

ABSTRACT
An emerging hypothesis linking arsenic toxicity involves altered epigenetic mechanisms, such as DNA methylation. In this study, we examined the relationship between parents’ arsenic exposure and DNA methylation in tissues obtained from 28 infants with spina bifida from Bangladesh. We analyzed arsenic in parents’ toenails using inductively coupled plasma mass spectrometry (ICP-MS). DNA methylation was measured in infants’ dural tissue, buccal swabs, and whole blood using the Illumina Infinium MethylationEPIC BeadChip. We performed epigenome-wide association analyses (EWAS) and tested differentially methylated regions (DMRs). In EWAS, DNA methylation at cg24039697 in dural tissue was positively associated (β = 0.59, p = 7.6 × 10−9) with father’s toenail arsenic concentrations, adjusting for covariates. We did not identify any CpG sites related to father’s arsenic exposure in the other tissues, or any CpG sites related to mother’s arsenic exposure. Gene ontology analysis identified many biological pathways of interest, including the Wnt signaling pathways. We identified several DMRs across the tissues related to arsenic exposure that included probes mapping to genes that have previously been identified in studies of neural tube defects. This study emphasizes the potential impact of arsenic exposure in fathers, often understudied in epidemiological studies, on DNA methylation in a unique neurological tissue specific to spina bifida.
KEYWORDS: Arsenic, epigenetics, folate, neural tube defects, Bangladesh, environmental epidemiology
Introduction
Environmental exposure to arsenic remains a global public health problem. Arsenic can be found naturally; human exposure can occur through inhalation of arsenic dusts in air, consumption of contaminated food and water, and in certain occupational settings (i.e., in jobs that involve pesticide application or carpentry positions that utilize arsenic-containing compounds). Arsenic-contaminated groundwater is the predominant exposure source impacting countries such as the United States, India, China, Chile, and Bangladesh [1]. In Bangladesh, tubewells (15–50 m deep) were installed throughout the country to avoid consuming pathogen-contaminated surface waters, but many of these wells were later found to contain high concentrations of arsenic [2]. Roughly 16.7% of Bangladesh’s population have household drinking water with arsenic concentrations above the 10 μg/L World Health Organization guideline [3]. Previous studies have shown relationships between chronic exposure to arsenic and skin lesions, diabetes, and other adverse health conditions [4].
An emerging hypothesis linking arsenic to detrimental health outcomes involves epigenetic mechanisms, which regulate gene expression but do not alter the DNA sequence [5]. Epigenetic mechanisms include histone modifications, non-coding RNAs, and DNA methylation, with the latter mechanism most frequently examined. DNA methylation involves the addition of a methyl group to a cytosine ring at its 5-carbon position by DNA methyltransferases and it predominately occurs in a CpG dinucleotide [6]. Human studies of the impact of arsenic, including prenatal exposure, on patterns of global or site-specific DNA methylation have been the focus of numerous reviews [7–13].
Most of the analyses of the relationship between arsenic exposure and gene-specific DNA methylation have been conducted using blood samples (i.e., leukocyte DNA), with only a few reported studies assessing DNA methylation in target tissue, such as placenta and exfoliated urothelial cells [11]. The examinations undertaken in the present study assess DNA methylation profiles in dural tissue of children with spina bifida. Dural tissue surrounds the brain and spinal cord and protrudes in spina bifida, a birth defect caused by incomplete closure of the neural plate, the embryonic precursor to the brain and spinal cord, which fails to close around three to four weeks of gestation. Spina bifida has been associated with detrimental health outcomes in surviving children, including permanent spinal cord damage and genitourinary disorders [1,14]. Arsenic-induced neural tube defects have been observed in chicks [15], hamsters [16], mice [17], and rats [18]. In human epidemiological studies, arsenic levels measured in water and biological samples have shown either null relationships [19–24] or higher arsenic levels observed in neural tube defect cases [25]. Recently, we reported a significant and positive association between fathers’ arsenic exposure and risk of spina bifida, a spinal neural tube defect, in a case-control study from Bangladesh that was initiated in 2016 [26]. In the current study, we utilized data from this study population to examine whether parents’ arsenic exposure is associated with DNA methylation in dural tissue, whole blood, and buccal swab samples collected from infants from Bangladesh with spina bifida. To our knowledge, our study is the first to characterize DNA methylation profiles in dural tissue, a target nervous system tissue collected at surgical closure of the neural tube defect, and to assess the association with arsenic exposure in parents, particularly fathers who are often understudied in epigenetic and exposure research.
Materials and methods
Study participants
Study participants included a subset of infants with spina bifida and their parents who participated in a case-control study from December 2016 through December 2022 in the National Institute of Neurosciences & Hospital (NINS&H) in Dhaka, Bangladesh [26]. We used samples from 28 cases for whom whole blood, buccal swab, and dural tissue were available. The Bangladesh Medical Research Council (BMRC IRB registration number 006 23 08 2016; reference number BMRC/NREC/2016-2019/1651; approved 10 November 2016) and the Human Research Committees at Boston Children’s Hospital (protocol number IRB-P00019768; approved 22 August 2015) and NINS&H (ERC-NINS number 2016/07/10, approved 1 August 2016) approved all study protocols, and informed consent was obtained from parents prior to enrollment. All procedures were performed in compliance with relevant laws and institutional guidelines.
Folate assessment
Parents were interviewed by trained staff members at the hospital visit to gather information on their drinking water source, education, occupation, and medical history, including maternal use of folic acid supplements. Fasting blood samples were collected from mothers via venipuncture at the time of the infant’s hospital visit. Mothers were instructed to fast for a minimum of 8 hours prior to sample collection. Blood was collected in a tube without anticoagulant, which was centrifuged to separate serum from clot. Aliquots from the tube were processed to assess serum folate levels. Serum folate was measured using a Chemiluminescent Microparticle Immunoassay (CMIA) assay on the ARCHITECT plus ci4000 (Abbott Company, Abbott Park, IL, USA), adhering to the manufacturer’s instructions. Folate was measured at NINS&H.
Toenail metal concentrations
A detailed description of the measurement of toenail metals can be found in Tindula et al. [26]. Briefly, we collected toenails from mothers and fathers at the hospital visit and they were analyzed by the Dartmouth Trace Element Analysis Core using Inductively Coupled Plasma – Mass Spectrometry (ICP-MS) [27]. Quality control procedures included the incorporation of certified reference material, blanks, and spiked controls in the experimental runs.
Biospecimen collection
At the time of surgery to close the spina bifida defect, we collected whole blood, buccal swab, and dural tissue samples from infants. For the buccal swab collection, study staff rinsed the infant’s mouth and waited at least one hour to begin sample collection. A T-swab was inserted into the infant’s mouth and rubbed firmly for one minute against the inside of the cheek or under the lower or upper lip before being placed in a BuccalFix Tube containing 0.5 mL BuccalFix Stabilization Buffer. This process was repeated with a second swab. Collected samples were stored at 4°C until shipped to Boston, MA.
Staff collected blood from infants in a PAXgene Blood DNA Tube (IVD; PreAnalytiX GmbH, Hombrechtikon, Switzerland) via venipuncture. After inverting the capped tube 8–10 times, the tube was transferred to NINS&H freezers and within 72 hours to the Bangabandhu Sheikh Mujib Medical University (BSMMU), where it was stored at −80°C until shipped to Boston for long-term storage.
