

Abstract
Acute myeloid leukemia (AML) is a hematological malignancy characterized by clonal expansion and differentiation arrest of the myeloid progenitor cells, which leads to the accumulation of immature cells called blasts in the bone marrow and peripheral blood. Mutations in the receptor tyrosine kinase FLT3 occur in 30% of normal karyotype patients with AML and are associated with a higher incidence of relapse and worse survival. Targeted therapies against FLT3 mutations using small-molecule FLT3 tyrosine kinase inhibitors (TKIs) have long been investigated, with some showing favorable clinical outcomes. However, major setbacks such as limited clinical efficacy and the high risk of acquired resistance remain unresolved. FLT3 signaling, mutations, and FLT3 inhibitors are topics that have been extensively reviewed in recent years. Strategies to target FLT3 beyond the small molecule kinase inhibitors are expanding, nevertheless they are not receiving enough attention. These modalities include antibody-based FLT3 targeted therapies, immune cells mediated targeting strategies, and approaches targeting downstream signaling pathways and FLT3 translation. Here, we review the most recent advances and the challenges associated with the development of therapeutic modalities targeting FLT3 beyond the kinase inhibitors.
Keywords: FLT3, AML, FLT3-ITD, scFv, Monoclonal antibody, CAR-T, microRNAs
1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy characterized by clonal expansion of myeloid progenitors in the bone marrow and peripheral blood (Saultz & Garzon, 2016). Recurrent cytogenetic and genetic alterations contribute to the development and the heterogeneity of AML, determine the risk stratification, and guide the therapeutic decision (Daver, Schlenk, Russell, & Levis, 2019; Döhner et al., 2017; Ley et al., 2013).
About 25–30% of patients newly diagnosed with AML present with mutations in the FMS-like tyrosine kinase 3 (FLT3), with ~25% presenting with the FLT3 internal tandem duplication (ITD), and the other 5–10% having the FLT3 tyrosine kinases domain (TKD) mutations. The ITD is the most frequently observed somatic mutation in AML patients and acts as a gain of function mutation. The FLT3-ITD fragments consist of 3–400 base pairs mapped to the sequence coding for the juxtamembrane domain. These fragments are either duplicated or inserted in direct orientation (Small, 2006; Thiede et al., 2002). FLT3-TKD is a point mutation that mainly occurs in codon 835 or 836 (Smith et al., 2012; Thiede et al., 2002). Activating mutations involving different residues mutated to other amino acids were also found either in TKD1 or TKD2 on activation loops within the kinase domain (Rummelt et al., 2020; Small, 2006; Smith et al., 2012; Takahashi, 2011; Zhang et al., 2014).
In normal human hematopoiesis, FLT3 expression is restricted to immature hematopoietic progenitors and hematopoietic stem cells (HSC) (Rosnet et al., 1996), and reduces as cells differentiate into mature myeloid cells (Ghiaur & Levis, 2017). In humans, the FLT3 gene is located on chromosome 13q12 and spans 24 exons. The FLT3 membrane-bound glycosylated protein consists of 993 amino acids and is about 160 kDa (Markovic, MacKenzie, & Lock, 2005). When FLT3 ligand (FL) binds to its receptor; FLT3, it synergizes with other growth factors such as KIT ligand, thrombopoietin, and interleukin-3, to expand the hematopoietic stem progenitor cells (HSPC) (Small, 2006). Binding of FL to its receptor induces receptor autophosphorylation at tyrosine residues resulting in signal transduction and activation of downstream signaling pathways (Dosil, Wang, & Lemischka, 1993). FLT3 has a cytoplasmic domain that interacts with phosphoinositol-3-kinase, RAS GTPase, phospholipase C-γ, SHC, GRB2, and the SRC family tyrosine kinases, leading to the activation of AKT and MAPK pathways which consequently increase cell proliferation and promote survival (Takahashi, 2011).
FLT3 mutant protein is constitutively active even in the absence of its ligand; hence it activates the signaling cascade of FLT3, thereby promoting cell proliferation and decreasing cell apoptosis. Unlike FLT3 wild type (WT), FLT3-ITD mutations have also been shown to cause activation in signal transducer and activator of transcription 5 (STAT5) signaling pathways (Grundler, Miething, Thiede, Peschel, & Duyster, 2005; Mizuki et al., 2003). Patients presenting with the FLT3 mutations have a higher relapse rate, worse overall survival (OS) rate, and shorter disease-free survival (DFS) compared with patients with FLT3-WT (Mrózek, Marcucci, Paschka, Whitman, & Bloomfield, 2007; Whitman et al., 2001; Whitman et al., 2008). Therefore, once first complete remission (CR) is achieved, an early allogeneic hematopoietic stem cell transplant (HSCT) is recommended for patients with FLT3-ITD mutations (Cornelissen et al., 2012; Döhner et al., 2017).
2. FLT3 inhibitors and their limitations
2.1. FLT3 inhibitors
With the high frequency of FLT3 mutations in de novo AML and the adverse prognostic impact of FLT3-ITD mutations, mutant FLT3 is considered an appropriate therapeutic target for leukemia-directed therapies (Ley et al., 2013). The inhibition of FLT3 has been a widely investigated strategy for the treatment of AML with FLT3 mutations. Several small-molecule tyrosine kinase inhibitors have been developed to combat oncogenic FLT3 signaling in AML (Daver et al., 2019). Based on the drug effectiveness and the target specificity, the FLT3 inhibitors are grouped into two generations: first- and second-generation inhibitors.
First-generation FLT3 inhibitors, tyrosine kinase inhibitors such as sunitinib, sorafenib, lestaurtinib, and midostaurin, are relatively non-specific for FLT3 receptors and present with a broad range of off-target effects. On the other hand, second-generation FLT3 inhibitors such as quizartinib, crenolanib, and gilteritinib, have narrow kinome activity as they are more selective and effective for inhibiting FLT3 (Wander, Levis, & Fathi, 2014).
FLT3 inhibitors are further classified into type I and type II inhibitors based on the mechanism of action in which they interact with the FLT3 receptor (Daver et al., 2019). The binding mechanism of type I inhibitors such as lestaurtinib, midostaurin, gilteritinib, and crenolanib is not affected by receptor conformation (i.e. can bind an active or inactive receptor) as they bind to FLT3 gatekeeper domain close to the activation loop or the ATP binding pocket of this enzyme. Thus, type I FLT3 inhibitors are considered effective against FLT3-ITD and FLT3-TKD (Ke et al., 2015). Type II inhibitors such as sorafenib and quizartinib bind adjacent to the ATP binding domain in the hydrophobic region when the protein is in an inactive conformational state. Therefore, type II inhibitors have a superior effect against FLT3-ITD mutations compared with FLT3-TKD mutations, which they confer resistance (Ambinder and Levis, 2021).
Midostaurin (PKC412), is the first FLT3 inhibitor approved to treat newly diagnosed FLT3-mutated AML patients in the United States (US). Using midostaurin together with traditional chemotherapy has shown to improve the OS rate in AML patients (Stone et al., 2017). Gilteritinib is a second-generation type I FLT3 inhibitor approved by the FDA in 2018. This drug has a single agent activity and is used to treat adult patients who have relapsed/refractory (R/R) FLT3-mutated AML (Perl et al., 2019). Quizartinib is currently approved for use in Japan due to its effectiveness in improving the OS in patients with R/R FLT3-ITD mutations (Cortes et al., 2019). Due to some safety concerns and the minimal survival benefit achieved with this drug, quizartinib is currently not approved for use in the US and European Union (Garcia-Horton & Yee, 2020). FLT3 inhibitors such as tandutinib, crenolanib, and cabozantinib are presently undergoing clinical trials or still in the process of development (Hou & Tien, 2020).
2.2. Limited clinical efficacy
Although few FLT3 inhibitors exert some single-agent activity, such as gilteritinib, the majority pose limited antileukemic activity when used as monotherapy. To achieve better efficacy, FLT3 inhibitors are tested in combination with standard chemotherapy regimens and other modalities. The addition of midostaurin to chemotherapy has significantly improved patients’ survival, yet the 4-year OS rate observed in patients treated with this drug was only 63.7% (Stone et al., 2017). In a study investigating the incorporation of midostaurin with standard care therapy following allogeneic HSCT, it was observed that patients with FLT3-ITD AML benefited clinically from this therapy. Nonetheless, the addition of midostaurin did not significantly increase the estimated 24 months OS rate, nor did it extend the median time to relapse from the time of transplant (Maziarz et al., 2020). Gilteritinib is the only FDA-approved FLT3 inhibitor to be used as a single agent in R/R FLT3 mutant AML. However, results from the phase 3 clinical trial (NCT02421939) showed that while the drug indeed prolonged OS, only 37% of the patients treated with gilteritinib survived more than one year (Perl et al., 2019).
