

Abstract
Introduction
Faced to the growing development of collecting systematic molecular analyses in relapsed pediatric cancers to transform their targeted matched therapies, this study aimed to assess the clinical and therapeutic indications of systematic diagnostic genomic explorations performed in pediatric solid cancers to determine which type of screening and if it afford at relapse time an accurate targeted strategy.
Methods
A total of 280 patients less than 22 years, referred at the University Hospitals of Strasbourg for a newly diagnosed solid tumor from January 2015 to December 2021, were prospectively genomically investigated since diagnosis. Using 7 different molecular tests going from single-gene methods (IHC, FISH, RT-PCR, Sanger sequencing, droplet digital PCR) to largescale analyses (Next-Generation sequencing, RNAsequencing and FoundationOne®CDx), we explored retrospectively the molecular findings in those pediatric solid tumors (except hematolymphoid cancers) to improve diagnosis, prognosis assessment and relapse therapeutics.
Results
One hundred and ninety-eight patients (71%) underwent molecular biology (MB) at diagnosis. Thirty-eight different histologies were grouped into cerebral tumors (30%), sarcomas (26%, bone and soft tissues), various blastomas (27%), and other entities (17%). Over a median 40-month follow-up, the overall survival rate of patients was 85% and the relapse rate 28%. Of the 326 analyses carried out, 245 abnormalities (single nucleotide variations: 50%, fusions: 25%, copy number alteration: 20%) concerning 70 oncogenes were highlighted. The overall clinical impact rate was 84%. Broad-spectrum analyses had a higher therapeutic impact (57%) than the targeted analyses (28%). 75% of broad-spectrum tests found an actionable variant conducting 23% of patients to receive rapidly a matched targeted therapy since first relapse.
Conclusion
Our experience highlighted the clinical utility of molecular profiling of solid tumors as soon as at diagnosis in children to expect improving access to innovative agents at relapse.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12885-024-13034-7.
Keywords: Next-generation sequencing, Pediatric brain tumors, Solid malignancies, Precision medicine, Targeted therapies
Introduction
Pediatric cancers represent worldwide a significant health burden and remain one of the leading causes of death during pediatric and adolescent period [1–3]. Nevertheless, they benefited during the last decades from clear improvements in the clinical and therapeutic management, and from molecular technologies investigating oncogenic checkpoints to understand their development process and how to develop individualized treatments based on those actionable genomic lesions [4–7]. Current and precise genomic data will then become crucial since diagnosis to develop effective cancer trials to reduce the burden of childhood and adolescent cancer relapse frequency, optimized current frontline therapy using adequate disease-prognostic biomarkers and gene target matched treatments (TMT), and subsequently improve long-term outcomes [4]. Beyond these medical and health challenges, systematic comprehensive genomic profiling might also contribute to decrease economic and societal burden of pediatric cancers.
The first pediatric precision medicine initiatives were developed subsequently to the personalized medicine approach already validated in refractory adult cancers [8–10]. These previous standardized, high-throughput molecular profiling or targeted genomic analyses were the tribute confirming the real need to implement predictive biomarker–based stratification and maximize clinical efficacy when using TMT. As for adults, the pediatric precision medicine programs are focusing on high-risk oncological situations or relapsed cancers associated with reduced life expectancy [4–6, 11–21]. In those largescale sequencing studies, 50% to 70% of primary pediatric tumors exhibited a potentially targetable genetic event [14–21]. Around 30% of the patients included in such programs had a TMT proposal, which seems to affect positively the patients’ response rate or outcomes. Only two initiatives (e.g., INFORM and PRISM trials) [18, 21, 22] were starting to include a proportion of patients since their diagnosis. Nevertheless, the broad-spectrum technology approaches are for now little performed in routine clinical practice at diagnosis due to their high cost and late results. More routinely suitable techniques are currently used, such as Next-Generation Sequencing (NGS), methylation profiling or specific gene panels [23, 24].
In France, subsequently to the adult and pediatric molecular screening and TMT trials (e.g., MOSCATO-01, SHIVA and MAPPYACTS) [8, 9, 20, 25] was developed the Plan France Médecine Génomique 2025 (PFMG 2025), which proposed systematic tumor molecular profiling to all children and adolescent with refractory malignancies. French health authorities and the operational committee of the PFMG 2025 asked the French “Société Française des Cancers de l’Enfant” (SFCE) to define the pediatric pre-indications of largescale genomic assessments at diagnosis. This evolving diagnostic list notably include now solid tumors like neuroblastomas, osteosarcomas and some central nervous system (CNS) tumors.
In this preliminary context, the primary objective of our study was to assess the clinical impact (change in diagnosis, prognosis, or therapeutic outcomes) of systematic molecular analyses performed prospectively in a cohort of diagnostic solid tumors in children, adolescent and young adults until 22 years from the French region of Alsace. Using different types of molecular techniques, our study aimed to describe retrospectively the molecular strategies according to the tumor type and the molecular findings of the pediatric solid tumors in our center during the seven-year program. Based on those early molecular screenings, we were able rapidly at the first relapse to propose TMT and evaluate their impact.
Material and methods
Study design and ethics
The study was an observational and single-institutional study, conducted in the pediatric onco-hematology department of the University Hospitals of Strasbourg (UHS). It was approved by the UHS ethics committee (CE-2022–8661) and was conducted in accordance with the ethical principles of the Declaration of Helsinki. A written and informed consent for all patients and their parents was obtained prior to genomic analyses and data collection. We included systematically all pediatric patients (under 18 years of age, as defined by the World Health Organization (WHO)) and the young adults less than 22 years referred in our UHS center, newly diagnosed between January 2015 and December 2021 and bearing a solid tumor. The exclusion criteria were hematolymphoid malignancies, extracranial benign tumors and patients lost to follow-up prior to December 2021.
Data collection
Useful patient data (demographic characteristics, histological data, molecular analyses when performed, treatments over time, and clinical outcomes) were extracted retrospectively from the UHS electronic medical files and were entered into the study database. The database comprised for each patient 41 items overlapping all clinical, diagnostic, molecular and therapeutic characteristics. An unfavorable patient prognosis was defined at diagnosis by an expected overall survival probability < 30% as for the Australian PRISM program [14, 21, 22]. The survival rates were based on contemporary literature and recent outcome establishment in the latest protocols. An altered performance status was characterized by a Lansky score ≤ 70% (WHO data). The duration of follow-up was defined until patient death or December 2022 (aiming at least a 12-month follow-up for the latest included patients).
Tumor samples
All patients had a proven histological diagnosis, and indeterminate cases were reviewed by national networks of experienced pathologists in the dedicated cancer subgroups (for example, RENOCLIP-LOC (French network of neurooncology histological reviewing) for brain tumors or RESOS (French network of bone tumor histological reviewing) for bone sarcomas) [26, 27]. Tumor samples were obtained by biopsy or resection of the primary tumor or metastases, and sometimes blood or marrow samples were needed (e.g., neuroblastomas). Genomic analyses were performed on formalin-fixed paraffin embedded (FFPE) or fresh-frozen tissue. The percentage of tumor cells was collected, with a pejorative threshold defined when less than 70%.
Molecular analyses and data integration
Genomic analyses were performed at diagnosis according to trial instructions, national and/or UHS center guidelines, and they were repeated if recurrence. Analyses were grouped as follows: a) targeted panel for single gene sequencing (Sanger Sequencing (for targeted mutation search), reverse transcription (RT)-qPCR (for targeted fusion search), digital droplet Polymerase Chain Reaction (ddPCR), protein testing (immunohistochemistry (IHC), and fluorescent in situ hybridization (FISH) testing), b) broad-panel sequencing by Next Generation Sequencing (NGS) performed locally with small panels (from 13 fusions up to 52 genes’ mutations) or largest panels that are FoundationOne®CDx analyses (FO, Foundation Medicine®, Inc.) (from 311 to 406 genes), Comparative Genomic Hybridization (CGH)-array and RNAsequencing (RNAseq).
