

Abstract
Chimeric antigen receptor (CAR) T cells from allogeneic donors promise “off-the-shelf” availability by overcoming challenges associated with autologous cell manufacturing. However, recipient immunologic rejection of allogeneic CAR-T cells may decrease their in vivo lifespan and limit treatment efficacy. Here, we demonstrate that the immunosuppressants rapamycin and tacrolimus effectively mitigate allorejection of HLA-mismatched CAR-T cells in immunocompetent humanized mice, extending their in vivo persistence to that of syngeneic humanized mouse-derived CAR-T cells. In turn, genetic knockout (KO) of FKBP prolyl isomerase 1A (FKBP1A), which encodes a protein targeted by both drugs, was necessary to confer CD19-specific CAR-T cells (19CAR) robust functional resistance to these immunosuppressants. FKBP1AKO 19CAR-T cells maintained potent in vitro functional profiles and controlled in vivo tumor progression similarly to untreated 19CAR-T cells. Moreover, immunosuppressant treatment averted in vivo allorejection permitting FKBP1AKO 19CAR-T cell-driven B cell aplasia. Thus, we demonstrate that genome engineering enables immunosuppressant treatment to improve the therapeutic potential of universal donor-derived CAR-T cells.
Keywords: immunotherapy, CAR-T cells, universal donor, immunosuppressants, gene editing, base editing
Graphical abstract
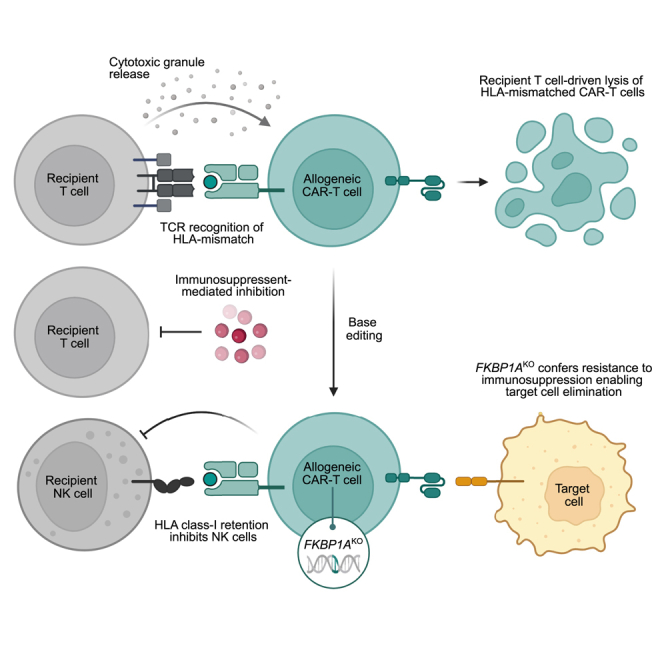
Immunogenicity of allogeneic CAR-T cell products may limit in vivo persistence and clinical efficacy. Here, Maldini and colleagues leverage the immunosuppressants rapamycin and tacrolimus to abrogate host immunologic rejection of HLA-mismatched CAR-T cells, which in turn necessitates genetic disruption of FKBP1A in allogeneic CAR-T cells to confer drug resistance.
Introduction
Allogeneic chimeric antigen receptor (CAR) T cell therapies promise to deliver breakthroughs in the treatment of cancer. The development of universal donor-derived CAR-T cells could provide “off-the-shelf” availability, which simplifies patient delivery, reduces wait time to treatment, and eliminates manufacturing failure inherent to autologous cell therapies.1,2,3 Despite these advantages, HLA mismatch between allogeneic donor and recipient patients may represent a barrier that limits the efficacy of allogeneic CAR-T cell therapy. Inactivation of the T cell receptor (TCR) in allogeneic CAR-T cells can prevent graft-versus-host disease (GVHD),4,5,6 but the expression of mismatched HLA alleles renders allogeneic cells susceptible to recipient T cell-mediated rejection.7 Notably, although genetic disruption of β-2-microglobulin (B2M) and class II transcriptional activator (CIITA) effectively ablate HLA surface expression and abrogate recognition by recipient T cells,8,9 HLA class I-deficient cells are now susceptible to missing self-recognition and lysis by NK cells.10 Thus, HLA surface expression on allogeneic CAR-T cells represents a quandary necessitating alternative immune-modulatory strategies to overcome rejection.
Immunosuppressants are routinely used in transplant medicine to mitigate the intensity of an immune reaction. For example, immunosuppressants may condition the recipient to accept a transplanted organ from an unrelated donor11,12 and similarly limit GVHD-induced organ tissue damage from transplanted allogeneic hematopoietic stem cells.13,14 Two common immunosuppressants, rapamycin (RPM) and tacrolimus (TAC), are structural analogs both of which bind the immunophilin FKBP prolyl isomerase 1A (FKBP1A).15 However, these drugs differentially regulate immune responses either by inhibiting mTOR16,17,18 or calcineurin-induced NFAT activation,19 respectively. The downstream consequences of these drug-FKBP1A interactions attenuate immune cell proliferation, response to IL-2, and pro-inflammatory cytokine secretion.20,21,22,23 Given that RPM and TAC bind a common intracellular protein, disruption of the gene encoding FKBP1A in allogeneic CAR-T cells may confer upon them drug resistance while simultaneously enabling immunosuppressant treatment to avert recipient allorejection of the CAR-T cells.
Here, we leveraged stringent humanized mouse models to interrogate a strategy that combines immunosuppressant treatment to inhibit alloreactivity and genome engineering to confer allogeneic CAR-T cell functional resistance to suppression. To do so, we first established that RPM and TAC treatment reduced recipient-driven immunologic rejection of HLA-mismatched CAR-T cells. Next, we demonstrated that FKBP1A-disrupted CD19-specific CAR-T cells maintained functional immune responses that controlled tumor progression and induced B cell aplasia in vivo amid immunosuppressant therapy. Furthermore, we confirmed that the in vivo persistence of allogeneic CAR-T cells required concurrent immunosuppressant treatment, whereafter drug treatment interruption the recipient immune response cleared allogeneic CAR-T cells. Together, these findings highlight an effective approach to overcome allorejection of “off-the-shelf” CAR-T cells and provides a means of controlling the duration of the CAR-T cell therapy.
Results
HLA class I expression dictates susceptibility of allogeneic cells to rejection by T cells or NK cells
HLA-mismatched allogeneic cells elicit responses by the recipient immune system.7 Thus, we obscured host T cell recognition by ablating HLA class I (HLA-I) and HLA class II (HLA-II) surface expression using base editing to knockout (KO) B2M and CIITA, respectively (Figures 1A–1C). HLA-deficient T cells were invisible to alloreactive T cells and resisted elimination in mixed leukocyte assays (Figures 1D and 1E). To confirm these findings in vivo we employed humanized immune system (HIS) mice that are capable of rejecting HLA-mismatched cells24,25 (Figure S1). HIS mice were co-infused with an equal amount of HLA+ and HLA-deficient (B2MKOCIITAKO) T cells from an unrelated human donor (Figure 1F). These allogeneic T cells were also base edited to disrupt TCR expression preventing GVHD (Figure S2) and engineered to express a non-targeting CD4-based CAR (4CAR) with a molecular tag facilitating ex vivo identification by flow cytometry (Figure S3). After transfer into HLA-mismatched HIS mice, allogeneic HLA+ 4CAR-T cells were rapidly eliminated from circulation (Figures 1G and 1H) and tissue (Figure 1I), whereas HLA-deficient 4CAR-T cells persisted for the study duration (Figures 1G–1I).
Figure 1.

HLA-I expression influences susceptibility of allogeneic T cells to recipient T cell or NK cell-driven rejection
(A) Generation of allogeneic HLA-I- and HLA-II-deficient T cells using base editing to knock out (KO) B2M and CIITA, respectively. (B and C) Histograms indicate HLA-I and HLA-II surface expression (B) and frequency of on-target A>G nucleotide conversion by next-generation sequencing (C) in T cells base edited with B2M- and CIITA-specific sgRNAs and ABE8.20m mRNA. Symbols represent independent donors. (D and E) Mixed leukocyte assay as described in the materials and methods. FACS plots (D) and summarized data (E) for frequency of allogeneic B2MKOCIITAKO and HLA+ T cells after culture with HLA-mismatched alloreactive T cells at different effector-to-target (E/T) ratios. Symbols represent allogeneic cells from independent experiments in duplicate. (F–I) Allogeneic CD4-based CAR-T cells (4CAR) co-expressed GFP and were base edited to disrupt T cell receptor expression (TCRKO), or TCRKO, B2MKO, and CIITAKO. 4CAR-T cells (5 × 106) of each type were infused into HLA-mismatched humanized immune system (HIS) mice (BLT-NSG; n = 6) (F). (G and H) FACS plots indicate frequency (G) and summarized data show concentration (H) of peripheral HLA+ and B2MKOCIITAKO 4CAR-T cells. (I) Total 4CAR-T cells from individual mouse splenic tissue 60 days post-infusion. (J) NK cell cytotoxicity assay as described in materials and methods. Percentage specific lysis of allogeneic B2MKO T cells and CIITAKO T cells at different E/T ratios. Symbols represent mean of four independent NK cell donors in duplicate. (K) Percentage change in CD107a+ NK cells after stimulation with B2MKO T cells or CIITAKO T cells from unmodified T cell control. Symbols indicate NK cells from three independent donors in duplicate. (L–N) Allogeneic TCRKO and TCRKOB2MKO 4CAR-T cells (5 × 106) were infused into huNK mice (n = 5) or NSG-IL-15tg mice (n = 5) (L). (M and N) FACS plots indicate frequency (M) and summarized data shows concentration (N) of peripheral B2MKO 4CAR-T cells in recipient mice. (H and N) Dotted and bold lines indicate individual mice or mean, respectively. Statistical significance was calculated by paired Student’s t test (I) and Wilcoxon rank-sum test (J and K). Error bars show ±SEM, and sample sizes indicate biologically independent animals.
Although the absence of HLA surface expression mitigated T cell-driven allorejection, we found that disruption of HLA-I, not HLA-II, sensitized allogeneic T cells to in vitro NK cell-mediated lysis (Figure 1J) and elicited robust NK cell degranulation (Figure 1K). Yet, HIS mice did not readily reject HLA-deficient T cells (Figure 1H) and, congruent with previous reports,26,27,28 our data indicated that their NK cell compartment is underdeveloped relative to adult humans. For example, the degree of NK cell reconstitution in peripheral blood of HIS mice is markedly less than in humans (Figure S4A), and NK cells from HIS mice required priming with recombinant IL-15 protein to become functionally competent and reject HLA-I-deficient 4CAR-T cells (Figures S4B–S4E). However, repetitive IL-15 treatment of HIS mice induced systemic toxicity and weight loss, which prevented longitudinal assessment of 4CAR-T cell persistence (Figure S4F). Therefore, we assessed the fate of HLA-I-deficient 4CAR-T cells in a humanized NK cell (huNK) mouse model devoid of endogenous T cells.29 Here, primary human CD56+ NK cells were transplanted into NSG-IL-15 transgenic (tg) mice. After 2 weeks post-engraftment, allogeneic TCRKO HLA+ and TCRKO HLA-I– (B2MKO) 4CAR-T cells were co-infused into huNK mice and unreconstituted NSG-IL-15tg control mice (Figure 1L). In contrast to HIS mice, huNK mice rapidly rejected HLA-I– 4CAR-T cells, while HLA+ 4CAR-T cells resisted elimination (Figures 1M and 1N). Together, these data support that the status of HLA-I expression on allogeneic T cells largely dictates susceptibility to either host T cell- or NK cell-mediated rejection.