In a study to assess the efficacy of sorafenib versus placebo when added to standard therapy to previously untreated younger adult patients with AML (18–60 years), 3-year event-free survival was observed in 40% of patients on sorafenib compared to 22% in the placebo group. Administration of sorafenib presented with adverse effects like fever, cardiac event, rash, etc (Röllig et al., 2015). An improved toxicity profile is needed before it can be approved for treatment, particularly in older patients. The addition of Lestaurtinib with first-line standard chemotherapy in newly diagnosed younger patients with FLT3 mutated AML showed no statistically significant difference in OS or relapse-free survival (RFS) (UK trial ISRCTN17161961, ISRCTN55675535) (Knapper et al., 2017). Another clinical trial (NCT00079482) was conducted to investigate the combination of salvage chemotherapy with lestaurtinib on AML patients in the first relapse. This study reported no significant improvement in the response or OS of AML patients in their first relapse after administration of lestaurtinib. This finding was attributed to the pharmacokinetics complexity and off-target toxicities of the drug (Levis et al., 2011). Quizartinib was also compared with salvage chemotherapy in an open clinical trial (QuANTUM-R NCT02039726). A longer median OS of 6.2 months was noted with this drug compared to the 4.7 months with chemotherapy. However higher treatment-emergent deaths in the quizartinib group were observed (Cortes et al., 2019).
Despite their limitations (Table 1), most TKIs are more effective when combined with other chemotherapies. Thus, several clinical trials are aiming to investigate the efficacy of FLT3 inhibitors in combination with induction-consolidation therapy or with azacitidine in newly diagnosed patients with FLT3 mutated AML (NCT02236013) (NCT02752035) (NCT03135054) (NCT01892371).
Table 1.
Clinical trials of different FLT3 inhibitors including the FDA approved TKIs (midostaurin and gilteritinib)a.
| Drug | aChemical structure | Clinical trial ID | Sample size (year age) | Intervention/Treatment | Efficacy | Phase | Ref. |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Midostaurin |
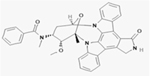
|
NCT00651261 | 717 (18–59) | Cytarabine and daunorubicin plus, midostaurin versus placebo in newly diagnosed AML patients | Median OS: 74.7 months, Midostaurin group; 25.6 months, Placebo group Median EFS: 8.2 months (95% CI, 5.4 to 10.7), midostaurin group; 3.0 months (95% CI, 1.9 to 5.9), placebo group | III | (Stone et al., 2017) |
| Gilteritinib (ASP2215) |
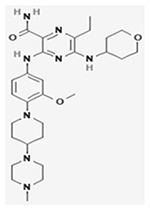
|
NCT02421939 | 371(18 and older) | Gilteritinib (ASP2215) versus salvage chemotherapy in R/R AML patients harboring FLT3 mutation | Median OS: 9.3 months, Gilteritinib; 5.6 months, Chemotherapy group Median EFS: 2.8 months, gilteritinib group; 0.7 months, chemotherapy group | III | (Perl et al., 2019) |
| Sorafenib |
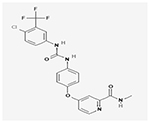
|
NCT00893373 | 276 (18–60) |
Sorafenib added to standard primary therapy in newly diagnosed AML patients | median EFS 9 months, placebo group versus 21 months, sorafenib group; 3-year-EFS of 22%, placebo group versus 40%, sorafenib group | II | (Rollig et al., 2015) |
| Lestaurtinib (CEP-701) |
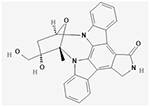
|
NCT00079482; | 224(18 and older); | Lestaurtinib (CEP-701) given in sequence with induction chemotherapy | 29 patients with CR/CRp in the lestaurtinib arm and 23 in the control arm; No difference in OS between the 2 arms; No overall clinical benefit | II | (Levis et al., 2011) |
| ISRCTN17161961and | (Knapper et al., 2017) | ||||||
| ISRCTN55675535 | 500(5 to 68) | in patients with relapsed AML expressing FLT3 activating mutations; patients with newly diagnosed AML or high-risk myelodysplastic syndrome | III | (Knapper et al., 2017) | |||
| Quizartinib |
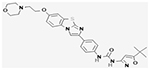
|
NCT02039726 | 367(18 and older) | Quizartinib Monotherapy vs. Salvage Chemotherapy | Median OS: 6·2 months (5·3–7·2), quizartinib group; 4·7 months (4·0–5·5), chemotherapy group | III | (Cortes et al., 2019) |
References of 2D structures in Table 1.
1. PubChem Identifier: CID 126565 (Lestaurtinib)
2. PubChem Identifier: CID 216239 (Sorafenib)
3. PubChem Identifier: CID 24889392 (Quizartinib)
4. PubChem Identifier: CID 49803313 (Gilteritinib)
6. PubChem Identifier: CID 9829523 (Midostaurin)
2.3. Resistance to FLT3 inhibitors
Even though FLT3 inhibitors exert clinical activity in AML, their effectiveness is short-lived and lasts only for a few months (Alvarado et al., 2014; Piloto et al., 2007). Different resistant mechanisms exert substantial challenges toward the limited long-term efficacy of FLT3 inhibitors (Ding et al., 2012). In addition to primary resistance seen in patients lacking response to FLT3 inhibitors, secondary resistance may emerge following the administration of FLT3 inhibitors (Alotaibi et al., 2021). Emergent mutations in the FLT3 target may develop and contribute to disease relapse. Resistant clones which were either absent at diagnosis or present at low frequencies are then acquired or expanded post TKIs treatment. A recent study has identified using targeted next-generation sequencing emergent mutations involving FLT3, epigenetic modifiers, RAS/MAPK pathway, and less frequently WT1 and TP53 in relapse bone marrow samples post treatment with FLT3 inhibitors (Alotaibi et al., 2021). Several studies showed that different point mutations developed after treatment with FLT3 inhibitors such as quizartinib, sorafenib, and midostaurin (Daver et al., 2015; Heidel et al., 2006; Smith et al., 2012; Zhang et al., 2014). These mutations can destabilize the binding ability of these inhibitors to FLT3 either by abrogating the inactive conformation, or the hinge segment’s conformation (Heidel et al., 2006; Smith et al., 2012). Some point mutations can elevate the phosphorylation levels of ERK, AKT, STAT5, and S6K, even when FLT3 is inhibited, resulting in suboptimal efficacy of FLT3 inhibitors (Ghiaur & Levis, 2017; Smith et al., 2012; Zhang et al., 2014). Upregulation of certain genes such as TESC (Man et al., 2014) and CCL5 (Waldeck et al., 2020) reportedly contributed to the resistance to FLT3 inhibitors. The activation of other signaling pathways such Janus kinase (JAK), receptor tyrosine kinase AXL, mTOR, AKT, and MAPK pathway were also identified as potential mechanisms of resistance (Lindblad et al., 2016; Park et al., 2015; Piloto et al., 2007; Rummelt et al., 2020).
2.4. FLT3-WT upregulation
FLT3 wild type (FLT3-WT) may also contribute to the reduced effectiveness of FLT3 inhibitors in newly diagnosed and/or relapsed AML patients (Kamezaki, Luchsinger, & Snoeck, 2014). Many patients with AML not only show upregulation in the FLT3-ITD but also in FLT3-WT. The phosphorylated and activated wild type FLT3 may also present a potential target in AML (Knapper et al., 2006; Takahashi, 2011). FLT3-WT and the activated FLT3-ITD share similar internalization and degradation patterns, irrespective of their different biogenesis profiles (Kellner et al., 2020). Additionally, FLT3-WT found in bone marrow stroma is sensitive to FLT3 ligand (FL) which mediates FLT3-WT activation, thereby promoting MAPK signaling pathway. Activation of this signaling pathway subsequently inhibits apoptosis and diminishes the effect of FLT3 inhibitors on blasts residing in the bone marrow. This, therefore, is considered a key mechanism that diminishes the impact of FLT3 inhibitors in clinical settings (Smith et al., 2004; Yang, Sexauer, & Levis, 2014). Nonetheless, the effect of FLT3 inhibitors on FLT3-WT is not completely understood (Kamezaki et al., 2014).
3. Other strategies to target the FLT3-ITD mutations in AML
Although midostaurin and gilteritinib have received FDA approval for treating patients with FLT3-ITD mutations (Dhillon, 2019; Stone, Manley, Larson, & Capdeville, 2018), these therapies are still limited since half of the patients die of their disease within five years. The current compiled knowledge of TKI’s limitations; presents the need for alternative therapeutic strategies.