For each analysis, the samples were prepared as recommended for the appropriate technique. Total DNA and/or RNA were extracted with the automated technique previously published [28]. The tumor slides were prepared, as recommended for IHC and/or FISH analyses, using a microtome (Microm HM 355S, Thermo Scientific™) to obtain a slide section of 4-µm thickness. The Sanger sequencing and RT-qPCR were the oldest molecular techniques performed to assess BRAF, KRAS, EGFR, IDH1 and IDH2 mutations or sarcoma and brain tumor fusions. Relayed by the ddPCR, this molecular assessment was based on the ultra-sensitive QX200 (BIO-RAD®, Hercules, CA, USA) technology, as described previously [29–31], and focused on brain tumor and sarcoma fusions, as well as copy number variations (CNV) of the following genes: CDKN2A, RB1, MET, EGFR and mutations of BRAF, EGFR, RAS genes. Using an automated pathology platform of analyses (Ventana Medical Systems, Inc., Tucson, AZ, USA, and a Leica automated Biosystems, Nanterre, France) [30], IHC and FISH focused on the presence of the following protein quantifications, gene’s mutations or fusions: β-catenin, INI1, H3K27m3, PTEN, CDKN2A, EGFR, ATRX, SDHb, BRAFV600E, IDHR132H, EWS fusions, ZFTA fusion, PAX-FOXO1 fusions.
Among the broad-spectrum molecular technologies, we had NGS approaches using both mutation and fusion screenings performed at Department of Cancer Molecular Genetics of UHS center, which was labelled by French National Institute of Cancer. For DNA assessments, using 2 panels detailed in supplementary tables 1, mutation screening was performed by Next Generation Sequencing (NGS) on a MiSeq Illumina platform using Tumor Hotspot MASTR Plus assay (Multiplicom-Agilent) (26 targeted genes) or custom panel (SureSelectXTHS- Agilent) (52 targeted genes). Sequencing data were aligned to human genome hg19 using BWA-MEM algorithm (Burrows-Wheeler Aligner-Maximal Exact Matches). Variants were called using three different variant callers: VarScan, GATK HaplotypeCaller, and GATK UnifiedGenotyper. The minimum coverage per base and variant allelic frequency were fixed at 300-fold and 4%, respectively. Data were visualized using the Integrative Genomics Viewer (Broad Institute). The RNA fusion screening on 13 targeted genes was performed on a MiSeq Illumina platform using Archer FusionPlex Lung panel v1.0 (ArcherDx®) according to the manufacturer protocol. Interpretation of sequencing data was performed using ArcherAnalysis V6.0.4. The hotspot coverage for fusion detection is detailed in supplementary Table 2.
The FoundationOne®CDx analyses are comprising three different tests: FoundationOne CDx assay, FoundationOne Liquid on liquid biopsy and FoundationOne Heme (using RNA sequencing performed usually on sarcoma samples). The FoundationOne CDx assays (324 targeted genes) and FoundationOne Heme tests (406 DNA screened genes associated to 31 introns of genes involved specific sarcoma rearrangements and 265 RNA targeted genes) were performed on FFPE samples [32]. The liquid biopsy adapted test was focusing on cell-free extracted DNA from plasmatic specimens [33]. The genes of those tests are detailed in supplementary tables 3 and known to be somatically altered in human cancers and validated as targets for therapies. RNA sequencing method as described previously [34] was performed on RNAs qualified by NanoDrop (ThermoFisher Scientific) and their quality was controlled (DV200 value cutoff > 30%) by TapeStation with Hs RNA Screen Tape (Agilent, Courtaboeuf, France). The libraries with the TruSeq RNA Access Library Prep Kit (Illumina, San Diego, USA) were prepared on 100 ng of total RNA and pooled at 4 nM with 1% PhiX. Sequencing was performed with a NextSeq 500/550 High Output V2 kit on an Illumina NextSeq 500 (Illumina) machine. The fusion transcripts were called with STAR-Fusion, FusionMap, FusionCatcher and EricScript. Expression profiles were extracted from fastq files with Kallisto and transformed as log2(TPM + 2) prior to quantile normalization using the Limma package v3.32.2 performed in the R environment v3.4.1. Genes with a coding sequence annotation (based on set GRCh38p5 annotation) and with a maximum expression value above 2 across all samples were considered. For sarcomas, a t-SNE analysis was done with a perplexity of 4, using the ConsensusClusterPlus v1.46.0 and Rtsne v0.15 R packages, respectively. A supervised clustering was performed using a Welsh t-test and a gene set enrichment analysis using gsea-3.0 with all the gene sets of MSigDB v6.2.
Complementary analyses, for medulloblastomas, as recommended by the French national CNS tumor committee and treatment trials, for each tumor, were assessed by CGH-array using Agilent arrays and nanoString targeted gene-expression profiling from frozen samples, as previously described [23, 35]. For the subgroup of neuroblastomas, upon the NMYC status, a CGH-array, as described in medulloblastomas, was performed to determine the numerical and segmental chromosomal aberrations [36].
Molecular findings and reports associated to clinical impact and flow
All analyses were considering tumor cellularity and led to a molecular report provided by a molecular geneticist. In case of targeted anatomopathological analyses, the results were integrated into the histological report. Depending on analysis, the molecular biology conclusions were considering somatic molecular aberrations including single nucleotide variants (SNVs), small insertions/deletions, CNVs, gene fusions, other structural variants, specific gene mutations, or overexpressed gene/proteins. The CGH profiles for neuroblastomas and medulloblastomas were systematically integrated to patient chart. The clinical impact of each molecular test was defined as a change in terms of diagnosis, prognosis, or therapeutics. Molecular biology results (MB) were classified into three categories: 1) informative MB discovering significant molecular abnormalities for the patients (mutation, fusions, SNVs), 2) informative MB highlighting only CNVs and numerical or segmental chromosomal aberrations, and 3) non-contributive MB (absence of any molecular alterations or CGH modifications). The molecular alterations were then categorized into “immediately actionable target” (in the case of an available reference targeted therapy "ready for routine use") or “potentially actionable target” (in the case of a validated or investigational agent targeting the concerned oncological pathway). The molecular event relevance was determined using currently available guidelines and databases (such as the catalogue of somatic mutations (COSMIC), cBioportal, OncoKB, etc.) or published works. According to the clinical context, constitutional mutations were searched and led to family genetic counseling.
Each case was reviewed by a Molecular Tumor Board (MTB) involving pathologists, biologists, and pediatric oncologists. Potential TMTs were discussed at every stage of disease management and decision to treat was at oncologist’s discretion, preferably within the context of a clinical trial or national recommended guidelines for each entity.
Statistical analyses
Two populations of patients (Fig. 1a) were defined: the overall population of eligible patients to describe the demographics of children treated for a solid tumor in the UHS, and the subgroup of patients with MB performed at diagnosis to assess the clinical impact of this genomic testing (Table 1). Descriptive analyses were carried out. Study parameters were described using mean and standard deviations or medians and interquartile ranges for continuous variables, and proportions for categorical variables. Normality of the distributions was tested using the Shapiro–Wilk or Kolmogorov–Smirnov test. There are no missing data for diagnoses or molecular biology results. For the other covariates, missing data were treated by simple deletion. Comparisons between categorical variables were carried out using Chi-squared test or Fisher’s exact test in the case of expected values in any of the cells of a contingency table were < 5. The significance level was set at 5% and tests were two-sides. Analyses were performed using R 4.2.1 software.
Fig. 1.

Flowchart describing the molecular biology in the cohort and their results (a), and its paired Sankey diagram (b). FISH, fluorescent in situ hybridization; IHC, immunohistochemistry; MB, molecular biology; NGS, next generation sequencing; PCR, polymerase chain reaction; RT, reverse transcription. Broad-spectrum procedures are represented in bold. *Methylome, whole exome, genomic profile, array-CGH, microsatellite status assessment (allelotyping)
Table 1.