Neurosurgeons provided two 1–2 g cube-shaped specimens from discarded dural tissue nearest the placode. One of the specimens was placed in a tube and covered in O.C.T. compound (Scigen Scientific, Gardena, CA). Similar to the blood samples, the tubes were initially transferred to NINS&H freezers and then within 72 hours to BSMMU and were stored at −80°C until shipped to Boston, MA.
DNA extraction
Whole blood, buccal swab, and dural tissue samples were shipped to Génome Québec (Montreal, Canada) for DNA isolation analysis. DNA was extracted from whole blood and buccal swab samples using the QIAsymphony DSP DNA Midi Kit (Qiagen, Valencia, CA) and processed using the QIAsymphony instrument. Dural tissue samples were treated with ethanol and water to remove the OCT gel and DNA was extracted from the tissue with the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) and the samples were processed with the QIAcube Connect instrument, according to the manufacturer’s protocol.
DNA methylation analysis
DNA was bisulfite converted using Zymo D5007 Bisulfite Conversion Kits (Zymo Research, Irvine, CA), whole genome amplified, enzymatically fragmented, purified, and applied to Illumina Infinium MethylationEPIC BeadChips (Illumina, San Diego, CA), according to the manufacturer’s instructions. DNA from the three tissue types were randomized across chips and plates to prevent batch effects. The EPIC BeadChips measured DNA methylation at a total of 865,859 CpG sites. Probe signal intensities were extracted by Illumina GenomeStudio software.
The ewastools package [28] was used initially to determine if any samples failed based on control metrics of the control probes. All samples passed the threshold and none were identified as failures. We subsequently verified that all of the samples had average detection p-values less than 0.05. Normalization of the methylation data involved background correction with dye-bias normalization using the preprocessNoob function [29] from the minfi package [30], followed by stratified quantile normalization using the preprocessQuantile function, also included in minfi. Probes were filtered in the following order: (1) sites with detection p-values greater than 0.01 in at least one sample; (2) sites mapped to the X and Y chromosomes; (3) sites with annotated probe SNPs; and (4) cross‐reactive probes identified by Chen et al. [31] and Pidsley et al. [32]. A total of 680,819 CpG sites remained for downstream analysis.
Repeat measures for five of the individuals were included in the analysis and correlation between the samples was high (complete pairwise Pearson correlation > 0.99 for all duplicates). For repeat samples, the duplicates with the highest call rates were retained in the analysis. Methylation beta values were logit transformed to the M‐value scale to better adhere to modeling assumptions [33]. To reduce the impact of DNA methylation outliers, methylation values greater or less than three times the interquartile range for a given CpG site were removed prior to statistical analysis.
Statistical analysis
All toenail arsenic concentrations were log2-transformed prior to statistical analysis to account for the right-skewed distributions and to reduce the influence of outliers. We initially performed differentially methylated position (DMP) analysis to determine the relationship between toenail arsenic (mother’s or father’s) as the exposure and DNA methylation in samples collected from infants (whole blood, buccal swab, or dural tissue) as the outcome. Specifically, we used limma linear models with empirical Bayes variance shrinkage [34]. Each of the six models (2 parental arsenic exposures, 3 sample types from infants with DNA methylation data) were adjusted for infant sex, infant age at sample collection (in days), mother’s serum folate levels (ng/mL), and surrogate variables, which are surrogates for unmodeled factors, calculated using the SmartSVA package to account for technical batch effects and tissue heterogeneity [35]. Since DNA methylation is cell-type dependent and reference panels of cell types are unavailable for dural tissue, we utilized SmartSVA, a surrogate variable analysis (SVA) with quicker processing time and enhanced false positive control compared to conventional SVA [35], as a method for reference-free adjustment of cell mixtures in the DMP analysis. To account for multiple testing, for each model we adjusted p‐values using the Benjamini – Hochberg false discovery rate (FDR) threshold for significance of 0.05 [36]. We performed gene ontology (GO) analysis on the results of the differentially methylated position analysis using the methylgometh function in the methylGSA package [37] in R, extracting ontology terms with p-values less than 0.05.
Additionally, we tested differentially methylated regions (DMRs) using the combp function [38] in the ENmix package [39], which is a modified method of the comb-p [40] DMR analysis. Regional gene ontology analysis of the results of the DMR analysis was performed using the ‘GO’ and ‘KEGG’ collections, specified in the goregion function in the missMethyl package [41] in R. We extracted terms where greater than 2 genes were included in the GO term and when the p-value for over-representation of the term was less than 0.05.
We performed a lookup analysis to determine whether the DMP and DMR results in the current study validated findings in recent EWAS [42,43] of the relationship between arsenic and DNA methylation. All analyses were performed using version 17.0 of STATA [44] and version 4.2.2 of the R statistical computing software [45].
Results
Parent’s Arsenic and Infant’s DNA methylation differentially methylated position analysis
Characteristics of the study participants are presented in Table 1. Of the 28 infants with samples sent for analysis, 25 were included in the whole blood DNA methylation analysis and 27 were in the dural tissue and buccal swab analyses. Toenail levels were significantly and positively correlated between mothers and fathers (Pearson correlation coefficients using log2-transformed arsenic concentrations; dural tissue analysis: 0.63; whole blood analysis: 0.76; buccal swab analysis: 0.63). We performed differentially methylated position analysis for a total of 6 models: two exposures examined (mother’s or father’s toenail arsenic) and three infant tissues (dural tissue, whole blood, and buccal swab) (Supplemental Figure S1). The top 20 sites for each model are displayed in Supplemental Table S1. After adjusting for infant age at collection, infant sex, maternal serum folate levels, and surrogate variables, none of the CpG sites remained significant in the maternal arsenic and infant dural tissue model after adjusting for multiple testing using the Benjamini – Hochberg FDR threshold (Supplemental Figure S2). In the models testing the relationship between paternal toenail arsenic levels and infant dural tissue DNA methylation, only one CpG site (cg24039697, chromosome 10) was significant after adjusting for multiple hypothesis testing (β = 0.59, p = 7.6 × 10−9) (Figures 1 and 2).
Table 1.
Characteristics of the study participants, including infants with spina bifida from a case-control study in Bangladesh, for each of the tissue DNA methylation statistical models (mean (SD) or count (%)).
| Characteristic | Dural Tissue Model (n = 27) | Whole Blood Model (n = 25) | Buccal Swab Model (n = 27) |
|---|---|---|---|
| Infants | |||
| Age at sample collection (days) | 174.7 (94.5) | 167.3 (96.5) | 178.1 (94.6) |
| Sex (% male) | 17 (63.0) | 14 (56.0) | 17 (63.0) |
| Birthplace (% home birth) | 10 (37.0) | 9 (36.0) | 10 (37.0) |
| Mothers | |||
| Age (years) | 25.9 (5.2) | 25.2 (5.3) | 25.9 (5.2) |
| Toenail arsenic (μg/g) | 1.4 (1.8) | 1.4 (1.8) | 1.4 (1.8) |
| Serum folate (ng/mL) | 9.2 (5.0) | 8.7 (5.0) | 9.2 (5.0) |
| Fathers | |||
| Age (years) | 33.9 (7.2) | 33.1 (7.4) | 33.9 (7.2) |
| Toenail arsenic (μg/g) | 1.7 (1.8) | 1.6 (1.8) | 1.7 (1.8) |
Figure 1.
