen T.V.O., Kanter-Schlifke I., Kokaia M. Adeno-associated viral vector-induced overexpression of neuropeptide Y Y2 receptors in the hippocampus suppresses seizures. Brain. 2010;133:2778–2788. doi: 10.1093/BRAIN/AWQ219. [DOI] [PubMed] [Google Scholar]
- 41.Agostinho A.S., Mietzsch M., Zangrandi L., Kmiec I., Mutti A., Kraus L., Fidzinski P., Schneider U.C., Holtkamp M., Heilbronn R., Schwarzer C. Dynorphin-based “release on demand” gene therapy for drug-resistant temporal lobe epilepsy. EMBO Mol. Med. 2019;11 doi: 10.15252/EMMM.201809963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snowball A., Chabrol E., Wykes R.C., Shekh-Ahmad T., Cornford J.H., Lieb A., Hughes M.P., Massaro G., Rahim A.A., Hashemi K.S., et al. Epilepsy Gene Therapy Using an Engineered Potassium Channel. J. Neurosci. 2019;39:3159–3169. doi: 10.1523/JNEUROSCI.1143-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu Y., O’Neill N., Maffei B., Zourray C., Almacellas-Barbanoj A., Carpenter J.C., Jones S.P., Leite M., Turner T.J., Moreira F.C., et al. On-demand cell-autonomous gene therapy for brain circuit disorders. Science. 2022;378:523–532. doi: 10.1126/SCIENCE.ABQ6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walker M.C., Kullmann D.M. Optogenetic and chemogenetic therapies for epilepsy. Neuropharmacology. 2020;168 doi: 10.1016/J.NEUROPHARM.2019.107751. [DOI] [PubMed] [Google Scholar]
- 45.Whitebirch A.C., LaFrancois J.J., Jain S., Leary P., Santoro B., Siegelbaum S.A., Scharfman H.E. Enhanced excitability of the hippocampal CA2 region and its contribution to seizure activity in a mouse model of temporal lobe epilepsy. Neuron. 2022;110:3121–3138.e8. doi: 10.1016/J.NEURON.2022.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holmes G.L., Lenck-Santini P.P. Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 2006;8:504–515. doi: 10.1016/J.YEBEH.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 47.Bernasconi N., Bernasconi A., Andermann F., Dubeau F., Feindel W., Reutens D.C. Entorhinal cortex in temporal lobe epilepsy: a quantitative MRI study. Neurology. 1999;52:1870–1876. doi: 10.1212/WNL.52.9.1870. [DOI] [PubMed] [Google Scholar]
- 48.Bernasconi N., Bernasconi A., Caramanos Z., Antel S.B., Andermann F., Arnold D.L. Mesial temporal damage in temporal lobe epilepsy: a volumetric MRI study of the hippocampus, amygdala and parahippocampal region. Brain. 2003;126:462–469. doi: 10.1093/BRAIN/AWG034. [DOI] [PubMed] [Google Scholar]
- 49.Ren H., Shi Y.J., Lu Q.C., Liang P.J., Zhang P.M. The role of the entorhinal cortex in epileptiform activities of the hippocampus. Theor. Biol. Med. Model. 2014;11:14. doi: 10.1186/1742-4682-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Redwine J.M., Kosofsky B., Jacobs R.E., Games D., Reilly J.F., Morrison J.H., Young W.G., Bloom F.E. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: A magnetic resonance microscopy and stereologic analysis. Proc. Natl. Acad. Sci. USA. 2003;100:1381–1386. doi: 10.1073/PNAS.242746599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardoe H.R., Pell G.S., Abbott D.F., Jackson G.D. Hippocampal volume assessment in temporal lobe epilepsy: How good is automated segmentation? Epilepsia. 2009;50:2586–2592. doi: 10.1111/J.1528-1167.2009.02243.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valles A., Evers M.M., Stam A., Gonzalez M.S., Brouwers C., Tornero C.V., Broekmans S.A., Paerels L., Klima J., Bohuslavova B., et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci. Transl. Med. 2021;13 doi: 10.1126/SCITRANSLMED.ABB8920. [DOI] [PubMed] [Google Scholar]
- 53.Lévesque M., Biagini G., de Curtis M., Gnatkovsky V., Pitsch J., Wang S., Avoli M. Elsevier Ltd; 2021. The Pilocarpine Model of Mesial Temporal Lobe Epilepsy: Over One Decade Later, with More Rodent Species and New Investigative Approaches. [DOI] [PubMed] [Google Scholar]
- 54.Becker W.R., Ober-Reynolds B., Jouravleva K., Jolly S.M., Zamore P.D., Greenleaf W.J. High-Throughput Analysis Reveals Rules for Target RNA Binding and Cleavage by AGO2. Mol. Cell. 2019;75:741–755.e11. doi: 10.1016/j.molcel.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Godinho B., Khvorova A. The era of RNA interference medicines: the clinical landscape of synthetic gene silencing drugs. Saúde & Tecnologia. 2019;21:5–17. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the supplemental information.