Patient baseline demographics, epidemiology, molecular and clinical characteristics. (MB = molecular biology subgroup)
| Characteristics | ||
|---|---|---|
| Demographic | Number (%) | |
| Age at diagnosis (years) | Median (IQR) | 7 years (2-14) |
| <2 | 39 (20) | |
| 2 – 12 | 85 (43) | |
| 12 - 18 | 65 (33) | |
| 18 – 22 | 9 (5) | |
| Gender | Female | 104 (53) |
| Male | 94 (47) | |
| Oncological family history | Yes | 38 (20) |
| Cancer at diagnosis | Number (%) | |
| Symptomatic | Yes | 191 (96) |
| Lansky score | <70% | 36 (19) |
| ≥70% | 149 (81) | |
| Disease stage | Early | 150 (76) |
| Metastatic | 48 (24) | |
| Prognosis | Favorable | 79 (40) |
| Unfavorable | 119 (60) | |
| Tumor biomarkers | Yes | 69 (35) |
| Inclusion in a clinical trial | Yes | 132 (67) |
| Sampling | Number (%) | |
| Sample type | Tissue | 332 (89) |
| Blood | 31 (8) | |
| Bone marrow | 11 (3) | |
| Surgical technique | Biopsy | 93 (47) |
| Surgical resection | 100 (53) | |
| Preservation | FFPE | 235 (72) |
| Fresh Frozen | 80 (25) | |
| Ficoll | 11 (3) | |
| Tumor cellularity | <70% | 95 (38) |
| ≥70% | 157 (62) | |
| Tumor site | Primary tumor | 310 (91) |
| Metastatic site | 29 (9) | |
| Number of samples per patient | Mean (DS) | 1.2 (0.4) |
| Molecular biology tests | Number (%) | |
| Patients with MB tests performed at | diagnosis | 173 (87) |
| diagnosis and relapse | 25 (13) | |
| Time to MB after surgery (days) | Median (range) | 0 (0-449) |
| Number of MB tests per patient | Mean ±DS (range) | 1.5 ±0.7 (1 to 7) |
| 1 MB test | 123 (62) | |
| MB tests | 57 (29) | |
| ≥3 tests | 18 (9) | |
| Number of MB tests per sample | Mean (range) | 1,2 (1 to 2) |
| Molecular biology tests (n=334) | Number (%) | |
| Targeted molecular tests | Digital PCR | 37 (11) |
| FISH/IHC | 28 (8) | |
| RT-PCR | 28 (8) | |
| Sanger Seq | 7 (2) | |
| Broad-spectrum molecular tests | NGS | 121 (36) |
| RNA Seq | 38 (11) | |
| Foundation One CDx | 21 (7) | |
| Other various MB tests | * | 54 (16) |
| Performance (at least one observed variant) | ||
| Overall performance ** | Variant(s) | 245/376 (65) |
| Combined test performance *** | ||
| Targeted MB tests (n=100) | Observed variants | 64 (58) |
| Broad-spectrum MB tests (n=180) | Observed variants | 116 (74) |
| Positivity of each MB test | FISH/IHC | 22/28 (79) |
| RT-PCR | 17/28 (61) | |
| Sanger Seq | 2/7 (29) | |
| Digital PCR | 23/37 (62) | |
| NGS testing | 68/121 (56) | |
| Foundation One CDx | 19/21 (90) | |
| RNA Seq | 29/38 (76) | |
| Number of molecular variants per patient | Mean (range) | 1,2 (0 to 11) |
| 1 variant | 61 (70) | |
| 2 variants | 15 (17) | |
| ≥3 variants | 11 (13) | |
| Outcome | Number (%) | |
| Duration of patient follow-up (months) | Median (IQR) | 40 (21-59) |
| Relapse | Yes | 55 (28) |
| Death | Yes | 29 (15) |
| Matched therapy received | Yes | 45 (23) |
| Targeted therapy according to MB results | At diagnosis | 32 (71) |
| At relapse | 11 (25) | |
| According to protocol | 2 (4) | |
| Death despite matched therapy | Yes | 20/45 (44) |
| Constitutional exploration performed | Yes | 14 (8) |
| Positivity of germline exploration | Yes | 10/14 (71) |
*Other various MB tests: exome, methylome, genomic profile, microsatellite status
**Other various MB tests included
***Other various MB tests excluded
Results
Patients and tumors characteristics
From January 2015 to December 2021, an overall population of 280 newly diagnosed eligible children was reported in our study and described in the study and flowcharts of Fig. 1 (standard flowchart in a) and Sankey diagram in b)). Among these children, 198 (71%) had at least one diagnostic MB (MB subgroup) (Fig. 1 and Table 1) on surgically accessible diagnostic tumors focusing on all solid tumors except lymphomas.
The yearly number of pediatric cancerous patients included in the study was broadly stable over this period (an average of 40 cases per year), and 62 to 80% patients per year had MB at diagnosis (Fig. 2). One hundred and seventy-three patients had diagnostic genomics (light green proportions on Fig. 2) and 25 had both diagnostic and relapse biology assessments (dark green proportions on Fig. 2).
Fig. 2.

Percentage of molecular biology performed over the 7-year period of the study in 284 patients. In grey, the percentage of patients not having any molecular biology (MB) analyses in solid tumors where a surgery was performed. In light green, the percentage of patients benefiting from a diagnostic MB. In dark green, the patients where diagnostic and relapse molecular assessments were performed with our molecular methods
Few MB tests were only performed at relapse (n = 4, < 2% of patients) during this period in our center. In fact, during the same period of inclusion, 35 other relapsing patients were included from 2016 to 2020 into the MAPPYACTS trial [20] and, during 2021, seven solid tumor relapses were integrated in the FMG2025 program. None of those relapses’ results (exome and RNA sequencing were systematically performed) were integrated in our analysis. Another dedicated tumor board was decided the TMT for those relapsing patients.
Concerning the clinical overall population characteristics, the solid tumor types were comprising CNS cancers in 30% [glial (ependymomas and gliomas) and embryonal tumors, and other rarer entities], bone and soft tissue sarcomas in 26%, malignant blastomas in 27%, and other histopathology in 17% [comprising carcinomas, extracranial germ line tumors and teratoid and rhabdoid forms, neuroendocrine cancers, and malignant peripheral nervous system tumors (MPNST)] (Table 1 and Fig. 3a).
Fig. 3.

Tumor type and molecular tests’ distribution, as well as proportions of molecular analyses performed according to histology groups. a Analyses performed in the overall population of 284 patients. Each color is paired with specific histologies (e.g., blue color for Central Nervous System (CNS) tumors; light orange for sarcomas, light green for blastomas and violin for multiple rare entities). The proportion of tumors explored by Molecular Biology (MB) is detailed in each histological group. b Percentages of molecular tests and details on the histologies per tests. (MPNST = malignant peripheral nervous system tumors; DIPG = diffuse intrinsic pontine glioma; ATRT = atypical teratoïd and rhabdoid tumor)
A total of 38 different histologies were identified. We screened 71% of the entire patient population (198/280) and 85% for CNS tumors. Among the 82 tumors without molecular analyses were listed 24% of germ line tumors, 17% of nephroblastomas, 14% of osteosarcomas (all were classified as good responders to neoadjuvant chemotherapy) and all carcinomas. All patients of the MB subgroups and their paired sample characteristics are described in additional Table 1. Patients were female in 53%, and not older than 18 years in 95% [median age, interquartile range (IQR): 7 (2–14) years]. Of these 198 patients, 173 (87%) benefited from diagnostic MB only, and 25 (13%) were explored at diagnosis and relapse. Disease prognosis was unfavorable in 60% of the cases, and metastatic disease was present since diagnosis in 24%. Patients were included in multinational trials in 67% (n = 132), and 38 patients (20%) had a family history of cancer in first- or second-degree relatives.