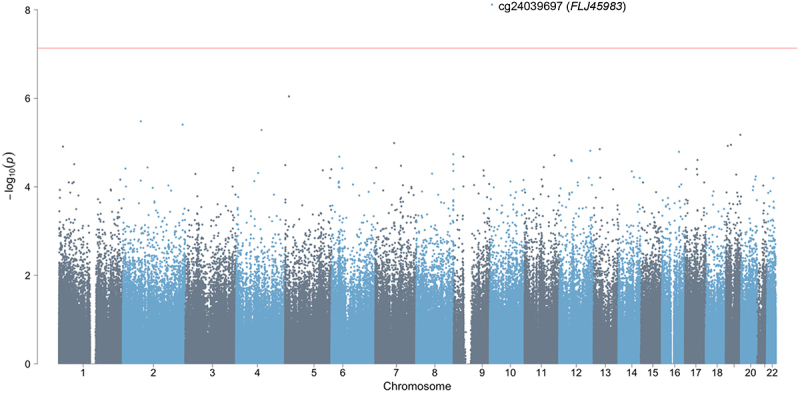
Manhattan plot of the epigenome-wide association between log2transformed father’s toenail arsenic concentrations (μg/g) and DNA methylation levels (M-values) in dural tissue samples from infants with spina bifida, adjusting for infant age at collection (days), infant sex, maternal serum folate levels (ng/mL), and surrogate variables. The red horizontal line represents the Bonferroni threshold.
Figure 2.
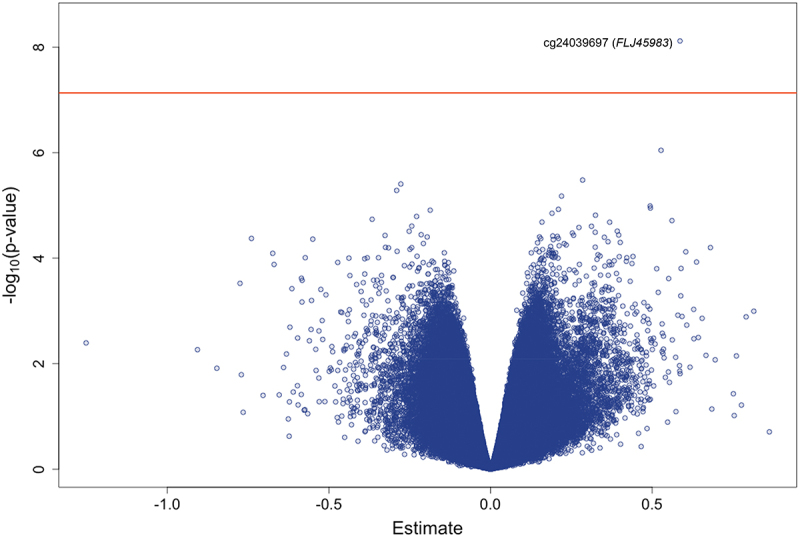
Volcano plot of the epigenome-wide association between log2transformed father’s toenail arsenic concentrations (μg/g) and DNA methylation levels (M-values) in dural tissue samples from infants with spina bifida, adjusting for infant age at collection (days), infant sex, maternal serum folate levels (ng/mL), and surrogate variables. The red horizontal line represents the Bonferroni threshold.
In the analyses testing the relationships between either mother’s or father’s arsenic and infant whole blood DNA methylation, no single CpG site was significant (Supplemental Figures S3 and S4). Similarly, there were no significant buccal swab CpG sites associated with either mother’s or father’s arsenic exposure, after accounting for multiple testing (Supplemental Figures S5 and S6).
Gene ontology analysis of the results of the DMP analysis across the 6 models included terms from diverse biological pathways (Supplemental Table S2). Of particular interest included the six Wnt signaling pathway gene ontology terms in the paternal arsenic and infant whole blood DNA methylation model.
Parent’s Arsenic and Infant’s DNA methylation differentially methylated region analysis
We performed differentially methylated regional analysis using the DMP output of the epigenome-wide association between log2transformed toenail arsenic concentrations (mother’s or father’s) and DNA methylation levels in dural tissue, whole blood, or buccal swab samples from infants, adjusting for covariates. The number of significant (Sidak p-value <0.05) DMRs for each of the 6 models ranged from 3 to 19, with the greatest number of DMRs observed in the analysis of the association between mother’s toenail arsenic and infant whole blood DNA methylation (Table 2 and Supplemental Table S3). A subset of the genes mapping to the DMRs overlapped across models (Figure 3). In the models assessing the association between mother’s or father’s arsenic exposure and infant whole blood DNA methylation, DMRs mapping to ABAT, ZBTB22, TAPBP, RABGAP1L, HOXC4, C20orf123, and OCSTAMP were reported in both models. DMRs mapping to both C8orf73 and MROH6 were observed in the models assessing parent’s arsenic exposure and dural tissue DNA methylation. Additional genes mapping to DMRs were found to overlap across different tissue models; specifically, APOC2 and APOC4-APOC2 in the mother’s arsenic and dural tissue or whole blood models, LOC102723376 in the mother’s arsenic and whole blood or buccal swab models, and C1orf69 in the father’s arsenic and whole blood or buccal swab models.
Table 2.
Differentially methylated regions (DMRs) of epigenome-wide associations between log2-transformed toenail arsenic concentrations (mother’s or father’s; μg/g) and DNA methylation levels (M-values) in dural tissue, whole blood, or buccal swab samples from infants with spina bifida, adjusting for infant age at collection (days), infant sex, maternal serum folate levels (ng/mL), and surrogate variables.
