

Abstract
Muscle wasting, a cardinal feature of cancer-associated cachexia (CAC), is a major clinical problem with few therapeutic options. In this Forum article we discuss cellular mechanisms of CAC, focusing on impaired muscle regeneration. We highlight muscle progenitor cell dysfunction and metabolism as two variables contributing to impaired regeneration in CAC.
Negative Consequences of Skeletal Muscle Loss in CAC
CAC is a widespread clinical problem with poorly standardized diagnostic protocols and no effective treatments. Defined as a loss of lean body/skeletal muscle mass with or without a loss in adipose/fat mass, CAC is often diagnosed at cancer diagnosis based on patient-reported weight data [1]. In the most severe cases, CAC progresses to a refractory stage, characterized by a 3 month life expectancy. CAC is a significant comorbidity of cancer, and is directly associated with quality of life, therapeutic response, and median survival. Mechanistically, nutritional changes (appetite, digestion, and anorexia), hyper-catabolism, and failed regenerative processes all contribute to progression of CAC. Despite efforts to improve diagnosis and treatment options, there has been little clinical advancement in treating this complex syndrome.
A major feature of CAC is elevated skeletal muscle catabolism, a state characterized by enhanced protein breakdown via proteasome and autophagic pathways. Activation of these pathways is linked to proinflammatory factors such as IL-6 and TNF-α, which can originate from the tumor as well as from host tissues. Previous work has shown that IL-6/STAT3 signaling leads to a complex multiorgan response, including elevated autophagy in mature muscle and cultured myotubes [2]. STAT3 signaling also plays a key role in myogenesis and muscle regeneration by regulating the proliferation rates of muscle progenitor cells [3]. In addition, cytokine-driven upregulation of E3 ubiquitin ligases, such as MuRF1 and FBXO32 (atrogin 1), has been shown by many groups to cause increased protein degradation and muscle atrophy in cell and rodent models. Although these pathways have been robustly characterized at a molecular level, resulting therapies targeting cytokine-mediated hyper-catabolism have been largely ineffective. Appetite stimulants, aimed to balance the highly catabolic state of CAC patients, had mixed effects: patients saw increases in weight but not in muscle function or survival. In addition to inflammation and proteolysis, recent literature suggests that disrupted muscle metabolism is a driver of CAC. In one study, non-targeted metabolomics profiling of myotubes exposed to CAC-inducing media revealed widespread changes in intracellular metabolism, including increased lipolysis and reactive oxygen species stress response. These metabolic changes were also reflected at a transcriptional level, as evidenced by elevated p38 signaling and increased expression of fatty acid oxidation enzymes. Furthermore, pharmacologic inhibition of fatty acid oxidation, using etomoxir, was able to prevent both myotube atrophy in vitro and weight loss in tumor-bearing mice [4]. Although still at the preclinical stage, this study highlights the importance of metabolic changes in CAC.
Key Contributions of Muscle Stem/Satellite Cells to Muscle Repair and Regeneration
Although therapies aimed at reversing the catabolic state of CAC have largely failed, enhancing muscle repair/regeneration represents an alternative strategy for limiting the progressive loss of muscle mass and function. Adult muscle has an impressive potential to regenerate after injury, and this is driven by resident adult stem cells, termed satellite cells (SCs). Like most adult stem cells, SCs are quiescent in their niche until an activation stimulus (e.g., myofiber damage) promotes entry into the cell cycle, proliferation, differentiation into mature myocytes, and eventually fusion into damaged myofibers. Seminal work by the groups of Kardon and Fan established that SCs are necessary for muscle regeneration postinjury [5,6]. Using genetic ablation of Pax7+ SCs in adult muscle, both groups showed severely limited muscle regeneration in animals lacking SCs, as evidenced by decreased myofiber cross-sectional area, decreased embryonic myosin heavy chain expression, and decreased ability of SC-ablated muscle fibers to engraft after transplant. Despite the clear role of SCs in regeneration post-injury, these and other studies report no change in muscle mass in uninjured, sedentary SC-ablated muscles. A similar model for SC ablation has also shown that SC ablation does not have an effect on the development or progression of age-associated muscle wasting (sarcopenia) [7]. These observations have led some groups to question the importance of SCs in other physiological contexts such as exercise, chronic illness, and cancer. Although work is still ongoing in many of these areas, emerging work suggests that SCs play a broader role in maintaining muscle mass and function outside experimental injury models.
A recent example of the expanding role for SCs in muscle maintenance comes from exercise studies in SC-ablated mice. Although voluntary wheel-running did not cause an accumulation of SCs or myonuclei in control or SC-ablated mice, the SC-ablated mice ran slower and shorter distances than control animals. SC-ablated animals also had lower grip strength and impaired coordination on balance beam and rotarod tests, suggesting that loss of SCs alone was able to alter muscle function [8]. Studies showing the importance of SCs in exercise and injury highlight the complex functions of this cell population, including communication with other mononuclear cell populations, as well as the many ways SCs could be important therapeutic targets in CAC.
