

Abstract
The NS5A nonstructural protein of hepatitis C virus (HCV) has been shown to inhibit the cellular interferon (IFN)-induced protein kinase R (PKR). PKR mediates the host IFN-induced antiviral response at least in part by inhibiting mRNA translation initiation through phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). We thus examined the effect of NS5A inhibition of PKR on mRNA translation within the context of virus infection by using a recombinant vaccinia virus (VV)-based assay. The VV E3L protein is a potent inhibitor of PKR. Accordingly, infection of IFN-pretreated HeLa S3 cells with an E3L-deficient VV (VVΔE3L) resulted in increased phosphorylation levels of both PKR and eIF2α. IFN-pretreated cells infected with VV in which the E3L locus was replaced with the NS5A gene (VVNS5A) displayed diminished phosphorylation of PKR and eIF2α in a transient manner. We also observed an increase in activation of p38 mitogen-activated protein kinase in IFN-pretreated cells infected with VVΔE3L, consistent with reports that p38 lies downstream of the PKR pathway. Furthermore, these cells exhibited increased phosphorylation of the cap-binding initiation factor 4E (eIF4E), which is downstream of the p38 pathway. Importantly, these effects were reduced in cells infected with VVNS5A. NS5A was also found to inhibit activation of the p38-eIF4E pathway in epidermal growth factor-treated cells stably expressing NS5A. NS5A-induced inhibition of eIF2α and eIF4E phosphorylation may exert counteracting effects on mRNA translation. Indeed, IFN-pretreated cells infected with VVNS5A exhibited a partial and transient restoration of cellular and viral mRNA translation compared with IFN-pretreated cells infected with VVΔE3L. Taken together, these results support the role of NS5A as a PKR inhibitor and suggest a potential mechanism by which HCV might maintain global mRNA translation rate during early virus infection while favoring cap-independent translation of HCV mRNA during late infection.
Chronic hepatitis C virus (HCV) infection has become a worldwide health problem, affecting an estimated 170 million people worldwide, including about 4 million Americans (1). Chronically infected patients often develop progressive liver disease, cirrhosis, hepatic failure, and hepatocellular carcinoma (36). There is no vaccine available against HCV, and current therapies, including the combination of alpha interferon (IFN-α) and ribavirin, are effective at viral eradication in only a small percentage of patients (26, 33, 34). However, the development of more effective anti-HCV therapeutic agents has been hampered by the lack of an efficient cell culture system and an adequate animal model for HCV infection and replication.
HCV is a hepacivirus belonging to the Flaviviridae family. The positive-sense single-stranded enveloped RNA genome is translated in an internal ribosomal entry site-dependent manner to generate a single polyprotein, which is proteolytically processed into at least nine proteins (40). Among these proteins, the NS5A nonstructural protein is receiving increasing attention as a potential target for anti-HCV therapy. The initial interest in NS5A stemmed from the observation that mutations within a discrete region of NS5A from certain HCV genotypes, termed the IFN sensitivity-determining region, correlated with increased sensitivity to IFN treatment (14, 15). Although the existence of the IFN sensitivity-determining region has been rather controversial (25), subsequent studies have shown that NS5A interacted with and inhibited protein kinase R (PKR), which is a key mediator of the host IFN antiviral response (18, 19, 20). PKR exerts its effects by phosphorylating the GTP-binding eukaryotic initiation factor 2 (eIF2) (11, 12). The eIF2 facilitates binding of the initiator Met-tRNA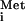 to the 40S ribosomal subunit during translation initiation. Phosphorylation of the α subunit of eIF2 (eIF2α) on Ser51 by PKR converts eIF2 into a competitive inhibitor of its guanine nucleotide exchange factor, eIF2B, resulting in the inhibition of general cellular protein synthesis and hence virus replication. Thus, NS5A-mediated inhibition of PKR may counteract the IFN-induced translational arrest, alluding to a possible mechanism by which HCV induces or sustains resistance to IFN treatment.
to the 40S ribosomal subunit during translation initiation. Phosphorylation of the α subunit of eIF2 (eIF2α) on Ser51 by PKR converts eIF2 into a competitive inhibitor of its guanine nucleotide exchange factor, eIF2B, resulting in the inhibition of general cellular protein synthesis and hence virus replication. Thus, NS5A-mediated inhibition of PKR may counteract the IFN-induced translational arrest, alluding to a possible mechanism by which HCV induces or sustains resistance to IFN treatment.
More recently, we have found that NS5A binds to a Src-homology 3 (SH3) domain of Grb2 (51). Grb2 is an adapter protein that mediates intracellular signaling by nucleating the formation of signal transduction complexes. The SH3 domains of Grb2 bind the nucleotide exchange factor Sos to trigger a series of signaling cascades in response to growth factor stimulation (10, 13). Stimulation with epidermal growth factor (EGF) induces the SH2 domain of Grb2 to bind to the EGF receptor, thereby recruiting Grb2 and Sos into a complex with the receptor and stimulating Sos to activate Ras. Activated Ras triggers the mitogen-activated protein kinase (MAPK) cascades, including the extracellular signal-regulated kinase (ERK) pathway (23, 57). ERKs phosphorylate a number of substrates, including the transcription factors E1k-1 and c-Jun, as well as the protein kinases Mnk1 and 3pK. Intriguingly, the phosphorylation of eIF4E by Mnk1 suggests a potential mechanism of translational control mediated through growth factor signaling (48, 49). Furthermore, the ERK pathway may play a role in IFN signaling and/or viral replication efficiency (28, 32), alluding to another possible mechanism of HCV resistance to IFN. In addition to NS5A, the E2 envelope protein of HCV has also been implicated as an inhibitor of PKR (54). The exact mechanism by which E2 inhibits PKR is not known, but inhibition may be mediated through E2 sequence homology with the autophosphorylation sites of PKR. Thus, HCV likely employs multiple strategies, including a two-pronged attack on PKR that incorporates the inhibitory functions of both NS5A and E2 proteins, to evade the IFN-induced antiviral response.
