

Abstract
Aims:
One unaddressed aspect of healing after myocardial infarction (MI) is how non-myocyte cells that survived the ischemic injury, keep withstanding additional cellular damage by stress forms typically arising during the post-infarction inflammation. Here we aimed to determine if cell survival is conferred by expression of a mitochondrial protein novel to the cardiac proteome, known as steroidogenic acute regulatory protein, (StAR/STARD1). Further studies aimed to unravel the regulation and role of the non-steroidogenic cardiac StAR after MI.
Methods and results:
Following permanent ligation of the left anterior descending coronary artery in mouse heart, timeline western blot analyses showed that StAR expression corresponds to the inflammatory response to MI. Following the identification of StAR in mitochondria of cardiac fibroblasts in culture, confocal microscopy immunohistochemistry (IHC) identified StAR expression in left ventricular (LV) activated interstitial fibroblasts, adventitial fibroblasts and endothelial cells. Further work with the primary fibroblasts model revealed that interleukin-1α (IL-1α) signaling via NF-κB and p38 MAPK pathways efficiently upregulates the expression of the Star gene products. At the functional level, IL-1α primed fibroblasts were protected against apoptosis when exposed to cisplatin mimicry of in vivo apoptotic stress; yet, the protective impact of IL-1α was lost upon siRNA mediated StAR downregulation. At the physiological level, StAR expression was nullified during post-MI inflammation in a mouse model with global IL-1α deficiency, concomitantly resulting in a 4-fold elevation of apoptotic fibroblasts. Serial echocardiography and IHC studies of mice examined 24 days after MI revealed aggravation of LV dysfunction, LV dilatation, anterior wall thinning and adverse tissue remodeling when compared with loxP control hearts.
Conclusions:
This study calls attention to overlooked aspects of cellular responses evolved under the stress conditions associated with the default inflammatory response to MI. Our observations suggest that LV IL-1α is cardioprotective, and at least one mechanism of this action is mediated by induction of StAR expression in border zone fibroblasts, which renders them apoptosis resistant. This acquired survival feature also has long-term ramifications on the heart recovery by diminishing adverse remodeling and improving the heart function after MI.
Keywords: Myocardial infarction, IL-1α, Cardiac fibroblasts, Apoptosis, StAR, Cardiac remodeling
1. Introduction
The first step toward healing after myocardial infarction (MI) is activation of the innate/sterile inflammatory response [1]. The predominant, yet not exclusive, trigger of the inflammatory response is the proinflammatory interleukin-1α (IL-1α) [2,3]. A result of the infarction ischemia is a passive release of the cytoplasmic and nuclear IL-1α content [4] from cardiomyocytes and possibly other cell types undergoing necrotic lysis in the injured tissue [2,3]. Also, IL-1α is only proximal to the MI-related release of additional intracellular substances and extracellular matrix (ECM) degradation products that can trigger the cardiac immune response, collectively called danger associated molecular patterns (DAMPs), or alarmins [1,5]. Immediate cell targets of IL-1α and DAMPs are resident interstitial fibroblasts in the infarct border zone that were spared during the ischemic demise; IL-1α binding to these cells results in a massive synthesis and secretion of more proinflammatory cytokines and chemokines, such as IL-6 and CCL2/MCP-1, which ignite the post-MI inflammation by recruiting immune cells to the site of the injury [1,6–8]. Concomitantly, resident fibroblasts of the infarct border zone are subjected to an additional mode of activation via the mechanical tension generated by loss of tissue volume due the lytic collapse of the necrotic cardiomyocytes. This stress dependent activation of the fibroblasts leads to proliferation, periostin (POSTN) expression, and early differentiation to smooth muscle α-actin (SMA) bearing myofibroblasts, which rapidly repopulate the infarct vacant space [9–11]. Finally, upon resolution of the inflammatory phase, fully differentiated myofibroblasts shape the nature of cardiac remodeling by deposition of collagen type I and other ECM components, so that the resulting scar tissue prevents left ventricular wall rupture [12–14].
An unaddressed aspect of the heart response to MI is the question how the border zone fibroblasts endure through the inflammatory response, when the injured tissue is infiltrated by neutrophils and proinflammatory monocytes/macrophages that release reactive oxygen species (ROS) required for their clearing activities [5,13,15]. We recently revealed that the post-MI injured left ventricular (LV) tissue expresses a protein new to the cardiac protein landscape, the steroidogenic acute regulatory protein, StAR [16]. The working hypothesis underlying the present study aimed to examine if StAR supports cell survival after MI. Originally, StAR discovery [17,18] unveiled a mechanism by which StAR activity provides cholesterol transfer to the inner membrane (IMM) of steroidogenic mitochondria, where the sterol serves as substrate for the synthesis of the first steroid by the IMM mitochondrial P450 enzyme complex of CYP11A1 [19]. In steroidogenic cells of the adrenal cortex and the gonads, the nuclear encoded Star gene is upregulated by trophic hormones and the cAMP signaling pathway [20,21]. In the heart, however, StAR expression cannot support steroidogenesis since CYP11A1 and 3βHSD [19,22], the two obligatory enzymes required for de novo steroid hormone synthesis from cholesterol, are missing [16,23]. We therefore hypothesized that cardiac StAR should have a new alternative activity to be harnessed for a non-steroidogenic function in the heart.
To identify the cell types that express StAR, we applied antibodies to cardiomyocytes-specific troponin I, to fibroblast-specific markers such as platelet derived growth factor receptor-α (PDGFRα) and periostin (POSTN), and endothelial cell marker, CD31/PECAM1 [24]. PDGFRα is required for the generation of the interstitial fibroblast population during development, and remains essential for cardiac fibroblast survival in adult mice [11,25,26]. Periostin is a 90 kDa hetero-functional matricellular protein [27,28] previously found instrumental for cardiac healing after MI [29,30].
Our in vitro experiments with cardiac fibroblasts revealed that IL-1α signaling is pivotal for StAR expression and function. Interleukin-1α, −1α, −33 and IL-18 comprise the leading members of the IL-1 gene family [31]. Interestingly, the modes of activation and release of IL-1α and IL-1β are profoundly different; in contrast to the passive release of IL-1α, secretion of IL-1β is highly regulated in the context of the innate and adaptive immune responses [32–35]. What unifies the two cytokines is their independent binding to a mutual receptor type I (IL-1R1) that in turn recruits a receptor accessory protein (IL-1R3) in order to activate downstream intracellular signaling. Relevant to this study are the two arms of the IL-1R1 signaling patterns, culminating in gene activation by NF-κB/AP-1 pathways, and activation of MAP kinase pathways (p38, JNK, ERK) [8,34,36,37].
Finally, we also examined the physiological impact of IL-1α absence on the recovery outcomes after MI in a global IL-1α deficient mouse model, which turned out to agree with the predicted centrality of IL-1α, and possibly StAR, in cardiac repair and remodeling after MI.
2. Materials and methods
For additional methods, see Supplemental Materials and Methods online.
2.1. Animals
BALB/c females (11–12 weeks old, weight 25–30 g) were used to study the wild-type mouse response to MI, and Sprague-Dawley male rats (200–250 g) were used for preparation of high yield cardiac fibroblast cultures; both rodents were obtained from Harlan Laboratories (Jerusalem, Israel) and maintained under a schedule of 12 h light, 12 h dark with food and water ad libitum. Animals were treated in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All experimentation on mice followed the appropriate analgesic and anesthesia guidelines and was approved by the Institutional Committee on Animal Care and Use (IACUC), The Alexander Silverman Institute of Life Sciences, The Hebrew University of Jerusalem. The Hebrew University is AAALAC approved. IL-1αKO: Use of global IL-1α knockout mice (IL-1αKO) was described previously [38–40]. Briefly, floxed IL-1αloxP mice on a C57BL/6NTac genetic background were generated by the Taconic Artemis Company, (Cologne, Germany) by introducing loxP sites flanking the coding exons 2–5 of the Il-1α gene. These mice were further crossed with commercially available Gt(ROSA)26Sortm16(cre)Arte mice that constitutively and ubiquitously express the Cre recombinase transgene under the control of the ROSA26 locus [41]. Deletion of Il1α by Cre induced excision of exons 2–5 was validated by PCR (Supplemental Fig. S8C–E). These mice do not exhibit evidence of spontaneous carcinogenesis, and their lifespan appears normal [32]. Mice were bred and kept at the Animal Facility of the Sheba Medical Center, Tel Hashomer, Israel. Animal care and studies were approved by the Institutional Committee on Animal Care and Use (IACUC) of the Sheba Medical Center.
2.2. Experimental myocardial infarction-
Based on the previously described MI procedure [16,42], 12 week old mice (BALB/c WT-females; C57Bl/6 IL-1αKO; C57Bl/6 loxP control males, 25–30 g) were anesthetized by inhalation of 2% isoflurane/ 98% O2. The isoflurane flow was monitored to maintain the heart rate at >400 bpm. For MI, the left coronary artery was permanently ligated at the lower border of the left atrium. Sham animals (control) were subjected to a similar surgery, passing the thread without ligating the artery. Artery occlusion was validated by visual bleaching distal to the occlusion site and, more importantly, by echocardiography 72 h after MI (Supplemental Table S1 online) using a small animal echocardiography system (Vevo 2100 Imaging System; VisualSonics) equipped with a 22 to 55-MHz linear-array transducer (MS550D MicroScan Transducer). Functional and remodeling echocardiography measurements were taken 7 days before surgery (baseline), and on d3 and d24 post-MI. To reduce the animal number to a minimum, data of sham animals were combined from both loxP and IL-1αKO, which showed no differences in baseline phenotype. The investigator performing the echocardiography measurements was blind to the genotype of the animals.
Use of C57Bl/6 males is widely accepted for work with genetically manipulated models. However, wild-type BALB/c females were the preferable model for the study of IL-1α/StAR roles after MI due to lower rate of infarct rupture and better cardiac remodeling compared with males and other strains of mice [43]. Nevertheless, our findings suggest that despite the use of different mouse strains and sex, the IL-1αKO studies on C57Bl/6 males background adequately addressed the central hypothesis of this study, owing to the use of loxP males to represent the response of normal C57Bl/6 mice to MI.