| Arsenic Exposure | DNA Methylation | Chr | Start | End | Sidak p | # of Probes | Genes | Average Regression Coefficienta |
|---|---|---|---|---|---|---|---|---|
| Maternal | Dural Tissue | |||||||
| 4 | 99,850,885 | 99,851,282 | 5.3E-06 | 8 | EIF4E | −0.31 | ||
| 8 | 144,654,887 | 144,655,169 | 5.3E-04 | 5 | C8orf73, MROH6 | −0.29 | ||
| 11 | 8,931,473 | 8,931,709 | 3.8E-06 | 4 | AKIP1, ST5, C11orf17 | −0.35 | ||
| 12 | 11,708,829 | 11,709,052 | 1.0E-03 | 3 | LINC01252 | 0.24 | ||
| 16 | 86,994,949 | 86,995,082 | 5.1E-04 | 3 | −0.20 | |||
| 17 | 48,042,779 | 48,043,064 | 9.0E-04 | 4 | 0.32 | |||
| 18 | 55,862,468 | 55,862,873 | 1.0E-03 | 7 | NEDD4L | 0.18 | ||
| 19 | 45,449,006 | 45,449,166 | 6.5E-03 | 5 | APOC2, APOC4-APOC2 | 0.17 | ||
| 20 | 62,327,968 | 62,328,095 | 1.1E-03 | 4 | TNFRSF6B, RTEL1-TNFRSF6B | −0.36 | ||
| Paternal | Dural Tissue | |||||||
| 2 | 241,564,613 | 241,564,828 | 4.7E-04 | 4 | GPR35 | −0.19 | ||
| 8 | 144,654,887 | 144,655,261 | 1.6E-12 | 6 | C8orf73, MROH6 | −0.31 | ||
| 8 | 144,659,831 | 144,659,884 | 8.6E-03 | 2 | NAPRT1 | −0.40 | ||
| Maternal | Whole Blood | |||||||
| 1 | 116,021,925 | 116,022,092 | 4.4E-03 | 3 | −0.11 | |||
| 1 | 174,843,523 | 174,843,972 | 4.0E-10 | 7 | RABGAP1L | −0.10 | ||
| 1 | 209,897,614 | 209,897,838 | 3.9E-03 | 3 | HSD11B1 | 0.07 | ||
| 2 | 10,571,959 | 10,572,110 | 4.9E-04 | 2 | −0.25 | |||
| 2 | 242,702,749 | 242,702,933 | 4.4E-03 | 3 | D2HGDH | −0.10 | ||
| 4 | 1,166,767 | 1,167,070 | 6.4E-05 | 8 | LOC100130872-SPON2, SPON2 | −0.07 | ||
| 4 | 153,099,743 | 153,099,834 | 3.7E-03 | 3 | 0.08 | |||
| 6 | 33,280,149 | 33,280,401 | 3.2E-02 | 7 | TAPBP | −0.08 | ||
| 6 | 33,282,736 | 33,283,294 | 2.7E-12 | 23 | ZBTB22, TAPBP | −0.08 | ||
| 10 | 81,904,244 | 81,904,332 | 4.7E-03 | 2 | PLAC9 | −0.22 | ||
| 10 | 101,825,029 | 101,825,186 | 1.8E-03 | 4 | CPN1 | −0.10 | ||
| 12 | 54,446,019 | 54,446,577 | 7.3E-09 | 10 | HOXC4 | 0.19 | ||
| 16 | 8,806,359 | 8,807,012 | 0.0E + 00 | 11 | ABAT | −0.25 | ||
| 18 | 10,862 | 11,156 | 3.1E-03 | 8 | LOC102723376 | −0.13 | ||
| 19 | 719,323 | 719,633 | 1.9E-04 | 3 | PALM | −0.15 | ||
| 19 | 45,449,006 | 45,449,166 | 4.4E-03 | 5 | APOC2, APOC4-APOC2 | 0.16 | ||
| 19 | 49,522,817 | 49,523,047 | 2.9E-04 | 3 | −0.37 | |||
| 20 | 43,935,222 | 43,935,552 | 4.5E-09 | 10 | MATN4, RBPJL | −0.14 | ||
| 20 | 45,179,226 | 45,179,414 | 1.7E-04 | 5 | C20orf123, OCSTAMP | 0.08 | ||
| Paternal | Whole Blood | |||||||
| 1 | 174,843,523 | 174,843,768 | 1.2E-05 | 5 | RABGAP1L | −0.10 | ||
| 1 | 228,361,582 | 228,361,642 | 1.2E-02 | 2 | C1orf69, IBA57 | −0.26 | ||
| 3 | 113,417,803 | 113,418,005 | 1.3E-03 | 3 | −0.10 | |||
| 6 | 33,282,736 | 33,283,294 | 1.1E-10 | 23 | ZBTB22, TAPBP | −0.09 | ||
| 6 | 33,560,953 | 33,561,450 | 3.3E-03 | 8 | C6orf227 | −0.23 | ||
| 6 | 76,203,225 | 76,203,676 | 7.3E-09 | 7 | FILIP1 | 0.18 | ||
| 9 | 114,557,226 | 114,557,447 | 1.5E-04 | 4 | C9orf84 | −0.19 | ||
| 10 | 4,868,328 | 4,868,399 | 7.7E-03 | 6 | AKR1E2 | 0.19 | ||
| 10 | 72,219,713 | 72,219,820 | 7.4E-04 | 3 | −0.16 | |||
| 12 | 54,413,101 | 54,413,559 | 9.3E-06 | 5 | HOXC6, HOXC5, HOXC4 | 0.12 | ||
| 12 | 54,446,019 | 54,446,577 | 5.5E-10 | 10 | HOXC4 | 0.23 | ||
| 14 | 54,418,728 | 54,418,881 | 7.7E-04 | 4 | BMP4 | 0.15 | ||
| 16 | 8,806,359 | 8,807,012 | 0.0E + 00 | 11 | ABAT | −0.28 | ||
| 19 | 7,983,877 | 7,984,172 | 1.1E-05 | 5 | SNAPC2 | −0.18 | ||
| 19 | 49,522,817 | 49,523,047 | 1.2E-05 | 3 | −0.42 | |||
| 20 | 45,179,226 | 45,179,414 | 4.2E-03 | 5 | C20orf123, OCSTAMP | 0.09 | ||
| Maternal | Buccal Swab | |||||||
| 6 | 33,288,180 | 33,288,636 | 1.9E-06 | 8 | DAXX | −0.16 | ||
| 13 | 114,138,225 | 114,138,358 | 2.6E-02 | 4 | DCUN1D2 | −0.16 | ||
| 16 | 2,879,998 | 2,880,168 | 1.1E-05 | 3 | ZG16B | −0.21 | ||
| 18 | 10,862 | 11,156 | 3.4E-05 | 8 | LOC102723376 | −0.16 | ||
| Paternal | Buccal Swab | |||||||
| 1 | 228,362,309 | 228,362,510 | 5.8E-04 | 3 | C1orf69 | −0.72 | ||
| 8 | 6,481,431 | 6,481,609 | 3.8E-05 | 3 | MCPH1, MCPH1-AS1 | −0.27 | ||
| 14 | 37,126,582 | 37,126,903 | 3.6E-07 | 4 | PAX9 | −0.43 | ||
| 15 | 86,313,834 | 86,313,974 | 2.1E-06 | 7 | MIR1276, KLHL25 | 0.19 | ||
| 21 | 46,654,108 | 46,654,236 | 1.7E-06 | 3 | −0.22 |
Abbreviations: chr, chromosome.
aThe average regression coefficient on the M-value scale was calculated by extracting the EWAS coefficients for each CpG site included in a DMR and then averaging the values across the CpG sites.
Figure 3.
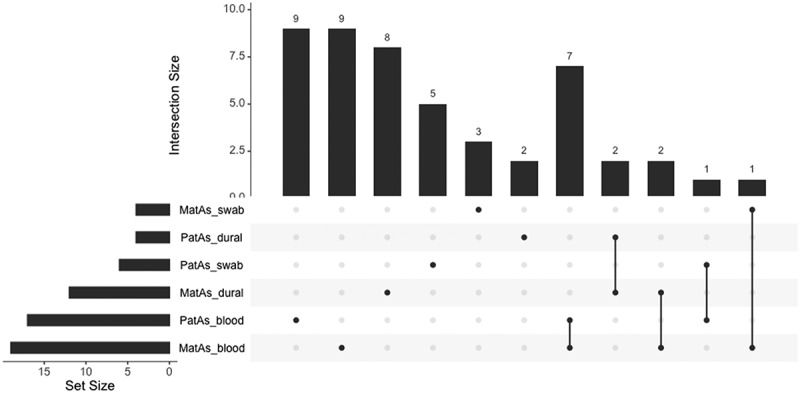
Upset plot of the genes mapping to the differentially methylated regions (DMRs) of the epigenome-wide association between log2transformed toenail arsenic concentrations (mother’s or father’s; μg/g) and DNA methylation levels (M-values) in dural tissue, whole blood, or buccal swab samples from infants with spina bifida, adjusting for infant age at collection (days), infant sex, mother’s serum folate levels (ng/mL), and surrogate variables.