The use of cell lines inducibly expressing NS5A to determine the relevance of NS5A-mediated inhibition of PKR in HCV resistance to IFN have led to conflicting results (16, 19, 20, 39). In this report, we investigated the role of NS5A in PKR inhibition in the setting of a virus infection by using an established recombinant vaccinia virus (VV)-based system. We found that expression of NS5A in this system perturbed PKR-mediated signaling cascades, including the p38 pathway, possibly resulting in opposing effects on mRNA translation. Our results provide for the first time evidence supporting the PKR inhibitory role of NS5A in virus-infected cells and highlight a potential mechanism by which HCV subverts cellular signaling pathways to favor cap-independent translation of its mRNA.
MATERIALS AND METHODS
Cells and virus.
HeLa S3 cells (ATCC CCL-2.2) were maintained in Dulbecco's minimal essential medium (Gibco/BRL) supplemented with 10% fetal bovine serum, penicillin-streptomycin (100 U/ml), and 2 mM l-glutamine and were grown at 37°C in 5% CO2. VV (Copenhagen strain VC-2) was propagated, and working stocks were generated as described by Tartaglia et al. (53). The generation of recombinant VV devoid of E3L (VVΔE3L) and recombinant VV expressing NS5A in place of E3L (VVNS5A) was previously described (3, 51). VV infection of HeLa S3 cells was performed according to the protocol described by Kibler et al. (30). Maintenance, NS5A induction, and EGF treatment of stable Tet-Off HeLa cells (Clontech) inducibly expressing NS5A were performed as previously described (51).
In vivo radioisotope labeling of VV-infected HeLa S3 cells.
At 2, 4, and 6 h postinfection, HeLa S3 cells untreated or pretreated with human type I IFN (400 IU/ml) for 24 h (Access) and infected with VV, VVΔE3L, or VVNS5A at multiplicity of infection of 10 were pulse-labeled with [35S]Met (50 μCi/ml) (NEN) for 30 min (3). Cell lysates were prepared and protein concentration was determined as previously described (51). Protein lysates (20 μg/sample) were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (14% gel) and visualized by autoradiography. Protein synthesis levels were also quantified by subjecting cell lysates (2 μg/sample) to trichloroacetic acid (TCA) precipitation followed by quantification with a Beckman Coulter LS6500 scintillation counter.
Radioimmunoprecipitation assays.
Radioimmunoprecipitation assays were performed as previously described (47). Briefly, 5 μl of antiserum against VV proteins (provided by Bertram Jacobs) was added to 10 μg of [35S]Met-labeled protein lysates prepared from VV-infected cells and incubated on ice with occasional agitation for 1 h. Protein A-agarose beads (20 μl) (Boehringer Mannheim) were added to the antiserum and lysate mixture and were incubated on ice for 2 h with occasional agitation. After incubation, the beads were washed three times with ice-cold phosphate-buffered saline buffer and resuspended in 20 μl of 2× SDS protein loading buffer (125 mM Tris-HCl [pH 6.8], 20% glycerol, 2% SDS, 2% β-mercaptoethanol, and 0.02% bromophenol blue). Bound proteins were removed by boiling the beads for 5 min. Proteins were resolved by SDS-PAGE (14% gel) and visualized by autoradiography.
Analysis of PKR autophosphorylation.
Cell lysates were prepared and protein concentration was determined as previously described (51). Lysates from VV-infected cells were subjected to SDS-PAGE (7.5% gel) followed by electroblotting to nitrocellulose filters (Schleicher & Schuell). The membranes were probed with a rabbit polyclonal antiserum specific for PKR (2), and detection was performed by using enhanced chemiluminescence (Amersham Pharmacia Biotech).
Determination of eIF2α, p38, and eIF4E phosphorylation.
For analysis of eIF2α, p38, and eIF4E phosphorylation, equal amounts of cell lysates (20 μg) were subjected to SDS-PAGE and electroblotting. Filter immunoblots were probed with antibodies specific to phosphorylated forms of eIF2α (Ser51; Research Genetics), p38 (Thr180/Tyr182; New England Biolabs), or eIF4E (Ser209; New England Biolabs). The blots were stripped and reprobed with a monoclonal antibody that recognizes eIF2α (a gift from the E. C. Henshaw laboratory) or with antibodies directed against p38 (Santa Cruz Biotech) or eIF4E (Transduction Laboratories) to estimate the amount of expressed proteins. The relative levels of protein phosphorylation were determined by quantifying the immunoblots with ImageQuant (version 5.1). The signals from the phospho-specific immunoblots were normalized against their individual control signals, and the ratio of phospho-specific signal to control signal was determined.
RESULTS
Transient expression of NS5A from a recombinant VV system.
VV is relatively resistant to the antiviral effects of IFN-α because the virus encodes a number of proteins that can interfere with components of the IFN signaling pathways (5, 37, 42, 44). One of these proteins is E3L, a double-stranded RNA (dsRNA)-binding protein that potently inhibits PKR, apparently by both sequestering dsRNA activators of PKR (8) and directly binding to the protein kinase (46). Accordingly, a recombinant VV lacking the E3L gene is exquisitely sensitive to IFN treatment (3, 4, 9). To use this system to characterize the PKR inhibitory function of NS5A, we inserted the NS5A coding sequence (obtained from an HCV genotype 1b isolate from a patient who did not respond to IFN therapy; see reference 18) into VVΔE3L such that its expression was under the control of the E3L promoter (51). This enabled us to examine the effects of NS5A expression within the context of virus infection and IFN treatment.