2.3. Primary cell cultures and cytokine treatments
Primary adult rat cardiac fibroblasts.
Cardiac fibroblasts were isolated from Sprague-Dawley rats (200–250 g males). The rationale for selecting the rat heart for preparation of high-crop primary fibroblast culture is detailed in Supplemental Results). To this aim, minced rat heart (~1 g) was washed in cold PBS/ 20 mM glucose and dissociated in Dulbecco’s Modified Eagle Medium (DMEM) containing collagenase-dispase (Sigma Aldrich 10,269,638,001), DNase I (Sigma-Aldrich D5025) and BSA (Sigma-Aldrich A4919) mixture of 3 mg/ml, 0.1 mg/ml and 10 mg/ml, respectively [16]. A typical yield of dissociated fibroblasts from one heart provides 107 live cells seeded onto forty 35 mm tissue culture wells (9.6 cm2, or proportionate equivalents of other well sizes), thereafter yielding 3–6 × 104 cells/cm2 after 5 and 6 days of growth, respectively. For additional protocol details, as well as description of agonist treatments and preparation of MEF fibroblast cultures, see Supplemental Materials and Methods online. Preparation of primary neonate rat cardiac cell culture was described before [16].
2.4. Western analyses, immunohisto- and immunocytochemistry (IHC, ICC)
The preparations of tissues and cells for western blot analyses and confocal microscopy immuno-detection of StAR [44] and other antigens are detailed in Supplemental Materials and Methods online.
2.5. Quantitative real-time PCR
See details in Supplemental Materials and Methods online.
2.6. Apoptosis assays
In this study we used several assays of apoptosis in cultured cells and in tissue sections, including: nuclear fragmentation [45], TUNEL, time-lapse morphological apoptosis, MTT viability assay, determination of cleaved caspase 9 levels and apoptosis index in vivo (see Supplemental Materials and Methods online).
2.7. RNAi
For expression knockdown by siRNA, primary cardiac fibroblasts were plated in 6-well tissue culture plates, and on day 4 the cells were transfected with 50 pmol of either negative control siRNA (siControl) or siStAR (Invitrogen) using Lipofectamine RNAiMAX transfection reagent (Invitrogen 13,778,075) according to the manufacturer’s instructions. The indicated treatments commenced 24 h later.
2.8. Statistical analyses
GraphPad Prism 7 and SPSS 25 software programs were used to test statistical significance. Data are presented as mean ± the standard error of the mean (SEM) of multiple (n ≥ 3) independent experiments. In vivo loxPloxP vs. IL-1αKO histograms are presented as mean ± 95% confidence interval (CI95) of n ≥ 3 mice from each group. Histograms depicting in vivo wild-type data are presented as mean ± SEM of n = 3 mice from each group. The same software programs were used to assess statistical significance and to run the Shapiro-Wilk normality test. Unpaired t-tests were used to compare two groups. Multiple group comparisons were done by ANOVA using Holm-Sidak’s posttest. Where indicated, a non-parametric Kruskal-Wallis using Dunn’s posttest was performed. Where relevant, mean values were compared using the Pearson correlation test. To analyze changes in echocardiography parameters of left ventricular (LV) remodeling and function over time, we used general linear model 2-way repeated-measures ANOVA, followed by Bonferroni posttest. Data were considered statistically significant when P < 0.05.
3. Results
3.1. Identification of StAR expressing cells
Since StAR expression has been observed in cultured adult and neonate rat cardiac fibroblasts (Supplemental Fig. S4B and S4D, respectively), we aimed to map StAR expression in the mouse heart shown before to express StAR after MI [16]. Transverse paraffin sections taken three days after MI from below the point of the left coronary artery occlusion, were examined by confocal microscopy. Unlike the unharmed myocardium of a sham-operated heart (Fig. 1A1), loss of troponin I content after MI provided visual evidence of the muscle injury that extends thoughout the thinning LV free wall (Fig. 1A2–3(. Yet, the injury pattern reveals three layers of the myocardium that remained viable, namely, two strips of subepicardium and sub-endocardium longitudinal myocytes (transversely sectioned), and core bundles of circumferential myocytes (Fig. 1A2–2a). Hence, the classical border zone (BZ) terminology [46] applies here to any interface between the viable myocytes and the dead ones throughout the entire length of the LV free wall infarct zone (IZ).
Fig. 1.

Immunohistochemical co-expression of StAR and fibroblast cell markers. Transverse heart sections were prepared on day 3 post-MI (12 weeks old BALB/c WT-females) and cellular distribution of StAR and fibroblast cell markers was studied by confocal microscopy. A1, Whole heart orientation image of sham operated heart, transversely sectioned and post-stain studied by scanning and image tiling. Note normal troponin I (Tropo) content and intact thickness of the left ventricle (LV) free wall. RV, right ventricle; Sep, septum. A 2–3, Orientation sections taken on d3 after MI, stained with either troponin and PDGFRα (PDGFR), or troponin and POSTN, respectively. Note a massive loss of troponin in the necrotic infarct zone (IZ) extending anterior-to-posterior throughout the entire LV free wall. Yet, as also shown in A2a, typical viable myocytes are noted in the sub-epicardium, sub-endocardium (both are longitudinal myocytes, lm, arrowheads) and a core strip of circumferential myocytes (arrows, cm), as well as sub-epicardium and sub-endocardium longitudinal cardiomyocytes (arrowheads, lm) you already mentioned lm earlier in the sentence. Other than (this is usually used to mean “except”, is that what you meant? Or did you mean “in addition to”?) the classically defined border zones [46] at the flanks of the injured LV (BZ in A3), our observations are consistent with a BZ definition anywhere in the LV where covering the interface between viable myocytes and necrotic infarct zone that is typically invaded by PDGFR+/POSTN+ activated fibroblasts (B-E series) and immune cells (Supplemental Fig. S3A2, B2). B, Troponin I (artificial white) and PDGFRα (PDGFR) staining of a typical border zone (BZ) region of interest (ROI) at the posterior side of the LV free wall. V, sub-endocardial vein. Ba-b, Higher magnification border zone ROI in two consecutive sections (10 mm apart) depicting elongated interstitial fibroblasts (color matched arrowheads) expressing both PDGFRα and StAR. m, troponin-emptied necrotic circumferential myocytes. Nuclear DAPI staining is blue in all images. This image series represents similar observations made in 2 additional hearts. @, staining artifact. C (ROI localization depicted in A3), Posterior sub-epicardial border zone (BZ) depicting the interface of viable, transversely sectioned myocytes (*), and troponin-emptied infarct zone (IZ) containing multiple activated fibroblasts noted by StAR and POSTN co-expression (Da-b, color matched arrows). Image Dc depicts a single cell example demonstrating StAR containing mitochondria and a distinct exstracellular localization of the matricellular POSTN protein. *, Intact troponin-stained, or lysed cardiomyocytes. DAPI staining denotes nuclei in all images. E1-4 (ROI localization depicted in A2-3), Consecutive sections depicting anterior border zone where the infarct injury (IZ) initiated. Massive StAR staining is noted in the epicardium cells (C2, short arrows) that also express PDGFRα (C3) and POSTN (C4). Arrowheads depict interstitial fibroblasts in between the sub-epicardium troponin+ longitudinal myocytes, and elongated fibroblasts in between necrotic myocytes (arrowheads). F1-4 (ROI localization depicted in A3), StAR is expressed in endocardial epithelial cell (short arrows in F2) that also co-express POSTN (F3, consecutive section to F1-2) and PDGFRα (F4). The three proteins are also expressed in the sub-endocardium border zone (BZ) and infarct zone (IZ) fibroblasts (arrowheads). *, Asterisks denote cardiomyocyte sectioned transversely or in the sagittal plane. G, shown are activated replicating fibroblasts (POSTN-positive) noted by Ki67 labeled nuclei (arrowheads) in between border zone (BZ) myocytes (*). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Elongated interstitial fibroblasts are readily noted in the border zone of the core myocytes at the posterior end of the infarct zone, where high magnification images show that StAR is co-expressed with the fibroblast marker PDGFRα (Fig. 1Ba–b). Despite the fact that fibroblasts have recently been shown to start expressing smooth muscle α-actin (αSMA) as early as post-MI d3 [9,47], in this study we prefer to adhere to the generalized term ‘fibroblasts’, a choice reasonably supported by the observation that primary heart fibroblasts are highly capapble of StAR expression prior to their spontaneous differentiation into myofibroblasts in culture (Supplemental Results and Fig. S4B1–5).
Similar characteristics also applied to fibroblasts in the posterior subepicardial border zone tissue that did not succumb to necrosis. In this instant, we also applied antiserum periostin (POSTN, Fig. 1A3) a known marker of activated fibroblasts and central for tissue remodeling [48]. Border zone POSTN+ fibroblasts are scattered in between sub-epicardium transversely sectioned viable myocytes, and also invade the infarct zone (Fig. 1C). Higher magnification revealed the colocalization of POSTN and StAR in the very same cells (Fig. 1D, Da–b), while the distinct mitochondrial localization of StAR and the extracellular localization of POSTN are demonstratable in some of the cells (Fig. 1D, 1Dc). Other than interstitial fibroblasts, the confocal IHC appraoach revealed additional minor populations of StAR-expressing cell types of potential physiological interest, including: (i) the anterior thickened cell layers of the epicardium (Fig. 1E2–4), and cells of the endocardial epithelium (Fig. 1F2–4), all of which express both PDGFRα and POSTN as well; (ii) StAR+:PDGFR+ fibroblasts that do not seem to become activated and do not express POSTN (Supplemental Fig. S1Da–c); (iii) adventitial fibroblasts in small arteries of the damaged LV (Supplemental Fig. S1Da–c and S1C2–3); (iv) StAR is expressed in endothelial cells of veins and arteries in the damaged LV, but not the vessels’ smooth muscle cells (Supplemental Fig. S1A–C); (v) interestingly, StAR+ adventitial fibroblasts and endothelial cells are noted in arteires of the RV septum, as well as the RV endocardial epithelium (Supplemental Fig. S1F1–3 and S1F1,4, respectively). The respective cell types in the RV free wall were devoid of StAR (Supplemental Fig. S1G); (vi) Understandably, StAR expression did not rise in sham operated hearts (Supplemental Fig. S2Fb), whereas POSTN is sensitive enough in its response to the minor myocardium injury to be seen localized close to the point of the needle passing (Supplemental Fig. S2E, Fa). Finally, as expected, no POSTN staining could be observed in MI operated hearts of POSTN deficient mice [29] (data not shown).