Regarding the analyzed samples summarized in Table 1, MB tests were mostly performed on tumor-tissue samples (89%) obtained by resection (51%) or biopsy (47%). Liquid biopsies (e.g., 8% of blood or 3% of marrow out of the total samples) were less frequent and mostly obtained in neuroblastomas (9%). FFPE specimens were the most frequent storage modality (72%). Tumor specimens came from primary sites in 91% of cases, with a satisfying tumor cellularity rate (≥ 70%) in 62% of the total samples. No significant correlations between the tumor cellularity and the type of surgical technique were observed (poor tumor cellularity concerned 43% of biopsies and 57% of resections, p = 0.6). Among the 198 patients with MB, 252 samples were obtained with a mean of 1.2 ± 0.4 sample per patient (range: 1–3). Sixteen patients were benefiting from a second analysis, 15 patients were finally explored by liquid biopsy, and 20 patients were needing a second biopsy or bone marrow analysis in case of medullary metastatic disease.
Molecular approach distribution and implementation
The median time between surgery and molecular biology was less than 1 month except for 29 patients (15%). These late testing was requested at tumor progression on first line standard therapy or for a poor prognostic evolution of the tumor during standard therapeutic management (e.g., poor chemoresponse or metastatic resistance after neoadjuvant chemotherapies). It was done one diagnostic sample. A total of 334 molecular tests were performed with a mean of 1.5 ± 0.7 test per patient (range: 1–7). Forty percent of patients had more than one MB. Finally, only 8 samples (2%) on the entire molecularly explored population were not assessed because of sequencing failure, mainly comprising osteosarcomas. These unsuccessful MB were due to technical issues, necrotic sampling, and/or a poor quality of DNA/RNA obtained from diagnostic samples usually of very small size and having decalcification process before DNA/RNA extraction (Fig. 1 and Table 1).
Considering broad-spectrum analyses (NGS/FoundationOne®CDx/RNAseq) (Fig. 1, Fig. 3b and Table 1), NGS was the most frequently performed (36%), followed by RNAseq (11%). Among the targeted gene/protein analyses (FISH/IHC, RT-PCR, Sanger sequencing or ddPCR), an average of 8% of each test (except Sanger sequencing) was used for molecular analyses. Their results were mostly confirmed by other techniques. Other various MB tests were carried out in few samples (54/334) including whole exome, methylome, and microsatellite status to determine notably the tumor mutational burden. Sarcomas, as well as CNS tumors, were benefiting from almost all techniques depending on the study time and the mutations/fusions’ discovery (Fig. 3b). In fact, in our cohort, a turning point was reached in 2017, where broad-spectrum analyses became more frequently performed comparatively to targeted analyses (6% versus 27% in 2016; 18% versus 7% in 2020; p < 0.05). This progression was based on the evolution of molecular knowledge and necessity over time to target multiple genes in pediatric cancers, which was supported by the DGOS and French National Institute of Cancer (INCa) through labelled infrastructures like our hospital platform of Cancer Molecular Genetics. The FoundationOne®CDx assays were mostly prescribed for rarer entities like hepatoblastoma, where NGS techniques were not contributive due to an unsuitable panel, or in case of non-accessible amount of DNA concentration in the tumor specimens needing liquid approaches to assess the molecular biology (e.g., samples with a high necrotic burden or in case of unavailable tumor tissues, n = 15).
The ddPCR was usually sufficient to capture rapidly the targeted gene CNVs like for RB1 or CNDKN2A or in case of specific mutation (for example, BRAFv600e) covering a small range of diagnoses. Ewing sarcomas and rhabdomyosarcomas (RMS) were, by the past, explored by RT-qPCR looking to a restricted panel of fusions most frequently present in those tumors and, only in case of negativity, the exploration was complemented by RNAseq. This diagnostic molecular assessment strategy was evolving during the period of the study. The final distribution of RT-qPCR and RNAseq represents 80% and 55%, respectively, in the entire population of sarcomas, leading now to a systematic first line RNAseq exploration (by either NGS Archer multiplex or larger RNAseq). For osteosarcomas, the highly necrotic sampling and the genomic DNA chaos led to combine NGS and FoundationOne®CDx assays to obtain quality results.
Considering the molecular implementation presented in Table 1, in Fig. 1 and Fig. 3b, MB tests highlighting molecular variants is reaching 70% of the MB subgroup. Individual test performance, illustrated by at least one observed variant, reached 90% for FoundationOne®CDx (19/21 tests), 76% for RNAseq (29/38), and 56% for NGS (68/121). As expected, broad-spectrum analyses showed a better ability to detect alterations than the targeted tests (74% versus 58% of positivity). Based on the histopathology, CNS tumors and Ewing sarcomas had a positivity rate in MB analyses higher than 75%, and medulloblastomas and neuroblastomas around 50%. However, NGS analyses were negative in 65% of RMS, when considering CNVs and/or fusion detections, in 65% of osteosarcomas, when considering SNVs or mutations, and in all nephroblastomas. Thanks to the CGH-array results of the 10 medulloblastomas and the 33 neuroblastomas, an adequate subgrouping as proposed in the international classification [36, 37] was achievable. These analyses provided the NMYC or MYC amplification research, which was positive in 27% of neuroblastomas. None of the medulloblastomas in our cohort had any MYC amplification.
Identification of somatic molecular events and their prioritization
Among the 326 molecular tests successfully performed, 245 molecular alterations were detected, with a mean of 1.2 molecular alteration per patient (range: 0–11) (Fig. 1a). Fifty-one patients had only one molecular abnormality, and 32 patients’ tumors were not bearing any rearrangements or mutations. Among the 245 observed pathological variants, 130 (53%) were SNVs, 62 (25%) were fusions, 47 (19%) were CNVs, and 6 (2%) were unspecified variants (variant of uncertain significance (VUS), dysmethylation). All mutation details are provided in Table 2. One hundred eighty-two significant abnormalities were identified in 70 different genes. Among these molecular aberrations, we selected the most common abnormalities that were 11 targetable anomalies, and 4 somatic events mostly involved as diagnostic tools and presented in Fig. 4a and b. All these 15 genes were present in more than 4 analyses. The actionable abnormalities were mostly in BRAF gene (n = 32) (e.g., BRAFv600e mutation in 15 cases and KIAA1549-BRAF fusion in 16 low-grade gliomas), NMYC CNV (n = 13), CTNNB1 mutations (n = 10), and SMARCB1 mutation (n = 10). More rarely, RB1, TP53, CDKN2A, ALK, NOTCH, IDH1/2 or PI3KCA were affected. Figure 4b also depicts the therapeutic actionability (concordance between each targetable oncogene and potential matched targeted therapies). Otherwise, non-targetable aberrations, like H3F3A K27M mutation or EWS-FLI1 fusions, as well as a global hypomethylation in tumors like DIPG or group A ependymomas, presented clear diagnostic or prognostic impact. BRAF, MEK, ALK, mTor and EZH2 inhibitors were mostly prescribed following those MB at relapse time (in light blue words on Fig. 4b abscissa).
Table 2.
Oncogene variants and actionability for the alterations in more than 2 patients-molecular biology subgroup

SNV single nucleotide variation, CNV copy number variations, CNS central nervous system, RMS rhabdomyosarcoma, TNGM malignant non germ line tumor, DIPG diffuse intrinsic pontine glioma, ATRT atypical teratoid rhabdoid tumor
Fig. 4.