Retention of HLA-I expression is necessary to broadly inhibit NK cell reactivity toward allogeneic T cells
Approaches to restore NK cell tolerance by engaging HLA-specific inhibitory receptors include forced expression of an invariant HLA single-chain (SC) molecule into B2MKO T cells (Figure 2A),30,31,32 or targeted disruption of HLA alleles.33,34 However, broad implementation of these strategies may be challenged by the heterogeneous expression pattern of HLA-specific inhibitory receptors governing NK cell activity35,36 (Figure 2B). This stochastic expression yields NK cell subsets either lacking or expressing one or more inhibitory killer cell Ig-like receptors (KIRs) (Figure S5), resulting in distinct NK cells that may react toward cells without the appropriate HLA/KIR ligand(s). Indeed, during co-culture allogeneic B2MKO T cells expressing an HLA-Bw4SC, HLA-C1SC, HLA-C2SC, or HLA-ESC molecule attenuated degranulation by NK cells that expressed the corresponding HLA-specific inhibitory receptor either KIR3DL1, KIR2DL2/L3, KIR2DL1, or NKG2A, respectively (Figures 2C and S6). However, these HLASC molecules failed to reduce the net frequency of responding CD107a+ total NK cells to levels observed by stimulation with unmodified (HLA-I+) allogeneic T cells (Figure 2D). Furthermore, HLASC molecules did not substantially protect B2MKO T cells from lysis by NK cells unlike unmodified T cells that were not eliminated (Figure 2E). These data suggest that the overall benefit of an HLASC was nullified due to the insufficient frequency of NK cell subsets expressing the corresponding HLA-specific inhibitory receptor, whereas broad inhibition by allogeneic T cells required the full complement of HLA-I alleles.
Figure 2.

HLA-I retention by allogeneic T cells broadly inhibits NK cell reactivity
(A) Schematic of allogeneic HLA-I-deficient (B2MKO) T cells expressing an HLA-I single-chain (HLASC) molecule that inhibits NK cells by engaging a cognate HLA-specific inhibitory receptor. (B) Bubble plot indicates frequency of CD56+ NK cells expressing the indicated HLA-specific inhibitory receptor from 14 independent human donors. (C and D) NK cells were stimulated with allogeneic HLA-I+ T cells, B2MKO T cells, or B2MKO T cells engineered to express one HLASC: HLA-Bw4SC (HLA-B∗57), HLA-C1SC (HLA-C∗01:02 or C∗07:02), HLA-C2SC (HLA-C∗04:01, C∗05:01, C∗06:02, or C∗18:01), or HLA-ESC (HLA-E∗01:03). (C) Summarized data indicate frequency of NK cell subsets expressing the indicated HLA-specific inhibitory receptor that were CD107a+ after stimulation with allogeneic HLA-I+ T cells, B2MKO T cells, or B2MKO T cells expressing the HLASC inhibitory ligand for the corresponding NK cell subset (i.e., KIR3DL1-HLA-Bw4SC, KIR2DL2/L3-HLA-C1SC, KIR2DL1-HLA-C2SC, NKG2A-HLA-ESC). Horizontal dashed line indicates average frequency of CD107a+ NK cells after stimulation with allogeneic HLA-I+ T cells. (D) Frequency of total CD107a+ NK cells after stimulation with the indicated target T cell population. Vertical dashed line indicates average frequency of CD107a+ NK cells in the absence of stimulation. (E) NK cell cytotoxicity assay as described in materials and methods. Percentage specific lysis of allogeneic HLA-I+ T cells, B2MKO T cells, or B2MKO T cells engineered to express the indicated HLASC molecule 48 h post-culture at different E/T ratios. (C–E) Symbols represent aggregated data from three to five independent NK cell donors in duplicate. Bars indicate mean and error bars show ±SEM. Statistical significance was calculated by Wilcoxon matched pairs signed rank test (C) and Kruskall-Wallace test with Dunn’s test for multiple comparisons (D and E).
Immunosuppression treatment mitigates in vivo rejection of allogeneic HLA-mismatched CAR-T cells
Given that complete retention of HLA-I surface expression was required to broadly inhibit NK cell reactivity toward allogeneic T cells, we investigated an alternative strategy where immunosuppressant treatment could be leveraged to avert rejection of HLA-mismatched CAR-T cells. We first determined if immunosuppressants prevented the in vitro priming of alloreactive T cells. To do so, we cultured peripheral blood mononuclear cells and CellTrace Violet (CTV)-labeled T cells from HLA-mismatched donors in the presence of RPM, TAC, or vehicle (VEH) control. As expected, the addition of immunosuppressants severely attenuated the proliferation of alloreactive CD4+ and CD8+ CTV-labeled T cells relative to VEH treatment (Figure S7). Next, we tested whether immunosuppressants alleviated in vivo rejection of allogeneic HLA-I+ 4CAR-T cells. Five cohorts of HIS mice derived from unrelated human donors were injected daily with RPM, TAC, or VEH for 2 weeks, and at 1 day post-treatment initiation all mice received an equal amount of allogeneic TCRKO HLA+ and TCRKO HLA-deficient 4CAR-T cells (Figure 3A). Syngeneic HIS mouse-derived 4CAR-T cells were also infused into mice in cohorts 3 and 4 to control for autologous cell persistence.
Figure 3.

Immunosuppressant treatment mitigates in vivo rejection of allogeneic HLA-I+ CAR-T cells
(A) Five HIS mouse cohorts (1–5) were allocated into groups receiving vehicle (VEH) (n = 25), rapamycin (RPM) (n = 14), or tacrolimus (TAC) (n = 13) daily for 2 weeks. At 1 day post-treatment, 5 × 106 allogeneic, TCRKO (HLA+) and TCRKOB2MKOCIITAKO (HLA-deficient) 4CAR-T cells were mixed and infused into HLA-mismatched mice. Mice in cohorts 3 and 4 also received an equal amount of syngeneic HIS mouse-derived 4CAR-T cells. (B) FACS plots indicate longitudinal frequency of peripheral allogeneic HLA+ and HLA-deficient 4CAR-T cells in VEH-, RPM-, or TAC-treated mice at 1, 7, and 13 days post-infusion. (C) Aggregate peripheral allogeneic HLA+ 4CAR-T cell persistence relative to HLA-deficient 4CAR-T cells from individual mice in cohorts 1–5 during the drug treatment interval. (D) Peripheral allogeneic HLA+ 4CAR-T cell persistence relative syngeneic HIS mouse-derived 4CAR-T cells from individual mice in cohorts 3–4 during the drug treatment interval. (E and F) Correlation between percentage change in allogeneic HLA+ 4CAR-T cells from 1 to 7 days post-infusion and contemporaneous trough plasma concentration of RPM in cohorts 1–4 (E) and TAC in cohorts 3–4 (F) at 7 days post-infusion. (G) Total splenic allogeneic HLA+ and HLA-deficient 4CAR-T cells from individual mice treated with VEH (n = 5) and TAC (n = 5) in cohort 5, 1 day post-drug withdrawal. (H and I) FACS plots indicate frequency (H) and summarized data show total (I) splenic allogeneic HLA+ and HLA-deficient 4CAR-T cells from individual mice treated with VEH (n = 4), RPM (n = 6), and TAC (n = 6) in cohorts 3 and 4, 30 days post-drug cessation. (J and K) Cumulative persistence of peripheral syngeneic 4CAR-T cells and allogeneic HLA+ 4CAR-T cells during the drug treatment interval (1–13 days post-infusion) (J) and post-drug cessation (22–42 days post-infusion) (K). For all data, symbols and sample sizes indicate biologically independent animals. Bars and lines represent mean and error bars show ±SEM. Statistical significance was calculated by Kruskall-Wallace test with Dunn’s test for multiple comparisons (C and D), Spearman correlation (E and F), Wilcoxon matched pairs signed rank test (G and I), and Wilcoxon rank-sum test (J and K). AUC, area under the curve; r, coefficient of correlation.
Both allogeneic 4CAR-T cell populations were equally detected in the peripheral blood from all groups at 1 day post-infusion but, thereafter, we observed rapid depletion of circulating HLA+ 4CAR-T cells and persistence of HLA-deficient 4CAR-T cells in VEH-treated mice. However, RPM and TAC treatment significantly mitigated rejection of allogeneic HLA+ 4CAR-T cells at 1 and 2 weeks post-treatment initiation (Figures 3B, 3C, and S8). We also observed that allogeneic HLA+ 4CAR-T cells from RPM-treated mice persisted to the same degree as syngeneic 4CAR-T cells within individual mice until 1 week post-T cell infusion, while allogeneic HLA+ 4CAR-T cells from TAC-treated mice persisted similarly to their syngeneic counterparts for the entire drug treatment interval (Figures 3D and S8). Moreover, the extent of circulating allogeneic HLA+ 4CAR-T cell preservation positively correlated with the contemporaneous trough plasma concentration of RPM and TAC (Figures 3E and 3F). Mice from cohort 5 were then euthanized at 1 day post-drug withdrawal to determine if TAC treatment inhibited allorejection in tissue. We detected equivalent levels of allogeneic HLA+ and HLA-deficient 4CAR-T cells from spleens of TAC-treated mice, whereas HLA+ 4CAR-T cells from VEH-treated mice were depleted (Figure 3G). Notably, following drug treatment cessation in mice from cohorts 3 and 4, allogeneic HLA+ 4CAR-T cells were swiftly eliminated from circulation (Figure S9), and when mice were euthanized 30 days after drug withdrawal these cells were nearly undetectable in tissue, indicating recovery of host immunologic rejection (Figures 3H and 3I). At the given dosages, TAC more than RPM improved cumulative allogeneic HLA+ 4CAR-T cell persistence during the drug treatment interval (Figure 3J) and post-drug withdrawal (Figure 3K). These data demonstrated that RPM and TAC treatment extended the in vivo lifespan of allogeneic HLA+ 4CAR-T cells upon transfer into HLA-mismatched HIS mice.
Genetic disruption of FKBP1A renders CAR-T cells resistant to RPM and TAC
To overcome RPM- and TAC-mediated impairment of CAR-T cell function, we engineered immunosuppressant-resistant CAR-T cells by disrupting the gene encoding FKBP prolyl isomerase 1A (FKBP1A), which is an intracellular binding partner of RPM and TAC.15 FKBP1A-specific single-guide RNAs (sgRNAs) (Table S1) were paired with mRNA encoding either an adenosine (ABE8.20m) or cytosine (BE4) base editor and electroporated into activated primary human T cells (Figure 4A). This screen identified an ABE8.20m-sgRNA (TSBTx1538) pair targeting an intron-exon splice junction that achieved a mean on-target genomic editing efficiency of 93.7% (Figure 4B) and reduced FKBP1A protein expression (Figure S10). Following activation, FKBPIAKO T cells treated with RPM retained phosphorylation of the S6 ribosomal protein, a downstream substrate of the PI3K/Akt/mTOR pathway (Figures 4C and 4D), and maintained calcineurin-induced NFAT-driven GFP expression after TAC treatment (Figures 4E and 4F) indicating that FKBP1AKO restores proximal signaling events inhibited by these immunosuppressants.
Figure 4.

FKBP1A-disrupted CAR-T cells resist rapamycin and tacrolimus immunosuppression in vitro
(A) Frequency of maximum on-target nucleotide conversion by next-generation sequencing (NGS) in T cells base edited with individual FKBP1A-specific sgRNAs complexed with mRNA encoding an adenosine (ABE8.20m) or cytosine (BE4) base editor. (B) Frequency of maximum on-target A>G nucleotide conversion by NGS in T cells base edited with TSBTx1538 sgRNA and ABE8.20m mRNA (FKBP1AKO). Symbols indicate independent donors. (C and D) FACS plots (C) and summarized data (D) indicate frequency of phosphorylated S6 protein in unmodified and FKBP1AKO T cells that were activated and treated with rapamycin (RPM) or DMSO vehicle (VEH) control. (E and F) FACS plots (E) and summarized data (F) indicate frequency of NFAT-driven GFP expression in reporter T cells that were unmodified or FKBP1AKO after treatment with tacrolimus (TAC) or VEH. (G) Percentage change in total CD19-specific CAR-T cells (19CAR) counts 1 week post-treatment with RPM or TAC relative to VEH. Symbols represent three independent donors in duplicate. (H and I) Intracellular cytokine expression was measured in unmodified and FKBP1AKO 19CAR-T cells after stimulation with JeKo-1 tumor cells in the presence of RPM, TAC, or VEH. FACS plots indicate frequency of IFN-γ+ and TNF-α+ 19CAR-T cells (H) and summarized data show percentage change in cytokine expression in RPM- and TAC-treated conditions relative to VEH (I). (J) Incucyte cytotoxicity assay as described in materials and methods. Tumor burden quantified as green calibrated units (GCUs) derived from the fluorescence intensity of GFP+ JeKo-1 tumors that were cultured in triplicate with either untransduced (UTD) T cells, unmodified 19CAR-T cells, or FKBP1AKO 19CAR-T cells at a 0.25:1 ratio. Solid lines represent mean GCU from images taken every 4 h, dotted lines show ±SEM, and vertical lines indicate redosing with VEH, RPM, or TAC. (D, F, and I) Symbols represent three independent donors in duplicate. For all data, lines and bars represent mean and error bars show ±SEM.
