

Abstract
Long non‐coding RNAs (lncRNAs) have emerged as significant contributors to the regulation of various biological processes, and their dysregulation has been linked to a variety of human disorders. Accurate prediction of potential correlations between lncRNAs and diseases is crucial for advancing disease diagnostics and treatment procedures. The authors introduced a novel computational method, iGATTLDA, for the prediction of lncRNA‐disease associations. The model utilised lncRNA and disease similarity matrices, with known associations represented in an adjacency matrix. A heterogeneous network was constructed, dissecting lncRNAs and diseases as nodes and their associations as edges. The Graph Attention Network (GAT) is employed to process initial features and corresponding adjacency information. GAT identified significant neighbouring nodes in the network, capturing intricate relationships between lncRNAs and diseases, and generating new feature representations. Subsequently, the transformer captures global dependencies and interactions across the entire sequence of features produced by the GAT. Consequently, iGATTLDA successfully captures complex relationships and interactions that conventional approaches may overlook. In evaluating iGATTLDA, it attained an area under the receiver operating characteristic (ROC) curve (AUC) of 0.95 and an area under the precision recall curve (AUPRC) of 0.96 with a two‐layer multilayer perceptron (MLP) classifier. These results were notably higher compared to the majority of previously proposed models, further substantiating the model’s efficiency in predicting potential lncRNA‐disease associations by incorporating both local and global interactions. The implementation details can be obtained from https://github.com/momanyibiffon/iGATTLDA.
Keywords: biocomputing, bioinformatics, data mining, diseases, medical computing, network theory (graphs)
The authors introduced a novel computational method, iGATTLDA, for the prediction of lncRNA‐disease associations. The model integrated Graph Attention Network (GAT) and transformer to capture complex relationships and interactions that conventional approaches may overlook. iGATTLDA attained an area under the receiver operating characteristic (ROC) curve (AUC) of 0.95 and an area under the precision recall curve (AUPRC) of 0.96 with a two‐layer multilayer perceptron (MLP) classifier, which surpassed the majority of previously proposed models.
1. INTRODUCTION
Long non‐coding RNAs (lncRNAs) refers to a class of non‐coding RNAs with a length exceeding 200 nucleotides [1]. Extensive research has unveiled the pivotal role played by lncRNAs in the progression and development of various human diseases. For instance, the expression of lncRNA EGOT in breast cancer significantly diminishes compared to non‐cancerous tissues [2]. In prostate cancer cells, lncRNA NEAT1 exhibits a marked upregulation relative to healthy prostate cells [3]. The upregulation of lncRNA MALAT1 in lung cancer correlates with an escalation in metastatic activity [4]. Due to the costly, time‐consuming, and labour‐intensive nature of traditional biological experiments, the demand for advanced computational models to accurately and efficiently predict associations between lncRNAs and diseases is increasingly evident. These models play a pivotal role in enhancing our understanding of disease mechanisms and expediting the discovery of disease biomarkers, thereby facilitating accurate and timely diagnosis, treatment, and therapeutic response [5, 6, 7].
In recent years, various computational models have been proposed for predicting the associations between lncRNAs and diseases [8, 9]. For instance, based on the random walk techniques, Sun et al. [10] proposed a computational model to predict potential lncRNA‐disease associations by implementing a random walk with restart on a lncRNA functional similarity network. The model was evaluated using leave‐one‐out cross validation (LOOCV) method, and achieved an AUC of 0.822. However, the model faced limitations in predicting lncRNAs related to new diseases without prior known associations. Chen et al. [11] introduced an improved random walk with restart model, named IRWRLDA, which integrates known associations with disease and lncRNA similarity information to predict lncRNA‐disease associations. With the ability to predict disease‐related lncRNAs without previously known associations, IRWRLDA achieved AUCs of 0.7242 and 0.7872 on two different datasets from the lncRNADisease database. Additionally, Hu et al. [12] proposed a bi‐random walk algorithm, called BiWalkLDA, which predicts lncRNA‐disease associations through the integration of similarity interaction profiles and gene ontology details. The algorithm obtained two scores by applying a random walk technique on the disease and lncRNA similarity networks, with their mean serving as the prediction score. The BiWalkLDA algorithm obtained AUCs of 0.8268, 0.8510 and 0.8473 for the three datasets, respectively.
Also, some methodologies have been proposed based on inductive matrix completion and bipartite graph for efficient prediction of lncRNA‐disease associations. For instance, Lu et al. [13] proposed an inductive matrix completion‐based method known as SIMCLDA to identify potential lncRNA‐disease associations. The model utilised the lncRNA Gaussian interaction profile (GIP) kernel and disease functional similarities, from which significant features were extracted through the principal component analysis (PCA) technique. As a result, they attained AUCs of 0.8237, 0.8526 and 0.8578 on three datasets, respectively. On the other hand, Ping et al. [14] proposed a bipartite network‐based prediction model utilising three publicly available datasets from LncRNADisease, Lnc2Cancer and MNDR databases. The model was evaluated with the LOOCV and obtained AUCs of 0.8825, 0.9004, and 0.9292 for the three datasets, respectively.
With the rapid advancement of computing technology and the availability of big data [15], advanced machine learning and deep learning‐based models have been proposed based on both classical and advanced deep learning frameworks [16, 17, 18, 19, 20, 21]. For instance, Wang et al. [22] introduced a deep forest‐based model, known as MLCDForest, for the prediction of potential lncRNA‐disease interactions. This model employed multi‐label classification and integrates multi‐grained scanning (MGS) and cascade forest (CS) techniques. In the MGS stage, transformed feature representations were categorised based on different forest models, while in the CS stage, a layer‐wise random forest approach was applied to generate more distinctive features. The CNNLDA model proposed by Xuan et al. [23] utilised a double convolution neural network (CNN) combined with attention mechanism for predicting lncRNA‐disease associations. The model integrated the diverse similarities between lncRNAs, miRNAs and diseases to construct feature matrices for learning the global and attention representations of lncRNA‐disease associations.
While the CNNLDA achieved an AUC of 0.952, Xuan et al. [24] further proposed a relatively advanced model called GCNLDA, based on graph convolutional network (GCN) to identify potential disease‐related lncRNAs. GCNLDA also generated a heterogeneous network with the help of known interactions between lncRNAs, diseases, and miRNAs. Topological information were then integrated using a graph convolution‐based autoencoder and an attention mechanism useful for identifying the most significant node features [25]. Additionally, CNN was utilised to focus on specific interactions for given lncRNA‐disease pairs. GCNLDA achieved an AUC of 0.959 on 450 diseases, which is relatively higher compared to other similar methods.