In all experiments, HeLa S3 cells were pretreated with human type I IFN (400 IU/ml) for 24 h and then washed with fresh medium prior to infection with different recombinant VV. Expression of NS5A was confirmed by immunoblot analysis of lysates derived from IFN-pretreated HeLa S3 cells infected with wild-type VV, VVΔE3L, or VVNS5A by using an antibody specific to NS5A. Using this analysis, NS5A was detected as early as 2 h postinfection (Fig. 1A), consistent with the fact that the early E3L promoter regulates NS5A expression. The immunoblot analysis also revealed multiple forms of NS5A, presumably due to differential phosphorylation (41, 52) or other forms of posttranslational modifications. A similar pattern of NS5A protein expression was observed in the absence of IFN treatment (data not shown).
FIG. 1.

NS5A reduces virus-induced PKR autophosphorylation and eIF2α phosphoryltion. (A) Expression of NS5A in VVNS5A-infected cells. Lysates (20 μg of protein) from HeLa S3 cells pretreated with IFN-α/β and mock-infected (lane A) or infected with wild-type (WT) VV (lane B), VVΔE3L (lane C), or VVNS5A (lanes D through F) at 2, 4, or 6 h postinfection (p.i.) were resolved by SDS-PAGE (14% gel). For mock infections and infections with wild-type VV or VVΔE3L, only samples from the 6-h time point are shown. NS5A was detected by Western blotting (WB) using an anti-NS5A antibody (ID Labs). Arrows indicate the various migrating forms of NS5A. Sizes are indicated in kilodaltons. (B) Transient inhibition of virus-induced PKR autophosphorylation by NS5A. Lysates (20 μg of protein) used for panel A were resolved by SDS-PAGE (7.5% gel). PKR (top panel) and actin (bottom panel) proteins were detected with an anti-PKR antibody and an anti-actin (ICN) antibody, respectively. Hyperphosphorylated PKR protein is denoted by an asterisk. (C) Reduced eIF2α phosphorylation in the presence of NS5A. Lysates were immunoblotted with an antibody specific to the Ser51-phosphorylated form of eIF2α (top panel) or an anti-eIF2α antibody to detect protein levels (bottom panel). The relative levels of eIF2α phosphorylation were determined by quantitative densitometry and normalized against the individual total protein amounts. The ratio of the phospho-specific signal to the total protein signal is indicated below the lanes for lysates from VVΔE3L- and VVNS5A-infected cells. ND, not detectable.
Inhibition of virus-induced phosphorylation of PKR by NS5A.
We first measured the activity of PKR in IFN-pretreated HeLa S3 cells infected with the various recombinant VV. Previous studies established that autophosphorylation of PKR correlates with its activation and that higher autophosphorylation leads to lower electrophoretic mobility (17, 27, 45, 46). We thus examined differential phosphorylation of PKR based on differential migration in SDS-PAGE (Fig. 1B). Mock-infected cells served as a negative control in which PKR was unphosphorylated and inactive. In lysates from IFN-pretreated wild-type VV-infected cells, PKR migrated at the same rate as that in lysates from mock-infected cells, indicating that in wild-type VV-infected cells PKR was unphosphorylated and inactive. In lysates from IFN-treated cells infected with VVΔE3L, PKR migrated at slower rates 4 h after virus infection, indicating the presence of phosphorylated and active PKR. In contrast, PKR migrated at faster rates in lysates from VVNS5A-infected cells, indicating that NS5A could inhibit the phosphorylation and activation of PKR (Fig. 1B, arrows). This effect was transient, as the phosphorylated forms of PKR reappeared at 6 h postinfection. The difference in PKR abundance is noteworthy since it was specific and highly reproducible. This dissimilarity may be due to the fact that PKR downregulates its own expression at the translational level by a mechanism that depends on its phosphorylation, such that PKR abundance is inversely proportional to its kinase activity (45, 46). Alternatively, the phosphorylated PKR forms may be degraded by an unknown mechanism, as is the case during poliovirus infection (6, 7). This assay was also performed on lysates from VV-infected cells not treated with IFN, and similar results were observed (data not shown).
Virus-induced phosphorylation of PKR substrate, eIF2α, is also inhibited by NS5A.
PKR exerts its antiviral effects in part by phosphorylating eIF2α on Ser51. To test whether the inhibition of PKR by NS5A affected the phosphorylation status of eIF2α, we measured the relative amount of eIF2α phosphorylation by using an antibody specific to the Ser51 phosphorylated form of the protein. In agreement with the results above, mock-infected or wild-type VV-infected cells produced no detectable level of eIF2α phosphorylation at Ser51 (Fig. 1C). As expected, in VVΔE3L-infected cells, there was a significant increase in phosphorylation of eIF2α by PKR. Infection with VVNS5A, however, clearly decreased the proportion of phosphorylated eIF2α compared with that of VVΔE3L-infected cells, while the protein level of eIF2α remained unaffected. On average, NS5A caused a two- to threefold decrease in eIF2α phosphorylation. Previous studies have established that as little as a 15% change in the phosphorylation levels of eIF2α can result in a dramatic impact on protein synthesis initiation (11). The different kinetics and levels of PKR activation and eIF2α phosphorylation at different time points postinfection are likely due to the presence of other eIF2α kinases that can also phosphorylate the initiation factor (12). Taken together, these results support the role of NS5A in counteracting PKR activity and subsequent phosphorylation of eIF2α, suggesting a potential mechanism by which NS5A evades the antiviral effects of IFN.