To evaluate whether FKBP1AKO confers T cells with functional resistance to immunosuppression, we stimulated CD19-specific CAR-T cells (19CAR) with JeKo-1 mantle cell tumors in the presence of RPM, TAC, or VEH. Under immunosuppression, both FKBP1AKO CD4+ and CD8+ 19CAR-T cells proliferated greater than unmodified 19CAR-T cells and, notably, expanded to a similar extent as their VEH-treated counterparts (Figure 4G). Furthermore, immunosuppressant treatment also drastically reduced the frequency of unmodified 19CAR-T cells producing GM-CSF, IL-2, IFN-γ, and TNF-α, while the magnitude of cytokine-positive FKBP1AKO 19CAR-T cells remained unchanged from VEH treatment (Figures 4H and 4I). FKBP1AKO 19CAR-T cells also overcame RPM and TAC inhibition to eradicate GFP+ JeKo-1 tumor cells in vitro with nearly the same kinetics as treatment with VEH. In contrast, immunosuppressants diminished the ability of unmodified 19CAR-T cells to control tumor growth (Figures 4J, S11, and S12).
In addition, we evaluated whether FKBP1AKO rendered 19CAR-T cells resistant to corticosteroids, a treatment option for patients experiencing adverse events following CAR-T cell therapy.37,38,39 Both dexamethasone and prednisone suppressed pro-inflammatory cytokine secretion and antigen-driven proliferation of FKBP1AKO 19CAR-T cells (Figure S13), indicating that FKBP1AKO does not interfere with steroid-mediated suppression. Collectively, these findings support that genetic ablation of FKBP1A renders CAR-T cells resistant to RPM- and TAC-induced inhibition.
FKBP1AKO 19CAR-T cells resist immunosuppression to control in vivo tumor progression
To evaluate whether FKBP1AKO 19CAR-T cells resisted immunosuppression in vivo, we transplanted luciferase-expressing JeKo-1 tumors into NSG mice and 1 week later initiated RPM, TAC, or VEH treatment daily for 2 weeks. One day post-treatment initiation, mice were infused with either FKBP1AKO or unmodified 19CAR-T cells, or untransduced (UTD) control T cells (Figure 5A). Both 19CAR-T cell populations eradicated tumors in VEH-treated mice relative to mice that received UTD T cells. TAC treatment attenuated the ability of unmodified 19CAR-T cells to control tumor outgrowth, whereas FKBP1AKO CAR-T cells potently suppressed tumor progression under TAC (Figures 5B and 5C). Moreover, during the TAC treatment interval FKBP1AKO 19CAR-T cells drastically reduced cumulative tumor burden equivalent to unmodified 19CAR-T cells in VEH-treated mice (Figure 5D), indicating that FKBP1AKO confers 19CAR-T cell resistance to TAC inhibition in vivo.
Figure 5.

FKBP1AKO 19CAR-T cells retain in vivo anti-tumor function in the presence of tacrolimus and rapamycin
(A) Schematic of drug treatment in xenograft tumor-bearing mouse model for (B)–(E). JeKo-1.Luc cells (5 × 105) were transplanted into recipient NSG mice (n = 72), then 7 days later mice initiated vehicle (VEH) (n = 24), rapamycin (RPM) (n = 24), or tacrolimus (TAC) (n = 24) treatment daily for 2 weeks. One day later, mice from each treatment group were infused with 1 × 106 untransduced (UTD) T cells (n = 8 per group), unmodified CD19-specific CAR-T cells (19CAR) (n = 8 per group), or FKBP1AKO 19CAR-T cells (n = 8 per group). (B) Representative longitudinal bioluminescent flux imaging of JeKo-1.Luc-bearing NSG mice treated with TAC and UTD, 19CAR, or FKBP1AKO 19CAR-T cells. (C) Longitudinal tumor burden (flux p/s) of T cell-treated mice that received VEH or TAC treatment. (D) Cumulative tumor burden of T cell-infused mice during the VEH or TAC treatment interval. (E) Longitudinal tumor burden of T cell-infused mice that received VEH or RPM treatment. (F and G) NSG mice were implanted with 5 × 105 JeKo-1.FKBP1AKO.Luc cells and 6 days later initiated VEH (n = 30) or RPM (n = 30) treatment daily for 2 weeks. One day after, mice from each treatment group were infused with either 1 × 106 UTD T cells (n = 10 per group), unmodified 19CAR-T cells (n = 10 per group), or FKBP1AKO 19CAR-T cells (n = 10 per group). (F) Longitudinal tumor burden of T cell-infused mice that received VEH or RPM treatment. (G) Cumulative tumor burden of T cell-infused mice during the VEH or RPM treatment interval. (C, E, and F) Vertical dotted line indicates T cell infusion. For all data, symbols and bars reflect means and error bars show ±SEM, except (D) and (G), where symbols represent individual mice. Sample sizes represent biologically independent animals. Statistical significance was calculated by Kruskall-Wallace test with Dunn’s test for multiple comparisons (D and G). AUC, area under the curve.
Unlike TAC treatment, RPM administration mitigated JeKo-1 growth in control mice, which obscured our ability to detect additive anti-tumor benefit in mice receiving FKBP1AKO over unmodified 19CAR-T cells (Figure 5E). This observation is consistent with B cell malignancies exhibiting constitutive PI3K/Akt/mTOR pathway activation40,41 and sensitivity to mTOR inhibitors.42,43 Indeed, RPM treatment attenuated protein phosphorylation of mTOR and downstream substrates S6 and 4EBP1, as well as inhibited in vitro proliferation of JeKo-1, Raji, and Nalm6 cancer cell lines (Figure S14). To overcome this obstacle, we generated FKBP1AKO JeKo-1 tumors that, when in the presence of RPM, exhibited comparable in vivo expansion kinetics to when they are treated with VEH (Figure 5F). Using FKBP1AKO JeKo-1 tumors, we now demonstrated that FKBP1AKO 19CAR-T cells, not unmodified 19CAR-T cells, decreased cumulative tumor burden within the RPM treatment interval to the same extent as in VEH-treated mice (Figure 5G). These data demonstrated that FKBP1AKO 19CAR-T cells effectively resist immunosuppression and exhibit robust in vivo anti-tumor activity.
HLA-mismatched FKBP1AKO 19CAR-T cells and TAC treatment overcome rejection to induce B cell aplasia in vivo
Finally, we assessed whether immunosuppressant treatment mitigates in vivo rejection of allogeneic HLA+ FKBP1AKO 19CAR-T cells to an extent that permits functional immune responses. Here, we leveraged the endogenous B cell compartment of HIS mice as an on-target population for 19CAR-T cells, where both the depth of B cell aplasia and the persistence of allogeneic cells could assess treatment efficacy. HIS mice from four HLA-mismatched cohorts (6–9) were allocated into groups that received UTD T cells and VEH treatment (group 1), HLA+ FKBP1AKO 19CAR-T cells and VEH treatment (group 2), HLA+ FKBP1AKO 19CAR-T cells and TAC treatment (group 3), and HLA-deficient 19CAR-T cells and VEH treatment (group 4) (Figure 6A). Given that allogeneic HLA-deficient T cells evade rejection in HIS mice (Figures 1G and 1H), these cells served as a benchmark to measure the effectiveness of TAC and allogeneic HLA+ 19CAR-T cell therapy (group 3). Upon infusion, we observed that TAC and FKBP1AKO 19CAR-T cell-treated mice (group 3) exhibited lower CD19+ B cell counts in peripheral blood (Figures 6B and 6C), spleen (Figure 6D), and bone marrow (Figure 6E) compared with VEH-treated mice (group 2), and to a near-equivalent level in mice treated with HLA-deficient 19CAR-T cells (group 4). We also measured a significant reduction in CD19 surface density on residual B cells (Figure 6F) and emergence of CD19dimCD22+ B cells (Figure 6G) from mice in group 3 compared with the group 2 control, indicating effective selection pressure by HLA+ FKBP1AKO 19CAR-T cells in the presence of TAC. Furthermore, allogeneic HLA+ FKBP1AKO 19CAR-T cells from TAC-treated mice (group 3) persisted in peripheral blood (Figures 6H and 6I) and spleen (Figure 6J) to a similar magnitude as HLA-deficient 19CAR-T cells (group 4). Together, these data demonstrate that TAC treatment confers allogeneic HLA+ FKBP1AKO CAR-T cells sufficient protection from immunologic rejection to deplete endogenous B cells.
Figure 6.

Allogeneic FKBP1AKO 19CAR-T cells induce B cell aplasia in tacrolimus-treated HIS mice
(A) Four independent cohorts (6–9) of huNCG HIS mice were evenly distributed into groups: group 1 (n = 16) received allogeneic HLA-deficient (B2MKOCIITAKO) UTD T cells and VEH treatment, group 2 (n = 16) received allogeneic HLA+ CD19-specific CAR-T cells (19CAR) and VEH treatment, group 3 (n = 15) received allogeneic HLA+FKBP1AKO 19CAR-T cells and TAC treatment, and group 4 (n = 16) received allogeneic HLA-deficient 19CAR-T cells and VEH treatment. All T cells were base edited to disrupt TCR expression. (B and C) Concentration (B) and FACS plots indicate frequency (C) of peripheral CD19+ B cells 6 days post-T cell infusion from mice in groups 1–4. (D and E) Total CD19+ B cells from individual mouse splenic (D) and bone marrow (E) tissue 10 days post-T cell infusion in groups 1–4. (F) Geometric median fluorescent intensity (MFI) of CD19 expression on peripheral CD22+ B cells 6 days post-T cell infusion from mice in groups 1–4. (G) Concentration of peripheral CD22+CD19dim B cells 6 days post-T cell infusion from mice in groups 1–4. (H) FACS plots indicate frequency of EGFR+ 19CAR-T cells from mice in groups 2–4, 10 days post-T cell infusion. (I and J) Peripheral concentration of 19CAR-T cells (I) and total splenic 19CAR-T cells (J) from mice in groups 2–4, 10 days post-T cell infusion. For all data, bars reflect mean, error bars show ±SEM, and symbols indicate biologically independent animals. Statistical significance was calculated by Wilcoxon rank-sum test (B, D, and E) and Kruskall-Wallace test with Dunn’s test for multiple comparisons (F, G, I, and J).
Discussion
Here, we describe a strategy where allogeneic, universal donor-derived CAR-T cells in the presence of immunosuppressant treatment persist and retain effector function in humanized mice. Allogeneic CAR-T cells were base edited to inactivate the T cell receptor complex preventing GVHD, and critically, FKBP1A, which conferred robust resistance to RPM and TAC suppression. Simultaneous immunosuppressant therapy was necessary to mitigate recipient immune-driven rejection of HLA-mismatched CAR-T cells, whereafter drug withdrawal allogeneic CAR-T cells were eliminated. This strategy demonstrates that genome engineering enables immunosuppressants to increase the therapeutic potential of allogeneic CAR-T cells by overcoming allorejection.
Reconstitution of the patient immune system following lymphodepleting chemotherapy accelerates rejection of allogeneic CAR-T cells, which may limit durable responses and overall treatment efficacy. Previous reports describe complex genome engineering strategies to limit alloreactivity. For example, simultaneous disruption of B2M and CIITA abrogate recognition by recipient CD8+ and CD4+ T cells, respectively.8,9 However, HLA deficiency necessitates additional intervention to evade or inhibit NK cell killing, including deletion of adhesion molecules (e.g., CD54 and CD58)44 and activating ligands (e.g., CD155),45 or overexpression of inhibitory ligands (e.g., HLA-E,30,31,32,45 HLA-G,30,46 CD47,46,47 PD-L146). Indeed, enhanced resistance to allorejection has been observed, but only after combining multiplex gene editing with forced expression of protective transgenes.30,31,32,45,46,47 Instead, single genetic disruption of FKBP1A with an adenosine base editor simplifies the genome engineering strategy, which may ultimately mitigate the extent of genomic abnormalities associated with CRISPR-Cas9-induced double-strand DNA breaks48,49,50 and alleviate lentiviral51 or AAV52 vector packaging constraints resulting from the delivery of multiple transgenes.