3.1. FLT3 antibodies
3.1.1. Monoclonal FLT3 antibodies
Monoclonal antibodies have the advantage of high specificity and affinity, which can be leveraged for an effective therapeutic strategy (Fig. 1). Several FLT3 antibodies have been developed and investigated in both preclinical settings (Hofmann et al., 2012; Li et al., 2004; Park et al., 2020; Piloto et al., 2005) and in early phase clinical trials (NCT00887926) (Table 2). The FLT3 antibodies (IMC-EB10 and IMC-NC7) have displayed strong anti-leukemic activity in preclinical models. Human anti-FLT3 antibody, IMC-EB10 (IgG1, κ), was constructed from the antigen-binding fragment (Fab) selected from a naïve human-antibody, phage-display library through immunopanning against soluble FLT3-Fc protein. Similarly, IMC-NC7 was also developed as an anti-FLT3 mAb (IgG1, κ), derived from a naïve human antibody phage display library based on its ability to strongly bind FLT3-Fc. Both antibodies showed strong and specific binding affinity to FLT3, and blocked FL interaction with FLT3, thereby preventing ligand-induced phosphorylation and activation of wild type FLT3 in both AML cell lines and primary blasts. Also, both antibodies were able to inhibit the phosphorylation of FL independent FLT3-ITD cell lines, but only IMC-NC7 was able to partially inhibit FLT3 phosphorylation in FLT3-ITD primary blast. This inhibition resulted in blocking the downstream FLT3-mediated MAPK pathway. Treatment with IMC-NC7 was more potent in blocking FLT3 ligand-mediated FLT3 activation than IMC-EB10. Both IMC-NC7 and IMC-EB10 were shown to strongly inhibit phosphorylation of FLT3 in wild type FLT3 primary blasts. The ability of the two antibodies to differentially block FLT3 ligand activation could be owing to the differences in the bound epitopes (Youssoufian, Rowinsky, Tonra, & Li, 2010). Though these antibodies had an effect on FLT3 signaling pathways, stand-alone treatment with these antibodies has a limited effect on cell proliferation and apoptosis. However, treatment with IMC-EB10 in the presence of natural killer (NK) cells induced antibody-dependent cell-mediated cytotoxicity (ADCC) in cells expressing FLT3-WT and ITD. Also, IMC-EB10, administered intraperitoneally at 400μg, extended the survival of mice in both FLT3-WT and FLT3-ITD models and proved to be therapeutically effective in vivo (Piloto et al., 2005), an effect that was not observed with IMC-NC7. This difference in the in vivo efficacy between these two antibodies is likely due to the fact that IMC-EB10 but not IMC-NC7 could induce ADCC. IMC-EB10 also reduced engraftment of primary leukemic blasts, whereas it did not affect the engraftment of normal human cord blood CD34+ cells known to express lower levels of FLT3 (Li et al., 2004; Piloto et al., 2005). Based on this, IMC-EB10 was tested in a dose-escalation Phase I clinical trial (ID NCT00887926) in 26 patients with R/R AML. However, this study was discontinued since the maximum tolerated dose of IMC-EB10 was below that needed to achieve meaningful efficacy.
Fig. 1.

Multiple therapeutic options acting on different targets on FLT3 receptors. FLT3 inhibitors are classified into type I and type II inhibitors based on the mechanism of action in which they interact with the FLT3 receptor. Type I inhibitors such as lestaurtinib, midostaurin, gilteritinib, and crenolanib are not affected by receptor conformation (i.e., can bind an active or inactive receptor) as they bind to FLT3 gatekeeper domain close to the activation loop or the ATP binding pocket of this kinase. Type II inhibitors such as sorafenib and quizartinib bind adjacent to the ATP binding domain in the hydrophobic region and this binding must be done when the protein is in an inactive conformational state. Human anti-FLT3 antibodies, IMC-EB10 and IMC-NC7 (IgG1, κ) were constructed from the antigen-binding fragment (Fab) region of a naive human-antibody for soluble FLT3-Fc. Both antibodies showed strong and specific binding affinity to FLT3 and blocked FL interaction with FLT3, thereby preventing ligand-induced phosphorylation and activation of FLT3 in both AML cell lines and primary blasts. FLT3, FMS-like tyrosine kinase; ITD, internal tandem duplication; JMD, juxtamembrane domain; TK, tyrosine kinase; TKD, tyrosine kinase domain; mAb, monoclonal antibody.
Table 2.
Summary of the antibodies targeting FLT3 and small molecules targeting FLT3 expression that have been developed and in clinical trialsa.
| Class | Antibody/Small molecule | In-vivo/In-vitro model | Clinical Trial ID | Phase | Reference |
|---|---|---|---|---|---|
|
| |||||
| monoclonal antibody | IMC-EB10 | Yes/Yes | NCT00887926 | I | (Piloto et al., 2005) |
| IMC-NC7 | Yes/Yes | (Piloto et al., 2005) | |||
| Fc-optimized antibody | BV10SDIEM | Yes/Yes | NCT02789254 | I/II | (Hofmann et al., 2012) |
| 4G8SDIEM (FLYSYN) | Yes/Yes | (Hofmann et al., 2012) | |||
| scFv-ELP fusion protein | a-FLT3-A192 | Yes/Yes | (Park et al., 2020) | ||
| FLT3 bispecific antibody | 4G8 X UCHT1 | No/Yes | (Durben et al., 2015) | ||
| Anti-FLT3-CD3 immunoglobulin G (IgG)-based bispecific antibody | 7370 Anti-FLT3-CD3 | Yes/Yes | (Yeung et al., 2020) | ||
| FLT3 targeted CART Cell | anti-FLT3-scFv-CD28-CD3ζ | Yes/Yes | NCT03904069 | I | (Chen et al., 2017) |
| FLT3 CAR-T AMG 553 | Yes/Yes | ||||
| FLT3 Ligand-based CART Cell | FLT3L CAR-T | Yes/Yes | (Wang et al., 2018) | ||
| Yes/Yes | (Jetani, Garcia-Cadenas, Nerreter, Goetz, 2018, Jetani, Garcia-Cadenas, Nerreter, Thomas, 2018) | ||||
| Maternal embryonic leucine zipperkinase (MELK) inhibitors |
aOTS167
|
Yes/Yes | NCT02795520 | I/II | (Eisfelder et al., 2021) |
| Natural compound inhibitor of eIF4A |
aSilvestrol
|
Yes/Yes | (Alachkar et al., 2013) | ||
| NEDD8-activating enzyme inhibitor |
aMLN4924 (Pevonedistat)
|
Yes/Yes |
NCT00911066
NCT03268954 NCT02610777 NCT02782468 NCT04266795 NCT03772925 NCT04090736 NCT03013998 |
I III II I II I III I/II |
(Swords et al., 2015) |
| T-LAK cell-originated protein kinase (TOPK) |
aOTS514
|
Yes/Yes | (Alachkar et al., 2015) | ||
| Dual FLT3/TOPK inhibitor |
aHSD1169
|
No/Yes | (Dayal et al., 2018) | ||
| Deubiquitinating enzyme (USP10) |
aHBX19818 |
Yes/Yes | (Weisberg et al., 2017) | ||
| Inhibitor |
aP22077
|
||||
References of 2D structures in Table 2.
1. Dayal, N., Opoku-Temeng, C., Hernandez, D. E., Sooreshjani, M. A., Carter-Cooper, B. A., Lapidus, R. G., & Sintim, H. O. (2018). Dual FLT3/TOPK inhibitor with activity against FLT3-ITD secondary mutations potently inhibits acute myeloid leukemia cell lines. Future Med Chem, 10, 823–835. https://doi.org/10.4155/fmc-2017-0298.
2. PubChem Identifier: CID 11787114 (Silvestrol)
3. PubChem Identifier: CID 135398499 (Otssp167)
4. PubChem Identifier: CID 16720766 (Pevonedistat)
5. PubChem Identifier: CID 46931953 (P22077)
6. PubChem Identifier: CID 73349008 (HBX19818)
7. PubChem Identifier: CID 92044487 (OTS514 hydrochloride)
3.1.2. Fc-optimized FLT3 antibodies
Another approach to target FLT3 is based on Fc-optimization of antibodies that is achieved by recombinant antibody technology. This process involves modifying the amino acid sequence of the human immunoglobulin G1, Fc region, and adding a C-terminus tag to facilitate antibody detection. Two versions of the FLT3 antibodies were generated, BV10SDIEM and 4G8SDIEM (Hofmann et al., 2012). Antibody BV10SDIEM recognizes domain 2, whereas 4G8SDIEM recognizes domain 4 of FLT3. Both antibodies were active in vitro at even 1 ng/ml. Compared with the parent BV10 and 4G8 antibodies, the modified antibodies induced ADCC up to 100 times more. On a side note, because EB10 was not used at a concentration lower than 10 μg/ml, it is difficult to compare the ADCC activity between these antibodies. 4G8SDIEM antibody had better binding ability than BV10SDIEM and induced higher ADCC in NALM-16 cells cultured with peripheral blood mononuclear cells (PBMCs). However, this effect was far less pronounced on leukemic blasts isolated from patient’s peripheral blood that were used as targets for allogeneic and autologous PBMCs. This was speculated to be due to the resistance of primary cells to antibody-mediated killing rather than the lower expression of FLT3. Notably, the effect of 4G8SDIEM therapy on FLT3 expression was transient and did not cause a reduction in FLT3 surface expression below 50%. This effect is beneficial to prevent antigenic shift because 4G8SDIEM relies solely on ADCC, where the lack of FLT3 surface expression due to internalization will prevent 4G8SDIEM therapeutic activity (Hofmann et al., 2012). The binding affinity of 4G8SDIEM to normal bone marrow precursor cells and dendritic cells was very low, and treatment with this antibody did not induce toxicity or ADCC to the cells (Hofmann et al., 2012). 4G8SDIEM treatment for compassionate use in a patient with AML (complex karyotype with 45, XY, inv. (3)(q21q26), −7, 9q-, FAB M0) with relapse after haploidentical and unrelated donor stem cell transplantation (SCT) resulted in potent NK cell activation and rapid decrease of leukemic blasts from the peripheral blood and slight decrease in the bone marrow four days after treatment with 10 mg dose. However, this effect was only transient, and treatment with 4G8SDIEM did not reduce leukemic burden in the proliferative phase of AML, which was speculated to be due to unfavorable NK cells to leukemia cells ratios in the bone marrow (Salih et al., 2013). Nevertheless, this antibody is currently in phase 1 clinical trial (NCT02789254) for AML patients with minimal residual disease (MRD) in morphological complete remission. The rationale is that this antibody may have better therapeutic benefit in cases with high effector to target cell ratios.