Another GNN‐based model proposed by Lan et al. [26] called GANLDA, was used to predict disease‐related lncRNAs. The model utilised Graph Attention Network (GAT) for feature embeddings on the heterogeneous network, followed by PCA for noise‐reduction, resulting in an AUC of 0.8834 in 10‐fold cross‐validation [27] and 0.8581 in denovo test. In another study, Shi et al. [28] introduced a model based on a heterogeneous network for predicting lncRNA‐disease associations by integrating lncRNA similarity, lncRNA‐disease and lncRNA‐miRNA associations. Restart random walk technique was employed to sample strong correlation neighbours of a fixed size for every node before applying type‐based neighbour aggregation and heterogeneous graph neural networks to capture each pair’s embedding information. An attention mechanism was equally integrated to identify neighbours’ contribution to specific nodes, resulting in a high AUC of 0.9786. However, the model was notably limited by disregarding significant features within the global network perspective.
On the other hand, Liang et al. [29] proposed a model based on graph autoencoder, named GraLTR‐LDA. This model generates both homogeneous and heterogeneous graphs using lncRNAs and disease similarity features. An attention mechanism was integrated to extract embedded features from their respective graphs before employing a learning‐to‐rank approach to predict the diseases order corresponding to the subject lncRNAs.
Despite the significant success and discoveries achieved by previous techniques, predicting lncRNA‐disease associations remain challenging due to the complexity of the respective biological interactions. Many classical methods rely on basic similarity measures, overlooking the complex inter‐dependencies and interactions within the molecular domain. Therefore, this study proposed the iGATTLDA model to predict disease‐related lncRNAs. The model utilised lncRNA and disease similarity matrices to generate a heterogeneous network, leveraging GAT and transformer techniques with a double attention mechanism to handle local and global attention, respectively, to extract new node representations. The integration of these techniques is especially significant as it captures significant features across the entire network. GAT captures substantial features within the local network perspective, while the transformer contributes by capturing long‐range dependencies, thus ensuring the global network perspective is incorporated. Mini‐batches were employed to generate sub‐graphs in three heterogeneous networks for model training, facilitating feature learning from multiple perspectives within the network. Finally, a multilayer perceptron (MLP) classifier was implemented to predict lncRNA‐disease associations based on the low‐dimensional data. In this context, the iGATTLDA emerges as an innovative and promising methodology, effectively utilising both local and global feature similarities.
2. MATERIALS AND METHODS
2.1. Datasets
The datasets utilised in this study were sourced from the research conducted by Shi et al. [30], comprising lncRNA functional similarity, disease similarity and lncRNA‐disease associations. The dataset includes 240 lncRNAs and 412 diseases, covering 2697 experimentally confirmed associations derived from three widely recognised and extensively used databases that is, LncRNADisease [31] Lnc2Cancer [32] and GeneRIF [33]. For clarity of representation, details regarding the association between lncRNAs and diseases were organised into an adjacency matrix , where l n and d n refers to the number of lncRNAs and diseases, respectively.
2.2. LncRNA and disease similarities
The lncRNA functional similarity was calculated using the approach outlined by Chen et al. [34]. In this method, the similarities between LncRNAs were represented by the similarity of lncRNA‐related diseases. For instance, considering two lncRNAs L i and L j , associated with sets of diseases D i = {d i1,d i2, d im } and D j = {d j1,d j2, d jn }, respectively, their functional similarity can be computed as shown in Eq. 1
| (1) |
where FS is the functional similarity of lncRNAs L i and L j , DS is the disease semantic similarity for two given diseases, d(ix) ∈ D i and d(jy) ∈ D j , calculated based on the method proposed by Wang et al. [35]. The denominators m and n refer to the number diseases in groups D i and D j , respectively.
In the end, we obtained a 240 × 240 matrix representing the functional similarity of lncRNAs and a 412 × 412 matrix representing disease similarity. Figure 1 provides insights into the existing relationships between different lncRNAs and diseases based on their respective similarity profiles. These matrices served as the initial feature representations used for training the iGATTLDA model.
FIGURE 1.
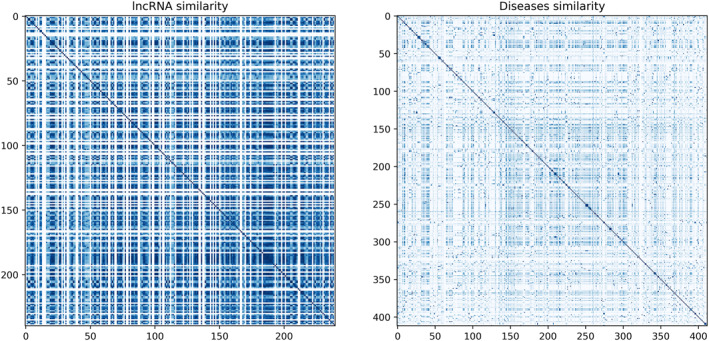
Heatmaps illustrating lncRNAs and diseases similarity matrices. The colour intensity represents the degree of similarity, with darker shades indicating higher similarity values and vice versa.
2.3. The heterogeneous network
Heterogeneous networks are intricate network architectures that encompass a diverse range of nodes and edges, making them significant for depicting and scrutinising complex associations [36, 37, 38, 39, 40]. They are capable of capturing both direct and indirect relationships between different types of nodes, hence facilitating identification of potential associations, which may otherwise be disregarded by classical techniques [30]. In the context of lncRNA‐disease associations, they can reveal underlying biological mechanisms and pathways, leading to the discovery of novel biomarkers and therapeutic targets [41, 42]. As a result, this study utilised a heterogeneous network to represent lncRNA‐disease associations given the presence of two node types; lncRNAs L = {l1,l2,,l m } and diseases D = {d1,d2,,d n }. The network was generated with the integration of confirmed lncRNA‐disease associations, lncRNA functional similarity (LS) and disease similarity (DS) matrices. Known associations were initially presented in an adjacency matrix in which A ij = 1 when an association exists between lncRNA i and disease j, and 0 otherwise. The heterogeneous network G = (V,E) was generated based on A and can be represented as shown in Eq. 2.
| (2) |
where V and E are the nodes and edges, respectively, LS and DS are the initial features through which new node embeddings are obtained by the GAT, AT specifically captures a different perspective of A for a comprehensive view of the known associations.