Subversion of the p38 MAPK signaling pathway by NS5A during viral infection.
Recently, it has been suggested that PKR might act upstream of the p38 MAPK pathway (24, 29, 59). We thus examined whether the p38 pathway is affected by NS5A in the VV system. As shown in Fig. 2A, in mock- and wild-type VV-infected cells pretreated with IFN, there was no detectable p38 activation, as indicated by the absence of phosphorylation at Thr180/Tyr182. VVΔE3L-infected cells exhibited a significant increase in p38 activation despite the presence of comparable protein levels of p38 in all samples, consistent with the scenario where PKR is activated in the absence of E3L. Importantly, the activation of p38 was significantly decreased (approximately twofold on average) in VVNS5A-infected cells, presumably at least in part due to NS5A inhibition of PKR.
FIG. 2.

Inhibition of p38 activation and eIF4E phosphorylation by NS5A expression in VV-infected cells. (A) Virus-induced p38 activation in HeLa S3 cells infected with different recombinant VV at various time points postinfection (p.i.). Lysates (20 μg of protein) were fractionated by SDS-PAGE. The activation state of p38 was then determined by Western blotting (WB) using antibody specific for the dually phosphorylated activated form of p38 (Thr180/Tyr182) (top panel) or the total p38 protein kinase (bottom panel). The position of activated p38 is indicated by an arrow. The relative levels of p38 phosphorylation were determined by quantitative densitometry and normalized against the individual total protein amounts, and the phospho-specific signal/total protein signal ratios are indicated below the lanes for lysates from VVΔE3L- and VVNS5A-infected cells. ND, not detectable; WT, wild type. (B) Inhibition of Ser209 phosphorylation of eIF4E by NS5A expression in VV-infected cells. Phosphorylation levels of eIF4E at Ser209 were measured by Western blotting of the lysates used for Fig. 1 by using a phospho-specific antibody (top panel), while protein levels of eIF4E were determined by using an anti-eIF4E antibody (bottom panel). The relative levels of eIF4E phosphorylation were determined by quantitative densitometry and normalized against the individual total protein amounts. The ratio of the phospho-specific signal to total protein signal is indicated below the lanes.
An important substrate of p38 is MAPK-interacting protein kinase 1 (Mnk1), which in turn phosphorylates the translation initiation factor eIF4E. Phosphorylation of eIF4E on residue Ser209 by Mnk1 can enhance the cap-binding activity of the translation initiation factor (48, 49). We thus examined the effect of NS5A on eIF4E phosphorylation in recombinant VV-infected HeLa S3 cells pretreated with IFN. In mock- and wild-type VV-infected HeLa S3 cells a low basal level of eIF4E phosphorylation was detectable (Fig. 2B). A slight increase in eIF4E phosphorylation was observed in wild-type VV-infected cells, possibly due to the activation of MAPK pathways by the virus-encoded EGF homologue. Consistent with the p38 activation profile described above, a much higher level of eIF4E phosphorylation was observed in VVΔE3L-infected cells. As predicted, the presence of NS5A reversed this effect by approximately two- to fivefold, indicating that NS5A can subvert the PKR-p38 pathway. The inhibition of p38 and eIF4E phosphorylation by NS5A is specific, since we did not observe an inhibition of the unrelated AKT cell survival pathway in VVNS5A-infected cells (data not shown).
NS5A inhibits EGF-induced phosphorylation of p38 and eIF4E.
To confirm our data and examine the effect of NS5A on the p38-eIF4E pathway outside the context of virus infection, we compared the levels of EGF-induced activation of p38 and eIF4E in an inducible stable NS5A-expressing Tet-Off HeLa cell line (51). In accordance with the results that NS5A could affect the p38 MAPK-eIF4E pathway during viral infection, and in agreement with a previous study that NS5A can interact with the Grb2 adaptor molecule and perturb the downstream MAPK pathway (51), we found that cells stably expressing NS5A were indeed more refractory to EGF-induced activation of p38 than control cells lacking NS5A (Fig. 3A). Likewise, NS5A expression also inhibited EGF-induced phosphorylation of eIF4E (Fig. 3B). Importantly, the protein levels of both p38 and eIF4E were expressed to comparable levels in the cells. Inducible expression of NS5A in these cells was confirmed by Western blot analysis (data not shown). Taken together with the data obtained from the VV infection studies, these results suggest NS5A, whether expressed from a tetracycline-regulated promoter or as a virus-encoded protein, has the capability to inhibit the p38 signaling pathway and downregulate eIF4E phosphorylation. Furthermore, these results support the significance of our findings, demonstrating the ability of a single viral protein to regulate different translation initiation factors in different settings by modulating different cellular pathways.
FIG. 3.

Inhibition of EGF-induced phosphorylation of p38 and eIF4E in an inducible stable NS5A-expressing HeLa cell line system. Tet-Off HeLa cells cultured in the presence (−NS5A) or in the absence of tetracycline (+NS5A) were mock- or EGF-treated (20 ng/ml for 2 min), washed, and incubated for 4 h prior to lysis. The activation state of p38 (A) and Ser209 phosphorylation of eIF4E (B) were then determined by immunoblotting using antibody directed against the phosphorylated forms of the proteins as described in the legend to Fig. 2. Total protein levels were determined as described in the legend to Fig. 2. WB, Western blotting.
NS5A inhibits IFN-induced translational arrest during virus infection.