We first demonstrated that genetic disruption of HLA surface expression on allogeneic CAR-T cells effectively prevented recognition by recipient T cells but sensitized them to NK cell-driven attack due to missing self-recognition. We observed that reconstitution of HLA-I single-chain molecules into B2MKO T cells conferred marginal protection against NK cell lysis. This effect was likely due to the insufficient frequency of NK cell subsets expressing the corresponding inhibitory KIR or NKG2A. Instead, allogeneic T cells that fully maintained HLA-I surface expression resisted rejection by NK cells, which we also observed in vivo using huNK mice. Given the extensive diversity of HLA and KIR alleles between unrelated donor-recipient pairs,36 this finding suggests that allogeneic HLA-I+ CAR-T cells are best capable to broadly inhibit recipient NK cells by reducing the probability of KIR/KIR-ligand mismatch. Moreover, it may be highly advantageous to pre-select allogeneic donors based on HLA genotype. In this way, donors that express KIR-ligands, HLA-Bw4, HLA-C1, and HLA-C2 alleles, are poised to inhibit diverse populations of KIR+ NK cells.
To protect HLA+ CAR-T cells after infusion into HLA-mismatched HIS mice, we co-administered immunosuppressants to mitigate allorejection. RPM and TAC interact with FKBP1A to inhibit mTOR16,17,18 or calcineurin activation,19 respectively, and are routinely used to prevent GVHD in the allogeneic hematopoietic stem cell transplant (HSCT) setting.13,14 During the treatment interval, TAC over RPM improved the persistence of allogeneic HLA+ CAR-T cells to a level equivalent to syngeneic HIS mouse-derived CAR-T cells. Their degree of persistence correlated with the contemporaneous trough plasma concentration of TAC (1–3 ng mL−1), which was lower than the drug target exposure in humans post-HSCT (5–20 ng mL−1).13 Unlike TAC, RPM treatment significantly inhibited B cell tumor progression, which is consistent with RPM analogs exhibiting single-agent activity in patients with aggressive lymphomas.53,54 Together, this suggests an opportunity to augment therapeutic efficacy by leveraging anti-rejection and anti-tumor benefits of these immunosuppressants with allogeneic FKBP1AKO 19CAR-T cells.
The optimal duration of allogeneic CAR-T cell persistence will likely depend on the disease indication. However, transient treatment with RPM or TAC, not long-term exposure, may be sufficient to facilitate meaningful CAR-T cell-driven responses while minimizing side effects associated with immunosuppression. In the autologous CAR-T cell setting, deeper initial remissions and higher peak CAR-T cell expansion are associated with improved patient outcomes.55 These clinical observations combined with the kinetics of both cellular immune reconstitution and CAR-T cell expansion could inform the duration of immunosuppressant treatment. For example, patient CD8+ T cells recover at a median time of 21 days post-lymphodepletion,56 meanwhile peak CAR-T cell expansion generally occurs by 3 weeks post-infusion.37,57,58 These findings imply that transient immunosuppressant treatment, possibly 3–4 weeks, around the time of immune reconstitution would be critical to inhibit allorejection, thereby permitting unhindered expansion of allogeneic CAR-T cells to drive deep initial responses. Utilizing short-term immunosuppressant treatment to maximize the initial response may also be advantageous against autoimmune diseases, where transient peripheral engraftment of 19CAR-T cells can induce remission of systemic lupus erythematosus.59 During this treatment interval, patients would likely be monitored for trough drug concentrations to avoid increased risk of allograft rejection or toxicity if drug levels were too low or high, respectively. Furthermore, CAR-T cell patients are infection prone60 and immunosuppressant therapy may exasperate this risk, which could necessitate enhanced monitoring or even prophylaxis to manage these complications.
In these studies, we used a 2-week drug treatment interval as proof-of-principle, although allogeneic CAR-T cells were infused into lymphoreplete mice without prior conditioning that would otherwise be performed in humans. Despite pre-existing endogenous T cells, HLA-mismatched FKBP1AKO 19CAR-T cells coupled with TAC induced B cell aplasia to the same extent as their HLA-deficient counterparts. Although not directly evaluated in our study, immunosuppressant-mediated suppression of host anti-tumor immune responses could be an undesired consequence given that endogenous immunity can contribute to favorable outcomes in the post-CAR-T cell therapy setting.61,62 However, immunosuppressants may compensate for the loss of endogenous T cell and NK cell responses by exerting anti-tumor activity through indirect and direct pathways. For example, TAC may remodel the tumor microenvironment and remove tumor-support mechanisms by preventing formation of neutrophil extracellular traps,63 mitigating M2 polarization of pro-fibrotic macrophages,64,65 and destabilizing regulatory T cell identity.66,67,68 Meanwhile, single-agent use of the rapalog temsirolimus in patients with renal cell carcinoma improved survival outcomes compared with treatment with IFN-α,69,70 which exhibits potent immune-stimulatory activity. Furthermore, steroid-resistant CAR-T cells with concurrent dexamethasone treatment induced localized tumor regression and necrosis in glioblastoma patients.71 Together, these findings provide examples where treatment with immunosuppressants alone or in combination with CAR-T cells exhibit meaningful clinical responses in the absence of endogenous immunity. Careful consideration should be given to utilizing these drugs in combination with FKBP1AKO CAR-T cells for a given disease, where their combined contribution may outweigh the anti-tumor benefit of endogenous immune responses.
Immunosuppressant treatments and cellular immunotherapies have coincided at an increasing rate, highlighting the practical utility of their interaction for diverse applications.71,72,73,74 We demonstrated that FKBP1AKO CAR-T cells are resistant to RPM and TAC, and both drugs mitigate allorejection in a stringent humanized small-animal model. Importantly, this demonstration may afford healthcare providers flexibility to develop patient-specific treatment plans or adjust drug selection and treatment duration in response to the patient’s condition. In particular, drug cessation potentially could be leveraged to manage treatment-related adverse events, prevent long-term B cell aplasia, or reduce the risk of aberrant clonal outgrowth that may arise from genome engineering.75,76 Collectively, these findings indicate that immunosuppressant treatment extends the in vivo lifespan of HLA-mismatched CAR-T cells offering promise that allogeneic cell therapies can achieve autologous cell-like persistence and associated clinical responses.
Materials and methods
Ethics
BLT (bone marrow, liver, thymus) humanized mice were produced using anonymous human fetal tissue (gestational age of 17–19 weeks) acquired from healthy donors with informed consent by Advanced Bioscience Resources and used under approved protocols from Partners Healthcare Institutional Review Board. BLT mouse experiments were conducted at the Ragon Institute of Mass General, MIT and Harvard, and were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC) under the protocol 2021N000279. NSG, NSG-IL-15tg, and humanized CD34+ NCG mouse studies were performed at the Charles River Accelerator and Development Lab (CRADL) and were approved by the CRADL IACUC under protocol 2021-1298. Animal studies were performed following the instructions in the NIH Guide for the Care and Use of Laboratory Animals.
Humanized immune system mice
BLT humanized mice were generated from female (aged 6–8 weeks) NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory) as described previously.77 In brief, NSG mice were anesthetized and whole-body irradiated (2 Gy), and then implanted with 1 mm3 fragments of human fetal liver and thymus tissue beneath the murine kidney capsule. Subsequently, 1 × 105 autologous fetal liver-derived CD34+ stem cells were intravenously injected within 6 h of transplantation. Beam Therapeutics obtained female NOD-Prkdcem26Cd52IL2rgem26CD22/NjuCrl (NCG) CD34+ humanized (huNCG) mice from Charles River and female (aged 6–8 weeks) NSG-Tg(IL15)1Sz/SzJ (NSG-IL15tg) mice from Jackson Laboratory. huNCG mice were generated from female (aged 4–6 weeks) NCG mice that were myeloablated and then intravenously infused with human umbilical cord blood-derived CD34+ stem cells from a qualified source. huNK mice were generated by supplementing the water of NSG-IL-15tg with Baytril (Bayer) for 1 week followed by whole-body irradiation (2 Gy). Primary human CD56+ NK cells (5 × 106) were then intravenously injected 24 h later. At the Ragon Institute and CRADL, mice were housed in microisolator cages and fed autoclaved food and water. Animal rooms were maintained at 72°F ± 2°F, 30%–70% relative humidity, and were on a 12:12 h light/dark cycle. Human reconstitution was assessed from 12 to 17 weeks post-transplant in BLT and huNCG mice, and 2–3 weeks post-transplant in huNK mice. Mice were included in studies when ≥25% of blood cells in the lymphocyte gate were human CD45+ in BLT and huNCG mice, and when human CD56+ cells achieved 10 cells μL−1 blood in huNK mice.
In vivo drug formulation
RPM (Thermo Fisher Scientific) and TAC (Cayman Chemical) were reconstituted in DMSO (Sigma-Aldrich) at 10 and 25 mg mL−1, respectively, and 0.22 μm sterile filtered. RPM was diluted to 0.15 mg mL−1 and TAC was diluted to 1.5 mg mL−1 using sterile-filtered VEH solution comprising a 1:1 ratio of 5% (v/v) polyethylene glycol (Sigma-Aldrich) and 5% (v/v) Tween 80 (Sigma-Aldrich). PEG-Tween solution served as VEH control and percent volume DMSO was normalized across all treatments.
HIS mouse-driven allorejection model
For the study described in Figure S1, BLT mice (n = 3) were intravenously injected with 5 × 106 syngeneic BLT mouse-derived CD4-based CAR-T cells (4CAR) and 5 × 106 allogeneic human donor-derived 4CAR-T cells. For Figures 1F–1I, BLT mice (n = 6) were infused with an allogeneic human donor-derived T cell product comprising 5 × 106 TCRKO 4CAR-T cells and 5 × 106 TCRKOB2MKOCIITAKO 4CAR-T cells. All mice were bled through puncture of the retro-orbital sinus 1 day post-infusion and weekly thereafter until necropsy at day 21 (Figure S1) or day 60 (Figures 1F–1I) when various tissues were collected to analyze 4CAR-T cells.
For Figure 3, BLT mice were used in cohort 2 (n = 12) and cohort 5 (n = 13), and huNCG mice were used in cohort 1 (n = 10), cohort 3 (n = 12), and cohort 4 (n = 10). HIS mice from each cohort were allocated into groups via matched distribution based on human T cell reconstitution (cells μL−1 blood) using StudyLog software (Studylog Systems) and received daily intraperitoneal injections of RPM (1 mg kg−1), TAC (10 mg kg−1), or VEH control for 14 days. Each mouse cohort represented an independent study where mice in cohort 1 received VEH (n = 5) or RPM (n = 5); cohort 2 received VEH (n = 5) or RPM (n = 7); cohort 3 received VEH (n = 4), RPM (n = 4), or TAC (n = 4); cohort 4 received VEH (n = 4), RPM (n = 3), or TAC (n = 3); and cohort 5 received VEH (n = 7) or TAC (n = 6). One day post-drug treatment initiation, HIS mice were intravenously infused with a unique allogeneic human donor-derived T cell product comprising 5 × 106 TCRKO 4CAR-T cells and 5 × 106 TCRKOB2MKOCIITAKO 4CAR-T cells. Mice in cohorts 3 and 4 were also infused with 5 × 106 syngeneic HIS mouse-derived 4CAR-T cells. Persistence of 4CAR-T cells was monitored at 1 day post-T cell infusion and approximately weekly thereafter via blood draws from the retro-orbital sinus or submandibular vein. HIS mice in cohort 5 were necropsied 24 h after final drug treatment and tissues were collected to analyze 4CAR-T cells.
For Figure 6, huNCG mice were used in cohort 6 (n = 16), cohort 7 (n = 12), cohort 8 (n = 24), and cohort 9 (n = 12) and were evenly distributed into four groups via matched distribution based on human T cell reconstitution. HIS mice in group 1 received VEH and 5 × 106 TCRKOB2MKOCIITAKO UTD T cells, group 2 received VEH and 5 × 106 TCRKOFKBP1AKO CD19-specific CAR-T cells (19CAR), group 3 received TAC and 5 × 106 TCRKOFKBP1AKO 19CAR-T cells, and group 4 received VEH and 5 × 106 TCRKOB2MKOCIITAKO 19CAR-T cells. HIS mice were treated daily with TAC (10 mg kg−1) or VEH for 11 days via intraperitoneal injections and at 1 day post-drug treatment initiation mice were intravenously infused with T cells derived from an allogeneic human donor. Endogenous B cell aplasia and persistence of 19CAR-T cells was monitored at 7 and 11 days post-drug treatment via blood draw from the submandibular vein and tissue collection at necropsy.