In the given set of 240 lncRNAs and 412 diseases, G contained a total of 652 nodes and 2697 edges. During data processing, the graph edges were split into training, validation and testing sets by the PyTorch geometrics’ RandomLinkSplit function, where 80% of the edges were allocated for training, 10% for validation, and 10% for testing, while negative edges were generated at a rate of 0.1, meaning for every 10 positive edges, approximately 1 negative edge was generated. Afterwards, a batch size of 96 samples represented as 3 × 32 for flexibility was used for training. The mini batches consisting of 20 neighbours for every two entities, that is, lncRNA and disease entities, were generated for model training at a negative sampling ratio of 0.2 at each training batch. The model was eventually validated with the validation data in the training phase and later tested on the testing data.
2.4. Deep learning model
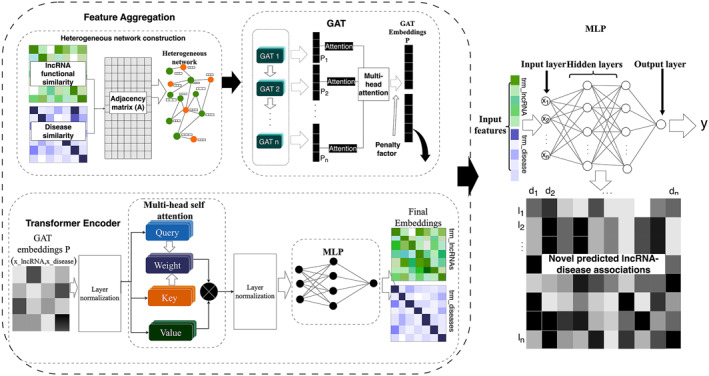
Based on the Graph Attention Network (GAT) and the transformer architecture, a novel deep learning model for the prediction of lncRNA‐disease associations has been proposed in this study, named the iGATTLDA as illustrated in Figure 2. By combining the strengths of local and global attention mechanisms, these two cutting‐edge approaches aim to improve the prediction of lncRNA‐disease associations by enriching the node features. GAT captures local interactions, while the transformer takes care of the global dependencies by comparing a given node to non‐neighbouring nodes. The approach enhances the model’s capacity in capturing short and long‐range dependencies, leading to improved identification of feature significance, as illustrated in Eq. 3, where Trm(.) refers to the transformer operation on the GAT output to capture global features for a given node i.
| (3) |
FIGURE 2.
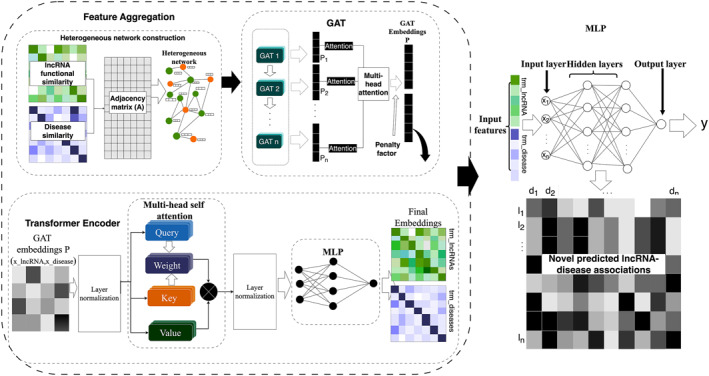
An illustration of the proposed iGATTLDA model for the prediction of lncRNA‐disease associations based on the attention‐based GAT and transformer, leading to the prediction of novel associations by the MLP classifier.
GAT is a GNN framework with an attention mechanism for learning feature embeddings on graph structured data sets through feature aggregation [43]. Based on GAT, iGATTLDA assigns an attention coefficient to each neighbouring node for indication of the neighbour’s feature significance before updating the subject node features. Therefore, an attention score for neighbouring node j to node i can be calculated as shown in Eq. 4.
| (4) |
where e ij is the attention score for the neighbouring node j to node i, a is the learnable weight vector, W and h are the weight matrix and feature embeddings, respectively, while denotes the feature vectors concatenation operation. The attention coefficients for a given set of neighbouring nodes j ∈ N i are then normalised with a SoftMax function to obtain a normalised attention coefficient, as shown in Eq. 5 for simplified node comparison [43].
| (5) |
To generate new node embeddings for node i, a corresponding attention coefficient is multiplied to its neighbour state, essentially incorporating a linear combination of the node feature vectors weighted with each node importance. This process enhances the significant features while suppressing the least significant ones, and can be represented as shown in Eq. 6, where is the new representation of node i, σ is the non‐linear activation function, while α ij is the attention coefficient incorporated to the new node embeddings.
| (6) |
GAT was implemented in three layers with multi‐head attention (4 heads) incorporated for iGATTLDA to selectively attend to different neighbours based on their significance in various attention heads as described in Eq. 7, where M refers to the number of attention heads.
| (7) |
A Rectified Linear Unit (ReLU) activation function was applied after every GAT layer for non‐linearity, after which a penalty factor λ was employed to the final embedding layer p through a linear layer, in which it is automatically determined based on the learnt parameters during the forward pass, hence it controls node contribution as shown in Eq. 8, serving as the first input of the iGATTLDA model. The λ value ranges between 0 and 1, where 0 or 1 indicates that p is primarily influenced by either LS or DS, respectively. Therefore, having λ between 0 and 1 facilitates a trade‐off between the contributions of LS and DS feature embeddings.
| (8) |
Consequently, the GAT‐based node embeddings were subjected to the transformer, an attention‐based deep learning architecture widely adopted in Natural Language Processing (NLP) given its exceptional sequence modelling capabilities [44]. A standard transformer attention mechanism treats each element within the input sequence as being related to three specific vectors: query (Q), key (K), and value (V) vectors [27], which facilitates the computation of attention scores by determining the extent to which each element in the sequence should focus on other elements. Therefore, the model examines the importance of each K in comparison to the corresponding Q, and the attention weights are subsequently applied to the corresponding V to produce the final output. The transformer utilised by iGATTLDA calculates the attention scores for the input vectors Q, K and V as shown in Eq. 9, where , , and , d k is the dimension of K which aids weight normalisation.
| (9) |
The transformer’s multi‐head attention capability was utilised for computation of different attention weights for each embedding which are combined to generate new node embeddings, specifically focussing on global node dependencies [27]. The attention mechanism is simultaneously performed on each Q, K and V, where each attention function is in charge of a single subspace in the output at various locations. Different attention function results are eventually merged for linear transformation to generate the ultimate output. With and , , utilising d k = d v , multi‐head attention was computed as demonstrated in Eqs. 10 and 11, where the attention scores from multiple attention heads h are concatenated.
| (10) |
| (11) |
The transformer contains an MLP classifier as seen in Figure 2, which is responsible for transforming the rich feature embeddings from the transformer into useable format by introducing non‐linearity, dimensionality reduction, optimisation based on the tasks and feature aggregation. Furthermore, an additional MLP classifier with two fully connected layers was implemented for the prediction of lncRNA‐disease associations by processing data for extraction of edge features in the lncRNA and disease nodes based on the edge label index. Extracted features are concatenated to form the edge features, which are passed through the MLP classifier for prediction of potential associations.