A prediction from the inhibition of eIF2α phosphorylation through PKR downregulation by NS5A (Fig. 1) is maintenance of cellular protein synthesis during virus infection. However, the decrease in eIF4E phosphorylation (Fig. 2 and 3), possibly due to NS5A-mediated inhibition of the PKR-p38 pathway, would also argue for a reduction of cap-dependent translation of cellular mRNAs. We therefore examined the consequences of such opposing effects on mRNA translation in VVNS5A-infected cells. HeLa S3 cells pretreated with IFN were infected with wild-type or recombinant VV and pulse-labeled with [35S]Met at 2, 4, or 6 h postinfection. As previously reported (3, 4, 43), IFN pretreatment had little effect on global protein synthesis in wild-type VV-infected cells (Fig. 4A), as confirmed by quantification of protein translation levels in VV-infected cells by TCA precipitation followed by scintillation counting (data not shown). In contrast, cells infected with VVΔE3L displayed a significant decrease in global protein synthesis, an effect largely attributed to the activation of PKR and phosphorylation of eIF2α (3, 4, 57). On average, VVΔE3L infection caused an approximately 10-fold decrease in protein translation levels (data not shown) compared with those of mock- and wild-type VV-infected cells. As predicted, NS5A partially and transiently restored global protein synthesis in VVNS5A-infected cells, consistent with the notion that the restoration is a net result of dephosphorylation of eIF2α and eIF4E. On average, NS5A expression resulted in an approximately sixfold increase in protein translation levels, with the most significant effect observed at 4 h postinfection (an approximately 18-fold increase).
FIG. 4.

NS5A partially reverses the translational arrest phenotype observed in VVΔE3L-infected cells. (A) Profiles of global protein synthesis in recombinant VV-infected cells. HeLa S3 cells were pretreated with IFN-α/β and mock-infected or infected with wild-type (WT) VV, VVΔE3L, or VVNS5A. Cells were pulse-labeled with [35S]Met and harvested at 2, 4, and 6 h postinfection (p.i.) Lysates (20 μg of protein) were resolved by SDS-PAGE and detected by autoradiography. The protein translation levels were also quantified by subjecting lysates to TCA precipitation followed by scintillation counting. (B) Profiles of viral protein synthesis in recombinant VV-infected cells. The lysates used for panel A were immunoprecipitated with a rabbit polyclonal antiserum against total VV proteins, and precipitated proteins were subjected to SDS-PAGE and autoradiography.
We next investigated whether NS5A could rescue viral protein synthesis from the IFN-induced translational arrest phenotype observed in VVΔE3L-infected cells. Cell lysates were prepared from wild-type and recombinant VV-infected HeLa S3 cells that were pulse-labeled with [35S]Met at 4 h postinfection. Viral proteins were then immunoprecipitated from these lysates using a rabbit polyclonal antiserum raised against VV proteins. The immunoprecipitated proteins were resolved by SDS-PAGE and visualized by autoradiography. While efficient viral protein synthesis could be detected in HeLa S3 cells infected with wild-type VV, there was a significant decrease in viral protein synthesis in VVΔE3L-infected cells (Fig. 4B). As predicted, viral protein synthesis was partially restored by NS5A expression in VVNS5A-infected cells. These assays were also performed with VV-infected cells not treated with IFN, and a similar, although less significant, effect of NS5A expression on global and viral protein translation was seen (data not shown). Thus, these observations also suggest that the effect of reduced eIF2α phosphorylation may offset the effect of reduced eIF4E phosphorylation during NS5A expression. It therefore appears that NS5A can functionally replace E3L as an inhibitor of PKR and restore global protein synthesis during virus infection.
The ability of NS5A to interact with Grb2 is not required for restoration of protein synthesis during virus infection.
We previously showed that NS5A also binds Grb2, an adapter protein that mediates signaling by nucleating the formation of signal transduction complexes in response to growth factor stimulation. An effect of NS5A interaction with Grb2 is an inhibition of downstream MAPK activation by EGF (51). A proline-rich motif present in the C-terminal region of NS5A is required for Grb2 binding and ERK1/2 inhibition (51; H. Nakao et al., unpublished data). Because Grb2 also mediates signaling to the p38 pathway, we cautioned that the observed protein synthesis restoration by NS5A might also involve downregulation of p38 via the interaction of NS5A with Grb2. To test this, we examined the ability of a recombinant VV that expresses a NS5A variant containing point mutations within the proline-rich motif (VVPro3) to restore global protein synthesis during virus infection. As shown in Fig. 5, expression of the Grb2 binding-deficient NS5A protein restored mRNA translation to an extent similar to that of wild-type NS5A, as confirmed by quantification of protein translation levels as described above (data not shown). Thus, the rescue is not dependent on Grb2-mediated pathways in this system but is likely due to NS5A-mediated inhibition of PKR.
FIG. 5.

NS5A-mediated reversal of the translation arrest phenotype observed in VVΔE3L-infected cells is independent of its ability to bind Grb2. Analyses of global protein synthesis in HeLa S3 cells pretreated with IFN-α/β and infected with a recombinant VV expressing a Grb2 binding-defective form of NS5A (VVPro3) were performed as described in the legend to Fig. 4. WT, wild type; p.i., postinfection.