Exploratory gene ontology analysis of the DMR results identified numerous biological pathways overlapping between different parental arsenic and infant DNA methylation models (Supplemental Tables S4 and S5). For example, in the regional gene ontology analysis using the ‘GO’ collection of pathways, 112 gene ontology terms, which were associated with at least two of the genes mapping to a DMR and had a p-value less than 0.05, were found in both models assessing parent’s arsenic and infant whole blood DNA methylation. The next two models with the greatest overlap in gene ontology terms were (1) mother’s arsenic and infant dural tissue and (2) mother’s arsenic and infant whole blood, with 64 overlapping terms across an array of biological pathways. In the gene ontology analysis using KEGG pathways, gene ontology terms related to metabolism were found between different analysis models. For instance, the ‘cholesterol metabolism’ term was found in the models for both (1) mother’s arsenic and infant dural tissue and (2) mother’s arsenic and infant whole blood. Additional metabolism terms (i.e., Butanoate metabolism; beta-Alanine metabolism; Propanoate metabolism; and Alanine, aspartate and glutamate metabolism), as well as the terms ‘Valine, leucine and isoleucine degradation’ and ‘antigen processing and presentation,’ were observed in both the mother’s and father’s arsenic models related to infant whole blood DNA methylation.
Lookup analysis
In comparing the results from the current EWAS, specifically the significant and positive association between DNA methylation at cg24039697 in dural tissue and father’s toenail arsenic concentrations (β = 0.59, p = 7.6×10−9), we were not able to validate the finding in recent EWAS of arsenic exposure and DNA methylation [42,43]. DMP results for the current EWAS models for the overlapping probes that were identified as significantly associated with arsenic exposure from the analysis by Bozack and colleagues can be found in Supplemental Tables S6 and S7. We were not able to replicate any of the DMRs identified in the previous studies in our current analysis.
Discussion
In this study, we examined the relationship between parent’s arsenic exposure and DNA methylation in multiple tissues collected from infants with spina bifida, including dural tissue, a neurological tissue that is difficult to obtain from living patients. We found that arsenic concentration in father’s toenails was associated with dural tissue DNA methylation at the CpG site, cg24039697, after adjusting for multiple hypothesis testing and relevant covariates (p = 7.6 × 10−9). Gene ontology analysis of the CpG site-specific results identified biological pathways relevant to neural tube defects; in particular, the Wnt signaling pathways highlighted from the results of the father’s arsenic and infant whole blood model. In addition, regional analysis for each parent arsenic and infant DNA methylation model identified numerous DMRs, with the total number of DMRs for a given model ranging from 3 to 19. Gene ontology analysis of the regional results spanned an array of biological pathways, including terms related to different metabolic pathways. The findings from this study are critical as they address the knowledge gap of the relationship between arsenic exposure in parents, which remains a public health concern in many areas of the world, and tissue-specific DNA methylation in infants, including an understudied nervous system tissue.
Increasing evidence from studies in humans have demonstrated relationships between arsenic exposure and epigenetic alterations, including global or site-specific DNA methylation, and the associations have been examined in numerous reviews [7–13]. Challenges with synthesizing studies of the impact of arsenic exposure on DNA methylation, and their lack of consistency in specific differential methylation signatures, may be a result of differences in timing, dose, and assessment matrices of arsenic exposure [7], as well as differences in aspects of the DNA methylation assessment, including platforms used, targeted regions assessed, and tissue type. The majority of EWAS examining the impact of arsenic exposure have characterized DNA methylation in blood samples [7].
In the current study, we identified novel CpG site-specific and regional DNA methylation associations in infant dural tissue, a tissue collected during spina bifida surgery and potentially an important target tissue in understanding the relationships between environmental exposures and spina bifida pathogenesis. In the analysis of father’s toenail arsenic and infant dural tissue DNA methylation in the current study, cg24039697 was positively and significantly associated with arsenic exposure. This CpG site has been previously observed to be differentially methylated related to stages of osteoarthritis (subchronal bone DNA methylation) [46], pre-pregnancy maternal underweight status (cord blood DNA methylation) [47], and pancreatic ductal adenocarcinoma (pancreatic ductal adenocarcinoma and nontransformed pancreata DNA methylation) [48]. The site, cg24039697, is located on chromosome 10 within the gene body of FLJ45983, a gene with unknown function previously observed to have increased methylation in tissues collected from patients with breast cancer [49]. Another study reported greater DNA methylation of FLJ45983 in buccal cells among NICU infants experiencing serious neonatal morbidities [50].
In the regional analysis, we observed associations of arsenic exposure in fathers and mothers with infant DNA methylation in dural tissue at three and nine DMRs, respectively. The models with the greatest number of observed DMRs corresponded to the analyses of (1) mother’s arsenic and infant whole blood DNA methylation and (2) father’s arsenic and infant whole blood DNA methylation, with 19 and 16 DMRs, respectively. Genes mapping to DMRs were found to overlap across different models. Of the 19 unique genes mapping to DMRs in the model of mother’s arsenic and infant whole blood DNA methylation and the 17 unique genes from the model of father’s arsenic and infant whole blood DNA methylation, seven genes (ABAT, ZBTB22, TAPBP, RABGAP1L, HOXC4, C20orf123, and OCSTAMP) were reported in both models. Differentially methylated regions within the ABAT gene have previously been associated with glyphosate exposure [51] and autism spectrum disorder [52]. DNA methylation at CpG sites or regions within the TAPBP gene has been associated with factors such as mother’s pre-pregnancy obesity [53] and night shift work [54]. DNA methylation at TAPBP and ZBTB22, another overlapping gene between the models, has been linked to maternal sensitivity [55]. Additional genes found to overlap across different tissue models included (1) C8orf73 and MROH6 in the parent’s arsenic and infant’s dural tissue models, (2) APOC2 and APOC4-APOC2 in the mother’s arsenic and dural tissue or whole blood models, (3) LOC102723376 in the mother’s arsenic and whole blood or buccal swab models, and (4) C1orf69 in the father’s arsenic and whole blood or buccal swab models.
A subset of the genes mapping to the DMRs found in the current study have previously been associated with birth defects, including spina bifida. A study by Rochtus et al. [56], assessing DNA methylation in leukocytes in patients with spina bifida (i.e., myelomeningocele) compared to controls, observed significant hypomethylation of CpG sites within the ABAT gene in DNA methylation analysis using the Illumina 450K BeadChip array. In validation analysis in a larger cohort of cases and controls using the Sequenom EpiTYPER, the authors were not able to replicate the significant methylation difference between the groups in the ABAT gene. Additionally, CpG sites within the RABGAP1L gene have been shown to be differentially methylated in infants born with congenital Zika virus microcephaly compared to controls, with lower methylation observed in cases [57]. The gene, HOXC4, along with other members of the Hox family of genes important in neural tube closure, have previously been shown by Yu and colleagues to be upregulated in a retinoic acid induced mouse neural tube defect model and in retinoic acid treated embryonic stem cells [58]. Additionally, the authors compared mRNA levels in brain tissues collected from fetuses with neural tube defects (i.e., anencephaly, spina bifida, spina bifida and hydrocephaly, and encephalocele) with those in age-matched controls. They observed that HOXC4 expression was significantly upregulated compared to controls only in the tissues collected from patients with anencephaly.