3. RESULTS AND DISCUSSION
The implementations of the iGATTLDA model were conducted on an intel core i7‐10700 CPU with 16 GB RAM, with the Python programming language, PyTorch geometric library and the Jupyter Notebook environment. Through the Scikit‐Learn Python library, popular evaluation metrics that is, the AUC and AUPRC were employed to establish the capacity of iGATTLDA in efficiently predicting potential associations based on the AUC and AUPRC evaluation metrics. Furthermore, other evaluation techniques such as the calibration curve, learning curve, cumulative gain curve, lift curve and the histogram of predicted probabilities were further evaluated for detailed analysis of the prediction performance.
3.1. Prediction performance
The MLP classifier utilised GAT and transformer‐based node embeddings as input features to predict potential lncRNA‐disease associations. The 10‐fold cross‐validation [29] was used to evaluate the model’s performance on training data. After full training on the entire dataset, the model was tested on independent validation and test sets. The predictive performance of the iGATTLDA model was demonstrated using an area under the receiver operating characteristic (ROC) curve (AUC) and an area under the precision‐recall curve (AUPRC). AUC is a commonly used performance evaluation metric for binary classification, assessing the trade‐off between true and false positive rates while quantifying the model’s discriminating power between different classes. An AUC value closer to 1 indicates a good model performance and vice‐versa [45]. AUPRC is also useful when handling imbalanced datasets, in which the positive class may have smaller samples than the negative class. Unlike the AUC, which equally considers true negatives and false positives, the AUPRC is based on precision and recall values, whereby smaller improvements in either precision or recall can facilitate substantial gains in performance. Therefore, these two metrics were selected given their suitability to the nature of our data, which also had issues of class imbalances. During training, weights and biases were adjusted to minimise the loss. Eventually, the iGATTLDA model achieved an AUC of 0.95 and AUPRC of 0.96 with 2000 training epochs, a learning rate of 0.001 and the Adam optimiser, as depicted in Figure 3.
FIGURE 3.
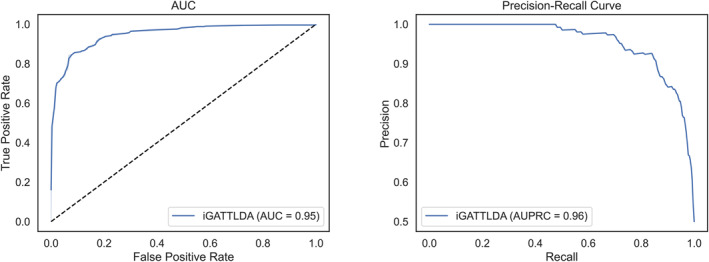
An illustration showing the AUC and AUPRC used to evaluate the prediction performance of the proposed iGATTLDA model in the prediction of lncRNA‐disease associations, which highlights the model’s reliability and effectiveness in distinguishing true positives from false positives.
For further evaluation, the performance of iGATTLDA was compared with similar models, namely SIMCLDA [13], IRWRLDA [11], GANLDA [26], GCNLDA [24] and HGNNLDA [28], each implemented using different methods. Specially, SIMCLDA and IRWRLDA were based on matrix completion and random‐walk with restart, respectively, while the GANLDA, GCNLDA and HGNNLDA were based on different GNN architectures. Despite the distinct implementation approaches, GANLDA, GCNLDA and HGNNLDA were closely related to the iGATTLDA model, as they were implemented on GNN architectures with the help of lncRNA and disease association data presented in heterogeneous networks. In the comparison, iGATTLDA model demonstrated superior performance with an AUC of 0.95 and AUPRC of 0.96. SIMCLDA achieved an average AUC of 0.8447 across three datasets, IRWRLDA obtained 0.7242 and 0.7872 on two different datasets, respectively, GANLDA produced 0.8834 and 0.8581 in 10‐fold CV and denovo test, respectively. GCNLDA attained 0.959, while HGNNLDA exhibited the highest compared performance at 0.9786. However, both GCNLDA and HGNNLDA had relatively lower AUPRCs compared to that of the iGATTLDA model as demonstrated in Figure 4.
FIGURE 4.
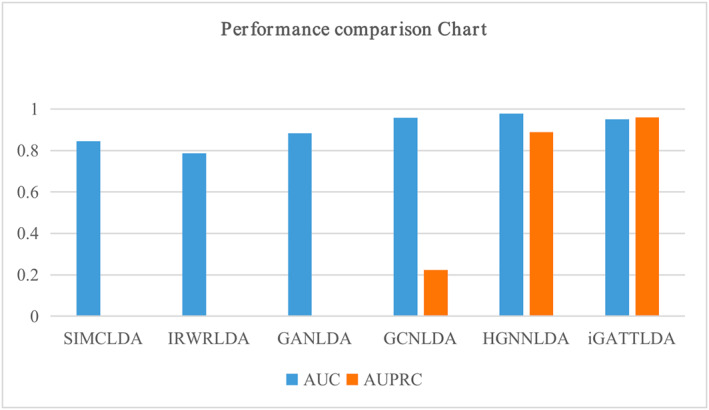
A demonstration of the prediction performance of the proposed iGATTLDA model in comparison to other similar models previously proposed.