DISCUSSION
In the present report, we used a recombinant VV-based assay to study the PKR inhibitory effects of NS5A within the context of virus infection for the first time. In the absence of a robust HCV cell culture infection system, it is necessary to use surrogate systems such as the vaccinia system described herein. Consistent with the notion that E3L is a potent inhibitor of PKR, infection of IFN-treated cells with VVΔE3L resulted in increased phosphorylation of PKR and eIF2α and a marked arrest in protein synthesis initiation (Fig. 1 and 4). Importantly, expression of NS5A in VVNS5A-infected cells decreased the phosphorylation of PKR and eIF2α and alleviated the translational block in a partial and transient manner. Stimulation of protein synthesis by NS5A was likely directly caused by inhibition of the PKR pathway rather than the Grb2 pathway (51; H. Nakao et al., unpublished) since the PxxP Grb2 binding-deficient NS5A mutant stimulated protein synthesis to the same degree as the wild type. We are currently constructing a VV containing a PKR binding-deficient NS5A mutant to prove this point unequivocally. Despite the need for this and other mutants to pinpoint precise mechanisms, we should stress that we have utilized four separate VV in the present study to provide the necessary controls and to prove that the effects are specific to the HCV NS5A. In all experiments, we compared the results to both wild-type and isogenic VV lacking the E3L gene. We also included an additional virus containing NS5A with the described proline mutations. It is also relevant to note that our present results build upon previous reports from our laboratory and others, which have detailed the PKR inhibition by wild-type and mutant NS5A in a “nonviral” setting (18, 19, 20). These data are also consistent with reports from several independent laboratories that have shown that NS5A, expressed in trans, can confer IFN resistance to otherwise IFN-sensitive viruses (39, 50).
Perhaps most importantly, our results highlight a previously unknown effect of NS5A: the inhibition of p38 activation during VV infection or EGF stimulation (Fig. 2 and 3). This is supportive of previous reports that p38 lies downstream of the PKR signaling pathway (24, 29, 59) and our previous finding that NS5A can perturb the MAPK pathway by targeting Grb2 (51; H. Nakao et al., unpublished). Relevant to translational regulation, p38 phosphorylates Mnk1 protein kinase, which in turn phosphorylates the translation initiation factor eIF4E (48, 49). Indeed we can now demonstrate that reductions in p38 activity caused by HCV NS5A also decreased 4E phosphorylation levels, perhaps having a negative impact on cap-dependent mRNA translation. Curiously, however, p38 is best known for its role in the stress-activated transcriptional regulatory pathways (35). At this point one might only speculate that HCV encodes a mechanism to downregulate this pathway to enhance and/or sustain infection and negate the ability of the infected cell to properly respond to stress-mediated signaling.
One unanswered question relates to why only a transient and partial restoration of cellular and viral protein synthesis by NS5A occurs. There is always the possibility that the lack of total rescue of protein synthesis late after infection was simply due to the massive activation of PKR and resultant eIF2α phosphorylation. This suggests that NS5A simply cannot fully compensate for the loss of E3L. The latter could be due to the inherently different properties of E3L and NS5A. In fact, the VV system mainly has been used to study only dsRNA-binding inhibitors, including the reovirus S4 protein (3), the porcine group C rotavirus NSP3 protein (31), the Escherichia coli RNase III protein (47), and the cellular TAR RNA-binding protein (38). In this regard, it is interesting that NS5A, which has not been shown to possess dsRNA-binding activity, is capable of downregulating PKR in the VV system. E3L, by virtue of its ability to bind dsRNA, can probably inhibit other PKR-independent IFN-stimulated dsRNA-dependent pathways (4). Indeed, a recent study showed that VV E3L could function as an inhibitor of the IFN-induced 2-5A synthetase enzyme (43). Thus, it may not be surprising that NS5A may not fully substitute for E3L.
We rather favor an alternative explanation for the incomplete restoration of mRNA translation by NS5A. We suggest that NS5A may exert counteracting effects on mRNA translation during viral infection. While NS5A-mediated inhibition of PKR could function to ensure the availability of functional eIF2α to maintain global mRNA translation, it could also lead to dramatic decreases in eIF4E phosphorylation and function, thereby incapacitating cap-dependent translation of cellular mRNAs. Since translation of HCV mRNA occurs via an internal ribosomal entry site-dependent, cap-independent mechanism (55, 58), it is tempting to speculate that NS5A-mediated disruption of the PKR-p38-Mnk1-eIF4E pathway during HCV infection may ultimately favor the translation of viral mRNAs over host mRNAs and therefore contribute to the replication and pathogenesis of HCV. This scenario is reminiscent of the strategies utilized by poliovirus that encodes dual mechanisms to inhibit PKR activity (6, 7, 21, 22) and disrupts cap-dependent translation (56). Indeed, eukaryotic viruses in general display an impressive array of strategies to ensure the efficient and selective translation of viral mRNAs (for a comprehensive and recent review see reference 22). Unfortunately, complete examination of translational regulatory strategies employed by HCV awaits the development of an in vitro infection system.
ACKNOWLEDGMENTS
We are grateful to Teri Shors and Rick Ferguson for technical support. We also thank Marcus J. Korth for editorial assistance, Michael J. Gale, Jr., for helpful discussion, and Haruhisa Nakao for the stable NS5A-expressing HeLa cell lines.
This work was supported by grants from the National Institutes of Health (AI-22646, RR-00166, and AI-41629) to M.G.K.