Gene ontology analysis of the differentially methylated position and region analyses revealed diverse biological pathways. Of particular relevance, given the assessment of DNA methylation in samples collected from infants with neural tube defects, were the Wnt signaling pathways in the DMP gene ontology analysis of the father’s arsenic and infant whole blood DNA methylation model. Wnt ligands bind to receptors and co-receptors to initiate signal transduction pathways that are either β-catenin-dependent (canonical) or β-catenin-independent (non-canonical) [59]. Wnt signaling is involved in embryonic developmental processes, such as neurulation [59]. Neural tube closure is dependent on both convergent extension and neural-plate apical constriction, processes that are connected by the non-canonical Wnt/planar cell polarity (PCP) pathway [60,61]. In our study, gene ontology terms related to regulation and cell-cell signaling of the Wnt pathway were observed for the DMP analysis of father’s arsenic and infant whole blood, an intriguing finding given the exploratory nature of the gene ontology analysis and the relevance of Wnt signaling in neural tube development.
One of the limitations of the current study was the small sample size, which limited our power to detect associations between parent’s arsenic and infant DNA methylation. This was reflected in mostly null findings in individual CpG analyses. However, regional analyses increase statistical power [62] and provided some evidence of associations across tissues, but these findings must be interpreted with caution as a higher type I error rate is possible with these methods [63]. Enrollment and tissue collection was limited to infants undergoing surgery at a hospital in order to close the neural tube defect. Since only case children were included in the analysis and we did not collect the same biological samples from controls, we were not able to discern epigenetic differences between individuals with and without neural tube defects.
One of the strengths of the study included the characterization of arsenic in toenail samples collected from both mothers and fathers. Toenail samples reflect exposures from the past 3–18 months [64,65]. Due to the ubiquitous exposure of individuals to arsenic contaminated water in Bangladesh, we expect exposure to be relatively stable over time. Although we did not have repeat toenail measures in the current analysis, a previous study of 52 women from Bangladesh with toenails collected during the first prenatal visit and within two weeks after birth demonstrated a moderate and significant correlation (correlation coefficient of 0.49) between the measurements [66]. Since we were able to collect exposure information from fathers, our study adds to the growing body of literature demonstrating relationships between exposure and health status of fathers and child DNA methylation. For instance, DNA methylation in children has been associated with father’s obesity [67–69], adverse childhood experiences [70], and nutrition [71]. Another strength included the ability to leverage DNA methylation data from multiple tissue types collected from infants with spina bifida to explore tissue-specific effects of parental arsenic exposure. Our study assessed DNA methylation in dural tissue, a unique and understudied tissue collected from infants with spina bifida at the time of surgical closure of the neural tube defect, with findings suggesting associations at both the CpG and regional level related to arsenic exposure in parents.
In conclusion, we measured arsenic, a chemical with widespread exposure due to contaminated drinking water in Bangladesh, in toenail samples from parents of infants with spina bifida, a severe birth defect with lifelong health consequences in surviving children. We reported a novel and significant association between father’s toenail arsenic and infant dural tissue methylation at cg24039697 (FLJ45983), a CpG site on chromosome 10. Multiple DMRs were reported for each of the analysis models, with the majority observed in the parent’s arsenic and infant’s whole blood analyses. Gene ontology analysis of the EWAS DMP results for all parent’s arsenic and infant’s tissue methylation models highlighted several pathways involved in early development and neurology. Of particular interest were the Wnt signaling pathways, which are involved in neural tube closure.
We plan to extend the current analysis in a larger group of participants and to explore the relationship of arsenic and genetic, epigenetic, and nutritional factors on DNA methylation patterns in samples collected from infants with spina bifida. In our previous findings [26] we observed that father’s arsenic exposure was associated with increased risk of neural tube defects in participants of the case-control study. The results from the current analysis expand on our previous study and indicate potential methylation differences in target tissue of spina bifida cases following father’s arsenic exposure. We are interested in exploring potential mechanisms that could mediate the influence of father’s exposure on child disease risk and the potential ameliorating effects of father’s folic acid supplementation.
Supplementary Material
Acknowledgments
We are grateful to the field staff at the National Institute of Neurosciences and Hospital (NINS&H) and study participants for their contributions. Parental toenail metal analysis was carried out at the Dartmouth Trace Element Core Facility, which was established by grants from the National Institutes of Health (NIH) and National Institute of Environmental Health Sciences (NIEHS) Superfund Research Program (P42 ES007373) and the Norris Cotton Cancer Center at Dartmouth Hitchcock Medical Center. DNA methylation analysis was performed at the Centre d’expertise et de services Génome Québec, with help from François Bacot and Valerie Catudal.
Funding Statement
This work was supported by the National Institute of Environmental Health Sciences [NIEHS R01 ES026317, R21 ES030784, P30 ES000002, R01 ES034713], the National Institute of Mental Health [NIMH T32 MH112510] and the Eunice Kennedy Shriver National Institute of Child Health and Human Development [NICHD R13 hD100191]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding sources were not involved in the study design; in the collection, analysis and interpretation of the data; in the writing of the report; and in the decision to submit the article for publication.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that supports our research are available from Boston Children’s Hospital upon reasonable request. Given agreements with the Bangladesh Medical Research Council (BMRC), requests for data sharing require their additional review. However, the authors, specifically the corresponding author, invite any and all communication pertaining to the data and are happy to present the data in a setting where the oversight committees have granted permission.
Ethics approval
The Bangladesh Medical Research Council and the Human Research Committees at Boston Children’s Hospital and the National Institute of Neurosciences & Hospital approved all study protocols, which were conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from parents prior to enrollment.