3.2. Performance interpretation
Previous research has indicated that lncRNAs play crucial roles in the stages of the cell cycle through different mechanisms, contributing significantly to key biological processes such as epigenetic regulation, cell differentiation and apoptosis, metabolic processes, cell cycle control, tissue developments, transcriptional and post‐transcriptional regulation [46, 47, 48, 49, 50]. Despite the evident involvement of lncRNAs in the development of human diseases, the prediction of lncRNA‐disease associations remains a complex task, requiring advanced computational models to enhance prediction efficiency. This study introduces a highly anticipated iGATTLDA model that aims to fully leverage local and global feature information to improve the prediction performance of disease‐related lncRNAs. iGATTLDA integrates two state‐of‐the‐art deep learning techniques (GAT and transformer), capturing complex interactions in a heterogeneous network through their double self‐attention mechanisms, which served as the primary factor for high prediction performance. At first, three GAT layers effectively capture the underlying graph structure in the association network and identify the most crucial interactions, thanks to their self‐attention mechanisms. Additionally, multi‐head attention was applied to enhance the ability to capture inherent interactions between nodes and edges. Subsequently, the Transformer is applied for its ability to handle long‐range dependencies, particularly from non‐neighbouring nodes. Similar to GAT, the transformer utilises a self‐attention mechanism to focus on specific lncRNA‐disease pairs for improved prediction. As a result, the iGATTLDA achieved remarkable performance with AUC and AUPRC of 0.95 and 0.96, respectively, underscoring the significance of fully utilising neighbourhood information within the entire network.
The iGATTLDA model was trained on three heterogeneous networks to capture diverse feature information from different perspectives. Each network edge feature embeddings were split into training, validation and test sets at the rate of 0.8, 0.1 and 0.1, respectively. The approach ensured model training and evaluation without data leakage. The embeddings were fed to the model in mini‐batches for computational efficiency, simultaneously addressing the common issue of over‐smoothing in GNN. The model underwent initial training with 10‐fold CV to enhance generalisation and was subsequently fine‐tuned with the entire training data before using the validation set for capturing more generalisable interactions. Throughout model training, a lower learning rate resulted in sub‐optimal performance with the optimal rate determined to be 0.001. This efficient training of the iGATTLDA model was evidenced by consistent loss curves that signifies a stable prediction performance (Figure 5). This was obtained with the Binary Cross‐Entropy Loss function as shown in Eq. 12, where y i and refer to the actual label and predicted probability, respectively, while N is the total number of samples. The figure included training loss curves under 10‐fold CV, alongside the learning curve, learning rate scheduler, and the calibration curve. The calibration curve was crucial for ensuring that predicted probabilities aligned well with actual probabilities, demonstrating that the iGATTLDA model achieved near perfect calibration. This suggests high reliability and suitability of the model for accurate prediction of lncRNA‐disease associations. Examining the learning curve, the performance of iGATTLDA increased in line with the rising learning curve before stabilising, suggesting that further training may not improve the model performance. Therefore, the model has learnt underlying data patterns as the learning process has converged to consistent levels.
| (12) |
FIGURE 5.
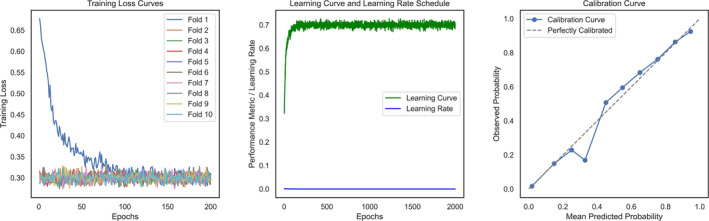
An illustration of iGATTLDA’s model training comprising the loss curves in 10‐fold cross‐validation, learning curve and learning rate schedule, and the calibration curve.
The model was further evaluated with the help of the cumulative gain and lift curves as illustrated in Figure 6, alongside the histogram of predicted probabilities. The cumulative gain curve clarifies how much of the positive edges iGATTLDA can capture using a given percentage of the prediction sample, while the lift curve measures the performance improvement of the iGATTLDA model compared to a randomly generated base model. For instance, iGATTLDA captures more than 80% of true interactions by investigating 50% of the total sample size and identifies interactions at almost twice the rate of a randomly generated model when considering 20% of the prediction sample. Note that the steeper the cumulative gain curve, the more effective the model is at identifying positive interactions early in the ranking. Also, the higher the lift value, the higher the chances of effectively identifying positive true interactions compared to a random model. Therefore, gain and lift curves contribute to our comprehension of how well the iGATTLDA model performs across different threshold values.
FIGURE 6.
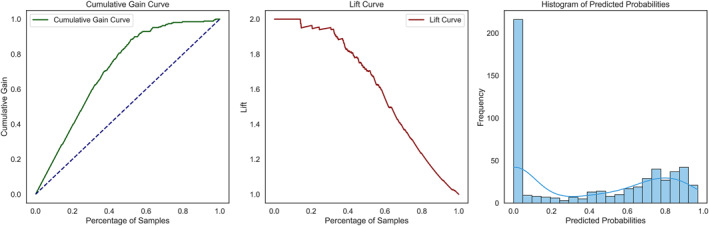
An illustration of the cumulative gain and lift curves, alongside the predicted probabilities histogram used for detailed evaluation of the iGATTLDA model.
A histogram facilitates a clear comprehension of the model’s confidence in predicting negative and positive interactions. For instance, close to 200 samples were confidently classified as negative samples, while there was an increment in the number of samples predicted as positive associations with probability values ranging between 0.4 and 1.0. The higher the probability value, the higher the confidence in predicting positive interactions.
Due to the MLP classifier’s capability to learn complex decision boundaries in high‐dimensional feature space, the classifier was employed to predict potential associations between lncRNAs and diseases. Additionally, the MLP classifier can adapt to different architectures by adjusting the number of neurons, layers, and activation functions as needed to match the complexity of the given classification task.
The evaluation and testing of the proposed model on independent datasets confirmed its robust performance. Although its AUC was slightly lower than that of GCNLDA [24] and HGNNLDA [28], our model outperformed them in terms of AUPRC. The AUPRC values for GCNLDA and HGNNLDA were 0.223 and 0.8891, respectively, while iGATTLDA achieved 0.96. This superiority can be attributed to our model’s utilisation of both local and global features, whereas the other two models paid less attention to the significance of global features. Furthermore, in comparison to these models, our model emphasised positive lncRNA‐disease interactions, and its high precision underscored its superior ability to select positive cases with accuracy. The introduction of transformer’s scalability and its effective modelling of dependencies over long sequences after GAT were the key contributions in this study, significantly enhancing the model’s applicability in handling large graphs with complex relationships.