When available, HLA typing was performed on genomic DNA isolated from whole blood of HIS mice and from purified allogeneic donor T cells (Table S2). Genomic DNA was isolated with QIAamp DNA Blood Mini Kit (QIAGEN) using the manufacturer’s instructions and submitted to Scisco Genetics for HLA class I typing using ScisGo-HLA-v6 next-generation sequencing technology.
NK cell rejection models
For the study described in Figure S4, BLT mice (n = 13) received intraperitoneal injections every 2–3 days of 2.5 μg recombinant human IL-15 (BioLegend) or sterile PBS (n = 6) for 6 total injections. Subsequently, IL-15-treated (n = 6) and PBS-treated (n = 4) mice were intravenously infused with an allogeneic human donor-derived T cell product comprising 5 × 106 TCRKO 4CAR-T cells and 5 × 106 TCRKOB2MKOCIITAKO 4CAR-T cells. Persistence of 4CAR-T cells was monitored at 1, 4, and 7 days post-T cell infusion via blood draw from the retro-orbital sinus. For the study described in Figures 1L–1N, huNK cell mice (n = 5) and unreconstituted NSG-IL-15tg control mice (n = 5) were intravenously infused with an allogeneic human donor-derived T cell product comprising 5 × 106 TCRKO 4CAR-T cells and 5 × 106 TCRKOB2MKOCIITAKO 4CAR-T cells. Persistence of 4CAR-T cells was monitored at 1, 4, and 7 days post-T cell infusion via blood draw from submandibular vein.
Anti-tumor efficacy model
For the study described in Figures 5A–5E, NSG mice were intravenously injected with 5 × 105 JeKo-1.Luc cells at day 0. On day 7, mice initiated daily intraperitoneal injections of VEH, RPM (1 mg kg−1), or TAC (10 mg kg−1) for 2 weeks. Mice from each treatment cohort were injected on day 8 with 1 × 106 UTD T cells, unmodified 19CAR-T cells, or FKBP1AKO 19CAR-T cells (n = 8 per group). For the study described in Figures 5F and 5G, NSG mice were intravenously injected with 5 × 105 JeKo-1.FKBP1AKO.Luc cells at day 0. On day 7, mice initiated daily intraperitoneal injections of VEH or RPM (1 mg kg−1) for 2 weeks. Mice from each treatment cohort were injected on day 8 with 1 × 106 UTD T cells, unmodified 19CAR-T cells, or FKBP1AKO 19CAR-T cells (n = 10 per group). For all studies, tumor burden was measured every 3–4 days post-tumor implant by bioluminescence imaging (IVIS Spectrum, Spectral Instruments Imaging) 30 min after intraperitoneally injecting mice with 150 mg kg−1 XenoLight D-Luciferin (PerkinElmer). The acquisition time was automatically determined by the instrument for each exposure, and quantification of flux from imaging datasets was performed with the Living Image Studio software (PerkinElmer). In brief, a constant region of interest was drawn over the mouse and flux was reported as total photon per second.
Mass spectrometry
For the study described in Figures 3E and 3F, plasma concentrations of RPM and TAC were determined using liquid chromatography-mass spectrometry (LC-MS). Mouse plasma from whole blood 1 week post-T cell infusion was isolated by centrifugation (2,200 × g, 5 min, 4°C) and then cryopreserved. Thawed samples were injected into an Agilent Infinity 1290 II liquid chromatography coupled with a ScieX 6500+ triple quad mass spectrometer. In brief, the sample plasma, calibration curve (CC), and quality control (QC) samples were prepared by aliquoting 5 μL per sample into a 96-well plate. CC standards (0.5, 1, 5, 25, 100, 500, 900, and 1,000 ng mL−1) and QC samples (1.5, 75, and 750 ng mL−1) were prepared in mouse plasma (BioIVT). Tolbutamide (10 ng mL−1) in acetonitrile was the internal standard and 100 μL was added per well except the wells containing 5 μL blank mouse plasma and 100 μL of acetonitrile. The plate was sealed and vortexed (1,650 rpm, 3 min), and subsequently centrifuged (3,500 rpm, 10 min) at room temperature. Fifty microliters of supernatant was transferred to a separate 96-well collection plate containing 50 μL water per well. The plate was vortexed (1,600 rpm, 1 min) and samples were injected into the LC-MS-tandem MS system for analysis.
The separation column was a Zorbax SB-Phenyl, 1.7 μm, 40 × 2.1 mm column (Agilent). The mobile phase was water containing 0.1% formic acid (mobile phase A) and methanol containing 0.1% formic acid (mobile phase B). The flow rate was 0.8 mL min−1 with an operating column temperature of 50°C. The gradient was from 25% to 80% B in 1 min, then 80%–98% B in 0.7 min followed by a 1 min hold, then 98%–80% B in 0.7 min followed by a 0.5 min hold, and finally brought back to 25% B in 0.5 min followed by 1.35 min of re-equilibration.
The MS instrument was operated in multiple reaction monitoring (MRM) mode and positive electrospray ion mode with the following ion source conditions: curtain gas, 35 psi; gas 1, 70 psi; gas 2, 80 psi; ion spray voltage, 5,500 V; and temperature, 500°C. The MRM transition and collision energy were m/z 936.6 > 409.3 and 74 V for RPM, m/z 826.6 > 616.4 and 48 V for TAC, and m/z 271.0 > 155.1 and 25 V for the internal standard, tolbutamide.
T cell generation, base editing, and lentiviral transduction
Healthy donor adult human leukopaks were obtained from HemaCare (Charles River). T cells were isolated using StraightFrom Leukopak CD4/CD8 MicroBead Kit (Miltenyi Biotec) following the manufacturer’s protocol and cryopreserved in CS10 (STEMCELL Technologies). Syngeneic HIS mouse-derived T cells were isolated from splenic tissue of a reconstituted huNCG mouse using CD2 Microbeads (Miltenyi Biotec) following the manufacturer’s protocol and cryopreserved in CS10. T cells were thawed and placed in culture at 106 cells mL−1 in complete medium comprising ImmunoCult XF T Cell Expansion Medium (STEMCELL Technologies), 1% penicillin-streptomycin, 2 mM GlutaMax, 25 mM HEPES Buffer (Life Technologies), and 5% CTS ImmuneCell SR (Thermo Fisher). Complete medium was complemented with 10 ng mL−1 human IL-15 (BioLegend) and 10 ng mL−1 IL-7 (BioLegend). T cells were stimulated with ImmunoCult Human CD3/CD28/CD2 T cell activator (STEMCELL Technologies) following the manufacturer’s instructions and incubated at 37°C, 5% CO2, and 95% humidity. Two days post-activation, T cells were counted, washed with sterile PBS, and resuspended in P3 buffer (Lonza) at 5 × 107 cells mL−1. T cells were electroporated with 1 μg sgRNA from Agilent and 2 μg mRNA encoding ABE8.20m or BE4 per 106 cells using the Lonza 4D Nucleofector system (program DH-102). Base editing with more than 1 sgRNA was achieved by adding 1 μg of the additional sgRNA(s) per 106 cells to the electroporation reaction. T cells then recovered in complete medium at 106 cells mL−1 and were transduced with 300 μL of concentrated lentiviral vector supernatant per 106 cells. Medium was exchanged every other day to adjust cell concentration to 5 × 105 cells mL−1 until day 10 when cells were cryopreserved in CS10. Sequences for the 20 nucleotide protospacers for CD3E, TRAC, B2M, CIITA, and FKBP1A can be found in Table S1. T cell products for in vivo studies were generated using TRAC sgRNA and BE4 mRNA (Figure S1); TRAC, B2M (TSBTx845), CIITA sgRNAs, and BE4 mRNA (Figures 1F–1I); CD3E, B2M (TSBTx760), CIITA sgRNAs, and ABE8.20m mRNA (Figures 1L–1N and 3); FKBP1A (TSBTx1538) sgRNA and ABE8.20m mRNA (Figure 5); and CD3E, B2M, CIITA, FKBP1A sgRNAs, and ABE8.20m mRNA (Figure 6). All T cell products for in vitro assays were generating using B2M, CIITA, FKBP1A sgRNAs, and ABE8.20m mRNA.
Plasmid construction
The transgene cassette comprising a CD4-based CAR and molecular tag comprising GFP or truncated NGFR, EGFR, or CD19 separated by an intervening T2A linker is described elsewhere.78,79 The CD19-specific CAR comprised the FMC63 single-chain variable fragment,80 CD8α hinge/transmembrane domain, and 4-1BB/CD3ζ intracellular domain, and was separated by a T2A linker to truncated EGFR. To generate the NFAT-GFP reporter plasmid, six NFAT response elements (GGAGGAAAAACTGTTTCATACAGAAGGCGT) were cloned upstream of the murine IFN-β promoter driving expression of GFP. The HLA single-chain molecules were cloned into an expression cassette upstream of a T2A linker and truncated EGFR. HLA single-chain molecules are fusion proteins consisting of B2M signal peptide, B2M mature chain, (G4S)3 linker, and the HLA extracellular, transmembrane, and cytoplasmic domains. HLA alleles include HLA-Bw4 group: HLA-B∗57:01; HLA-C1 group: HLA-C∗01:02 and C∗07:02; HLA-C2 group: HLA-C∗04:01, C∗05:01, C∗06:02, and C∗18:01; and HLA-E∗01:03. HLA-E single chain comprised an HLA-G∗01 signal sequence-derived peptide (VMAPRTLFL) between the signal peptide and B2M chain. HLA amino acid sequences are from the IPD-IMGT/HLA database. All gene fragments were codon optimized (IDT), and then custom synthesized and cloned by GenScript into a self-inactivating (SIN) lentiviral vector.
Lentivirus production
Lentiviral particles were generated using packaging expression vectors from Aldevron: VSV glycoprotein (pALD-VSV-G), HIV Rev (pALD-Rev), and HIV Gag/Pol (pALD-GagPol). The packaging plasmids and the appropriate SIN transfer vector were transfected into HEK293T cells using Lipofectamine 2000 (Life Technologies). At 24 h post-transfection, the supernatant was collected, filtered through a 0.45-μm syringe-driven filter, mixed with PEG-it Virus Precipitation Solution (System Biosciences) and stored at 4°C overnight following the manufacturer’s instructions. After incubation, the virus solution was concentrated by centrifugation for 30 min at 1,500 × g, 4°C. The supernatant was aspirated, and the virus pellet was resuspended in 600 μL of complete medium and stored at −80°C.
Next-generation sequencing of genomic DNA
Genomic DNA (gDNA) samples were prepared to determine base editing efficiency as described previously.81 In brief, 5 × 105 cells were lysed using QuickExtract DNA Extraction Solution (Lucigen) according to the manufacturer’s protocol. Two microliters of gDNA was added to a 25 μL polymerase chain reaction (PCR) containing Q5 High-Fidelity DNA Polymerase (New England Biolabs) and 0.5 μM forward and reverse primers. Primer sequences for gDNA amplification are listed in Table S1. PCR products were then amplified using unique Illumina barcoding primer pairs, and then the resulting product was purified using solid-phase reversible immobilization beads (Beckman Coulter) and quantified using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). Barcoded amplicons were sequenced on an Illumina MiSeq instrument according to the manufacturer’s instructions.
Base editor mRNA production
mRNA production for adenosine (ABE8.20m)82 and cytosine (BE4)81 base editors was performed as described previously. In brief, editors were cloned into a plasmid comprising a T7 promoter, 5′ UTR, Kozak sequence, open reading frame encoding the editor, 3′ UTR, and poly(A) tail. Plasmids were linearized using BbsI-HF (New England Biolabs) and purified using DNA Clean and Concentrate Columns (Zymo Research). Linearized plasmid served as template for in vitro transcription with HiScribe T7 High-Yield RNA Synthesis Kit (New England BioLabs) following the manufacturer’s instructions, except CleanCap AG (TriLink Biotechnologies) was used for cotranscriptional capping. mRNA was purified using lithium chloride precipitation.
Tumor cell line generation
JeKo-1 (CRL-3006), Raji (CCL-86), and Nalm6, clone G5 (CRL-3273), were obtained from the ATCC. Cell lines were transduced with a lentiviral vector encoding both GFP and Click Beetle Green luciferase (Luc). Nalm6 cells were also transduced with a lentiviral vector encoding iRFP670. To generate JeKo-1.FKBP1AKO.Luc cells, parental JeKo-1.Luc cells were electroporated with 1 μg TSBTx1538 sgRNA and 2 μg ABE8.20m mRNA using the Lonza 4D Nucleofector system (SF buffer, program DJ-105). To generate Nalm6.CD19KO.iRFP670 cells, parental Nalm6.iRFP670 cells were electroporated with 1 μg TSBTx3773 sgRNA and 2 μg ABE8.20m mRNA using the Lonza 4D Nucleofector system (SF buffer, program CV-104). All tumor cells were sorted on GFP or iRFP670 positivity, or CD19-negative surface expression using the Aria Phusion (BD Bioscience) to obtain a clonal population. Single cell clones were analyzed by next-generation sequencing to confirm FKBP1A disruption.