Author contributions
Conceptualization: G.T., A.C., M.M.; Data curation: G.T., S.K.B.; Formal analysis: G.T.; Funding acquisition: G.T., A.C., M.M.; Methodology: G.T., D.C.C., B.L., L.L., A.C., M.M.; Project administration: S.K.M., S.M.E., D.M.A., J.I., S.K.B., M.M.; Supervision: D.C.C., B.C.W., B.L., L.L., A.C., M.M.; Visualization: G.T.; Writing-original draft: G.T., A.C., M.M.; Writing – review & editing: S.K.M., S.M.E., D.M.A., J.I., S.K.B., B.C.W, D.C.C, B.L., L.L.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2416345
References
- [1].Mazumdar M. Does arsenic increase the risk of neural tube defects among a highly exposed population? A new case–control study in Bangladesh. Birth Defects Res. 2017;109(2):92–14. doi: 10.1002/bdra.23577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ahmad SA, Khan MH, Haque M. Arsenic contamination in groundwater in Bangladesh: implications and challenges for healthcare policy. Risk Manag Healthc Policy. 2018;11:251–261. doi: 10.2147/RMHP.S153188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bangladesh Bureau of Statistics (BBS) and UNICEF Bangladesh . Bangladesh multiple indicator cluster survey 2019, Progotir Pathey: survey findings report. Dhaka, Bangladesh: Bangladesh Bureau of Statistics (BBS); 2019. [Google Scholar]
- [4].Naujokas MF, Anderson B, Ahsan H, et al. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect. 2013;121(3):295–302. doi: 10.1289/ehp.1205875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Foley DL, Craig JM, Morley R, et al. Prospects for epigenetic epidemiology. Am J Epidemiol. 2009;169(4):389–400. doi: 10.1093/aje/kwn380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655 [DOI] [PubMed] [Google Scholar]
- [7].Argos M. Arsenic exposure and epigenetic alterations: recent findings based on the illumina 450K DNA methylation array. Curr Environ Health Rep. 2015;2(2):137–144. doi: 10.1007/s40572-015-0052-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21(2):243–251. doi: 10.1097/MOP.0b013e32832925cc [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bailey KA, Fry RC. Arsenic-associated changes to the epigenome: what are the functional consequences? Curr Environ Health Rep. 2014;1(1):22–34. doi: 10.1007/s40572-013-0002-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chakraborty A, Ghosh S, Biswas B, et al. Epigenetic modifications from arsenic exposure: a comprehensive review. Sci Total Environ. 2022;810:151218. doi: 10.1016/j.scitotenv.2021.151218 [DOI] [PubMed] [Google Scholar]
- [11].Martin EM, Fry RC. Environmental influences on the epigenome: exposure- associated DNA methylation in human populations. Annu Rev Public Health. 2018;39(1):309–333. doi: 10.1146/annurev-publhealth-040617-014629 [DOI] [PubMed] [Google Scholar]
- [12].Ray PD, Yosim A, Fry RC. Incorporating epigenetic data into the risk assessment process for the toxic metals arsenic, cadmium, chromium, lead, and mercury: strategies and challenges. Front Genet. 2014;5:201. doi: 10.3389/fgene.2014.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vaiserman A, Lushchak O. DNA methylation changes induced by prenatal toxic metal exposure: an overview of epidemiological evidence. Environ Epigenet. 2021;7(1):dvab007. doi: 10.1093/eep/dvab013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Greene ND, Copp AJ. Neural tube defects. Annu Rev Neurosci. 2014;37(1):221–242. doi: 10.1146/annurev-neuro-062012-170354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Han ZJ, Song G, Cui Y, et al. Oxidative stress is implicated in arsenic-induced neural tube defects in chick embryos. Int J Dev Neurosci. 2011;29(7):673–680. doi: 10.1016/j.ijdevneu.2011.06.006 [DOI] [PubMed] [Google Scholar]
- [16].Carpenter SJ. Developmental analysis of cephalic axial dysraphic disorders in arsenic-treated hamster embryos. Anat Embryol (Berl). 1987;176(3):345–365. doi: 10.1007/BF00310189 [DOI] [PubMed] [Google Scholar]
- [17].Hill DS, Wlodarczyk BJ, Finnell RH. Reproductive consequences of oral arsenate exposure during pregnancy in a mouse model. Birth Defects Res B Dev Reprod Toxicol. 2008;83(1):40–47. doi: 10.1002/bdrb.20142 [DOI] [PubMed] [Google Scholar]
- [18].Beaudoin AR. Teratogenicity of sodium arsenate in rats. Teratology. 1974;10(2):153–157. doi: 10.1002/tera.1420100211 [DOI] [PubMed] [Google Scholar]
- [19].Brender JD, Suarez L, Felkner M, et al. Maternal exposure to arsenic, cadmium, lead, and mercury and neural tube defects in offspring. Environ Res. 2006;101(1):132–139. doi: 10.1016/j.envres.2005.08.003 [DOI] [PubMed] [Google Scholar]
- [20].Jin L, Zhang L, Li Z, et al. Placental concentrations of mercury, lead, cadmium, and arsenic and the risk of neural tube defects in a Chinese population. Reprod Toxicol. 2013;35:25–31. doi: 10.1016/j.reprotox.2012.10.015 [DOI] [PubMed] [Google Scholar]
- [21].Mazumdar M, Ibne Hasan MO, Hamid R, et al. Arsenic is associated with reduced effect of folic acid in myelomeningocele prevention: a case control study in Bangladesh. Environ Health. 2015;14(1):34. doi: 10.1186/s12940-015-0020-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ozel S, Ozyer S, Aykut O, et al. Maternal second trimester blood levels of selected heavy metals in pregnancies complicated with neural tube defects. J Matern Fetal Neonatal Med. 2019;32(15):2547–2553. doi: 10.1080/14767058.2018.1441280 [DOI] [PubMed] [Google Scholar]
- [23].Sanders AP, Desrosiers TA, Warren JL, et al. Association between arsenic, cadmium, manganese, and lead levels in private wells and birth defects prevalence in North Carolina: a semi-ecologic study. BMC Public Health. 2014;14(1):955. doi: 10.1186/1471-2458-14-955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang B, Zhu Y, Yan L, et al. Association of maternal chronic arsenic exposure with the risk of neural tube defects in Northern China. Environ Int. 2019;126:222–227. doi: 10.1016/j.envint.2019.02.016 [DOI] [PubMed] [Google Scholar]
- [25].Demir N, Basaranoglu M, Huyut Z, et al. The relationship between mother and infant plasma trace element and heavy metal levels and the risk of neural tube defect in infants. J Matern Fetal Neonatal Med. 2019;32(9):1433–1440. doi: 10.1080/14767058.2017.1408064 [DOI] [PubMed] [Google Scholar]
- [26].Tindula G, Mukherjee SK, Ekramullah SM, et al. Parental metal exposures as potential risk factors for spina bifida in Bangladesh. Environ Int. 2021;157:106800. doi: 10.1016/j.envint.2021.106800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].O’Brien KM, White AJ, Sandler DP, et al. Do post-breast cancer diagnosis toenail trace element concentrations reflect prediagnostic concentrations? Epidemiology. 2019;30(1):112–119. doi: 10.1097/EDE.0000000000000927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Heiss JA, Just AC. Identifying mislabeled and contaminated DNA methylation microarray data: an extended quality control toolset with examples from GEO. Clin Epigenetics. 2018;10(1):73. doi: 10.1186/s13148-018-0504-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Triche TJ Jr., Weisenberger DJ, Van Den Berg D, et al. Low-level processing of illumina infinium DNA methylation BeadArrays. Nucleic Acids Res. 2013;41(7):e90. doi: 10.1093/nar/gkt090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aryee MJ, Jaffe AE, Corrada-Bravo H, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the illumina infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17(1):208. doi: 10.1186/s13059-016-1066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11(1):587. doi: 10.1186/1471-2105-11-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):1–25. doi: 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- [35].Chen J, Behnam E, Huang J, et al. Fast and robust adjustment of cell mixtures in epigenome-wide association studies with SmartSVA. BMC Genomics. 2017;18(1):413. doi: 10.1186/s12864-017-3808-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- [37].Ren X, Kuan PF. methylGSA: a Bioconductor package and shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics. 2019;35(11):1958–1959. doi: 10.1093/bioinformatics/bty892 [DOI] [PubMed] [Google Scholar]