REFERENCES
- 1.Alter M J H. Epidemiology of hepatitis C. Hepatology. 1997;26:62–65. doi: 10.1002/hep.510260711. [DOI] [PubMed] [Google Scholar]
- 2.Barber G N, Tomita J, Garfinkel M S, Meurs E, Hovanessian A, Katze M G. Detection of protein kinase homologues and viral RNA-binding domains utilizing polyclonal antiserum prepared against a baculovirus-expressed dsRNA-activated 68,000-Da protein kinase. Virology. 1992;191:670–679. doi: 10.1016/0042-6822(92)90242-h. [DOI] [PubMed] [Google Scholar]
- 3.Beattie E, Denzler K L, Tartaglia J, Perkus M E, Paoletti E, Jacobs B L. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol. 1995;69:499–505. doi: 10.1128/jvi.69.1.499-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beattie E, Paoletti E, Tartaglia J. Distinct patterns of IFN sensitivity observed in cells infected with vaccinia K3L− and E3L− mutant viruses. Virology. 1995;210:254–263. doi: 10.1006/viro.1995.1342. [DOI] [PubMed] [Google Scholar]
- 5.Beattie E, Tartaglia J, Paoletti E. Vaccinia virus-encoded eIF-2α homolog abrogates the antiviral effect of interferon. Virology. 1991;183:419–422. doi: 10.1016/0042-6822(91)90158-8. [DOI] [PubMed] [Google Scholar]
- 6.Black T L, Barber G N, Katze M G. Degradation of the interferon-induced 68,000-M(r) protein kinase by poliovirus requires RNA. J Virol. 1993;67:791–800. doi: 10.1128/jvi.67.2.791-800.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black T L, Safer B, Hovanessian A, Katze M G. The cellular 68,000-Mr protein kinase is highly autophosphorylated and activated yet significantly degraded during poliovirus infection: implications for translational regulation. J Virol. 1989;63:2244–2251. doi: 10.1128/jvi.63.5.2244-2251.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang H W, Watson J C, Jacobs B L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci USA. 1992;89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang H W, Uribe L H, Jacobs B L. Rescue of vaccinia virus lacking the E3L gene by mutant of E3L. J Virol. 1995;69:6605–6608. doi: 10.1128/jvi.69.10.6605-6608.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chardin P, Cussac D, Maignan S, Ducruix A. The Grb2 adaptor. FEBS Lett. 1995;369:47–51. doi: 10.1016/0014-5793(95)00578-w. [DOI] [PubMed] [Google Scholar]
- 11.Clemens M J. Protein kinases that phosphorylate eIF2 and eIF2B, and their role in eukaryotic cell translational control. In: Hershey J W B, Mathews M B, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 139–172. [Google Scholar]
- 12.Dever T E. Translation initiation: adept at adapting. Trends Biochem Sci. 1999;24:398–403. doi: 10.1016/s0968-0004(99)01457-7. [DOI] [PubMed] [Google Scholar]
- 13.Downward J. The GRB2/Sem-5 adaptor protein. FEBS Lett. 1994;338:113–117. doi: 10.1016/0014-5793(94)80346-3. [DOI] [PubMed] [Google Scholar]
- 14.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Maruno F, Sato C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 15.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murankami T, Yamamoto C, Izumi N, Marumo F, Sato C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b. J Clin Investig. 1995;96:224–230. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francois C, Duverlie G, Rebouillat D, Khorsi H, Castelain S, Blum H E, Gatignol A, Wychowski C, Moradpour D, Meurs E F. Expression of hepatitis C virus proteins interferes with the antiviral action of interferon independently of PKR-mediated control of protein synthesis. J Virol. 2000;74:5587–5596. doi: 10.1128/jvi.74.12.5587-5596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galabru J, Hovanessian A G. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 18.Gale M, Jr, Korth M J, Tang N M, Tan S L, Hopkins D A, Dever T E, Polyak S J, Gretch D R, Katze M G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 19.Gale M, Jr, Blakely C M, Kwieciszewski B, Tan S L, Dossett M, Korth M J, Polyak S J, Gretch D R, Katze M G. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gale M, Jr, Kwieciszewski B, Dossett M, Nakao H, Katze M G. Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J Virol. 1999;73:6506–6516. doi: 10.1128/jvi.73.8.6506-6516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gale M, Jr, Katze M G. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 22.Gale M, Jr, Tan S L, Katze M G. Translational control of viral gene expression in eukaryotes. Microbiol Mol Biol Rev. 2000;64:239–280. doi: 10.1128/mmbr.64.2.239-280.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrington T P, Johnson G L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 24.Goh K C, deVeer M J, Williams B R. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000;19:4292–4297. doi: 10.1093/emboj/19.16.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herion D, Hoofnagle J H. The interferon sensitivity determining region: all hepatitis C virus isolates are not the same. Hepatology. 1997;25:769–771. doi: 10.1002/hep.510250346. [DOI] [PubMed] [Google Scholar]
- 26.Hoofnagle J H. Therapy of acute and chronic viral hepatitis. Adv Intern Med. 1994;39:241–275. [PubMed] [Google Scholar]
- 27.Hovanessian A G. The double stranded RNA-activated protein kinase induced by interferon: dsRNA-PK. J Interferon Res. 1989;9:641–647. doi: 10.1089/jir.1989.9.641. [DOI] [PubMed] [Google Scholar]
- 28.Ihle J N. STATs and MAPKs: obligate or opportunistic partners in signaling. Bioessays. 1996;18:95–98. doi: 10.1002/bies.950180204. [DOI] [PubMed] [Google Scholar]
- 29.Iordanov M S, Paranjape J M, Zhou A M, Wong J, Williams B R G, Meurs E F, Silverman R H, Magun B E. Activation of p38 mitogen-activated protein kinase and c-Jun NH2-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R, and alternative pathways. Mol Cell Biol. 2000;20:617–627. doi: 10.1128/mcb.20.2.617-627.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kibler K V, Shors T, Perkins K B, Zeman C C, Banaszak M P, Biesterfeldt J, Langland J O, Jacobs B L. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol. 1997;71:1992–2003. doi: 10.1128/jvi.71.3.1992-2003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langland J O, Pettiford S, Jiang B, Jacobs B L. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon-induced protein kinase PKR. J Virol. 1994;68:3821–3829. doi: 10.1128/jvi.68.6.3821-3829.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leaman D W, Leung S, Li X, Stark G R. Regulation of STAT-dependent pathways by growth factors and cytokines. FASEB J. 1996;10:1578–1588. [PubMed] [Google Scholar]