While the challenge of capturing long‐range dependencies in lncRNA‐disease associations persists, there is an urgent need for continuous research and refinement of graph‐structured data to improve the prediction of complex biological interactions [51, 52]. Despite the successful utilisation of the transformer in capturing long‐range dependencies, a primary limitation occurred in the uncertainties involved when handling large and complex graphs, where the impact of attention weights in the iGATTLDA model may diminish with increasing distance between graph elements, leading to inadequate performance of the transformer as a result of insufficient feature aggregation in the global network perspective [53]. Therefore, future studies can address this limitation by developing tailored algorithms through the incorporation of domain‐specific knowledge with specialised adaptive attention mechanisms that dynamically adjust the weights based on the input graph data. For instance, an algorithm capable of learning how to assign higher attention weights to significant nodes while equally suppressing the attention weights for the less significant ones regarded as irrelevant. Additionally, these algorithms can enhance the stability and effectiveness of attention weight’s through the incorporation of significant regularisation and optimisation strategies. This may include the prevention of over‐reliance on specific attention weights through the incorporation of drop‐out regularisation and the introduction of specialised loss functions, which can enable the model to focus on specific elements deemed to be relevant and reduce the effect of irrelevant or noisy network information which may affect the prediction performance.
3.3. Case study
To further evaluate the performance of the iGATTLDA model in predicting lncRNA‐disease associations, we focused on three common diseases: colon, lung and stomach cancers. These diseases were selected due to their prevalence, and these three cancers contribute significantly to the global cancer burden [54]. For instance, lung cancer is recognised as a leading cause of cancer deaths and the most diagnosed cancer globally [55], while colon and stomach cancers consistently rank among the top cancers by incidence [54, 56]. Therefore, the prediction of lncRNAs associated with these diseases can play a crucial role in disease diagnosis, treatment and prevention.
Here, we selected lncRNAs associated with these diseases and disconnected these associations from the graph. Then the new data was used to train the model and predict these disconnected associations. The predicted probabilities were ranked in descending order, and the top 10 predicted lncRNAs were selected for validation. This validation was conducted using the LncRNADisease and Lnc2Cancer v3.0 databases, which contain experimentally confirmed lncRNA‐disease associations. Table 1 demonstrates that out of the top 10 predicted lncRNAs, 9, 8, and 6 were verified to be associated with colon, lung, and stomach cancers, respectively, based on either the LncRNADisease or Lnc2Cancer databases. These findings further confirmed the reliability and effectiveness of the iGATTLDA model in the identification of potential lncRNA‐disease associations.
TABLE 1.
The top 10 lncRNA candidates predicted by the iGATTLDA model for colon, lung and stomach cancers.
| Colon cancer | Lung cancer | Stomach cancer | |||
|---|---|---|---|---|---|
| LncRNA | Evidence | LncRNA | Evidence | LncRNA | Evidence |
| LINC‐ROR | LncRNADisease, lnc2cancer 3.0 | CRNDE | lnc2cancer 3.0 | KCNQ1OT1 | lnc2cancer 3.0 |
| PRNCR1 | LncRNADisease, lnc2cancer 3.0 | NEAT1 | LncRNADisease, lnc2cancer 3.0 | TUSC7 | LncRNADisease, lnc2cancer 3.0 |
| HCP5 | lnc2cancer 3.0 | DLEU2 | lnc2cancer 3.0 | IL12A‐AS1 | Unverified |
| SNHG1 | Unverified | TUSC7 | LncRNADisease, lnc2cancer 3.0 | NEAT1 | LncRNADisease, lnc2cancer 3.0 |
| TUSC8 | lnc2cancer 3.0 | LINC00629 | Unverified | NRON | Unverified |
| ZEB1‐AS1 | lnc2cancer 3.0 | LINC00963 | lnc2cancer 3.0 | MIR137HG | Unverified |
| FENDRR | lnc2cancer 3.0 | GACAT2 | lnc2cancer 3.0 | LINC01133 | lnc2cancer 3.0 |
| XIST | LncRNADisease, lnc2cancer 3.0 | LINC00942 | lnc2cancer 3.0 | LINC00473 | lnc2cancer 3.0 |
| BCYRN1 | lnc2cancer 3.0 | LINC00473 | LncRNADisease, lnc2cancer 3.0 | CAHM | Unverified |
| HULC | LncRNADisease, lnc2cancer 3.0 | MCHR2‐AS1 | Unverified | GACAT2 | lnc2cancer 3.0 |
4. CONCLUSION
Considering the expensive and time‐consuming nature of traditional biological experiments, along with the significant roles played by lncRNAs in the development of human diseases, there is an increasingly urgent demand for computational techniques. By leveraging advanced computational resources and the available datasets, computer‐based solutions can effectively enhance the identification of potential lncRNA‐disease associations, and further contribute to improvements in disease diagnosis, treatment and prevention‐based efforts through an improved understanding of disease biomarkers and molecular mechanisms.
The proposed iGATTLDA model offers an effective solution for predicting lncRNA‐disease associations. The model fully harnesses the capabilities of GAT and Transformer and successfully combines the attention mechanisms of GAT with the self‐attention techniques of Transformer which allows the model to capture both local and global significant feature information from the heterogeneous network for a comprehensive understanding of the graph data. Therefore, the iGATTLDA model significantly contributes to the development of reliable prediction solutions for lncRNA‐disease associations with case studies further confirming the model’s ability in identifying potential associations. Additionally, the model’s prediction performance has profound implications in medical practices and future studies. For instance, the model can significantly aid timely disease diagnosis for improved patient outcomes by identifying disease biomarkers, especially on diseases that are presently difficult to detect at the early stages. Also, the findings of this study can serve as a guiding principle for future studies by identifying potential and previously unknown associations, which can undergo further investigation.
AUTHOR CONTRIBUTIONS
Biffon Manyura Momanyi: Conceptualisation; methodology; software; writing—original draft preparation; investigation. Sebu Aboma Temesgen: Formal analysis; investigation. Tian‐Yu Wang: Formal analysis; investigation. Hui Gao: Validation; resources; investigation; formal analysis. Ru Gao: Resources; supervision; funding acquisition. Hua Tang: Project administration; funding acquisition. Li‐Xia Tang: Supervision; funding acquisition; project administration.
All authors have read the final manuscript and agree to be accountable for the contents of the submitted work.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACKNOWLEDGEMENT
This work was supported by the National Natural Science Foundation of China (62172343), the Sichuan Science and Technology Program (2022YFS0614) and Medico‐Engineering Cooperation Funds from UESTC (ZYGX2021YGLH202).