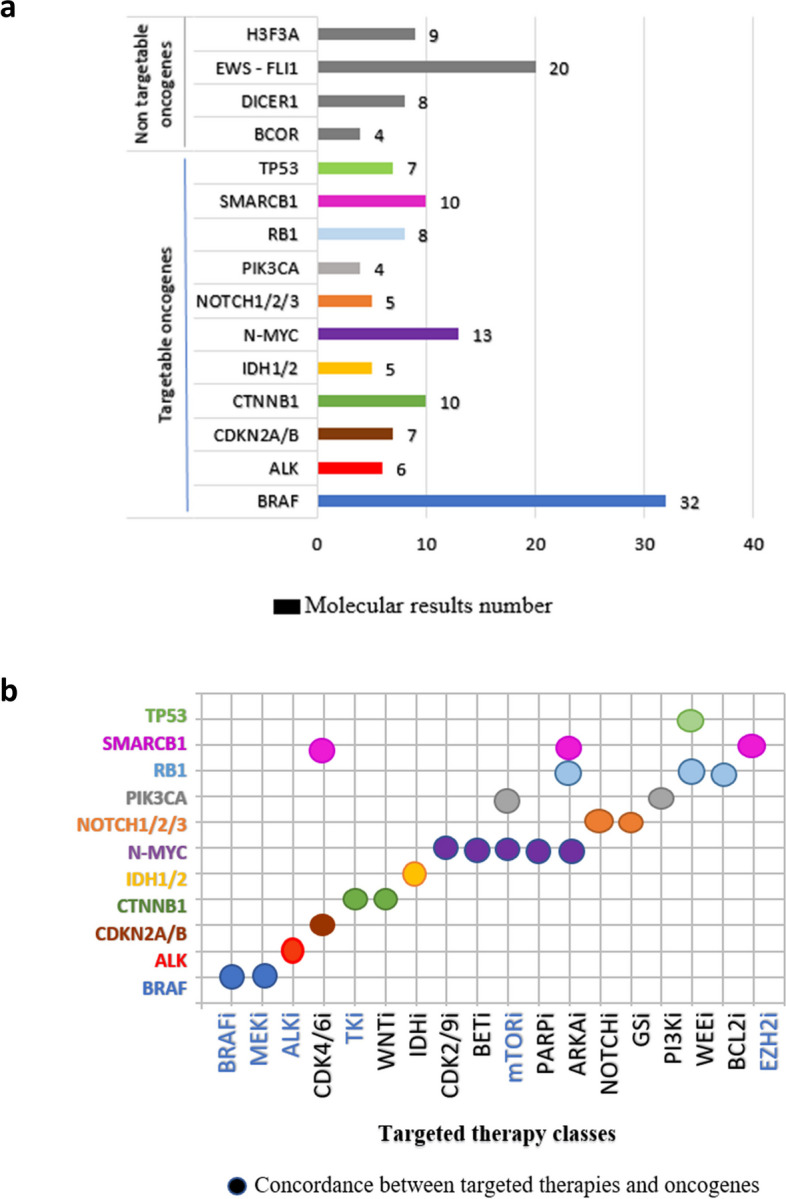
Distribution of the most frequent altered oncogenes and their potential therapeutic actionability. a Distribution of the most frequent and common reportable altered oncogenes and their potential therapeutic actionability (actionable in several color types and non-targetable in grey bars). b Correlation graph between the molecular targets and their matched treatment classes. In light blue words on abscissa, are pinpointed the inhibitors prescribed in the cohort at relapse time to target the identified molecular abnormalities
No new mutational signature or pathway involvement linked to a currently known TMT was identified with diagnostic screening in our cohort. We did not analyze the episodical abnormalities in depth in tumors’ RNAseq. Thirty-four somatic mutations (e.g., DICER1, RB1, VHL, BLM) led to genetic counseling. Based on patient and familial past-history, constitutional oncogenetic appropriate screening was performed in 14/198 patients (7%, Table 1), where 10 patients were bearing a constitutional abnormality leading to patient and familial genetic counseling and follow-up.
Clinical implementation and impact
The overall rate of the MB testing clinical impact was 84%. The analyses confirmed the diagnosis in 59% of cases, modified the prognosis in 48%, and underlined a potential therapeutic actionability in 52% (Fig. 5a). Targeted and broad-spectrum MB tests had similar diagnostic and prognostic performances, but therapeutic actionability reached 57% in largescale approaches versus 28% in targeted tests (p < 0.05) (Fig. 5b).
Fig. 5.

Diagnostic, prognostic, and therapeutic impacts according to the different molecular analyses (targeted versus broad-spectrum analyses). Clinical impact according to the entire explored population in (a), based on the extension of the molecular analyses (targeted versus broad-spectrum analyses) in (b) and impact among the broad-spectrum biology in c). This impact was calculated on the analysis of 288 procedures, after exclusion of the other various tests (e.g., methylome, whole exome, genomic profile, array-CGH, microsatellite status assessment)
The overall clinical impact was similar between the three largescale tests (RNAseq, versus FoundationOne®CDx versus NGS panel) (Fig. 5c). By detailing performances, RNAseq had a better diagnostic performance, FoundationOne®CDx a better prognostic performance, and therapeutic actionability was similar for NGS and FoundationOne®CDx testing (around 65%).
Regarding therapeutic actionability on the total 139 significant variants, “immediately actionable” variants were present in 47% of the cases and “potentially actionable” in 53% that accounted for a mean of 0.7 targetable variant per patient, with a total of 139 variants.
According to histologies, the CNS tumors were characterized by the highest range of targetable abnormalities with an immediate actionability in 90% of the cases (Fig. 6a). On the other hand, considering the low actionability, osteosarcoma molecular profiling and genomic implementation did not reach the TMT impact expected with such analyses. Osteosarcoma results mostly showed an absence of rearrangements excepted for certain types of CNVs. The same observation was made in Ewing sarcoma cohort where, despite the fusion, no other target was usually diagnosed. Only two patients (one melanoma and one grade II sarcoma) benefited in first-line therapy of a BRAF inhibitor and a mTOR inhibitor, respectively.
Fig. 6.

Therapeutic actionability of targetable variants according to histology classes and the use at relapse time. a Summary of detailed variants in each tumor types. Actionable targets are in bold, and the targets used at relapse time are in red (CNS, central nervous system; RMS, rhabdomyosarcoma; gglNB, ganglioneuroblastoma). b and (c) Comparative results at diagnosis and relapse times in 25 patients explored with the same MBs
Having metastatic cancer increased by 1.8 time the probability to have a clinical impact even if it was not statistically significant (p = 0.22). The rare relapses analyzed in this cohort (e.g., 25 cases) showed identical rearrangements than diagnostic analyses except for seven cases (28%) (Fig. 6b and c): one low-grade glioma where an additional abnormality on EGFR gene was present at recurrence, 4 neuroblastomas where ALK mutations became detectable in 2 cases, NRAS mutation in one case and a CDKN2A deletion evidenced in one patient, and 2 Ewing sarcomas, where a TP53 mutation and a FGFR3 mutation were respectively and additionally detected to the standard fusion. The small number of analyses cannot be contributive to significantly interpret a potential tumor heterogeneity across recurrences and/or the selection of a specific and resistant tumor subclone.
Outcomes and therapeutics
Based on a median follow-up of 40 months (IQR: 21–59), 28% of patients (55 cases) experienced one relapse, 2.5% a progression during first-line therapy (5 cases) and 15% deceased during the study period. An unfavorable initial prognosis multiplied by 3 (95% CI: 1–7) the relapse risk (p < 0.05) and by 24 (4–989) the death risk (p < 0.05). Ninety-one percent of diagnoses with an unfavorable prognosis benefited from molecular screening upon diagnosis, while tumors with a favorable prognosis were explored in 62% of cases. Deaths and recurrences were mainly associated to CNS tumors (Table 1). After national or local multidisciplinary MTB (comprising the treating pediatric oncologists, early phase trial oncologists, molecular pathologists, disease-specific pediatric tumor oncologists/biologists and bioinformaticians) discussion, a total of 45 patients (23%) received a relapse strategy, 73% of them according to results of MB analyses performed at diagnosis, 22% according to the diagnostic/relapse results, and 5% in line with clinical trials’ instructions (Fig. 6a and b/c where we detailed 25 patients where MB was done locally at diagnosis and recurrence, Table 2). In these 45 patients, 19 different TMTs were administered subsequently to diagnostic screening results. Eight major oncogenes or signaling pathway (BRAF, PTEN, RAS/MAPK/mTOR pathway, PDGFRA, EGFR, VEGFR, ALK/ROS1, and IDH1) were targeted by inhibitors: in 26% by a tyrosine kinase inhibitor (TKi), in 21% a mTOR inhibitor (mTORi), in 16% a VEGF inhibitor (VEGFi), in 16% a MEK inhibitor (MEKi), in 7% ALK/ROS1 inhibitors and 7% of BRAF plus MEK inhibitors. Despite early treatment administration at recurrence, death occurred rapidly in 20 patients (44%) (survival: 2–6 months). The remaining 25 patients were defined by a 15-month median survival (11 to 36 months) pooling sarcomas, neuroblastomas, brain tumors (e.g., medulloblastomas, high-grade gliomas and low-grade gliomas) and rare entities. When considering the comparison between TMT strategy and chemotherapies (done at relapse because of recommendations and/or no available gene deregulation to target), the outcome was a 12-month median survival (3–36 months) for the group of TMTs and a 13-month median survival (2–36 months) in patients with chemotherapies. For the TMT group, at 4 months, we had a majority of stable disease (75%), 15% of progressive disease and 10% of partial response. In the chemotherapy group, almost the same proportions were observed and only two patients are still in persistent complete remission.