To elucidate the exact timing of StAR expression following MI, we conducted time-dependent western blot analysis of the injured LV StAR content (Fig. 2A1–3). The immediate rise of StAR attaining a peak level on day 3 and fading out by day 14 after MI, suggested a concurrent expression during the inflammatory response. Additional blots of the same tissue extracts also revealed the expression pattern of POSTN and proliferating cell nuclear antigen (PCNA) reporting the status of cell proliferation (Fig. 2B1–2). The relative dynamic changes of StAR, POSTN and PCNA could be compared after normalization as shown in Fig. 2C. First, none of the three proteins were expressed without the MI trigger. As expected, MI triggers immediate fibroblast proliferation (Fig. 1G) known to occur at the onset of the fibroblasts’ activation by the arising mechanical tension, together with POSTN expression [9]. Fig. 2C also reveals that the conspicuous IHC staining of POSTN on day 3 reflected no more than 30% of its maximal levels on d7, which is expected given the role of this matricellular protein in scar formation [30,49]. We were able to rule out the possibility that StAR or PCNA are of neutrophil origin, although these cells massively invade the infarct border zone at the onset of the the inflammatory response (Supplemental Fig. S3).
Fig. 2.
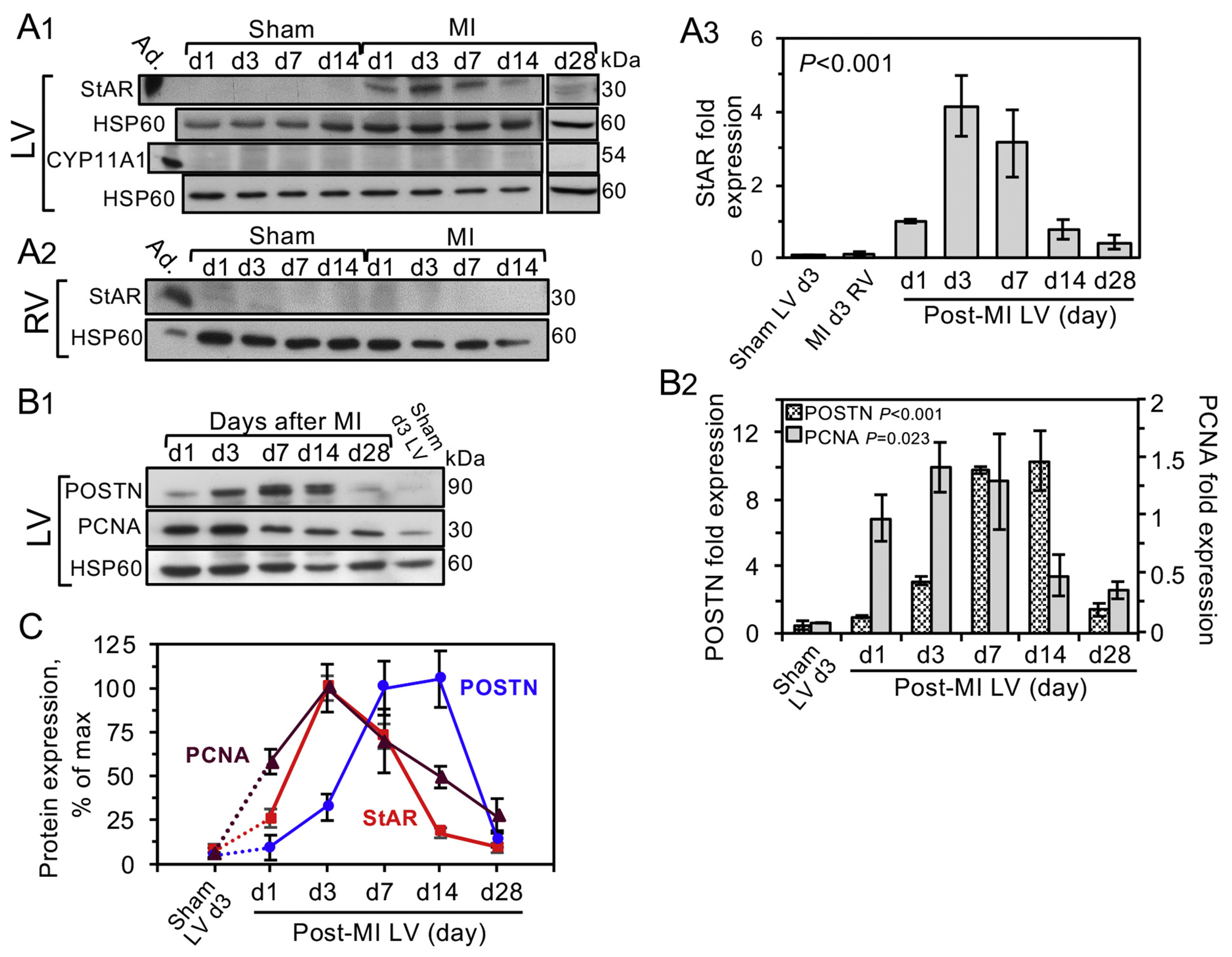
Timeline of StAR expression coincides with the inflammatory response.
A1, On the indicated days (d1-28) following MI or sham operation (12 weeks old BALB/c WT-females), the free walls of the left ventricle (LV) and right ventricle A2, RV) were extracted and analyzed by western blot (20 μg/lane) using antisera to StAR or CYP11A [16,42]. Mitochondrial heat shock protein 60 (HSP60) was used as loading marker. Adrenal extract (Ad.,1 μg) was used as positive StAR control. A3, Western blot quantitation of StAR shows the mean ± SEM of 3 independent experiments. B1-B2, The same extracts of the LV free wall used in A1 were used to determine temporal expression of POSTN and cell replication marker PCNA after MI. B1 is a representative blot and B2 shows the mean ± SEM of 3 independent experiments. Extract pools of 3 hearts were used in each time point. Significance of the western signal was examined by one-way ANOVA and Holm-Sidak post hoc analyses. C, StAR, POSTN and PCNA time-dependent expression, all shown as % of maximal expression. Why does the graph go above 100%?
3.2. Interleukin-1α induces StAR expression in cultured cells
The immediate onset of StAR expression after MI, as well as the disappearance of the protein by the end of the inflammatory response, could imply that StAR induction depends on injury associated signals known to trigger the inflammatory response within minutes after coronary artery ligature [5,32]. To screen for StAR expression in response to several DAMPs/alarmins and proinflammatory cytokines/chemokines, we used a primary cardiac fibroblast culture model (Supplemental Methods and Supplemental Fig. S4), which revealed that IL-1α is by far the most effective signal for StAR induction (Fig. 3A–B); within 3–24 h of exposure to IL-1α in serum-free medium, StAR mRNA and protein levels rose 6–10 fold, reaching maximal efficacy at 0.5–1.0 ng/ml (Fig. 3D1–2, E1–2). By contrast, other alarmins, such as HMGB1, hyalronan fragment (O-HA6), S1008/9 and ATP known to activate their cognate RAGE, TLRs2/4 and P2RX7 receptors, respectively [5], as well as acute phase pro- and anti-inflammatory cytokines (TNFα, IL-6, TGFβ) and chemokines (CCL2/MCP-1), all appeared ineffective in elevating StAR above baseline level, even when tested at concentrations of up to 100 ng/ml (Fig. 3A). Also, cytokines of the IL-1 family, i.e., IL-33 and IL-18, were no more than weak activators of StAR expression at 100 ng/ml (Fig. 3B). Less obvious is the weak response to IL-1β, which appears to be no better than a partial activator of StAR expression (Fig. 3C), possibly resulting from the lower affinity of IL-1β to the shared IL-1α/β receptor 1 (IL-1R1) [reviewed 36, 32].
Fig. 3.