Momanyi, B.M. , et al.: iGATTLDA: Integrative graph attention and transformer‐based model for predicting lncRNA‐Disease associations. IET Syst. Biol. 18(5), 172–182 (2024). 10.1049/syb2.12098
Contributor Information
Ru Gao, Email: msrancd@outlook.com.
Hua Tang, Email: huatang@swmu.edu.cn.
Li‐Xia Tang, Email: lixiatang@uestc.edu.cn.
DATA AVAILABILITY STATEMENT
Our Expects Data Policy requires a Data Availability Statement, even if no data are available, so please enter one in the space below. Sample statements can be found here. Please note that this statement will be published alongside your manuscript, if it is accepted for publication.
The data that support the findings of this study are openly available in LncRNADisease at http://www.rnanut.net/lncrnadisease/, and Lnc2Cancer at http://www.bio‐bigdata.net/lnc2cancer.
REFERENCES
- 1. Mercer, T.R. , Dinger, M.E. , Mattick, J.S. : Long non‐coding RNAs: insights into functions. Nat. Rev. Genet. 10(3), 155–159 (2009). 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- 2. Broadbent, H.M. , et al.: Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet. 17(6), 806–814 (2008). 10.1093/hmg/ddm352 [DOI] [PubMed] [Google Scholar]
- 3. Pasmant, E. , et al.: ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. Faseb. J. 25(2), 444–448 (2011). 10.1096/fj.10-172452 [DOI] [PubMed] [Google Scholar]
- 4. Gutschner, T. , et al.: The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 73(3), 1180–1189 (2013). 10.1158/0008-5472.can-12-2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu, J. , et al.: A novel probability model for LncRNA⁻Disease association prediction based on the naïve bayesian classifier. Genes 9(7), 345 (2018). 10.3390/genes9070345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao, C. , et al.: webTWAS: a resource for disease candidate susceptibility genes identified by transcriptome‐wide association study. Nucleic Acids Res. 50(D1), D1123–D1130 (2022). 10.1093/nar/gkab957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin, Q. , et al.: DUNet: a deformable network for retinal vessel segmentation. Knowl. Base Syst. 178, 149–162 (2019). 10.1016/j.knosys.2019.04.025 [DOI] [Google Scholar]
- 8. Xie, G. , et al.: DHOSGR: lncRNA‐disease association prediction based on decay high‐order similarity and graph‐regularized matrix completion. Curr. Bioinf. 18(1), 92–104 (2023). 10.2174/1574893618666221118092849 [DOI] [Google Scholar]
- 9. Zeng, X. , et al.: Predicting disease‐associated circular RNAs using deep forests combined with positive‐unlabeled learning methods. Briefings Bioinf. 21(4), 1425–1436 (2020). 10.1093/bib/bbz080 [DOI] [PubMed] [Google Scholar]
- 10. Sun, J. , et al.: Inferring novel lncRNA‐disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 10(8), 2074–2081 (2014). 10.1039/c3mb70608g [DOI] [PubMed] [Google Scholar]
- 11. Chen, X. , et al.: IRWRLDA: improved random walk with restart for lncRNA‐disease association prediction. Oncotarget 7(36), 57919–57931 (2016). 10.18632/oncotarget.11141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu, J. , et al.: A novel algorithm based on bi‐random walks to identify disease‐related lncRNAs. BMC Bioinf. 20((Suppl 18)), 569 (2019). 10.1186/s12859-019-3128-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu, C. , et al.: Prediction of lncRNA‐disease associations based on inductive matrix completion. Bioinformatics 34(19), 3357–3364 (2018). 10.1093/bioinformatics/bty327 [DOI] [PubMed] [Google Scholar]
- 14. Ping, P. , et al.: A novel method for LncRNA‐disease association prediction based on an lncRNA‐disease association network. IEEE ACM Trans. Comput. Biol. Bioinf 16(2), 688–693 (2019). 10.1109/tcbb.2018.2827373 [DOI] [PubMed] [Google Scholar]
- 15. Zeng, X. , et al.: Deep generative molecular design reshapes drug discovery. Cell Reports Medicine 4(12), 100794 (2022). 10.1016/j.xcrm.2022.100794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, Y. , et al.: SBSM‐pro: support bio‐sequence machine for proteins. arXiv preprint, 10275 (2023). arXiv:2308 [Google Scholar]
- 17. Qi, R. , Zou, Q. : Trends and potential of machine learning and deep learning in drug study at single‐cell level. Research 6, 0050 (2023). 10.34133/research.0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen, L. , Yu, L. , Gao, L. : Potent antibiotic design via guided search from antibacterial activity evaluations. Bioinformatics 39(2), btad059 (2023). 10.1093/bioinformatics/btad059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan, K. , et al.: sAMPpred‐GAT: prediction of antimicrobial peptide by graph attention network and predicted peptide structure. Bioinformatics 39(1), btac715 (2023). 10.1093/bioinformatics/btac715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang, Y. , Pang, Y. , Liu, B. : IDP‐Seq2Seq: identification of intrinsically disordered regions based on sequence to sequence learning. Bioinformatics 36(21), 5177–5186 (2021). 10.1093/bioinformatics/btaa667 [DOI] [PubMed] [Google Scholar]
- 21. Li, H. , Pang, Y. , Liu, B. : BioSeq‐BLM: a platform for analyzing DNA, RNA, and protein sequences based on biological language models. Nucleic Acids Res. 49(22), e129 (2021). 10.1093/nar/gkab829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang, W. , et al.: MLCDForest: multi‐label classification with deep forest in disease prediction for long non‐coding RNAs. Briefings Bioinf. 22(3) (2021). 10.1093/bib/bbaa104 [DOI] [PubMed] [Google Scholar]
- 23. Xuan, P. , et al.: Dual convolutional neural networks with attention mechanisms based method for predicting disease‐related lncRNA genes. Front. Genet. 10, 416 (2019). 10.3389/fgene.2019.00416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xuan, P. , et al.: Graph convolutional network and convolutional neural network based method for predicting lncRNA‐disease associations. Cells 8(9), 1012 (2019). 10.3390/cells8091012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang, W. , et al.: Tumor origin detection with tissue‐specific miRNA and DNA methylation markers. Bioinformatics 34(3), 398–406 (2018). 10.1093/bioinformatics/btx622 [DOI] [PubMed] [Google Scholar]
- 26. Lan, W. , et al.: GANLDA: graph attention network for lncRNA‐disease associations prediction. Neurocomputing 469, 384–393 (2022). 10.1016/j.neucom.2020.09.094 [DOI] [Google Scholar]