Despite the varying necessity of SCs in injury, homeostasis, aging, and exercise, the published literature shows that disrupted myogenesis can contribute to CAC. In mice bearing C26 tumors (a model commonly used to induce CAC in mice), Guttridge and colleagues observed an increase in Pax7 protein in whole muscle and increased numbers of Pax7+ cells per myofiber. Elevated Pax7 indicates activation of SCs in response to tumor-derived stimuli; however, persistent Pax7 is sufficient to block myogenesis and drive muscle wasting. Haploinsufficiency of Pax7, or overexpression of the downstream regulatory factor, MyoD, was able to increase myofiber cross-sectional area and muscle mass in tumor-bearing mice [9]. These results underscore the importance of myogenesis in CAC. In addition, in agreement with previous studies showing an auxiliary role for other cell types in the regeneration process, tumor-derived factors can significantly alter the local muscle immune microenvironment. Normal immune cell dynamics during muscle repair involve early neutrophil infiltration, followed by M1 and then M2 macrophage infiltration. Disrupting this progression likely exacerbates CAC-associated SC defects, thus further impairing myogenesis and accelerating CAC progression [10,11]. Together, these studies highlight SCs as a dynamic cell population that requires more robust investigation in the context of CAC.
Metabolic Regulation of Muscle Satellite Cells
Considering the roles of both disrupted myogenesis and muscle metabolism in CAC, an emerging field of interest is metabolic regulation of SC function. Studies in embryonic stem cells showing a relationship between metabolic status and propensity to differentiate have paved the way for rigorous investigation of metabolic regulation of adult stem cell populations, including several studies on SCs. First, environmental nutrients, such as glucose, are able to modulate the degree of myoblast differentiation in vitro through AMPK signaling and SIRT1 activity. Ultimately, these studies show that SCs have defined pathways for nutrient sensing that directly affect myogenesis [12]. Given metabolic changes in substrate utilization in cachectic patients, it is likely that SCs sense and respond to systemic metabolic changes associated with CAC. Later studies took this a step further by documenting a switch to glycolytic metabolism when SCs activate and proliferate. Interestingly, this study shows that MyoD expression is modulated by metabolism-induced epigenetic changes, establishing a dependence of myogenic regulatory factor (MRF) expression on the metabolic state of SCs [13]. In many cancer patients there are lowered serum glucose levels as a result of elevated tumor-associated glucose consumption rates. Although this phenomenon is not unique to patients with CAC, the effects likely limit the metabolic transitions necessary for myogenesis [14]. Last, Wüst et al. have demonstrated a concerted mechanism for preventing misexpression of myogenic regulatory factors and for shifting metabolism to favor a more oxidative output later in differentiation. These studies identified microRNAs capable of restricting Mef2A expression, thus promoting metabolic maturation during differentiation, further highlighting the important regulatory role of metabolism in stem cell function [15]. Rigorous work will be necessary to determine whether disrupted SC metabolism is the result of tumor-derived stimuli and/or of globally disrupted host metabolism. Regardless, metabolism is emerging as an important regulator of myogenesis in CAC. We posit that tumor-induced metabolic changes disrupt the ability of SCs to aid in repletion of damaged/atrophied muscle in CAC patients. Therefore, we are optimistic about possible therapeutics targeting disrupted metabolism, with the potential that these interventions could be minimally invasive, well-tolerated, and highly effective at minimizing muscle mass loss (Figure 1).
Figure 1. Consequences of Disrupted Metabolism in Cancer-Associated Cachexia (CAC).