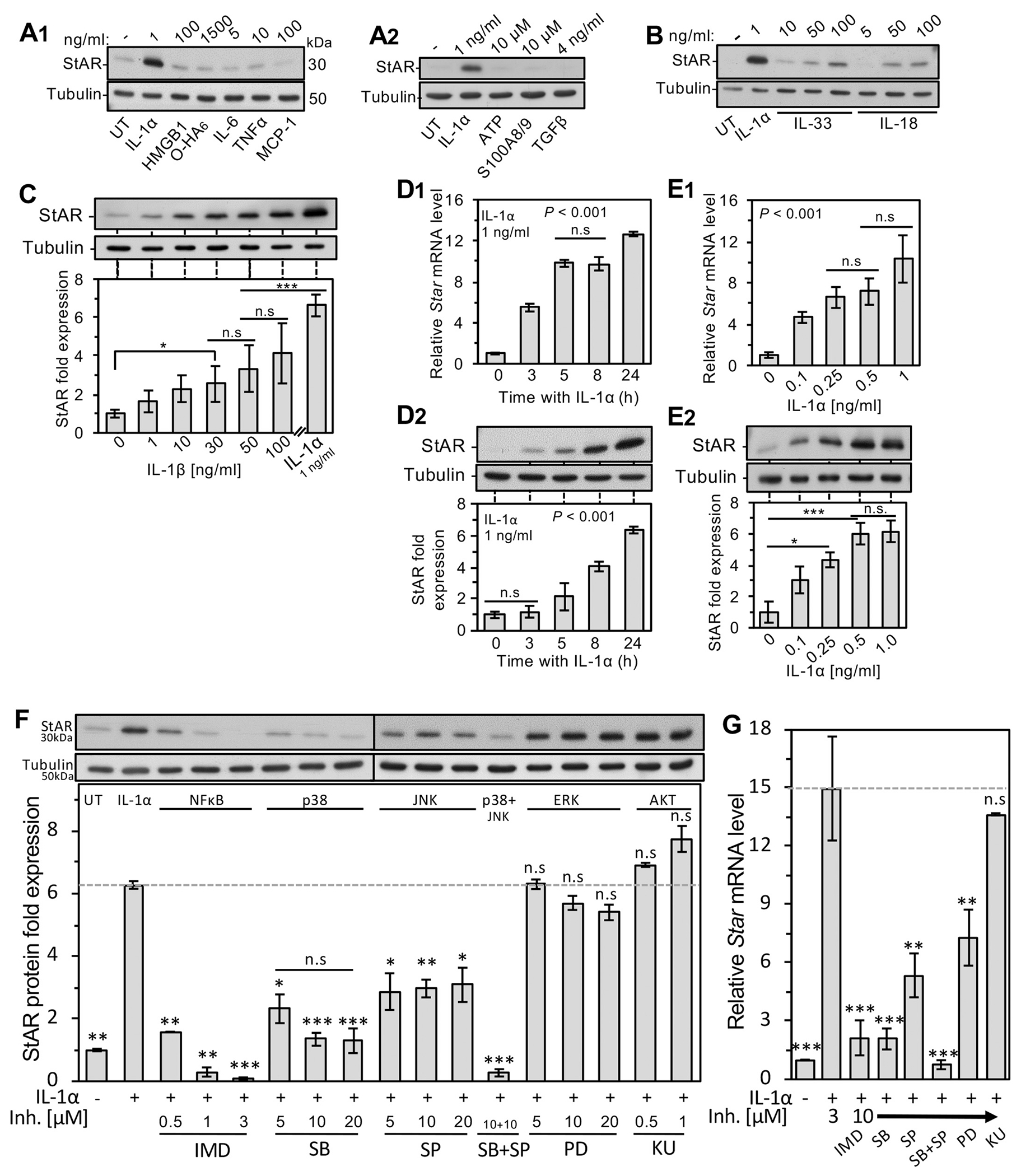
Selective induction of StAR by IL-1α signaling in primary adult rat cardiac fibroblasts. A1,2 &B, After 5 days in culture, the growth medium was switched to serum-free, and the cells were treated for 24 h with the indicated DAMPs/alarmins, cytokines of the IL-1 gene family and cytokines/chemokines known to rise upon the onset of the inflammatory response to MI: IL-1α, HMGB1, hyaluronic acid oligosaccharide O-HA6, IL-6, TNFα, MCP-1, ATP, S100A8/9, TGFβ, IL-33 and IL-18. Shown is a representative western blot analysis (10 μg/lane) using tubulin as loading marker. C, Dose response of StAR protein to IL-1β compared with IL-1α (1 ng/ml) after a 24 h treatment. D1, Time dependent Star mRNA RT-qPCR response to IL-1α. Normalizing transcript was the ribosomal protein l19 mRNA. D2, Time dependent western analysis of StAR induction by IL-1α. E1, Expression of Star mRNA and protein (E2) in response to a 24 h incubation with graded doses of IL-1α assessed by RT-qPCR and western blot analyses, respectively. Bar histograms depict the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001; n.s, not-significant. UT, no addition basal response. F-G, IL-1α induced StAR/Star is regulated by NF-κB/MAPKs pathways. After 5 days’ growth in culture, the culture medium was switched to serum-free, and the following signaling inhibitors were added at the indicated concentrations 1 h prior to addition of IL-1α (1 ng/ml): IMD-0354 (IMD) inhibitor of NF-κB signaling; SB-202190 (SB), pan-inhibitor of p38 MAP kinase (p38); (SP-600125, (SP), the pan-JNK inhibitor; PD-98059 (PD) ERK inhibitor; KU-63794 (KU), inhibitor of the AKT pathway. After overnight incubation, protein and RNA were harvested and further examined by western blot analyses and RT-qPCR, respectively. F, Upper panel depicts representative western blot using antisera to StAR and tubulin loading marker. Lower panel shows quantitative results of StAR protein levels in the absence (UT) or presence of IL-1α and the pathway inhibitors at the indicated concentration. G, Similar experimental layout was applied for RT-qPCR determination of Star mRNA relative levels normalized by the ribosomal protein l19 mRNA. Bar values represent mean ± SEM of three individual experiments. *P < 0.05, **P < 0.01, ***P < 0.001; n.s, not-significant when compared to IL-1α treatment alone.
Interestingly, the regulation of the Star gene, extensively studied before in endocrine cells downstream of GPCR/cAMP pathways [20,21], is highly responsive in cardiac fibroblasts to NF-κB/p38 MAPK signaling (Fig. 3F–G), both known to act downstream of IL-1α binding to the IL-1 type I receptor (IL-1R1) [8]. To demonstrate the latter, Star transcript and StAR protein were studied using inhibitors of several suspected pathways suggested before [37], including a classical NFκB pathway inhibitor IMD-0354 (IMD) and the inhibitor of p38 MAPK (SB203580, SB), both of which profoundly inhibited Star gene products (Fig. 3F–G). In contrast, Star mRNA and protein were only partially inhibited by the inhibitor of JNK, SP600125, and remained unaffected in the presence of the inhibitors of the ERK pathway (PD032590), and in the presence of the KU-0063794 (KU) dual inhibitor of the AKT activators, i.e., mTORC1/2 complexes.
3.3. StAR is anti-apoptotic
Since StAR is a new player of the inflammatory response protein landscape, and the clearing functions of the neutrophils infiltrating the damaged myocardium include the release of proapoptotic reactive oxygen species, ROS [15], we considered the possibility that IL-1α impact includes StAR mediated antiapoptotic protection of the fibroblasts during inflammation. To first examine this hypothesis in the primary fibroblast culture, cells were exposed to increasing doses of cisplatin known to cause mitochondria-dependent cell death by several mechanisms downstream of nuclear and mitochondrial DNA damage [50]. Cisplatin alone did not induce StAR expression (Fig. 4B inset), and 50–100 μM cisplatin caused apoptosis identified by TUNEL positive nuclear fragmentation in up to 30–40% of the cells (Figs 4A1, A3, and 4B, respectively), which is consistent with the general notion that cardiac fibroblasts are relatively resistant to apoptosis [51]. Treatment of the cells with IL-1α (0.5–1 ng/ml) prior to the addition of cisplatin markedly reduced the apoptotic rate down to 4–6% (Figs 4A2 and 4B), which is also below the basal apoptosis rate (10–13%) we normally observed under serum-free medium conditions (Fig. 4B). Moreover, the anti-apoptotic impact of IL-1α correlated linearly (R2 = −0.979, P < 0.005) with the amount of StAR induced by the cytokine, i.e., longer incubation with IL-1α resulted in higher StAR expression and proportionally decreasing apoptosis rates (Fig. 4C). Interestingly, unlike IL-1α, at 1 ng/ml IL-1β did not protect against apoptosis (Figs. 4B), which is consistent with the inability of IL-1β to upregulate StAR at this concentration (Figs. 3C).
Fig. 4.
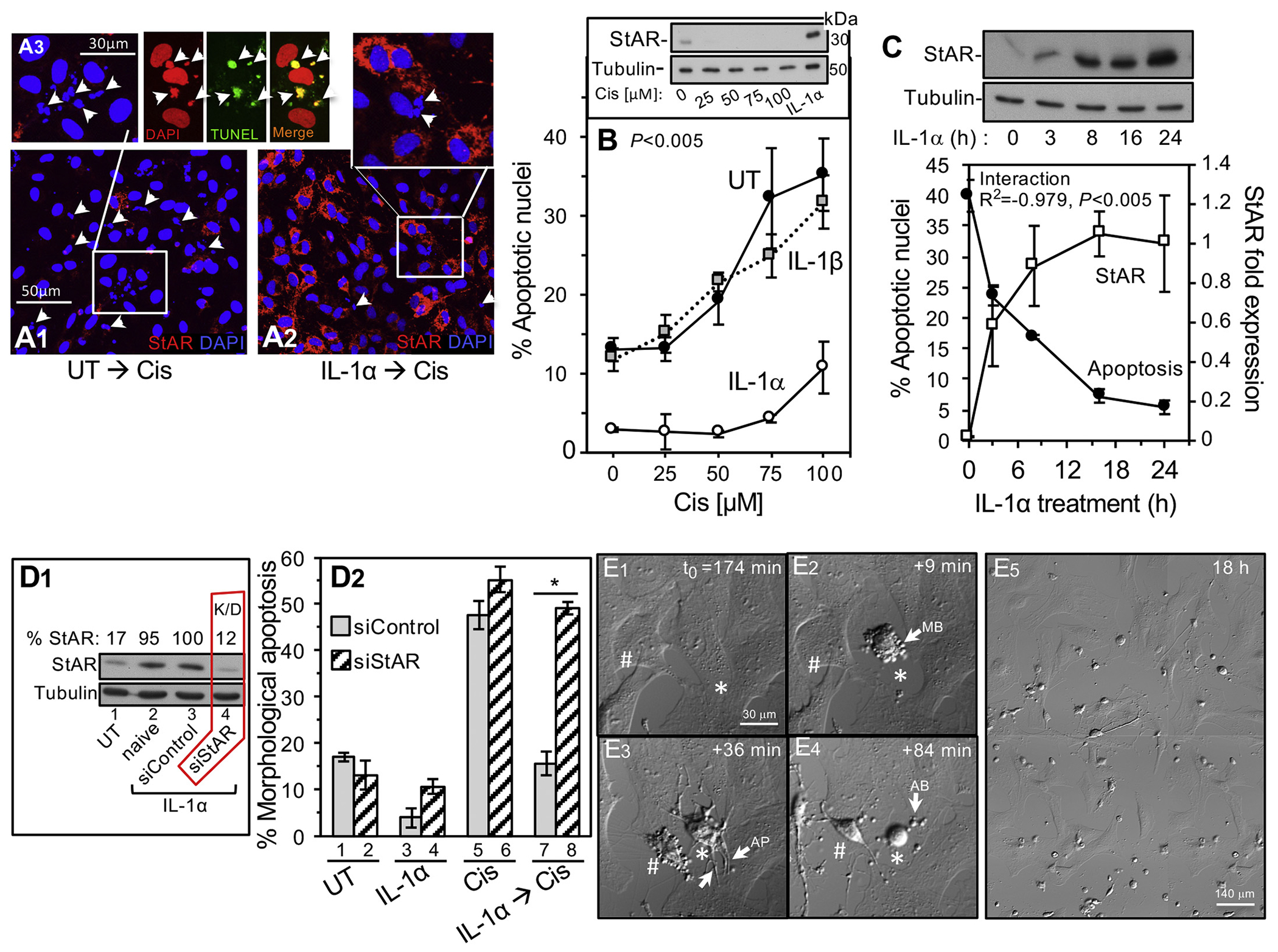