Mixed leukocyte reaction
Alloreactive T cells were generated in Figures 1D and 1E by culturing HLA-A∗02– CD3+ T cells (effectors) with HLA-A∗02+ CD3-depleted PBMCs (targets) from a separate donor in duplicate. CD3 selections were performed using CD3 MicroBeads (Miltenyi Biotec) following the manufacturer’s protocol. Effector and target cells were mixed at a 1:1 ratio in complete medium supplemented with 300 U mL−1 IL-2 (Sartorius) for 7–10 days. Activated HLA-A∗02- effector T cells were then cultured at different ratios with PBMC donor-matched HLA-A∗02+ T cells that were unmodified HLA-A∗02+ (on-target) or HLA-deficient B2MKOCIITAKO (off-target) and labeled with CellTrace Far Red (Thermo Fisher Scientific). On- and off-target T cells were combined at 1:1 ratio before adding effector T cells to measure the relative frequency of target T cells within the same well 48 h post-culture using flow cytometry.
For the study described in Figure S7, CellTrace Violet (Thermo Fisher Scientific) CD3+ T cells (effectors) and CD3-depleted PBMCs (targets) from HLA-mismatched donors were cultured in triplicate at a 1:1 ratio in complete medium supplemented with 300 U mL−1 IL-2. Cells were cultured in the presence of RPM at 10−2, 10−3, and 10−4 μg mL−1, TAC at 100, 10−1, and 10−2 μg mL−1, and DMSO VEH control. Effector T cells cultured in the absence of target cells served as an unstimulated control. Frequency of dividing effector CD8+ and CD4+ T cells was measured at 5 and 7 days post-stimulation by flow cytometry.
NK cell cytotoxicity and degranulation assays
Human NK cells were isolated using StraightFrom Leukopak REAlease CD56 MicroBead Kit (Miltenyi Biotec) or CD56 MicroBeads (Miltenyi Biotec). NK cells were primed for 3 days in complete medium with 5 ng mL−1 IL-15 and 300 U mL−1 IL-2. Primed NK cells were then cultured at different ratios with allogeneic T cells that were unmodified HLA+ (off-target) or HLA-deficient B2MKOCIITAKO (on-target). On- and off-target T cells were combined at a 1:1 ratio before adding NK cells to measure the relative change in frequency of target T cells compared with control wells in the absence of NK cells. Specific lysis of on-target T cells was measured 48 h post-culture by flow cytometry using the formula: specific lysis (%) = 100 – (100 × (% survival in the presence of NK cells/% survival in the absence of NK cells)). NK cell degranulation was measured by culturing 105 primed NK cells with 105 on-target or off-target allogeneic T cells, or alone. Anti-CD107a antibody was added at the start of stimulation followed by the addition of 1X Monensin Solution and 1X Brefeldin A (BioLegend) 1 h later. Cells were incubated for 6 h total before analysis by flow cytometry.
CAR-T cell cytotoxicity assays
Cryopreserved UTD T cells, unmodified 19CAR-T cells, and FKBP1AKO 19CAR-T cells were thawed and rested overnight at 37°C, 5% CO2. To perform the Incucyte cytotoxicity assay, a 96-well flat-bottom plate (Corning) was coated with 0.01% poly-L-ornithine solution (Sigma-Aldrich) for 30 min, and then decanted and left to dry for 30 min. Afterward, 2.5 × 104 JeKo-1.FKBP1AKO.GFP+ tumor cells were seeded into the 96-well plate and cultured in triplicate with rested T cells at 0:1, 1:1, 0.25:1, and 0.125:1 effector-to-target cell (E/T) ratios. RPM (100 nM), TAC (100 ng mL−1), and VEH (DMSO) control were added at the start of culture and then again at 48, 96, and 144 h post-culture. Samples were transferred to an Incucyte SX5 Live-Cell Analysis System (Sartorius) and imaged every 4 h to assess GFP fluorescence intensity.
To perform the VITAL cytotoxicity assay,83 2.5 × 104 Nalm6.CD19WT.GFP+ cells and 2.5 × 104 Nalm6.CD19KO.iRFP670+ cells were seeded per well of a 96-well flat-bottom plate. UTD T cells, unmodified 19CAR-T cells, and FKBP1AKO 19CAR-T cells were added at 0:125:1, 0.06:1, 0.03:1, 0.015:1, and 0:1 E/T ratios. RPM (100 nM), TAC (100 ng mL−1), and VEH (DMSO) control were added at the start of culture. Samples were incubated at 37°C, 5% CO2 for 48 h before flow cytometric analysis to measure the frequency of viable GFP+ and iRFP670+ Nalm6 tumor cells. Specific lysis of Nalm6.GFP+ cells was measured using the formula: specific lysis (%) = 100 – (100 × (% survival in the presence of T cells/% survival in the absence of T cells)).
CAR-T cell cytokine production assay
Cryopreserved unmodified and FKBP1AKO 19CAR-T cells were thawed into complete medium and rested overnight at 106 cells mL−1 in the incubator at 37°C, 5% CO2. T cells were pre-treated overnight with RPM (100 nM), TAC (100 ng mL−1), dexamethasone (10 μg mL−1; Sigma-Aldrich), prednisone (1 μg mL−1; Sigma-Aldrich), or VEH (DMSO) control prior to assay set-up. The following day, T cells were washed, counted and 105 cells were seeded in duplicate into a 96-well flat-bottom plate alone or with 105 JeKo-1.GFP+ tumor cells in the presence of drug treatments. Anti-CD107a antibody was added at the start of stimulation followed by the addition of 1X Monensin Solution and 1X Brefeldin A (BioLegend) 1 h later. Cells were incubated for 6 h total before analysis by flow cytometry.
CAR-T cell and tumor proliferation assays
Cryopreserved unmodified and FKBP1AKO 19CAR-T cells were thawed into complete medium and rested overnight at 106 cells mL−1 in the incubator at 37°C, 5% CO2. The following day, T cells were washed, counted, and 104 cells were seeded in triplicate into a 96-well flat-bottom plate alone or with 104 JeKo-1.GFP+ tumor cells. Complete medium was supplemented with RPM (100 nM), TAC (100 ng mL−1), dexamethasone (10 μg mL−1), prednisone (1 μg mL−1), or VEH (DMSO) control and refreshed on days 2, 4, and 6 post-culture. On day 7, samples were analyzed by flow cytometry and 19CAR-T cells were enumerated using CountBright Counting Beads (Thermo Fisher Scientific) following the manufacturer’s protocol.
Raji, Nalm6, and JeKo-1 tumor cells were cultured in complete R10 medium comprising RPMI 1640 Medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 2 mM GlutaMax, and 25 mM HEPES buffer (Life Technologies) at 2.5 × 104 cells mL−1 and seeded into a 24-well tissue culture plate. Tumor cells were plated in triplicate per treatment condition. RPM was added at 10 and 0.1 μg mL−1, and DMSO served as VEH control. Cells were incubated at 37°C, 5% CO2, and viable cell counts were determined using the NucleoCounter NC200 (ChemoMetec) at 2 and 4 days post-culture.
Flow cytometry
Cultured cells were washed and stained in 50 μL of 1X PBS containing 2 mM EDTA and 2% fetal calf serum, and whole blood was stained directly with anti-human antibodies from BioLegend: CD45 (HI30), CD3 (OKT3), CD2 (RPA-2.10), CD4 (OKT4), CD8 (SK1), CD56 (5.1H11), CD107a (H4A3), CD19 (HIB19), CD22 (HIB22), HLA-A2 (BB7.2), HLA-DR (L243), NGFR (ME20.4), EGFR (AY13), NKG2A (S19004C), LIR-1 (GHI/75), HLA-ABC (W6/32), KIR2DL1 (HP-DM1), KIR2DL2/L3 (DX27), KIR3DL1 (DX9), KIR2DL4 (mAB33), and KIR2DL5 (UP-R1). Live cells were discriminated by staining negative for Fixable Viability Dye eFluor780 (eBioscience). CountBright Counting Beads were used according to the manufacturer’s instructions to determine the cell concentration from whole blood and tissue. Intracellular cytokines were detected using Cell Fixation & Cell Permeability Kit (Invitrogen) following the manufacturer’s protocol with anti-human antibodies from BioLegend: TNF-α (MAb11), IFN-γ (4S.B3), IL-2 (MQH-17H12), and GM-CSF (BVD2-21C11). Phosphorylated intracellular proteins were detected using BD Cytofix Fixation Buffer and BD Phosflow Perm Buffer III (BD Biosciences) according to the manufacturer’s instructions with anti-human antibodies from BD Biosciences: mTOR-pS2448 (O21-404), S6-pS235/pS236 (N7-548), and 4EBP1-pT36/pT45 (M31-16). Flow cytometry data were acquired on the MACSQuant Analyzer 16 (Miltenyi Biotec) and FACSymphony A3 Cell Analyzer (BD Biosciences). Data were analyzed using FlowJo software v.10 (Tree Star).
mTOR and calcineurin downstream assays
To evaluate downstream mTOR activity in T cells, phosphorylation of S6 was detected by incubating 2 × 105 T cells overnight in complete medium supplemented with RPM (1 nM) or DMSO VEH control at 37°C, 5% CO2. Subsequently, T cells were washed and stained with Fixable Viability Dye eFlour780 according to the manufacturer’s protocol. T cells were then seeded into a 96-well flat-bottom plate with RPM (1 nM) or DMSO and stimulated with Immunocult T cell Activator (1:40 dilution) or unstimulated for 1 h at 37°C, 5% CO2, prior to analysis by flow cytometry. To detect calcineurin activity in T cells, 2 × 105 T cells expressing an NFAT-GFP reporter were cultured in complete medium supplemented with TAC (100 ng mL−1) or DMSO VEH control. T cells were then stimulated using an Immunocult T Cell Activator (1:40 dilution) or unstimulated and cultured overnight at 37°C, 5% CO2, prior to analysis by flow cytometry. For both assays, T cells were unmodified or base edited to disrupt FKBP1A. To evaluate mTOR activity in B cell tumor lines, phosphorylation of mTOR, S6, and 4EBP1 was detected by incubating 1 × 105 Raji, Nalm6, and JeKo-1 cells overnight in R10 medium in a 96-well flat-bottom plate at 37°C, 5% CO2. Tumor cells were treated with either RPM (10 mg mL−1) or DMSO VEH control. The following day, tumor cells were prepared for flow cytometric analysis.
Protein detection
Unmodified and FKBP1AKO T cells (1–5 × 106) were washed with PBS and lysates were prepared using cold RIPA Buffer (Thermo Fisher Scientific) with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) according to the manufacturer’s protocol. Supernatants were collected and stored at −80°C. Protein was quantified using Pierce 660nm Protein Assay Reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. Detection and quantification of FKBP1A and GAPDH control was determined using ProteinSimple Jess (Biotechne). In brief, protein samples were diluted to 0.5 mg mL−1 using Sample Buffer, combined with Fluorescent Master Mix from EZ Standard Pack 5 (Biotechne), and then denatured at 95°C, 5 min. For FKBP1A protein detection, rabbit anti-human FKBP12 monoclonal antibody (Abcam) was diluted 1:100 in antibody diluent, combined with anti-rabbit secondary HRP antibody, chemiluminescent substrate, and peroxide from Biotechne. For GAPDH protein detection, mouse anti-human GAPDH monoclonal antibody (Cell Signaling Technologies) was diluted 1:100 in antibody diluent and combined with anti-mouse secondary NIR (Biotechne). All samples including the 2–40 kDa separation module (Biotechne) were loaded onto the capillary cartridge (Biotechne) according to the instructions. The plate was analyzed on the Jess, where chemiluminescence measured FKBP1A and fluorescence measured GAPDH. ProteinSimple Compass for SW software was used to quantify area under the curve for total protein signal.