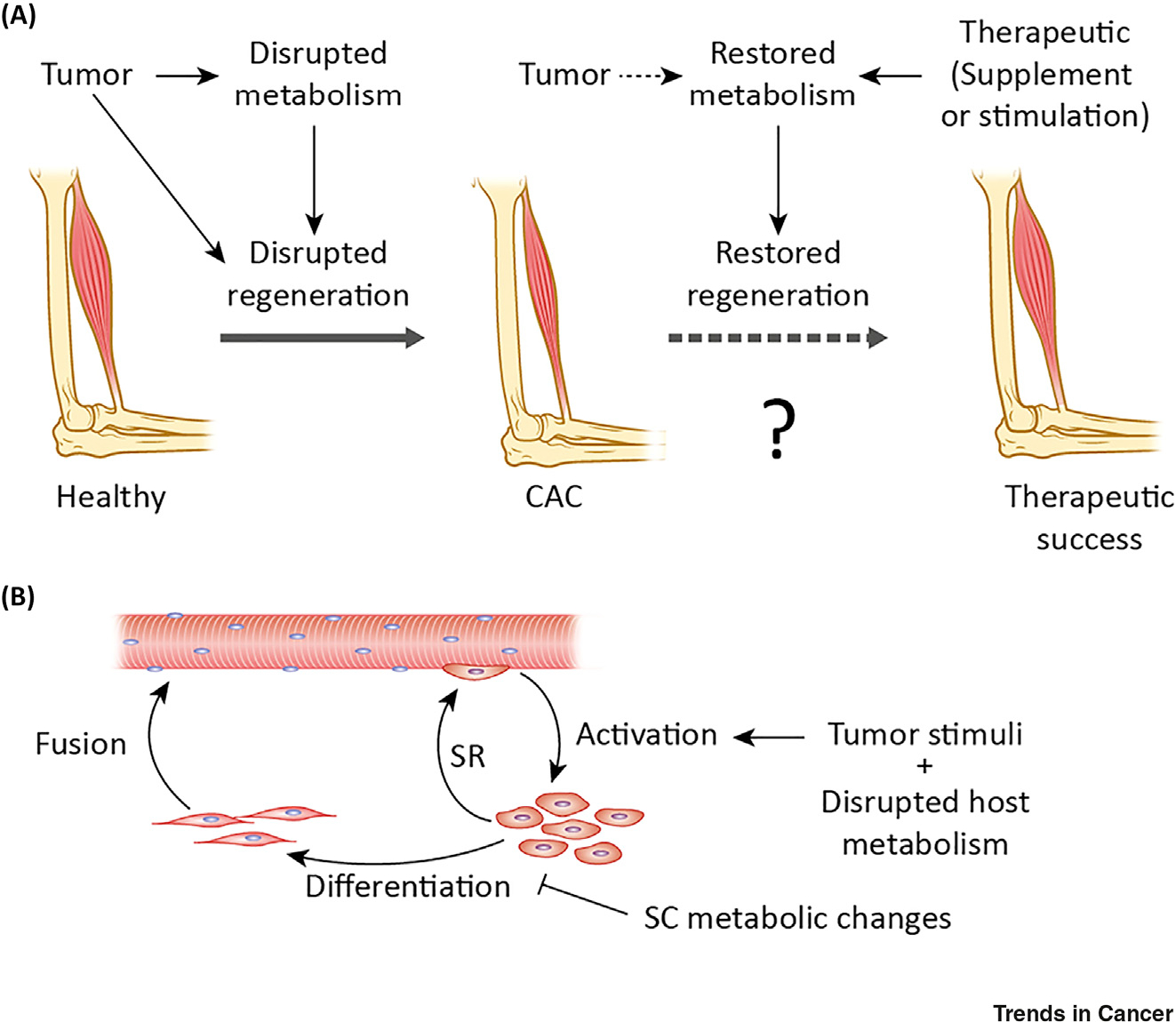
(A) Depiction of muscle loss in CAC and potential recovery using therapies targeting metabolic processes through supplementation or stimulation of specific metabolic pathways. Tumors disrupt both metabolism and muscle regeneration, leading to a net loss of muscle mass. (B) Depiction of myogenesis showing activation of satellite cells (SCs), their self-renewal (SR), differentiation into myocytes, and myocyte fusion. We hypothesize that disrupted SC metabolism prevents normal progression through myogenesis.
As highlighted above, metabolic SC regulation is an incompletely understood but powerful contributor to myogenesis. With continued advances in technical methods, including metabolite profiling and single-cell RNA sequencing, it will be exciting to explore the relationship between metabolic cues and myogenesis, thus identifying ways to enhance tissue repair and alleviate loss of muscle mass and function in CAC.
References
- 1.Fearon K, et al. (2013) Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 10, 90–99 [DOI] [PubMed] [Google Scholar]
- 2.Pettersen K, et al. (2017) Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 7, 2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zimmers TA, et al. (2016) STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 54, 28–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukawa T, et al. (2016) Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 22, 666–671 [DOI] [PubMed] [Google Scholar]
- 5.Lepper C, et al. (2011) An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138, 3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy MM, et al. (2011) Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fry CS, et al. (2015) Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 21, 76–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson JR, et al. (2015) Reduced voluntary running performance is associated with impaired coordination as a result of muscle satellite cell depletion in adult mice. Skelet. Muscle 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He WA, et al. (2013) NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. 123, 4821–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogan KA, et al. (2017) Tumor-derived cytokines impair myogenesis and alter the skeletal muscle immune microenvironment. Cytokine 107, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tidball JG (2017) Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 17, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fulco M, et al. (2008) Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryall JG, et al. (2015) The NAD+-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 16, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yand Q,J., et al. (2018) Serum and urine metabolomics study reveals a distinct diagnostic model for cancer cachexia. J. Cachexia. Sarcopenia Muscle 9, 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wüst S, et al. (2018) Metabolic maturation during muscle stem cell differentiation is achieved by miR-1/133a-mediated inhibition of the Dlk1–Dio3 mega gene cluster. Cell Metab. 27, 1026–1039 [DOI] [PubMed] [Google Scholar]