StAR is essential for the protective effect of IL-1α treatment against cisplatin induced fibroblast cell death. A1-A3, Following 5 days of primary adult rat cardiac fibroblasts growth, the cells were treated with IL-1α (1 ng/ml), and cisplatin (Cis, 75 μM) was added 24 h later. Shown are confocal microscopy images depicting DAPI stained nuclei and mitochondrial StAR. A1, Note very low basal StAR staining and fragmented nuclei (arrowheads) after exposure to cisplatin in cells without IL-1α treatment (UT). A3 triad, Fragmented nuclei (arrowheads) are apoptotic as indicated by TUNEL staining. A2, IL-1α treatment results in ample StAR expression and markedly reduced number of apoptotic nuclei. B, Apoptotic cell death response (fragmented nuclei assay) to graded cisplatin doses after 24 h with IL-1α (1 ng/ml) or IL-1β (1 ng/ml). Plots are mean ± SEM of three independent experiments. Two-way ANOVA statistical analysis was used and the P-value represents UT vs. IL-1α. Inset, representative western blot analysis showing that graded doses of cisplatin alone (24 h) do not elevate StAR levels; IL-1α treatment served as positive control. C, Cells were switched to serum-free medium, and after IL-1α treatment (1 ng/ml) for the indicated time periods, cisplatin was added for 24 h to sister plates used to determine the apoptotic rates and StAR levels (upper panel representative western blot). D, StAR knockdown by siRNA oligonucleotide (siStAR) transfection abrogates the anti-apoptotic activity of IL-1α. D1, Representative western blot depicting StAR expression in day 5 cells treated as follows: Lane 1, naive cells without treatment other than incubation for 24 h in serum-free medium (UT); Lane 2, IL-1α treated (24 h) naive cells; Lanes 3–4, cells transfected with siControl oligos or siStAR (K/D) oligos, respectively, and then treated with IL-1α. D2, Following 18 h incubation with cisplatin (75 μM), apoptosis rates were assessed by apoptotic cell morphology (E1-5 and Supplemental Figs. S6) in cells pre-transfected with either siControl or siStAR oligonucleotide: UT, no treatment; IL-1α, cytokine alone; Cis, cisplatin alone; cisplatin added after IL-1α treatment. All results are mean ± SEM of three independent experiments. *P < 0.05. E series, Live cell imaging of apoptosis dependent cell shape changes. E1-4, Shown is the progress of cell disassembly during apoptosis in day 5 fibroblasts plated in IBIDI microscope fit plates (response to 75 μM cisplatin, see Supplemental Methods). Cell images acquired in DIC were recorded every 4 min in four tiled fields (~350–450 cells) and percent count of dead cells was finalized after 18 h incubation (E5). E1-4 depict time-dependent typical morphological changes of two cells (* #) undergoing apoptosis starting 174 min after addition of cisplatin. As expected [76], a sequence of cell contraction and surface membrane blebbing appeared first (MB in E2 arrow), followed by growth of apoptotic membrane protrusions (AP in E3 arrows), and finally, generation of apoptotic bodies (arrow, AB in E4).
To find whether StAR is essential for halting apoptosis downstream of IL-1α signaling, we downregulated (K/D) StAR expression by siRNA and assessed cell death by two complementary assays. First, cisplatin cytotoxicity was assessed in live cells by counting apoptotic cells with a distinct morphology throughout 18 h of time lapse cinematography (Fig. 4D2, Supplemental Methods and Supplemental Fig. S5). As expected, in control cells pre-treated with either siControl or siStAR oligos without IL-1α priming, cisplatin alone caused 50–55% apoptosis (Fig. 4D2 Bars 5–6). Also expected was the reduced rate of apoptosis) 15%(in siControl transfected cells induced with IL-1α to express StAR (Bar 7). However, StAR protein knockdown (Fig. 4D1) completely abrogated the protective effect of IL-1α back to the 50% apoptosis rates observed in cells without the cytokine. Similar results were noted when StAR K/D annulled the protective effect of IL-1α against serum-free driven apoptosis (Fig. 4D2 Bars 3–4). Finally, the impact of StAR K/D was also assessed by metabolic vitality assay, MTT, often applied to determine rates of cell proliferation or cell death [52]. Indeed, the mitochondrial metabolic activities faithfully reflected the pro-apoptotic impact of cisplatin, which was halted by IL-1α pre-treatment evidently dependent on StAR expression (Supplemental Fig. S5A1–3). Collectively, these results suggest that StAR is essential for the anti-apoptotic impact of IL-1α signaling in cardiac fibroblasts/myofibroblasts.
3.4. Loss of StAR expression in IL-1α deficient mice
The apparent exclusiveness of StAR induction by IL-1α in culture experiments called for confirmation of these understandings in vivo, using a mouse model globally deficient for IL-1α expression [39]. Since IL-1α and IL-1β bind to the same receptor IL-1R1 [31], and IL-1β expression was shown to elevate after MI together with IL-1α [53,54], we examined the MI-dependent changes of the cytokine transcripts in the LV free wall. The basal level of IL-1α in sham operated loxP control mice apparently reflects the cytokine reservoir available for release upon ischemic necrosis of the injured cells due to MI (Fig. 5A). Following MI, the LV expression of both IL-1 transcripts elevated 4-fold in the loxP mice, suggesting that IL-1α and IL-1β exist at equimolar LV levels. As expected, the global deletion of exons 2–5 of the Il1a gene [38,39] resulted in total loss of the Il1a transcript in the IL-1αKO heart tissue. Importantly, the post-MI rise of the Il1b transcript is sustained in the IL-1αKO heart, suggesting an IL-1α independent regulation of the post-MI expression of IL-1β, known to rise in both cardiomyocytes and CFs; IL-1α is not expressed in the CFs [2].
Fig. 5.
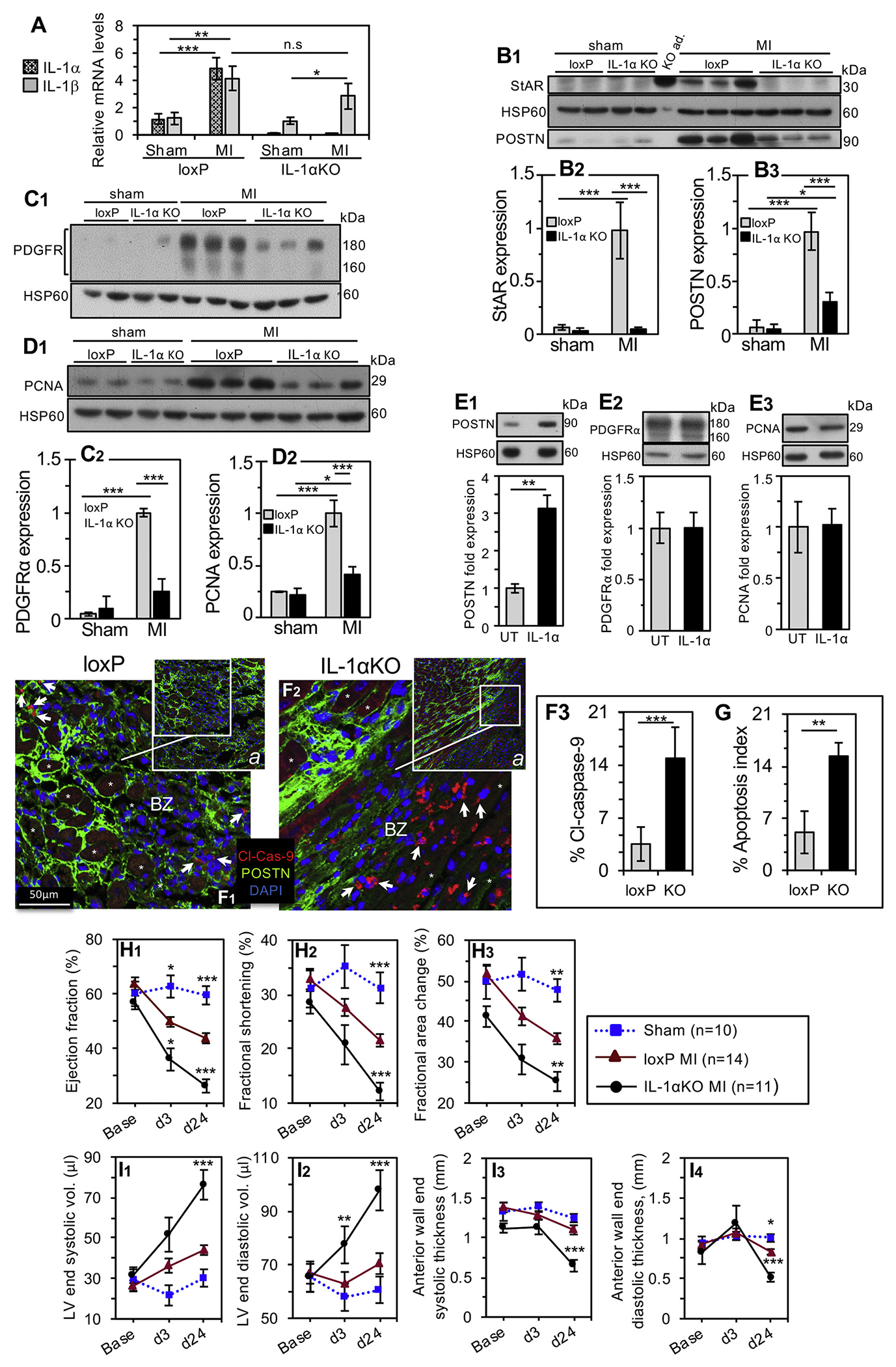
Adverse heart function and remodeling in mice globally deficient for IL-1α.