3.1.3. FLT3 scFv fusion protein
Recently, our group has developed a fusion protein designed to target FLT3. This fusion protein consists of an scFv antibody conjugated to elastin like polypeptide (ELP). Fusion of an ELP to an scFv addresses many of the stability and pharmacokinetic concerns seen with scFvs’ alone. The fusion protein, named FLT3-A192, exhibited antileukemic activity on AML cell lines and achieved inhibition of FLT3 mediated downstream signaling kinases (Park et al., 2020). In vivo, FLT3-A192 showed a reduction in leukemia burden and improved survival in NOD scid gamma (NSG) mice. Unlike FLT3 targeting mAbs, FLT3-A192 targets the liver and spleen, which means it may also be a substrate for reticuloendothelial uptake and retention in the bone marrow (Park et al., 2020). Retention of FLT3-A192 in the bone marrow enables it to target the peripheral blood and bone marrow cells, which are the sites of leukemogenesis in AML. Targeting FLT3 using an scFv and ELP fusion peptide resulted in an excellent pharmacokinetic profile and efficacy in preclinical models.
3.1.4. Bispecific antibodies
As a potential therapeutic strategy, the recruitment of T cells was tested by constructing Fabsc-antibodies with FLT3 X CD3 specificity. The anti-CD3 component binds to CD3 on the cell surface of T cells to recruit them toward the leukemic cells. This antibody consists of a Fab region, a linker CH2 domain that contains modifications to attenuate FcR-binding, and a single-chain antibody with a second specificity. This antibody was constructed using 4G8 and BV10 antibodies targeting FLT3 and linked to three C-terminal effector antibodies targeting TCR/CD3-complex: OKT3, UCHT1, and BMA031 (Durben et al., 2015). The bispecific antibody utilizing FLT3-antibody 4G8 and the CD3-antibody UCHT1 Fabsc-molecule appeared to be superior to all other comparisons in terms of their abilities to induce T-cell-mediated cytotoxicity. PBMCs from AML patients containing leukemic blasts and autologous T cells at physiological ratios were treated for three days with 10 ng/ml of 4G8 X UCHT1. A significant increase in activation and proliferation of T cells and a significant decrease in leukemic blasts was observed after treatment. When compared with the Fc-optimized FLT3 antibody that failed to reduce blasts, the 4G8 X UCHT1 antibody demonstrated T-cell–mediated killing that is far more superior than that induced by NK cells. However, unlike the Fc-optimized FLT3 antibody, treatment of normal bone marrow cells with 0.1 μg/ml of 4G8 X UCHT1 antibody activated CD4+ and CD8+ T cells exerted toxic effects by reducing CD34-positive precursor cells. Furthermore, 4G8 X UCHT1 antibody had a short half-life of ~3 h (Durben et al., 2015).
To overcome the shortcomings mentioned above, another anti-FLT3 CD3 IgG-based bispecific antibody was engineered. This antibody was generated against domain 4 (similar to 4G8) of FLT3 and paired with anti-CD3 antibody (2B4) called 7370 Anti-FLT3 CD3 bispecific antibody. This antibody demonstrated specific binding to human T cells and AML cell lines exhibiting high and low FLT3 expression with picomolar (pM) affinity for the FLT3 arm and a 500-fold difference in the affinities between the FLT3 and CD3 arm, which will beneficially prevent non-desirable T cell activation and makes it superior to the 4G8 X UCHT1 antibody. The bispecific antibody impressively has a half-life of about six days in mice and cynomolgus monkeys. This bispecific antibody also redirected the cytotoxic and cytokine-secreting function of human T cells against AML cells when used at effector: target ratio of 1:1 at pM potency. Treatment also induced CD25, CD69, 41BB, and PD1 on T cells with kinetics reflective of antigen and T cell receptor (TCR)-mediated T cell activation. Furthermore, 7370 anti-FLT3 bispecific IgG induced cytotoxicity against autologous blasts by activating AML patient T cells; however, nanomolar concentrations are required for significant killing (Yeung et al., 2020). Consistent with the in vitro efficacy, the bispecific antibody effectively redirected human T cells against AML cells (MV4–11, MOLM-13, and EOL-1) in vivo with an effect dependent on the expression of FLT3 antigen on AML cells irrespective of FLT3 mutational status. Treatment of cynomolgus monkeys with two weekly high doses (3 mg/kg) of 7370 Anti-FLT3 CD3 bispecific antibody completely depleted FLT3+ dendritic cells (DCs), CD1c + and CD123 + DC subsets, but after 2 weeks these populations were recovered to baseline levels, and the monkeys exhibited very low hematological toxicities. However, it should be noted that, since monkeys can develop anti-drug antibodies to human antibodies after a few doses, the researchers were unable to assess long-term dosing effects to better predict the impact on healthy cells in clinical settings (Yeung et al., 2020).
3.2. FLT3 targeting T cell therapy
Another alternative therapy involves autologous T cells to target the FLT3-ITD mutation. T cell therapies can be divided into two broad groups, chimeric antigen receptor (CAR) T cell and engineered T cell receptor T cell therapies. Both of these approaches share a similar process: removal of T cells from the afflicted patient, genetic modification of the T cells with a specialized receptor to target a cancer antigen, expansion of T cells, and re-engrafting them into the patient for cancer cell targeting and killing.
3.2.1. Chimeric antigen receptor T cells
CARs consist of an extracellular antigen-recognition domain, typically composed of an antibody single-chain variable fragment (scFv) or a receptor-ligand linked to an intracellular signaling domain (Sadelain, Brentjens, & Rivière, 2013). The function of the extracellular domain permits T cells to recognize specific antigen while the signaling domains stimulate T-cell proliferation, cytolysis, and cytokine secretion to eliminate the target cells (Jackson, Rafiq, & Brentjens, 2016). Currently, the efficacy of FLT3 targeted CAR T cells (Fig. 2A) has been demonstrated both in vitro and in vivo. In a preclinical model, second-generation anti-FLT3 CARs were tested by chromium release assay for targeted killing, and by the enzyme-linked immunosorbent assay (ELISA) to determine the secretion of both IFN-gamma and IL-2, which showed specific cytotoxicity against FLT3-ITD positive leukemia cell lines MV4–11 and MOLM-13 in vitro. The same group also tested the in vivo capabilities by treating a MOLM-13 xenograft mouse model with the FLT3 CAR T cells for three-weekly treatments. A survival rate of 100% for the treated mice compared to the appropriate controls was observed (Chen et al., 2017). Another in vivo experiment was conducted to further reiterate the lack of CAR T cell activity against the HSCs derived from cord blood (Chen et al., 2017). An assessment of safety for the FLT3 CAR T cells was also carried out via coculture with normal HSCs isolated from cord blood cells, which demonstrated no off-target toxicity. Besides, the efficacy of targeting FLT3 and CD33 with CAR T cells were compared in another study that determined the former treatment induced less toxicity to HSCs/multipotent progenitors and equivalent toxicity to multipotent/common myeloid progenitors (Chien et al., 2016).
Fig. 2.

Summarized illustration of FLT3 targeting CAR T-Cells (A) FLT3 CAR T-cell targeting FLT3 antigen; (B) FLT3L CAR T-Cell is a second-generation CAR T cells where the extracellular domain is the FLT3 ligand (FL) while 4–1BB and CD3ζ are the intracellular signal domains; (C) Bispecific FLT3 and WT-1 CAR T-cell that target the FLT3-ITD mutation and WT-1 mutation which has been demonstrated to be associated with FLT3-ITD mutations.
In the design of the second-generation CAR T cells, the extracellular domain is the FLT3 ligand (FL) (Fig. 2B), while 4–1BB and CD3ζ are the intracellular signal domains. The in vitro results looked promising, as coculture with FLT3 positive cell lines such as MV4–11, MOLM-13, REH, and THP-1 cells demonstrated T cell activation, up-regulation of CD107a, increased levels of interferon-gamma and tumor necrosis factor-alpha, and targeted cell killing. Similarly, the cytotoxic effect of FLT3L CAR-T cells to primary AML cells was also observed. This group also evaluated the in vivo function of these CAR T cells by intravenously inoculating MV4–11 cells into NSG mice and then treating them with CAR T cells on days 7 and 14. This study demonstrated that the median survival time of the CAR-T treatment group was significantly prolonged compared with the control group (Wang et al., 2018).