Discussion
In line with the recent high throughput sequencing pre-indications studied by PFMG program and recent publications [9, 14–18, 21, 22], we wanted to assess our past diagnostic molecular explorations’ practices and their clinical impact in all pediatric solid tumors except for hematolymphoid cancers. Based on baseline demographic, clinical outcome and therapeutics, our cohort overall survival rate was 85% at 3.3 years even with 60% of unfavorable prognosis cancers which is similar to those published by INCa (80% at 5 years for all pediatric malignancies in high-income countries) [37]. To improve those rates, the personalized medicine approach has already been validated in the population of refractory or recurrent tumors [6, 12, 14, 18–22]. To envision this precision medicine approach for all kids bearing a cancer, routine NGS testing was previously shown to play an important role in diagnosis, risk stratification, and prognostication and to have a therapeutic impact in more than 50% of pediatric solid cancers [38–41], reaching 69% in our smaller cohort where we predominantly performed this technique. Apart from similar results for the therapeutic impact of molecular profiling in children with solid tumors, all MB included, our study provides as expected from those previous publications complementary information on diagnosis, prognosis, but also on overall clinical impact including therapeutics. As we have only diagnostic tumors, where we mixed 60% unfavorable-estimated cases and 40% of favorable situations, the final numbers might be different based on these differences on the cohorts themselves. Nevertheless, we were able to screen efficiently the high-risk tumors and provided a high rate of screening with contributive results similar to the studies focusing on relapses’ screening. As already demonstrated in those previous studies, since CNS tumors have poor prognosis with fewer effective therapeutic first-line options, they gained from the integration of broad-spectrum panels into routine diagnostic molecular explorations able to detect largely all tumoral variants. We were able to evidence in those high-risk CNS tumors such as high-grade gliomas several targetable variants associated with potential therapeutic targets [20–22, 25, 38, 39], leading to a higher rate of MTB recommendations and a significant therapeutic impact in more than 50% of the children when anticipating the strategy since diagnosis. Consistently with previous TMT strategies [16–21], we reported that 75% of broad-spectrum MB tests highlighted actionable variants associated to a clinical utility at diagnosis around 80% but with a therapeutic impact twice higher than targeted tests (57% versus 28%, p < 0.05). However, molecular testing recommendations might be adapted to histological groups. Currently, some histologies do not require systematic broad-spectrum explorations at diagnosis as sufficient molecular information can be provided by a less expensive and faster targeted technique notably in Ewing sarcomas, retinoblastomas or rhabdomyosarcomas [15, 41, 42].
Focusing on molecular variant, we detected 53% of SNVs, 25% of fusions, and 20% of CNVs differing from other studies that targeted a tumor class or specific MB tests. Agreed with data from the MAPPYACTS trial or other precision medicine programs, the most frequent abnormalities concerned the RAS/MAPK pathway in 13% [12, 14, 18–22, 43]. TMT based on genomic findings were administered in 23% of our tested cohort since first relapse, which is a higher proportion than previously reported exclusively in relapsing cohorts and almost similar to recent initiatives including both diagnostic high-risk patients and relapses [16, 18–21]. This TMT strategy was as efficient as the standard chemotherapies usually proposed with similar median survivals. TKi, mTORi, VEGFi and MEKi represented 80% of the prescribed targeted therapies in our cohort in line with the international practices and current available compounds. Based on that, our aim of an early molecular profile of pediatric solid tumors would offer therapeutic contingency plans in the event of a first relapse to improve patient survival, while offering an accurate diagnosis. These strategies seem to be more carefully thought out right from the diagnosis stage to limit first-line treatment toxicity and afford precision medicine according to initial diagnostic risk assessment [6, 22]. Even if high-risk neuroblastomas are now one of validated pre-indication for targeted therapy since diagnosis (with anti-GD2 monoclonal antibodies), as well as midline and hemispheric high-grade gliomas or BRAFv600e-mutated gliomas, an extent of these pre-indications is expected in the future using more and more MEKi, TKi or immunotherapy in single or combination approaches [34, 44, 45]. Those diagnostic largescale biology tests might also afford using at an early stage those targeted therapies to decrease tumor resistance mechanisms and can also help the discovery of relevant relapsing biomarkers to evaluate the level of intra-tumoral heterogeneity resulting from the clonal evolution of tumor. They can also help to detect mutations involved in germline predisposition syndromes (e.g., the following genes in our study: NF1, NF2, BLM, VHL, APC, RB1, TP53, DICER1) that led to family genetic counseling and possible appropriate follow-up.
For now, repeated explorations on new samples had been done at the time of each relapse to ensure the absence of resistant molecular abnormalities and optimize the MTB discussions to decide accurately whether molecular abnormalities should be targeted. In this context, the development of liquid biopsies gives hope due to a less invasive follow-up [20, 33, 46, 47]. This non-invasive genomic profiling might be useful for recurrent pathologies (difficult surgical access and surgery-related morbidity) and an opportunity to follow the minimal residual disease. MAPPYACTS trial suggests a 75% correlation between the detection of actionable alterations in circulating-free DNA and tumor tissues [20, 46]. This correlation was at 100% in our study, but a limited number of patients was concerned.
Our study revealed also our limitations and biases, as it was a single-institutional experience and non-randomized design, where we analyzed together different pediatric solid tumors and different molecular biology tests. This is the reason why categorizations of histologies and MB tests were used. However, this heterogeneity made it difficult to interpret some final proportions notably the constitutional disease percentages and the final responses to targeted therapies even such strategies might replace accurately second line chemotherapies with the same median survivals. At last, there is a wide molecular heterogeneity between histology with molecular profiles ranging from “rich” to “silent”. The poor genomic results in osteosarcomas and the non-informative NGS testing in entities like hepatoblastomas suggest the potential necessity of specific panels by histology or the use of systematic broader sequencing technique in all patients like in the PFMG2025 plan (exome and RNAseq) or using dedicated epigenetic approaches in sarcomas to pick up the specific targets [47–51].
Conclusion
In our pediatric cohort, we reported a significant impact of largescale molecular profiling at diagnosis able to modify prognosis and treatment strategies at relapse time. Our findings tend to encourage the implementation of broad-spectrum sequencing analyses, particularly in high-risk tumors and validated its feasibility in routine oncology clinical care at early steps of the pediatric cancerous disease.
Supplementary Information
Acknowledgements
The authors would like to thank all the patients who participated in this study, but also all technicians in the Centre de Ressources Biologiques and in the Department of Cancer Molecular Genetics at University Hospitals of Strasbourg.