A. Three days after sham or MI surgery, LV free wall tissues were extracted from male IL-1αKO mice and loxP control animals, and the mRNA levels of IL-1α and IL-1β cytokines were analyzed by RT-qPCR. The ribosomal protein l19 mRNA was used as normalizing transcript. Bar histograms are mean ± SEM, loxP sham n = 4, MI n = 7; IL-1αKO sham n = 7, MI n = 9. Statistical significance was determined using t-test, * P < 0.05; ** P < 0.01; *** P < 0.001, n.s, not-significant. B1, Three days after sham or MI surgery, LV free wall tissues were extracted from IL-1αKO mice and loxP control animals, and the protein levels of StAR and POSTN, PDGFRα (C1) and PCNA (D1) were analyzed by western blot (20 μg/lane). Mitochondrial heat shock protein 60 (HSP60) was used as loading marker. Adrenal gland extract from an IL-1αKO mouse served as positive control for StAR (KO ad.). Panels B2, B3, C2 and D2 show the relative expression levels of StAR, POSTN, PDGFRα and PCNA, respectively. Bar histograms represent the mean ± CI95, (loxP: sham n = 3, MI n = 4; IL-1αKO: sham n = 3, MI n = 6), t-test *P < 0.05, ***P < 0.001 versus loxP MI. E1-3, The response to IL-1α treatment (1 ng/ml, 24 h) in day 5 primary cardiac fibroblasts was assessed by western blot analyses depicting POSTN, PDGFRα and PCNA expression. UT, untreated control cells. Representative western blots and bar histograms are mean ± SEM, n = 3, t-test **P < 0.005. F-G, In vivo apoptosis rates. F1-2, Representative sections of day 3 post-MI loxP control and IL-1αKO hearts depicting cytoplasmic labeling (arrowheads) of cleaved-caspase-9 (Cl-Cas-9) and fragmented nuclei in border zone (BZ) fibroblasts. *, denotes injured cardiomyocytes. F3, Percent of cleaved-caspase-9-bearing cells. G, Other animal cohort was used for the alternative apoptosis assay of aggregated apoptotic mitochondria (apoptosis index), visualized by HSP60 and TOM20 markers depicted in troponin I depleted border zone ROIs (Supplemental Results, Fig. S7 and S6). In both apoptosis analyses, data are shown as the geometric mean ± SD of 4 non-sequential fields from 3 mice in each group, t-test **P < 0.01, ***P < 0.001. H1 through I4, IL-1α deficiency impairs cardiac function and remodeling. Baseline 2D echocardiography was performed prior to left coronary artery ligation (Base) and on d3 and d24 after MI or sham surgery using IL-1αKO and loxP mice. The heart function was determined by LV ejection fraction (H1), LV fractional shortening (H2) and change in LV ejection fraction (H3). Remodeling was examined by the LV end-systolic (I1) and end-diastolic (I2) volumes, as well as the thickness of the LV anterior wall (end-systolic, I3, and end-diastolic, I4). Sham operated animals were pooled (loxP n = 5; IL-1αKO n = 5) to reduce animal number. All results are presented as mean ± SEM. Statistical analyses included a 2-way repeated-measure analysis of variance with Bonferroni posttest. *P < 0.05, **P < 0.01, ***P < 0.001 vs. loxP MI on the same day.
Next, we characterized the impact of IL-1α loss on StAR, POSTN, PDGFRα and proliferating cell nuclear antigen (PCNA) levels in vivo. We also gained additional insight by testing the fibroblasts’ responses to IL-1α in primary cells culture (Fig. 5E1–3). As for StAR, western blot analyses show that on day 3 post-MI, StAR expression was upregulated up to 15-fold in the left ventricular free wall of loxP control mice (Fig. 5B1,2), which faithfully recapitulated the fibroblasts’ response to IL-1α in culture (Fig. 3). By contrast, no StAR rise was noted in MI operated hearts of the IL-1αKO mice. Similar to StAR’s rise, a robust induction was also noted for POSTN, PDGFRα and PCNA expression in the MI control mice (Fig. 5B3, C2 and D2, respectively), which was substantially reduced by 70% in the IL-1αKO animals. Noteworthy is the different response to IL-1α in culture, where the cytokine generates no more than a modest 3-fold response in POSTN expression (Fig. 5E1), suggesting that additional DAMPs other than IL-1α are probably involved in the tour-de-force regulation of this matricellular protein in vivo. Also, IL-1α did not induce cell proliferation in culture (Fig. 5E3), which is consistent with the notion that the IL-1 cytokines are not mitogenic [55]; instead, post-MI cardiac fibroblasts are induced to proliferate by mechanically stretched paracrine-activated cardiomyocytes [56]; consistent with this in vivo notion are the cultured fibroblasts that become mitotically-activated (Supplemental Fig. S4A) by the mechanical tension generated by their adherence to the rigid tissue culture plastic substrate [57]. The latter activation of the fibroblasts in culture can also explain the high IL-1α-independent PDGFRα expression in vitro (Fig. 5E2), whereas the observed induction of PDGFRα by MI (Fig. 5C2) is consistent with similar findings by others [58,59].
The fact that, following MI, IL-1α deficient mice display only attenuated cell proliferation and a low level of fibroblast cell markers (Fig. 5D2, C2, B3), could result, at least in part, from increased cell death in the absence of StAR. To address this possibility, we examined apoptosis rates by two IHC approaches; first, counting border zone cells labeled with cleaved caspase-9 showed a 4-fold increase of apoptotic cells in IL-1αKO over loxP control hearts (Fig. 5F1–3). Secondly, we calculated apoptosis indices (Fig. 5G) based on perinuclear accumulation of apoptotic mitochondria as described in Supplemental Fig. S6D1–3 and Supplemental Results; this approach, too, corroborated a 3.5-fold increase of apoptosis in the heart of the IL-1αKO mice.
3.5. The impact of IL-1α absence on cardiac remodeling and function
To examine the impact of IL-1α absence on cardiac remodeling and function, we performed serial 2D echocardiography prior to surgery and on post-MI days 3 and 24. There was no significant difference in baseline cardiac function between sham and MI operated animals and post-MI d3 ejection fraction values validated successful infarction (Supplemental Table S1 online). Clearly, the deterioration of cardiac contractility markers, as evidenced by d24 measurements of LV ejection fraction, fractional shortening and fractional area change (Fig. 5G1, G2 and G3, respectively) was significantly worse in animals lacking IL-1α (IL-1αKO) compared with loxP control mice. Moreover, adverse remodeling evidenced by increase of LV end-systolic volume, LV end-diastolic volume (Fig. 5H1 and H2, respectively) and reduced LV wall thickness (Fig. 5H3 and H4, respectively), all attested to worsening LV dilatation observed in IL-1αKO mice compared with loxP control animals. These functional findings were consistant with morphological differences observed between loxP and IL-1αKO hearts examined after staining by troponin I, Picro-Sirius Red or H&E (Supplemental Fig.S8 and Supplemental Results). Collectively, representative heart sections suggested that 24 days after MI, typical IL-1αKO hearts exhibit worse remodeling in the form of a thinner LV free wall enriched with dense deposits of collagen.
4. Discussion
This study unraveled an overlooked aspect of the heart response to ischemic injury, whereby StAR, a vital mitochondrial protein indispensable for steroid hormone synthesis, is highly expressed in non-steroidogenic interstitial fibroblasts, adventitial fibroblasts and endothelial cells of the injured myocardium after MI. While presently focused on the cardiac fibroblasts, this study shows that the expression of StAR is selectively governed by IL-1α signaling and no other DAMPs/alarmins.
This understanding is supported both in culture model of primary cardiac fibroblasts, as well as in vivo using an IL-1αKO mouse model, where global deficiency of IL-1α results in total loss of the StAR response. It therefore seems that the regulation of the Star gene in cardiac fibroblasts is redirected to acquire a new response to NF-κB and p38 MAPK signaling that replaces the classical GPCR/cAMP/PKA/CREB pathway known for the steroidogenic activation of the Star gene [20,21].
The new antiapoptotic activity of cardiac StAR calls for further studies addressing the mechanism of StAR action in fibroblasts, possibly by impacting on cellular cholesterol homeostasis. In this regard, intracellular cholesterol mobilization has often been connected to questions of apoptosis and tumor biology [60], where elevation and depletion of cellular, or mitochondrial, cholesterol levels seemed to associate with chemotherapy resistance [61] or potentiation of apoptosis, respectively [62–64]. Most relevant in this regard are recent bioinformatics analyses (RNAseq) showing a 30-fold score increase of pathways controlling cholesterol biosynthesis, identified in MI day 3 fibroblasts [65]; such a score change is understandable in view of the need for cellular cholesterol to sustain proliferative activity, whilst cholesterol is probably required for the antiapoptotic activity of StAR as well.
The IL-1αKO mouse model, which shows no StAR presence after MI, provides compelling evidence attesting to the predominant role of IL-1α in StAR upregulation under physiological circumstances. Moreover, the absence of IL-1α and StAR resulted in a several-fold increase of apoptotic fibroblasts confined to the border zone area at the peak time of the inflammatory response. Hence, these in vivo results confirmed our in vitro predictions in full. Less straightforward is the interpretation of the long term effects of IL-1α deficiency on cardiac dysfunction and adverse remodeling, which were exacerbated in the heart of the IL-1αKO mouse. Unlike the fall of cell survival upon siRNA knockdown of StAR in CFs’ culture, StAR loss in vivo cannot be held responsible for the adverse infarct repair since it is secondary to IL-1α ablation. Therefore, the question of StAR contribution to cardiac remodeling and function after MI, must await the future availability of a model that allows conditional fibroblast-specific ablation of cardiac StAR, independent of the IL-1α status.