Furthermore, combining CAR T cells with relevant kinase inhibitors may also decrease potential immunotherapeutic resistance leading to leukemia relapse. One study provided evidence for the efficacy of combining anti-FLT3 CAR T cells with crenolanib, a TKI that has also demonstrated efficacy against FLT3-ITD mutations. The group demonstrated that pretreatment with crenolanib increased FLT3 expression on the cell surface, which leads to enhanced T cell recognition of AML cell lines both in vitro and in vivo (Jetani, García-Cadenas, Nerreter, Goetz, 2018). This is also supported by the synergistic effects of combining midostaurin or quizartinib with CAR T cells against FLT3-ITD positive cell lines (MV4–11 and MOLM-13 cells) observed in vitro, and the improved response rate in the mice treated with the TKI and the CAR T cells compared to those with either treatment alone (Jetani, Garcia-Cadenas, Nerreter, Thomas, 2018).
Future developments in CAR T cell technology may also increase the efficacy of these therapies. One such technology is the bispecific CAR T cell, a T cell with two CARs that can recognize different targets. Therefore, a CAR T cell can target the FLT3 and another target such as WT-1, which has been demonstrated to be associated with FLT3-ITD mutations (Fig. 2C) (Summers et al., 2007). By targeting multiple mutations, it is possible to address a phenomenon termed “antigen escape” in which patients relapse with a phenotypically similar disease due to lack of surface expression of a specific target. The efficacy of bispecific CAR T cells by targeting both CD19 and CD20 in acute lymphoblastic leukemia provided solid evidence for this therapeutic strategy (Zah, Lin, Silva-Benedict, Jensen, & Chen, 2016).
The fourth-generation T cell redirected for universal cytokine-mediated killing T cells (TRUCKs) includes a transgenic cytokine expression cassette in the CAR constructs, typically an interleukin-12 (IL-12) signaling domain (Li et al., 2018). The introduction of a transgenic cytokine initiates universal cytokine-mediated killing toward cancer cells that are invisible to CAR-T cells via the recruitment of additional immune cells (Li et al., 2018). While this approach has not been explored against FLT3, theoretically, the principle of TRUCKs can be applied to AML as the immunosuppressive environment associated with this disease is well established (Krönig et al., 2014; Shenghui et al., 2011; Wang et al., 2005).
CAR-T-cell therapy has been proven to be a potentially effective treatment of various B cell malignancies. Due to this initial success, many groups are currently designing strategies to obtain similarly successful results in the treatment of other types of blood cancers. Various research groups have shown that the FLT3-ITD mutation, which is one of the most common mutations in AML patients, is an effective T cell target both in vitro and in vivo; hence this receptor is a major focus. Currently, the clinical trial (NCT03904069) is testing the safety, efficacy, and tolerability of CAR T cells in patients with relapsed/refractory AML.
3.2.2. Endogenous T cell receptors
The feasibility of the adoptive transfer of human T-cells with antitumor-specific T cell receptor (TCR) α and β chains has been demonstrated previously (Vonderheide & June, 2014). Neoantigens are formed by a variety of nonsynonymous genetic alterations, including single-nucleotide variants (SNV), insertions and deletions, gene fusions, frameshift mutations, and structural variants (Buonaguro, Petrizzo, Tornesello, & Buonaguro, 2011). Targeting tumor antigens can reduce the risk of “on-target, off-tumor” toxicity but is challenging in practice (Fig. 3A). Despite the complexity and the cost of this approach, there have been several relatively successful attempts to use engineered T cell receptors to target cancer neoantigens like ERBB2 interacting protein (ERBB2IP) mutation in a patient with metastatic cholangiocarcinoma (Tran et al., 2014) and targeting a mutation in SLC3A2 for a patient suffering from ER-positive and HER2-negative breast cancer (Zacharakis et al., 2018). The fact that FLT3-ITD mutations can vary from approximately three to hundreds of nucleotides, combined with the individual variation associated with patient’s leukocyte antigen (HLA) alleles implies that engineered T cell receptors may need to be personalized for every patient. A research group sequenced the FLT3-ITD mutation in a patient with AML and identified five peptides with high combined prediction scores which were then selected to be used as targets for T cells. These five peptides were tested in vitro for T cell activation via coculture with FLT3-ITD transduced K562 cells. The in vitro responder populations containing peptide-reactive T cells were also able to recognize primary patient AML cells. The researchers isolated the top 19 TCR clones, 10 of which demonstrated activation against the FLT3-ITD mutation via interferon-gamma secretion. The study also validated the presence of the same TCR clones corresponding to the in vitro activated T cells in blood and bone marrow samples from the same patient. The identified T cells also demonstrated cytotoxicity via increased granzyme B secretion compared with control groups (Graf et al., 2007).
Fig. 3.

Summarized illustration of TCR engineered T cell approaches (A) targeting FLT3 neoantigen peptides; (B) Specific peptide enhanced affinity receptors (SPEARs) technology designed to modify the hypervariable complementary determining regions that bind to the displayed neoantigen peptide while simultaneously displaying no alloreactivity; (C) Hybrid murine-human TCR for better pairing and TCR/CD3 stability.
An alternative method that might be considered is to utilize murine-human hybrid T cell receptors to improve pairing and TCR/CD3 stability (Fig. 3C). Researchers replaced the original human constant regions of the TCR with murine ones. They observed preferential pairing of murine constant regions with themselves, less mispairing with the endogenous human TCR chains, and increased stability of the TCR/CD3ζ complex, thus leading to increased sensitivity to cancer cells (Cohen, Zhao, Zheng, Rosenberg, & Morgan, 2006). In addition to using the murine constant regions, another option to consider would be adding a single cysteine to each receptor subunit to incorporate an additional disulfide bond. By implementing this, one group demonstrated that cysteine-modified receptors were more highly expressed on the surface of human lymphocytes compared with their wild-type counterparts and able to mediate higher levels of cytokine secretion and specific lysis when co-cultured with specific tumor cell lines (Cohen et al., 2007). By adding these two modifications, the FLT3-ITD TCR may have improved cytotoxicity.
There are other technologies that have the potential to enhance the ability of engineered T cell receptors to target the FLT3-ITD mutation. One such technology includes specific peptide enhanced affinity receptors (SPEARs) (Fig. 3B), which is designed to modify the hypervariable complementary determining regions that bind to the displayed neoantigen peptide while simultaneously displaying no alloreactivity. Currently, clinical trials using this technology are ongoing in non-small cell lung cancer, melanoma, urothelial cancer, and head and neck cancer (NCT02989064, NCT02592577, NCT03132792, NCT03132922, NCT04044859, NCT04044768). Thus, it would be theoretically possible to apply the same technology for FLT3-ITD as a target with a promising prospect.
3.3. Targeting FLT3 expression
Although kinase inhibition has been the main approach to target FLT3 in AML, strategies focusing on targeting the expression of FLT3 by interfering with its transcription, translation, or protein degradation have been extensively explored in both preclinical and clinical studies. (Alachkar et al., 2013; Ghiaur & Levis, 2017).
Inhibiting the translation initiation and depletion of FLT3 was reported with the use of maternal embryonic leucine zipper kinase (MELK) inhibitors. MELK phosphorylates the eukaryotic translation initiation factor 4B (eIF4B) at Ser406 and regulates protein synthesis during mitosis (Wang et al., 2016). Also, the MELK inhibitor (OTS167) blocks FLT3 expression by inhibiting phosphorylation of eukaryotic initiation factor 4E–binding protein 1 (4E-BP1) and eukaryotic translation initiation factor 4B (eIF4B), leading to blocking FLT3 translation. The combination of OTS167 with TKIs results in synergistic induction of cell death in FLT3 mutant cell lines and prolonged survival in a FLT3 mutant AML xenograft mouse model (Eisfelder et al., 2021). MELK inhibitor is currently under clinical investigation in early phase clinical trials in AML (NCT02795520). Inhibitors of mRNA translations such as the natural compound silvestrol have also proven to pose a potent antileukemia activity against FLT3-ITD+ AML. It was determined that the IC50 of silvestrol was lower in FLT3-ITD than in FLT3-WT AML cells and primary blasts. Silvestrol proved to significantly induce apoptosis in both FLT3-ITD and FLT3-WT cells (Alachkar et al., 2013). It has been reported that silvestrol alters the interaction between eIF4F translation complexes with capped mRNA, leading to inhibition of protein synthesis of certain proteins, including FLT3, that depend on this mechanism for their translation (Cencic et al., 2009) (Lucas et al., 2009) (Bordeleau et al., 2008). Silvestrol completely abolished total FLT3 protein and the surface expression when measured by western blot or flow cytometry, respectively. Silvestrol treatment also resulted in a significant reduction in miR-155 expression (Alachkar et al., 2013), a microRNA that is upregulated in FLT3-ITD positive AML (Faraoni et al., 2012; Whitman et al., 2010). miR-155 promotes FLT3-ITD-induced myeloid expansion by reducing the growth-inhibitory effects of the interferon (IFN) response (Wallace et al., 2017). Therefore, modulating the expression or function of miR-155 may present an effective strategy to target FLT3-ITD function. Indeed, pharmacological targeting of miR-155 with an inhibitor against NEDD8-activating enzyme (MLN4924, Pevonedistat) appears to have a preferential activity on cells harboring FLT3-ITD and in turn expressing higher miR-155 (Khalife et al., 2015). Pevonedistat has shown feasibility in phase 1 clinical trials (Swords et al., 2015) and is currently undergoing clinical testing in combination with other targeted therapies (NCT03268954, NCT02610777, NCT02782468, NCT04266795, NCT03772925, NCT04090736, NCT03013998).