- [38].Xu Z, Xie C, Taylor JA, et al. ipDMR: identification of differentially methylated regions with interval P-values. Bioinformatics. 2021;37(5):711–713. doi: 10.1093/bioinformatics/btaa732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu Z, Niu L, Li L, et al. ENmix: a novel background correction method for illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44(3):e20. doi: 10.1093/nar/gkv907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pedersen BS, Schwartz DA, Yang IV, et al. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28(22):2986–2988. doi: 10.1093/bioinformatics/bts545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32(2):286–288. doi: 10.1093/bioinformatics/btv560 [DOI] [PubMed] [Google Scholar]
- [42].Bozack AK, Boileau P, Wei L, et al. Exposure to arsenic at different life-stages and DNA methylation meta-analysis in buccal cells and leukocytes. Environ Health. 2021;20(1):79. doi: 10.1186/s12940-021-00754-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bozack AK, Domingo-Relloso A, Haack K, et al. Locus-specific differential DNA methylation and urinary arsenic: an epigenome-wide association study in blood among adults with low-to-moderate arsenic exposure. Environ Health Perspect. 2020;128(6):67015. doi: 10.1289/EHP6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].StataCorp . Stata statistical software: release 17. StataCorp LLC. College Station (TX); 2021. [Google Scholar]
- [45].R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2022. [Google Scholar]
- [46].Zhang Y, Fukui N, Yahata M, et al. Identification of DNA methylation changes associated with disease progression in subchondral bone with site-matched cartilage in knee osteoarthritis. Sci Rep. 2016;6(1):34460. doi: 10.1038/srep34460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sharp GC, Lawlor DA, Richmond RC, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the avon longitudinal study of parents and children. Int J Epidemiol. 2015;44(4):1288–1304. doi: 10.1093/ije/dyv042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nones K, Waddell N, Song S, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135(5):1110–1118. doi: 10.1002/ijc.28765 [DOI] [PubMed] [Google Scholar]
- [49].Piotrowski A, Benetkiewicz M, Menzel U, et al. Microarray-based survey of CpG islands identifies concurrent hyper- and hypomethylation patterns in tissues derived from patients with breast cancer. Genes Chromosomes & Cancer. 2006;45(7):656–667. doi: 10.1002/gcc.20331 [DOI] [PubMed] [Google Scholar]
- [50].Everson TM, O’Shea TM, Burt A, et al. Serious neonatal morbidities are associated with differences in DNA methylation among very preterm infants. Clin Epigenetics. 2020;12(1):151. doi: 10.1186/s13148-020-00942-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lucia RM, Huang WL, Pathak KV, et al. Association of glyphosate exposure with blood DNA methylation in a cross-sectional study of postmenopausal women. Environ Health Perspect. 2022;130(4):47001. doi: 10.1289/EHP10174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nardone S, Sams DS, Zito A, et al. Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb Cortex. 2017;27(12):5739–5754. doi: 10.1093/cercor/bhx250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martin CL, Jima D, Sharp GC, et al. Maternal pre-pregnancy obesity, offspring cord blood DNA methylation, and offspring cardiometabolic health in early childhood: an epigenome-wide association study. Epigenetics. 2019;14(4):325–340. doi: 10.1080/15592294.2019.1581594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Clarkson-Townsend DA, Everson TM, Deyssenroth MA, et al. Maternal circadian disruption is associated with variation in placental DNA methylation. PLoS One. 2019;14(4):e0215745. doi: 10.1371/journal.pone.0215745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dall’ Aglio L, Rijlaarsdam J, Mulder RH, et al. Epigenome-wide associations between observed maternal sensitivity and offspring DNA methylation: a population-based prospective study in children. Psychol Med. 2022;52(13):2481–2491. doi: 10.1017/S0033291720004353 [DOI] [PubMed] [Google Scholar]
- [56].Rochtus A, Winand R, Laenen G, et al. Methylome analysis for spina bifida shows SOX18 hypomethylation as a risk factor with evidence for a complex (epi)genetic interplay to affect neural tube development. Clin Epigenetics. 2016;8(1):108. doi: 10.1186/s13148-016-0272-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Anderson D, Neri J, Souza CRM, et al. Zika virus changes methylation of genes involved in immune response and neural development in Brazilian babies born with congenital microcephaly. J Infect Dis. 2021;223(3):435–440. doi: 10.1093/infdis/jiaa383 [DOI] [PubMed] [Google Scholar]
- [58].Yu J, Wang L, Pei P, et al. Reduced H3K27me3 leads to abnormal hox gene expression in neural tube defects. Epigenet & Chromatin. 2019;12(1):76. doi: 10.1186/s13072-019-0318-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhao T, McMahon M, Reynolds K, et al. The role of Lrp6-mediated Wnt/β-catenin signaling in the development and intervention of spinal neural tube defects in mice. Dis Model Mech. 2022;15(6). doi: 10.1242/dmm.049517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chen Z, Lei Y, Cao X, et al. Genetic analysis of Wnt/pcp genes in neural tube defects. BMC Med Genomics. 2018;11(1):38. doi: 10.1186/s12920-018-0355-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nishimura T, Honda H, Takeichi M. Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell. 2012;149(5):1084–1097. doi: 10.1016/j.cell.2012.04.021 [DOI] [PubMed] [Google Scholar]
- [62].Mallik S, Odom GJ, Gao Z, et al. An evaluation of supervised methods for identifying differentially methylated regions in illumina methylation arrays. Brief Bioinform. 2019;20(6):2224–2235. doi: 10.1093/bib/bby085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lent S, Cardenas A, Rifas-Shiman SL, et al. Detecting differentially methylated regions with multiple distinct associations. Epigenomics. 2021;13(6):451–464. doi: 10.2217/epi-2020-0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gutierrez-Gonzalez E, Garcia-Esquinas E, de Larrea-Baz NF, et al. Toenails as biomarker of exposure to essential trace metals: a review. Environ Res. 2019;179:108787. doi: 10.1016/j.envres.2019.108787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Signes-Pastor AJ, Gutiérrez-González E, García-Villarino M, et al. Toenails as a biomarker of exposure to arsenic: a review. Environ Res. 2021;195:110286. doi: 10.1016/j.envres.2020.110286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Huyck KL, Kile ML, Mahiuddin G, et al. Maternal arsenic exposure associated with low birth weight in Bangladesh. J Occup Environ Med. 2007;49(10):1097–1104. doi: 10.1097/JOM.0b013e3181566ba0 [DOI] [PubMed] [Google Scholar]
- [67].Noor N, Cardenas A, Rifas-Shiman SL, et al. Association of periconception paternal body mass index with persistent changes in DNA methylation of offspring in childhood. JAMA Netw Open. 2019;2(12):e1916777. doi: 10.1001/jamanetworkopen.2019.16777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Soubry A, Murphy SK, Wang F, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int J Obes. 2015;39(4):650–657. doi: 10.1038/ijo.2013.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Soubry A, Schildkraut JM, Murtha A, et al. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a newborn epigenetics study (NEST) cohort. BMC Med. 2013;11(1):29. doi: 10.1186/1741-7015-11-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Merrill SM, Moore SR, Gladish N, et al. Paternal adverse childhood experiences: associations with infant DNA methylation. Dev Psychobiol. 2021;63(6):e22174. doi: 10.1002/dev.22174 [DOI] [PubMed] [Google Scholar]
- [71].Pauwels S, Truijen I, Ghosh M, et al. The effect of paternal methyl-group donor intake on offspring DNA methylation and birth weight. J Dev Orig Health Dis. 2017;8(3):311–321. doi: 10.1017/S2040174417000046 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports our research are available from Boston Children’s Hospital upon reasonable request. Given agreements with the Bangladesh Medical Research Council (BMRC), requests for data sharing require their additional review. However, the authors, specifically the corresponding author, invite any and all communication pertaining to the data and are happy to present the data in a setting where the oversight committees have granted permission.