An optimal immune response is essential for the downstream events of tissue repair after myocardial injury [1,6,14]. Therefore, if the post-MI release of IL-1α is indeed pivotal for the onset of the inflammatory response [2], the present observation of worsened LV function in the absence of IL-1α is understandable. Moreover, even though pharmacological approaches used in recent clinical and preclinical trials have resulted in some inconsistent outcomes regarding IL-1α’s cardiovascular effects [66–68], pharmacological studies in animal models suggested that IL-1α is cardioprotective, long known as capable of reducing ischemic injury in rodents [69–71]. Hence, deficiency of cardioprotective IL-1α in the IL-1αKO mice, can explain the presently observed adverse results of MI, particularly when injurious IL-1β [72,73] remains high to freely interact with the receptor in the IL-1αKO heart. Yet, the present results are apparently inconsistent with notions previously reached using other genetic approaches to target IL-1α signaling after MI. For example, MI in mice with global [3], or fibroblast-specific ablation of the IL-1 receptor 1 (IL1R1), resulted in rather improved cardiac remodeling and function [54], effects which were also achieved by down-sizing the population of activated fibroblasts [59]. However, a distinct difference exists between the different mouse models, which may explain the inconsistent outcomes upon loss of IL-1α signaling. For one, contrary to the IL-1αKO model, IL-1R1 deficiency should have also resulted in loss of the IL-1β response due to shared binding of the two IL-1 cytokines to IL-1RI; this in itself could predict improvement of left ventricular remodeling in the IL-1R1KO mice, since the compromising activity IL-1β [72,73] was prevented. This study made additional IHC observations of potential interest with respect to their physiological implications: (a) Expression of StAR was observed in border zone fibroblasts of the sub-epicardium, sub-endocardium and in core fibroblasts juxtaposing the free wall circumferential myocytes that did not succumb to necrosis. It is tempting to suggest that these triple layered myocytes/fibroblasts complexes provide an optimal scaffold for deposition of matricellular POSTN that probably prevents early phase rupture of the injured free wall. Furthermore, the structured scar tissue observed 24 days after MI, follows the cellular pattern of day 3 MI survivors, i.e., epicardial and endocardial myocyte layers flanking the fibrotic tissue in the free wall center. (b) The expression of antiapoptotic StAR in adventitial fibroblasts and endothelial cells of blood vessels could be advantageous under stress [74] by protecting these cells toward their subsequent role in post-MI neovascularization [75]. (c) Finally, interesting was also the visual expression of StAR and POSTN in blood vessel cells and close-by interstitial fibroblasts of the unharmed septum, remote from the infarction territory. It is tempting to assume that Star and Postn genes might respond to humoral transmission of low level IL-1α reaching the septum tissue remote from the infarct injury.
5. Conclusions
We propose a working hypothesis by which post-MI IL-1α binding to fibroblasts and endothelial cells in charge of the infarct repair, generates a dual impact (Supplemental Fig. S9): on one hand, previous findings revealed that IL-1α triggers the post-MI cardiac inflammation by promoting the fibroblasts’ own release of proinflammatory mediators that upscale the immune response [2,3,5,13,54]. Then, by virtue of the very same signaling, IL-1α upregulates the expression of StAR that protects the fibroblasts from falling victims to apoptotic stress generated by the clearing activities of the immune cells recruited to the infarct site by the fibroblasts’ own proinflammatory activity. Altogether, the present studies suggest that undisturbed cardiac IL-1α signaling is cardioprotective and instrumental for the improvement of heart remodeling and function, a process in which StAR probably plays a pivotal role.
Supplementary Material
Acknowledgements
We thank Mujahed Musa of the Hebrew University Authority for Biological and Biomedical Models for his valuable preparation of the histological slides for IHC. We also thank Dr. Sara Eimerl and Tiki Sasson for their assistance.
Funding
This study was funded by the Israel Science Foundation grant 1100/17 and the Ines Mandl Research Foundation, New York, USA (JO); NIH R01 HL135657 (SJC).
Abbreviations:
- StAR
steroidogenic acute regulatory protein
- Interleukins IL-1α/β
6, 33, 18
- HMGB1
the high mobility group box 1
- O-HA6
hyaluronic acid oligosaccharide
- TNFα
tumor necrosis factor α
- MCP-1
monocyte chemoattractant protein 1/chemokine C—C motif ligand 2 (MCP-1)
- IMD-0354 (IMD)
IκB kinase-β inhibitor of NFκB signaling
- SB-202190
pan-inhibitor of p38 mitogen activated protein kinase (p38 MAPK)
- SP-600125 (SP)
pan inhibitor of c-Jun N-terminal protein kinase mitogen-activated protein kinase (JNK MAPK)
- PD-98059 (PD)
inhibitor of the mitogen-activated protein kinase kinase (ERK)
- KU-63794
dual inhibitor of mammalian target of rapamycin complex 1 and 2 (TORC1/C2) activators of the AKT pathway
- HSP60
mitochondrial heat-shock protein 60, chaperon
- αSMA
smooth muscle α actin
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PCNA
proliferating cells nuclear antigen
Footnotes
Disclosures
The authors have no potential conflict of interest to declare.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2020.10.013.
References
- [1].Epelman S, Liu PP, Mann DL, Role of innate and adaptive immune mechanisms in cardiac injury and repair, Nat. Rev. Immunol 15 (2015) 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lugrin J, Parapanov R, Rosenblatt-Velin N, Rignault-Clerc S, Feihl F, Waeber B, et al. , Cutting edge: IL-1α is a crucial danger signal triggering acute myocardial inflammation during myocardial infarction, J. Immunol 194 (2015) 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. , Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling, Am. J. Pathol 173 (2008) 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Voronov E, Dinarello CA, Apte RN, Interleukin-1α as an intracellular alarmin in cancer biology, Semin. Immunol 38 (2018) 3–14. [DOI] [PubMed] [Google Scholar]
- [5].Turner NA, Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs), J. Mol. Cell. Cardiol 94 (2016) 189–200. [DOI] [PubMed] [Google Scholar]
- [6].Humeres C, Frangogiannis NG, Fibroblasts in the infarcted, remodeling, and failing heart, JACC. Basic to Transl. Sci 4 (2019) 449–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen C-J, Kono H, Golenbock D, Reed G, Akira S, Rock KL, Identification of a key pathway required for the sterile inflammatory response triggered by dying cells, Nat. Med 13 (2007) 851–856. [DOI] [PubMed] [Google Scholar]
- [8].Turner NA, Effects of interleukin-1 on cardiac fibroblast function: relevance to post-myocardial infarction remodelling, Vasc. Pharmacol 60 (2014) 1–7. [DOI] [PubMed] [Google Scholar]
- [9].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, et al. , Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart, J. Clin. Invest 128 (2018) 2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. , Genetic lineage tracing defines myofibroblast origin and function in the injured heart, Nat. Commun 7 (2016) 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moore-Morris T, Cattaneo P, Guimarães-Camboa N, Bogomolovas J, Cedenilla M, Banerjee I, et al. , Infarct fibroblasts do not derive from bone marrow lineages, Circ. Res 122 (2018) 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Myofibroblastmediated mechanisms of pathological remodelling of the heart, Nat. Rev. Cardiol 10 (2013) 15–26. [DOI] [PubMed] [Google Scholar]
- [13].Frangogiannis NG, The inflammatory response in myocardial injury, repair, and remodelling, Nat. Rev. Cardiol 11 (2014) 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Davis J, Molkentin JD, Myofibroblasts: trust your heart and let fate decide, J. Mol. Cell. Cardio 70 (2014) 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hori M, Nishida K, Oxidative stress and left ventricular remodelling after myocardial infarction, Cardiovasc. Res 81 (2008) 457–464. [DOI] [PubMed] [Google Scholar]
- [16].Anuka E, Yivgi-Ohana N, Eimerl S, Garfinkel B, Melamed-Book N, Chepurkol E, et al. , Infarct-induced Steroidogenic acute regulatory protein: a survival role in cardiac fibroblasts, Mol. Endocrinol 27 (2013) 1502–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Clark BJ, Wells J, King SR, Stocco DM, The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR), J. Biol. Chem 269 (1994) 28314–28322. [PubMed] [Google Scholar]
- [18].Lin D, Sugawara T, Strauss JF, Clark BJ, Stocco DM, Saenger P, et al. , Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis, Science. 267 (1995) 1828–1831. [DOI] [PubMed] [Google Scholar]
- [19].Miller WL, Auchus RJ, The molecular biology, biochemistry, and physiology of human Steroidogenesis and its disorders, Endocr. Rev 32 (2011) 81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yivgi-Ohana N, Sher N, Melamed-Book N, Eimerl S, Koler M, Manna PR, Stocco DM, Orly J, Transcription of steroidogenic acute regulatory protein in the rodent ovary and placenta: alternative modes of cyclic adenosine 3’, 5’-monophosphate dependent and independent regulation, Endocrinology. 150 (2009) 977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Selvaraj V, Stocco DM, Clark BJ, Current knowledge on the acute regulation of steroidogenesis†, Biol. Reprod 99 (2018) 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Payne AH, Hales DB, Overview of Steroidogenic enzymes in the pathway from cholesterol to active steroid hormones, Endocr. Rev 25 (2004) 947–970. [DOI] [PubMed] [Google Scholar]
- [23].Gomez-Sanchez CE, Gomez-Sanchez EP, Cardiac Steroidogenesis—new sites of synthesis, or much ado about nothing? J. Clin. Endocrinol. Metab 86 (2001) 5118–5120. [DOI] [PubMed] [Google Scholar]
- [24].Pusztaszeri MP, Seelentag W, Bosnian FT, Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues, J. Histochem. Cytochem 54 (2006) 385–395. [DOI] [PubMed] [Google Scholar]