Abbreviations
- TMT
Gene target matched treatments
- PFMG 2025
Plan France Médecine Génomique 2025
- SFCE
Société Française des Cancers de l’Enfant
- CNS
Central nervous system
- UHS
University Hospitals of Strasbourg
- WHO
World Health Organization
- FFPE
Formalin-fixed paraffin embedded
- ddPCR
Digital droplet polymerase chain reaction
- CNV
Copy number variation
- NGS
Next generation sequencing
- IHC
Immunohistochemistry
- FISH
Fluorescent in situ hybridization
- CGH
Comparative genomic hybridization
- RNAseq
RNAsequencing
- SNV
Single nucleotide variant
- MB
Molecular biology results
- MTB
Molecular tumor board
- INCa
French national institute of cancer
- VUS
Variant of uncertain significance
Authors’ contributions
J.S, D.R, E.G, F.L and N.E.W designed the research and J.S, D.R, E.G, M.O.B and N.E.W wrote the manuscript. J.S, D.R, E.G, B.L, N.W, M.K, J.M.P, V.L, A.O, F.V, M.D and N.E.W completed the research. N.E.W., J.S, M.O.B, D.R., E.G and F.L analyzed data. N.E.W, B.L, N.W, S.J, A.S, A.S, G.B and F.V contributed to clinical data and samples. All authors approved the research and the manuscript.
Funding
The financial support was provided by the Foundation of Strasbourg University and Clinical Research Department of University Hospitals of Strasbourg.
Data availability
The data generated in this study are available upon request from the corresponding author.
Declarations
Ethics approval and patient consent to participate
The study was approved by the UHS ethics committee (CE-2022–8661) and was conducted in accordance with the ethical principles of the Declaration of Helsinki. Written and informed consent was provided for all patients prior to data collection.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Smith MA, Altekruse SF, Adamson PC, Reaman GH, Seibel NL. Declining childhood and adolescent cancer mortality. Cancer. 2014;120(16):2497–506. 10.1002/cncr.28748. Epub 2014 May 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poulalhon C, Goujon S, Marquant F, Faure L, Guissou S, Bonaventure A, et al. Factors associated with 5- and 10-year survival among a recent cohort of childhood cancer survivors (France, 2000–2015). Cancer Epidemiol. 2021;73: 101950. 10.1016/j.canep.2021.101950. (Epub 2021 Jun 29). [DOI] [PubMed] [Google Scholar]
- 3.Steliarova-Foucher E, Colombet M, Ries LAG, Moreno F, Dolya A, Bray F, et al. International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol. 2017;18(6):719–31. 10.1016/S1470-2045(17)30186-9. (Epub 2017 Apr 11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–7. 10.1038/nature25480. (Epub 2018 Feb 28PMID: 29489754). [DOI] [PubMed] [Google Scholar]
- 5.Ma X, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, Zhou X, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371–6. 10.1038/nature25795. Epub 2018 Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forrest SJ, Geoerger B, Janeway KA. Precision medicine in pediatric oncology. Curr Opin Pediatr. 2018;30(1):17–24. 10.1097/MOP.0000000000000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartmaier RJ, Albacker LA, Chmielecki J, Bailey M, He J, Goldberg ME, et al. High-throughput genomic profiling of adult solid tumors reveals novel insights into cancer pathogenesis. Cancer Res. 2017;77(1):2464–75. 10.1158/0008-5472.CAN-16-2479. (Epub 2017 Feb 24). [DOI] [PubMed] [Google Scholar]
- 8.Belin L, Kamal M, Mauborgne C, Plancher C, Mulot F, Delord JP, et al. Randomized phase II trial comparing molecularly targeted therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer: cross-over analysis from the SHIVA trial. Ann Oncol. 2017;28(3):590–6. 10.1093/annonc/mdw666. [DOI] [PubMed] [Google Scholar]
- 9.Massard C, Michiels S, Ferté C, Le Deley MC, Lacroix L, Hollebecque A, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7(6):586–95. 10.1158/2159-8290.CD-16-1396. Epub 2017 Apr 1. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research Network. Demicco E. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171(4):950-965.e28. 10.1016/j.cell.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malbari F. Pediatric Neuro-Oncology. Neurol Clin. 2021;39(1):829–45. 10.1007/s11060-020-03416-9. Epub 2020 Feb 5. [DOI] [PubMed] [Google Scholar]
- 12.Packer RJ. Childhood brain tumors: accomplishments and ongoing challenges. J Child Neurol. 2008;23(10):1122–7. 10.1177/0883073808320758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg T, Bandopadhayay P. Molecular genetics of pediatric brain tumours and opportunities for precision medicine - a focus on infant tumours. Curr Opin Neurol. 2022;35(6):772–8. 10.1097/WCO.0000000000001110. (Epub 2022 Oct 4). [DOI] [PubMed] [Google Scholar]
- 14.Rapport F, Smith J, O’Brien TA, Tyrell VJ, Mould EV, Long JC, et al. Development of an implementation and evaluation strategy for the Australian “Zero Childhood Cancer” (Zero) Program: a study protocol. BMJ Open. 2020;10(6):e034522. 10.1136/bmjopen-2019-034522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiemenz MC, Ostrow DG, Busse TM, Buckley J, Maglinte DT, Bootwalla M, et al. OncoKids: A comprehensive next-generation sequencing panel for pediatric malignancies. J Mol Diagn. 2018;20(6):765–76. 10.1016/j.jmoldx.2018.06.009. (Epub 2018 Aug 20). [DOI] [PubMed] [Google Scholar]
- 16.Allen CE, Laetsch TW, Mody R, Irwin MS, Lim MS, Adamson PC, et al. Target and agent prioritization for the Children’s Oncology Group-National Cancer Institute Pediatric MATCH trial. J Natl Cancer Inst. 2017;109(5):djw274. 10.1093/jnci/djw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med. 2020;26(1):1742–53. 10.1038/s41598-023-30395-4. [DOI] [PubMed] [Google Scholar]
- 18.Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalized medicine for high-risk pediatric cancer patients - The INFORM pilot study. Eur J Cancer. 2016;65:91–101. 10.1016/j.ejca.2016.06.009. Epub 2016 Jul 29. [DOI] [PubMed] [Google Scholar]
- 19.Langenberg KPS, Meister MT, Bakhuizen JJ, Boer JM, van Eijkelenburg NKA, Hulleman E, et al. Implementation of pediatric precision oncology into clinical practice: the individualized therapies for children with cancer program “iTHER.” Eur J Cancer. 2022;175:311–25. 10.1016/j.ejca.2022.09.001. Epub 2022 Sep 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berlanga P, Pierron G, Lacroix L, Chicard M, Adam de Beaumais T, Marchais A, et al. The European MAPPYACTS trial: Precision medicine program in pediatric and adolescent patients with recurrent malignancies. Cancer Discov. 2022;12(5):1266–81. 10.1158/2159-8290.CD-21-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau LMS, Khuong-Quang DA, Mayoh C, Wong M, Barahona P, Ajuyah P, et al. Precision-guided treatment in high-risk pediatric cancers. Nat Med. 2024;30(7):1913–22. 10.1038/s41591-024-03044-0. Epub 2024 Jun 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayle A, Belcaid L, Aldea M, Vasseur D, Peyraud F, Nicotra C, et al. Clinical utility of circulating tumor DNA sequencing with a large panel: a National Center for Precision Medicine (PRISM) study. Ann Oncol. 2023;S0923–7534(23):00046–7. 10.1016/j.annonc.2023.01.008. Epub 2023 Jan 25. [DOI] [PubMed] [Google Scholar]
- 23.Dufour C, Foulon S, Geoffray A, Masliah-Planchon J, Figarella-Branger D, Bernier-Chastagner V, et al. Prognostic relevance of clinical and molecular risk factors in children with high-risk medulloblastoma treated in the phase II trial PNET HR+5. Neuro Oncol. 2021;23(7):1163–72. 10.1093/neuonc/noaa301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DE, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. 10.1038/s41467-020-20603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harttrampf AC, Lacroix L, Deloger M, Deschamps F, Puget S, Auger N, et al. Molecular screening for cancer treatment optimization (MOSCATO-01) in pediatric patients: a single-institutional prospective molecular stratification trial. Clin Cancer Res. 2017;23(20):6101–12. 10.1158/1078-0432.CCR-17-0381. Epub 2017 Jul 21. [DOI] [PubMed] [Google Scholar]