Statistical analysis
Comparison of matched samples were performed using two-sided non-parametric Wilcoxon matched pairs signed rank test. Comparison of unmatched samples were performed using two-sided Student’s t test, non-parametric Wilcoxon rank-sum test, or Kruskal-Wallace test followed by Dunn’s test for multiple comparisons. Bivariate correlations were performed using two-sided Spearman’s rank correlation. Area under the curve calculations were performed using either cell concentration per microliter of blood or frequency of cells. All statistical analyses were performed using GraphPad Prism version 9.3.0 (GraphPad).
Data and code availability
Plasmids encoding ABE8.20m and BE4 base editors used in this work are available from Addgene. The 20 nucleotide protospacer sequences and primers for the amplification of on-target amplicons can be found in Table S1. Amino acid sequences of HLA alleles used in this work are available from the IPD-IMGT/HLA database (https://www.ebi.ac.uk/ipd/imgt/hla/).
Acknowledgments
We thank J. Decker, A. Cozier, M. Patel, C. Lazzara, L. Young, and L. Barrera for next-generation sequencing and computational support. We acknowledge L. Hardy and H. Kromer for single-cell sorting, and W. Schmidt, N. Mastrangelo, J. Bynoe, and E. Chiang for facilitating institutional interactions between Beam Therapeutics and the Ragon Institute. This study was supported by internal funding from Beam Therapeutics and NIH grant R37 AI170189 (to T.M.A). BioRender.com was used to create some figures.
Author contributions
C.R.M. conceived and directed the work, designed and conducted experiments, performed analyses, and wrote the manuscript. A.C.M. and P.B.B. designed and conducted in vitro experiments to interrogate FKBP1A disruption in CAR-T cells and performed analyses. L.J.C. performed the huNK mouse study and NK cell phenotyping, and C.K. generated lentivirus and T cells for in vitro and in vivo experiments. A.J.C. and D.T.C. designed and supervised in vivo experiments, and F.M.M., M.L.W., J.J.R., V.D.V., and E.M. carried out all animal studies. F.L. and B.Y. were responsible for mass spectrometry and analysis. T.M.A. oversaw research. C.R.M., A.J.C., D.T.C., C.L.B., and T.M.A. edited the manuscript.
Declaration of interests
C.R.M., A.C.M., P.B.B., A.J.C., F.M.M., M.L.W., J.J.R., L.J.C., C.K., F.L., and B.Y. were employees of Beam Therapeutics when the work was conducted and are shareholders in the company. Beam Therapeutics has filed patent applications based on this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.06.022.
Supplemental information
References
- 1.Wang X., Borquez-Ojeda O., Stefanski J., Du F., Qu J., Chaudhari J., Thummar K., Zhu M., Shen L.B., Hall M., et al. Depletion of high-content CD14+ cells from apheresis products is critical for successful transduction and expansion of CAR T cells during large-scale cGMP manufacturing. Mol. Ther. Methods Clin. Dev. 2021;22:377–387. doi: 10.1016/j.omtm.2021.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen E.S., Stroncek D.F., Ren J., Eder A.F., West K.A., Fry T.J., Lee D.W., Mackall C.L., Conry-Cantilena C. Autologous lymphapheresis for the production of chimeric antigen receptor T cells. Transfusion (Paris) 2017;57:1133–1141. doi: 10.1111/trf.14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 4.Kernan N.A., Collins N.H., Juliano L., Cartagena T., Dupont B., O’Reilly R.J. Clonable T lymphocytes in T cell-depleted bone marrow transplants correlate with development of graft-v-host disease. Blood. 1986;68:770–773. doi: 10.1182/blood.v68.3.770.bloodjournal683770. [DOI] [PubMed] [Google Scholar]
- 5.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K., et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 6.Philip L.P.B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R., Lebuhotel C., et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 7.Anasetti C., Amos D., Beatty P.G., Appelbaum F.R., Bensinger W., Buckner C.D., Clift R., Doney K., Martin P.J., Mickelson E., et al. Effect of HLA Compatibility on Engraftment of Bone Marrow Transplants in Patients with Leukemia or Lymphoma. N. Engl. J. Med. 1989;320:197–204. doi: 10.1056/nejm198901263200401. [DOI] [PubMed] [Google Scholar]
- 8.Kagoya Y., Guo T., Yeung B., Saso K., Anczurowski M., Wang C.H., Murata K., Sugata K., Saijo H., Matsunaga Y., et al. Genetic ablation of HLA class I, class II, and the T-cell receptor enables allogeneic T cells to be used for adoptive T-cell therapy. Cancer Immunol. Res. 2020;8:926–936. doi: 10.1158/2326-6066.CIR-18-0508. [DOI] [PubMed] [Google Scholar]
- 9.Lee J., Sheen J.H., Lim O., Lee Y., Ryu J., Shin D., Kim Y.Y., Kim M. Abrogation of HLA surface expression using CRISPR/Cas9 genome editing: a step toward universal T cell therapy. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-74772-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kärre K., Ljunggren H.G., Piontek G., Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 11.Moes D.J.A., Guchelaar H.J., De Fijter J.W. Sirolimus and everolimus in kidney transplantation. Drug Discov. Today. 2015;20:1243–1249. doi: 10.1016/j.drudis.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Reichenspurner H. Overview of tacrolimus-based immunosuppression after heart or lung transplantation. J. Heart Lung Transpl. 2005;24:119–130. doi: 10.1016/j.healun.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 13.Sharma N., Zhao Q., Ni B., Elder P., Puto M., Benson D.M., Rosko A., Chaudhry M., Devarakonda S., Bumma N., et al. Effect of early post-transplantation tacrolimus concentration on the risk of acute graft-versus-host disease in allogenic stem cell transplantation. Cancers (Basel) 2021;13 doi: 10.3390/cancers13040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X., Sun H., Cassady K., Yang S., Chen T., Wang L., Yan H., Zhang X., Feng Y. The Addition of Sirolimus to GVHD Prophylaxis After Allogeneic Hematopoietic Stem Cell Transplantation: A Meta-Analysis of Efficacy and Safety. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.683263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber S.L. Chemistry and Biology of the Immunophilins and Their Immunosuppressive Ligands. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y., Chen H., Rhoad A.E., Warner L., Caggiano T.J., Failli A., Zhang H., Hsiao C.L., Nakanishi K., Molnar-Kimber K.L. A putative sirolimus (rapamycin) effector protein. Biochem. Biophys. Res. Commun. 1994;203:1–7. doi: 10.1006/bbrc.1994.2140. [DOI] [PubMed] [Google Scholar]
- 17.Brown E.J., Albers M.W., Shin T.B., Ichikawa K., Keith C.T., Lane W.S., Schreiber S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 18.Sabers C.J., Martin M.M., Brunn G.J., Williams J.M., Dumont F.J., Wiederrecht G., Abraham R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995;270:815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 19.Mattila P.S., Ullman K.S., Fiering S., Emmel E.A., McCutcheon M., Crabtree G.R., Herzenberg L.A. The actions of cyclosporin A and FK506 suggest a novel step in the activation of T lymphocytes. EMBO J. 1990;9:4425–4433. doi: 10.1002/j.1460-2075.1990.tb07893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terada N., Lucas J.J., Szepesi A., Franklin R.A., Domenico J., Gelfand E.W. Rapamycin blocks cell cycle progression of activated T cells prior to events characteristic of the middle to late G1 phase of the cycle. J. Cell. Physiol. 1993;154:7–15. doi: 10.1002/jcp.1041540103. [DOI] [PubMed] [Google Scholar]
- 21.Dumont F.J., Staruch M.J., Koprak S.L., Melino M.R., Sigal N.H. Distinct mechanisms of suppression of murine T cell activation by the related macrolides FK-506 and rapamycin. J. Immunol. 1990;144:251–258. doi: 10.4049/jimmunol.144.1.251. [DOI] [PubMed] [Google Scholar]
- 22.Tocci M.J., Matkovich D.A., Collier K.A., Kwok P., Dumont F., Lin S., Degudicibus S., Siekierka J.J., Chin J., Hutchinson N.I. The immunosuppressant FK506 selectively inhibits expression of early T cell activation genes. J. Immunol. 1989;143:718–726. doi: 10.4049/jimmunol.143.2.718. [DOI] [PubMed] [Google Scholar]
- 23.Kino T., Hatanaka H., Miyata S., Inamura N., Nishiyama M., Yajima T., Goto T., Okuhara M., Kohsaka M., Aoki H., et al. Fk-506, A Novel Immunosuppressant Isolated From A Streptomyces Ii. Immunosuppressive Effect Of Fk-506 In Vitro. J. Antibiot. 1987;40:1256–1265. doi: 10.7164/antibiotics.40.1256. [DOI] [PubMed] [Google Scholar]
- 24.Rong Z., Wang M., Hu Z., Stradner M., Zhu S., Kong H., Yi H., Goldrath A., Yang Y.G., Xu Y., Fu X. An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell. 2014;14:121–130. doi: 10.1016/j.stem.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao F., Ma L., Zhao M., Huang G., Mirenda V., Dorling A., Lechler R., Lombardi G. Ex vivo expanded human regulatory T cells delay islet allograft rejection via inhibiting islet-derived monocyte chemoattractant protein-1 production in CD34+ stem cells-reconstituted NOD-scid IL2rγnull mice. PLoS One. 2014;9 doi: 10.1371/journal.pone.0090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huntington N.D., Legrand N., Alves N.L., Jaron B., Weijer K., Plet A., Corcuff E., Mortier E., Jacques Y., Spits H., Di Santo J.P. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009;206:25–34. doi: 10.1084/jem.20082013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaur K., Jewett A. Differences in Tumor Growth and Differentiation in NSG and Humanized-BLT Mice; Analysis of Human vs. Humanized-BLT-Derived NK Expansion and Functions. Cancers (Basel) 2022;15 doi: 10.3390/cancers15010112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalberer C.P., Siegler U., Wodnar-Filipowicz A. Human NK cell development in NOD/SCID mice receiving grafts of cord blood CD34+ cells. Blood. 2003;102:127–135. doi: 10.1182/blood-2002-07-2024. [DOI] [PubMed] [Google Scholar]
- 29.Katano I., Nishime C., Ito R., Kamisako T., Mizusawa T., Ka Y., Ogura T., Suemizu H., Kawakami Y., Ito M., Takahashi T. Long-term maintenance of peripheral blood derived human NK cells in a novel human IL-15- transgenic NOG mouse. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-17442-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo Y., Xu B., Wu Z., Bo J., Tong C., Chen D., Wang J., Wang H., Wang Y., Han W. Mutant B2M-HLA-E and B2M-HLA-G fusion proteins protects universal chimeric antigen receptor-modified T cells from allogeneic NK cell-mediated lysis. Eur. J. Immunol. 2021;51:2513–2521. doi: 10.1002/eji.202049107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W., Zhu X., Xu Y., Chen J., Zhang H., Yang Z., Qi Y., Hong J., Li Y., Wang G., et al. Simultaneous editing of TCR, HLA-I/II and HLA-E resulted in enhanced universal CAR-T resistance to allo-rejection. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.1052717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jo S., Das S., Williams A., Chretien A.S., Pagliardini T., Le Roy A., Fernandez J.P., Le Clerre D., Jahangiri B., Chion-Sotinel I., et al. Endowing universal CAR T-cell with immune-evasive properties using TALEN-gene editing. Nat. Commun. 2022;13 doi: 10.1038/s41467-022-30896-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torikai H., Mi T., Gragert L., Maiers M., Najjar A., Ang S., Maiti S., Dai J., Switzer K.C., Huls H., et al. Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci. Rep. 2016;6 doi: 10.1038/srep21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu H., Wang B., Ono M., Kagita A., Fujii K., Sasakawa N., Ueda T., Gee P., Nishikawa M., Nomura M., et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell. 2019;24:566–578.e7. doi: 10.1016/j.stem.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Purdy A.K., Campbell K.S. Natural killer cells and cancer: Regulation by the killer cell ig-like receptors (KIR) Cancer Biol. Ther. 2009;8:2211–2220. doi: 10.4161/cbt.8.23.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichise H., Nagano S., Maeda T., Miyazaki M., Miyazaki Y., Kojima H., Yawata N., Yawata M., Tanaka H., Saji H., et al. NK Cell Alloreactivity against KIR-Ligand-Mismatched HLA-Haploidentical Tissue Derived from HLA Haplotype-Homozygous iPSCs. Stem Cell Rep. 2017;9:853–867. doi: 10.1016/j.stemcr.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/nejmoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S., Deng B., Yin Z., Pan J., Lin Y., Ling Z., Wu T., Chen D., Chang A.H., Gao Z., et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10 doi: 10.1038/s41408-020-0280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lakomy T., Akhoundova D., Nilius H., Kronig M.N., Novak U., Daskalakis M., Bacher U., Pabst T. Early Use of Corticosteroids following CAR T-Cell Therapy Correlates with Reduced Risk of High-Grade CRS without Negative Impact on Neurotoxicity or Treatment Outcome. Biomolecules. 2023;13 doi: 10.3390/biom13020382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sebestyén A., Sticz T.B., Márk Á., Hajdu M., Timár B., Nemes K., Nagy N., Váradi Z., Kopper L. Activity and complexes of mTOR in diffuse large B-cell lymphomas - A tissue microarray study. Mod. Pathol. 2012;25:1623–1628. doi: 10.1038/modpathol.2012.141. [DOI] [PubMed] [Google Scholar]