- 27. Huang, X. , et al.: Tabtransformer: tabular data modeling using contextual embeddings. (2020)
- 28. Shi, H. , et al.: Heterogeneous graph neural network for lncRNA‐disease association prediction. Sci. Rep. 12(1), 17519 (2022). 10.1038/s41598-022-22447-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liang, Q. , et al.: LncRNA‐disease association identification using graph auto‐encoder and learning to rank. Briefings Bioinf. 24(1) (2023). 10.1093/bib/bbac539 [DOI] [PubMed] [Google Scholar]
- 30. Shi, C. , et al.: A survey of heterogeneous information network analysis. IEEE Trans. Knowl. Data Eng. 29(1), 17–37 (2016). 10.1109/tkde.2016.2598561 [DOI] [Google Scholar]
- 31. Bao, Z. , et al.: LncRNADisease 2.0: an updated database of long non‐coding RNA‐associated diseases. Nucleic Acids Res. 47(D1), D1034–d1037 (2019). 10.1093/nar/gky905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ning, S. , et al.: Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 44(D1), D980–D985 (2016). 10.1093/nar/gkv1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu, Z. , Cohen, K.B. , Hunter, L. : GeneRIF quality assurance as summary revision. Pac Symp Biocomput, 269–280 (2007). 10.1142/9789812772435_0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen, X. , et al.: Constructing lncRNA functional similarity network based on lncRNA‐disease associations and disease semantic similarity. Sci. Rep. 5(1), 11338 (2015). 10.1038/srep11338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang, J.Z. , et al.: A new method to measure the semantic similarity of GO terms. Bioinformatics 23(10), 1274–1281 (2007). 10.1093/bioinformatics/btm087 [DOI] [PubMed] [Google Scholar]
- 36. Khandekar, A. , et al.: LTE‐advanced: heterogeneous networks. In: 2010 European Wireless Conference (EW). IEEE; (2010) [Google Scholar]
- 37. Zhao, X. , Zhao, X. , Yin, M. : Heterogeneous graph attention network based on meta‐paths for lncRNA‐disease association prediction. Briefings Bioinf. 23(1) (2022). 10.1093/bib/bbab407 [DOI] [PubMed] [Google Scholar]
- 38. Feng, J. , et al.: Microbe‐bridged disease‐metabolite associations identification by heterogeneous graph fusion. Briefings Bioinf. 23(6) (2022). 10.1093/bib/bbac423 [DOI] [PubMed] [Google Scholar]
- 39. Zhu, H. , Hao, H. , Yu, L. : Identifying disease‐related microbes based on multi‐scale variational graph autoencoder embedding Wasserstein distance. BMC Biol. 21(1), 294 (2023). 10.1186/s12915-023-01796-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li, H. , Liu, B. : BioSeq‐Diabolo: biological sequence similarity analysis using Diabolo. PLoS Comput. Biol. 19(6), e1011214 (2023). 10.1371/journal.pcbi.1011214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao, X. , et al.: BPLLDA: predicting lncRNA‐disease associations based on simple paths with limited lengths in a heterogeneous network. Front. Genet. 9, 411 (2018). 10.3389/fgene.2018.00411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ren, L. , et al.: MetaboliteCOVID: a manually curated database of metabolite markers for COVID‐19. Comput. Biol. Med. 167, 107661 (2023). 10.1016/j.compbiomed.2023.107661 [DOI] [PubMed] [Google Scholar]
- 43. Velickovic, P. , et al.: Graph attention networks.1050(20): p. 10–48550 (2017) [Google Scholar]
- 44. Tunstall, L. , Von Werra, L. , Wolf, T. : Natural Language Processing with Transformers, Revised Edition. O’Reilly Media, Incorporated: (2022) [Google Scholar]
- 45. Hajian‐Tilaki, K. : Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Caspian J Intern Med 4(2), 627–635 (2013) [PMC free article] [PubMed] [Google Scholar]
- 46. Managadze, D. , et al.: Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol Evol 3, 1390–1404 (2011). 10.1093/gbe/evr116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moran, V.A. , Perera, R.J. , Khalil, A.M. : Emerging functional and mechanistic paradigms of mammalian long non‐coding RNAs. Nucleic Acids Res. 40(14), 6391–6400 (2012). 10.1093/nar/gks296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ning, L. , et al.: Development and application of ribonucleic acid therapy strategies against COVID‐19. Int. J. Biol. Sci. 18(13), 5070–5085 (2022). 10.7150/ijbs.72706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang, Y. , et al.: P450Rdb: a manually curated database of reactions catalyzed by cytochrome P450 enzymes. J. Adv. Res. (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou, H. , et al.: Identify ncRNA subcellular localization via graph regularized k‐local hyperplane distance nearest neighbor model on multi‐kernel learning. IEEE ACM Trans. Comput. Biol. Bioinf 19(6), 3517–3529 (2022). 10.1109/tcbb.2021.3107621 [DOI] [PubMed] [Google Scholar]
- 51. Wang, Y. , et al.: Retrosynthesis prediction with an interpretable deep‐learning framework based on molecular assembly tasks. Nat. Commun. 14(1), 6155 (2023). 10.1038/s41467-023-41698-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin, J. , et al.: iDNA‐ABF: multi‐scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 23(1), 1–23 (2022). 10.1186/s13059-022-02780-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang, R. , et al.: DeepBIO: an automated and interpretable deep‐learning platform for high‐throughput biological sequence prediction, functional annotation and visualization analysis. Nucleic Acids Res. 51(7), 3017–3029 (2023). 10.1093/nar/gkad055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bray, F. , et al.: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6), 394–424 (2018). 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 55. Barta, J.A. , Powell, C.A. , Wisnivesky, J.P. : Global epidemiology of lung cancer. Ann Glob Health 85(1) (2019). 10.5334/aogh.2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mattiuzzi, C. , Lippi, G. : Current cancer epidemiology. J Epidemiol Glob Health 9(4), 217–222 (2019). 10.2991/jegh.k.191008.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Our Expects Data Policy requires a Data Availability Statement, even if no data are available, so please enter one in the space below. Sample statements can be found here. Please note that this statement will be published alongside your manuscript, if it is accepted for publication.
The data that support the findings of this study are openly available in LncRNADisease at http://www.rnanut.net/lncrnadisease/, and Lnc2Cancer at http://www.bio‐bigdata.net/lnc2cancer.