Another approach to target FLT3 focuses on FLT3-ITD signaling pathways. T-LAK cell-originated protein kinase (TOPK) is highly expressed in many malignancies, AML cell lines, and primary AML blasts. TOPK inhibitors resulted in inhibition of STAT5, a downstream target of the FLT3 signaling pathway (Alachkar et al., 2015; Dayal et al., 2018). The loss of function approach by knocking down TOPK resulted in a significant decrease in cell viability by enhancing apoptosis in AML cells. Furthermore, treatment with TOPK inhibitor, OTS514, showed a dose-dependent decrease in cell viability in FLT3 mutant AML blasts. In xenograft mouse models, treatment with TOPK inhibitor reduced leukemia burden and prolonged mouse survival compared with respective control (Alachkar et al., 2015). Another group has presented a novel compound named HSD1169 to achieve the dual effect against mutant FLT3 and TOPK in TKI-resistant cell lines. This novel compound contains a chemical scaffold (8,9,10,11-tetrahydro-3H-pyrazolo[4,3-a] phenanthridine), which aids in the inhibition of mutant FLT3 AML cell lines (Dayal et al., 2018).
Inducing FLT3 protein degradation is another alternative approach to modulate FLT3 levels. This has been demonstrated with the inhibition of USP10, a deubiquitinating enzyme that is required to stabilize FLT3. Targeting USP10 showed preclinical efficacy in FLT3-ITD positive models of AML, including cell lines, primary patient blasts, and FLT3-driven leukemia mouse models (Weisberg et al., 2017).
4. Ongoing challenges and future potentials
The suboptimal efficacy of TKIs in AML, along with the dismal clinical outcome of patients with FLT3 mutations, suggests the need to explore more effective strategies. Leveraging other features of the FLT3 target besides its constitutive kinase activity for the development of therapeutic approaches may overcome the challenges associated with TKIs. Whether it is mutated or upregulated, being a cell surface receptor makes it a suitable target for antibody or T cell mediated immunotherapeutic modalities. Monoclonal antibodies, Fc-optimized, and T cell-based approaches have the advantage of high specificity and affinity, which can be advantageous over some TKIs therapeutic strategies that are less specific for FLT3 and may inhibit a broad range of off-target kinases. Considering that the antileukemia activity of FLT3 immunotherapies or strategies targeting FLT3 expression is independent of FLT3 mutational status, they have the potential to circumvent resistance mechanisms associated with the use of TKIs, particularly those involving the upregulation of FLT3 wild type. FLT3 inhibitors promote the surface expression of FLT3-ITD or TKD (D835Y) mutant that often sequester intracellularly in the perinuclear endoplasmic reticulum ER. Indeed, TKIs-induced FLT3 surface expression is significantly higher in FLT3 mutants than FLT3-WT-expressing cells (Reiter et al., 2018). Samples of patients with FLT3 mutated AML also showed higher surface expression of FLT3 after sorafenib treatment than at the time of diagnosis (Reiter et al., 2018). In addition, it was demonstrated that TKIs treatment increased T cell-mediated cytotoxicity (TCMC) in the FLT3-ITD-positive AML cell lines and primary blasts. These observations suggest a potential benefit of the combined use of TKIs with FLT3-directed immunotherapy with the rationale that TKIs treatment will upregulate FLT3 antigen to be recognized by immunotherapy (Reiter et al., 2018). Despite the advances in the development of modalities targeting FLT3 beyond the TKIs and their promising antileukemia activities, the majority of these strategies are still in preclinical or phase I-II clinical testing, and more research is needed to establish their full potential. Efforts focusing on the identification of the optimal target domain of the FLT3 receptor are needed for the development of a more effective FLT3 antibody with favorable physicochemical properties and pharmacokinetic profiles. In addition, efforts are needed to address challenges associated with engineering FLT3 CAR T cell strategies to improve their safety, efficacy, and manufacturing feasibility. Moreover, targeting the expression of FLT3 with MELK inhibitor or enhancing FLT3 degradation with USP10 comprises a promising area for therapeutic optimization and potential synergism with TKIs (Eisfelder et al., 2021; Weisberg et al., 2017). In addition, targeting FLT3 degradation induces neoantigen-reactive T cells and may induce antileukemia mediated T cell activity. Clinical trials designed to investigate combinatorial strategies will be crucial to position these strategies within the space of AML treatment.
5. Concluding remarks
Due to its role in the proliferation and survival of the AML blasts, FLT3 is a key target in treating AML. Although TKIs present a plausible therapeutic strategy, the outcome remains unsatisfactory as relapse, lack of specificity, dismal monotherapy efficacy, and the emergence of resistance all pose clinical challenges. To this date, midostaurin and gilteritinib are the only TKIs that have received FDA approval for treating AML patients with FLT3-ITD. Yet, they are still therapeutically limited, even with promising advancement, since half of these patients die of their disease within five years. Thus, incorporating more than one drug has the benefit of filling the gaps in treatment protocols, yet the toxic adverse effects present an additional challenge. Altogether, the need for alternative therapeutic strategies beyond the TKIs is high.
Targeting FLT3 through alternative approaches such as monoclonal antibodies, T cell-based therapies, or with small-molecule inhibitors targeting FLT3 expression levels or signaling pathways are all strategies that have demonstrated effectiveness in preclinical studies. While early to conclude their values in the clinical settings, thus far, several approaches appeared to be safe and feasible. In this commentary, we argue that alternative modalities beyond the kinase inhibitors are expanding for the treatment of FLT3 mutant AML. These strategies may potentially hold the promise to cure this type of leukemia.
Acknowledgment
We acknowledge the University of Southern California School of Pharmacy Seed Fund. This work was also supported by Ming Hsieh foundation, and the Norris Cancer Center Pilot funding support to HA.
All authors have read the journal’s authorship agreement.
Abbreviations:
- ADCC
Antibody-dependent cell-mediated cytotoxicity
- AML
Acute myeloid leukemia
- AKT
Protein kinase B
- ELP
Elastin like polypeptide
- Fab
Antigen-binding fragment
- AXL
Tyrosine-protein kinase receptor UFO
- ATP
Adenosine triphosphate
- CAR
Chimeric antigen receptor
- CRc
Composite complete remission
- DC
Dendritic cell
- eIF4B
eukaryotic translation initiation factor 4B
- ELISA
Enzyme-linked immunosorbent assay
- ER
Endoplasmic reticulum
- ERBB2IP
ERBB2 interacting protein
- FLT3
FMS-like tyrosine kinase 3
- FL
FLT3 ligand
- HSC
Hematopoietic stem cells
- HSPC
Hematopoietic stem progenitor cells
- IFN
Interferon
- ITD
Internal tandem duplication
- mAb
Monoclonal antibody
- MELK
Maternal embryonic leucine zipper kinase
- MHC
Major histocompatibility complex
- MRD
Minimal residual disease
- miRNA
microRNA
- NSG
NOD scid gamma mice
- TKD
Tyrosine kinase domain
- OS
overall survival
- DFS
disease-free survival
- CR
complete remission
- HSCT
Hematopoietic stem cell transplant
- R/R
Refractory/Relapsed
- FDA
Food and Drug Administration
- RFS
Relapse-free survival
- TKI
Tyrosine kinase inhibitor
- TESC
Tescalcin
- CCL5
C-C Motif Chemokine Ligand 5
- JAK
Janus kinase
- MAPK
mitogen-activated protein kinase
- PBMC
Peripheral blood mononuclear cells
- scFv
Single-chain variable fragment
- SNV
Single-nucleotide variants
- TCMC
T cell-mediated cytotoxicity
- TCR
T-Cell receptor
- TOPK
T-LAK cell-originated protein kinase
- TRUCKs
T cell redirected for universal cytokine-mediated killing T cells
- WT
Wild type
Footnotes
Associate editor: Beverly Teicher.
Declaration of Competing Interest
The authors have declared that no conflict of interest exists.