- 33.Lindsay K L. Therapy of hepatitis C: overview. Hepatology. 1997;26:71S–77S. doi: 10.1002/hep.510260713. [DOI] [PubMed] [Google Scholar]
- 34.Moradpour D, Blum H E. Current and evolving therapies for hepatitis C. Eur J Gastroenterol Hepatol. 1999;11:1199–1202. doi: 10.1097/00042737-199911000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Obata T, Brown G E, Yaffe M B. MAP kinase pathways activated by stress: the p38 MAPK pathway. Crit Care Med. 2000;28:N67–N77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- 36.Okuda K. Hepatitis C and hepatocellular carcinoma. J Gastroenterol Hepatol. 1998;13:294–298. doi: 10.1111/j.1440-1746.1998.tb01896.x. [DOI] [PubMed] [Google Scholar]
- 37.Paez E, Esteban M. Resistance of vaccinia virus to interferon is related to an interference phenomenon between the virus and the interferon system. Virology. 1984;134:12–28. doi: 10.1016/0042-6822(84)90268-x. [DOI] [PubMed] [Google Scholar]
- 38.Park H, Davies M V, Langland J O, Chang H W, Nam Y S, Tartaglia J, Paoletti E, Jacobs B L, Kaufman R J, Venkatesan S. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci USA. 1994;91:4713–4717. doi: 10.1073/pnas.91.11.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polyak S J, Paschal D M, Mcardle S, Gale M J, Jr, Moradpour D, Gretch D R. Characterization of the effects of hepatitis C virus nonstructural 5A protein expression in human cell lines and on interferon-sensitive virus replication. Hepatology. 1999;29:1262–1271. doi: 10.1002/hep.510290438. [DOI] [PubMed] [Google Scholar]
- 40.Reed K E, Rice C M. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 41.Reed K E, Xu J, Rice C M. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J Virol. 1997;71:7187–7197. doi: 10.1128/jvi.71.10.7187-7197.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice A P, Kerr I M. Interferon-mediated, double-stranded RNA-dependent protein kinase is inhibited in extracts from vaccinia virus-infected cells. J Virol. 1984;50:229–236. doi: 10.1128/jvi.50.1.229-236.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rice A P, Roberts W K, Kerr I M. 2-5A accumulates to high levels in interferon-treated, vaccinia virus-infected cells in the absence of any inhibition of virus replication. J Virol. 1984;50:220–228. doi: 10.1128/jvi.50.1.220-228.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivas C, Gil J, Melkova Z, Esteban M, Diaz-Guerra M. Vaccinia virus E3L protein is an inhibitor of the interferon (IFN)-induced 2-5A synthetase enzyme. Virology. 1998;243:406–414. doi: 10.1006/viro.1998.9072. [DOI] [PubMed] [Google Scholar]
- 45.Romano P R, Green S R, Barber G N, Mathews M B, Hinnebusch A G. Structural requirements for double-stranded RNA binding, dimerization, and activation of the human eIF-2 alpha kinase DAI in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:365–378. doi: 10.1128/mcb.15.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romano P R, Zhang F, Tan S L, Garcia-Barrio M T, Katze M G, Dever T E, Hinnebusch A G. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol Cell Biol. 1998;18:7304–7316. doi: 10.1128/mcb.18.12.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shors T, Jacobs B L. Complementation of deletion of the vaccinia virus E3L gene by the Escherichia coli RNase III gene. Virology. 1997;227:77–87. doi: 10.1006/viro.1996.8319. [DOI] [PubMed] [Google Scholar]
- 48.Sonenberg N. mRNA 5′ cap-binding protein eIF4E and control of cell growth. In: Hershey J, Mathews M, Sonenberg N, editors. Translational control. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 245–270. [Google Scholar]
- 49.Sonenberg N, Gingras A C. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol. 1998;10:268–275. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- 50.Song J, Fujii M, Wang F, Itoh M, Hotta H. The NS5A protein of hepatitis C virus partially inhibits the antiviral activity of interferon. J Gen Virol. 1999;80:879–886. doi: 10.1099/0022-1317-80-4-879. [DOI] [PubMed] [Google Scholar]
- 51.Tan S L, Nakao H, He Y, Vijaysri V, Neddermann P, Jacobs B L, Mayer B J, Katze M G. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc Natl Acad Sci USA. 1999;96:5533–5538. doi: 10.1073/pnas.96.10.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanji Y, Kaneko T, Satoh S, Shimotohno K. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J Virol. 1995;69:3980–3986. doi: 10.1128/jvi.69.7.3980-3986.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tartaglia J, Perkus M E, Taylor J, Norton E K, Audonnet J C, Cox W I, Davis S W, van der Hoeven J, Meignier B, Riviere M, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188:217–232. doi: 10.1016/0042-6822(92)90752-b. [DOI] [PubMed] [Google Scholar]
- 54.Taylor D R, Shi S T, Romano P R, Barber G N, Lai M M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 55.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ventoso I, MacMillan S E, Hershey J W, Carrasco L. Poliovirus 2A proteinase cleaves directly the eIF-4G subunit of eIF-4F complex. FEBS Lett. 1998;435:79–83. doi: 10.1016/s0014-5793(98)01027-8. [DOI] [PubMed] [Google Scholar]
- 57.Vojtek A B, Der C J. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:9925–9928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 58.Wang C, Sarnow P, Siddiqui A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J Virol. 1993;67:3338–3344. doi: 10.1128/jvi.67.6.3338-3344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams B R G. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]