- 26.Pages M, Uro-Coste E, Colin C, Meyronet D, Gauchotte G, Maurage CA, et al. The implementation of DNA methylation profiling into a multistep diagnostic process in pediatric Neuropathology: A 2-year real-world experience by the French Neuropathology Network. On Behalf Of The Renoclip-Loc Network. Cancers (Basel). 2021;13(6):1377. 10.3390/cancers13061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galant C, Bouvier C, Larousserie F, Aubert S, Audard V, Brouchet A, et al. Histological diagnosis of bone tumors: Guidelines of the French committee of bone pathologists reference network on bone tumors (RESOS). Bull Cancer. 2018;105(4):368–74. 10.1016/j.bulcan.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 28.Blandin AF, Durand A, Litzler M, Tripp A, Guérin É, Ruhland E, et al. Hypoxic Environment and Paired Hierarchical 3D and 2D Models of Pediatric H3.3-Mutated Gliomas Recreate the Patient Tumor Complexity. Cancers (Basel). 2019;11(12):1875. 10.3390/cancers11121875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schultz E, Pencreach E, Rimelen V, Palama A, Holder E, Gantzer J, Bender L. Acquired L718V/ TP53 co-mutation and discordant molecular pattern between plasmatic and cerebrospinal fluid in a bone and meningeal metastatic L858R+ non-small cell lung cancer: a case report. Ann Transl Med. 2023;11(5):223. 10.21037/atm-22-3861. Epub 2023 Mar 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geyer L, Wolf T, Chenard MP, Cebula H, Schott R, Noel G, et al. p16 immunohistochemical expression as a surrogate assessment of CDKN2A alteration in gliomas leading to prognostic significances. Cancers (Basel). 2023;15(5):1512. 10.3390/cancers15051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Appay R, Fina F, Macagno N, Padovani L, Colin C, Barets D, et al. Duplications of KIAA1549 and BRAF screening by Droplet Digital PCR from formalin-fixed paraffin-embedded DNA is an accurate alternative for KIAA1549-BRAF fusion detection in pilocytic astrocytomas. Mod Pathol. 2018;31(10):1490–501. 10.1038/s41379-018-0050-6. Epub 2018 May 25. [DOI] [PubMed] [Google Scholar]
- 32.Ali SM, Collier KA, Cherian MA, Noonan AM, Sardesai S, VanDeusen J, Wesolowski R, Williams N, Lee CN, Shapiro CL, Macrae ER, Ramaswamy B, Lustberg MB. Prospective decision analysis study of clinical genomic testing in metastatic breast cancer: impact on outcomes and patient perceptions. JCO Precis Oncol. 2019;3:PO.19.00090. 10.1200/PO.19.00090. eCollection 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cahn F, Revon-Riviere G, Min V, Rome A, Filaine P, Pelletier A, Abed S, Gentet JC, Verschuur A, André N. Blood-derived liquid biopsies using foundation one® liquid CDx for children and adolescents with high-risk malignancies: a monocentric experience. Cancers (Basel). 2022;14(11):2774. 10.3390/cancers14112774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karanian M, Pissaloux D, Gomez-Brouchet A, Chevenet C, Le Loarer F, Fernandez C, et al. SRF-FOXO1 and SRF-NCOA1 fusion genes delineate a distinctive subset of well-differentiated rhabdomyosarcoma. Am J Surg Pathol. 2020;44(5):607–16. 10.1097/PAS.0000000000001464. [DOI] [PubMed] [Google Scholar]
- 35.Northcott PA, Shih DJ, Remke M, Cho YJ, Kool M, Hawkins C, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012;123(4):615–26. 10.1007/s00401-011-0899-7. Epub 2011 Nov 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambros IM, Tonini GP, Pötschger U, Gross N, Mosseri V, Beiske K, et al. Age Dependency of the prognostic impact of tumor genomics in localized resectable MYCN-non amplified neuroblastomas. J Clin Oncol. 2020;38(31):3685–97. 10.1200/JCO.18.02132. (Epub 2020 Sep 9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Defossez G, Le Guyader-Peyrou S, Uhry Z, Grosclaude P, Colonna M, Dantony E, et al. Estimations nationales de l’incidence et de la mortalité par cancer en France métropolitaine entre 1990 et 2018. Volume 1 – Tumeurs solides. Saint-Maurice (Fra) : Santé publique France, 2019. 372 p. https://www.ecancer.fr/Expertises-et-publications/Catalogue-des-publications/Complements-Volume-1-Tumeurs-solides-Estimations-nationales-de-l-incidence-et-de-la-mortalite-par-cancer-en-France-metropolitaine-entre-1990-et-2018.
- 38.Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019;11(1):32. 10.1186/s13073-019-0644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.George SL, Izquierdo E, Campbell J, Koutroumanidou E, Proszek P, Jamal S, et al. A tailored molecular profiling program for children with cancer to identify clinically actionable genetic alterations. Eur J Cancer. 2019;121:224–35. 10.1016/j.ejca.2019.07.027. Epub 2019 Sep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barsan V, Paul M, Gorsi H, Malicki D, Elster J, Kuo DJ, Crawford J. Clinical impact of next-generation sequencing in pediatric neuro-oncology patients: a single-institutional experience. Cureus. 2019;11(12):e6281. 10.7759/cureus.6281. [DOI] [PMC free article] [PubMed]
- 41.Gutiérrez-Jimeno M, Alba-Pavón P, Astigarraga I, Imízcoz T, Panizo-Morgado E, García-Obregón S, et al. Clinical value of NGS genomic studies for clinical management of pediatric and young adult bone sarcomas. Cancers (Basel). 2021;13(21):5436. 10.3390/cancers13215436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ludwig JA, Meyers PA, Dirksen U. Ewing’s sarcoma. N Engl J Med. 2021;384(15):1476. 10.1056/NEJMc2102423. [DOI] [PubMed] [Google Scholar]
- 43.Benezech S, Saintigny P, Attignon V, Pissaloux D, Paindavoine S, Faure-Conter C, et al. Tumor molecular profiling: pediatric results of the ProfiLER study. JCO Precis Oncol. 2020;4:785–95. 10.1200/PO.20.00023. [DOI] [PubMed] [Google Scholar]
- 44.Schreck KC, Grossman SA, Pratilas CA. BRAF Mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers (Basel). 2019;11(9): 1262. 10.3390/cancers11091262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eckstein OS, Allen CE, Williams PM, Roy-Chowdhuri S, Patton DR, Coffey B, et al. Phase II study of selumetinib in children and young adults with tumors harboring activating mitogen-activated protein kinase pathway genetic alterations: arm E of the NCI-COG Pediatric MATCH Trial. J Clin Oncol. 2022;40(20):2235–45. 10.1200/JCO.21.02840. Epub 2022 Apr 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiser DA, West-Szymanski DC, Fraint E, Weiner S, Rivas MA, Zhao CWT, et al. Progress toward liquid biopsies in pediatric solid tumors. Cancer Metastasis Rev. 2019;38(4):553–71. 10.1007/s10555-019-09825-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the individualized Cancer Therapy (iCat) study. JAMA Oncol. 2016;2(5):608–15. 10.1001/jamaoncol.2015.5689. [DOI] [PubMed] [Google Scholar]
- 48.Wu CC, Livingston JA. Genomics and the immune landscape of osteosarcoma. Adv Exp Med Biol. 2020;1258:21–36. 10.1007/978-3-030-43085-6_2. [DOI] [PubMed] [Google Scholar]
- 49.Groves A, Cooney TM. Epigenetic programming of pediatric high-grade glioma: pushing beyond proof of concept to clinical benefit. Front Cell Dev Biol. 2022;10:1089898. 10.3389/fcell.2022.1089898. eCollection 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mack SC, Hubert CG, Miller TE, Taylor MD, Rich JN. An epigenetic gateway to brain tumor cell identity. Nat Neurosci. 2016;19(1):10–9. 10.1038/nn.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mack SC, Northcott PA. Genomic analysis of childhood brain tumors: methods for genome-wide discovery and precision medicine become mainstream. J Clin Oncol. 2017;35(21):2346–54. 10.1200/JCO.2017.72.9921. Epub 2017 Jun 22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.