- 41.Vajpayee N., Thakral C., Gopaluni S., Newman N., Gajra A. Activation of mammalian target of rapamycin in diffuse large B-cell lymphoma: A clinicopathological study. Leuk. Res. 2012;36:1403–1409. doi: 10.1016/j.leukres.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 42.Gottschalk A.R., Boise L.H., Thompson C.B., Quintáns J. Identification of immunosuppressant-induced apoptosis in a murine B-cell line and its prevention by bcl-x but not bcl-2. Proc. Natl. Acad. Sci. USA. 1994;91:7350–7354. doi: 10.1073/pnas.91.15.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muthukkumar S., Ramesh T.M., Bondada S. Rapamycin, a potent immunosuppressive drug, causes programmed cell death in B lymphoma cells. Transplantation. 1995;60:264–270. doi: 10.1097/00007890-199508000-00010. [DOI] [PubMed] [Google Scholar]
- 44.Hammer Q., Perica K., van Ooijen H., Mbofung R., Momayyezi P., Varady E., Martin K.E., Pan Y., Jelcic M., Groff B., et al. Genetic ablation of adhesion ligands averts rejection of allogeneic immune cells. bioRxiv. 2023 doi: 10.1101/2023.10.09.557143. Preprint at. [DOI] [PubMed] [Google Scholar]
- 45.Wang B., Iriguchi S., Waseda M., Ueda N., Ueda T., Xu H., Minagawa A., Ishikawa A., Yano H., Ishi T., et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 2021;5:429–440. doi: 10.1038/s41551-021-00730-z. [DOI] [PubMed] [Google Scholar]
- 46.Han X., Wang M., Duan S., Franco P.J., Kenty J.H.-R., Hedrick P., Xia Y., Allen A., Ferreira L.M.R., Strominger J.L., et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2019;116:10441–10446. doi: 10.1073/pnas.1902566116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu X., Manner K., DeJesus R., White K., Gattis C., Ngo P., Bandoro C., Tham E., Chu E.Y., Young C., et al. Hypoimmune anti-CD19 chimeric antigen receptor T cells provide lasting tumor control in fully immunocompetent allogeneic humanized mice. Nat. Commun. 2023;14:2020. doi: 10.1038/s41467-023-37785-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 50.Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sweeney N.P., Vink C.A. The impact of lentiviral vector genome size and producer cell genomic to gag-pol mRNA ratios on packaging efficiency and titre. Mol. Ther. Methods Clin. Dev. 2021;21:574–584. doi: 10.1016/j.omtm.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grieger J.C., Samulski R.J. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J. Virol. 2005;79:9933–9944. doi: 10.1128/JVI.79.15.9933-9944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hess G., Herbrecht R., Romaguera J., Verhoef G., Crump M., Gisselbrecht C., Laurell A., Offner F., Strahs A., Berkenblit A., et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2009;27:3822–3829. doi: 10.1200/JCO.2008.20.7977. [DOI] [PubMed] [Google Scholar]
- 54.Witzig T.E., Reeder C.B., LaPlant B.R., Gupta M., Johnston P.B., Micallef I.N., Porrata L.F., Ansell S.M., Colgan J.P., Jacobsen E.D., et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25:341–347. doi: 10.1038/leu.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cappell K.M., Kochenderfer J.N. Long-term outcomes following CAR T cell therapy: what we know so far. Nat. Rev. Clin. Oncol. 2023;20:359–371. doi: 10.1038/s41571-023-00754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galli E., Fresa A., Bellesi S., Metafuni E., Maiolo E., Pansini I., Frioni F., Autore F., Limongiello M.A., Innocenti I., et al. Hematopoiesis and immune reconstitution after CD19 directed chimeric antigen receptor T-cells (CAR-T): A comprehensive review on incidence, risk factors and current management. Eur. J. Haematol. 2024;112:184–196. doi: 10.1111/ejh.14052. [DOI] [PubMed] [Google Scholar]
- 57.Shah B.D., Ghobadi A., Oluwole O.O., Logan A.C., Boissel N., Cassaday R.D., Leguay T., Bishop M.R., Topp M.S., Tzachanis D., et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398:491–502. doi: 10.1016/S0140-6736(21)01222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munshi N.C., Anderson L.D., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 59.Mackensen A., Müller F., Mougiakakos D., Böltz S., Wilhelm A., Aigner M., Völkl S., Simon D., Kleyer A., Munoz L., et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022;28:2124–2132. doi: 10.1038/s41591-022-02017-5. [DOI] [PubMed] [Google Scholar]
- 60.Korell F., Schubert M.-L., Sauer T., Schmitt A., Derigs P., Weber T.F., Schnitzler P., Müller-Tidow C., Dreger P., Schmitt M. Infection Complications after Lymphodepletion and Dosing of Chimeric Antigen Receptor T (CAR-T) Cell Therapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia or B Cell Non-Hodgkin Lymphoma. Cancers (Basel) 2021;13 doi: 10.3390/cancers13071684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Avanzi M.P., Yeku O., Li X., Wijewarnasuriya D.P., van Leeuwen D.G., Cheung K., Park H., Purdon T.J., Daniyan A.F., Spitzer M.H., Brentjens R.J. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018;23:2130–2141. doi: 10.1016/j.celrep.2018.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alizadeh D., Wong R.A., Gholamin S., Maker M., Aftabizadeh M., Yang X., Pecoraro J.R., Jeppson J.D., Wang D., Aguilar B., et al. IFNγ Is Critical for CAR T Cell-Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021;11:2248–2265. doi: 10.1158/2159-8290.CD-20-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu L., Cai M. Tacrolimus Maintains the Balance of Neutrophil Extracellular Traps by Inducing DNA Methylation of Neutrophils to Reduce Immune Rejection. Life (Basel) 2023;13 doi: 10.3390/life13122253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kannegieter N.M., Hesselink D.A., Dieterich M., Kraaijeveld R., Rowshani A.T., Leenen P.J.M., Baan C.C. The Effect of Tacrolimus and Mycophenolic Acid on CD14+ Monocyte Activation and Function. PLoS One. 2017;12 doi: 10.1371/journal.pone.0170806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu B., Jiang Q., Chen R., Gao S., Xia Q., Zhu J., Zhang F., Shao C., Liu X., Li X., et al. Tacrolimus ameliorates bleomycin-induced pulmonary fibrosis by inhibiting M2 macrophage polarization via JAK2/STAT3 signaling. Int. Immunopharmacol. 2022;113 doi: 10.1016/j.intimp.2022.109424. [DOI] [PubMed] [Google Scholar]
- 66.Gallon L., Traitanon O., Yu Y., Shi B., Leventhal J.R., Miller J., Mas V., L X., Mathew J.M. Differential Effects of Calcineurin and Mammalian Target of Rapamycin Inhibitors on Alloreactive Th1, Th17, and Regulatory T Cells. Transplantation. 2015;99:1774–1784. doi: 10.1097/TP.0000000000000717. [DOI] [PubMed] [Google Scholar]
- 67.Miroux C., Morales O., Ghazal K., Othman S.B., de Launoit Y., Pancré V., Conti F., Delhem N. In vitro effects of cyclosporine A and tacrolimus on regulatory T-cell proliferation and function. Transplantation. 2012;94:123–131. doi: 10.1097/TP.0b013e3182590d8f. [DOI] [PubMed] [Google Scholar]
- 68.Furukawa A., Wisel S.A., Tang Q. Impact of Immune-Modulatory Drugs on Regulatory T Cell. Transplantation. 2016;100:2288–2300. doi: 10.1097/TP.0000000000001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hudes G., Carducci M., Tomczak P., Dutcher J., Figlin R., Kapoor A., Staroslawska E., Sosman J., McDermott D., Bodrogi I., et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 70.Faes S., Demartines N., Dormond O. Mechanistic Target of Rapamycin Inhibitors in Renal Cell Carcinoma: Potential, Limitations, and Perspectives. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.636037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown C.E., Rodriguez A., Palmer J., Ostberg J.R., Naranjo A., Wagner J.R., Aguilar B., Starr R., Weng L., Synold T.W., et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro. Oncol. 2022;24:1318–1330. doi: 10.1093/neuonc/noac024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Amini L., Wagner D.L., Rössler U., Zarrinrad G., Wagner L.F., Vollmer T., Wendering D.J., Kornak U., Volk H.-D., Reinke P., Schmueck-Henneresse M. CRISPR-Cas9-Edited Tacrolimus-Resistant Antiviral T Cells for Advanced Adoptive Immunotherapy in Transplant Recipients. Mol. Ther. 2021;29:32–46. doi: 10.1016/j.ymthe.2020.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y., Fang H., Wang G., Yuan G., Dong R., Luo J., Lyu Y., Wang Y., Li P., Zhou C., et al. Cyclosporine A-resistant CAR-T cells mediate antitumour immunity in the presence of allogeneic cells. Nat. Commun. 2023;14:8491. doi: 10.1038/s41467-023-44176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jonnalagadda M., Brown C.E., Chang W.C., Ostberg J.R., Forman S.J., Jensen M.C. Efficient selection of genetically modified human T cells using methotrexate-resistant human dihydrofolate reductase. Gene Ther. 2013;20:853–860. doi: 10.1038/gt.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fraietta J.A., Nobles C.L., Sammons M.A., Lundh S., Carty S.A., Reich T.J., Cogdill A.P., Morrissette J.J.D., DeNizio J.E., Reddy S., et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shah N.N., Qin H., Yates B., Su L., Shalabi H., Raffeld M., Ahlman M.A., Stetler-Stevenson M., Yuan C., Guo S., et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 2019;3:2317–2322. doi: 10.1182/bloodadvances.2019000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brainard D.M., Seung E., Frahm N., Cariappa A., Bailey C.C., Hart W.K., Shin H.-S., Brooks S.F., Knight H.L., Eichbaum Q., et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J. Virol. 2009;83:7305–7321. doi: 10.1128/JVI.02207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leibman R.S., Richardson M.W., Ellebrecht C.T., Maldini C.R., Glover J.A., Secreto A.J., Kulikovskaya I., Lacey S.F., Akkina S.R., Yi Y., et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. Plos Pathog. 2017;13 doi: 10.1371/journal.ppat.1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maldini C.R., Claiborne D.T., Okawa K., Chen T., Dopkin D.L., Shan X., Power K.A., Trifonova R.T., Krupp K., Phelps M., et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med. 2020;26:1776–1787. doi: 10.1038/s41591-020-1039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nicholson I.C., Lenton K.A., Little D.J., Decorso T., Lee F.T., Scott A.M., Zola H., Hohmann A.W. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol. Immunol. 1997;34:1157–1165. doi: 10.1016/s0161-5890(97)00144-2. [DOI] [PubMed] [Google Scholar]
- 81.Diorio C., Murray R., Naniong M., Barrera L., Camblin A., Chukinas J., Coholan L., Edwards A., Fuller T., Gonzales C., et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140:619–629. doi: 10.1182/blood.2022015825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.-J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 83.Hermans I.F., Silk J.D., Yang J., Palmowski M.J., Gileadi U., McCarthy C., Salio M., Ronchese F., Cerundolo V. The VITAL assay: a versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J. Immunol. Methods. 2004;285:25–40. doi: 10.1016/j.jim.2003.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Plasmids encoding ABE8.20m and BE4 base editors used in this work are available from Addgene. The 20 nucleotide protospacer sequences and primers for the amplification of on-target amplicons can be found in Table S1. Amino acid sequences of HLA alleles used in this work are available from the IPD-IMGT/HLA database (https://www.ebi.ac.uk/ipd/imgt/hla/).