- [25].Tallquist MD, Molkentin JD, Redefining the identity of cardiac fibroblasts, Nat. Rev. Cardiol 14 (2017) 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD, Platelet-derived growth factor receptor-α is essential for cardiac fibroblast survival, Am. J. Physiol. Heart Circ. Physiol 317 (2019) H330–H344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kudo A, Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell, Cell. Mol. Life Sci 68 (2011) 3201–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Conway SJ, Izuhara K, Kudo Y, Litvin J, Markwald R, Ouyang G, et al. , The role of periostin in tissue remodeling across health and disease, Cell. Mol. Life Sci 71 (2014) 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, et al. , Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular Remodeling, Circ. Res 101 (2007) 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, et al. , Periostin is essential for cardiac healingafter acute myocardial infarction, J. Exp. Med 205 (2008) 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Garlanda C, Dinarello CA, Mantovani A, The Interleukin-1 family: Back to the future, Immunity. 39 (2013) 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dinarello CA, Immunological and inflammatory functions of the Interleukin-1 family, Annu. Rev. Immunol 27 (2009) 519–550. [DOI] [PubMed] [Google Scholar]
- [33].Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. , Inflammasome-dependent IL-1β release depends upon membrane permeabilisation, Cell Death Differ. 23 (2016) 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Risbud MV, Shapiro IM, Role of cytokines in intervertebral disc degeneration: pain and disc content, Nat. Rev. Rheumatol 10 (2014) 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Broz P, Dixit VM, Inflammasomes: mechanism of assembly, regulation and signalling, Nat. Rev. Immunol 16 (2016) 407–420. [DOI] [PubMed] [Google Scholar]
- [36].Garlanda C, Riva F, Bonavita E, Gentile S, Mantovani A, Decoys and regulatory “receptors” of the IL-1/toll-like receptor superfamily, Front. Immunol 4 (2013) 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weber A, Wasiliew P, Kracht M, Interleukin-1 (IL-1) pathway, Sci. Signal 3 (2010) cml. [DOI] [PubMed] [Google Scholar]
- [38].Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, et al. , Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice, Gut. 63 (2014) 598–609. [DOI] [PubMed] [Google Scholar]
- [39].Almog T, Kandel Kfir M, Levkovich H, Shlomai G, Barshack I, Stienstra R, et al. , Interleukin-lα deficiency reduces adiposity, glucose intolerance and hepatic denovo lipogenesis in diet-induced obese mice, BMJ Open Diabetes Res. Care. 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Uri-Belapolsky S, Shaish A, Eliyahu E, Grossman H, Levi M, Chuderland D, et al. , Interleukin-1 deficiency prolongs ovarian lifespan in mice, Proc. Natl. Acad. Sci. U. S. A Ill (2014) 12492–12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Otto C, Fuchs I, Kauselmann G, Kern H, Zevnik B, Andreasen P, et al. , GPR30 does not mediate estrogenic responses in reproductive organs in mice, Biol. Reprod 80 (2009) 34–41. [DOI] [PubMed] [Google Scholar]
- [42].Naftali-Shani N, Levin-Kotler L-P, Palevski D, Amit U, Kain D, Landa N, et al. , Left ventricular dysfunction switches mesenchymal stromal cells toward an inflammatory phenotype and impairs their reparative properties via toll-like Receptor-4, Circulation. 135 (2017) 2271–2287. [DOI] [PubMed] [Google Scholar]
- [43].van den Borne SWM, van de Schans VAM, Strzelecka AE, Vervoort-Peters HTM, Lijnen PM, Cleutjens JPM, et al. , Mouse strain determines the outcome of wound healing after myocardial infarction - PubMed, Cardiovasc. Res 84 (2009) 273–282. [DOI] [PubMed] [Google Scholar]
- [44].Granot Z, Geiss-Friedlander R, Melamed-Book N, Eimerl S, Timberg R, Weiss AM, et al. , Proteolysis of normal and mutated steroidogenic acute regulatory proteins in the mitochondria: the fate of unwanted proteins, Mol. Endocrinol 17 (2003) 2461–2476. [DOI] [PubMed] [Google Scholar]
- [45].Verstraeten SL, Albert M, Paquot A, Muccioli GG, Tyteca D, Mingeot-Leclercq M-P, Membrane cholesterol delays cellular apoptosis induced by ginsenoside Rh2, a steroid saponin, Toxicol. Appl. Pharmacol 352 (2018) 59–67. [DOI] [PubMed] [Google Scholar]
- [46].Olivetti G, Ricci R, Beghi C, Guideri G, Anversa P, Response of the border zone to myocardial infarction in rats, Am. J. Pathol 125 (1986) 476–483. [PMC free article] [PubMed] [Google Scholar]
- [47].Tallquist MD, Cardiac fibroblast diversity, Annu. Rev. Physiol 82 (2020) 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Conway SJ, Izuhara K, Kudo Y, Litvin J, Markwald R, Ouyang G, et al. , The role of periostin in tissue remodeling across health and disease, Cell. Mol. Life Sci 71 (2014) 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schwanekamp JA, Lorts A, Sargent MA, York AJ, Grimes KM, Fischesser DM, et al. , TGFBI functions similar to periostin but is uniquely dispensable during cardiac injury, PLoS One 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dasari S, Bernard Tchounwou P, Cisplatin in cancer therapy: molecular mechanisms of action, Eur. J. Pharmacol 740 (2014) 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mayorga M, Bahi N, Ballester M, Cornelia JX, Sanchis D, Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death, J. Biol. Chem 279 (2004) 34882–34889. [DOI] [PubMed] [Google Scholar]
- [52].Berridge MV, Herst PM, Tan AS, Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction, Biotechnol. Annu. Rev 11 (2005) 127–152. [DOI] [PubMed] [Google Scholar]
- [53].Guillen I, Blanes M, Gomez-Lechon MJ, Castell JV, Cytokine signaling during myocardial infarction: sequential appearance of IL-1β and IL-6, Am. J. Phys. Regul. Integr. Comp. Phys 269 (1995) R229–R235. [DOI] [PubMed] [Google Scholar]
- [54].Bageghni SA, Hemmings KE, Yuldasheva NY, Maqbool A, Gamboa-Esteves FO, Humphreys NE, et al. , Fibroblast-specific deletion of IL-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction, JCI Insight 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Souders CA, Bowers SLK, Baudino TA, Cardiac fibroblast: the renaissance cell, Circ. Res 105 (2009) 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD, Mechanical regulation of cardiac fibroblast profibrotic phenotypes, Mol. Biol. Cell 28 (2017) 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA, Myofibroblasts and mechano-regulation of connective tissue remodelling, Nat. Rev. Mol. Cell Biol 3 (2002) 349–363. [DOI] [PubMed] [Google Scholar]
- [58].Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ, Flynn ER, Freeman TC, Garrett MR, DeLeon-Pennell KY, Lindsey ML, Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis, Basic Res. Cardiol 114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, et al. , Targeted ablation of Periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice, Circ. Res 118 (2016) 1906–1917. [DOI] [PubMed] [Google Scholar]
- [60].Ribas V, García-Ruiz C, Fernández-Checa JC, Mitochondria, cholesterol and cancer cell metabolism, Clin. Transl. Med 5 (2016) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, et al. , Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma, Cancer Res. 68 (2008) 5246–5256. [DOI] [PubMed] [Google Scholar]
- [62].Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR, Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells, Clin. Cancer Res. 5 (1999) 2223–2229. [PubMed] [Google Scholar]
- [63].Calleros L, Sanchez-Hernandez I, Baquero P, Toro MJ, Chiloeches A, Oncogenic Ras, but not V600EB-RAF, protects from cholesterol depletion-induced apoptosis through the PI3K/AKT pathway in colorectal cancer cells, Carcinogenesis. 30 (2009) 1670–1677. [DOI] [PubMed] [Google Scholar]
- [64].Calleros L, Lasa M, Rodríguez-Álvarez FJ, Toro MJ, Chiloeches A, RhoA and p38 MAPK mediate apoptosis induced by cellular cholesterol depletion, Apoptosis. 11 (2006) 1161–1173. [DOI] [PubMed] [Google Scholar]
- [65].Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ, Flynn ER, Freeman TC, et al. , Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis, Basic Res. Cardiol 114 (2019) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Panahi M, Papanikolaou A, Torabi A, Zhang JG, Khan H, Vazir A, et al. , Immunomodulatory interventions in myocardial infarction and heart failure: a systematic review of clinical trials and meta-analysis of IL-1 inhibition, Cardiovasc. Res 114 (2018) 1445–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hartman MHT, Groot HE, Leach IM, Karper JC, van der Harst P, Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure, Trends Cardiovasc. Med 28 (2018) 369–379. [DOI] [PubMed] [Google Scholar]
- [68].Van Tassell B, Lipinski MJ, Appleton D, Trankle CR, Kadariya D, Abouzaki NA, et al. , Effects of Interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction on recurrent ischemic events: results from the VCUART3 study, Eur. Heart J 40 (2019) 3990. [Google Scholar]
- [69].Maulik N, Engelman RM, Wei Z, Lu D, Rousou JA, Das DK, Interleukin-1α preconditioning reduces myocardial ischemia reperfusion injury, in: Circulation, 1993, pp. 387–394. [PubMed] [Google Scholar]
- [70].Nogae C, Makino N, Hata T, Nogae I, Takahashi S, Suzuki KI, et al. , Interleukin 1α-induced expression of manganous superoxide dismutase reduces myocardial reperfusion injury in the rat, J. Mol. Cell. Cardiol 27 (1995) 2091–2099. [DOI] [PubMed] [Google Scholar]
- [71].Brown JM, White CW, Terada LS, Grosso MA, Shanley PF, Mulvin DW, et al. , Interleukin 1 pretreatment decreases ischemia/reperfusion injury, Proc. Natl. Acad. Sci 87 (1990) 5026–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Harouki N, Nicol L, Remy-Jouet I, Henry JP, Dumesnil A, Lejeune A, et al. , The IL-1β antibody Gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure, JACC Basic to Transl. Sci 2 (2017) 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. , Antiinflammatory therapy with Canakinumab for atherosclerotic disease, N. Engl. J. Med 377 (2017) 1119–1131. [DOI] [PubMed] [Google Scholar]
- [74].Irani K, Oxidant Signaling in vascular cell growth, death, and survival, Circ. Res 87 (2000) 179–183. [DOI] [PubMed] [Google Scholar]
- [75].Manavski Y, Lucas T, Glaser SF, Dorsheimer L, Günther S, Braun T, et al. , Clonal expansion of endothelial cells contributes to ischemia-induced neovascularization, Circ. Res 122 (2018) 670–677. [DOI] [PubMed] [Google Scholar]
- [76].Atkin-Smith GK, Poon IKH, Disassembly of the dying: mechanisms and functions, Trends Cell. Biol 27 (2017) 151–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.