References
- Alachkar H, Mutonga M, Malnassy G, Park JH, Fulton N, Woods A, … Nakamura Y (2015). T-LAK cell-originated protein kinase presents a novel therapeutic target in FLT3-ITD mutated acute myeloid leukemia. Oncotarget 6(32), 33410–33425. 10.18632/oncotarget.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alachkar H, Santhanam R, Harb JG, Lucas DM, Oaks JJ, Hickey CJ, … Marcucci G (2013). Silvestrol exhibits significant in vivo and in vitro antileukemic activities and inhibits FLT3 and miR-155 expressions in acute myeloid leukemia. Journal of Hematology & Oncology 6, 21. 10.1186/1756-8722-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, Loghavi S, Kadia TM, DiNardo CD, … Daver N (2021). Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Blood Cancer Discovery 2(2), 125–134. 10.1158/2643-3230.bcd-20-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado Y, Kantarjian HM, Luthra R, Ravandi F, Borthakur G, Garcia-Manero G, … Cortes JE (2014). Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer 120(14), 2142–2149. 10.1002/cncr.28705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambinder AJ, & Levis M (2021). Potential targeting of FLT3 acute myeloid leukemia. Haematologica 106, 671–681. 10.3324/haematol.2019.240754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, … Pelletier J (2008). Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. The Journal of Clinical Investigation 118(7), 2651–2660. 10.1172/jci34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonaguro L, Petrizzo A, Tornesello ML, & Buonaguro FM (2011). Translating tumor antigens into cancer vaccines. Clinical and Vaccine Immunology 18(1), 23–34. 10.1128/cvi.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencic R, Carrier M, Galicia-Vázquez G, Bordeleau ME, Sukarieh R, Bourdeau A, … Pelletier J (2009). Antitumor activity and mechanism of action of the cyclopenta [b]benzofuran, silvestrol. PLoS One 4(4), Article e5223. 10.1371/journal.pone.0005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Mao H, Zhang J, Chu J, Devine S, Caligiuri MA, & Yu J (2017). Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia 31, 1830–1834. 10.1038/leu.2017.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CD, Sauter CT, Ishii K, Nguyen SM, Shen F, Tasian SK, … Fry TJ (2016). Preclinical development of FLT3-redirected chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia. Blood 128(22), 1072. 10.1182/blood.V128.22.1072.1072. [DOI] [Google Scholar]
- Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, & Morgan RA (2007). Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Research 67(8), 3898–3903. 10.1158/0008-5472.can-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, & Morgan RA (2006). Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Research 66(17), 8878–8886. 10.1158/0008-5472.can-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen JJ, Gratwohl A, Schlenk RF, Sierra J, Bornhäuser M, Juliusson G, … Ossenkoppele GJ (2012). The European LeukemiaNet AML working party consensus statement on allogeneic HSCT for patients with AML in remission: An integrated-risk adapted approach. Nature Reviews. Clinical Oncology 9(10), 579–590. 10.1038/nrclinonc.2012.150. [DOI] [PubMed] [Google Scholar]
- Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, … Levis MJ (2019). Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. The Lancet Oncology 20(7), 984–997. 10.1016/s1470-2045(19)30150-0. [DOI] [PubMed] [Google Scholar]
- Daver N, Cortes J, Ravandi F, Patel KP, Burger JA, Konopleva M, & Kantarjian H (2015). Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 125(21), 3236–3245. 10.1182/blood-2014-10-605808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daver N, Schlenk RF, Russell NH, & Levis MJ (2019). Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 33(2), 299–312. 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal N, Opoku-Temeng C, Hernandez DE, Sooreshjani MA, Carter-Cooper BA, Lapidus RG, & Sintim HO (2018). Dual FLT3/TOPK inhibitor with activity against FLT3-ITD secondary mutations potently inhibits acute myeloid leukemia cell lines. Future Medicinal Chemistry 10(7), 823–835. 10.4155/fmc-2017-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S (2019). Gilteritinib: First global approval. Drugs 79(3), 331–339. 10.1007/s40265-019-1062-3. [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, … DiPersio JF (2012). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481(7382), 506–510. 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, … Bloomfield CD (2017). Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129(4), 424–447. 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosil M, Wang S, & Lemischka IR (1993). Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Molecular and Cellular Biology 13(10), 6572–6585. 10.1128/mcb.13.10.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durben M, Schmiedel D, Hofmann M, Vogt F, Nübling T, Pyz E, … Jung G (2015). Characterization of a bispecific FLT3 X CD3 antibody in an improved, recombinant format for the treatment of leukemia. Molecular Therapy 23(4), 648–655. 10.1038/mt.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfelder BJ, Saygin C, Wynne J, Colton MW, Fischietti M, Beauchamp EM, … Stock W (2021). OTS167 blocks FLT3 translation and synergizes with FLT3 inhibitors in FLT3 mutant acute myeloid leukemia. Blood Cancer Journal 11(3), 48. 10.1038/s41408-021-00433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraoni I, Laterza S, Ardiri D, Ciardi C, Fazi F, & Lo-Coco F (2012). MiR-424 and miR-155 deregulated expression in cytogenetically normal acute myeloid leukaemia: Correlation with NPM1 and FLT3 mutation status. Journal of Hematology & Oncology 5, 26. 10.1186/1756-8722-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Horton A, & Yee KW (2020). Quizartinib for the treatment of acute myeloid leukemia. Expert Opinion on Pharmacotherapy 21(17), 2077–2090. 10.1080/14656566.2020.1801637. [DOI] [PubMed] [Google Scholar]
- Ghiaur G, & Levis M (2017). Mechanisms of resistance to FLT3 inhibitors and the role of the bone marrow microenvironment. Hematology/Oncology Clinics of North America 31(4), 681–692. 10.1016/j.hoc.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf C, Heidel F, Tenzer S, Radsak MP, Solem FK, Britten CM, … Wölfel T (2007). A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood 109(7), 2985–2988. 10.1182/blood-2006-07-032839. [DOI] [PubMed] [Google Scholar]
- Grundler R, Miething C, Thiede C, Peschel C, & Duyster J (2005). FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood 105(12), 4792–4799. 10.1182/blood-2004-11-4430. [DOI] [PubMed] [Google Scholar]
- Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, … Fischer T (2006). Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 107(1), 293–300. 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Große-Hovest L, Nübling T, Pyż E, Bamberg ML, Aulwurm S, … Jung G (2012). Generation, selection and preclinical characterization of an fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 26(6), 1228–1237. 10.1038/leu.2011.372. [DOI] [PubMed] [Google Scholar]
- Hou HA, & Tien HF (2020). Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. Journal of Biomedical Science 27 (1), 81. 10.1186/s12929-020-00674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson HJ, Rafiq S, & Brentjens RJ (2016). Driving CAR T-cells forward. Nature Reviews. Clinical Oncology 13(6), 370–383. 10.1038/nrclinonc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetani H, García-Cadenas I, Nerreter T, Goetz R, Sierra J, Bonig H, … Hudecek M (2018). FLT3 inhibitor treatment increases FLT3 expression that exposes FLT3-ITD+ AML blasts to elimination by FLT3 CAR-T cells. Blood 132(Supplement 1), 903. 10.1182/blood-2018-99-118171.30006329 [DOI] [Google Scholar]
- Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, … Hudecek M (2018). CAR T-cells targeting FLT3 have potent activity against FLT3(−)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 32(5), 1168–1179. 10.1038/s41375-018-0009-0. [DOI] [PubMed] [Google Scholar]
- Kamezaki K, Luchsinger LL, & Snoeck HW (2014). Differential requirement for wild-type Flt3 in leukemia initiation among mouse models of human leukemia. Experimental Hematology 42(3), 192–203 e191 10.1016/j.exphem.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke YY, Singh VK, Coumar MS, Hsu YC, Wang WC, Song JS, … Hsieh HP (2015). Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Scientific Reports 5, 11702. 10.1038/srep11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner F, Keil A, Schindler K, Tschongov T, Hünninger K, Loercher H, … Müller JP (2020). Wild-type FLT3 and FLT3 ITD exhibit similar ligand-induced internalization characteristics. Journal of Cellular and Molecular Medicine 24(8), 4668–4676. 10.1111/jcmm.15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalife J, Radomska HS, Santhanam R, Huang X, Neviani P, Saultz J, … Marcucci G (2015). Pharmacological targeting of miR-155 via the NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia 29 (10), 1981–1992. 10.1038/leu.2015.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, & Burnett AK (2006). The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: The induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood 108(10), 3494–3503. 10.1182/blood-2006-04-015487. [DOI] [PubMed] [Google Scholar]
- Knapper S, Russell N, Gilkes A, Hills RK, Gale RE, Cavenagh JD, … Burnett AK (2017). A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood 129(9), 1143–1154. 10.1182/blood-2016-07-730648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönig H, Kremmler L, Haller B, Englert C, Peschel C, Andreesen R, & Blank CU (2014). Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. European Journal of Haematology 92(3), 195–203. 10.1111/ejh.12228. [DOI] [PubMed] [Google Scholar]
- Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, … Smith BD (2011). Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 117(12), 3294–3301. 10.1182/blood-2010-08-301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, … Eley G (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England Journal of Medicine 368(22), 2059–2074. 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li W, Huang K, Zhang Y, Kupfer G, & Zhao Q (2018). Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. Journal of Hematology & Oncology 11(1), 22. 10.1186/s13045-018-0568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li H, Wang MN, Lu D, Bassi R, Wu Y, … Hicklin DJ (2004). Suppression of leukemia expressing wild-type or ITD-mutant FLT3 receptor by a fully human anti-FLT3 neutralizing antibody. Blood 104(4), 1137–1144. 10.1182/blood-2003-07-2585. [DOI] [PubMed] [Google Scholar]
- Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, Kabir NN, … Kazi JU (2016). Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 35(39), 5119–5131. 10.1038/onc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas DM, Edwards RB, Lozanski G, West DA, Shin JD, Vargo MA, … Grever MR (2009). The novel plant-derived agent silvestrol has B-cell selective activity in chronic lymphocytic leukemia and acute lymphoblastic leukemia in vitro and in vivo. Blood 113(19), 4656–4666. 10.1182/blood-2008-09-175430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man CH, Lam SS, Sun MK, Chow HC, Gill H, Kwong YL, & Leung AY (2014). A novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AML. Blood 123(16), 2530–2539. 10.1182/blood-2013-07-512194. [DOI] [PubMed] [Google Scholar]
- Markovic A, MacKenzie KL, & Lock RB (2005). FLT-3: A new focus in the understanding of acute leukemia. The International Journal of Biochemistry & Cell Biology 37(6), 1168–1172. 10.1016/j.biocel.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Maziarz RT, Levis M, Patnaik MM, Scott BL, Mohan SR, Deol A, … Fernandez HF (2020). Midostaurin after allogeneic stem cell transplant in patients with FLT3-internal tandem duplication-positive acute myeloid leukemia. Bone Marrow Transplantation. 10.1038/s41409-020-01153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuki M, Schwable J, Steur C, Choudhary C, Agrawal S, Sargin B, … Serve H (2003). Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 101(8), 3164–3173. 10.1182/blood-2002-06-1677. [DOI] [PubMed] [Google Scholar]
- Mrózek K, Marcucci G, Paschka P, Whitman SP, & Bloomfield CD (2007). Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: Are we ready for a prognostically prioritized molecular classification? Blood 109(2), 431–448. 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, … Caligiuri MA (2015). Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 29(12), 2382–2389. 10.1038/leu.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Vaikari VP, Lam AT, Zhang Y, MacKay JA, & Alachkar H (2020). Anti-FLT3 nanoparticles for acute myeloid leukemia: Preclinical pharmacology and pharmacokinetics. Journal of Controlled Release 324, 317–329. 10.1016/j.jconrel.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, … Levis MJ (2019). Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. The New England Journal of Medicine 381(18), 1728–1740. 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- Piloto O, Levis M, Huso D, Li Y, Li H, Wang MN, … Small D (2005). Inhibitory anti-FLT3 antibodies are capable of mediating antibody-dependent cell-mediated cytotoxicity and reducing engraftment of acute myelogenous leukemia blasts in nonobese diabetic/severe combined immunodeficient mice. Cancer Research 65(4), 1514–1522. 10.1158/0008-5472.can-04-3081. [DOI] [PubMed] [Google Scholar]
- Piloto O, Wright M, Brown P, Kim KT, Levis M, & Small D (2007). Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 109(4), 1643–1652. 10.1182/blood-2006-05-023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter K, Polzer H, Krupka C, Maiser A, Vick B, Rothenberg-Thurley M, … Greif PA (2018). Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia 32(2), 313–322. 10.1038/leu.2017.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, … Ehninger G (2015). Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. The Lancet Oncology 16(16), 1691–1699. 10.1016/s1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- Rosnet O, Bühring HJ, deLapeyrière O, Beslu N, Lavagna C, Marchetto S, … Birnbaum D (1996). Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta Haematologica 95(3–4), 218–223. 10.1159/000203881. [DOI] [PubMed] [Google Scholar]
- Rummelt C, Gorantla SP, Meggendorfer M, Charlet A, Endres C, Döhner K, … von Bubnoff N (2020). Activating JAK-mutations confer resistance to FLT3 kinase inhibitors in FLT3-ITD positive AML in vitro and in vivo. Leukemia. 10.1038/s41375-020-01077-1. [DOI] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R, & Rivière I (2013). The basic principles of chimeric antigen receptor design. Cancer Discovery 3(4), 388–398. 10.1158/2159-8290.cd-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih HR, Hofmann M, Grosse-Hovest L, Nuebling T, Bamberg M, Aulwurm S, … Jung G (2013). Elimination of minimal residual disease (MRD) in AML patients with a novel fc-optimized FLT3 antibody (4G8-SDIEM). Blood 122(21), 1454. 10.1182/blood.V122.21.1454.1454. [DOI] [Google Scholar]
- Saultz JN, & Garzon R (2016). Acute myeloid leukemia: A concise review. Journal of Clinical Medicine 5(3). 10.3390/jcm5030033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, & Xi X (2011). Elevated frequencies of CD4+ CD25+ CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. International Journal of Cancer 129(6), 1373–1381. 10.1002/ijc.25791. [DOI] [PubMed] [Google Scholar]
- Small D (2006). FLT3 mutations: Biology and treatment. Hematology. American Society of Hematology. Education Program, 178–184. 10.1182/asheducation-2006.1.178. [DOI] [PubMed] [Google Scholar]
- Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, … Small D (2004). Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 103(10), 3669–3676. 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, … Shah NP (2012). Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 485(7397), 260–263. 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, … Döhner H (2017). Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. The New England Journal of Medicine 377(5), 454–464. 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RM, Manley PW, Larson RA, & Capdeville R (2018). Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Advances 2(4), 444–453. 10.1182/bloodadvances.2017011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers K, Stevens J, Kakkas I, Smith M, Smith LL, Macdougall F, … Fitzgibbon J (2007). Wilms’ tumour 1 mutations are associated with FLT3-ITD and failure of standard induction chemotherapy in patients with normal karyotype AML. Leukemia 21, 550–551 author reply 552 10.1038/sj.leu.2404514. [DOI] [PubMed] [Google Scholar]
- Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, … Medeiros BC (2015). Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: A phase 1 study. British Journal of Haematology 169(4), 534–543. 10.1111/bjh.13323. [DOI] [PubMed] [Google Scholar]
- Takahashi S (2011). Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. Journal of Hematology & Oncology 4, 13. 10.1186/1756-8722-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, … Illmer T (2002). Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99(12), 4326–4335. 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, … Rosenberg SA (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344(6184), 641–645. 10.1126/science. 1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH, & June CH (2014). Engineering T cells for cancer: Our synthetic future. Immunological Reviews 257(1), 7–13. 10.1111/imr.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck S, Rassner M, Keye P, Follo M, Herchenbach D, Endres C, … von Bubnoff N (2020). CCL5 mediates target-kinase independent resistance to FLT3 inhibitors in FLT3-ITD-positive AML. Molecular Oncology 14(4), 779–794. 10.1002/1878-0261.12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Kagele DA, Eiring AM, Kim CN, Hu R, Runtsch MC, … O’Connell RM (2017). miR-155 promotes FLT3-ITD-induced myeloproliferative disease through inhibition of the interferon response. Blood 129(23), 3074–3086. 10.1182/blood-2016-09-740209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wander SA, Levis MJ, & Fathi AT (2014). The evolving role of FLT3 inhibitors in acute myeloid leukemia: Quizartinib and beyond. Therapeutic Advances in Hematology 5(3), 65–77. 10.1177/2040620714532123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng J, Liu J, Yao J, He Y, Li X, … Huang S (2005). Increased population of CD4(+)CD25(high), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. European Journal of Haematology 75(6), 468–476. 10.1111/j.1600-0609.2005.00537.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Begley M, Li Q, Huang HT, Lako A, Eck MJ, … Zhao JJ (2016). Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proceedings of the National Academy of Sciences of the United States of America 113 (35), 9810–9815. 10.1073/pnas.1606862113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, … Wang J (2018). Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. Journal of Hematology & Oncology 11(1), 60. 10.1186/s13045-018-0603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, … Buhrlage SJ (2017). Inhibition of USP10 induces degradation of oncogenic FLT3. Nature Chemical Biology 13(12), 1207–1215. 10.1038/nchembio.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, … Caligiuri MA (2001). Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: A cancer and leukemia group B study. Cancer Research 61(19), 7233–7239. [PubMed] [Google Scholar]
- Whitman SP, Maharry K, Radmacher MD, Becker H, Mrózek K, Margeson D, … Bloomfield CD (2010). FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: A Cancer and leukemia group B study. Blood 116(18), 3622–3626. 10.1182/blood-2010-05-283648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman SP, Ruppert AS, Radmacher MD, Mrózek K, Paschka P, Langer C, … Bloomfield CD (2008). FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 111(3), 1552–1559. 10.1182/blood-2007-08-107946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Sexauer A, & Levis M (2014). Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. British Journal of Haematology 164(1), 61–72. 10.1111/bjh.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung YA, Krishnamoorthy V, Dettling D, Sommer C, Poulsen K, Ni I, … Djuretic I (2020). An optimized full-length FLT3/CD3 Bispecific antibody demonstrates potent anti-leukemia activity and reversible hematological toxicity. Molecular Therapy 28 (3), 889–900. 10.1016/j.ymthe.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssoufian H, Rowinsky EK, Tonra J, & Li Y (2010). Targeting FMS-related tyrosine kinase receptor 3 with the human immunoglobulin G1 monoclonal antibody IMC-EB10. Cancer 116(4 Suppl), 1013–1017. 10.1002/cncr.24787. [DOI] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, … Feldman SA (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nature Medicine 24(6), 724–730. 10.1038/s41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zah E, Lin MY, Silva-Benedict A, Jensen MC, & Chen YY (2016). ADDENDUM: T cells expressing CD19/CD20 Bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunology Research 4(7), 639–641. 10.1158/2326-6066.cir-16-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Gao C, Konopleva M, Chen Y, Jacamo RO, Borthakur G, … Andreeff M (2014). Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clinical Cancer Research 20(9), 2363–2374. 10.1158/1078-0432.ccr-13-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]